
Home » Posts tagged 'PHASE 3' (Page 17)
Tag Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AVOSENTAN

AVOSENTAN
N-[6-Methoxy-5-(2-methoxyphenoxy)-2-(4-pyridyl)pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]-amide,
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide,
Endothelin ETA Receptor Antagonists
M.Wt: 479.51
Formula: C23H21N5O5S
Roche (Originator)
CAS No.: 290815-26-8
- RO 67-0565
- SPP 301
- UNII-L94KSX715K
PHASE 3
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=spp301+OR+Avosentan
SPP-301 is an oral, once-daily, second-generation endothelin ETA receptor antagonist which had been in phase III clinical development at Speedel for the treatment of diabetic nephropathy. In December 2006, the company reported that the phase III trial had been stopped based on the recommendation from the trial’s Data Safety Monitoring Board (DSMB) to stop the trial following incidence of a significant imbalance in fluid retention in patients in the study arms. Speedel reported that the compound will be evaluated for potential new clinical development for the treatment of diabetic kidney disease and other indications.
Originally developed by Roche and specifically optimized for improved liver safety, SPP-301 was licensed to Speedel in October 2000. In 2003, Speedel exercised its option to license from Roche all rights to SPP-301, including exclusive worldwide rights for the full development and commercialization of the ETA antagonist. SPP-301 has fast track designation and has undergone a special protocol assessment (SPA) by the FDA. Speedel had been studying the drug for the treatment of hypertension.
AVOSENTAN
290815-26-8 CAS
PATENTS
2. WO 2004078104
3. WO 2005113543
4. WO 2007031501
5. WO 2008077916
Dutzler R, Ernstb B, Hediger MA, Keppler D, Mohr P, Neidhart W, Märki HP.Chimia (Aarau). 2010;64(9):662-6.
………………………
INTRODUCTION
-
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide corresponding to the formula

is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2.
-
Own investigations have shown that there exist two distinct crystalline forms, hereinafter referred to as form A and form B, as well as a number of further solvates, in particular the methanol, ethanol, isopropanol, dichloromethane, acetone, methyl ethyl ketone and tetrahydrofuran solvates.
-
It was further surprisingly found that the thermodynamically stable crystalline form – form B – can be prepared under controlled conditions and that said form B can be prepared with a reliable method in an industrial scale, which is easy to handle and to process in the manufacture and preparation of formulations.

………………..

4,6-Dichloro-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine (described in EP 0 799 209) can be transformed to the intermediate of formula (III)—according to scheme 1—on reaction with an appropriate sulfonamide of formula (II), wherein R1 is as defined in claim 1, in a suited solvent such as DMSO or DMF at room temperature or at elevated temperature and in the presence of a suited base such as potassium carbonate.


EXAMPLE 1
[0064] a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
[0065] C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
[0066] Preparation of the starting material:
[0067] b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide ( CHLORO STARTING MATERIAL) as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
……………………………….
http://www.google.com/patents/US6417360
EXAMPLE 1
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
Preparation of the starting material:
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
…………………….
http://www.google.com/patents/EP0799209B1
SYNTHESIS OF
4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine
A BASIC STARTING MATERIAL FOR AVOSENTAN
- Preparation of the starting material
-
- b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hours 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- c) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
- d) A suspension of 154.6 g of 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCl3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2Cl2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2Cl2. The combined CH2Cl2 extracts are dried with MgSO4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2Cl2remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine, melting point 178-180°C.
…………………………
http://www.google.com/patents/WO2000052007A1
Preparation of the starting material:
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide IE THE 6 CHLORO COMPD
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 1 .66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40°C until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60 °C for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as yellow crystals. Melting point 177-179 °C. ISN mass spectrum, m/e 482.2 (M-l calculated for C22Hi8ClN5O5Sι: 482).

………………………………………………………………………………………….
NEXT

Example 1AVOSENTAN
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl- pyrimidin-4-yl] -amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg SO , concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120 °C to give the desired 5-methyl-pyridine-2-sulfonic acid [6- methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as white crystals. Melting point 225-226 °C. ISN mass spectrum, m/e 478.2 (M-l calculated for
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
…………………………………………….

IS DESCRIBED IN
http://www.google.com/patents/EP2331513A1?cl=en
ALSO

-
Diabetic nephropathy is the principle cause of end stage renal disease in the western world. It is a major cause of morbidity and mortality in Type-I Diabetes, but is an increasing problem in Type-II Diabetes and because the incidence of this is five times that of Type-I Diabetes, it contributes at least 50% of diabetics with end stage renal disease.
-
The initial stage of subtle morphologic changes in the renal glomeruli is followed by microalbuminuria. This is associated with a modestly rising blood pressure and an increased incidence of cardiovascular disease. There follows a continued increase in urinary protein excretion and declining glomerular filtration rate. Diabetic nephropathy has many possible underlying pathophysiological causes including metabolic, glycosylation of proteins, haemodynamics, altered flow/pressure in glomeruli, the development of hypertension and cytokine production; all of these are associated with the development of extracellular matrix and increased vascular permeability leading to glomerular damage and proteinuria.
| WO2005113543A1 * | May 12, 2005 | Dec 1, 2005 | Alexander Bilz | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO2007031501A2 * | Sep 11, 2006 | Mar 22, 2007 | Speedel Pharma Ag | Pyridylsulfonamidyl-pyrimidines for the prevention of blood vessel graft failure |
| WO2008077916A1 * | Dec 21, 2007 | Jul 3, 2008 | Ovidiu Baltatu | Pharmaceutical composition using aliskiren and avosentan |
| EP1454625A1 * | Mar 6, 2003 | Sep 8, 2004 | Speedel Development AG | Pyridylsulfonamidyl-pyrimidines for the treatment of diabetic nephropathies |
| EP1595880A1 * | May 13, 2004 | Nov 16, 2005 | Speedel Pharma AG | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| EP1938812A1 * | Dec 22, 2006 | Jul 2, 2008 | Speedel Pharma AG | Pharmaceutical composition using aliskiren and avosentan |
| US6951856 | Jul 10, 2001 | Oct 4, 2005 | Actelion Pharmaceuticals Ltd. | Arylethene-sulfonamides |
| US7402587 | May 12, 2005 | Jul 22, 2008 | Speedel Pharma Ag | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
| EP0713875A1 * | Nov 13, 1995 | May 29, 1996 | F. Hoffmann-La Roche AG | Sulfonamides |
| EP0897914A1 * | Aug 10, 1998 | Feb 24, 1999 | F. Hoffmann-La Roche Ag | Process for the preparation of 2,5-disubstitued pyridines |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html

APREMILAST, … ORALLY ACTIVE PDE4 INHIBITOR

APREMILAST
PDE4 inhibitor
N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl]-4-acetylaminoisoindolin-l,3-dione,
(S)—N-{2-[1-(3-ethoxy-4-methoxy-phenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
Molecular Formula: C22H24N2O7S Molecular Weight: 460.50016
608141-41-9 CAS NO
Celgene (Originator)
CC-10004 (apremilast) is an oral compound that is being studied in multiple Phase III clinical trials for the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. We successfully completed our early stage studies, demonstrating clinical activity and tolerability and meeting safety endpoints in a placebo controlled proof-of mechanism trial in moderate-to-severe psoriasis and psoriatic arthritis. With the initiation of six multi-center international clinical trials, we are advancing the clinical development of CC-10004.
CC-10004, , Apremilast (USAN), SureCN302992, Apremilast (CC-10004), QCR-202,
- Apremilast
- CC 10004
- CC-10004
- CC10004
- UNII-UP7QBP99PN
- CLINICAL TRIALS….http://clinicaltrials.gov/search/intervention=Apremilast+OR+CC-10004
Apremilast is an orally available small molecule inhibitor of PDE4 being developed byCelgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
 APREMILAST
APREMILAST
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.
“Apremilast is Celgene’s lead oral phosphodiesterase IV inhibitor and anti-TNF alpha agent in phase III clinical studies at Celgene for the oral treatment of moderate to severe plaque-type psoriasis and for the oral treatment of psoriatic arthritis.
Early clinical development is also ongoing for the treatment of acne, Behcet’s disease, cutaneous sarcoidosis, prurigo nodularis, ankylosing spondylitis, atopic or contact dermatitis and rheumatoid arthritis. No recent development has been reported for research for the treatment of skin inflammation associated with cutaneous lupus erythematosus.
In 2011, Celgene discontinued development of the compound for the management of vision-threatening uveitis refractory to other modes of systemic immunosuppression due to lack of efficacy.
Celgene had been evaluating the potential of the drug for the treatment of asthma; however, no recent development has been reported for this research. The drug candidate is also in phase II clinical development at the William Beaumont Hospital Research Institute for the treatment of chronic prostatitis or chronic pelvic pain syndrome and for the treatment of vulvodynia (vulvar pain).
In 2013, orphan drug designations were assigned to the product in the U.S. and the E.U. for the treatment of Behcet’s disease.
Celgene Corp has been boosted by more impressive late-stage data on apremilast, an oral drug for psoriatic arthritis, this time in previously-untreated patients.
The company is presenting data from the 52-week PALACE 4 Phase III study of apremilast tested in PsA patients who have not taken systemic or biologic disease modifying antirheumatic drugs (DMARDs) at the American College of Rheumatology meeting in San Diego. The results from the 527-patient trial show that at week 16, patients on 20mg of the first-in-class oral inhibitor of phosphodiesterase 4 (PDE4) achieved an ACR20 (ie a 20% improvement in the condition) response of 29.2% and 32.3% for 30mg aapremilast, compared with 16.9% for those on placebo.
After 52 weeks, 53.4% on the lower dose and 58.7% on 30mg achieved an ACR20 response. ACR50 and 70 was reached by 31.9% and 18.1% of patients, respectively, for apremilast 30mg. The compound was generally well-tolerated and discontinuation rates for diarrhoea and nausea were less than 2% over 52 weeks.
Commenting on the data, Alvin Wells, of the Rheumatology and Immunotherapy Center in Franklin, Wisconsin, noted that apremilast demonstrated long-term safety and tolerability and significant clinical benefit in treatment-naive patients. He added that “these encouraging results suggest that apremilast may have the potential to be used alone and as a first-line therapy”. Celgene is also presenting various pooled data from the first three trials in the PALACE programme which, among other things, shows that apremilast significantly improves swollen and tender joints.
Treatment for PSA, which affects about 30% of the 125 million people worldwide who have psoriasis, currently involves injectable tumour necrosis factor (TNF) inhibitors, notably AbbVie’s Humira (adalimumab) and Pfizer/Amgen’s Enbrel (etanercept), once patients have not responded to DMARDs (at least in the UK). While the biologics are effective, the side effect profile can be a concern, due to the risk of infection and tuberculosis and many observers believe that apremilast will prove popular with patients and doctors due to the fact that it is oral, not injectable.
Apremilast was filed for PsA with the US Food and Drug Administration in the first quarter and will be submitted on both sides of the Atlantic for psoriasis before year-end. The European filing will also be for PsA.
Apremilast impresses for Behcet’s disease
Celgene has also presented promising Phase II data on apremilast as a treatment for the rare inflammatory disorder Behcet’s disease. 71% of patients achieved complete response at week 12 in clearing oral ulcers
 APREMILAST
APREMILAST
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06.
- Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor.
Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, Blease K, Leisten J, Shirley MA, Tang Y, Babusis DM, Chen R, Stirling D, Muller GW.
J Med Chem. 2009 Mar 26;52(6):1522-4. doi: 10.1021/jm900210d.
- Therapeutics: Silencing psoriasis.Crow JM.Nature. 2012 Dec 20;492(7429):S58-9. doi: 10.1038/492S58a. No abstract available.
- NMR…http://file.selleckchem.com/downloads/nmr/S803401-Apremilast-HNMR-Selleck.pdf
- WO 2003080049
- WO 2013126495
- WO 2013126360
- WO 2003080049
- WO 2006065814
- US2003/187052 A1 …..MP 144 DEG CENT
- US2007/155791
-
J. Med. Chem., 2008, 51 (18), pp 5471–5489DOI: 10.1021/jm800582j
-
J. Med. Chem., 2011, 54 (9), pp 3331–3347DOI: 10.1021/jm200070e

…………………………………………
INTRODUCTION
2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione is a PDE4 inhibitor that is currently under investigation as an anti-inflammatory for the treatment of a variety of conditions, including asthma, chronic obstructive pulmonary disease, psoriasis and other allergic, autoimmune and rheumatologic conditions. S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.

Existing methods for synthesizing (S)-aminosulfone 1 involve resolution of the corresponding racemic aminosulfone by techniques known in the art. Examples include the formation and crystallization of chiral salts, and the use of chiral high performance liquid chromatography. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). In one example, as depicted in Scheme 1 below, (5)-aminosulfone 1 is prepared by resolution of racemic aminosulfone 3 with N-Ac-L-Leu. Racemic aminosulfone 3 is prepared by converting 3-ethoxy-4-methoxybenzonitrile 4 to enamine intermediate 5 followed by enamine reduction and borate hydrolysis. This process has been reported in U.S. Patent
Application Publication No. 2010/0168475.

