
Home » Posts tagged 'PHASE 3' (Page 15)
Tag Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Saracatinib, AZD0530 in phase 3 for Ovary Cancer,

Saracatinib
NCGC00241099, cas 379231-04-6
893428-71-2 (trihydrate)
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methyl-1-piperazinyl)ethoxy]-5-[(tetrahydro-2H-pyran-4-yl)oxy]-4-quinazolinamine
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydropyran-4-yloxy)quinazolin-4-amine
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline
AZD0530
C27H32ClN5O5
542.03
AstraZeneca Pharmaceuticals LP
| Astrazeneca Ab, Astrazeneca Uk Ltd, |
Saracatinib (AZD0530) is a highly selective, orally available, dual-specific Src/Abl kinase inhibitor with IC50 of 2.7 and 30 nM for c-Src and Abl kinase, respectively.Saracatinib (AZD0530) demonstrated potent antimigratory and antiinvasive effects in vitro, and inhibited metastasis in a murine model of bladder cancer. Antiproliferative activity of AZD0530 in vitro varied between cell lines (IC50=0.2 ~10 mM).
c-Src, Bcr–Abl, Yes1, Lck.target
AZD0530 is orally available 5-, 7-substituted anilinoquinazoline with anti-invasive and anti-tumor activities. AZD0530 is a dual-specific inhibitor of Src and Abl, protein tyrosine kinases that are overexpressed in chronic myeloid leukemia cells. This agent binds to and inhibits these tyrosine kinases and their effects on cell motility, cell migration, adhesion, invasion, proliferation, differentiation, and survival. Specifically, AZD0530 inhibits Src kinase-mediated osteoclast bone resorption.
AZD-0530 is a highly selective, dual-specific small molecule Src/Abl kinase inhibitor currently in phase II/III clinical trials at AstraZeneca for the treatment of ovarian cancer. Phase II clinical trials are also under way at the company for the treatment of solid tumors and hematological neoplasms. The Mayo Clinic is developing AZD-0530 in phase II clinical studies for the treatment of metastatic pancreas cancer.
Additional phase II trials are under way at the National Cancer Institute (NCI) for the treatment of colorectal cancer, prostate cancer, breast cancer, lung cancer, stomach cancer, soft tissue sarcoma, stage II or IV melanoma and thymic malignancies. A phase II trial for pancreatic cancer has been suspended. Src and Abl kinase are highly expressed in various human tumor types. No recent development has been reported for research into the treatment of head and neck cancer.
Phase II study of Saracatinib (AZD0530) for for the treatment of patients with hormone receptor-negative metastatic breast cancer : Nine patients were treated on study. After a median of 2 cycles (range 1-3), no patient had achieved CR, PR, or SD >6 months. The median time to treatment failure was 82 days (12-109 days).The majority (89%) of patients discontinued saracatinib because of disease progression. One patient acquired potentially treatment-related grade 4 hypoxia with interstitial infiltrates and was removed from the study. Common adverse events included fatigue, elevated liver enzymes, nausea, hyponatremia, dyspnea, cough, and adrenal insufficiency. CONCLUSIONS: These efficacy results were not sufficiently promising to justify continued accrual to this study. Based on this series, saracatinib does not appear to have significant single-agent activity for the treatment of patients with ER(-)/PR(-) MBC. (source: Clin Breast Cancer. 2011 Oct;11(5):306-11.)
Phase II study of Saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: Saracatinib has insufficient activity as a single agent in patients with advanced gastric adenocarcinoma to warrant further investigation. Further development in gastric cancer would require rational drug combinations or identification of a tumor phenotype sensitive to Src inhibition. (source: Invest New Drugs. 2011 Mar 12. [Epub ahead of print]).
Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Nine patients were enrolled. All patients had received prior radiotherapy and six patients had received prior chemotherapy for recurrent or metastatic disease. The most common adverse event was fatigue. Eight patients had progression of disease by response evaluation criteria in solid tumors (RECIST) within the first eight-week cycle and one patient was removed from the study after 11 days due to clinical decline with stable disease according to the RECIST criteria. Median overall survival was six months. The study was closed early due to lack of efficacy according to the early stopping rule. CONCLUSION: Single-agent saracatinib does not merit further study in recurrent or metastatic HNSCC. (source: Anticancer Res. 2011 Jan;31(1):249-53.)
893428-72-3 Saracatinib difumarate
893428-73-4 also
Saracatinib (AZD0530) is a Src inhibitor for c-Src with IC50 of 2.7 nM.
…………………………….

http://www.google.com/patents/WO2001094341A9?cl=en

………………….
WO 2006064217
http://www.google.fm/patents/EP1871769A2?cl=en
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline which compound is disclosed as Compound No. 73 within the Table in Example 14 of International Patent Application WO 01/94341. That compound is described herein by way of the Formula I

and as AZD0530, the code number by which the compound is known.
AZD0530 is an inhibitor of the Src family of non-receptor tyrosine kinase enzymes and, thereby, is a selective inhibitor of the motility of tumour cells and a selective inhibitor of the dissemination and invasiveness of mammalian cancer cells leading to inhibition of metastatic tumour growth. In particular, the compound AZD0530 is an inhibitor of c-Src non-receptor tyrosine kinase and should be of value as an anti-invasive agent for use in the containment and/or treatment of solid tumour disease in the human or animal body. The route for preparing the compound of the Formula I that is disclosed in International Patent Application WO 01/94341 involves the reaction of the compound 4-(6-chloro-2,3-methylenedioxyanilino)-7-hydroxy-5-tetrahydropyran-4-yloxyquinazoline with an alkylating agent to form the 2-(4-methylpiperazin-l-yl)ethoxy side-chain at the 7-position. The product of the reaction is disclosed in WO 01/94341 in the form of a dihydrochloride salt and in the form of a free base.
Example 14 4-(6-chloro-2,3-methylenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline (route 4)
Under an atmosphere of nitrogen gas, l-(2-hydroxyethyl)-4-methylpiperazine (13.93 g) was added to a stirred mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-fluoro- 5-tetrahydropyran-4-yloxyquinazoline (12.9 g), sodium te/t-pentoxide (9.87 g) and 1 ,2-diethoxyethane (37.5 ml). Water (1.34 g) and 1,2-diethoxyethane (25 ml) were added and the resultant reaction mixture was stirred and heated to 86°C for 18 hours. The reaction mixture was cooled to 5O0C and, under vacuum distillation at approximately 60 millibar pressure, approximately 50 ml of reaction solvent was distilled off. The reaction mixture was neutralised to pH 7.0 to 7.6 by the addition of a mixture of concentrated aqueous hydrochloric acid (36%, 10 ml) and water (84 ml) at a rate that kept the temperature of the reaction mixture at a maximum of 6O0C. With the temperature of the reaction mixture being kept at 6O0C, the reaction mixture was extracted with ethyl acetate (225 ml). The organic solution was washed with water (50 ml). Water (25 ml) was added and, with the temperature being kept at 6O0C, the mixture was stirred for 10 minutes, then allowed to stand for 30 minutes and the aqueous layer was separated. The organic layer was concentrated to a volume of about 100 ml by distillation of solvent at about 9O0C under atmospheric pressure. The residual mixture was cooled during 1 hour to 450C and held at that temperature for 2 hours to allow crystallisation of product. The mixture was warmed briefly to 550C and then cooled during 4 hours to 180C and held at that temperature for 1 hour. The crystalline precipitate was isolated by filtration and washed in turn with water (17 ml) and with tø’t-butyl methyl ether (17 ml). There was thus obtained 4-(6-chloro-2,3-πiethylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a trihydrate (11 g; 88% purity by HPLC using Method B, retention time 7.3 minutes); NMR Spectrum: (CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,lH), 6.05 (s, 2H), 6.55 (s, IH), 6.75 (d, IH), 6.85 (s, IH), 7.0 (d, IH), 8.55 (s, IH), 9.25 (s, IH).
A portion (10 g) of the material so obtained was placed on a filter and dried at ambient temperature in a stream of dry nitrogen gas. The resultant material was dissolved at 6O0C in dry isopropanol (140 ml) whilst maintaining a dry nitrogen atmosphere. The solution was allowed to cool to ambient temperature and to stand under a dry nitrogen atmosphere for 2 days. The resultant crystalline solid was isolated by filtration under a dry nitrogen atmosphere. The material (8 g) so obtained was a crystalline anhydrous form of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l -yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline, m.p. 142 to 1440C.
Example 15
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (200 ml) and water (10 ml) was heated to 75°C. A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (40 ml) was heated to 😯0C. A portion (80 ml) of the warmed solution of the quinazoline compound was added to the fumaric acid solution whilst the temperature was maintained at 750C. The resultant mixture was stirred at 750C for 75 minutes. The remainder of the quinazoline compound solution was added during 1 hour whilst the temperature was maintained at 750C. Isopropanol (50 ml) was added and the resultant mixture was stirred at 750C for 7 hours. The mixture was cooled slowly over at least 25 minutes to 5O0C and was stirred at that temperature for 6 hours. The mixture was cooled slowly over at least 20 minutes to 2O0C and was stirred at that temperature for 18.5 hours. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro- 2,3-methylenedioxyanilino)-7-[2-(4-methylρiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (37.0 g); m.p. 233-2370C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H)5 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H)3 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)3 6.6 (s, 4H)5 6.83 (d5 IH)3 6.84 (d, IH)5 6.91 (d3 IH)5 7.05 (d, IH)3 8.33 (s, IH)3 9.18 (s, IH).
Example 16
4-(6-chloro-2,3-methyIenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yIoxyquinazolme difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (210 ml) and water (30 ml) was heated to 4O0C and the mixture was filtered. The filter was washed with isopropanol (20 ml) and the washings were added to the warm filtrate. The resultant solution was warmed to 75°C.
A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (20 ml) was heated to 700C and the resultant mixture was filtered. A portion (110 ml) of the fumaric acid solution was added to the warmed solution of 4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin- 1 -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline whilst the temperature was maintained at 75°C. Seed crystals of 4-(6-chloro-
253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (0.02 g) were added and the resultant mixture was stirred at 750C for 1 hour. The remainder of the fumaric acid solution was added during 1 hour whilst the temperature was maintained at 750C and the resultant mixture was stirred at 750C for 14 hours.
The mixture was cooled slowly over at least 2 hours to 200C and was stirred at that temperature for 1 hour. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difumarate salt (35.8 g); m.p. 234-237°C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H)5 2.1-2.17 (m5 2H)5 2.33 (s5 3H)5 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)5 6.6 (s, 4H), 6.83 (d, IH)5 6.84 (d, IH)5 6.91 (d, IH)5 7.05 (d, IH)5 8.33 (s5 IH)5 9.18 (s5 IH).
Example 17 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline sesquifumarate salt
A mixture of 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difurnarate (0.15 g) and water (20 ml) was warmed using a heat gun to obtain a solution. The sample was allowed to evaporate slowly at ambient temperature to a volume of about 3 ml under a flow of air for 24 hours whereupon a precipitate had started to form. The mixture was placed in a refridgerator at 4°C for 2 days. The resultant precipitate was isolated by filtration and washed with water. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a sesquifumarate tetrahydrate salt (0.084 g) which was characterised using XRPD5 DSC5 TGA5 FTIR and solution NMR techniques.
………………..
A simplified process for the manufacture of AZD0530, a potent SRC kinase inhibitor
Org Process Res Dev 2011, 15(3): 688
http://pubs.acs.org/doi/abs/10.1021/op100161y

Process research and development of a synthetic route towards a novel SRC kinase inhibitor is described. The Medicinal Chemistry route was very long and suffered from extensive use of chlorinated solvents and chromatography. A number of steps in the Medicinal Chemistry route were also unattractive for large-scale use for a variety of reasons. The route was modified to produce a shorter synthetic scheme that started from more readily available materials. By using the modified route, the title compound was manufactured on kilogram scale without recourse to chromatography and in significantly fewer steps. The scaled synthesis required two Mitsunobu couplings, which were developed and scaled successfully. An interesting hydrazine impurity was identified in the second Mitsunobu coupling; a mechanism for its formation is proposed, and a method for its control is described. The formation and control of some other interesting impurities are also described.
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
Final Purification of N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
AZD0530 difumarate (4.234 kg at 89% w/w, 4.87 mol) was refluxed in a mixture of IPA (10.L) and water (10.L, Fresenius). A solution was not obtained, so further IPA (450 mL) and water (450 mL, Fresenius) were added, and the mixture was refluxed. The resulting solution was cooled to 68 °C and screened over 3.5 min through a 20 μm in-line filter into a vessel preheated to 65 °C. IPA(20.4 L) at 65 °C was added via the first vessel and in-line filter, and the resulting solution was stirred at 65 °C for 2 h. Crystallisation was evident after 20 min. The mixture was allowed to self-cool to ambient temperature overnight before filtering and washing the cake with a mixture (prescreened through a 20 μm membrane) of water (640 mL) and IPA (5.76 L). The cake was washed with IPA (6.4 L, prescreened) and MTBE (6.4 L, prescreened) and dried to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (2.865 kg, at 95.2% w/w, 3.52 mol, 72% yield). Spectroscopic analysis was in agreement with the reported data…………Ford, J. G.; McCabe, J. F.; O’Kearney-McMullan, A.; O’Keefe, P.; Pointon, S. M.; Powell, L.; Purdie, M.; Withnall, J. WO/2006/064217, 2006.
……………………….
SEE…………N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor
J Med Chem 2006, 49(22): 6465

…………..
1: Hannon RA, Finkelman RD, Clack G, Iacona RB, Rimmer M, Gossiel F, Baselga J, Eastell R. Effects of Src kinase inhibition by saracatinib (AZD0530) on bone turnover in advanced malignancy in a Phase I study. Bone. 2012 Jan 8. [Epub ahead of print] PubMed PMID: 22245630.
2: Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011 Oct;11(5):306-11. Epub 2011 May 3. PubMed PMID: 21729667; PubMed Central PMCID: PMC3222913.
3: Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, Macalpine K, Wang L, Wright JJ, Oza AM. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2011 Mar 12. [Epub ahead of print] PubMed PMID: 21400081.
4: Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, Stambuk H, Haque S, Pfister DG. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011 Jan;31(1):249-53. PubMed PMID: 21273606.
5: Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I, Walsh W, McIntosh L, Seymour L. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2010 Dec 18. [Epub ahead of print] PubMed PMID: 21170669.
6: Dalton RN, Chetty R, Stuart M, Iacona RB, Swaisland A. Effects of the Src inhibitor saracatinib (AZD0530) on renal function in healthy subjects. Anticancer Res. 2010 Jul;30(7):2935-42. PubMed PMID: 20683035.
7: Arcaroli JJ, Touban BM, Tan AC, Varella-Garcia M, Powell RW, Eckhardt SG, Elvin P, Gao D, Messersmith WA. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010 Aug 15;16(16):4165-77. Epub 2010 Aug 3. PubMed PMID: 20682712.
8: Morrow CJ, Ghattas M, Smith C, Bönisch H, Bryce RA, Hickinson DM, Green TP, Dive C. Src family kinase inhibitor Saracatinib (AZD0530) impairs oxaliplatin uptake in colorectal cancer cells and blocks organic cation transporters. Cancer Res. 2010 Jul 15;70(14):5931-41. Epub 2010 Jun 15. PubMed PMID: 20551056; PubMed Central PMCID: PMC2906706.
9: Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, Eastell R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010 Mar;25(3):463-71. PubMed PMID: 19775203.
10: Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009 Jun 15;15(12):4138-46. Epub 2009 Jun 9. PubMed PMID: 19509160.
11: Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, Slingerland JM. Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin Cancer Res. 2009 May 15;15(10):3396-405. PubMed PMID: 19451593.
12: Lara PN Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, Posadas E, Stadler W, Gandara DR. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009 Mar;20(3):179-84. PubMed PMID: 19396016; PubMed Central PMCID: PMC3225398.
13: Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Plé PA, Bermingham A, Holdgate GA, Ward WH, Hennequin LF, Davies BR, Costello GF. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009 Jun;3(3):248-61. Epub 2009 Feb 7. PubMed PMID: 19393585.
14: de Vries TJ, Mullender MG, van Duin MA, Semeins CM, James N, Green TP, Everts V, Klein-Nulend J. The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol Cancer Res. 2009 Apr;7(4):476-88. PubMed PMID: 19372577.
15: Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009 Jun;94(6):2199-203. Epub 2009 Mar 17. PubMed PMID: 19293266; PubMed Central PMCID: PMC2690419.
16: Purnell PR, Mack PC, Tepper CG, Evans CP, Green TP, Gumerlock PH, Lara PN, Gandara DR, Kung HJ, Gautschi O. The Src inhibitor AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells. J Thorac Oncol. 2009 Apr;4(4):448-54. PubMed PMID: 19240653; PubMed Central PMCID: PMC2716757.
17: Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, Ottmann OG. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. BMC Cancer. 2009 Feb 13;9:53. PubMed PMID: 19216789; PubMed Central PMCID: PMC2654659.
18: Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008 Oct 23;27(49):6365-75. Epub 2008 Aug 4. PubMed PMID: 18679417.
Src inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo.
Simpkins et al. Clin Cancer Res. 2012 Aug 15. PMID: 22896656.
Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo.
Liu et al. Int J Cancer. 2012 May 24. PMID: 22623106.
Common PIK3CA mutants and a novel 3′ UTR mutation are associated with increased sensitivity to saracatinib.
Arcaroli et al. Clin Cancer Res. 2012 May 1;18(9):2704-14. PMID: 22553375.
Phase I study of saracatinib (AZD0530) in combination with paclitaxel and/or carboplatin in patients with solid tumours.
Kaye et al. Br J Cancer. 2012 May 22;106(11):1728-34. PMID: 22531637.
Taiho’s Colon Cancer Drug Ups OS in Phase 3

