
Home » Posts tagged 'phase 2' (Page 11)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.
AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
![]()
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref………. Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
See all Bboroles at………http://apisynthesisint.blogspot.in/p/borole-compds.html
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
Ataciguat

Ataciguat HMR-1766
Hoechst Marion Roussel De Gmbh
5-Chloro-2-[[(5-chloro-2-thienyl)sulfonyl]amino]-N-[4-(4-morpholinylsulfonyl)phenyl]benzamide
C21H19Cl2N3O6S3
UNII-QP166M390Q;
576.49306 g/mol
A guanylate cyclase activator potentially for the treatment of aortic valve stenosis.
![]()
CAS No. 254877-67-3
- Originator sanofi-aventis
- Developer Mayo Clinic; National Center for Advancing Translational Sciences; Sanofi; sanofi-aventis
- Class Anthranilic acids; Benzamides; Cardiovascular therapies; Chlorobenzenes; Morpholines; Small molecules; Sulfonamides; Thiophenes
- Mechanism of Action Guanylate cyclase stimulants
- 30 Jun 2015 Mayo Clinic plans a phase II trial for Aortic valve stenosis in USA (NCT02481258)
- 29 Jan 2014 Phase-I clinical trials in Aortic valve stenosis in USA (PO)
- 01 Jan 2010 Discontinued – Phase-II for Peripheral arterial occlusive disorders in Austria, Canada, France, Germany, Italy, Poland, Portugal, Russia, South Africa and USA (PO) prior to 2010
SYNTHESIS

The Intermediates hown above is used in next step shown below

Paper
Organic Letters (2013), 15(7), 1638-1641
http://pubs.acs.org/doi/abs/10.1021/ol400411v
http://pubs.acs.org/doi/suppl/10.1021/ol400411v/suppl_file/ol400411v_si_001.pdf

The Ru(II)-catalyzed intermolecular o-C–H amidation of arenes in N-benzoylated sulfoximine with sulfonyl azides is demonstrated. The reaction proceeds with broad substrate scope and tolerates various functional groups. Base hydrolysis of the amidation product provides the anthranilic acid derivatives and methylphenyl sulfoximine (MPS) directing group. This method is successfully employed for the synthesis of HMR 1766.
PATENT
WO 2009043495
http://www.google.com/patents/WO2009043495A1?cl=en
Patent
http://www.google.com/patents/WO2008124505A2?cl=en
HMR-1766 (ataciguat sodium, see patent publication WO2000002851)

PATENT
http://www.google.com/patents/WO2000002851A1?cl=en
| Patent | Submitted | Granted |
|---|---|---|
| TRA COMBINATION THERAPIES [US2007238674] | 2007-10-11 | |
| sGC STIMULATORS OR sGC ACTIVATORS ALONE AND IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF CYSTIC FIBROSIS [US2013035340] | 2011-02-03 | 2013-02-07 |
| SOLUBLE GUANYLATE CYCLASE (SGC) MODULATORS FOR TREATMENT OF LIPID RELATED DISORDERS [US2013123354] | 2013-01-08 | 2013-05-16 |
| Novel combination [US2005059660] | 2004-07-29 | 2005-03-17 |
| SGC STIMULATORS OF SGC ACTIVATORS IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF ERECTILE DYSFUNCTION [US2014288079] | 2014-03-18 | 2014-09-25 |
| Patent | Submitted | Granted |
|---|---|---|
| novel use of activators and stimulators of soluble guanylate cyclase for the prevention or treatment of renal disorders [US2010016305] | 2010-01-21 | |
| HETEROARYL-SUBSTITUTED PIPERIDINES [US8119663] | 2009-12-10 | 2012-02-21 |
| Use of soluble guanylate cyclase activators for the treatment of Raynaud’s Phenomenon [US2009215769] | 2009-08-27 | |
| Use of Activators of Soluble Guanylate Cyclase for Promoting Wound Healing [US2009221573] | 2009-09-03 | |
| Use of Suluble Guanylate Cyclase Acitvators for Treating Acute and Chronic Lung Diseases [US2009286781] | 2009-11-19 | |
| Use of Activators of Soluble Guanylate Cyclase for Treating Reperfusion Damage [US2009298822] | 2009-12-03 | |
| HETEROCYCLIC DERIVATIVE AND USE THEREOF [US2011028493] | 2011-02-03 | |
| SUBSTITUTED PIPERIDINES [US8202862] | 2010-12-02 | 2012-06-19 |
| METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS [US2009022729] | 2009-01-22 | |
| sGC STIMULATORS [US2014323448] | 2014-04-29 | 2014-10-30 |



/////////
C1COCCN1S(=O)(=O)C2=CC=C(C=C2)NC(=O)C3=C(C=CC(=C3)Cl)NS(=O)(=O)C4=CC=C(S4)Cl
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics

| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
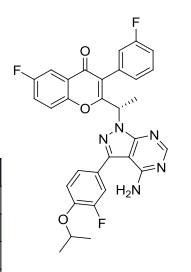
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)

Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.

REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
DRL 17822 from Reddy US Therapeutics/Dr Reddy’s



CAS 920493-71-6 and CAS 898911-09-6



DRL 17822
MW 603.6045, MFC30 H31 F6 N7
| Molecular Formula: | C30H31F6N7 |
|---|---|
| Molecular Weight: | 603.604459 g/mol |
Cas 898911-09-6, 1454689-50-9
3-([[3,5-Bis(trifluoromethyl)benzyl](2-methyl-2H-tetrazol-5-yl)amino]methyl)-N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amine
3-Quinolinemethanamine, 2-[bis(cyclopropylmethyl)amino]-N-[[3,5-bis(trifluoromethyl)phenyl]methyl]-8-methyl-N-(2-methyl-2H-tetrazol-5-yl)-
3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine
(3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
Reddy US Therapeutics (Innovator)