CH2CI2, NaOH

Scheme 1
The procedure for preparing an enantiomerically enriched or enantiomerically pure aminosulfone, such as compound 1, may be inefficient because it involves the resolution of racemic aminosulfone 3. Thus, a need exists as to asymmetric synthetic processes for the preparation of an enantiomerically enriched or enantiomerically pure aminosulfone, particularly for manufacturing scale production. Direct catalytic asymmetric hydrogenation of a suitable enamine or ketone intermediate is of particular interest because it eliminates the need for either classic resolution or the use of stoichiometric amount of chiral auxiliary, and thus, may be synthetically efficient and economical.
……………………………………….
SYNTHESIS OF KEY INTERMEDIATE
Example 1
Synthesis of 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine

[00232] A slurry of dimethylsulfone (85 g, 903 mmol) in THF (480 ml) was treated with a
1.6M solution of n-butyllithium in hexane (505 ml, 808 mmol) at 0 – 5 °C. The resulting mixture was agitated for 1 hour then a solution of 3-ethoxy-4-methoxybenzonitrile (80 g, 451 mmol) in THF (240 ml) was added at 0 – 5 °C. The mixture was agitated at 0 – 5 °C for 0.5 hour, warmed to 25 – 30 °C over 0.5 hour and then agitated for 1 hour. Water (1.4 L) was added at 25 – 30 °C and the reaction mass was agitated overnight at room temperature (20 – 30 °C). The solid was filtered and subsequently washed with a 2: 1 mixture of water :THF (200 ml), water (200 ml) and heptane (2 x 200 ml). The solid was dried under reduced pressure at 40 – 45 °C to provide the product as a white solid (102 g, 83% yield); 1H NMR (DMSO-d6) δ 1.34 (t, J=7.0 Hz, 3H), 2.99 (s, 3H), 3.80 (s, 3H), 4.08 (q, J=7.0 Hz, 2H), 5.03 (s, 1H), 6.82 (s, 2H), 7.01 (d, J=8.5 Hz, 1H), 7.09 – 7.22 (m, 2H).
Example 2
Synthesis of (R)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine

[00233] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (36 mg, 0.074 mmol) and (i?)-l-[(5)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (40 mg, 0.074 mmol) in 25 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. The mixture was evaporated and the residue was purified by chromatography on a CI 8 reverse phase column using a water-acetonitrile gradient. The appropriate fractions were pooled and evaporated to -150 mL. To this solution was added brine (20 mL), and the resulting solution was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and evaporated to provide the product as a white crystalline solid (1.4 g, 70% yield); achiral HPLC (Hypersil BDS C8, 5.0 μπι, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 9.11 (99.6%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 7.32 (97.5%), 8.26 (2.47%); 1H NMR (DMSO-de) δ 1.32 (t, J= 7.0 Hz, 3H), 2.08 (s, 2H), 2.96 (s, 3H), 3.23 (dd, J= 3.6, 14.4 Hz, 1H), 3.41 (dd, J= 9.4, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 7.0 Hz, 2H), 4.26 (dd, J= 3.7, 9.3 Hz, 1H), 6.89 (s, 2H), 7.02 (s, 1H); 13C NMR (DMSO-d6) δ 14.77, 41.98, 50.89, 55.54, 62.03, 63.68, 111.48, 111.77, 118.36, 137.30, 147.93, 148.09. Example 3
Synthesis of (6 -l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine N-Ac-L-Leu salt

[00234] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (17 mg, 0.037 mmol) and (5)-l-[(i?)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (20 mg, 0.037 mmol) in 10 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. Ecosorb C-941 (200 mg) was added and the mixture was stirred at ambient temperature for 3 h. The mixture was filtered through Celite, and the filter was washed with additional trifluoroethanol (2 mL). Then, the mixture was heated to 55 °C, and a solution of N- acetyl-L-leucine (1.3 g, 7.5 mmol) was added dropwise over the course of 1 h. Stirring proceeded at the same temperature for 1 h following completion of the addition, and then the mixture was cooled to 22 °C over 2 h and stirred at this temperature for 16 h. The crystalline product was filtered, rinsed with methanol (2 x 5 mL), and dried under vacuum at 45 °C to provide the product as a white solid (2.6 g, 80% yield); achiral HPLC (Hypersil BDS Cg, 5.0 μιη, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 8.57 (99.8%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 8.35 (99.6%); 1H NMR (DMSO-<¾) δ 0.84 (d, 3H), 0.89 (d, J= 6.6 Hz, 3H), 1.33 (t, J= 7.0 Hz, 3H), 1.41 – 1.52 (m, 2H), 1.62 (dt, J= 6.7, 13.5 Hz, 1H), 1.83 (s, 3H), 2.94 (s, 3H), 3.28 (dd, J= 4.0, 14.4 Hz, 1H), 3.44 (dd, J= 9.1, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 6.9 Hz, 2H), 4.18 (q, J= 7.7 Hz, 1H), 4.29 (dd, J= 4.0, 9.1 Hz, 1H), 5.46 (br, 3H), 6.90 (s, 2H), 7.04 (s, 1H), 8.04 (d, J= 7.9 Hz, 1H); Anal. (C20H34N2O7S) C, H, N. Calcd C, 53.79; H, 7.67; N 6.27. Found C, 53.78; H, 7.57; N 6.18.
SUBSEQUENT CONVERSION
S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.
……………………………………
 APREMILAST
APREMILAST
GENERAL SYNTHESIS AND SYNTHESIS OF APREMILAST




(apremilast)
[0145] Preparation of 3-Ethoxy-4-methoxybenzonitrile (Compound 2). 3-Ethoxy-
4-methoxybenzaldehyde (Compound 1, 10.0 gm, 54.9 mmol, Aldrich) and hydroxylamine hydrochloride (4.67 gm, 65.9 mmol, Aldrich) were charged to a 250 mL three-necked flask at room temperature, followed by the addition of anhydrous acetonitrile (50 mL). The reaction mixture was stirred at room temperature for thirty minutes and then heated to reflux (oil bath at 85 °C). After two hours of reflux, the reaction mixture was cooled to room temperature, and added 50 mL of deionized water. The mixture was concentrated under reduced pressure to remove acetonitrile and then transferred to a separatory funnel with an additional 80 mL of deionized water and 80 mL dichloromethane. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed successively with water (80 mL) and saturated sodium chloride (80 mL). The organic layer was dried over anhydrous sodium sulfate (approximately 20 gm). The organic layer was filtered and concentrated under reduced pressure to give a yellow oil. Purification by silica gel chromatography (0 to 1 % MeOH/DCM ) afforded 3-Ethoxy-4-methoxybenzonitrile
(Compound 2) as a white solid (7.69 gm, 79 % yield). MS (ESI positive ion) m/z 178.1 (M + 1). HPLC indicated >99% purity by peak area. 1H-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 3.83 (s, 3H), 4.05 (q, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.40 (dd, J = 2.0 Hz, 1H).
[0146] Preparation of l-(3-Ethoxy-4-methoxyphenyi)-2-
(niethylsulfonyl)ethanamine (Compound 3). Dimethyl sulfone (2.60 gm, 27.1 mmol, Aldrich) and tetrahydrofuran (10 mL, Aldrich) were charged to a 250 mL three-necked flask at room temperature. The mixture was cooled to 0 – 5 °C, and the solution gradually turned white. n-Butyllithium (10.8 mL, 27.1 mmol, 2.5 M solution in hexanes, Aldrich) was added to the flask at a rate such that the reaction mixture was maintained at 5 – 10 °C. The mixture was stirred at 0 – 5 °C for one hour, turning light-yellow. 3-Ethoxy-4-methoxybenzonitrile (Compound 2, 4.01 gm, 22.5 mmol) in tetrahydrofuran (8 mL) was then charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for another 15 minutes. After warming to room temperature, the reaction mixture was stirred for another 1.5 hours and then transferred to a second 250 mL three-necked flask containing a suspension of sodium borohydride (1.13 gm, 29.3 mmol, Aldrich) in
tetrahydrofuran (1 1 mL), maintained at – 5 – 0 °C for 30 minutes. Trifluoroacetic acid (“TFA,” 5.26 mL, 68.3 mmol, Aldrich) was charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for 40 minutes and an additional 17 hours at room temperature. The reaction mixture was then charged with 2.7 mL of deionized water over five minutes at room temperature. The mxiture was stirred at room temperature for 15 hours. Aqueous NaOH (10 N, 4.9 mL) was charged to the flask over 15 minutes at 45 °C. The mixture was stirred at 45 °C for two hours, at 60 °C for 1.5 hours, and at room temperature overnight. After approximately 17 hours at room temperature the mixture was cooled to 0 °C for thirty minutes and then concentrated under reduced pressure. The residual material was charged with deionized water (3 mL) and absolute ethanol (3 mL) and stirred at 0 – 5 °C for 2 hours. The mixture was filtered under vacuum, and the filtered solid was washed with cold absolute ethanol (3 x 5 mL), followed by deionized water until the pH of the wash was about 8. The solid was air dried overnight, and then in a vacuum oven at 60 °C for 17 hours to afford Compound 3 as a white solid (4.75 gm, 77 %). MS (ESI positive ion) m/z 274.1 (M + 1). Ή-NMR (500 MHz, DMSO-c¾): δ ppm 1.32 (t, J = 7.0 Hz, 3H), 2.08 (bs, 2H), 2.95 (s, 3H), 3.23 (dd, J = 4.0 Hz, 1H), 3.40 (dd, J = 9.5 Hz, 1H), 3.72 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 4.25 (dd, J = 3.5 Hz, 1H), 6.88 (s, 2H), 7.02 (s, 1H).
[0147] Preparation of 4-Nitroisobenzofuran-l,3-dione (Compound 5). Into a 250 mL round bottom flask, fitted with a reflux condenser, was placed 3-nitrophthalic acid (21.0 gm, 99 mmol, Aldrich) and acetic anhydride (18.8 mL, 199 mmol, Aldrich). The solid mixture was heated to 85 °C, under nitrogen, with gradual melting of the solids. The yellow mixture was heated at 85 °C for 15 minutes, and there was noticeable thickening of the mixture. After 15 minutes at 85 °C, the hot mixture was poured into a weighing dish, and allowed to cool. The yellow solid was grinded to a powder and then placed on a cintered funnel, under vacuum. The solid was washed with diethyl ether (3 x 15 mL), under vacuum and allowed to air dry overnight, to afford 4-nitroisobenzofuran-l ,3-dione, Compound 5, as a light-yellow solid (15.8 gm, 82 %). MS (ESI positive ion) m/z 194.0 (M + 1). TLC: Rf = 0.37 (10% MeOH/DCM with 2 drops Acetic acid) Ή-NMR (500 MHz, DMSO-i¾: δ ppm 8.21 (dd, J = 7.5 Hz, 1H), 8.39 (dd, J = 7.5 Hz, 1H), 8.50 (dd, J = 7.5 Hz, 1 H).
[0148] Preparation of 2-(l-(3-Ethoxy-4-methoxyphenyI)-2-
(methylsulfonyl)ethyl)-4-nitroisoindoline-l,3-dione (Compound 6). Into a 2 – 5 mL microwave vial was added 4-nitroisobenzofuran-l ,3-dione (Compound 5, 0.35 gm, 1.82 mmol), the amino-sulfone intermediate (Compound 3, 0.50 gm, 1.82 mmol) and 4.0 mL of glacial acetic acid. The mixture was placed in a microwave at 125 °C for 30 minutes. After 30 minutes the acetic acid was removed under reduced pressure. The yellow oil was taken up in ethyl acetate and applied to a 10 gm snap Biotage samplet. Purification by silica gel chromatography (0 to 20 % Ethyl Acetate/Hexanes) afforded Compound 6 as a light-yellow solid (0.67 gm, 82 %). MS (ESI positive ion) m/z 449.0 (M + 1). TLC: Rf = 0.19
(EtOAc:Hexanes, 1 : 1). HPLC indicated 99% purity by peak area. Ή-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 2.99 (s, 3H), 3.73 (s, 3H), 4.02 (m, 2H), 4.21 (dd, J = 5.0 Hz, 1H), 4.29 (dd, J = 10.0 Hz, 1H), 5.81 (dd, J = 5.0 Hz, 1H), 6.93 (d, J – 8.5 Hz, 1H), 7.00 (dd, J = 2.0 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 8.07 (t, J = 15.5 Hz, 1H), 8.19 (dd, J = 8.5 Hz, 1H), 8.30 (dd, J = 9.0 Hz, 1H).
[0149] Preparation of 4-Amino-2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsulfonyl)ethyl)isoindoline-l,3-dione (Compound 7). Compound 6 (0.54 gm, 1.20 mmol) was taken up in ethyl acetate / acetone (1 : 1 , 24 mL) and flowed through the H-cube™ hydrogen reactor using a 10 % Pd/C CatCart™ catalyst cartridge system (ThalesNano, Budapest Hungary). After eluting, the yellow solvent was concentrated under reduced pressure to give Compound 7 as a yellow foam solid (0.48 gm, 95 %). MS (ESI positive ion) m/z 419.1 (M + 1). 1H-NMR (500 MHz, DMSO-<¾): δ ppm 1.31 (t, J = 7.0 Hz, 3H), 2.99 (s, 3H), 3.72 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 4.34 (m, 1H), 5.71 (dd, J = 5.5 Hz, 1H), 6.52 (bs, 2H), 6.92-6.98 (m, 3H), 7.06 (bs, 1 H), 7.42 (dd, J = 7.0 Hz, 1H).
[0150] Preparation of N-(2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsuIfonyl)ethyl)-l,3-dioxoisoindolin-4-yl)acetamide (Apremilast, Compound 8).
Into a 2-5 mL microwave vial was placed Compound 7 (0.18 gm, 0.43 mmol), acetic anhydride (0.052 mL, 0.53 mmol) and acetic acid (4 mL). The microwave vial was placed into a Biotage microwave and heated to 125 °C for 30 minutes. The solvents were removed under reduced pressure and the residue was purified by silica gel chromatography (0 to 5% MeOH/DCM) to afford apremilast (Compound 8) as a yellow oil (0.14 gm, 71%). HPLC indicated 94.6% purity by peak area.
1H-NMR (500 MHz, DMSO-c 6): δ ppm 1.31 (t, 3H), 2.18 (s, 3H), 3.01 (s, 3H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 4,14 (dd, J = 4.0 Hz, 1H), 4.33 (m, 1H), 5.76 (dd, J = 3.0 Hz, 1H), 6.95 (m, 2H), 7.06 (d, J = 1.5 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 9.72 (bs, 1H).
……………………..
SYNTHESIS
5. EXAMPLES
Certain embodiments provided herein are illustrated by the following non-limiting examples.
5.1 PREPARATION OF (+)-2-[l-(3-ETHOXY-4-METHOXYPHENYL)-2- METHANESULFONYLETHYLJ-4- ACETYL AMINOISOINDOLIN-1,3- DIONE (APREMILAST)