TAS-102 (nonproprietary names: trifluridine and tipiracil hydrochloride)
Taiho’s Colon Cancer Drug Ups OS in Phase 3
Taiho Pharmaceutical Co. Ltd. announced results from its global Phase 3 RECOURSE trial on its oral combination anticancer drug TAS-102 in refractory metastatic colorectal cancer (mCRC). Read more…
FULL STORY
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.[1]
References
- “New Drug for Colorectal Cancer Shows Promise in Phase II Trial”. 28 Aug 2012.
- “Novel Drug TAS-102 Makes Headway in Refractory Colorectal Cancer”. 4 Oct 2011.
- “Phase II study of TAS-102 for pretreated metastatic colorectal cancer”. 29 Aug 2012.
- “A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA.”. Sep 2004.

Trifluridine
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
A Cochrane Systematic Review showed that trifluridine was a more effective treatment than idoxuridine or vidarabine, significantly increasing the relative number of successfully healed eyes in 14 days.[1]
References
- Wilhelmus KR (2010). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. Cochrane Database Syst Rev 12: CD002898. doi:10.1002/14651858.CD002898.pub4. PMID 21154352.
External links
- Costin D, Dogaru M, Popa A, Cijevschi I (2004). “Trifluridine therapy in herpetic in keratitis”. Rev Med Chir Soc Med Nat Iasi 108 (2): 409–12. PMID 15688823.
- Kuster P, Taravella M, Gelinas M, Stepp P (1998). “Delivery of trifluridine to human cornea and aqueous using collagen shields.”. CLAO J 24 (2): 122–4. PMID 9571274.
- O’Brien W, Taylor J (1991). “Therapeutic response of herpes simplex virus-induced corneal edema to trifluridine in combination with immunosuppressive agents.”. Invest Ophthalmol Vis Sci 32 (9): 2455–61. PMID 1907950.
- “Trifluridine Ophthalmic Solution, 1%” (PDF). Retrieved 2007-03-24.


Fig 4. Open Babel bond-line chemical structure with annotated hydrogens. Click to toggle size.
Spectrum Plot

Fig 5. 1H NMR spectrum of C10H11F3N2O5 in CDCL3 at 400 MHz

Tipiralacil, also known as TPI, is a thymidine phosphorylase inhibitor (TPI). Tipiracil is one of the active components in TAS-102, which is an anticancer drug candidate currently in clinical trials. TAS-102 consists of the cytotoxin Trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil. Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
183204-72-0 (Tipiracil -HCl); 183204-74-2(Tipiracil ).

|
References |
1: Peters GJ, Bijnsdorp IV. TAS-102: more than an antimetabolite. Lancet Oncol. 2012 Dec;13(12):e518-9. doi: 10.1016/S1470-2045(12)70426-6. PubMed PMID: 23182191.
2: Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, Tsuji Y, Moriwaki T, Esaki T, Hamada C, Tanase T, Ohtsu A. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012 Oct;13(10):993-1001. doi: 10.1016/S1470-2045(12)70345-5. Epub 2012 Aug 28. PubMed PMID: 22951287.
3: Sobrero A. TAS-102 in refractory colorectal cancer: caution is needed. Lancet Oncol. 2012 Oct;13(10):959-61. doi: 10.1016/S1470-2045(12)70376-5. Epub 2012 Aug 28. PubMed PMID: 22951286.
4: Doi T, Ohtsu A, Yoshino T, Boku N, Onozawa Y, Fukutomi A, Hironaka S, Koizumi W, Sasaki T. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012 Jul 24;107(3):429-34. doi: 10.1038/bjc.2012.274. Epub 2012 Jun 26. PubMed PMID: 22735906; PubMed Central PMCID: PMC3405214.
5: Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011 May;2(3):393-397. Epub 2011 Mar 21. PubMed PMID: 22977515; PubMed Central PMCID: PMC3440718.
6: Shintani M, Urano M, Takakuwa Y, Kuroda M, Kamoshida S. Immunohistochemical characterization of pyrimidine synthetic enzymes, thymidine kinase-1 and thymidylate synthase, in various types of cancer. Oncol Rep. 2010 May;23(5):1345-50. PubMed PMID: 20372850.
7: Temmink OH, Bijnsdorp IV, Prins HJ, Losekoot N, Adema AD, Smid K, Honeywell RJ, Ylstra B, Eijk PP, Fukushima M, Peters GJ. Trifluorothymidine resistance is associated with decreased thymidine kinase and equilibrative nucleoside transporter expression or increased secretory phospholipase A2. Mol Cancer Ther. 2010 Apr;9(4):1047-57. doi: 10.1158/1535-7163.MCT-09-0932. Epub 2010 Apr 6. PubMed PMID: 20371715.
8: Bijnsdorp IV, Kruyt FA, Fukushima M, Smid K, Gokoel S, Peters GJ. Molecular mechanism underlying the synergistic interaction between trifluorothymidine and the epidermal growth factor receptor inhibitor erlotinib in human colorectal cancer cell lines. Cancer Sci. 2010 Feb;101(2):440-7. doi: 10.1111/j.1349-7006.2009.01375.x. Epub 2009 Sep 29. PubMed PMID: 19886911.
9: Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010 May 15;126(10):2457-68. doi: 10.1002/ijc.24943. PubMed PMID: 19816940.
10: Bijnsdorp IV, Kruyt FA, Gokoel S, Fukushima M, Peters GJ. Synergistic interaction between trifluorothymidine and docetaxel is sequence dependent. Cancer Sci. 2008 Nov;99(11):2302-8. doi: 10.1111/j.1349-7006.2008.00963.x. Epub 2008 Oct 18. PubMed PMID: 18957056.
11: Overman MJ, Kopetz S, Varadhachary G, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff P, Xiong H, Abbruzzese JL. Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest. 2008 Oct;26(8):794-9. doi: 10.1080/07357900802087242. PubMed PMID: 18798063.
12: Overman MJ, Varadhachary G, Kopetz S, Thomas MB, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff PM, Xiong H, Abbruzzese JL. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008 Oct;26(5):445-54. doi: 10.1007/s10637-008-9142-3. Epub 2008 Jun 5. PubMed PMID: 18528634.
13: Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007 Jun;98(6):779-89. Epub 2007 Apr 18. Review. PubMed PMID: 17441963.
14: Temmink OH, Hoebe EK, van der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin-induced cytotoxicity to colorectal cancer cells. Br J Cancer. 2007 Jan 29;96(2):231-40. PubMed PMID: 17242697; PubMed Central PMCID: PMC2360012.
Buparlisib in phase 3 for Breast tumor; Hematological neoplasm; Solid tumor

Buparlisib
5-[2,6-Di(4-morpholinyl)-4-pyrimidinyl]-4-(trifluoromethyl)-2-pyridinamine.
5-[2,6-Di(morpholin-4-yl)pyrimidin-4-yl]-4-(trifluoromethyl)pyridin-2-amine
5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine
944396-07-0
Chemical Formula: C18H21F3N6O2
Mass: 410.16781
NVP-BKM-120, BKM-120;
Novartis AG phase 3 for breast cancer
Phosphoinositide 3-kinase inhibitor
Buparlisib, also known as BKM120, is an orally bioavailable specific oral inhibitor of the pan-class I phosphatidylinositol 3-kinase (PI3K) family of lipid kinases with potential antineoplastic activity. PI3K inhibitor BKM120 specifically inhibits class I PIK3 in the PI3K/AKT kinase (or protein kinase B) signaling pathway in an ATP-competitive manner, thereby inhibiting the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate and activation of the PI3K signaling pathway. This may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis. Dysregulated PI3K signaling may contribute to tumor resistance to a variety of antineoplastic agents.
NVP-BKM-120 is an oral selective phosphatidylinositol 3-kinase (PI3K) inhibitor in phase III clinical development at Novartis for the treatment of breast cancer in combination with fulvestrant in postmenopausal women with hormone receptor-positive HER2-negative locally advanced or metastatic breast cancer which progressed on or after aromatase inhibitor treatment.
Early clinical development at Novartis Oncology, a division of Novartis, is also ongoing for the treatment of solid tumors, advanced endometrial carcinoma, non-small cell lung cancer (NSCLC), bladder cancer, gastrointestinal stromal cancer and for the treatment of metastatic castration-resistant prostate cancer.
Novartis is conducting phase II clinical trials for the treatment of follicular lymphoma, diffuse large B-cell lymphoma, mantle cell lymphoma and squamous cell carcinoma of head and neck.
The University of Kansas is evaluating the compound in phase I clinical trials for the treatment of advanced colorectal cancer in combination with irinotecan, while additional phase I trials are ongoing at the Dana-Farber Cancer Institute for the treatment of renal cell carcinoma. The Dana-Farber Cancer Institute is also conducting phase II clinical trials for the oral treatment of recurrent glioblastoma and preclinical studies for the treatment of ovarian cancer. Novartis is also conducting early clinical studies for the treatment of metastatic melanoma
pyrimidine derivative 5-(2,6-Di- 4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine (Compound A, see below), its hydrates, its salts and hydrates and solvates of its salts, to said specific solid forms thereof, to pharmaceutical compositions containing said solid forms, to processes for the preparation of pharmaceutical compositions containing said solid forms, to methods of using said solid forms and to pharmaceutical compositions for the therapeutic treatment of warm-blooded animals, especially humans. Background of the invention
WO 2007/084786 (priority date: January 20, 2006) describes certain pyrimidine derivatives having PI3 inhibiting properties, their use as pharmaceuticals and manufacturing processes thereof. One pyrimidine derivative disclosed in WO 2007/084786 is the selective
phosphatidylinositol 3-kinase inhibitor compound 5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine, hereinafter referred to as “Compound A” or “the compound of formula A”.

Compound A is described in WO 2007/084786 in free form and as the hydrochloric acid salt. The manufacturing process for preparing Compound A is described in Example 10 of this document. The manufacturing processes described therein are, although suitable, regarded as disadvantageous for commercial production.
Due to the high potency of pyrimidine derivatives, in particular PI3K inhibitors, there is a need for improved manufacturing methods of such compounds. In particular there is a need to provide processes that fulfill one or more of the following criteria: scalable, safer; simpler; higher yielding and more economical when compared to known.
…………………………………….
WO 2007084786
http://www.google.com/patents/WO2007084786A1?cl=en
Example 10
Preparation of 4-(“trifluoromethyπ-5-(2,6-dimorpholmoρyrirnidin-4-yπpyridin-2- amine
c

[0388] To a slurry of 2-moφholino-4,6-dichloropyrimidine (prepared as in
Method 22, 2.0 g, 8.54 mmol) in NMP (14 mL), triethylamine (1.43 mL, 10.25 mmol) was added. The heterogeneous mixture was stirred for 15 minutes, then treated with morpholine (0.75 mL, 8.54 mmol). Upon refluxing at 85 0C under argon for 2 hours, the solution was cooled, then added to EtOAc (160 mL). The organic solution was washed with 25 mL of NaHCO3(sat.) (2 x), water (2 x) and brine, dried over Na2SO4, filtered and concentrated. The crude material was dissolved in 200 mL EtOAc and filtered through a SiO2 pad, further eluting with EtOAc, yielding 2.2 g (93%) of 2,4-dimorpholino-6- chloropyrimidine as an off-white solid. LCMS (m/z): 285.0 (MH+), 1H NMR (CDCl3): δ 5.86 (s, IH), 3.71-3.76(m, 12H), 3.52-3.56(m, 4H).
[0389] 4-(trifluoromethyl)-5-(2,6-dimoφholmopyrimidin-4-yl)pyridin-2-amine 8