Treatment of Atherosclerosis Therapy Lipoprotein Disorders,
CETP inhibitor (dyslipidemia/atherosclerosis/cardiovascular diseases), Dr Reddy’s
Selective inhibitor of cholesteryl ester transfer protein (CETP)
- 30 Jun 2012Dr Reddy’s Laboratories completes a phase II trial in Hypercholesterolaemia in Italy, Poland and Ukraine (NCT01388816)
- 09 Mar 2012Dr Reddy’s Laboratories completes enrolment in its phase II trial for Hypercholesterolaemia in Italy, Poland, and Ukraine (NCT01388816)
- 02 Sep 2011Phase-II clinical trials in Hypercholesterolaemia in Ukraine (PO)
CLINICAL TRIALS…..Type II Hyperlipidemia PHASE 2…………https://clinicaltrials.gov/ct2/show/NCT01388816
Cardiovascular disease is a leading cause of death worldwide. Among cardiovascular disorders, coronary heart disease (CHD) caused by atherosclerosis is the most common cause of morbidity and mortality. Prevention, stabilization and regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events.
LDL-C lowering agents, primarily the statins, are the current mainstay in the pharmacologic management of dyslipidemia. However even with stain use, residual CHD risk from dyslipidemia remains. Epidemiologic and observational studies have shown that HDL-C is also a strong independent predictor of CHD, suggesting that raising HDL-C levels might afford clinical benefit in the reduction of cardiovascular risk.
Presently only niacin is approved by the FDA for HDL-C elevation and can raise HDL-C levels by 20-30%. However its use can be limited by a high incidence of flushing and, less commonly, by elevation of blood glucose and potential hepatic toxicity.
Cholesteryl ester transfer protein (CETP) inhibitors are being explored for their ability to elevate HDL-C. A small molecule CETP inhibitor, torcetrapib, has been demonstrated to elevate HDL-C by 60-100%. However, a large clinical trial (ILLUMINATE) where it increased HDL-C by a mean of 72% compared to baseline was halted as it failed to show benefit. Post-hoc analysis of this study implicated an off-target increase in blood pressure as potentially counteracting any anti-atherosclerotic benefits. Post-hoc subgroup analysis showed that patients in the highest HDL-C quartile had a 57% reduction in the risk of cardiovascular events.
Increased blood pressure appears to be specifically related to torcetrapib as two other small molecule CETP inhibitors, anacetrapib and dalcetrapib, have not shown this in clinical trials and have been well tolerated. DRL-17822 has also not shown elevation of blood pressure in either animals or in normal volunteers.
This study will investigate the efficacy and tolerability of DRL-17822 as dyslipidemia monotherapy in patients with Type II hyperlipidemia.
Hyperlipidemia or an elevation in serum lipids is associated with an increase incidence of cardiovascular disease and atherosclerosis. Primary hyperlipidemia is a term used to describe a defect in lipoprotein metabolism. The lipoproteins commonly affected are low density lipoprotein (LDL) cholesterol, which transports mainly cholesterol, and very low density lipoprotein-cholesterol (VLDL-cholesterol), which transports mainly triglycerides (TG). Most subjects with hyperlipidemia have a defect in LDL metabolism, characterized by raised cholesterol, LDL-C levels, with or without raised triglyceride levels; such subjects are termed hypercholesterolemic (Fredrickson Type II). Familial hypercholesterolemia (FH) is caused by any one of a number of genetically-determined defects in the LDL receptor, which is important for the entry of cholesterol into cells. The condition is characterized by a reduced number of functional LDL receptors, and is therefore associated with raised serum LDL-C levels due to an increase in LDL.

It is reasonably known in the art that the likelihood of cardiovascular disease can be decreased, if the serum lipids, and in particular LDL-C, can be reduced. It is further known that the progression of atherosclerosis can be retarded or the regression of atherosclerosis can be induced if serum lipids can be lowered. In such cases, individuals diagnosed with hyperlipidemia or hypercholesteremia should consider lipid-lowering therapy to retard the progression or induce the regression of atherosclerosis for purposes of reducing their risk of cardiovascular disease, and in particular coronary artery disease.
Cholesteryl ester-transfer protein (CETP) is an important player in metabolism of lipoproteins, such as, for example, a high density lipoprotein (HDL). CETP is a 70 kDa plasma glycoprotein that is physically associated with HDL particles. It facilitates the transport of cholesteryl ester from HDL to apolipoprotein B-containing lipoproteins. This transfer is accompanied by transfer of triglycerides in the opposite direction. Thus, a decrease in CETP activity can result in an increase in the level of HDL cholesterol and a decrease in the level of very low density lipoprotein (VLDL) and low density lipoprotein (LDL). CETP can therefore simultaneously affect the concentrations of pro-atherogenic (for example, LDL) and anti-atherogenic (for example, HDL) lipoproteins.
Several CETP inhibitors are currently in various clinical phases of development for treating various aforementioned disorders. In spite of having various advantages, CETP inhibitors are proven to be difficult to formulate for oral administration. CETP inhibitors are of a highly lipophilic nature and have extremely low solubility in water. Due to their poor solubility, bioavailability of conventional oral compositions is very poor. The lipophilic nature of CETP inhibitors not only leads to low solubility but also tends to poor wettability, further reducing their tendency to be absorbed from the gastrointestinal tract. In addition to the low solubility, CETP inhibitors also tend to have significant, “food effect”, where a significant difference in rate and amount of drug absorption is observed when the drug is administered with or without a meal. This “food effect”, often complicates the dosing regimen and may require high dosing to achieve the desired therapeutic effect, resulting in potentially unwanted side effects.
Several attempts have been made to improve the solubility of CETP inhibitors, but have generally ended up with limited success. At the outset, most methods aimed at enhancing aqueous concentration and bioavailability of low-solubility drugs only offer moderate improvements. References describing improving the dissolution of poorly soluble drugs include: U.S. Patent Nos. 5,456,923, 5,993,858, 6,057,289, 6,096,338, 6,267,985, 6,280,770, 6,436,430, 6,451,339, 6,531,139, 6,555,558, 6,638,522, 6,962,931 and 7,374,779.
PATENT
WO 2014128564
https://www.google.co.in/patents/WO2014128564A2?cl=en
WO-2014076568
http://www.google.com/patents/WO2014076568A2?cl=en
EXAMPLES
In the following Examples 1-17, various compositions in accordance with the present application were prepared comprising 3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine as the CETP inhibitor.:
EXAMPLE 1 :

1. 3-(((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)methyl)- N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amineand hydroxypropyl methyl cellulose acetate succinate were mixed together in given solvent mixture to form clear solution.
2. To the solution of step I, Polyoxyl 35 castor oil and talc were added to form a homogenous suspension.
3. The suspension of step 2 was sprayed over inert sugar spheres and dried.
4. The drug layered spheres of step 3 were coated with dispersion made from given seal layer ingredients.
5. The coated spheres of step 4 were formulated further as capsule dosage form.
PATENT
WO 2013046045
https://www.google.co.in/patents/WO2013046045A1?cl=en
PATENT
WO 2013024358
PATENT
WO 2007075194
https://www.google.co.in/patents/WO2007075194A1?cl=en
Syntheis construction