5.1.1 Preparation of 3-aminopthalic acid
10% Pd/C (2.5 g), 3-nitrophthalic acid (75.0 g, 355 mmol) and ethanol (1.5 L) were charged to a 2.5 L Parr hydrogenator under a nitrogen atmosphere. Hydrogen was charged to the reaction vessel for up to 55 psi. The mixture was shaken for 13 hours, maintaining hydrogen pressure between 50 and 55 psi. Hydrogen was released and the mixture was purged with nitrogen 3 times. The suspension was filtered through a celite bed and rinsed with methanol. The filtrate was concentrated in vacuo. The resulting solid was reslurried in ether and isolated by vacuum filtration. The solid was dried in vacua to a constant weight, affording 54 g (84%> yield) of 3-aminopthalic acid as a yellow product. 1H-NMR (DMSO-d6) δ: 3.17 (s, 2H), 6.67 (d, 1H), 6.82 (d, 1H), 7.17 (t, 1H), 8-10 (brs, 2H). 13C-NMR(DMSO-d6) δ: 112.00, 115.32, 118.20, 131.28, 135.86, 148.82, 169.15, 170.09.
5.1.2 Preparation of 3-acetamidopthalic anhydride
A I L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 3-aminophthalic acid (108 g, 596 mmol) and acetic anhydride (550 mL). The reaction mixture was heated to reflux for 3 hours and cooled to ambient temperature and further to 0-5. degree. C. for another 1 hour. The crystalline solid was collected by vacuum filtration and washed with ether. The solid product was dried in vacua at ambient temperature to a constant weight, giving 75 g (61% yield) of 3-acetamidopthalic anhydride as a white product. 1H-NMR (CDCI3) δ: 2.21 (s, 3H), 7.76 (d, 1H), 7.94 (t, 1H), 8.42 (d, 1H), 9.84 (s, 1H).
5.1.3 Resolution of 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)- ethyl-2-amine
A 3 L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine (137.0 g, 500 mmol), N-acetyl-L-leucine (52 g, 300 mmol), and methanol (1.0 L). The stirred slurry was heated to reflux for 1 hour. The stirred mixture was allowed to cool to ambient temperature and stirring was continued for another 3 hours at ambient temperature. The slurry was filtered and washed with methanol (250 mL). The solid was air-dried and then dried in vacuo at ambient temperature to a constant weight, giving 109.5 g (98% yield) of the crude product (85.8% ee). The crude solid (55.0 g) and methanol (440 mL) were brought to reflux for 1 hour, cooled to room temperature and stirred for an additional 3 hours at ambient temperature. The slurry was filtered and the filter cake was washed with methanol (200 mL). The solid was air-dried and then dried in vacuo at 30°C. to a constant weight, yielding 49.6 g (90%> recovery) of (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine-N-acety 1-L-leucine salt (98.4% ee). Chiral HPLC (1/99 EtOH/20 mM KH2P04 @pH 7.0, Ultron Chiral ES-OVS from Agilent Technologies, 150 mm.times.4.6 mm, 0.5 mL/min., @240 nm): 18.4 min (S-isomer, 99.2%), 25.5 min (R-isomer, 0.8%)
5.1.4 Preparation of (+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl] -4-acetylaminoisoindolin- 1 ,3-dione
A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer,
thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), 3-acetamidophthalic anhydride (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50°C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x
2), saturated aqeous NaHC03 (250 mL.times.2), brine (250 mL.times.2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL.times.2). The product was dried in vacuo at 60°C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 APREMILAST with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 @pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., @240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%).
1H-NMR (CDC13) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H).
13C-NMR(DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………………………………..
NMR
1H-NMR (CDCl3) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………….

aReagents and conditions: (a) LiN(SiMe3)2, then Me2SO2/n-BuLi/BF3Et2O, −78 °C; (b) N-Ac-l-leucine, MeOH; (c) HOAc, reflux.
……………………
SARCOIDOSIS
Sarcoidosis is a disease of unknown cause. Sarcoidosis is characterized by the presence of granulomas in one or more organ systems. The most common sites of involvement are the lungs and the lymph nodes in the mediastinum and hilar regions. However, sarcoidosis is a systemic disease and a variety of organ systems or tissues may be the source of primary or concomitant clinical manifestations and morbidity. The clinical course of sarcoidosis is extremely variable, and ranges from a mild or even asymptomatic disease with spontaneous resolution to a chronic progressive disease leading to organ system failure and, in 1-5% of cases, death. See Cecil
Textbook of Medicine, 21st ed. (Goldman, L., Bennett, J. C. eds), W. B. Saunders Company, Philadelphia, 2000, p. 433-436.
While the cause of sarcoidosis is unknown, a substantial body of information suggests that immune mechanisms are important in disease pathogenesis. For example, sarcoidosis is
characterized by enhanced lymphocyte and macrophage activity. See Thomas, P.D. and
Hunninghake, G.W., Am. Rev. Respir. Dis., 1987, 135: 747-760. As sarcoidosis progresses, skin rashes, erythema nodosum and granulomas may form. Granulomas or fibrosis caused by sarcoidosis can occur throughout the body, and may affect the function of vital organs such as the lungs, heart, nervous system, liver or kidneys. In these cases, the sarcoidosis can be fatal. See
http://www.nlm.nih.gov/medlineplus/sarcoidosis.html (accessed November 12, 2009).
Moreover, a variety of exogenous agents, both infectious and non-infectious, have been hypothesized as a possible cause of sarcoidosis. See Vokurka et ah, Am. J. Respir. Crit. Care Med., 1997, 156: 1000-1003; Popper et al, Hum. Pathol, 1997, 28: 796-800; Almenoff et al, Thorax, 1996, 51 : 530-533; Baughman et al., Lancet, 2003, 361 : 1111-1118. These agents include mycobaceria, fungi, spirochetes, and the agent associated with Whipple’s disease. Id.
Sarcoidosis may be acute or chronic. Specific types of sarcoidosis include, but are not limited to, cardiac sarcoidosis, cutaneous sarcoidosis, hepatic sarcoidosis, oral sarcoidosis, pulmonary sarcoidosis, neurosarcoidosis, sinonasal sarcoidosis, Lofgren’s syndrome, lupus pernio, uveitis or chronic cutaneous sarcoidosis.
As the lung is constantly confronted with airborne substances, including pathogens, many researchers have directed their attention to identification of potential causative transmissible agents and their contribution to the mechanism of pulmonary granuloma formation associated with sarcoidosis. See Conron, M. and Du Bois, R.M., Clin. Exp. Allergy, 2001, 31 : 543-554; Agostini et al, Curr. Opin. Pulm. Med. , 2002, 8: 435-440.
Corticosteroid drugs are the primary treatment for the inflammation and granuloma formation associated with sarcoidosis. Rizatto et al. , Respiratory Medicine, 1997, 91 : 449-460. Prednisone is most often prescribed drug for the treatment of sarcoidosis. Additional drugs used to treat sarcoidosis include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, minocycline, doxycycline and chloroquin. TNF-a blockers such as thalidomide and infliximab have been reported to be effective in treating patients with sarcoidosis. Baughman et al, Chest, 2002, 122: 227-232; Doty et al, Chest, 2005, 127: 1064-1071. Antibiotics have also been studied for the treatment of sarcoidosis, such as penicillin antibiotics, cephalosporin antibiotics, macrolide antibiotics, lincomycin antibiotics, and tetracycline antibiotics. Specific examples include minocycline hydrochloride, clindamycin, ampicillin, or clarithromycin. See, e.g., U.S. Patent Publication No. 2007/0111956.
There currently lacks a Food and Drug Administration-approved therapeutic agent for the treatment of sarcoidosis, and many patients are unable to tolerate the side effects of the standard corticosteroid therapy. See Doty et al, Chest, 2005, 127: 1064-1071. Furthermore, many cases of sarcoidosis are refractory to standard therapy. Id. Therefore, a demand exists for new methods and compositions that can be used to treat patients with sarcoidosis.
……………..
PATENTS
|
8-15-2012
|
PROCESSES FOR THE PREPARATION OF AMINOSULFONE COMPOUNDS
|
|
|
11-4-2011
|
HETEROCYCLIC COMPOUNDS AS PHOSPHODIESTERASE INHIBITORS
|
|
|
5-27-2011
|
Nanosuspension of a Poorly Soluble Drug via Microfluidization Process
|
|
|
5-28-2010
|
METHODS AND COMPOSITIONS USING PDE4 INHIBITORS FOR THE TREATMENT AND MANAGEMENT OF CANCERS
|

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Palbociclib
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
recent studies have identified a number of selective CDK4 inhibitors that, as discussed above, may prove useful in treating cancer—either as anti-cancer agents or as chemoprotective agents—and in treating cardiovascular disorders, such as restenosis and atherosclerosis, diseases caused by infectious agents, and autoimmune disorders, including rheumatoid arthritis. For a disclosure of these selective CDK4 inhibitors, see commonly assigned International Patent Application PCT/IB03/00059, filed Jan. 10, 2003 (the ‘059 application), which is herein incorporated by reference in its entirety for all purposes.
The ‘059 application discloses a particularly potent and selective CDK4 inhibitor, 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one:

In standard enzyme assays the compound of Formula 1 exhibits IC50 concentrations for CDK4 and CDK2 inhibition (at 25° C.) of 0.011 μM and >5 μM, respectively. For a discussion of standard CDK4 and CDK2 assays for IC50 determinations, see D. W. Fry et al., J. Biol. Chem. (2001) 16617-16623.
Though the compound of Formula 1 is a potent and selective CDK4 inhibitor, its use in pharmaceutical products presents challenges. For example, the free base has poor water solubility (9 μg/mL) and exhibits low bioavailability in animal studies. A di-HCl salt of the compound of Formula 1 appears to exhibit adequate water solubility. However, moisture uptake studies reveal that, even at low relative humidity (10% RH), the di-HCl salt absorbs water in an amount greater than about 2% of its mass, making it unsuitable for use in a solid drug product. A mono-HCl salt of the compound of Formula 1 is marginally hygroscopic, absorbing more than 2% of its mass at a relative humidity above 80%. However, the process for preparing the mono-HCl salt yields partially crystalline drug substance, indicating potential problems with process scale-up. Other salt forms of the compound of Formula 1 are thus needed.
Pfizer’s breast cancer drug Palbociclib (PD-0332991), a first in the class oral inhibitor of cyclin-dependent kinases (CDK) 4 and 6, is widely seen by investors as Pfizer’s most valuable compound in late-stage development. The FDA awarded Palbociclib “breakthrough therapy designation” in April 2013 based on the preliminary phase 2 data showing palbociclib, combined with Novartis’ drug,Femara (Letrozole), stopped breast tumors progression for more than two years as compared with 7.5 months with letrozole alone. The phase 3 trial started in February 2013 and estimated final completion date is March 2016. Leerink Swann analyst Seamus Fernandez forecasts palbociclib could become a $5 billion drug, with potential for $3 billion in first-line metastatic breast cancer alone.
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases.
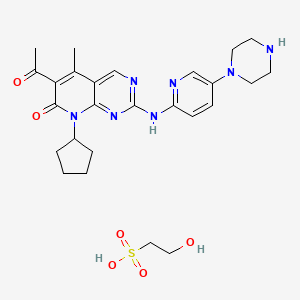
6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (also referred to as “Compound 1”),

as well as its intermediates. Compound 1 is described in U.S. Pat. No. 6,936,612, the disclosure of which is hereby incorporated in its entirety. This compound is a protein kinase inhibitor and represents a synthetic, small molecule inhibitor capable of modulating cell cycle control.
A method of preparing Compound 1 is disclosed as Example 36 of U.S. patent application Ser. No. 6,936,612. Methods of preparing the isethionate salt forms of Compound 1 are disclosed in Examples 1-13 of WO 2005/005426. These methods are for synthesis of small quantities of the salt forms of Compound 1 and are not designed for commercial scale-up. Therefore, a preparation of the salt forms for CDK inhibitor 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride which is cost-efficient, scaleable and productive is highly desirable.