[0390] Argon gas was bubbled through a heterogeneous mixture of 2,4- dimoφholino-6-chloropyrimidine (4.1 g, 14.3 mmol) and 4-(trifluoromethyl)-5-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)pyridm-2-amine (16.5 g, 57.3 mmol) in 1,2- dimethoxyethane and 2M Na2Cθ3 (3:1) for 20 minutes. 1,1′-
Bis(diphenylphosphino)ferrocene palladium (IT) chloride (292 mg, 0.36 mmol) was added and the high pressure glass vessel containing the mixture was sealed. The reaction mixture was then heated at 900C for 15 hours, cooled and diluted with EtOAc (300 mL). The organic solution was washed with 300 mL of a mixture of water: Na2Cθ3(sat.):NH4θH(conc.) = 5:4:1, then NH4Cl(sat), and brine (2x), dried over Na2SO4, filtered and concentrated. The crude material was purified by SiO2 chromatography (50- 90% EtOAc/hexanes with 0.1% TEA) resulting in 5.62 g (95%) of 4-(trifluoromethyl)-5- (2,6-dimorpholinopyrimidin-4-yl)pyridin-2-amine as an off-white solid.
LCMS (m/z): 411.3 (MH+);
1H NMR (CDCl3): δ 8.27 (s, IH), 6.78 (s, IH), 5.97 (s, IH), 4.77 (bs, 2H), 3.59-3.80(m, 12H), 3.58-3.61(m, 4H).
…………….
WO2012044727 or equi as below
http://www.google.com/patents/EP2621908A2?cl=en
Example 1: 4,4′-(6-Chloropyrimidine-2,4-diyl)di[morpholine] (3) U 2011/053808
63
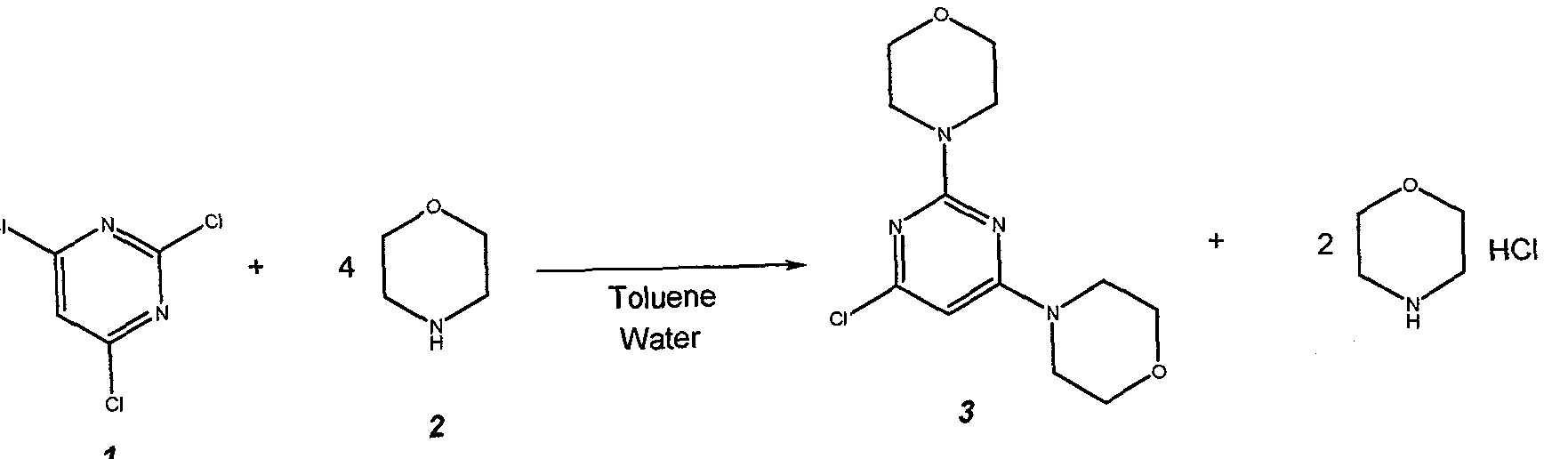
Prepare a solution of 22 g (0.12 mol) of 2,4,6-trichloropyrimidine 1 , in 95.2 g (110 mL) of toluene and charge it to the 25 mL addition funnel. Charge a nitrogen-flushed 500 mL round bottom 4- neck flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet with 62.7 g (63 mL, 0.72 mol) of morpholine 2, 95.2 g (110 mL) of toluene and 44 g (44 mL) of water. Add the toluene solution of 1 over 10 minutes. Heat the reaction mixture to 83 ± 3 °C. Stir at 83 ± 3 °C for 2 h. Check the progress of the reaction. Cool to 30 + 3 °C. Transfer the 2-phase mixture to a 1L separatory funnel.
Separate the phases. Wash the organic phase (top) twice with 200 mL (2 x 100 mL) of warm (30 °C) water. Separate the phases after each wash. Transfer the organic (top) phase back to the 500 mL reaction flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet. Stir and add 50.0 mL of 10.0 N aqueous hydrochloric acid solution. Heat the solution to 53 ± 3 °C and stir for 12 – 18 h. Check the progress of the reaction. Cool to 22 + 3 °C. Transfer the 2-phase mixture to a 1 L separatory funnel. Separate the phases. Transfer the aqueous (bottom) phase to a 500 mL round bottom 4-neck flask equipped with a cooling bath, thermocouple, addition funnel, pH probe, mechanical stirrer and nitrogen inlet / outlet. Stir and cool to 0 ± 3 °C. Add 85.0 g of 25% aqueous sodium hydroxide solution by drops over 30 minutes, maintaining a batch temperature of 10 ± 10 °C throughout the addition. Warm to 20 ± 3 °C and stir for 30 minutes. Isolate the solids by vacuum filtration. Wash the cake with 3 x 100 mL of water. Dry the solids (55°C, 30 mbar) for 24 hours to afford 30.9 g (91.9% yield) of 3 as a white crystalline solid.
Example 2:
4,4′-[6-(4>4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] (4)

Charge a nitrogen-flushed 2 L round bottom 4-neck flask that equipped with a condenser, heating mantle, thermocouple, rubber septum, mechanical stirrer and nitrogen inlet / outlet with 100.0 g (0.351 mol) of 4,4′-(6-chloropyrimidine -2,4-diyl)di[morpholine] 3 and 943 g (1200 mL) of acetonitrile. Stir and heat to 60 + 3 °C. Hold this solution at 60 + 3 °C for charge to batch. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 115.9 g (0.457 mol) of bis(pinacolato)- diboron, 51.7 g (0.527 mol) of potassium acetate, 12.9 g (0.014 mol) of tris(dibenzylideneacetone) – dipalladium(O), 7.9 g (0.029 mol) of tricyclohexylphosphine and 393 g (500 mL) of acetonitrile. Stir and heat the slurry to 84 ± 3 °C (reflux). Collect 00 mL of distillate. Transfer the warm 3 acetonitrile solution via peristaltic pump to the 3 L reactor containing the reaction mixture over 30 minutes and continue collecting distillate. Wash the 2 L flask and transfer lines with 79 g (100 mL) of acetonitrile and transfer the wash to the batch. Maintain distillation at 84 ± 3 °C and collect an additional 900 mL of distillate (batch volume ~ 1100 mL). Check the progress of the reaction 2 h from the start of the addition of 3. Cool the reaction mixture to 70 ± 3 °C and charge 693 g (800 mL) of toluene over 1-2 min. The batch will cool upon the addition of the toluene. Further cool the reaction mixture to 50 ± 3 °C. Charge to a clean 1 L flask, 347 g (400 mL) of toluene and warm it to 50 °C. This will be used as the cake wash. Filter the reaction mixture through a 15 g pad of Celite 545. Wash the filter cake with the warm (50 °C) toluene (400 mL) and collect this wash separately from the batch. This wash will be charged to the distillation residue later in the process. Transfer the filtrate back to the 3 L reactor. Concentrate the batch (25 °C to 40 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached.
Charge toluene cake wash held in reserve (~400 mL) and continue to concentrate the batch (37 °C to 43 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached. Check for complete removal of acetonitrile using the described Process Steering Control. Warm to 50 °C and stir for 15 min. Add 164 g (240 mL) of heptane over 30 minutes maintaining 50 °C throughout the addition. Stir the resulting suspension for 1 h. Cool the slurry to 23 ± 3 °C over 1 h and hold at this temperature for at least 1 h. Blanket the filtering funnel used for isolation of the product with nitrogen (to avoid moisture) and quickly filter the solids. Wash the filter cake twice with a mixture of 22 g (25 mL) of toluene and 51 g (75 mL) of heptane. Dry the solids at 50 °C, 35 mbar for 16 h to afford 4.4 g (72.7% corrected yield) of 4 as a sandy, beige solid. Example 3: 5-Bromo-4-(trifluoromethyl)pyridin-2-amine (4a)

4b 4a
Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 112.14 g (0.63 mol) of N-bromosuccinimide (NBS) and 645 g (725 mL) of tetrahydrofuran. Stir and cool the slurry to -5 ± 3 °C. Charge a nitrogen- flushed 1 L round bottom 4-neck flask that equipped with a thermocouple, mechanical stirrer and nitrogen inlet / outlet with 97.26 g (0.6 mol) of 2-amino-4-(trifluoromethyl)pyridine, 4b and 511 g (575 mL) of tetrahydrofuran. Stir to dissolve the 4b. Transfer the 4b solution to the addition funnel on the reactor and add the solution to the NBS slurry over 2 h maintaining an internal temperature of 0 ± 3 °C throughout the addition. Rinse the 1 L flask and addition funnel with 44 g (50 mL) of tetrahydrofuran and add the wash to the reaction mixture. Warm the solution to 20 + 3 °C over 30 minutes. Check for completeness of the reaction. Quench by charging a solution of 24.6 g of sodium thiosulfate pentahydrate dissolved in 475 mL of water over 10 minutes, maintaining a batch temperature of 20 ± 3 °C throughout the addition. Stir for 1 h after the quench. Concentrate (internal temp = 25 °C, 50 mbar) to remove tetrahydrofuran. Add 379 g (500 mL) of fert-butyl methyl ether. Stir and warm the resulting solution/suspension to 30 ± 3 °C and stir for 15 minutes. Separate the phases. Wash the extract four times with a solution of 32 g of sodium chloride dissolved in 768 g (768 mL) of water (4 x 200 mL per wash), separating the phases after each wash. Finally, wash the extract with 150 g (150 mL) of water. Separate the phases. Charge 152 g (200 mL) of terf-butyl methyl ether. Partially concentrate (57 ± 3 °C) to a volume of 350 mL. Cool to 50 °C and add 265 g (350 mL) of ferf-butyl methyl ether. Resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 265 g (350 mL) of fe/f-butyl methyl ether. Again, resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 103 g (150 mL) of terf-butyl methyl ether to raise the batch volume to 500 mL. Charge 1026 g (1500 mL) of heptane over 15 minutes maintaining 45 ± 3 °C throughout the addition. Slowly increase the vacuum and concentrate (internal temp = 40 °C to 50 °C) to a batch volume of 1000 mL. Release the vacuum and seed the batch. Resume the distillation, further increase the vacuum (slowly) and concentrate (internal temp = 25 °C to 40 °C) to a batch volume of 500 mL. Stir the resulting suspension at 0 °C for 30 min. Filter the solids. Wash the filter cake with 68 g (100 mL) of cold (0 °C) heptane (containing 30 ppm Octastat). Dry the solids (40 °C, 50 mbar) for 16 h to afford 109.8 g (78.0% yield) 4a as an orange solid.
Example 4: 5-(2,6-Di-4-morpholinyl^^yrimidinyl)-^trifluoromethylpyridin-2-ami^ (5)

Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 190.9 g (0.456 mol) of 4,4′-[6-(4,4,5,5- tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1,1′-bis(di-ferf-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of thf. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1 – 2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction. Cool to 22 ± 3 °C. Separate the phases. Partially concentrate the THF (25 °C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0. 25N aqueous N-acetyl-L- cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of 1 N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous HCI solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 ± 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
Alternative procedure:
Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 90.9 g (0.456 mol) of
4,4′[6(4,4,5,5tetramethyl1 ,3,2 dioxaborolan2yl)pyrimidine2,4diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1 ,1′-bis(di-fert-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of tetrahydrofuran. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1-2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction . Cool to 22 + 3 °C. Separate the phases. Partially concentrate the THF (25 C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0.125N aqueous N- acetyl-L-cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 + 3 °C C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 + 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N- acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous hydrochloric acid solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 + 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
…………..
Improved process for manufacturing 5-(2,6-di-4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine
Improved process for the preparation of buparlisib, an oral PI3K inhibitor Novartis is developing for the treatment of solid tumors, including breast cancer and hematological tumors. In January 2014, a phase III development was ongoing and Novartis expected to file for regulatory approval for breast cancer in 2015. Buparlisib was originally claimed in WO2007084786, protection for which expires in both the US and Europe in January 2027. Also see WO2012044727 for a more recent process case.
Burger, M.T.; Pecchi, S.; Wagman, A.; et al.
Discovery of BKM120, a pan class I PI3 kinase inhibitor in phase I/II clinical trials
240th ACS Natl Meet (August 22-26, Boston) 2010, Abst MEDI 489
Vu, A.T.; Morris, J.; Malhotra, S.V.
Efficient and improved synthesis of a PI3K inhibitor anticancer agent
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst ORGN 115
Tafenoquine…..GSK Launches Phase 3 Malaria Drug Trials

Tafenoquine
N-[2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinolin-8-yl]pentane-1,4-diamine
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”


Tafenoquine succinate, Etaquine, SB-252263, WR-238605
in phase 2
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
Tafenoquine is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[1][2]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax (and also Plasmodium ovale) that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[3]
Like primaquine, tafenoquine causes haemolysis in people with G-6-P deficiency.[1] Indeed the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[4]
Synonyms

………………..
US 4431807

Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.

Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
………………..
US 4617394

Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
……………….
US 6479660; WO 9713753

Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.

Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.

Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
//////////////////////
J Med Chem 1989,32(8),1728-32

Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.
- Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis 33 (12): 1968–74. doi:10.1086/324081. JSTOR 4482936.PMID 11700577.
- Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet 355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID 10885356.
- Elmes NJ, Nasveld PE, Kitchener SJ, Kocisko DA, Edstein MD (November 2008). “The efficacy and tolerability of three different regimens of tafenoquine versus primaquine for post-exposure prophylaxis of Plasmodium vivax malaria in the Southwest Pacific”. Transactions of the Royal Society of Tropical Medicine and Hygiene 102 (11): 1095–101.doi:10.1016/j.trstmh.2008.04.024. PMID 18541280.
- Nasvelda P, Kitchener S. (2005). “Treatment of acute vivax malaria with tafenoquine”. Trans R Soc Trop Med Hyg 99 (1): 2–5. doi:10.1016/j.trstmh.2004.01.013. PMID 15550254.
- Peters W (1999). “The evolution of tafenoquine–antimalarial for a new millennium?”. J R Soc Med 92 (7): 345–352.PMID 10615272.
- J Med Chem 1982,25(9),1094
|
8-3-2007
|
Methods and compositions for treating diseases associated with pathogenic proteins
|
|
|
12-6-2006
|
Process for the preparation of quinoline derivatives
|
|
|
3-14-2002
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
4-2-1998
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
4-18-1997
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
12-20-1996
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
12-15-1993
|
Use of interferon and a substance with an antimalarial activity for the treatment of malaria infections
|
|
|
10-15-1986
|
4-methyl-5-(unsubstituted and substituted phenoxy)-2,6-dimethoxy-8-(aminoalkylamino) quinolines
|
Binimetinib in phase 3 for for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600