Example 1
Synthesis of (3-{[3,5-bis trifluoromethyl-benzyl )-(2-cyclopropyImethyI-2H- tetrazole -5-yl)-amino]-methyl-}-8-methyI-quinolme-2-yl)-bis- cyclopropylmethyl-amine Step (i): Synthesis of 2~chloro-8-methyl-quinoline-3-carbaldehyde
DMF (1.22 g, 16.7 mmol) was taken in a flask equipped with a drying tube and POCl3 (7.32 g, 46.7 mmol) was added dropwise with stirring at 0° C. To this solution, TV-o-Tolyl acetamide (1.00 g, 6.7 mmol) was added and the solution was refluxed for 6 h at 90° C. The excess POCl3 was distilled off, water was added to the residue and this was stirred at room temperature for 10 min. The solid was filtered and dried under vacuum..This crude compound was purified over silica gel (100-200 mesh) using 6% ethyl acetate and petroleum ether to give the product as a yellowish solid (yield: 78%). 1H NMR (CDCl3, 200 MHz): δ 10.5 (s, IH)5 8.71 (s, IH), 7.83- 7.79 (m, IH), 7.74- 7.70 (m, IH), 7.56-7.49 (m, IH), 2.79 (s, 3H); m/z (EI-MS): 206 (M+, 100%). Step (ϋ): Synthesis of 2-(bis(cyclopropylmethyl)amino)-8-methylquinoline-3- carbaldehyde:
2-Chloro-8-methyl-quinoline-3-carbaldehyde (.115 g, 0.559 mmol), and potassium carbonate (0.231 g, 1.67 mmol) were put in a 25 mL two necked RB flask. To this, 3 mL of DMF was added followed by dropwise addition of bis- cyclopropylmethyl amine (0.083 g, 0.67 mmol). The reaction mixture was refluxed for 2 h and was cooled to RT. It was then poured on crushed ice (10 mL) and extracted with EtOAc (3 x 10 mL). The organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated under vacuum to give a yellow colored oil (0.081 g, 50%).
1H NMR (CDCl3, 400 MHz): δ 10.5 (s, IH), 8.71 (s, IH), 7.83- 7.79 (m, IH),
7.74-7.70 (m, IH), 7.56-7.49 (m, IH), 3.55-3.47 (m, 4H), 2.79 (s, 3H), 1.73-1.72
(m, 2H), 1.70-1.46 (m, 4H), 1.20-1.11 (m, 4H); m/z (ES-MS ): 295 (M+H-I5
100%); IR (neat, cm“1): 3385, 2948, 1691.
Step (iii): Synthesis of 3-((3,5-bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine
2-(Bis(cyclopropylmethyl)amino)-8-methylquinoline-3-carbaldehyde (0.081 g, 0.39 mmol), 3,5-bis-trifluoromethylbenzylamine (0.096 g, 0.39 mmol) and acetic acid (0.047 g, 0.78 mmol) were put in a 25 mL RB flask. To this, 2 rnL of methanol was added and stirred at RT for 15 min. Sodium cyanoborohydri.de (0.075 g, 0.77 mmol) was added portionwise and stirring was continued at RT for another 1 h. Methanol was removed from the reaction mixture under vacuum, water was added to this crude and was extracted with ethyl acetate (3 x 50 mL). The organic layer was washed with saturated NaHCO3 solution, brine and dried over sodium sulphate. The solvent was evaporated and the crude residue was purified by column chromatography over silica gel (100-200 mesh) eluting with 4% ethyl acetate in petroleum ether to give the title amine (0.142 g, yield: 99%). 1R NMR (CDCl3, 400 MHz): δ 7.89-7.86 (m, IH), 7.80 (m, IH), 7.75-7.74 (m, IH), 7.60-7.40 (m, 3H), 7.30-7.26 (m,lH), 4.12 (s, 2H), 3.88 (s, 2H), 3.24-3.22 (m, 4H), 2.72 (s, 3H), 0.99-0.92 (m, 2H), 0.44-0.35 (m, 4H), 0.11-0.05 (m, 4H); m/z (EI-MS ): 522 (M++l, 100%); IR (neat, cm“1): 3357, 2929, 2851.
Step (iv): Synthesis of N-(3,5-bis(trifluoromethyl)benzyl)-N-((2- (bis(cyclopropylmethyl)amino)-8-methylqumolin-3-yl)methyl)cyanamide
To a solution of 3-((3,5~bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine (0.176 g , 0.33 mmol ), obtained in step (iii) , in MeOH (4 mL) under N2 atmosphere was added sodium bicarbonate (0.056 g, 0.67 mmol ) followed by the addition of cyanogen bromide (0.063 g, 0.60 mmol). The reaction mixture was stirred at RT for 2 h. The solvent was removed under vacuum to give the crude residue which was dissolved in water, extracted with ethyl acetate and dried over sodium sulphate. The solvent was evaporated and concentrated in vacuo to afford N-(3,5-bis(trifluoromethyl)benzyl)- N-((2-(bis(cyclopropylmethyl)amino)-8-methylquinolin-3-yl)methyl)cyanamide (0.118 g, 64%).
1H NMR (CDCl3, 400 MHz ): δ 8.07 (s, IH) , 7.82 (s, IH), 7.70 (s, 2H), 7.56-7.55 (m, IH), 7.50-7.49 (m, IH), 4.49 (s, 2H), 4.23 (s, 2H), 3.17 -3.15 (m, 4H), 2.71 (s, 3H), 0.097-0.085 (m, 2H), 0.405-0.401 (m, 4H), 0.385-0.381 (m, 4H); m/z (ES- MS): 547 (M++l, 100%); IR(KBr ,Cm“1 ) : 2273, 1280.
Step (v): Synthesis of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
7V-(3,5-Bis(tiifluoromethyl)benzyl)-N-((2-(bis(cyclopropylmethyl)amino)- 8-methylqumolin-3-yl)methyl)cyanamide (0.118 g, 0.216 mmol), sodium azide (0.70 g 1.08 mmol) and ammonium chloride (.058 g, 1.08 mmol) were put in a RB flask under N2atmosphere. To this reaction mixture, DMF (2 mL) was added and was refluxed for 1 h. The reaction mixture was cooled to RT and ice was added to this and extracted with ethylacetate (3×10 mL). The combined organic layer was washed with brine, dried over sodium sulphate and then concentrated under vacuum to afford of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine as a yellow solid (0.125 g, 99%).
1H NMR (CDCl3, 400 MHz ): δ 7.99 (s, IH) , 7.79 -7.74 (m, 4H ), 7.41-7.40 (m,
IH ), 7.33-7.31 (m, IH), 4.99 (s, 2H), 4.80 (s, 2H), 3.68 (s, 4H), 2.16 (s, IH) 1.56-
1.06 (m, HH); m/z (ES-MS): 578 (M++l, 100%); IR (KBr , cm“1) 3680 , 2922 ,
1660 , 1616.
METHYLATION SHOULD GIVE THE PRODUCT
Scheme 1


PATENT
WO 2006073973
http://www.google.co.in/patents/WO2006073973A2?cl=en
Example 47
Synthesis of [2-(bis-cycIopropylmethyI-amino)-8-methyl-quinolin-3-ylmethyI]-(3,5- bis-trifluoromethyl-benzyl)-carbamic acid methyl ester
Step (i): Synthesis of bis-cyclopropylmethyl-amine
(i) a. Synthesis of cyclopropanecarboxylic acid cyclopropylmethyl-amide:

Cyclopropyl carboxylic acid (1.0 g, 11.63 mmol) was added to a 50 mL two neck round bottom flask, along with DCM (25 mL). This mixture was cooled to 0° C, EDCI (4.15 g, 13.95 mmol) was added portionwise to the mixture with stirring under nitrogen atmosphere, and the temperature was maintained for 0.5 h. After this time, hydroxybenzotriazole (1.88 g, 13.95 mmol) was added to the 0° C mixture which was stirred for 10 min, then triethylamine (1.7 g, 11.63 mmol) was added, and stirring of the mixture was continued at the same temperature for another 0.5 h. Then, cyclopropylmethylamine (0.825 g, 11.63 mmol) was added, and the reaction was allowed to reach RT, and stirring was continued overnight. The solvent was then removed in vacuo, and the crude residue was purified by passing through a column over 60-120 silica gel, eluting with dichloromethane, to afford the title compound (1.6 g), yield: 87%. 1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.17-3.16 (m, 2H), 1.00-0.80 (m, 4H), 0.77-0.67 (m, 2H), 0.56-0.43 (m, 2H), 0.24-0.16 (m, 2H) m/z (CI-MS): 139 (M+, 100%) (i) b. Synthesis of bis-cyclopropylmethyl-amine

To a suspension of lithium aluminum hydride (1.3 g, 9.35mmol) in 10 mL dry ether, a solution of N-cyclopentenoyl-ethylamine (1.7 g, 13.3 mmol) in dry ether (10 mL) was added under a nitrogen atmosphere. This reaction was stirred at RT for 8 h and the reaction mixture was then quenched with saturated sodium sulfate solution, filtered, and the precipitate was washed with diethyl ether. The filtrate was concentrated to afford the title amine (0.8 g), yield: 69%.
1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.16-3.09 (m, 2H), 2.50-2.4 (m, 2H), 0.56-0.43 (m, 4H), 0.24-0.21 (m, 3H), 0.21-0.13 (m, 3H) m/z (ES-MS): 139 (M^+14, 100%)
Step (ii): Synthesis of [2-(bis-cyclopropylmethyl-amino)-8-methyl-quinolin-3-ylmethyl]- (S^-bis-trifluoromethyl-benzy^-carbamicacid methyl ester