USAN (zz-153)
PALBOCICLIB ISETHIONATE
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
MOLECULAR FORMULA C24H29N7O2 . C2H6O4S
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
- PD 0332991-0054
- PF-00080665-73
- UNII-W1NYL2IRDR

SYNTHESIS

:WO2008032157
……………………………….
http://www.google.com/patents/US7781583



COMPARATIVE EXAMPLE 1A Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
COMPARATIVE EXAMPLE 1B Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1A), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
COMPARATIVE EXAMPLE 1C Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in 2005-0059670A1) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 2A Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg (6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6 L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-70° C. and held for 30 hours after which some solids precipitated. Water was added and the reaction cooled to 25° C. over 2 hrs. The resulting slurry was filtered, washed and dried at 45° C. to give 1.2 kg (79% crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (2)
60.0 g of 20% Pd(OH)2/C, 1213.1 g (3.9 moles) of intermediate 2a, and isopropanol were charged and stirred in a Parr reactor, then purged under gas, followed by removal of the catalyst under pressure. The filtrates were concentrated in vacuo at ˜20° C. leaving 917 g of dry brown powder (crude yield ˜84%).
EXAMPLE 3 Preparation of 2-Chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one

EXAMPLE 3A Preparation of 5-bromo-2-chloro-4-cyclopentyl-aminopyrimidine
To 1 g (0.004 mol) of 5-bromo-2,4-dichloropyrimidine in ethanol was added 1.5 kg (0.018 mol) cyclopentylamine under nitrogen. The mixture was stirred at 25° C. for 2 hrs. Water was added to precipitate the product, and the solid was recrystallized using hexane 4:1 to give a white crystalline product (3A).
EXAMPLE 3 Preparation of 2-Chloro-8-Cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one
41.5 g (0.15 mol) of 5-bromo-2-chloro-4-cyclopentylaminopyrimidine 3a and 32.3 g (0.375 mol) of crotonic acid were mixed in 100 L of THF and 105 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred, evacuated and refilled with nitrogen three times, after which 860 mg (0.0022 mol) palladium dichloride dibenzonitrile complex and 685 mg (0.0022 mol) tri-ortho-tolylphosphine were added and the resulting slurry degassed an additional three times. The mixture was then heated and stirred at 70° C. for 16 hrs, after which 35 ml acetic anhydride was added and the mixture stirred for an additional 1.5 hrs. The mixture was cooled and diluted with 100 ml MTBE and then extracted with 1NHCl, then aqueous sodium bicarbonate and brine. The organic phase was dried over magnesium sulfate, filtered, concentrated in vacuo, and recrystallized from IPA to yield 31.2 g (68%) of crude product (3).
EXAMPLE 4 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 4A Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidine-7-one
10 g (0.04 mol) of intermediate 3 and 13 g (0.16 mol) of sodium acetate were mixed with 50 ml of glacial acetic acid and 12 g (0.08 mol) bromine under nitrogen. The solution was heated to 50° C. and stirred for 35 hrs, then cooled to room temperature. Sodium bisulfite solids were added until the bromine color disappeared, then quenched, filtered and washed to provide a solid which was subsequently dissolved in 500 ml hot IPA, filtered hot, and cooled. The resulting crystals were further filtered, and dried in vacuo at 65° C. to yield 8 g (61%) of crude product (4A).
EXAMPLE 4 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
3.78 g (2.10 equiv; 13.6 mmoles) of intermediate 1, 25 ml toluene and lithium bis(trimethylsilyl)amide in 1 M THF (13.6 mmoles; 13.6 mL; 12.1 g) were mixed for 10 min under nitrogen to form a dark solution. In a separate beaker the intermediate 4a (1.00 equiv, 6.47 mmoles; 2.50 g) was slurried in toluene then added to the mixture containing 1 and stirred for 30 min, after which the combined mixture was quenched with 25 ml 1 M sodium bicarbonate and then filtered. Alternatively, the combined mixture can be quenched with ammonium chloride. The filter cake was washed with toluene, then acetone, then water and dried at 60° C. to give 3.5 g (92%) of a grey-yellow solid 4.
EXAMPLE 5 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cycloentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester

768 g (1.3 mol) of intermediate 4, was mixed with 395 g (3.9 mol) of butyl vinyl ether, 4.7 L of n-butanol, and 275 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred and placed under ca. 50 tore vacuum and then refilled with nitrogen; this was repeated 2 more times. To this degassed solution was added 22 g (0.03 mol) Bis-(diphenylphosphinoferrocene)palladium dichloride dichloromethane complex and the resulting slurry was degassed an additional three times as described above. The mixture was then heated and stirred at 95° C. for 20 hrs. The resulting thin red slurry was diluted with 4 L branched octane’s and cooled to about 5° C. after which 1 L saturated aq. potassium carbonate was added and the mixture was filtered and rinsed with 500 ml branched octanes. After drying for 16 hrs at 45° C., 664 g (83%) of gray-solid product (5) was obtained. In addition, column chromatography can be used to further purify the crude product.
EXAMPLE 6 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one

11.6 g (1.00 eq, 19.2 mmol) of intermediate 5, water (10.1 equiv; 193 mmoles; 3.48 mL; 3.48 g) and methanol (3.62 moles; 146 mL; 116 g) were combined and heated to 55-60° C. Isethionic acid was added slowly until a clear solution was obtained; 3.3 g isethionic acid solution was necessary to reach this end point. The resulting clear orange solution was filtered through paper and rinsed through with 20 ml methanol, after which the filtrate was reheated to 55-60° C. and the remaining isethionic acid was added (a total of 9.93 g was added). The reaction mixture precipitated and thickened for 6 hours, after which it was cooled and held at 30-35° C. while triethylamine (2.92 g; 28.8 mmoles) was added slowly as a 10% solution in methanol over 12 hrs. About halfway through the addition of triethylamine, desired polymorphic seeds were added to help formation of the desired polymorph. The resulting slurry was cooled and held at 5° C. for 15 minutes and the crystals were filtered and washed with methanol. The solid product was dried in vacuo at 55° C. to obtain 11 g of yellow crystals of the title compound.

……………………………………………………………………
http://www.google.com/patents/US7345171
EXAMPLES
The following examples are intended to be illustrative and non-limiting, and represent specific embodiments of the present invention.
Example 1 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
Example 2 Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2.3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
Example 3 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in Example 2) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
Example 4 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
To a slurry of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (7.0 g, 15.64 mmol, prepared as in Example 3 following contact with NaOH) dispersed in 250 mL of water was added drop-wise 30 mL of a 0.52 M solution of isethionic acid in MeOH (15.64 mmol) to a pH of 5.2. The solution was filtered through a glass filter (fine) and the clear solution was freeze-dried to give 9.4 g of the amorphous salt. The amorphous salt (3.16 g) was mixed with 25 mL of MeOH and after almost complete dissolution a new precipitate formed. Another 25 mL of MeOH was added and the mixture was stirred at 46° C. to 49° C. for four hours. The mixture was slowly cooled to 32° C. and put in a cold room (+4° C.) overnight. A sample was taken for PXRD, which indicated formation of Form B. The mixture was filtered and the precipitate was dried overnight at 50° C. in a vacuum oven. This furnished 2.92 g of the mono-isethionate salt of the compound of Formula 1 in 92% yield. HPLC-99.25%, PXRD-Form B, CHNS, H-NMR were consistent with the structure.
Example 5 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
MeOH (100 mL) was placed in a 250 mL flask equipped with a mechanical stirrer, thermocouple/controller, condenser, and heating mantle and preheated to 35° C. An amorphous isethionate salt (2 g, prepared as in Example 4) was slowly added in three even portions with a 25 min to 30 min interval between the additions. The reaction mixture was stirred overnight at 35° C. and subsequently cooled. A sample was filtered and examined by PXRD. It was pure Form B. The whole reaction mixture was then used as Form B seeds in a larger scale experiment.
Example 6 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
MeOH (50 mL) was placed in a 250 mL flask equipped with a magnetic stirrer, condenser, thermocouple/controller, and heating mantle, and preheated to 40° C. An amorphous isethionate salt (1 g, prepared as in Example 4) was slowly added in three even portions with 30 min interval between the portions and then stirred overnight at 40° C. The reaction was monitored by in-situ Raman spectroscopy. The sample was taken, filtered and analyzed by PXRD. It was pure Form B by PXRD and Raman spectroscopy. The mixture was cooled to 25° C. at a rate of 3° C./h, cooled to −10° C., filtered, and vacuum dried to furnish 0.85 g of the Form B crystalline product.
Example 7 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
The free base (Formula 1, 0.895 mg, 2 mmol) was mixed with 10 mL of MeOH and seeded with 33 mg of a mono-isethionate salt of the compound of Formula 1 (Form B). Then 5.6 mL of a 0.375 M solution of isethionic acid in MeOH (2.1 mmol) was added in 10 even portions over 75 min time period. The mixture was stirred for an additional hour and a sample was taken for PXRD analysis. It confirmed formation of crystalline Form B. The mixture was stirred at RT overnight and another PXRD was taken. There was no change in the crystal form. The mixture was cooled in a refrigerator at −8° C. overnight, filtered, and dried at 50° C. in a vacuum oven to give 1.053 g (91.8% of theory) of the above-named compound (Form B). HPLC—99.8%, CHNS, H-NMR, IR are consistent with the structure, PXRD-Form B.
Example 8 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form A)
An amorphous isethionate salt (47 mg, prepared as in Example 4) was mixed with 4 mL of EtOH in a 15 mL flask equipped with a magnetic stirrer, thermocouple and condenser. The mixture was heated to reflux, which resulted in the formation of a nearly clear solution. After refluxing for 10-15 min, the mixture became cloudy. It was slowly cooled to 50° C. and was seeded at 69° C. with Form A. The mixture was held at 50° C. for 5 h and was allowed to cool to RT overnight. The mixture was subsequently cooled to 1° C. with an ice bath, held for 1.5 h, filtered, washed with 0.5 mL of cold EtOH, air-dried, and then dried in a vacuum oven at 70° C. overnight to furnished 38.2 mg of a fine crystalline material. The crystalline material was found to be mono-isethionate salt Form A by PXRD. H-NMR was consistent for the mono-isethionate salt and indicated the presence of residual EtOH ca. 5.9 mol % or 0.6 wt %.
Example 9 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form D)
An amorphous isethionate salt (9.0 g, prepared as in Example 4) was mixed with 300 mL of MeOH, stirred and heated to 63.8° C. (at reflux). To the slightly cloudy mixture was added two 50-mL portions of MeOH. The hot mixture was filtered into a 2-L flask equipped with a mechanical stirrer. The mixture was briefly heated to reflux and then cooled to 60° C. IPA (100 mL) was added to the mixture. The mixture was again heated to 60° C. and an additional 110 mL of IPA was added. A precipitate started to form at 59.7° C. The mixture was reheated to 67.5° C., cooled to 50° C., and held overnight. A sample was taken the next morning for PXRD analysis. The mixture was cooled to 25° C. at a rate of 3° C./h and another PXRD sample was taken when the mixture reached 28° C. The mixture was allowed to cool to RT overnight. A precipitate was collected and dried in a vacuum oven at 65° C. and 30 Torr. The procedure produced 7.45 g (82.8% yield) of the crystalline compound (Form D by PXRD analysis). Previously analyzed samples were also Form D. HPLC showed 98.82% purity and CHNS microanalysis was within +/−0.4%. A slurry of isethionate salt Form A, B, and D in MeOH yielded substantially pure Form B in less than three days.
Example 10 Preparation of isethionic acid (2-hydroxy-ethanesulfonic acid)
A 5-L, four-necked, round-bottomed flask, equipped with mechanical stirrer, thermocouple, gas sparger, and an atmosphere vent through a water trap was charged with 748 g (5.05 mol) of sodium isethionate (ALDRICH), and 4 L of IPA. The slurry was stirred at RT. An ice bath was used to keep the internal temperature below 50° C. as 925 g (25.4 mol) of hydrogen chloride gas (ALDRICH) was sparged into the system at a rate such that it dissolved as fast as it was added (as noted by lack of bubbling through the water trap). Sufficient HCl gas was added until the system was saturated (as noted by the start of bubbling through the water trap). During the addition of HCl, the temperature rose to 45° C. The slurry was cooled to RT and filtered over a coarse-fritted filter. The cake was washed with 100 mL of IPA and the cloudy filtrate was filtered through a 10-20μ filter. The resulting clear, colorless filtrate was concentrated under reduced pressure on a rotary evaporator, while keeping the bath temperature below 50° C. The resulting 1.07 kg of clear, light yellow oil was diluted with 50 mL of tap water and 400 mL of toluene and concentrated under reduced pressure on a rotary evaporator for three days, while keeping the bath temperature below 50° C. The resulting 800 g of clear, light yellow oil was diluted with 500 mL of toluene and 250 mL of IPA and concentrated under reduced pressure on a rotary evaporator for 11 days, keeping the bath temperature below 50° C. The resulting 713 g of clear, light yellow oil was titrated at 81 wt % (580 g, 91.1% yield) containing 7.9 wt % water and 7.5 wt % IPA.
Example 11 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A 5-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (300 g, 0.51 mol, prepared as in Example 2), butyl vinyl ether (154 g, 1.54 mol, ALDRICH), n-butanol (1.5 L, ALDRICH), and diisopropyl ethylamine (107 mL, 0.62 mol, ALDRICH). The slurry was placed under approximately 50 Torr vacuum and then refilled with nitrogen 3 times. To this was added 8.3 g (0.01 mol) bis-(diphenylphosphinoferrocene) palladium dichloride dichloromethane (JOHNSON MATTHEY, Lot 077598001) and the resulting slurry was purged an additional three times as described above. The mixture was then heated to 95° C. and stirred for 20 h. The resulting thin red slurry was diluted with 2 L of heptane and cooled to approximately 5° C. At this temperature, 400 mL saturated aqueous potassium carbonate was added and the mixture was filtered and rinsed with 250 mL of heptane. After drying in an oven for 16 h at 45° C., 231.7 g (75% yield) of the title compound was obtained as a yellow solid.
Example 12 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-Pyrido[2,3-d]pyrimidin-7-one (Form B)
A 22-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (725 g, 1.20 mol, prepared as in Example 11) and MeOH (14 L). The slurry was stirred at RT as it was charged with a solution of isethionic acid (530 g, 4.20 mol, prepared as in Example 10), MeOH (1.5 L), and water (70 mL, 3.89 mol). The resulting slurry was heated to 55° C. over 30 minutes and then stirred at 55° C. for 30 minutes. A solution of 175 g (1.73 mol) of Et3N (ALDRICH) in 200 mL of MeOH was charged to the slurry as it was cooled to 30° C. The slurry was held at 30° C. as a solution of 128 g (1.26 mol) of Et3N in 2 L of MeOH was added dropwise over 6 hours. The resulting slurry was sampled to determine crystal form (Form B). The slurry was cooled and held at 5° C. for 15 minutes and was subsequently filtered through a coarse-fritted filter. The resulting filter cake was washed with multiple washes of 200 mL of cold MeOH. The solid product was dried at 55° C. under vacuum to yield 710 g (91% yield) of the title compound as yellow crystals.