Binimetinib
Array BioPharma Inc;PHASE 3 Cancer, ovary (serous)
Novartis PHASE 3 Melanoma
AGARRY-162
ARRY-438162
MEK-162
MEK-1 protein kinase inhibitor; MEK-2 protein kinase inhibitor
Liver injury; Melanoma; Noonan syndrome; Ovary tumor; Solid tumor
Growth factor-mediated proliferative signals are transmitted from the extracellular environment to the nucleus through several pathways, including the RAS/RAF/ MEK pathway. The RAS/RAF/MEK kinase signal transduction pathway is activated through initial extracellular binding and stimulation of tyrosine receptor kinases (RTKs) by their respective cognate ligands. Upon autophosphorylation of specific tyrosine residues in the cytosolic domain of RTKs, the Grb2-Sos complex translocates to the plasma membrane, and converts the inactive RAS’GDP to active RAS’GTP. The interaction between the Grb2 docking protein and the activated kinases or the phosphorylated receptor associated proteins is mediated by the Src Homology (SH2) domain of the signaling protein that recognizes specific phosphotyrosine sequences. RAS undergoes a conformational change upon guanosine 5 ‘-triphosphate (GTP) binding and causes the recruitment of RAF- 1 to the cytoplasmic membrane where it is phosphorylated by several kinases and simultaneous disphosphorylated at key residues by protein phosphatase-2B. Activated RAF phosphorylates the mitogen- activated protein kinase kinase (MEK) on two serine residues in the activation loop, which results in the activation of this protein kinase. MEK then phosphorylates and activates extracellular signal-regulated kinase (ERK), allowing its translocation to the nucleus where it phosphorylates transcriptional factors permitting the expression of a variety of genes.
The RAS/RAF/MEK signal transduction pathway is deregulated, often through mutations that result in ectopic protein activation, in roughly 1/3 of human cancers. This deregulation in turn results in a wide array of cellular changes that are integral to the etiology and maintenance of a cancerous phenotype including, but not limited to, the promotion of proliferation and evasion of apoptosis (Dhillon et al., Oncogene, 2007, 26: 3279-3290).
Accordingly, the development of small molecule inhibitors of key members of the RAS/ RAF/ MEK signal transduction pathway has been the subject of intense effort within the pharmaceutical industry and oncology community.
MEK is a major protein in the RAS/ RAF/ MEK pathway, which signals toward cell proliferation and survival, and frequently activated in tumors that have mutations in the RAS or RAF oncogenes or in growth receptor tyrosine kinases. MEK is a key player in the RAS/RAF/MEK pathway as it is downstream of RAS and RAF. Despite being only rarely mutated in cancer (Murugan et al., Cell Cycle, 2009, 8: 2122-2124; Sasaki et al., J. Thorac. Oncol., 2010, 5: 597-600), inhibitors of the MEK1 and MEK2 proteins have also been targeted for small molecule inhibition owing to their central position within the RAS/ RAF/ MEK signal transduction pathway signaling cascade (Fremin and Meloche, J. Hematol.
Oncol., 2010, 3:8). Recently a potent MEK inhibitor failed to demonstrate efficacy in clinical trials in patients with advanced non-small cell lung cancer (Haura et al., Clin. Cancer Res., 2010, 16: 2450-2457). The reason for failure in this trial is not clear.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (hereinafter, “Compound A”) is a benzimidazole compound that is a known potent and selective inhibitor of the MEK1 and MEK2 proteins, and useful in the treatment of hyperproliferative diseases, particularly cancer, in mammals. For example, in a recently published Phase I study of 28 patients suffering from unresectable, locally advanced or metastatic biliary cancer and who had received < 1 prior systemic therapy, oral Compound A treatment (60 mg twice daily) resulted in 1 complete regression, 1 partial regression and 11 stable disease diagnoses after at least 6 weeks of treatment (Finn et al., J. Clin. Oncol. 30, 2012 (Supplement 4, 2012 Gastrointestinal Cancers Symposium, Abstract No. 220). Compound A has also been demonstrated to be effective in the treatment of patients with either BRAFV600 or NRAS-mutant melanoma (Ascierto et al., J. Clin. Oncol. 30, 2012 (Supplement, 2012 ASCO Annual Meeting, Abstract No. 8511).
The compound, as well as a process for its preparation, is disclosed in PCT Pub. No. WO 03/077914
MEK-162, a potent, orally active MEK1/2 inhibitor, is in phase III clinical trials at Array BioPharma and licensee Novartis for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600. Phase III studies are also under way at Array BioPharma for the treatment of low grade serous carcinomas of the ovary, fallopian tube or primary peritoneum following at least one prior platinum-based chemotherapy regimen and no more than three lines of prior chemotherapy regimens. Novartis and Array BioPharma are also conducting phase II clinical studies for the treatment of locally advanced and unresectable or metastatic malignant cutaneous melanoma, harboring BRAFV600E mutations; in BRAF mutated melanoma in combination with AMG-479 and for the treatment of Noonan’s syndrome, and in non-small cell lung cancer harboring KRAS or EGFR mutation and in combination with erlotinib. MEK-162 is being evaluated in phase I/II as first line treatment of advanced biliary tract carcinoma and for the treatment of adult patients with mutant or wild-type RAS metastatic colorectal cancer. The product is in early clinical trials at Array Biopharma for the treatment of biliary cancer.
According to Array, MEK-162 may also provide broad therapeutic benefits in the treatment of chronic degenerative diseases. However, a phase II trial for the treatment of stable rheumatoid arthritis (RA) did not meet its primary endpoint. Based on these data, the company focused development of MEK-162 solely in oncology.
In 2010, MEK-162 was licensed to Novartis by Array BioPharma for worldwide development. In 2013, orphan drug designation was assigned in Japan for the treatment of malignant melanoma with NRAS or BRAF V600 mutation.
WO-2014063024 DEALS WITH Preparation, crystalline forms, and formulations comprising binimetinib. Binimetinib is a MEK-1/2 inhibitor originally claimed in WO03077914, which Array and Novartis are developing for the treatment of cancer, including melanoma, low-grade serous ovarian cancer, and other solid tumors, as well as Noonan syndrome hypertrophic cardiomyopathy and hepatic impairment. See also WO2014018725 for the most recent filing on the agent
//////////////////////////
WO 03/077914
http://www.google.com/patents/WO2003077914A1?cl=en
Schemes 1-4.
Scheme 1


Scheme la

Scheme 2

Scheme 3

17 18
Scheme 4

25
Scheme 5


General synthetic methods which may be referred to for preparing some of the compounds of the present invention are provided in PCT published application number WO 00/42022 (published July 20, 2000). The foregoing patent application is incorporated herein by reference in its entirety.
similar ie chloro instead of fluoro
Example 52

6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide (lOcc) Step A: 3-Chloro-2,4-difluoro-5-nitro-benzoic acid 2a
3-Chloro-2,4-difluoro-benzoic acid la (3.00 g, 15.6 mmol) is added to a stirred solution of concentrated H2SO4 (16 mL) and fuming nitric acid (0.85 mL, 20.3 mmol). After 3 hours a precipitate forms. The yellow slurry is poured onto ice water (100 mL). The aqueous mixture is extracted with diethyl ether (3x). The organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 3.50 g (95%) of clean desired product as a pale yellow solid.
Step B: 4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a
Ammonium hydroxide solution (6.88 g, -30% in water, 58.9 mmol) is added to a solution of 3-chloro-2,4-difluoro-5-nitro-benzoic acid 2a (3.5 g, 14.7 mmol) in water (16 mL) at 0 °C with stirring. Upon completion of the ammonium hydroxide addition the reaction mixture is warmed to room temperature. After 5 hours the reaction mixture is cooled to 0 °C and concentrated HCl is carefully added until the pH of the reaction mixture is near zero. The solid is collected by filtration and washed with water and diethyl ether. The solids are transferred to a round bottom flask as a solution in MeOH and EtOAc and concentrated under reduced pressure to give 2.96 g of a yellow solid. The filtrate is partitioned between diethyl ether and water and the organic layer is washed with brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure to give 0.65 g of product. Recovered a total of 3.61 g (104%) of pure desired product, that is carried forward without further purification.
Step C: 4~Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a
To a stirred solution of 4-amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a (3.61 g, 15.4 mmol) in THF (30 mL) and MeOH (10 mL), TMS diazomethane (9.23 mL, 2.0 M solution in hexanes, 18.5 mmol) is added. After completion of reaction, the reaction mixture is concentrated via rotary evaporation with acetic acid in the trap. The recovered oily solid is triturated with diethyl ether to provide 1.51 g of a yellow solid. The filtrate is concentrated and triturated with diethyl ether to give an additional 0.69 g of yellow solid. A total of 2.20 g (57%) of pure desired product is recovered.
Step D: 4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c
4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a (2.20 g, 8.84 mmol) is suspended in MeOH (9.4 mL) and aniline (3.22 mL, 35.4 mmol) is added. The reaction mixture is heated to reflux with stirring under a nitrogen atmosphere. After 19 hours, the reaction is complete. Distilled water (3.22 mL) is added to the reaction mixture and refluxing is continued for one hour. The reaction mixture is cooled to 0 °C in an ice bath for 20 minutes. The reaction mixture is filtered and washed with 3:10 distilled water/MeOH (65 mL total) and then with MeOH. The solid is dissolved with CH2C12 and concentrated under reduced pressure to give 2.40 g (84%) of pure desired product. MS APCI (-) m/z 320.3 (M-l) detected.
Step E: 4, 5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b
4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c (0.50 g, 1.55 mmol) is dissolved into 2:1 EtOH/MeOH (15.5 mL). Saturated aqueous NH4C1 (15 mL), Zn powder (1.02 g, 15.6 mmol), and THF (10 mL) are added. After stirring for 20 hours, the reaction mixture is diluted with CH C12/THF and water. The organic layer is washed with water (3x). The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure. The solids are triturated with ether to give 0.32 g (70%) clean desired product. Step F: 7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c
4,5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b (0.32 g, 1.09 mmol) and formamidine acetate (72 mg, 1.64 mmol) in EtOH (36 mL) are heated, with stirring, to 80 °C. After 44 hours, the reaction mixture is cooled to room temperature and diluted with EtOAc and washed with water (3x), saturated NaHCO3, and brine. The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 0.33 g (99%) clean desired product as a solid. MS APCI (+) m/z 302.3 (M+l) detected.
Step G: 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g
7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c (0.327 g, 1.08 mmol) is dissolved into DMF (16 mL) and NBS (0.193 g, 1.08 mmol) is added. After one hour, the reaction mixture is quenched by the addition of saturated aqueous NaHSO3. The reaction mixture is then partitioned between EtOAc/THF and water. The organic layer is washed with water and brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure. The recovered solid is triturated with ether to give 0.225 g (54%) pure desired product. MS ESI (+) m/z 382, 384 (M+, Br pattern) detected.
Step H: 6-(4-Bromo-2-chloro-phenylamino)- 7 -chloro-3H-benzoimidazole-5 -carboxylic acid methyl ester lOdd 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g (0.225 g, 0.591 mmol) is dissolved in DMF (2 mL) and NCS (79 mg, 0.591 mmol) is added. After the NCS is in solution concentrated HCl (0.005 mL, 0.059 mmol) is added. After 2 hours, sodium bicarbonate, water and NaHSO3 are added to the reaction mixture. Solids are filtered and washed with water and ether to give 0.141 g (57%) of clean desired product as a tan solid. MS APCI (-) m/z 414, 416 (M-, Br pattern) detected.
Step I: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester lOdd (0.141 g, 0.34 mmol), potassium carbonate (0.141 g, 1.02 mmol), and iodomethane (0.063 mL, 1.02 mmol) are dissolved in dimethylformamide (3 mL). After 20 hours, the reaction mixture is diluted with EtOAc and washed with water (3x), potassium carbonate, and brine. The organic layer is dried (Na2SO4) and concentrated to a brown oil. The N3 and Nl alkylated regioisomers are separated by flash chromatography (EtOAc). The recovery of the N3 alkylated regioisomer is 20.4 mg (28%). MS ESI (+) m/z 428, 430 (M+, Br pattern) detected.
Step J: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10 ff
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee (21 mg, 0.048 mmol) is dissolved into 2:1 THF/water (1.2 mL) and NaOH (0.190 mL, 1.0 M aqueous solution, 0.190 mmol) is added. After stirring for 4 hours the reaction is diluted with water and acidified to pH 2 by addition of 1.0 M HCl. The mixture is then extracted with 3:1 EtOAc/THF (3x), dried (Na2SO ) and concentrated to give quantitative yield of desired prodcut as a white solid. MS APCI (+) m/z 414, 416 (M+, Br pattern) detected.
Step K: 6-(4-Bromo-2’chloro-phenylamino)- 7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy) -amide lOgg
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid lOff (32 mg, 0.077 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.010 mL, 0.092 mmol), HOBt (13 mg, 0.093 mmol), triethylamine (0.011 mL, 0.077 mmol), and EDCI (19 mg, 0.10 mmol) are dissolved into dimethylformamide (1.0 mL) and allowed to stir under a nitrogen atmosphere at room temperature for 24 hours. The reaction mixture is diluted with EtOAc, washed with water (3x), 10% potassium carbonate (2x), saturated ammonium chloride, brine, dried (Na2SO4), and concentrated under reduced pressure to give 39 mg of 85% pure material. MS APCI (-) m/z 497, 501 (M-, Br pattern) detected.
Step L: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide lOcc
Hydrochloric acid (0.78 mL, 1.0 M aqueous solution, 0.78 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H- benzoimidazole-5-carboxylic acid lOgg (2-vinyloxy-ethoxy)-amide (39 mg, 0.078 mmol) in MeOH (1 mL). After one hour, the reaction mixture is neutralized to pH 7 and concentrated under reduced pressure. The solids are dissolved in EtOAc, washed with brine, dried (Na SO4), and concentrated under reduced pressure. Flash chromatography (20:1 CH2Cl2/MeOH) provides 9 mg (23%) of pure product: MS APCI (+) m/z 473, 475 (M+, Br pattern) detected; 1H NMR (400 MHz, CDC13) δ 8.30 (s, IH), 8.08 (s, IH), 7.57
(d, IH), 7.15 (dd, IH), 6.21 (d, IH), 3.97 (s, 3H) 3.86 (m, 2H), 3.57 (m, 2H).
actual is below
Example 18
The following compounds are prepared by methods similar to those described in
Example 10 by using methyl ester 8d and the appropriate alkylating agent (Step A) and
the appropriate hydroxylamine (Step C):

/////////////////////
COMPD A
Example 1. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-

Compound 1 Compound 3
In an inertized (N2) reaction vessel at internal temperature 20°C and under exclusion of humidity and air, Compound 1 (1.0 eq.) and Compound 2 (1.2 eq.) are reacted in the presence of cesium carbonate (2.4 eq.), tris(dibenzylidenaceton) dipalladium(O) (0.035 eq.) and Xantphos (0.07 eq.) in a mixture of toluene and 1 ,4-dioxane at internal temperature of 99°C. After 8 hours, the mixture is cooled to internal temperature of 60°C.
Subsequently, dimethylformamide (DMF), filter aid (CEFOK) and activated charcoal (EKNS) are added, and the mixture is stirred and cooled to internal temperature of 35 °C. The solids are filtered off and washed with a mixture of dimethylformamide and toluene. To the filtrate, which contains the product Compound 3, is introduced at internal temperature of
25 °C hydrogen chloride gas (CLC) whereupon the HQ salt of Compound 3 crystallizes. The palladium residue mainly remains in solution. After warming to 60 °C and cooling to 0°C, the solids are filtered using a centrifuge and are washed with a mixture of toluene and dimethylformamide.
The damp Compound 3 HC1 salt is charged to a reactor (equipped with pH probe) together with dimethylformamide and is heated to 60°C. By adding a 4 wt% of aqueous tripotassium phosphate solution, the pH is adjusted to a pH range of 6.8-7.6 (with a target of pH 7.2) while Compound 3 crystallizes as free base. After cooling to 22°C and stirring, the solids are filtered using a centrifuge and are washed with drinking water. The moist solids are dried at 50 °C under vacuum to give dry, crude Compound 3.
In order to remove residual palladium, dry, crude Compound 3 is dissolved in dimethylformamide at internal temperature of 60°C and stirred together with Smopex-234 (commercially available from Johnson Matthey) and activated charcoal for 90 minutes. The solids are filtered off at internal temperature of 60°C and are washed with
dimethylformamide. To the filtrate are added drinking water and Compound 3 seed crystals. More drinking water is added while Compound 3 crystallizes. After cooling to internal temperature of 20 °C, the solids are filtered using a centrifuge and are washed with a mixture of deionized water and dimethylformamide and with deionized water. The moist solids are dried at 50°C under vacuum, providing 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3).
Example 2. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide
A. “One-pot” Synthesis

Compound 3 Intermediate 1
t-Bu-O. /\ ^ H2
(Compound 4)

Compound 5
In an inertized reaction vessel at internal temperature 20-25 °C under nitrogen, 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3, 1.0 eq.) is added to a mixture of DMF and THF. To this slurry, a solution of potassium trimethylsilanolate (1.05 eq.) in THF is added to the mixture at internal temperature of 25 °C over a period of about 40 minutes, and the resulting mixture is stirred for about 1 hour, providing a potassium salt solution of Intermediate 1. A THF/methanol mixture is then sequentially distilled off from the mixture at 85-120°C during about 2 hours.
The potassium salt solution is then added to a suspension of CDI (1.25 eq.) and imidazole hydrochloride (1.40 eq.) in THF at internal temperature of 25 °C over a period of about 1 hour. The resulting mixture is then stirred for approximately 1 hour at 50°C, and the following imidazolide intermediate

The imidazolide intermediate is not further isolated.
Subsequently, 1.2 eq. of 0-(2-tert-butoxyethyl)hydroxylamine (Compound 4, CAS No. 1023742-13-3, available from suppliers such as Huhu Technology, Inc.®) is added over a period of about 30 minutes at 50°C and stirred for 1.5 hours. Demineralized water is then added at 50°C, producing a precipitate. After cooling to 20°C and stirring for about 3-16 hours, the slurry is filtered off, washed with THF/ demineralized water (1 :2) in 2 portions and with demineralized water in three portions, and dried at 50°C / <70 mbar for about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
B. A synthesis method with isolation of the intermediate of step a) from the reaction mixture of step a) prior to the reaction of step b)
Alternatively, 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5 -carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) can be made by the synthesis method as shown below. Compound 3, which is a methyl ester, is first converted to a carboxylic acid, which is then isolated by a crystallization to form Compound
6. Compound 6 is then coupled with Compound 4 to form Compound 5 as monohydrate.
The crystallization step in this method removes starting materials such as Compound 1, process impurities, and the dba ligand from the prior catalyst before the coupling reaction with Compound 4, and at the same time maintains the overall yield of the synthesis.