The title compound was synthesized by using the same procedure as in Example 35, except using o-tolyl acetanilide in step (i) instead of acetanilide and bis- cyclopropylmethyl amine in step (iii), which yielded the desired product as a light yellow, viscous liquid (0.05 g), yield:40%, of purity 98.8% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt12.719 min).
1H NMR (CDCl3, 400 MHz): d 7.7 (s, IH), 7.68-7.44 (m, 3H), 7.27-7.24 (m, 2H), 4.78- 4.65 (m, 2H), 4.47-4.4 (m, 2H), 3.8 (s, 3H), 3.16-3.14 (d, J=7Hz, 2H), 2.7 (s, 3H), 1.55 (s, 3H), 1.01-0.9(m, IH), 0.38-0.34 (m, 4H), 0.07-0.05 (m, 4H); m/z (CI-MS): 579 (M+, 100%)
Example 57
Synthesis of (3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
The title compound was prepared as an oil by following the same synthetic procedures as in Example 52, except using {3-[(3,5-bis-trifluoromethyl-benzylamino)- methyl]-8-methyl-quinolin-2-yl}-bis-cyclopropylmethyl-amine in step (i) instead of {3- [(3,5-bis-trifluoromethyl-benzylamino)-methyl]-quinolin-2-yl}-cyclopentylmethyl-ethyl- amine (0.07 g), yield: 52%.
Purity: 95.53% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt 9.538 min).
IR (neat, cm4) 3079, 2925, 1582;
1H NMR (CDCl3, 400 MHz): d 7.82 (s, IH), 7.69-7.67 (m, 2H), 7.44-7.41 (m, IH), 7.23- 7.2 (m, 3H), 4.91 (s, 2H), 4.65 (s, 2H), 4.21 (s, 3H), 3.29 -3.19 (m, 4H)5 2.71 (s, 3H), 1.01-1.00 (m, 2H), 0.99-0.83 (m, 2H), 0.39-0.34 (m, 3H), 0.08-0.07 (m, 3H). m/z (ES-MS): 604 (M++!, 100%)
Dr. Reddy’s announces start of Phase II study with the CETP inhibitor, DRL-17822 in dyslipidemia patients
Hyderabad, India, September 02, 2011: Dr Reddy’s Laboratories (NYSE: RDY) announced the initiation of dosing with DRL-17822 in patients with diagnosis of type II dyslipidaemia. DRL-17822, is a selective, orally bioavailable inhibitor of cholesteryl ester transfer protein (CETP), for the treatment and/or prevention of dyslipidaemia, atherosclerosis and associated cardiovascular disease.
The current study is being conducted under a CTA in a number of countries in Europe. The objective of the study is to evaluate the efficacy and safety of DRL-17822 in patients with Type-II dyslipidemia. This is a randomized, double blind, placebo controlled, parallel group study in 160 subjects. The primary outcome measure is to assess the elevation in HDL cholesterol and reduction in LDL cholesterol from baseline to end of treatment compared to placebo. Three doses (50, 150 & 300 mg) of DRL-17822 given once daily for 4 weeks will be evaluated during this study.
Three human Phase I studies with DRL-17822 had already been conducted in Europe, where DRL-17822 was shown to be safe and well tolerated. In these studies, the proof of mechanism had been demonstrated by dose-dependent inhibition of plasma CETP activity as well as by significant increase in HDL cholesterol & decrease in LDL cholesterol levels.
Cardiovascular disease is a leading cause of death among men and women worldwide. Among cardiovascular disorders, coronary heart disease (CHD), caused by atherosclerosis is the most common cause of morbidity and mortality. Stabilization and/or regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events. Low-density lipoprotein cholesterol lowering agents, primarily the statins, are the current mainstay in the pharmacological management of dyslipidaemia. However, significant residual cardiovascular risk remains despite use of statins.
Epidemiological and observational studies demonstrate that reduced high density lipoprotein cholesterol levels are a strong, independent predictor of CHD, suggesting that raising HDL cholesterol levels might afford clinical benefit in the reduction of cardiovascular risk. One approach to raise HDL level has been inhibition of CETP activity. Currently it is believed that, raising HDL cholesterol and lowering LDL cholesterol through CETP inhibition would lead to a significant benefit in terms of CHD risk reduction.
Dr. K. Anji Reddy, Founder Chairman, Dr. Reddy’s Laboratories added, “We are committed to delivering products of differentiated value in this area of high global clinical unmet need. We are excited to continue to advance our CETP program and look forward to the data from our Phase II study. This class of therapy could transform the treatment of CHD and DRL 17822 is in a position to be one of the front-running products in the class”.
Disclaimer
This press release includes forward-looking statements, as defined in the U.S. Private Securities Litigation Reform Act of 1995. We have based these forward-looking statements on our current expectations and projections about future events. Such statements involve known and unknown risks, uncertainties and other factors that may cause actual results to differ materially. Such factors include, but are not limited to, changes in local and global economic conditions, our ability to successfully implement our strategy, the market acceptance of and demand for our products, our growth and expansion, technological change and our exposure to market risks. By their nature, these expectations and projections are only estimates and could be materially different from actual results in the future.
About Dr. Reddy’s
Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: www.drreddys.com
For more information please contact:
Investors and Financial Analysts:
Kedar Upadhye at kedaru@drreddys.com / +91-40-66834297
Raghavender R at raghavenderr@drreddys.com / +91-40-49002135
Milan Kalawadia (USA) at mkalawadia@drreddys.com / +1 908-203-4931
Media:
S Rajan at rajans@drreddys.com / +91-40- 49002445
| WO2005011634A1 * | Jul 21, 2004 | Feb 10, 2005 | William John Curatolo | Dosage forms providing controlled release of cholesteryl ester transfer protein inhibitors and immediate release of hmg-coa reductase inhibitors |
| WO2006073973A2 * | Dec 28, 2005 | Jul 13, 2006 | Reddy Us Therapeutics Inc | Novel benzylamine derivatives as cetp inhibitors |
| WO2006129167A1 * | May 22, 2006 | Dec 7, 2006 | Pfizer Prod Inc | PHARMACEUTICAL COMPOSITIONS OF CHOLESTERYL ESTER TRANSFER PROTEIN INHIBITORS AND HMG-CoA REDUCTASE INHIBITORS |
| WO2007128568A1 * | May 8, 2007 | Nov 15, 2007 | Novartis Ag | Bicyclic derivatives as cetp inhibitors |
| EP0298666A2 * | Jul 1, 1988 | Jan 11, 1989 | American Home Products Corporation | Spray dried ibuprofen compositions |
| EP1741424A2 * | Jul 27, 1998 | Jan 10, 2007 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailabilty |
| US5456923 | Dec 23, 1993 | Oct 10, 1995 | Nippon Shinyaku Company, Limited | Method of manufacturing solid dispersion |
| US5474989 | Mar 1, 1994 | Dec 12, 1995 | Kurita Water Industries, Ltd. | Drug composition |
| US5985326 | May 30, 1996 | Nov 16, 1999 | Icos Corporation | Method of producing a solid dispersion of a poorly water soluble drug |
| US6350786 | Sep 7, 1999 | Feb 26, 2002 | Hoffmann-La Roche Inc. | Stable complexes of poorly soluble compounds in ionic polymers |
| US6548555 | Jan 31, 2000 | Apr 15, 2003 | Pfizer Inc | Basic drug compositions with enhanced bioavailability |
| US6638522 | Dec 10, 1999 | Oct 28, 2003 | Pharmasolutions, Inc. | Microemulsion concentrate composition of cyclosporin |
| US6730679 | Mar 20, 1997 | May 4, 2004 | Smithkline Beecham Corporation | Pharmaceutical formulations |
| US7008640 | Jul 16, 2001 | Mar 7, 2006 | Yamanouchi Pharmaceutical Co., Ltd. | Pharmaceutical composition for oral use with improved absorption |
| US7034013 | Mar 19, 2002 | Apr 25, 2006 | Cydex, Inc. | Formulations containing propofol and a sulfoalkyl ether cyclodextrin |
| US7037528 | Jun 5, 2001 | May 2, 2006 | Baxter International Inc. | Microprecipitation method for preparing submicron suspensions |
| US7078057 | Dec 19, 2000 | Jul 18, 2006 | Kerkhof Nicholas J | Process for producing nanometer particles by fluid bed spray-drying |
| US7081255 | Aug 14, 2002 | Jul 25, 2006 | Janssen Pharmaceutica, N.V. | Antifungal compositions with improved bioavailability |
| US8030359 | Feb 9, 2007 | Oct 4, 2011 | Merck Sharp & Dohme Corp. | Polymer formulations of CETP inhibitors |
| US20060178514 | Dec 28, 2005 | Aug 10, 2006 | Anima Baruah | Novel benzylamine derivatives as CETP inhibitors |
| Reference | ||
|---|---|---|
| 1 | * | EINFAL T ET AL: “Methods of amorphization and investigation of the amorphous state“, ACTA PHARMACEUTICA 20130901 CROATIAN PHARMACEUTICAL SOCIETY DEU, vol. 63, no. 3, 1 September 2013 (2013-09-01), pages 305-334, XP002721717, ISSN: 1330-0075 |
| 2 | * | KAI TOSHIYA ET AL: “Oral absorption improvement of poorly soluble drug using solid dispersion technique“, CHEMICAL AND PHARMACEUTICAL BULLETIN (TOKYO), vol. 44, no. 3, 1996, pages 568-571, XP002721716, ISSN: 0009-2363 |
| 3 | * | KIM TAE-WAN ET AL: “Characterization of dual layered pellets for sustained release of poorly water-soluble drug“, CHEMICAL & PHARMACEUTICAL BULLETIN (TOKYO), vol. 55, no. 7, July 2007 (2007-07), pages 975-979, XP002721715, ISSN: 0009-2363 |
| 4 | * | KIM TAE-WAN ET AL: “Modified release of coated sugar spheres using drug-containing polymeric dispersions.“, ARCHIVES OF PHARMACAL RESEARCH JAN 2007, vol. 30, no. 1, January 2007 (2007-01), pages 124-130, XP002721714, ISSN: 0253-6269 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014128564A2 * | Feb 21, 2014 | Aug 28, 2014 | Dr. Reddy’s Laboratories Ltd. | Pharmaceutical compositions of cetp inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Novel benzylamine derivatives and their utility as cholesterol ester-transfer protein inhibitors [US2007015758] | 2007-01-18 | |
| Novel benzylamine derivatives as CETP inhibitors [US2006178514] | 2006-08-10 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
EP-2919765-A2 |
2015-09-23 |
EN
|
|
||
|
2.
US-20150216866-A1 |
2015-08-06 |
|
|||
|
3.
US-9040558-B2 |
2015-05-26 |
|
|||
|
4.
WO-2014128564-A2 |
2014-08-28 |
EN
|
|
||
|
5.
WO-2014076568-A2 |
2014-05-22 |
EN
|
|
||
|
6.
US-20140134235-A1 |
2014-05-15 |
|
|||
|
7.
US-20070015758-A1 |
2007-01-18 |
|
SEE
http://circ.ahajournals.org/cgi/content/meeting_abstract/122/21_MeetingAbstracts/A13981
////////
Cn1nc(nn1)N(Cc2cc5cccc(C)c5nc2N(CC3CC3)CC4CC4)Cc6cc(cc(c6)C(F)(F)F)C(F)(F)F
CC1=CC=CC2=CC(=C(N=C12)N(CC3CC3)CC4CC4)CN(CC5=CC(=CC(=C5)C(F)(F)F)C(F)(F)F)C6=NN(N=N6)C
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.

Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
Zydus gets USFDA nod for clinical trials of Saroglitazar

November 19, 2015
New Delhi: Zydus Cadila has received US health regulator’s nod to initiate phase II clinical trials of Saroglitazar, its new drug for treating high fat levels in body due to diabetes, obesity, and sedentary habits.
“United States Food and Drug Administration (USFDA) has endorsed company’s plan to initiate a phase II clinical trial of Saroglitazar in patients with severe hypertriglyceridemia,” Zydus Cadila said in a statement.
http://www.medicaldialogues.in/zydus-gets-usfda-nod-for-clinical-trials-of-sarolitazar/

//////////////
AMG-319

AMG-319
N-((1S)-1-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine, WO2008118468
(S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine
CAS 1608125-21-8
Chemical Formula: C21H16FN7
Exact Mass: 385.14512
Phosphoinositide-3 kinase delta inhibitor
AMGEN, PHASE 2

PI3K delta isoform selective inhibitor that is being investigated in human clinical trials for the treatment of PI3K-mediated conditions or disorders, such as cancers and/or proliferative diseases
Useful for treating PI3K-mediated disorders such as acute myeloid leukemia, myelo-dysplastic syndrome, myelo-proliferative diseases, chronic myeloid leukemia, T-cell acute lymphoblastic leukemia, B-cell acute lymphoblastic leukemia, non-Hodgkins lymphoma, B-cell lymphoma, or breast cancer.
Amgen is developing AMG-319, a small molecule PI3K-δ inhibitor, for treating lymphoid malignancies and solid tumors including, head and neck squamous cell carcinoma.
AMG-319 is a highly selective, potent, and orally bioavailable small molecule inhibitor of the delta isoform of the 110 kDa catalytic subunit of class IA phosphoinositide-3 kinases (PI3K) with potential immunomodulating and antineoplastic activities. PI3K-delta inhibitor AMG 319 prevents the activation of the PI3K signaling pathway through inhibition of the production of the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3), thus decreasing proliferation and inducing cell death. Unlike other isoforms of PI3K, PI3K-delta is expressed primarily in hematopoietic lineages. The targeted inhibition of PI3K-delta is designed to preserve PI3K signaling in normal, non-neoplastic cells.
PATENT
http://www.google.com/patents/WO2008118468A1?cl=en
PATENT
WO2013152150
http://www.google.com/patents/WO2013152150A1?cl=en
PATENT
Example 4: Method of making N-((lSM-(7-fluoro-2-(2-pyridinyl)- 3-quinolinyl)ethyl)-9H-purin-6-amine
N-((l S)- 1 -(7-Fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine (4) is synthesized in four steps beginning with (S)-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine hydrochloride (1). A nucleophilic aromatic substitution between coupling partners 1 and purine 5 affords the penultimate intermediate 2. Cleavage of the p-methoxybenzyl (PMB) group leads to the isolation of the desired butyl acetate solvate 3. A crystalline form change is induced through an aqueous-acetone recrystallization to afford the target hydrate 4.
Synthetic Scheme
Step 1. Preparation of PMB protected pyridylpurinamine tosylate (2)
(S)- 1 -(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine is prepared similar to that described in US20130267524. The (S)-l-(7-fluoro-2-(pyridin-2-
yl)quinolin-3-yl)ethanamine hydrochloride (1) is coupled to PMB-chloropurine (5, prepared similar to that described in J. Med. Chem. 1988,31, 606-612) in the presence of K2CO3 in IPA. Upon reaction completion the K2CO3 is removed via filtration and the product is crystallized by the addition of /?-toluenesulfonic acid (pTSA). Isolation of the PMB-protected pyridylpurinamine tosylate (2) is conducted via filtration.