1)Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;
2)Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4; Journal of Medicinal Chemistry,2005,48(7),2371-2387;
3)Erdman, David Thomas et al;Preparation of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157
4)Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997
5)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381
6)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.
7)Phase III Study Evaluating Palbociclib (PD-0332991), a Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”;ClinicalTrials.gov number:NCT01864746;currently recruiting participants(as of January 2, 2013)
8)A Randomized, Multicenter, Double-Blind Phase 3 Study Of PD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For Advanced Disease;ClinicalTrials.gov number:NCT01740427;currently recruiting participants(as of January 2, 2013)
9)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy;ClinicalTrials.gov number:NCT01942135;currently recruiting participants(as of January 2, 2013)
| US6936612 | Jan 16, 2003 | Aug 30, 2005 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2005005426A1 | Jun 28, 2004 | Jan 20, 2005 | Vladimir Genukh Beylin | Isethionate salt of a selective cdk4 inhibitor |
| US20030229026 * | Dec 18, 2000 | Dec 11, 2003 | Al-Awar Rima Salim | Agents and methods for the treatment of proliferative diseases |
| US20040006074 * | Dec 2, 2002 | Jan 8, 2004 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| US20040048915 * | Sep 24, 2001 | Mar 11, 2004 | Engler Thomas Albert | Methods and compounds for treating proliferative diseases |
| US20050222163 * | Mar 30, 2005 | Oct 6, 2005 | Pfizer Inc | Combinations of signal transduction inhibitors |
| US20070027147 * | Dec 3, 2004 | Feb 1, 2007 | Takashi Hayama | Biarylurea derivatives |
| WO2008032157A2 * | Aug 27, 2007 | Mar 20, 2008 | David Thomas Erdman | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2010075074A1 | Dec 15, 2009 | Jul 1, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2012098387A1 | Jan 17, 2012 | Jul 26, 2012 | Centro Nacional De Investigaciones Oncológicas (Cnio) | 6, 7-ring-fused triazolo [4, 3 – b] pyridazine derivatives as pim inhibitors |
| US7781583 | Sep 10, 2007 | Aug 24, 2010 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
| US7855211 | Dec 15, 2009 | Dec 21, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| US8247408 * | Oct 9, 2006 | Aug 21, 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755 | Feb 9, 2010 | Sep 25, 2012 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |

old info
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)

| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Empagliflozin

Empagliflozin
BI-10773
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
M.Wt: 450.91
: C23H27ClO7
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral treatment of adults with type 2 diabetes (Phase III, expected to conclude by year’s end); Oral treatment of adults with type 2 diabetes plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. Empagliflozin is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra. The Empagliflozin phase III clinical trial program will include about 14,500 patients. The program consists of twelve ongoing international phase III clinical trials, including a large cardiovascular outcomes trial.
Empagliflozin is a novel SGLT2 inhibitor that is described for the treatment or improvement in glycemic control in patients with type 2 diabetes mellitus, for example in WO 05/092877, WO 06/117359, WO 06/120208, WO 2010/092126, WO 2010/092123, WO 2011/039107, WO 2011/039108. The use of a SGLT2 inhibitor in a method for treating obesity is described in WO 08/116,195
WO2005/092877
WO2006/117359 MP 149 DEG CENT
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment oftype 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in theproximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
As of December 2013, empagliflozin is in phase III clinical trials.[2]

When taken in dosages of 10 or 25 mg once a day, the incidence of adverse events was similar to placebo. However, there was a higher frequency of genital infections at both the 10 mg and the 25 mg dosages.
1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene of the formula

as described for example in WO 2005/092877. Methods of synthesis are described in the literature, for example WO 06/120208 and WO 2011/039108. According to this invention, it is to be understood that the definition of empagliflozin also comprises its hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. An advantageous crystalline form of empagliflozin is described in WO 2006/117359 and WO 2011/039107 which hereby are incorporated herein in their entirety. This crystalline form possesses good solubility properties which enables a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline form is physico-chemically stable and thus provides a good shelf-life stability of the pharmaceutical composition. Preferred pharmaceutical compositions, such as solid formulations for oral administration, for example tablets, are described in WO 2010/092126,

http://www.google.com/patents/WO2011039108A2
Example 1 : Synthesis of the fluoride VIII.1
Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N-dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) and fluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a
fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2- propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
Example 2: Synthesis of the ketone VII.1
To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-ie f-butanolate solution (20%) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2-Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with ie f.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8%) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
Example 3: Synthesis of the iodide V.1
To a solution of ketone VII.1 (217,4kg) and aluminium chloride (AICI3; 81 ,5kg) in toluene (366,8kg) is added 1 ,1 ,3,3-tetramethyldisiloxane (TMDS, 82,5kg) within 1 hr 30 min
(temperature: 18-26°C). After completion of the addition, the mixture is stirred for additional 1 hr at a temperature of 24°C. Then the conversion is determined via HPLC analysis.
Subsequently the reaction mixture is treated with acetone (15,0kg), stirred for 1 hr 5 min at 27°C temperature and the residual TMDS content is analyzed via GC. Then a mixture of water (573kg) and concentrated HCI (34kg) is added to the reaction mixture at a temperature of 20 to 51 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature:
51 °C). The stirrer is switched off and the mixture is left stand for 20 min (temperature: 52°C). The phases are separated and solvent is distilled off from the organic phase at 53-73°C temperature under reduced pressure. Toluene (52,8kg) and ethanol (435,7kg) are added to the residue at 61 to 70°C temperature. The reaction mixture is cooled to 36°C temperature and seeding crystals (0,25kg) are added. Stirring is continued at this temperature for 35 min. Then the mixture is cooled to 0 to 5°C and stirred for additional 30 min. The product is collected on a centrifuge, washed with ethanol (157kg) and dried at 15 to 37°C under reduced pressure. 181 kg (82,6%) of product are obtained as colourless solid. The identity of the product is determined via the HPLC retention time.
Example 4: Synthesis of the lactone IV.1
A suspension of the D-(+)-gluconic acid-delta-lactone IVa.1 (42,0kg), tetrahydrofuran (277,2kg), 4-methylmorpholine (NMM; 152,4kg) and 4-dimethylaminopyridine (DMAP;
1 ,44kg) is treated with chlorotrimethylsilane (TMSCI; 130,8kg) within 50 min at 13 to 19°C. After completion of the addition stirring is continued for 1 hr 30 min at 20 to 22°C and the conversion is determined via HPLC analysis. Then n-heptane (216,4kg) is added and the mixture is cooled to 5°C. Water (143kg) is added at 3 to 5°C within 15 min. After completion of the addition the mixture is heated to 15°C and stirred for 15 min. The stirrer is switched off and the mixture is left stand for 15 min. Then the phases are separated and the organic layer is washed in succession two times with water (143kg each). Then solvent is distilled off at 38°C under reduced pressure and n-heptane (130kg) is added to the residue. The resulting solution is filtered and the filter is rinsed with n-heptane (63kg) (filter solution and product solution are combined). Then solvent is distilled off at 39 to 40°C under reduced pressure. The water content of the residue is determined via Karl-Fischer analysis (result: 0,0%).
1 12,4kg of the product is obtained as an oil (containing residual n-heptane, which explains the yield of >100%). The identity of the product is determined via infrared spectrometry.
Example 5a: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (267kg) in tetrahydrofuran (429kg) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (472kg) at -21 to -15°C temperature within 1 hr 50 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 3,45%. Then the lactone IV.1 (320kg) is added at -25 to -18°C temperature within 1 hr 25 min. The resulting mixture is stirred for further 1 hr 30 min at -13 to -18°C. On completion the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (938L; concentration: 10 %-weight) is added to the reaction mixture of a volume of about 2500L at -13 to 19°C within 1 hr 25 min.
The solvent is partially distilled off from the reaction mixture (residual volume: 1816-1905L) at 20 to 30°C under reduced pressure and 2-methyltetrahydrofuran (532kg) is added. Then the stirrer is switched off and the phases are separated at 29°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 2 to 3. Then solvent is distilled off from the organic phase at 30 to 33°C under reduced pressure and methanol (1202kg) is added followed by the addition of a solution of 1 ,25N HCI in methanol (75kg) at 20°C (pH = 0). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 20 to 32°C under reduced pressure and addition of methanol (409kg).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. 2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of 111.1 is greater or equal to 97,0% to 3,0%.
In this particular case both criteria are met. Triethylamin (14kg) is added (pH = 7,4) and solvent is distilled off under reduced pressure, acetonitrile (835kg) is added and further distilled under reduced pressure. This procedure is repeated (addition of acetonitrile: 694kg) and methylene chloride (640kg) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,27%).
The reaction mixture is then added within 1 hr 40 min at 10 to 19°C to a preformed mixture of AICI3 (176kg), methylene chloride (474kg), acetonitrile (340kg), and triethylsilane (205kg). The resulting mixture is stirred at 18 to 20°C for 70 min. After completion of the reaction, water (1263L) is added at 20 to 30°C within 1 hr 30 min and the mixture is partially distilled at 30 to 53°C under atmospheric pressure and the phases are separated. Toluene (698kg) is added to the organic phase and solvent is distilled off under reduced pressure at 22 to 33°C. The product is then crystallized by addition of seeding crystals (0,5kg) at 31 °C and water (267kg) added after cooling to 20°C. The reaction mixture is cooled to 5°C within 55 min and stirred at 3 to 5°C for 12 hrs. Finally the product is collected on a centrifuge as colourless, crystalline solid, washed with toluene (348kg) and dried at 22 to 58°C. 21 1 kg (73%) of product are obtained. The identity of the product is determined via the HPLC retention time.
Example 5b: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (30g) in tetrahydrofuran (55ml_) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (53g) at -14 to -13°C temperature within 35 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 0,35%. Then the lactone IV.1 (36g) is added at -15 to -6°C temperature within 15 min. The resulting mixture is stirred for further 1 hr at -6 to -7°C. On completion, the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (105mL;
concentration: 10 %-weight) is added to the reaction mixture at -15 to 10°C within 30 min. The solvent is partially distilled off from the reaction mixture (residual volume: 200mL) at 20 to 35°C under reduced pressure and 2-methyltetrahydrofuran (71 mL) is added. Then the mixture is stirred for 25min at 30°C. Then the stirrer is switched off and the phases are separated at 30°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 3. Then solvent is distilled off from the organic phase at 35°C under reduced pressure and methanol (126ml_) is added followed by the addition of a solution of 1 ,25N HCI in methanol (10,1 ml_) at 25°C (pH = 1 -2). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 35°C under reduced pressure and addition of methanol (47ml_).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. In this particular case the ratio is 99,6% : 0,43%.
2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of III.1 is greater or equal to 97,0% to 3,0%. In this particular case the ratio is 98,7% : 1 ,3%.
Triethylamin (2,1 mL) is added (pH = 9) and solvent is distilled off at 35°C under reduced pressure, acetonitrile (120ml_) is added and further distilled under reduced pressure at 30 to 35°C. This procedure is repeated (addition of acetonitrile: 102ml_) and methylene chloride (55ml_) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,04%).
The reaction mixture is then added within 1 hr 5 min at 20°C to a preformed mixture of AICI3 (19,8g), methylene chloride (49ml_), acetonitrile (51 mL), and triethylsilane (23g). The resulting mixture is stirred at 20 to 30°C for 60 min. After completion of the reaction, water (156mL) is added at 20°C within 25 min and the mixture is partially distilled at 55°C under atmospheric pressure and the phases are separated at 33°C. The mixture is heated to 43°C and toluene (90mL) is added and solvent is distilled off under reduced pressure at 41 to 43°C. Then acetonitrile (1 OmL) is added at 41 °C and the percentage of acetonitrile is determined via GC measurement. In this particular case, the acetonitrile percentage is 27%- weight. The product is then crystallized by addition of seeding crystals (0,1 g) at 44°C and the mixture is further stirred at 44°C for 15min. The mixture is then cooled to 20°C within 60min and water (142mL) is added at 20°C within 30min. The reaction mixture is cooled to 0 to 5°C within 60 min and stirred at 3°C for 16 hrs. Finally the product is collected on a filter as colourless, crystalline solid, washed with toluene (80mL) and dried at 20 to 70°C. 20, 4g (62,6%) of product are obtained. The identity of the product is determined via the HPLC retention time.
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
[1]. Grempler R, Thomas L, Eckhardt M et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83-90.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
[2]. Thomas L, Grempler R, Eckhardt M et al. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012 Jan;14(1):94-6.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
[3]. Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab. 2012 Jul;14(7):601-7.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
[4]. Macha S, Rose P, Mattheus M et al. Lack of drug-drug interaction between empagliflozin, a sodium glucose cotransporter-2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2012 Oct 24. doi: 10.1111/dom.12028. [Epub ahead of print]
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin’s anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
[5]. Sarashina A, Koiwai K, Seman LJ et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Empagliflozin, a Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor, in Healthy Japanese Subjects. Drug Metab Pharmacokinet. 2012 Nov 13. [Epub ahead of print]
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
…………………………….
formula A