6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-memy acid In an inertized (N2) reaction vessel at internal temperature of 60°C, Compound 3 (1.0 eq.) is dissolved in DMF and stirred with a fiber, which is sold under the trademark
SMOPEX 234, and activated charcoal for the removal of palladium to not more than 100 ppm. The fiber and activated charcoal are removed by filtration at 60°C and washed with DMF.
The filtrate (containing Compound 3) is transferred to a second inertized (N2) reaction vessel and cooled to an internal temperature of 30°C. A thin suspension can form at this point of time. 30% sodium hydroxide (1.1 eq.) and water (for rinsing) are added, and the resulting reaction mixture is vigorously stirred for 3 hours at an internal temperature of 30 °C. The methyl ester is saponified. Conversion is checked by an IPC (HPLC). As soon as the IPC criterion is met, a filter aid, which is sold under the trademark HYFLO, is added. The mixture is stirred for 15 minutes and then filtered at 30°C via a plate filter and polish filter to a third reaction inertized (N2) vessel.
An aqueous HC1 solution 7.5 % is added to the clear filtrate in the third vessel at an internal temperature of 30 °C until a pH value of 8 is reached. Then the solution is seeded at an internal temperature of 30°C with Compound 6, and an aqueous HC1 solution 7.5 % is added under vigorous stirring until a pH value of pH 2.8 is reached. The product gradually crystalizes. The suspension is cooled over 60 min to an internal temperature of 25 °C and
water is added. The suspension is stirred for at least 4 hours at an internal temperature of 25°C.
The resulting solid is collected by centrifugation or filtration. The filter cake is first washed with DMF/water 1 :1 (w/w) and then with water, discharged and dried in a vacuum at 50°C. The water content is controlled by IPC. The crystalline product Compound 6 is discharged as soon as the IPC criterion is met.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid- (2-tert-butoxyethoxy) – amide
An inertized (N2) reaction vessel is charged with Compound 6 (1.0 eq.), DMF, and
THF at room temperature. The suspension is heated to 25 °C under stirring with flow of nitrogen. After CDI (1.13 eq.) is added, the suspension can get thinner and slight evolution of gases can be observed. After the suspension finally becomes a solution, it is then monitored by IPC (HPLC).
As soon as the IPC (HPLC) criterion is met, the reaction mixture is heated to 50°C over 20 minutes and imidazole hydrochloride (0.3 eq.) is added, forming a solution of
Intermediate 2.
To the solution of Intermediate 2, Compound 4 (1.3 eq.) is added over 60 minutes at internal temperature of 50°C under stirring at a speed of 300 rpm with flow of nitrogen. As soon as the IPC (HPLC) criterion is met, the mixture is cooled to 20-25 °C over 30 minutes. The mixture is then stored at ambient temperature overnight under nitrogen without stirring. DMF is added to the mixture followed by heating it to 50 °C over 30 minutes. Complete conversion of Intermediate 2 to Compound 5 is confirmed by IPC (HPLC).
Water is added to the mixture at internal temperature of 50 °C over 20 minutes. Then the solution is seeded with Compound 5. After stirring at 50 °C for 60 minutes, more water is added to the suspension at 50 °C over 90 minutes. After vigorous stirring, the suspension is cooled to 20 °C over 2 hours and filtered. The filter cake is washed twice with THF/water (v/v: 1 :2) at 20 °C, and twice with water at 20 °C. Finally, the filter cake is dried at 50 °C under vacuum to provide 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
Example 3. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A)

Compound 5 Compound A
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) monohydrate is added in 3 portions to a premixed solution of Acetonitrile and excess Phosphoric acid (85 % aqueous solution) at internal temperature 20-25 °C. After stirring for about 15 minutes, the suspension is heated to internal temperature 50-53 °C. The suspension is maintained at this temperature for 6 hours, cooled to internal temperature 20-25 °C. The mixture is then heated to internal temperature 35-37°C and diluted with Ethanol- Water (3 :1 v/v). EKNS and CEFOK are added, the reaction mixture is stirred approximately 15 minutes and filtered over a funnel coated with CEFOK. The filtrate is cooled to approximately 30°C. 3 N aqueous potassium hydroxide (ΚΟΗ) is added to the cooled filtrate over a period of 90 minutes until a pH- value of about 8.1 is reached. The suspension is heated to internal temperature 60-63 °C, stirred at this temperature for a period of about 2 hours, cooled to 20-23 °C over a period of about 45 minutes, filtered over a funnel, and dried at 50°C pressure <100 mbar over a period of about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) as a white powder.
Example 4. Preparation of Crystallized 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) In a dry vessel at room temperature, Compound A is added to a premixed solvent solution of methanol/THF/water (35/35/30 w/w). The suspension is heated to internal temperature 53-55°C, and the resulting solution is hot filtered by deep and membrane filtration (via a paper filter and PTFE membrane) at internal temperature 53-56°C. The clear solution is stirred and cooled to 47-48°C, and the seed crystals suspension (i.e., seed crystals of crystallized Compound A in water, 10% m/m) is added (0.2 to 0.5% of crystallized Compound A expected yield mass). After about 20 minutes, water is slowly added within 25 hours (33.3% within 15 hours and 66.6% within 10 hours with at least 10 minute stirring after addition of water) to obtain a final ratio of methanol THF/water (20/20/60 w/w). After the water is added, the suspension is cooled down to internal temperature 3-5 °C within 10 hours and stirred for 0.5 hours. The white suspension is filtered over a sinter glass nutsche (75 ml, diameter = 6 cm, pore 3) suction filter and washed once with ice cold methanol/THF/water (15/15/70 w/w at 2-4 °C), and two times with ice cold water (2-4 °C). Drying takes place in a vacuum oven dryer at 20°C for 10 hours, and then at 40°C for 10 hours, and then at 60°C for at least 12 hours with pressure < lOmbar, providing crystallized Compound A.
Example 5. Pharmaceutical Composition
Crystallized Compound A is formulated as indicated in Table 1 :
Table 1

* The weight of the drug substance is taken with reference to the dried substance (100%) on the basis of assayed value. The difference in weight is adjusted by the amount of lactose monohydrate.
** The Opadry II is combined with the sterile water to make a 12% w/w Opadry II (85F) film coat suspension, which is then sprayed onto the core tablet.
*** Removed during processing
Upon mixing of the tablet core components, the pharmaceutical composition is converted into a tablet form by direct compression. The formed tablet may be further coated with the tablet coating provided above.
Selumetinib.司美替尼 .. phase III trial in patients with KRAS mutation-positive NSCLC

Selumetinib司美替尼
6-(4-bromo-2-chloroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide
5-(4-Bromo-2-chlorophenylamino)-4-fluoro-1-methyl-1H-benzimidazole-6-carbohydroxamic acid 2-hydroxyethyl ester
6-(4-bromo-2-chloro- phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide
943332-08-9 (sulfate (1:1) salt) IS THE DRUG
Non-small-cell lung cancer (NSCLC) is the most common type of lung cancer. In October, AstraZeneca began a phase III trial of selumetinib in patients with KRAS mutation-positive NSCLC. AstraZeneca has also partnered with Roche Molecular Systems to develop a device to detect these mutations.
Selumetinib (AZD6244) is a drug being investigated for the treatment of various types of cancer, for example non-small cell lung cancer (NSCLC).
The gene BRAF is part of the MAPK/ERK pathway, a chain of proteins in cells that communicates input from growth factors. Activating mutations in the BRAF gene, primarily V600E (meaning that the amino acid valine in position 600 is replaced by glutamic acid), are associated with lower survival rates in patients with papillary thyroid cancer. Another type of mutation that leads to undue activation of this pathway occurs in the gene KRAS and is found in NSCLC. A possibility of reducing the activity of the MAPK/ERK pathway is to block the enzyme MAPK kinase (MEK), immediately downstream of BRAF, with the drug selumetinib. More specifically, selumetinib blocks the subtypes MEK1 and MEK2 of this enzyme.[1]
Selumetinib is a novel, selective, non-ATP-competitive inhibitor of MEK1/2 currently in phase III clinical development at AstraZeneca for the oral treatment of non-small lung cancer with KRAS mutation. Additional phase II trials are under way at both AstraZeneca and Array BioPharma for the treatment of other oncological indications, including colorectal cancer, thyroid cancer and malignant melanoma. AstraZeneca is conducting phase I/II clinical trials for the treatment of Kaposi’s sarcoma (AIDS-related) in combination with highly active anti-retroviral therapy (HAART). Also, phase I trials are ongoing at the companies targeting several solid tumors, including skin, pancreatic, colon, lung and breast tumors. The National Cancer Institute (NCI) is also evaluating selumetinib for the treatment of thyroid cancer, ovary cancer, myeloid leukemia, glioma, multiple myeloma, metastatic uveal melanoma, sarcoma, pancreatic cancer, plexiform neurofibromas and for the treatment of recurrent or persistent endometrial cancer. Additional early clinical trials are under way at the Massachusetts General Hospital for the treatment of cancers with BRAF mutations. No recent development has been reported for phase II clinical trials for the treatment of metastatic pancreatic cancer.

In addition to thyroid cancer, BRAF-activating mutations are prevalent in melanoma (up to 59%), colorectal cancer (5–22%), serousovarian cancer (up to 30%), and several other tumor types.[2]
KRAS mutations appear in 20 to 30% of NSCLC cases and about 40% of colorectal cancer.[1]
. The National Cancer Institute (NCI) is also evaluating selumetinib for the treatment of thyroid cancer, ovary cancer, myeloid leukemia, glioma, multiple myeloma, metastatic uveal melanoma, sarcoma, pancreatic cancer, plexiform neurofibromas and for the treatment of recurrent or persistent endometrial cancer. Additional early clinical trials are under way at the Massachusetts General Hospital for the treatment of cancers with BRAF mutations. No recent development has been reported for phase II clinical trials for the treatment of metastatic pancreatic cancer.
A Phase II clinical trial about selumetinib in NSCLC has been completed in September 2011;[3] one about cancers with BRAF mutations is ongoing as of June 2012.[4]
Selumetinib appears to efficiently target cancers with overactivation of MEK and associated cell signaling pathways. According to laboratory studies, selumetinib has an effect on human tumors at nanomolar concentrations. Potential advantages of selumetinib over marketed therapies include improved efficacy linked to a novel mechanism and ease of use based on the drug candidate’s oral formulation.
In 2013, AstraZeneca acquired exclusive worldwide rights to selumetinib from Array BioPharma.
AZD6244 (Selumetinib)
6-(4-Bromo-2- chloro-ρhenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide, or “Compound 1”, is exemplified in WO 03/077914 and possesses the following structural formula:


…………………………..
http://www.google.com/patents/US20030232869
Example 10

6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (29c)
Step A. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a and 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-1-methyl-1H-benzoimidazole-5-carboxylic acid methyl ester
A solution of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3H-benzoimidazole-5-carboxylic acid methyl ester 8b (150 mg, 0.38 mmol), iodomethane (28 μL, 0.45 mmol) and potassium carbonate (78 mg, 0.56 mmol) in dimethylformamide (1.5 mL) is stirred at 75° C. for one hour. The reaction mixture is diluted with ethyl acetate, washed with saturated aqueous potassium carbonate (2×), brine, and dried (Na2SO4). Flash column chromatography (20:1 methylene chloride/ethyl acetate) provides 56 mg (36%) of the more mobile 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a as a white solid. 19F NMR (376 MHz, CD3OD)-133.5 (s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected. Also isolated is 54 mg (35%) of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-1-methyl-1H-benzoimidazole-5-carboxylic acid methyl ester as a white solid. 19F NMR (376 MHz, CD3OD)-139.9 (s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected.
Step B. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid 10c
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a (56 mg, 0.14 mmol) is dissolved into 2:1 THF/water (3 mL) and NaOH (0.55 mL, 1.0 M aqueous solution, 0.55 mmol) is added. After stirring for two hours the reaction is reduced to one quarter initial volume via rotary evaporation and the remainder diluted to 50 mL with water. The aqueous solution is acidified to pH 2 by the addition of 1.0 M aqueous HCl and extracted with 1:1 tetrahydrofuran/ethyl acetate (3×), dried (Na2SO4) and concentrated under reduced pressure to provide 43 mg (79%) pure carboxylic acid as an off white solid. MS ESI (+) m/z 397, 398 (M+, Br pattern) detected.
Step C: 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid 10c (2.00 g, 5.0 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.776 g, 7.5 mmol), HOBt (0.88 g, 6.5 mmol), triethylamine (1.61 mL, 2.3 mmol) and EDCI (1.3 g, 6.5 mmol) are dissolved in dimethylformamide (52 mL) and stirred at room temperature for 48 hours. The reaction mixture is diluted with ethyl acetate, washed with water (3×), saturated potassium carbonate (2×), saturated ammonium chloride (2×), brine, dried (Na2SO4) and concentrated under reduced pressure to an off-white solid. Trituration of the solid with diethyl ether provides 2.18 g (90%) desired product as an off-white solid. MS ESI (+) m/z 483, 485 (M+ Br pattern) detected.
Step D: 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide 29c
Hydrochloric acid (14 mL, 1.0 M aqueous solution, 14 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a (2.18 g, 4.50 mmol) in ethanol (50 mL) and the reaction mixture allowed to stir for 24 hours. The reaction mixture is concentrated to dryness by rotary evaporation and the solids partitioned between 3:1 ethyl acetate/tetrahydrofuran and saturated potassium carbonate. The aqueous phase is extracted with 3:1 ethyl acetate/tetrahydrofuran (3×), the combined organics dried (Na2SO4), and concentrated to provide 2.11 g (100%) 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide as an off-white solid. MS ESI (+) m/z 457, 459 (M+, Br pattern) detected. 1H NMR (400 MHz, MeOH-d4) δ8.26 (s, 1H), 7.78 (s, 1H), 7.57 (d, 1H), 7.24 (dd, 1H), 6.40 (dd, 1H), 3.86 (s, 3H), 3.79 (m, 2H), 3.49 (m, 2H). 19F NMR (376 MHz, MeOH-d4)-133.68 (s).
…………
http://www.google.com/patents/WO2003077914A1?cl=en
Scheme 1
Scheme la
Scheme 2
Scheme 3
17 18
Scheme 4
25
Scheme 5
Example 1 and in this Example 9 by using the appropriate carboxylic acid and the appropriate hydroxylamine:

Example 10

6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (29c)Step A. 6-(4-Bromo-2-chloro-phenylamino)- 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester 9a and 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-l- methyl-lH-benzoimidazole-5-carboxylic acid methyl ester
A solution of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3H-benzoimidazole-5-
carboxylic acid methyl ester 8b (150 mg, 0.38 mmol), iodomethane (28 μL, 0.45 mmol)
and potassium carbonate (78 mg, 0.56 mmol) in dimethylformamide (1.5 mL) is stirred at
75 °C for one hour. The reaction mixture is diluted with ethyl acetate, washed with saturated aqueous potassium carbonate (2x), brine, and dried (Na SO ). Flash column chromatography (20:1 methylene chloride/ethyl acetate) provides 56 mg (36%) of the
more mobile 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-
5-carboxylic acid methyl ester 9a as a white solid. 19F NMR (376 MHz, CD3OD) -133.5
(s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected. Also isolated is 54 mg (35%)
of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-l-methyl-lH-benzoimidazole-5- carboxylic acid methyl ester as a white solid. 19F NMR (376 MHz, CD3OD) -139.9 (s).
MS APCI (+) m/z 412, 414 (M+, Br pattern) detected.
Step B. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10c
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester 9a (56 mg, 0.14 mmol) is dissolved into 2:1 THF/water (3 mL ) and NaOH (0.55 mL, 1.0 M aqueous solution, 0.55 mmol) is added. After stirring for two hours the reaction is reduced to one quarter initial volume via rotary evaporation and the remainder diluted to 50 mL with water. The aqueous solution is acidified to pH 2 by the addition of 1.0 M aqueous HCl and extracted with 1 : 1 tetrahydrofuran/ethyl acetate (3x), dried (Na2SO4) and concentrated under reduced pressure to provide 43 mg (79%) pure carboxylic acid as an off white solid. MS ESI (+) m/z 397, 398 (M+, Br pattern) detected.
Step C: 6-(4-Bromo-2-chloro-phenylamino)~ 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy)-amide 29a
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10c (2.00 g, 5.0 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.776 g, 7.5 mmol), HOBt (0.88 g, 6.5 mmol), triethylamine (1.61 mL, 2.3 mmol) and EDCI (1.3 g, 6.5 mmol) are dissolved in dimethylformamide (52 mL) and stirred at room temperature for 48 hours. The reaction mixture is diluted with ethyl acetate, washed with water (3x), saturated potassium carbonate (2x), saturated ammonium chloride (2x), brine, dried (Na2SO4) and concentrated under reduced pressure to an off-white solid. Trituration of the solid with diethyl ether provides 2.18 g (90%) desired product as an off- white solid. MS ESI (+) m/z 483, 485 (M+ Br pattern) detected.
Step D: 6-(4-Bromo-2-chloro-phenylamino)- 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy) -amide 29c
Hydrochloric acid (14 mL, 1.0 M aqueous solution, 14 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a (2.18 g, 4.50 mmol) in ethanol (50 mL) and the reaction mixture allowed to stir for 24 hours. The reaction mixture is concentrated to dryness by rotary evaporation and the solids partitioned between 3:1 ethyl acetate/tefrahydrofuran and saturated potassium carbonate. The aqueous phase is extracted with 3:1 ethyl acetate/tefrahydrofuran (3x), the combined organics dried (Na SO4), and concentrated to provide 2.11 g (100%) 6-(4-bromo-2-chloro- phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide as an off-white solid. MS ESI (+) m/z 457, 459 (M+, Br pattern) detected. 1H NMR (400 MHz, MeOH-c^) δ 8.26 (s, IH), 7.78 (s, IH), 7.57 (d, IH), 7.24 (dd, IH), 6.40 (dd, IH), 3.86 (s, 3H), 3.79 (m, 2H), 3.49 (m, 2H). 19F NMR (376 MHz, MeOH-d4) -133.68 (s).
………………
http://www.google.com/patents/EP1968948A2?cl=en
Example 1
Preparation of the Hydrogen sulfate salt of Compound 1

[0076] To a stirred suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fiuoro-3- methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (100 g, 0.206 mol) (obtainable as described in Example 10 of WO 03/077914, which is incorporated herein by reference and as described below) in 2-butanone (680 mL) and water (115 mL) at 0-5 0C was added sulfuric acid (12.3 mL, 0.226 mol) followed by water (5 mL) maintaining a temperature of 10 °C or lower. The stirred mixture was heated to 65 0C and held for 30 minutes before filtering to remove any extraneous matter. The filter was washed with a mixture of 2-butanone (85 mL) and water (15 mL). The combined filtrates were heated to 72 0C before adding 2-butanone (500 mL) maintaining a temperature of between 60-72 0C. The resulting mixture was distilled at atmospheric pressure (approximate distillation temperature 73-74°C) until 500 mL of distillate had been collected.
[0077] A second aliquot of 2-butanone (500 mL) was added, maintaining the temperature of the mixture above 70 0C. The resulting mixture was distilled again until 250 mL of distillate had collected. The mixture was cooled to 0-5 0C over approximately 1 hour. The resulting slurry was filtered, washed with 2-butanone (240 mL) and dried under reduced pressure at 50 0C, until a constant weight was achieved, to give 6-(4-bromo-2-chloro- phenylamino)-7-fiuoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)- amide hydrogen sulfate (103.5 g, 0.186 mol, 90% yield) as an off white crystalline solid.1H NMR (400 MHz, D6 DMSO) δ 3.58 (2H, t, CH2OH), 3.89 (2H, t, CH2ON), 3.99 (3H, s, CH3), 6.47 (IH, dd, ArH), 7.29 (IH, dd, ArH), 7.63 (IH, d, ArH), 7.91 (IH, s, ArH), 7.96 (3H, br, ROH, NH, SOH), 8.10 (IH, br, ArNH), 8.94 (IH, s, NCHN), 11.79 (IH, s, ONH). 13C NMR (100 MHz, D6 DMSO) δ 32.1 (CH3), 58.5 (CH2OH), 77.3 (CH2ON), 108.2 (CH), 109.6 (CBr), 115.8 (CH), 120.6 (CCl), 122.0 (C), 125.0 (CC=O), 129.4 (C), 130.5 (CH), 131.1 (CH), 132.3 (C), 140.6 (C), 145.8 (CF), 146.5 (CH), 164.2 (C=O). [0078] The results of the infrared analysis are shown in Figure 2. Spectral assignments axe summarized in Table 1.
Table 1
Wavenumber (cm“ ) Assignment 3,255 Includes the O-H stretching vibration of the primary alcohol group and the N-H stretching vibrations of the secondary aromatic amine and secondary amide groups.
3,200 – 2,700 Includes =C-H stretching vibrations of the aromatic ring and benzimidazole group and the aliphatic C-H stretching vibrations.
2,700 – 2,300 Includes the multiple NH+ stretching vibrations of the benzimidazole 1 : 1 sulfate salt group.
1,673 C=O stretching vibrations of the secondary amide group where
1,653 the carbonyl group is subject to different environmental effects such as hydrogen bonding.
1,640 – 1,370 Includes the C=C aromatic ring stretching vibrations, the C=C and C=N stretching vibrations of the benzimidazole group, the
O-H deformation vibration of the primary alcohol group and the aliphatic C-H deformation vibrations.
1,570 The CNH combination band of the secondary amide group.
1,506 Includes the CNH bending vibration of the secondary aromatic amine group.
1 ,213 The aryl C-F stretching vibration.
1,189 The asymmetric SO3 “ stretching vibration of the benzimidazole
1 : 1 sulfate salt group. 1,100 – 1,000 Includes the C-O stretching vibration of the primary alcohol group and the aryl C-Br stretching vibration. 1,011 The symmetric SO3 “ stretching vibration of the benzimidazole
1 :1 sulfate salt group. 920 – 600 Includes the C-H wag vibrations and C=C ring bending vibrations of the 1,2,4-trisubtituted aromatic ring and the benzimidazole group. 888 Includes the S-O(H) stretching vibration of the benzimidazole
1 : 1 sulfate salt group. Example IA
Preparation of the Hydrogen sulphate salt of Compound 1
[0079] Sulfuric acid (1.52 ml, 27.86 mmol) was added to a stirred suspension of 6-(4- bromo-2-chlorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2- hydroxyethoxy)-amide (1O g, 0.0214 mol) (obtainable as described in Example 10 of WO 03/077914, which is incorporated herein by reference and as described below) in tetrahydrofuran (THF) (62 ml) and water (8 ml) whilst maintaining a temperature of 10 0C or lower. The stirred mixture was heated to 65 0C and held for 30 minutes before filtering to remove any extraneous matter. THF (150 ml) was then added to the mixture maintaining the temperature above 60 0C. The mixture was then cooled to 0-5 0C over approximately 2 hour. The resulting slurry was filtered, washed with THF (30 ml) and dried under reduced pressure at 50 0C until a constant weight was achieved, to give 6-(4-bromo-2-chlorophenylamino)-7- fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethoxy)-amide hydrogen sulfate (9.81g, 0.17 mol, 82% yield) as an off white crystalline solid. The material was the same as that produced in Example 1 above.
References
- Troiani, T.; Vecchione, L.; Martinelli, E.; Capasso, A.; Costantino, S.; Ciuffreda, L. P.; Morgillo, F.; Vitagliano, D.; d’Aiuto, E.; De Palma, R.; Tejpar, S.; Van Cutsem, E.; De Lorenzi, M.; Caraglia, M.; Berrino, L.; Ciardiello, F. (2012). “Intrinsic resistance to selumetinib, a selective inhibitor of MEK1/2, by cAMP-dependent protein kinase a activation in human lung and colorectal cancer cells”. British Journal of Cancer 106 (10): 1648–1659.doi:10.1038/bjc.2012.129. PMC 3349172. PMID 22569000.
- Davies, H.; Bignell, G. R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M. J.; Bottomley, W.; Davis, N.; Dicks, E.; Ewing, R.; Floyd, Y.; Gray, K.; Hall, S.; Hawes, R.; Hughes, J.; Kosmidou, V.; Menzies, A.; Mould, C.; Parker, A.; Stevens, C.; Watt, S.; Hooper, S.; Wilson, R.; Jayatilake, H.; Gusterson, B. A.; Cooper, C.; Shipley, J. (2002). “Mutations of the BRAF gene in human cancer”. Nature 417 (6892): 949–954. doi:10.1038/nature00766. PMID 12068308.
- Jump up^ ClinicalTrials.gov NCT00890825 Comparison of AZD6244 in Combination With Docetaxel Versus Docetaxel Alone in KRAS Mutation Positive Non Small Cell Lung Cancer (NSCLC) Patients
- Jump up^ ClinicalTrials.gov NCT00888134 AZD6244 in Cancers With BRAF Mutations
- Journal of the American Chemical Society, 2013 , vol. 135, 35 p. 12994 – 12997
-
8-1-2013Identification of potent Yes1 kinase inhibitors using a library screening approach.Bioorganic & medicinal chemistry letters
| WEDGE S R ET AL: “AZD2171: A HIGHLY POTENT, ORALLY BIOAVAILABLE, VASCULAR ENDOTHELIAL GROWTH FACTOR RECEPTOR-2 TYROSINE KINASE INHIBITOR FOR THE TREATMENT OF CANCER“, CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, US, vol. 65, no. 10, 15 May 2005 (2005-05-15), pages 4389-4400, XP008066714, ISSN: 0008-5472, DOI: 10.1158/0008-5472.CAN-04-4409 | ||
| 52 | * | WEDGE STEPHEN R ET AL: “ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration“, CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, US, vol. 62, no. 16, 15 August 2002 (2002-08-15), pages 4645-4655, XP002425560, ISSN: 0008-5472 |
| 53 | WEDGE, S.R. ET AL.: ‘ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration‘ CANCER RES vol. 62, 2002, pages 4645 – 4655 |
- Ho, Alan L.; Grewal, Ravinder K.; Leboeuf, Rebecca; Sherman, Eric J.; Pfister, David G.; Deandreis, Desiree; Pentlow, Keith S.; Zanzonico, Pat B. et al. (2013). “Selumetinib-Enhanced Radioiodine Uptake in Advanced Thyroid Cancer”. New England Journal of Medicine 368 (7): 623–32. doi:10.1056/NEJMoa1209288. PMC 3615415.PMID 23406027.
|
1-30-2009
|
TOSYLATE SALT OF 6- (4-BR0M0-2-CHL0R0PHENYLAMIN0) -7-FLUORO-N- (2-HYDROXYETHOXY) -3-METHYL-3H-BENZIMI DAZOLE- 5 – CARBOXAMIDE , MEK INHIBITOR USEFUL IN THE TREATMENT OF CANCER
|
|
|
9-17-2008
|
N3 alkylated benzimidazole derivatives as MEk inhibitors
|
|
|
6-27-2007
|
N3 alkylated benzimidazole derivatives as MEK inhibitors
|
|
|
12-19-2003
|
N3 alkylated benzimidazole derivatives as MEK inhibitors
|
|
6-6-2012
|
METHOD OF TREATMENT USING N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
6-6-2012
|
COMPOSITIONS COMPRISING N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS AND METHODS OF USE THEREOF
|
|
|
5-16-2012
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
8-24-2011
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
7-6-2011
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
11-31-2010
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
8-18-2010
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
|
|
5-28-2010
|
COMBINATION THERAPY COMPRISING AZD2171 AND AZD6244 OR MEK-INHIBITOR II
|
|
|
10-2-2009
|
PHARMACEUTICAL COMPOSITION 271
|
|
|
8-19-2009
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
Dacomitinib in phase 3 for lung (non-small cell) (NSCLC) Cancer

Dacomitinib
(2E)-N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-methoxy-6-quinazolinyl}-4-(1-piperidinyl)-2-butenamide
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide
pf299804…… pfizer
EGFR (HER1; erbB1) Inhibitors
HER4 (erbB4) Inhibitors
HER2 (erbB2) Inhibitors
- Molecular formula:C24H25ClFN5O2
- Molecular mass:469.95
Dacomitinib (PF-00299804) is an experimental drug candidate under development by Pfizer for the treatment of non-small-cell lung carcinoma. It is a selective and irreversible inhibitor of EGFR.[1]
Dacomitinib has advanced to several Phase III clinical trials. The results of the first trials were disappointing, with a failure to meet the study goals,[2][3][4] Additional Phase III trials are ongoing.[2]
Dacomitinib is a HER (erbB) inhibitor in clinical trial development at Pfizer for the treatment of advanced non-small cell lung cancer (NSCLC) and for the treatment of relapsed/recurrent glioblastoma.
No recent development has been reported for research into the treatment of recurrent and/or metastatic head and neck squamous cell cancer. In 2012, Pfizer and SFJ Pharmaceuticals signed a codevelopment agreement for dacomitinib for the treatment of patients with locally advanced or metastatic NSCLC with activating mutations of epidermal growth factor receptor.
Substituted 4-phenylamino-quinazolin-6-yl-amides useful in the treatment of cancer have been described in the art, including those of U.S. Pat. No. 5,457,105 (Barker), U.S. Pat. No. 5,760,041 (Wissner et al.), U.S. Pat. No. 5,770,599 (Gibson), U.S. Pat. No. 5,929,080 (Frost), U.S. Pat. No. 5,955,464 (Barker), U.S. Pat. No. 6,251,912 (Wissner et al.), U.S. Pat. No. 6,344,455 (Bridges et al.), U.S. Pat. No. 6,344,459 (Bridges et al.), U.S. Pat. No. 6,414,148 (Thomas et al.), U.S. Pat. No. 5,770,599 (Gibson et al.), U.S. patent application 2002/0173509 (Himmelsbach et al.), and U.S. Pat. No. 6,323,209 (Frost).
Dacomitinib is a pan-human epidermal growth factor receptor (pan-HER) inhibitor developed by Pfizer, as ー small molecules targeting ffiR-1, HER-2 and HER-4 tyrosine kinase inhibitor by irreversibly binding to HER-l, HER-2, HER-4 and anti-tumor effect. Ni-line treatment of non-small cell lung cancer (NSCLC) display, Dacomitinib in non-small cell lung cancer Dinner erlotinib compared to some extend on progression-free survival and quality of life have mentioned the smell.
_4] Structural formula for Dacomitinib