Dry 100 L reactor under nitrogen. Set the temperature to 20 ± 5 °C. Charge (l S)-N-chloro-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine HCl salt (1) to the reactor. Then 9-(4-methoxybenzyl)-6-chloro-9H-purine (5) is added. Potassium carbonate is added to the reactor. Isopropyl alcohol is added to the reactor and the mixture is heated to 80 °C and stirred for 24 hours. Additional isopropyl alcohol is added to the reactor and the mixture is cooled to 20 °C. The mixture is filtered through Celite and the solid is washed with isopropyl alcohol and the isopropyl alcohol solutions containing 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine are collected.
The 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine isopropyl alcohol solution is heated to 50 °C. /^-Toluene sulfonic acid monohydrate is dissolved in isopropyl alcohol and added to the 9-(4-methoxybenzyl)-N-((S)-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine in portions. The mixture is slowly cooled to 20 ± 5 °C over 6 ± 2 hrs. The crystalline 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)- 9H-purin-6-amine toluene sulfonic acid salt is collected, rinsed with isopropyl alcohol and dried with vacuum.
Example 5: Method of Making the Crystalline Hydrate Form of N-((1S)-1- (7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine Step 1: Isolation of a Butyl Acetate (BuOAc) Solvate of N-((lS)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine (3)
To a 2 L jacketed reactor equipped with a condenser, a mechanical stirrer, and a bubbler, under an atmosphere of N2, was added N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9-(4-methoxybenzyl)-9H-purin-6-amine (2, 100.0 g, 0.148 mol), followed by acetic acid (AcOH; 240 mL) and 1 -dodecanethiol (71.1 mL, 0.295 mol). The vessel was evacuated and back-filled with nitrogen three times. Methanesulfonic acid (MSA; 28.7 mL, 0.443 mol) was added to the vessel over 10 minutes. Then, the reaction was heated to 80 °C and stirred for 20 hrs. The reaction was then cooled to ambient temperature, after which toluene (1000 mL) and water (700 mL) were sequentially added. The solution was then stirred for 30 minutes. The phases were separated by removing the organic phase, adding another charge of toluene (1000 mL) to the aqueous phase, and the mixture was stirred for another 30 minutes. After removing the organic phase again, the aqueous phase was charged to a jacketed 5 L reactor equipped with a mechanical stirrer followed by n-butyl acetate (1500 mL,) and heated to 50 °C. The aqueous phase was neutralized to pH 6.3 with 10 N NaOH (350 mL). The organic (BuOAc) phase was azeotropically dried to 600 ppm water, while keeping a constant volume. The dried organic phase was polish filtered at 50 °C to remove salts, which were subsequently washed with hot BuOAc (285 mL). The BuOAc was charged back into the 2 L jacketed reactor equipped with a mechanical stirred and distillation apparatus, and then concentrated to 54 mg/g of N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine in solution. The solution was then seeded with 1 wt% seed of the BuOAc solvate of N-((l S)- 1 -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The slurry was further concentrated to 300 mL total volume and cooled to ambient temperature over 1 hour. Heptane (460 mL) was added dropwise to the solution, and the solution was aged overnight. The supernatant concentration was checked, and determined to be 5.3 mg/g of N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The supernatant was filtered and the resulting solid cake was washed with 1 : 1 BuOAc:heptane (280 mL), followed by heptane (280 mL). The washed cake was then
allowed to dry on the filter. The BuOAc solvate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine was obtained as a white solid (59.5 g, 99.6 LCAP, 86.3 wt%, 90 % corrected yield). !H NMR (400 MHz, CDC13) δ 13.72 (s, 1H), 8.80 (s, 1H), 8.37 (s, 1H), 8.31 (s, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 18.8 Hz, 2H), 7.76 (t, J = 1 1.6 Hz, 2H), 7.39 (s, 1H), 7.31 (td, J = 8.7, 2.5 Hz, 1H), 6.15 (s, 1H), 4.06 (t, J = 6.7 Hz, 1H), 2.04 (s, 1H), 1.65 – 1.44 (m, 3H), 1.39 (dt, J = 14.9, 7.4 Hz, 1H), 1.33 – 1.20 (m, 2H), 0.93 (t, J = 7.4 Hz, 1H), 0.88 (t, J = 6.8 Hz, 1H); 13C NMR (101 MHz, CDC13) δ 152.28 (s), 148.46 (s), 138.10 (s), 137.22 (s), 135.58 (s), 129.47 (s), 124.80 (s), 123.53 (s), 1 13.24 – 1 13.09 (m), 1 12.89 (d, J = 20.3 Hz), 64.40 (s), 48.60 (s), 31.91 (s), 30.67 (s), 29.05 (s), 22.72 (s), 19.15 (s), 14.15 (s); IR: 3193, 3087, 2967, 2848, 1738, 1609, 1493, 1267, 1242, 1 143, 933, 874, 763, 677, 646, 627, 606, 581 , 559, 474 cm“1; exact mass m/z calcd for C2iH16FN7, (M + H)+386.1451 , found 386.1529; MP = 144 °C.
Step 2: Isolation of the Crystalline Hydrate of N-((lS)-l-(7-fluoro-2-(2-pyridinvn-3-quinolinyl)ethyl)-9H-purin-6-amine 4
To a 100 L reactor with its jacket set to 20 °C, 1.206 kg butyl acetate solvate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine 3 was charged, followed by 6.8 L of acetone and 6.8 L of water. The resulting mixture was stirred at 90 rpm under nitrogen for 13 minutes to ensure complete dissolution of all solids. During these charges, the reactor contents increased in temperature that maximized at 26 °C. The solution was then transferred to another clean 100 L reactor through a 5 μιη filter, and stirred at 85 rpm under nitrogen. The solution was heated to 45 °C, and water (14.8 L) was added to reach a water content (by Karl Fischer, KF) of 75 wt%. The reactor solution was assayed by HPLC and shown to contain 42 mg/g N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The solution was seeded with a slurry of 1 13 g of the crystalline hydrate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine in 1 L water, and the seed slurry was rinsed into the reactor with an additional 1 L water. The reactor contents were cooled to 0 °C over 16 h and held at that temperature for 1 h. The supernatant was then assayed, and found to contain 7.6 mg/g of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. Next, 10 L of water was added to the reactor over 38 min and aged for 1 h. The supernatant was assayed at 4.9 mg/g, and the solids were isolated by filtration. The solids were washed with an acetone/water solution (140 mL acetone in 2.7 L water), then 4 L water, and dried under nitrogen on the filter for 68 h. The crystalline hydrate of N-((l S)-l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine was isolated as an off-
white solid (1.12 kg, 616 ppm acetone, 3.73 wt% water, 99.56 LCAP, 95.88 wt%). This material was co-milled at 3900 rpm using a 0.024″ screen to yield an off-white powder (1.09 kg, 99.7 LCAP, 95.4 wt%, 75% yield). Calculated losses were 212 g (18%) to liquors, 5.5g (0.5%) to washes, and 23 g (2%) to fouling. ¾ NMR (400 MHz, DMSO) δ 12.86 (s, 1H), 8.69 (s, 1H), 8.64 (s, 1H), 8.27 (s, 1H), 8.10 (s, 1H), 8.06 – 7.91 (m, 4H), 7.76 (dd, J = 10.4, 2.4 Hz, 1H), 7.50 (ddd, J = 19.2, 9.5, 3.6 Hz, 2H), 6.03 (s, 1H), 3.38 (s, 2H), 1.63 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 163.58, 161.12, 158.36, 157.94, 151.99, 147.98, 146.49, 146.36, 136.82, 134.07, 130.24, 130.14, 124.69, 124.65, 123.30, 1 17.36, 1 17.1 1, 112.10, 1 1 1.90, 46.02, 22.01. HRMS m/z Calcd. for C2iH17FN7 (M + H): 386.15295. Found: 386.15161.
PAPER
1: Cushing TD, Hao X, Shin Y, Andrews K, Brown M, Cardozo M, Chen Y, Duquette J, Fisher B, Gonzalez-Lopez de Turiso F, He X, Henne KR, Hu YL, Hungate R, Johnson MG, Kelly RC, Lucas B, McCarter JD, McGee LR, Medina JC, San Miguel T, Mohn D, Pattaropong V, Pettus LH, Reichelt A, Rzasa RM, Seganish J, Tasker AS, Wahl RC, Wannberg S, Whittington DA, Whoriskey J, Yu G, Zalameda L, Zhang D, Metz DP. Discovery and in vivo evaluation of (S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and related PI3Kδ inhibitors for inflammation and autoimmune disease. J Med Chem. 2015 Jan 8;58(1):480-511. doi: 10.1021/jm501624r. Epub 2014 Dec 3. PubMed PMID: 25469863.
http://pubs.acs.org/doi/abs/10.1021/jm501624r