Example I

(5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone
38.3 ml oxalyl chloride and 0.8 ml of dimethylformamide are added to a mixture of
100 g of 5-bromo-2-chloro-benzoic acid in 500 ml dichloromethane. The reaction mixture is stirred for 14 h, then filtered and separated from all volatile constituents in the rotary evaporator. The residue is dissolved in 150 ml dichloromethane, the solution is cooled to -5 0C, and 46.5 g of anisole are added. Then 51.5 g of aluminum trichloride are added batchwise so that the temperature does not exceed 5 0C. The solution is stirred for another 1 h at 1 to 5 0C and then poured onto crushed ice. The organic phase is separated, and the aqueous phase is extracted another three times with dichloromethane. The combined organic phases are washed with aqueous 1 M hydrochloric acid, twice with aqueous 1 M sodium hydroxide solution and with brine. Then the organic phase is dried, the solvent is removed and the residue is recrystallised in ethanol. Yield: 86.3 g (64% of theory)
Mass spectrum (ESI+): m/z = 325/327/329 (Br+CI) [M+H]+
Example Il
4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene
A solution of 86.2 g (5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone and 101.5 ml triethylsilane in 75 ml dichloromethane and 150 ml acetonitrile is cooled to 1O0C. Then with stirring 50.8 ml of boron trifluoride etherate are added so that the temperature does not exceed 2O0C. The solution is stirred for 14 h at ambient temperature, before another 9 ml triethylsilane and 4.4 ml boron trifluoride etherate are added. The solution is stirred for a further 3 h at 45 to 5O0C and then cooled to ambient temperature. A solution of 28 g potassium hydroxide in 70 ml of water is added, and the resulting mixture is stirred for 2 h. Then the organic phase is separated off and the aqueous phase is extracted another three times with diisopropylether. The combined organic phases are washed twice with aqueous 2 M potassium hydroxide solution and once with brine and then dried over sodium sulfate. After the solvent has been removed the residue is washed in ethanol, separated again and dried at 6O0C. Yield: 50.0 g (61 % of theory)
Mass spectrum (ESI+): m/z = 310/312/314 (Br+CI) [M+H]+
Example III

4-(5-bromo-2-chloro-benzyl)-phenol
A solution of 14.8 g 4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene in 150 ml dichloromethane is cooled in an ice bath. Then 50 ml of a 1 M solution of boron tribromide in dichloromethane are added, and the solution is stirred for 2 h at ambient temperature. The solution is then cooled in an ice bath again, and saturated aqueous potassium carbonate solution is added dropwise. At ambient temperature the mixture is adjusted with aqueous 1 M hydrochloric acid to a pH of 1 , the organic phase is separated, and the aqueous phase is extracted another three times with ethyl acetate. The combined organic phases are dried over sodium sulphate, and the solvent is removed completely. Yield: 13.9 g (98% of theory) Mass spectrum (ESI ): m/z = 295/297/299 (Br+CI) [M-HV
Example IV

r4-(5-bromo-2-chloro-benzyl)-phenoxyl-tert-butyl-dimethyl-silane
A solution of 13.9 g 4-(5-bromo-2-chloro-benzyl)-phenol in 140 ml dichloromethane is cooled in an ice bath. Then 7.54 g tert-butyldimethylsilylchlorid in 20 ml dichloromethane are added followed by 9.8 ml triethylamine and 0.5 g 4- dimethylaminopyridine. The solution is stirred for 16 h at ambient temperature and then diluted with 100 ml dichloromethane. The organic phase is washed twice with aqueous 1 M hydrochloric acid and once with aqueous sodium hydrogen carbonate solution and then dried over sodium sulfate. After the solvent has been removed the residue is filtered through silica gel (cyclohexane/ethyl acetate 100:1 ). Yield: 16.8 g (87% of theory) Mass spectrum (El): m/z = 410/412/414 (Br+CI) [M]+
Example V

2.3.4.6-tetrakis-O-(trimethylsilyl)-D-glucopyranone
A solution of 20 g D-glucono-1 ,5-lactone and 98.5 ml Λ/-methylmorpholine in 200 ml of tetrahydrofuran is cooled to -5 0C. Then 85 ml trimethylsilylchloride are added dropwise so that the temperature does not exceed 5 0C. The solution is then stirred for 1 h at ambient temperature, 5 h at 35 0C and again for 14 h at ambient temperature. After the addition of 300 ml of toluene the solution is cooled in an ice bath, and 500 ml of water are added so that the temperature does not exceed 100C. The organic phase is then separated and washed in each case once with aqueous sodium dihydrogen phosphate solution, water and brine. The solvent is removed, the residue is taken up in 250 ml of toluene, and the solvent is again removed completely. Yield: 52.5 g (approx. 90% pure)
Mass spectrum (ESI+): m/z = 467 [M+H]+
Example Vl

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-(4-hvdroxybenzyl)-benzene
A solution of 4.0 g [4-(5-bromo-2-chloro-benzyl)-phenoxy]-te/Tf-butyl-dimethyl-silane in 42 ml dry diethyl ether is cooled to -800C under argon. 11.6 ml of a 1.7 M solution of te/if-butyllithium in pentane are slowly added dropwise to the cooled solution, and then the solution is stirred for 30 min at -80 0C. This solution is then added dropwise through a transfer needle, which is cooled with dry ice, to a solution of 4.78 g
2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone in 38 ml diethyl ether chilled to – 80 0C. The resulting solution is stirred for 3 h at -78 0C. Then a solution of 1.1 ml methanesulphonic acid in 35 ml of methanol is added and the solution is stirred for 16 h at ambient temperature. The solution is then neutralised with solid sodium hydrogen carbonate, ethyl acetate is added and the methanol is removed together with the ether. Aqueous sodium hydrogen carbonate solution is added to the remaining solution, and the resulting mixture is extracted four times with ethyl acetate. The organic phases are dried over sodium sulphate and evaporated down. The residue is dissolved in 30 ml acetonitrile and 30 ml dichloromethane and the solution is cooled to -10 0C. After the addition of 4.4 ml triethylsilane 2.6 ml boron trifluoride etherate are added dropwise so that the temperature does not exceed -5 0C. After the addition is complete the solution is stirred for another 5 h at -5 to -10 0C and then quenched by the addition of aqueous sodium hydrogen carbonate solution. The organic phase is separated, and the aqueous phase is extracted four times with ethyl acetate. The combined organic phases are dried over sodium sulfate, the solvent is removed, and the residue is purified by chromatography on silica gel (dichoromethane/methanol 1 :0->3:1 ). The product then obtained is an approx. 6:1 mixture of β/α which can be converted into the pure β-anomer by global acetylation of the hydroxy groups with acetic anhydride and pyridine in dichloromethane and recrystallization of the product from ethanol. The product thus obtained is converted into the title compound by deacetylation in methanol with aqueous 4 M potassium hydroxide solution. Yield: 1.6 g (46% of theory)
Mass spectrum (ESI+): m/z = 398/400 (Cl) [M+H]+
Preparation of the compound A:

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-r4-(<fS)-tetrahvdrofuran-3-yloxy)-benzyll- benzene
0.19 g (f?)-3-(4-methylphenylsulfonyloxy)-tetrahydrofuran are added to a mixture of 0.20 g 1-chloro-4-(β-D-glucopyranos-1-yl)-2-(4-hydroxybenzyl)-benzene and 0.29 g cesium carbonate in 2.5 ml dimethylformamide. The mixture is stirred at 75 0C for 4 h, before another 0.29 g caesium carbonate and 0.19 g (f?)-3-(4-methylphenyl- sulfonyloxy)-tetrahydrofuran are added. After an additional 14 h stirring at 75 0C the mixture is cooled to ambient temperature and brine is added. The resulting mixture is extracted with ethyl acetate, the combined organic extracts are dried over sodium sulfate, and the solvent is removed. The residue is purified by chromatography on silica gel (dichloromethane/methanol 1 :0 -> 5:1 ). Yield: 0.12 g (49% of theory) Mass spectrum (ESI+): m/z = 451/453 (Cl) [M+H] +
The US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat)
13 dec 2013
Genzyme’s Cerdelga NDA receives US FDA priority review designationpharmabiz.comThe US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat), an investigational oral therapy for adult patients with Gaucher disease
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
old article cut paste
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
Eliglustat tartrate (USAN)
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Chugai Pharma to commercialize Helsinn’s Anamorelin for anorexia-cachexia syndrome in Germany, France, Benelux, UK and Ireland

LUGANO, Switzerland, November 12, 2013 /PRNewswire/ —
Swiss Pharma Group Helsinn seals an alliance with Japanese company Chugai by granting exclusive distribution & licensing rights to commercialize its innovative phase III ghrelin receptor agonist in Germany, France, Benelux, UK andIreland
Swiss-based Helsinn group has granted Chugai Pharma Marketing Ltd., a wholly-owned subsidiary of Chugai Pharmaceutical Co., Ltd., exclusive commercialization rights to their innovative ghrelin receptor agonist, anamorelin, for the three major European pharma markets.
Anamorelin is a new first-in-class, oral, once daily drug, currently in phase III for the treatment of anorexia-cachexia in NSCLC, a detrimental multifactorial disorder that affects over 50% of people with cancer and in which systemic inflammation, reduced food intake and altered metabolism contribute to loss of muscle mass and reduction of body weight leading to reduced quality of life, functional impairment and decreased survival.
- Anamorelin is a synthetic orally active ghrelin receptor agonist which is under development for the management of non-small lung cancer associated cachexia/anorexia.
- Development status: Phase 3
- Appetite loss; Aging; Bone injury; Cachexia