[0005] U.S. patent US7772243 Dacomitinib first proposed a synthesis method, first, a fluorine-2_ _4_ amino acid and formamidine ring closure reaction to give 7 – fluoro-4 – quinazolinone, nitration and then successively chlorination reaction, to give 4 – chloro-7 – fluoro-6 – nitro-quinazoline; another aspect ー 3 – chloro-4 – amino-substituted on a fluoroaniline to give 3 – chloro – # – (3,4 – ni section yl methoxy)-4_ fluoro-aniline, obtained after the coupling of both an amino-protected N-(3 – chloro-4 – fluorophenyl)-7 – fluoro-6 – nitro-quinazoline -4 – amine, protected amino N-(3 – chloro-4 – fluorophenyl
Yl)-7_ fluoro-6 – nitro-quinazolin-4 – amine is of formula

Followed by a methoxy group, an amidation reaction and hydrogenation, the final deprotection ko under the action of trifluoroacetic acid to give the final product Dacomitinib. Throughout the reaction as follows:

synthesis
http://www.google.com/patents/CN103304492A?cl=en
Synthesis ー kind EGFR inhibitors Dacomitinib, synthetic route for

A synthetic method EGFR inhibitors Dacomitinib, concrete steps are as follows:
Step I, 7 – fluoro-4 – Synthesis of quinazolinone:

30 g (0.1934mol) 2 – fluoro-amino acid was dissolved in 250 ml _4_ formamide among the reaction was heated to 150 ° C for 6 inch, TLC plates to determine the point of completion of the reaction. The reaction was poured hot into 2000 ml of ice water, filtered, the filter cake was washed with water, vacuum dried at 50 ° C for 14 hours to give a pale brown solid powder 7 – fluoro-4 – quinazolinone, 28 g, yield 88%.
[0021] 2 walk 7 – fluoro-6 – nitro-4_ (hydrogen) _ Synthetic quinazolinones of:

Concentrated sulfuric acid (50 ml) and fuming nitric acid (50 ml) mixture was cooled with an ice bath to (TC hereinafter under stirring slowly added 25 g (0.1523mol) 7 – fluoro-4 – quinazolinone , the addition was complete, the reaction mixture was stirred at room temperature for I hour and then the reaction was heated to 110 ° C for 2 inch, TLC plates to determine the point of completion of the reaction the reaction was cooled to room temperature, 300 ml of ice water, the precipitated solid was stirred for 30 minutes , filtered, the filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give a yellow solid powder 7 – fluoro-6 – nitro-4 – (hydrogen) – quinazolinone, 26 g, yield 82%.
[0022] Step 3 6 – amino-7 – fluoro-4 – (hydrogen) – quinazolinone Synthesis:

24 g (0.1148mol) 7 – fluoro-6 – nitro _4_ (hydrogen) – quinazolinone was dissolved in 400 ml of methanol was added 2 g of palladium / carbon catalyst was added 8 ml of concentrated hydrochloric acid, and hydrogen was 2 small inch atmospheric reaction, TLC plates to determine the point of the reaction is complete. The catalyst was removed by suction filtration through celite, washed several fitness methanol, and the filtrate was concentrated by rotary evaporation to dryness to give 6 – amino-7_ fluoro-4 – (hydrogen) – quinazolinone, yellow powder, 20 g, yield 97%.
[0023] 4 walk, ⑶ -4 – (piperidin – Suites yl) -2 – butene acid methyl ester synthesis:

18 g (0.1006mol) 4 – bromo-methyl crotonate dissolved in 180 ml of methylene chloride ni added 27.9 g (0.2019mol) potassium carbonate, cooled to ice-bath (TC, was slowly added dropwise 10 ml (0.1012mol ) piperidine, (I reaction was stirred under a small inch TC, TLC plates to determine the point of completion of the reaction was concentrated by rotary evaporation to dryness, to give (E) -4 – (piperidin-1 – yl) – 2 – butenoic acid methyl Cool as a yellow solid, 17.1 g, yield 93%.
[0024] 5th walk, Buddhist) -4 – (piperidin-1 – yl) -2 – butene acid hydrochloride synthesis:

16 g (0.0873mol) of W) -4 – (piperidin-_1_ yl) -2 – butenyl acetate and 80 ml of concentrated hydrochloric acid was added to 250 ml of 1,4 – ni oxygen dioxane, heated under reflux 20 hours inch, TLC plate point the reaction was determined complete, the reaction solution was concentrated by rotary evaporation to dryness surplus was recrystallized from isopropanol to give a pale yellow solid, Buddhist) _4-(piperidin-1 – yl) -2 – butene acid hydrochloride, 14.5 g, yield 81%.
[0025] Step 6, (E) -4 – (piperazine Jie fixed -1 – yl) – 2 – butenyl chloride synthesis:

13 g (0.0632mol) of (K) ~ 4 ~ (piperidin-1 – yl) -2 – butene acid hydrochloride was dissolved in 750 ml of methylene chloride ni, 5 ml of DMF, was slowly added dropwise 8 ml ( 0.0933mol) of oxalyl chloride, the reaction was stirred at room temperature for I h, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation to give a pale yellow oil, Buddhist) _4-(piperidin-1 – yl) -2 – butyl allyl chloride, 11.8 g, yield 99%.
[0026] Step 7 (cargo) – # – (7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butene amide Synthesis:

11 g (0.0586mol) of the) -4 – (piperidin-1 – yl) – 2 – butenyl chloride ni chloride (50 ml) was slowly added dropwise to 6 – amino-1 – fluoro-4 – ( hydrogen) – quinazolinone (7 g, 0.0391mmol), three ko amine (14 ml) and the mixture was ni chloride (200 ml), the reaction mixture was stirred at room temperature for 2 hours the reaction inch, TLC determined the completion of reaction points board , was added 800 liters of halo ni halo chloroformate and 500 liters of burning the separated organic phase was washed with 500 liters of halo, halo and then with 500 liters of brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness was subjected to silica gel surplus Column chromatography (30% acid ko ko acetate / hexane) to give (M)-N-(7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butenamide, as a pale yellow solid, 12.3 g, yield 95%.
Step 8 [0027] (2 ^) – # – (7 – methoxy – 4 – oxo _3, 4_ ni hydrogen quinazolinyl _6_ yl)-4_ (piperidin-1 – yl) – Synthesis 2_ butenamide:

I ^ xN MeONa N. Under nitrogen atmosphere, to 100 ml of anhydrous methanol was slowly added 1.52 g of sodium metal (0.0661mol), stirred for 10 minutes to dissolve all of the sodium metal to the completion of the reaction, to obtain a freshly prepared solution of sodium methoxide, and the The sodium methoxide solution was added 11 g (0.0333mol) of (receive) (7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) 2_ butene-amide, the reaction was heated to reflux for 3 inch, TLC plates to determine completion of the reaction point, cooled to room temperature, acidified with 2N hydrochloric acid solution to pH = 3 ~ 4, and concentrated by rotary evaporation to dryness, the residue was washed with water beating, filtration, The filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give (article) – # – (7 – methoxy – 4 – oxo – ni hydrogen quinazolin-6 – yl) – 4_ (piperidin-1 – yl)-2_ butenamide yellow solid, 10.6 g, yield 93%.
[0028] Step 9, {W,-N-(4 – chloro-7 – methoxy-quinazoline _6_ yl)-4_ (piperidin _1_ yl)-amide <EMI butene 2_:

9 g (0.0263mol) of (receive) – # – (7 – methoxy _4_ oxo – ni hydrogen quinazolin-6 – yl)-4_ (piperidin-1 – yl) – 2_ butenamide were added to 40 ml of phosphorus oxychloride was heated under reflux for 2 inch, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation, ice water was added surplus, beating, filtered, the cake washed with washed with water, vacuum dried at 50 ° C in 14 hours to give {W,-N-(4 – chloro-7 – methoxy-quinazolin-6 – yl) -4 – (piperidin-1 – yl) – 2 – butene amide as a yellow solid, 7 g, yield 74%

(2E)-N-(4 – chloro-7 – methoxy-quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butene amide (6 g,
0.0166mol), 3 – chloro-4-fluoro-aniline (2.6 g, 0.0179mol) and three ko amine (2.6 ml, 0.0186mol) was added to 140 ml of isopropanol and the reaction was heated to reflux for 3 inch, TLC plates to determine the point completion of the reaction, cooled to room temperature, filtered, the filter cake washed with methanol, vacuum dried at 50 ° C in 14 hours to give the final product Dacomitinib, a yellow solid, 6.6 g, yield 84%.
/////////////////////////
synthesis
US7772243
http://www.google.com/patents/US7772243
Scheme 1, wherein the 4-position aniline group is represented a 4-fluoro-3-chloro aniline group.

4-Chloro-7-fluoro-6-nitroquinazoline (7) can be prepared by methods similar to those described in J.Med. Chem. 1996, 39, 918-928. Generally, 2-amino-4-fluoro-benzoic acid (1) can be reacted with formamidine (2) and acetic acid (3) in the presence of 2-methoxyethanol to provide 7-Fluoro-3H-quinazolin-4-one (4). The 7-fluoro-3H-quinazolin-4-one (4) can be nitrated to 7-fluoro-6-nitro-3H-quinazolin-4-one (5), which can be treated with thionyl chloride to yield 4-chloro-6-nitro-7-fluoro-3H-quinazoline (6). The 4-chloro-quinazoline compound (6) can be combined with a desirably substituted aniline, represented above by 4-fluoro-3-chloro-aniline, in the presence of a tertiary amine and isopropanol to provide the 4-anilino-6-nitro-7-fluoro-quinazoline (7).
The 4-anilino-6-nitro-7-fluoro-quinazoline (7) may be reacted with an alcohol of the formula R3OH, wherein R3 is as defined above, to yield the 7-alkoxylated compound (8). Reduction of the 6-nitro compound (8) provides the 6-amino analog (9).
The 6-position amino compound (9) may be reacted with a haloalkenoyl chloride (12), such as a 4-bromo-but-2-enoyl chloride, 5-bromo-pent-2-enoyl chloride, 4-chloro-but-2-enoyl chloride, or 5-chloro-pent-2-enoyl chloride, to provide an alkenoic acid[4-anilino]-7-alkoxylated-quinazolin-6-yl-amide (13). Haloalkenoyl chloride agents useful in this scheme may be prepared by methods known in the art, such as the treatment of a relevant haloalkenoic acid, represented by bromoalkenoic acid ester (10), with a primary alcohol, yielding the corresponding haloalkenoic acid (11), which may in turn be treated with oxalyl chloride to provide the desired haloalkenoyl chloride (12).
Finally, the quinazoline-6-alkanoic acid compound (13) may be treated with a cyclic amine, such as piperidine, piperazine, etc., to provide the desired final compound (14).
EXAMPLE 2
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide (Synthetic Route No. 1)

The title compound and other 7-methoxy analogs of this invention can be prepared as described in Example 1 by replacing the 2-fluoroethanol used in Example 1 with stoichiometric amount of methanol.
EXAMPLE 3 4-Piperidin-1 -yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxv -quinazolin-6-yl]-amide (Synthetic Route No. 2)
An alternative synthetic route for compounds of this invention involves preparing the 6-position substituent chain as a Het-alkenoyl chloride as depicted in Scheme 2, below.


It will be understood that other compounds within this invention may be prepared using Het-butenoyl halide, Het-pentenoyl halide and Het-hexenoyl halide groups of the formula:
wherein R4 is as described herein and halo represents F, Cl, Br or I, preferably Cl or Br. One specific group of these Het-alkenoyl halides includes those compounds in which halo is Cl or Br, R4 is —(CH2)m-Het, m is an integer from 1 to 3, and Het is piperidine or the substituted piperidine moieties disclosed above.
EXAMPLE 4 4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide (Synthetic Route No. 3)


3-Chloro-4-fluoro-phenylamine 15 (50.31, 345.6 mmole) and 3,4-Dimethoxy-benzaldehyde 16 (57.43 g, 345.6 mmole) were mixed in 500 ml of IPA and cooled in an ice-water. The glacial acetic acid was added (20.76 g, 345.6 mole) and then sodium cyanoborohydride in one portion. The reaction was stirred at room temperature (RT) for 24 hrs. 250 mL of 10% NaOH was added dropwise at RT after the reaction was completed. The mixture was stirred for ½ hr. The slurry was then filtered and washed with IPA and dried in vacuo. The mass weight 88.75 g (17, 87%).
Compounds 6 (3 g, 13.18 mmole) and 17 (3.9 g, 13.18 mmole) were combined in CH3CN (25 mL) and heated for one hr. Mass spectroscopy indicated no starting material. Saturated K2CO3 was added and the reaction was extracted 3× with EtOAc. The organic layers were combined, washed with brine and concentrated in vacuo to give 6.48 g of 7 (78.4%).
Compound 7 (72.76 g, 149.4 mmole) was added to a cool solution of NaOMe in 1.5 L of dry MeOH under N2. The cooling bath was removed and the mixture was heated to reflux and stirred for 1 hr. The reaction was cooled to room temperature and quenched with water until the product precipitated out. The solid was filtered and washed with water and hexanes. The product was slurred in refluxing EtOAc and filtered hot to provide 68.75 g of yellow soled 8 (73%).
Compound 8 (63.62 g, 127.5 mole) was hydrogenated using Raney/Ni as catalyst to obtain 43.82 g of 9 (100%). Oxalyl chloride (6.5 g, 51.18 mmole) was added slowly to a suspension of 13 (10.5 g, 51.2 mmole) in 200 ml of dichloromethane containing 8 drops of DMF, after the reaction become homogeneous, the solvent was removed and the residual light yellow solid was slurred in 200 ml of DMAC and 9 (20 g, 42.65 mmole) was added gradually as a solid. The reaction was stirred for 15 min. and poured slowly into 1N NaOH. The mixture was extrated 3× EtOAc. The combined organic layers were washed with brine, filtered and concentrated in vacuo to obtain 28.4 g (100%) 10.
Compound 10(13.07 g, 21.08 mmole) was dissolved in trifluoroacetic acid (TFA) (74 g, 649 mmole) and heated to 30° C. for 24 hrs. The reaction was cooled to RT and poured gradually into a cooled 1 N NaO H-brine solution. Precipitate formed and was filtered and washed with 3X water then dried. The precipitate was recrystallized from toluene to obtain pure 4-Piperidin-1-vl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino) -7-methoxv-puinazolin-6-yl]-amide (9.90 g, 89%).
Example 1 is similar but not same…caution
EXAMPLE 1 4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-(2-fluoro-ethoxy)-quinazolin-6-yl]-amide