Anamorelin HCl
| M. Wt | 583.16 |
|---|---|
| Formula | C31H43ClN6O3 |
| CAS No. | 861998-00-7 |
| Synonyms | RC 1291; RC1291; RC 1291-HCl; ONO-7643; ONO7643; ONO 7643. |
| Chemical Name | 2-amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazinecarbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride |
Phase 3-LY2439821 (ixekizumab) for psoriasis and psoriatic arthritis.
 |
|
|---|---|
http://www.ama-assn.org/resources/doc/usan/ixekizumab.pdf
USAN IXEKIZUMAB
PRONUNCIATION ix” e kiz’ ue mab
THERAPEUTIC CLAIM Treatment of autoimmune diseases
CHEMICAL NAMES
1. Immunoglobulin G4, anti-(human interleukin 17A) (human monoclonal LY2439821γ4-chain), disulfide with human monoclonal LY2439821 κ-chain, dimer
2. Immunoglobulin G4, anti-(human interleukin-17A (IL-17, cytotoxic
T-lymphocyte-associated antigen 8)); humanized mouse monoclonal LY2439821 des-Lys446-[Pro227]γ4 heavy chain {H10S>P,CH3107K>-} (133-219′)-disulfide with humanized mouse monoclonal LY2439821 κ light chain, dimer (225-225”:228-228”)-bisdisulfide
MOLECULAR FORMULA C6492H10012N1728O2028S46
MOLECULAR WEIGHT 146.2 kDa
SPONSOR Eli Lilly and Co.
CODE DESIGNATION LY2439821
CAS REGISTRY NUMBER 1143503-69-8
Ixekizumab is a humanized monoclonal antibody used in the treatment of autoimmune diseases.[1]
Ixekizumab was developed by Eli Lilly and Co.
Lilly’s Anti-IL-17 Monoclonal Antibody, Ixekizumab, Met Primary Endpoint in Phase II Study in Patients With Chronic Plaque Psoriasis – March 28, 2012
more info
Inflammation represents a key event of many diseases, such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis, asthma, multiple sclerosis,
atherosclerosis, cystic fibrosis, and sepsis. Inflammatory cells, such as neutrophils, eosinophils, basophils, mast cells, macrophages, endothelial cells, and platelets, respond to inflammatory stimuli and foreign substances by producing bioactive mediators. These mediators act as autocrines and paracrines by interacting with many cell types to promote the inflammatory response. There are many mediators that can promote inflammation, such as cytokines and their receptors, adhesion molecules and their receptors, antigens involved in lymphocyte activation, and IgE and its receptors. [0004] Cytokines, for example, are soluble proteins that allow for communication between cells and the external environment. The term cytokines includes a wide range of proteins, such as lymphokines, monokines, interleukins, colony stimulating factors, interferons, tumor necrosis factors, and chemokines. Cytokines serve many functions, including controlling cell growth, migration, development, and differentiation, and mediating and regulating immunity, inflammation, and hematopoiesis. Even within a given function, cytokines can have diverse roles. For example, in the context of mediating and regulating inflammation, some cytokines inhibit the inflammatory response (anti-inflammatory cytokines), others promote the inflammatory response (pro-inflammatory cytokines). And certain cytokines fall into both categories, i.e., can inhibit or promote inflammation, depending on the situation. The targeting of proinflammatory cytokines to suppress their natural function, such as with antibodies, is a well-established strategy for treating various inflammatory diseases.
Many inflammatory diseases are treated by targeting proinflammatory cytokines with antibodies. Most (if not all) of the anti-proinflammatory cytokine antibodies currently on the market, and those currently in clinical trials, are of the IgG class. See, for example, Nature Reviews, vol. 10, pp. 301-316 (2010); Nature Medicine, vol. 18, pp. 736-749 (2012); Nature Biotechnology, vol. 30, pp. 475-477 (2012); Anti-Inflammatory & Anti- Allergy Agents in Medicinal Chemistry, vol. 8, pp. 51-71 (2009);
FlOOO.com/Reports/Biology/content/1/70, F 1000 Biology Reports, 1 :70 (2009); mAbs 4: 1, pp. 1-3 (2012); mAbs 3: 1, pp. 76-99 (2011); clinicaltrials.gov (generally), and
clinicaltrialsregister.eu/ (generally). These IgG antibodies are administered systemically and thus are often associated with unwanted side effects, which can include one or more of, for example, infusion reactions and immunogenicity, hypersensitivity reactions,
immunosuppression and infections, heart problems, liver problems, and others. Additionally the suppression of the target cytokines at non-diseased parts of the body can lead to unwanted effects.
In an attempt to reduce side effects associated with systemic treatment and to eliminate the inconvenience and expense of infusions, an article proposed an oral anti-TNF therapy that could be useful in treating Crohn’ s disease. Worledge et al. “Oral Administration of Avian Tumor Necrosis Factor Antibodies Effectively Treats Experimental Colitis in Rats.” Digestive Diseases and Sciences 45(12); 2298-2305 (December 2000). This article describes immunizing hens with recombinant human TNF and an adjuvant, fractionating polyclonal yolk antibody (IgY, which in chickens is the functional equivalent to IgG), and administering the unformulated polyclonal IgY (diluted in a carbonate buffer to minimize IgY acid hydrolysis in the stomach) to rats in an experimental rodent model of colitis. The rats were treated with 600mg/kg/day of the polyclonal IgY. The uses of animal antibodies and polyclonal antibodies, however, are undesirable.
In a similar attempt to avoid adverse events associated with systemic administration, another group, Avaxia Biologies Inc., describes a topical (e.g., oral or rectal) animal-dervied polyclonal anti-TNF composition that could be useful in treating
inflammation of the digestive tract, such as inflammatory bowel disease. WO2011047328. The application generally states that preferably the polyclonal antibody composition is prepared by immunizing an animal with a target antigen, and the preferably the polyclonal antibody composition is derived from milk or colostrum with bovine colostrums being preferred (e.g., p. 14). The application also generally states that the animal derived polyclonal antibodies could be specific for (among other targets) other inflammatory cytokines (e.g., pp. 6-7). This application describes working examples in which cows were immunized with murine TNF and the colostrum was collected post-parturition to generate bovine polyclonal anti-TNF antibodies (designated as AVX-470). The uses of animal-derived antibodies and polyclonal antibodies, however, are undesirable.
IgA molecular forms have been proposed as treatments for various diseases, most notably as treatments for pollen allergies, as treatments against pathogens, and as treatments for cancer.
For example, one article describes anti-AmbCtl (a ragweed pollen antigen) humanized monomelic IgA and dimeric IgA antibodies made in murine cells (NSO and Sp2/0 cells). The dimeric IgA contains a mouse J-chain. The article proposes that the antibodies may be applied to a mucosal surface or the lower airway to inhibit entry of allergenic molecules across the mucosal epithelium and therefore to prevent the development of allergic response. Sun et al. “Human IgA Monoclonal Antibodies Specific for a Major Ragweed Pollen Antigen.” Nature Biotechnology 13, 779-786 (1995).
Several other articles propose the use of IgA antibodies as a defense against pathogens.
Two articles proposed the use of an anti-streptococcal antigen I II secretory IgA-G hybrid antibody. Ma et al. “Generation and Assembly of Secretory Antibodies in Plants.” Science 268(5211), 716-719 (May 1995); Ma et al. “Characterization of a
Recombinant Plant Monoclonal Secretory Antibody and Preventive Immunotherapy in Humans.” Nature Medicine 4(5); 601-606 (May 1998). The hybrid antibody contains murine monoclonal kappa light chain, hybrid Ig A-G heavy chain, murine J- Chain, and rabbit secretory component. The antibody was made by successive sexual crossing between four transgenic N. tabacum plants and filial recombinants to form plant cells that expressed all four protein chains simultaneously. The parent antibody (the source of the antigen binding regions, is identified as the IgG antibody Guy’s 13. The group proposes that although slgA may provide an advantage over IgG in the mucosal environment, such is not always the case (1998 Ma at p. 604, right column).
A related article identifies the anti-streptococcal antigen I/II secretory IgA-G hybrid antibody, which was derived from Guy’s 13 IgA, as CaroRx. Wycoff. “Secretory IgA Antibodies from Plants.” Current Pharmaceutical Design 10(00); 1-9 (2004). Planet Biotechnology Inc. This related article states that the CaroRx antibody was designed to block adherence to teeth of the bacteria that causes cavities. Apparently, the CaroRx antibody was difficult to purify; the affinity of Protein A for the murine Ig domain was too low and protein G was necessary for sufficient affinity chromatography. Furthermore, the article states that several other chromatographic media had shown little potential as purification steps for the hybrid slgA-G from tobacco leaf extracts. The article also indicates that the authors were unable to control for human-like glycosylation in tobacco, but that such was not a problem because people are exposed to plant glycans every day in food without ill effect.
WO9949024, which lists Wycoff as an inventor, Planet Biotechnology Inc. as the applicant, describes the use of the variable regions of Guy’s 13 to make a secretory antibody from tobacco. The application contains only two examples – the first a working example and the second a prophetic example. Working Example 1 describes the transient production of an anti-S. mutans SA I/III (variable region from Guy’s 13) in tobacco. The tobacco plant was transformed using particle bombardment of tobacco leaf disks. Transgenic plants were then screened by Western blot “to identify individual transformants expressing assembled human slgA” (p. 25). Prophetic Example 2 states that in a transformation system for Lemna gibba (a monocot), bombardment of surface-sterilized leaf tissue with DNA- coated particles “is much the same as with” tobacco (a dicot). The prophetic example also stops at screening by immunoblot analysis for antibody chains and assembled slgA, and states that the inventors “expect to find fully assembled slgA.” [0014] Another article proposed the use of an anti-RSV glycoprotein F IgA antibodies (mlgA, dlgA, and slgA). Berdoz et al. “In vitro Comparison of the Antigen-Binding and Stability Properties of the Various Molecular Forms of IgA antibodies Assembled and Produced in CHO Cells.” Proc. Natl. Acad. Sci. USA 96; 3029-3034 (March 1999). The slgA antibody was made in CHO cells sequentially transfected with chimeric heavy and light chains, human J-Chain, and human secretory component, respectively. Single clones were generated to express the mlgA (clone 22), the dlgA (clone F), and the slgA (clone 6) (p. 3031).
Still other articles proposed, for example: (1) anti-HSV mlgA made in maize (Karnoup et al. Glycobiology 15(10); 965-981 (May 2005)) (which states that at that time there had been little success in the application of IgA class antibodies to therapeutic use because of the difficulty in producing the dimeric form in mammalian cells at economic levels); (2) anti-C. difficile toxin A chimeric mouse-human monomeric and dimeric IgA made in CHO cells (Stubbe et al. Journal of Immunology 164; 1952-1960 (2000)); (3) anti-N. meningitidis chimeric IgA antibodies were produced in BHK cells cotransfected with human J-Chain and/or human secretory component (Vidarsson et al., Journal of Immunology 166; 6250-6256 (2001)); (4) mti-Pseudomonas aeruginosa 06 lipopolysaccharide chimeric mouse/human mlgAl made in CHO cells (Preston et al. Infection and Immunity 66(9); 4137- 4142 (September 1998)); (5) anti-Plasmodium mlgA made in CHO cells (Pleass et al. Blood 102(13); 4424-4429 (December 2003)) (which states that unlike their parental mouse IgG antibodies, the mlgA antibodies failed to protect against parasitic challenge in vivo); and (5) ^^-Helicobacter pylori urease subunit A slgA and dlgA (Berdoz et al. Molecular
Immunology 41(10); 1013-1022 (August 2004)). [0016] For a review article discussing passive and active protection against pathogens at mucosal surfaces, see Corthesy. “Recombinant Immunoglobulin A: Powerful Tools for Fundamental and Applied Research.” Trends in Biotechnology 20(2); 65-71 (February 2002).
Still other articles propose the use of IgA antibodies as a treatment for cancer.
For example, one article describes a Phase la trial of a muring anti-transferrin receptor IgA antibody (Brooks et al. “Phase la Trial of Murine Immunoglobulin A
Antitransferrin Receptor Antibody 42/6.” Clinical Cancer Research 1(11); 1259-1265 (November 1995)). Another article describes a human anti-Ep-CAM mIgA made in BHK (baby hamster kidney) cells (Huls et al. “Antitumor Immune Effector Mechanisms Recruited by Phase Display-Derived Fully Human IgGl and IgAl Monoclonal Antibodies.” Cancer Research 59; 5778-5784 (November 1999)). Still another article describes an anti-HLA Class II chimeric mIgA antibody made in BHK cells (Dechant et al. “Chimeric IgA Antibodies Against HLA Class II Effectively Trigger Lymphoma Cell Killing.” Blood 100(13); 4574- 4580 (December 2002)). Yet other articles describe anti-EGFR mIgA or dlgA antibodies made in CHO, including Dechant et al. “Effector Mechanisms of Recombinant IgA
Antibodies Against Epidermal Growth Factor Receptor.” Journal of Immunology 179; 2936- 2943 (2007), Beyer et al. “Serum- Free Production and Purification of Chimeric IgA
Antibodies.” Journal of Immunology 346; 26-37 (2009) (stating that as of 2009, IgA antibodies have not been commercially explored for problems including lack of production and purification methods), and Lohse et al. “Recombinant Dimeric IgA Antibodies Against the Epidermal Growth Factor Receptor Mediate Effective Tumor Cell Killing.” Journal of Immunology 186; 3770-3778 (February 2011).
For a review article on anti-cancer IgA antibodies, see Dechant et al. “IgA antibodies for Cancer Therapy. ” Critical Reviews in Oncology/Hematology 39; 69-77 (2001); states that compared with infectious diseases, the role of IgA in cancer immunotherapy is even less investigated).
IL17 and IFN-garama inhibition for the treatment of autoimmune inflammation
The IL-17 family of cytokines has been associated with the pathogenesis of autoimmune diseases and is generally blamed for the pathogenic symptoms of autoimmune inflammation. Overexpression of IL-17 is a hallmark for autoimmune diseases like rheumatoid arthritis, systemic lupus erythematomatosus, inflammatory bowel disease, multiple sclerosis, and psoriasis (Yao Z et. al., J Immunol, 155(12), 1995, 5483-6. Chang S H, et.al, Cytokine, 46, 2009, 7-11; Hisakata Yamada et.al, Journal of Inflamm. Res., 3, 2010, 33-44)).
The IL-17 cytokine family comprises six members, out of which IL-17 A and IL-17F are the best characterized. IL-17A and IL-17F exist as homo- as well as as heterodimers (IL-17AA, IL-17AF, IL-17FF). IL-17A and IL-17F are clearly associated with inflammation (Gaffen S H, Cytokine, 43, 2008, 402-407; Torchinsky M B et al, Cell. Mol. Life Sci., 67, 2010, 1407- 1421).
The secretion of IL-17 is predominantly caused by a specific subtype of T helper cells termed TH-17 cells. IL-23, TGFp and IL-6 were shown to be important factors leading to conversion of nai‘ve CD4+ T-cells to THl 7 cells. It was also reported that TGF and IL-6 potently induce in synergy THl 7 differentiation. Important transcription factors for the secretion of IL-17 from TH17 cells are RORyt and STAT3 (IvanovJ et.al. Cell 126, 2006, 1121-1133). IL-17 induces pro-inflammatory cytokines (IL-6, TNF- and IL-lb) and Chemokines (CXCL1,GCP-2,CXCL8 or IL-8,CINC,MCP-1). It increases the production of nitric oxide prostaglandin E2 and matrix-metalloproteinases. As a consequence of these events neutrophil infiltration, tissue damage and chronic inflammation occurs (PECK A et.al, Clin Immunol., 132(3), 2009, 295-304).
Before the recognition of the importance of IL-17 in autoimmune inflammation, IFN-gamma derived from THl cells was believed to be an important cytokine that drives autoimmune disorders (Takayanagi H et. al. Nature, 408, 2000, 600-605. Huang W. et. al. Arthritis Res. Ther., 5, 2002, R49-R59) The secretion of IFN-gamma is a key feature of the THl effector cell lineage and the secretion is regulated by the transcription factors T-bet and STAT4 (Bluestone JA et. al. Nat Rev Immunol, 11, 2009, 811-6). Infiltration of activated T-cells and elevation of M-CSF, IL-10 and TNF support this notion (Yamanda H et.al Ann. Rheu. Dis., 67, 2008, 1299-1304; Kotake S et.al. Eur. J. Immunol, 35, 2005, 3353-3363).
Recently, a more complex situation was proposed, where hybrid TH17/TH1 cells induced by IL-23 and IL-6 in concert with IL-1 secrete IL-17 and IFN-gamma. These cells are under the control of the transcription factors RORyt and T-bet, confirming the notion, that these are true hybrids of THl and THl 7 cells. It was also demonstrated that these double producing cells are the pathogenic species in IBD and EAE (Buonocore S et.al. Nature, 464, 2010, 1371-5; Ghoreshi K. et. al. Nature, 467, 2010, 967-971).
Compounds which target and suppress both IL-17 and IFN-gamma are predisposed for the treatment of autoimmune disorders.
The effectiveness of blocking IL-17 signaling as therapeutic treatment in autoimmune diseases has already been proven in clinical trials with e.g. monoclonal antibodies against IL- 17A (AIN457, secukinumab; Ly2439821,ixekizumab; RG4934) and/or the IL-17 receptor IL- 17RA (AMG827, brodalumab).
Positive results have been reported for the treatment of rheumatoid arthritis, psoriasis and uveitis (Hueber W et al, Sci. Transl. Med., 2, 2010, 52ra72, DOI: 10.1126/scitranslmed.3001107; van den Berg W B e/ al, Nat. Rev. Rheumatol, 5, 2009, 549-553), ankylosing spondylitis and spondyloarthritides (Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Secukinumab is currently under investigation in clinical trials for psoriatic arthritis, Behcet disease, uveitits, inflammatory bowel disease, Crohn’s disease, multiple sclerosis (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718; Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Brodalumab, Ixekizumab and RG4934 are currently in clinical trials for the treatment of rheumatoid arthritis, psoriasis and/or psoriatic arthritis (Kopf M et al, Nat. Rev. Drug Disc, 9, 2010, 703-718; clinicaltrials.gov; Medicines in development for skin diseases, 201 1, published by PhRMA, www .phrma. com) .
With regard to blocking of IFN-gamma signaling as therapeutic treatment in autoimmune diseases, the IFN-gamma-specific monoclonal antibody AMG811 is currently under clinical investigations for the treatment of systemic lupus erythematosus (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718).
Monoclonal antibody (mAbs) 2013