7-fluoro-6-nitro-4-chloroquinazoline (14.73,g, 65 mmol) was combined with 3-choro-4-fluoroaniline (9.49 g, 65 mmol) and triethylamine (10 mL, 72 mmol) in 150 mL of isopropanol. The reaction was stirred at room temperature for 1.5 hours, resulting in a yellow slurry. The solid was collected by filtration, rinsing with isopropanol and then water. The solid was dried in a 40° C. vacuum oven overnight to give 19.83 g (91%) of the product as an orange solid.
MS (APCI, m/z, M+1): 337.0
NaH (60% in mineral oil, 3.55 g, 88 mmol) was added, in portions, to a solution of 2-fluoroethanol (5.19 g, 80 mmol) in 200 mL THF. The reaction was stirred for 60 minutes at room temperature. To the reaction was added 7-fluoro-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (18.11 g, 54 mmol) as a solid, rinsing with THF. The reaction was heated to 65° C. for 26 hours. The reaction was cooled to room temperature and quenched with water. THF was removed in vacuo. The resulting residue was sonicated briefly in water then the solid collected by filtration. The solid was triturated with MeOH, filtered and dried in a 40° C. vacuum oven overnight to 12.63 g of the product. Additional product was obtained by concentrating the MeOH filtrate to dryness and chromatography eluting with 50% EtOAc/hex. The isolated material was triturated with MeOH (2×), filtered and dried. 3.90 g
Total yield: 16.53 g, 81%
MS (APCI, m/z, M+1): 381.0
7-(2-fluoroethoxy)-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (0.845 g, 2.2 mmol) in 50 mL THF was hydrogenated with Raney nickel (0.5 g) as the catalyst over 15 hours. The catalyst was filtered off and the filtrate was evaporated to give 0.77 g of product. (99%)
MS (APCI, m/z, M+1): 351.2
Methyl 4-bromocrotonate (85%, 20 mL, 144 mmol) was hydrolyzed with Ba(OH)2 in EtOH/H2O as described in J.Med.Chem. 2001, 44(17), 2729-2734.
MS (APCI, m/z, M−1): 163.0
To a solution of 4-bromocrotonic acid (4.17 g, 25 mmol) in CH2Cl2 (20 mL) was added oxalyl chloride (33 mL, 38 mmoL) and several drops of DMF. The reaction was stirred at room temperature for 1.5 hours. The solvent and excess reagent was removed in vacuo. The resulting residue was dissolved in 10 mL THF and added to a 0° C. mixture of 6-amino-7-(2-fluoroethoxy)-4-(3-chloro-4-fluoroaniline)quinazoline (5.28 g, 15 mmol) and triethylamine (5.2 mL, 37 mmol). The reaction was stirred at 0° C. for 1 hour. Water was added to the reaction and the THF removed in vacuo. The product was extracted into CH2Cl2 (400 mL). The organic layer was dried over MgSO4, filtered and concentrated. The crude material was chromatographed on silica gel eluting with 0-4% MeOH/CH2Cl2. An isolated gold foam was isolated. Yield: 4.58 g, 61%
MS (APCI, m/z, M−1): 497.1
Piperidine (0.75 mL, 6.7 mmol) was added to a solution of the above compound (3.35 g, 6.7 mmol) and TEA (2.80 mL, 20 mmol) in 10 mL DMA at 0° C. The reaction was stirred at 0° C. for 17 hours. Water was added to the reaction until a precipitate was evident. The reaction was sonicated for 40 minutes and the liquid decanted. The residue was dissolved in CH2Cl2, dried over MgSO4, filtered and concentrated. The material was chromatographed on silica gel eluting with 4-10% MeOH/CH2Cl2. The isolated residue was triturated with acetonitrile (2×) and collected by filtration. Impurity found: Michael addition of piperidine (2.2% in first trituration of acetonitrile). Additional material can be obtained from the acetonitrile filtrates.
Yield: 0.95 g, 27%
MS (APCI, m/z, M+1): 502.3
……………
US 20050250761 A1,
References
- “Dacomitinib”. NCI Drug Dictionary.
- Zosia Chustecka (January 27, 2014). “Dacomitinib Fails in Pretreated Non-small Cell Lung Cancer”. Medscape.
- “Blow to Pfizer as dacomitinib fails in lung cancer trials”. pmlive.com. 28th January 2014.
- “Pfizer Announces Top-Line Results From Two Phase 3 Trials Of Dacomitinib In Patients With Refractory Advanced Non-Small Cell Lung Cancer”. Pfizer Press Release. January 27, 2014.
- Tyrosine kinase inhibitors.17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(Phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides baring additional solubilizing functions
J Med Chem 2000, 43(7): 1380
- Dacomitinib Fact Sheet, Pfizer
| WO1996033980A1 * | 23 Apr 1996 | 31 Oct 1996 | Keith Hopkinson Gibson | Quinazoline derivatives |
| WO1997038983A1 * | 8 Apr 1997 | 23 Oct 1997 | Alexander James Bridges | Irreversible inhibitors of tyrosine kinases |
| WO2002050043A1 * | 12 Dec 2001 | 27 Jun 2002 | Boehringer Ingelheim Pharma | Quinazoline derivatives, medicaments containing said compounds, their utilization and method for the production thereof |
| WO2004069791A2 * | 3 Feb 2004 | 19 Aug 2004 | Hubert Gangolf Klemens Barth | Preparation of substituted quinazolines |
| US5760041 * | 21 Jan 1997 | 2 Jun 1998 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
Ozenoxacin in phase 3……topical formulation in the treatment of impetigo

1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster……http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H

Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.
- Ferrer Group. Key development projects. Available online: http://www.ferrergrupo.com/Innovation_Innovacion-Pipeline-de-proyectos-ENG (accessed on 15 April 2013).
- Yamakawa, T.; Mitsuyama, J.; Hayashi, K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J. Antimicrob. Chemother. 2002, 49, 455–465, doi:10.1093/jac/49.3.455.
- Ferrer Internacional. Efficacy and safety of ozenoxacin 1% cream versus placebo in the treatment of patients with impetigo. Available online: http://clinicaltrials.gov/ct2/show/NCT01397461 (accessed on 13 April 2013).
Amgen Drug Evolocumab Hits Endpoint of Cholesterol Reduction
 Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | PCSK9 |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 1256937-27-5 |
| ATC code | None |
| Chemical data | |
| Formula | C6242H9648N1668O1996S56 |
| Mol. mass | 141.8 kDa |
Evolocumab[1] is a monoclonal antibody designed for the treatment of hyperlipidemia.[2] Evolocumab is a fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9).

PCSK9 is a protein that targets LDL receptors for degradation and thereby reduces the liver’s ability to remove LDL-C, or “bad” cholesterol, from the blood.
Evolocumab, being developed by Amgen scientists, is designed to bind to PCSK9 and inhibit PCSK9 from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from the blood.

On 23 January 2014 Amgen announced that the Phase 3 GAUSS-2 (Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) trial evaluating evolocumab in patients with high cholesterol who cannot tolerate statins met its co-primary endpoints: the percent reduction from baseline in low-density lipoprotein cholesterol (LDL-C) at week 12 and the mean percent reduction from baseline in LDL-C at weeks 10 and 12. The mean percent reductions in LDL-C, or “bad” cholesterol, compared to ezetimibe were consistent with results observed in the Phase 2 GAUSS study.[3]
The GAUSS-2 trial evaluated safety, tolerability and efficacy of evolocumab in 307 patients with high cholesterol who could not tolerate effective doses of at least two different statins due to muscle-related side effects. Patients were randomized to one of four treatment groups: subcutaneous evolocumab 140 mg every two weeks and oral placebo daily; subcutaneous evolocumab 420 mg monthly and oral placebo daily; subcutaneous placebo every two weeks and oral ezetimibe 10 mg daily; or subcutaneous placebo monthly and oral ezetimibe 10 mg daily.
Safety was generally balanced across treatment groups. The most common adverse events (> 5 percent in evolocumab combined group) were headache (7.8 percent evolocumab; 8.8 percent ezetimibe), myalgia (7.8 percent evolocumab; 17.6 percent ezetimibe), pain in extremity (6.8 percent evolocumab; 1.0 percent ezetimibe), and muscle spasms (6.3 percent evolocumab; 3.9 percent ezetimibe).

Evolocumab, a PCSK9 inhibitor, was safe and effective at lowering low-density lipoprotein cholesterol (LDL-C) after one year of treatment, according to a study published online Nov. 19 inCirculation and presented simultaneously at the American Heart Association scientific session in Dallas.
The Open-Label Study of Long-term Evaluation Against LDL-C (OSLER) trial took place at 156 study centers around the world that participated in at least one of four phase 2 studies of between October 2011 and June 2012. Evolocumab is a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor made by Amgen.
Investigators led by Michael J. Koren, MD, of the Jacksonville Center for Clinical Research in Florida, randomized 1,104 participants in a 2:1 ratio to receive either evolocumab (420 mg every four weeks) plus standard-of-care therapy (based on guidelines for treatment of hypercholesterolemia) or evolocumab alone, which served as the control. After 12 weeks, lipid results were unblinded and investigators were able to adjust standard-of-care therapy in either group.
The main efficacy objective was to determine the effects of longer-term evolocumab therapy on cholesterol levels and the main safety endpoints included incidence of adverse events, serious adverse events and adverse events resulting in discontinuation of the drug.
Patients who received evolocumab for the first time in the OSLER study had an average LDL-C reduction of 52.3 percent at one year. Patients previously dosed with evolocumab in a prior trial and were in the evolocumab and standard-of-care group in OSLER had an average LDL-C reduction of 52.1 percent at the end of the study compared with 50.4 percent at baseline. Patients who terminated evolocumab when they entered OSLER had their LDL-C levels returned to around their baseline.
Adverse events occurred in 73.1 percent of the standard-of-care group and 81.4 percent of the evolocumab plus standard-of-care group. The researchers determined that 5.6 percent of adverse events were related to evolocumab. Serious adverse events occurred in 6.3 percent of the control group and 7.1 percent in the combination group.
The authors explained that their findings offer more insight into the use of this class of drugs to lower LDL-C in at-risk patients.
“Challenging patients such as those who fail to reach current lipid goals despite maximum doses of highly effective statin agents or those with well-documented statin intolerance are thus logical populations for treatment with PCSK9 inhibitors,” they concluded.

References
- World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108” (PDF). WHO Drug Information 26 (4).
- Statement On A Nonproprietary Name Adopted By The USAN Council – Evolocumab
- Estel Grace Masangkay, “Amgen Phase III GAUSS-2 Trial of Evolocumab (AMG 145) Meets Co-Primary Endpoints Of LDL Cholesterol Reduction”, Bioresearch Online (January 24 2014)

MEPOLIZUMAB….GSK to file severe asthma drug by year end
The first non-inhaled treatment for a difficult-to-treat form of severe asthma is getting closer to market after GlaxoSmithKline said it would initiate global filings for the drug at the end of this year, on the back of strong late-stage clinical data.
Mepolizumab – a monoclonal antibody that inhibits interleukin 5 – is being investigated as a treatment for severe eosinophilic asthma in patients who experience exacerbations despite high-dose oral or inhaled corticosteroids (ICS) and an additional controller such as long-acting beta-2 agonist.
Read more at: http://www.pharmatimes.com/Article/14-03-13/GSK_to_file_severe_asthma_drug_by_year_end.aspx#ixzz2vuANtYaK
Follow us: @PharmaTimes on Twitter
Mepolizumab (proposed trade name Bosatria) is a humanized monoclonal antibody that recognizes interleukin-5 (IL-5), and is used to treat certain kinds of asthma and white blood cell diseases.
 IL 5
IL 5
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | IL-5 |
Recent studies have concluded that mepolizumab may improve exacerbations in patients with severe eosinophilic asthma, an adult-onset asthma which represents less than 5% of all asthma.
IL-5 is a chemical messenger in the immune system that stimulates the growth of eosinophils. In eosinophilic asthma, eosinophils are present in the lungs. When mepolizumab was given to people with eosinophilic asthma, it eliminated eosinophils from the bloodstream,and reduced eosinophils in the lungs and bone marrow. Mepolizumab also reduced the number of asthma exacerbations, and reduced the need for corticosteroids.[1]Mepolizumab improved the quality of life, but the improvement was “not clinically meaningful,” according to a reviewer.[2] [3]
In a recent multi-centre, double-blinded, randomised, controlled trial study of Mepolizumab in severe eosinophilic asthma, Mepolizumab reduced the number of clinically significant exacerbations compared to a placebo. Additionally Mepolizumab reduced sputum and blood eosinophil counts and was shown to be safe for up to 12 months.[4]

Mepolizumab is also in development for the management of hypereosinophilic syndrome by GlaxoSmithKline (GSK) and has received orphan drug designation by the FDA.[5] Mepolizumab has been shown to reduce the need for corticosteroids and improve symptoms in FIP1L1/PDGFRA negative hypereosinophilic syndrome.[6]
UK pharma giant GlaxoSmithKline (LSE: GSK) says that a pivotal Phase III study of mepolizumab, an investigational IL-5 antagonist monoclonal antibody, met its primary endpoint of reduction in the frequency of exacerbations, in patients with severe eosinophilic asthma.
Mepolizumab could add £400 million ($668 million) to GSK’s revenue by 2021, according to estimates from Barclays reported by The Wall Street Journal. Analysts from Deutsche Bank forecast £300 million in mepolizumab sales by 2018 for the company, already a leader in the asthma treatment sector.

The study (MEA115588) evaluated the efficacy of two-dose regimens of mepolizumab in the treatment of patients with severe eosinophilic asthma. Patients remained on their current asthma maintenance therapy throughout the study and were randomized to receive either mepolizumab 75mg intravenous (IV), 100mg subcutaneous (SC), or placebo every four weeks.
For the primary end point, both mepolizumab treatment arms showed statistically significant reductions in the frequency of clinically significant exacerbations of asthma compared to placebo (75mg IV, 47%, p<0.001; 100mg SC, 53%, p<0.001).
Adverse events reported in the study were similar across all treatment groups. The most common reported adverse events across all treatment groups were nasopharyngitis, headache, upper respiratory tract infection and asthma. The frequency of adverse events was 83% in the placebo group, 84% in the mepolizumab 75mg IV and 78% in the mepolizumab 100mg SC group. The frequency of serious adverse events was 14% in the placebo group, 7% in the mepolizumab 75mg IV and 8% in the mepolizumab 100mg SC group.
Backs up earlier studies; regulatory filing mooted at year end
Dave Allen, head of GSK Respiratory Therapy Area Unit, R&D, said: “We are really pleased to have generated further positive data on mepolizumab, consistent with the findings from our earlier exacerbation study. We now have two studies showing a reduction in exacerbations in a specific group of patients with a severe form of asthma who continue to exacerbate despite treatment with high doses of their current maintenance therapies. This is very positive news for patients. For GSK it is exciting that this is the first non-inhaled treatment for severe asthma and we will be progressing towards global filings at the end of the year.”
In addition, a second Phase III study (MEA115575) designed to evaluate the use of mepolizumab 100mg SC, every four weeks in comparison to placebo in reducing daily oral corticosteroid use while maintaining asthma control also met its primary endpoint. The study showed that patients on mepolizumab 100mg SC were able to achieve greater reductions in their maintenance oral corticosteroid dose during weeks 20-24 compared to patients on placebo (p =0.008), while maintaining asthma control.
In this study adverse events were similar across treatment groups. The most common reported adverse events in the two treatment groups were headache, nasopharyngitis, bronchitis, sinusitis, fatigue and asthma. The frequency of adverse events was 92% in the placebo and 84% in the mepolizumab treatment group. Frequency of serious adverse events was 18% in the placebo group and 1% in the mepolizumab group.
Mepolizumab Useful in Refractory Eosinophilic Asthma, a Rare Subtype of Asthma
Eosinophil.
Eosinophil.Eeosinophilic form of asthma represents less than 5% of cases of adult-onset asthma and is difficult to treat.
Crystal structure of human IL-5. .
Mepolizumab reduced the number of blood and sputum eosinophils and allowed prednisone sparing in patients who had asthma with sputum eosinophilia despite prednisone treatment.
Mepolizumab therapy reduced exacerbations by 43% and improved Asthma Quality of Life Questionnaire (AQLQ) scores in patients with refractory eosinophilic asthma.
Eosinophils may have a role as important effector cells in the pathogenesis of severe exacerbations of asthma in patients with eosinophilic asthma.
Cytokine targets for immunomodulators for allergic disorders.
Mediators from Eosinophils
References
- Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009 Mar 5;360(10):973-84.
- Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009 Mar 5;360(10):985-93.
- Eosinophils in asthma – closing the loop or opening the door? Sally E. Wenzel, N Engl J Med. 2009 Mar 5;360(10):1026-7.
- Pavord, Ian D; Korn, Stephanie; Howarth, Peter; Bleecker, Eugene R; Buhl, Roland; Keene, Oliver N; Ortega, Hector; Chanez, Pascal (August 2012). “Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial”. The Lancet 380 (9842): 651–659. doi:10.1016/S0140-6736(12)60988-X.
- Phase III study of Bosatria (mepolizumab) showed disease control with reduced corticosteroid use in hypereosinophilic syndrome
- http://content.nejm.org/cgi/content/abstract/358/12/1215 Rothenberg et al 2008


{kind=link}