2013——-29 monoclonal antibody (mAbs) drugs are in Phase III clinical development.
While around 350 therapeutic mAbs are currently in clinical development globally, only 28 had entered active Phase 2/3 or Phase 3 studies as of January 2013, Additionally one mAb mixture was under evaluation in Phase III.
Historically, mAbs that target antigens relevant to cancer have comprised approximately 50% of the mAb clinical pipeline,
but in 2013 the picture has changed: 66% or 19 of the antibodies to watch in 2013 are for non-cancer indications.

The non-cancer mAbs include alirocumab (Regeneron; Sanofi, hypercholesterinemia);
AMG 145 (Amgen, hypercholesterinemia),
epratuzumab (UCB, SLE),
gantenerumab (Roche; Alzheimer’s disease),
gevokizumab (Xoma/Servier, Non-infectious uveitis),
itolizumab (Biocon, Plaque psoriasis), ixekizumab (Eli Lilly and Co., psoriasis),
lebrikizumab (Roche/Genentech, rheumatoid arthritis),
mepolizumab (GSK, Asthma, COPD etc.),
ocrelizumab (Roche/Genentech, multiple sclerosis),
reslizumab (Teva, Eosinophilic asthma), romosozumab (Amgen, Postmenopausal osteoporosis),
sarilumab (Regeneron; Sanofi, rheumatoid arthritis),
secukinumab (Novartis, rheuma, psoriasis),
sirukumab (Janssen R&D LLC, rheumatoid arthritis),
solanezumab (Eli Lilly and Co., Alzheimer’s disease),
tabalumab (Eli Lilly and Co., rheuma, SLE)
and
vedolizumab (Millenium, Ulcerative colitis; Crohn disease).
The mixture of actoxumab and bezlotoxumab (MK-3415A, Merck & Co.) is being evaluated in two Phase 3 studies as a treatment for Clostridium difficile infection.
The ten cancer mAbs are:
elotuzumab (Bristol-Myers Squibb, Abbott, multiple myeloma),
farletuzumab (Morphotek, ovarian cancer),
inotuzumab ozogamicin (Pfizer; UCB, ALL, NHL),
naptumomab estafenatox (Active Biotech, renal cell carcinoma),
necitumumab (ImClone LLC, NSCL),
nivolumab (Bristol-Myers Squibb, NSCL, renal cell carcinoma),
obinutuzumab (Roche/Genetech, Diffuse large B cell lymphoma, CLL, NHL),
onartuzumab (Roche/Genetech, NSCL cancer; gastric cancer),
racotumomab (CIMAB; Laboratorio Elea S.A.C.I.F. y A, NSCL),
and ramucirumab (ImClone LLC, Gastric; liver, breast, colorectal, NSCL cancers).
Antibody
ALIROCUMAB
ALIROCUMAB
http://www.ama-assn.org/resources/doc/usan/alirocumab.pdf
Immunoglobulin G1, anti-(human neural apoptosis-regulated proteinase 1) (human REGN727 heavy chain), disulfide with human REGN727 κ-chain, dimer
Immunoglobulin G1, anti-(human proprotein convertase subtilisin/kexin type 9
(EC=3.4.21.-, neural apoptosis-regulated convertase 1, proprotein convertase 9,
subtilisin/kexin-like protease PC9)); human monoclonal REGN727 des-448-
lysine(CH3-K107)-1 heavy chain (221-220′)-disulfide with human monoclonal
REGN727 light chain dimer (227-227”:230-230”)-bisdisulfide
Clinical Trials for Compound
| Number of clinical trials registered at clinicaltrials.gov | 30 |
Biological Sequence
| Description | Sequence |
| Alirocumab heavy chain | EVQLVESGGGLVQPGGSLRLSCAASGFTFNNYAMNWVRQAPGKGLDWVSTISGSGGTTNY ADSVKGRFIISRDSSKHTLYLQMNSLRAEDTAVYYCAKDSNWGNFDLWGRGTLVTVSSAS TKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGL YSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPS VFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNST YRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELT KNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQ GNVFSCSVMHEALHNHYTQKSLSLSPG |
| Alirocumab light chain | DIVMTQSPDSLAVSLGERATINCKSSQSVLYRSNNRNFLGWYQQKPGQPPNLLIYWASTR ESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQQYYTTPYTFGQGTKLEIKRTVAAPS VFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYS LSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC |
1245916-14-6 CAS
C6472H9996N1736O2032S42
Alirocumab is a human monoclonal antibody designed for the treatment of hypercholesterolemia.[1]
This drug was discovered by Regeneron Pharmaceuticals and is being co-developed by Regeron and Sanofi.
When the results from Phase II trials of Sanofi and Regeneron’s proprotein convertase subtilisin kexin 9 (PCSK9) inhibitor alirocumab were presented in March, they stunned even the company representatives working on the trials. “I’m still amazed by the reduction in low-density lipoprotein cholesterol (LDL-C) that we saw with our drug,” says Bill Sasiela, vice president of cardiovascular and metabolic research at Regeneron. The monoclonal antibody (mAb) reduced LDL-C levels by up to 73% in three mid-stage trials, irrespective of baseline LDL-C levels or background treatment, offering hope for millions of patients who can’t hit the recommended cholesterol targets with statins — the standard therapies for lowering LDL-C levels in patients with cardiovascular disease. Spurred on by these results, Sanofi and Regeneron geared up into Phase III trials of the first-in-class alirocumab (also known as REGN727 and SAR236553) over the summer, and initiated the latest and largest trial — an 18,000-patient outcomes study
It is a Proprotein convertase subtilisin/kexin type 9, (also known as PCSK9) inhibitor . Phase III trials showed a 47% reduction in LDL-C. There was a high rate of adverse events with 69% experiencing side effects (most common problem was infection).
About PCSK9 PCSK9 is known to be a determinant of circulating LDL levels, as it binds to LDL receptors resulting in their degradation so that fewer are available on liver cells to remove excess LDL-cholesterol from the blood. Moreover, traditional LDL-lowering therapies such as statins actually stimulate the production of PCSK9, which limits their own ability to lower LDL-cholesterol. Blocking the PCSK9 pathway is therefore a potentially novel mechanism for lowering LDL-cholesterol.
Alirocumab is an investigational, fully-human monoclonal antibody that targets and blocks PCSK9. It is administered via subcutaneous injection. By inhibiting PCSK9, a determinant of circulating LDL-C levels in the blood, alirocumab has been shown in pre-clinical studies to increase the number of LDL receptors on hepatocytes, thereby lowering LDL-C.
The investigational agent described above is currently under clinical development and its safety and efficacy have not been fully evaluated by any regulatory authority
References
- Statement On A Nonproprietary Name Adopted By The USAN Council – Alirocumab, American Medical Association.

PARIS and TARRYTOWN, N.Y., Oct. 16, 2013 /PRNewswire via COMTEX/ — Sanofi and Regeneron Pharmaceuticals, Inc. REGN -1.73% today announced that the Phase 3 ODYSSEY MONO trial with alirocumab, an investigational monoclonal antibody targeting PCSK9 (proprotein convertase subtilisin/kexin type 9), met its primary efficacy endpoint. The mean low-density lipoprotein-cholesterol (LDL-C, or “bad” cholesterol) reduction from baseline to week 24, the primary efficacy endpoint of the study, was significantly greater in patients randomized to alirocumab, as compared to patients randomized to ezetimibe (47.2% vs. 15.6%, p<0.0001). In the trial, which employed a dose increase (up-titration) for patients who did not achieve an LDL-C level of 70 milligrams/deciliter (mg/dL), the majority of patients remained on the initial low dose of alirocumab of 75 milligrams (mg). read at
Pipeline of selected PCSK9 inhibitors
| Drug name | Companies | Modality | Clinical phase |
|---|---|---|---|
| Alirocumab (also known as REGN727 and SAR236553) | Regeneron/Sanofi | Monoclonal antibody | III |
| AMG145 | Amgen | Monoclonal antibody | II |
| LGT209 | Novartis | Monoclonal antibody | II |
| RG7652 | Roche/Genentech | Monoclonal antibody | II |
| RN316 | Pfizer | Monoclonal antibody | II |
| BMS-962476 | Bristol-Myers Squibb | Adnectin | I |
| ALN-PCS | Alnylam | RNA interference | I |
| ISIS-405879/BMS-844421 | Isis/Bristol-Myers Squibb | Antisense | Discontinued |
| PCSK9, proprotein convertase subtilisin kexin 9. | |||

BAYER- sPRM (BAY 1002670) Vilaprisan is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids
http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
20,20,21,21,21-pentafluoro-17-hydroxy-11β-[4-
(methanesulfonyl)phenyl]-19-nor-17α-pregna-4,9-dien-3-one
progesterone receptor antagonist
BAY 1002670, vilaprisan
1262108-14-4
C27H29F5O4S, 544.574
http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
Bayer has also made good progress in the development of new treatment options for patients with gynecological diseases: sPRM (BAY 1002670) is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids. Based on promising early clinical data the initiation of a Phase III study is planned for mid-2014.
A selective progesterone receptor modulator (SPRM) is an agent that acts on the progesterone receptor. A characteristic that distinguishes such substances from receptor full agonists (such as progesterone) and full antagonists (such as mifepristone) is that their action differs in different tissues (agonist in some while antagonist in others). This mixed agonist/antagonist profile of action leads to selective stimulation or inhibition progesterone-like action in different tissues and furthermore raises the possibility of dissociation of desirable therapeutic effects from undesirable side effects in synthetic progesterone receptor drug candidates


amcrasto@gmail.com
email me if u like my posts

{kind=link}