The development and optimization of a series of quinolinylpurines as potent and selective PI3Kδ kinase inhibitors with excellent physicochemical properties are described. This medicinal chemistry effort led to the identification of 1 (AMG319), a compound with an IC50 of 16 nM in a human whole blood assay (HWB), excellent selectivity over a large panel of protein kinases, and a high level of in vivo efficacy as measured by two rodent disease models of inflammation.
(S)-N-(1-(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (1)
//////////
C[C@H](NC1=C2N=CNC2=NC=N1)C3=CC4=CC=C(F)C=C4N=C3C5=NC=CC=C5
LIK 066, Licogliflozin diprolinate
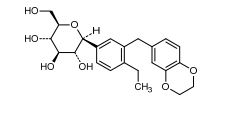
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
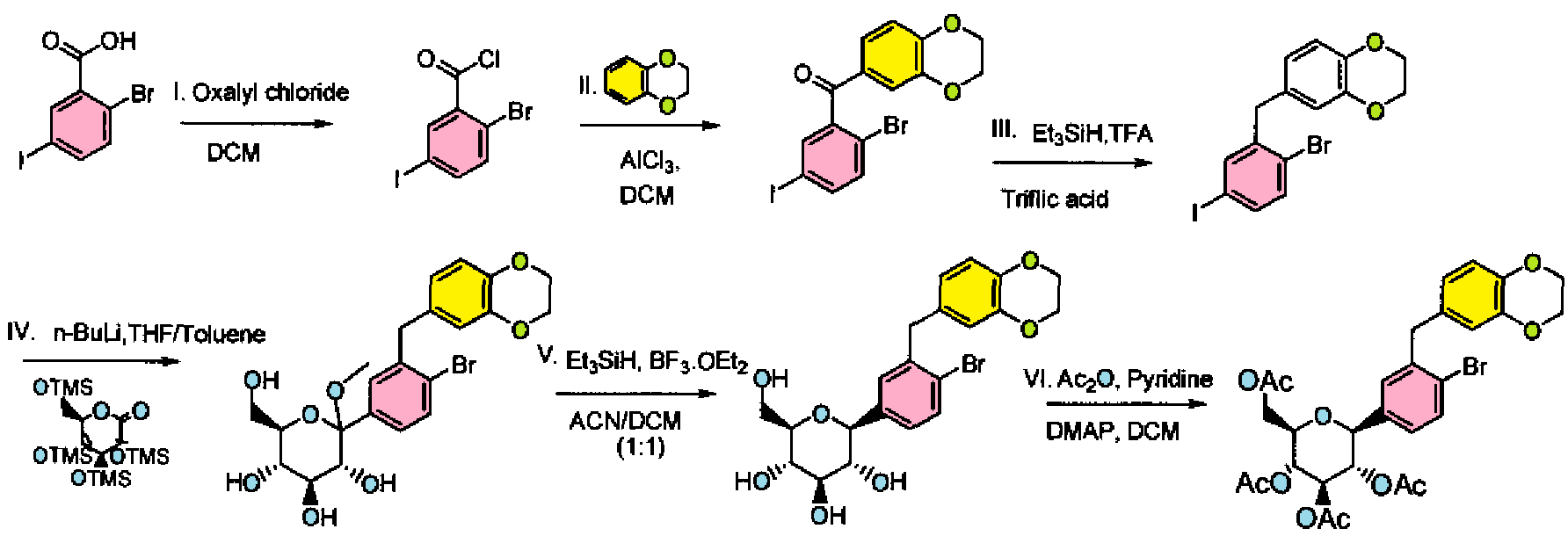
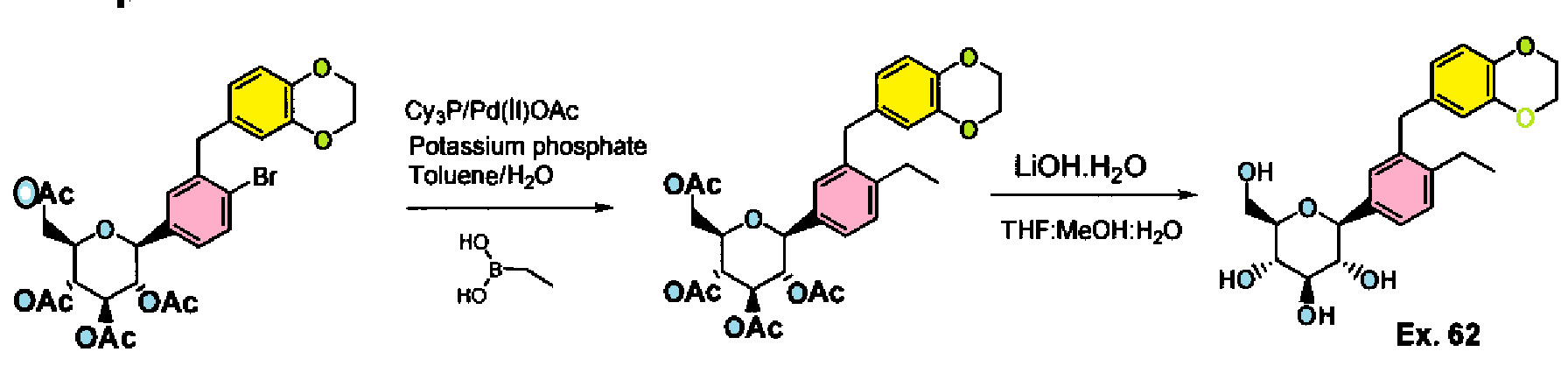
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
EV 077

EV-077
SER 150 (formerly EV-077)
Also known as: formerly EV-077-3201
EV-077-3201-2TBS
CAS 1384128-29-3
Evolva INNOVATOR
Oral thromboxane receptor antagonist and thromboxane synthase inhibitor
EV-077 is a small compound being developed for the treatment of complications of diabetes. In Phase 2. Outlicensed to Serodus in 2013.
In 2013, Serodus licensed the product candidate for the treatment of diabetic nephropathy and it is conducting phase II clinical trials on this research.
EV-077 is an oral, small molecule compound, belonging to a new structural class. Preclinical and early clinical studies indicate EV-077 has potential in reducing vascular inflammation by inhibiting the activity of prostanoids and isoprostanes – in particular in diabetes. Towards the end of 2011, the Russian Patent Office granted patent protection for EV-077 in the treatment of complications of diabetes for a term extending to 2026. Evolva has outlicenced EV-077 to Serodus in 2013. Serodus aims to bring EV-077 further through clinical development and at a future time point decide whether Serodus or a partner will conduct the final clinical trials.
EV-077 is in development as a potential pharmaceutical for the treatment of diabetic nephropathy and other diabetic complications. It is in Phase II clinical studies.
In 2013, Evolva out-licensed EV-077 to Serodus (Oslo, Norway). Serodus aims to bring EV-077 through Phase II and then decide whether or not to partner for the final clinical trials and commercialisation. Evolva is entitled to clinical and regulatory milestones as well as a single-digit royalty on sales. If Serodus sublicenses EV-077 then Evolva will receive up to 30% of Serodus’ total licensing income.
As of Q2 2015 Serodus continues active development of EV-077.
– See more at: http://www.evolva.com/ev-077/#sthash.4mgJ3E0f.dpuf
Patients with diabetes mellitus (DM) have increased propensity to generate thromboxane A2 (TXA2) and other eicosanoids which can contribute to their heightened platelet reactivity. EV-077 is a potent thromboxane receptor antagonist and thromboxane synthase inhibitor and thus represents an attractive therapy in patients with DM. However, the effects of EV-077 on pharmacodynamic (PD) profiles in patients with DM and coronary artery disease (CAD) while on antiplatelet therapy is poorly explored and represented the aim of this in vitro pilot investigation. Patients with DM and stable CAD (n = 10) on low-dose aspirin (81 mg/day) were enrolled and then switched to clopidogrel (75 mg/day) monotherapy for 7-10 days. PD assessments were conducted while on aspirin and on clopidogrel using light transmittance aggregometry following stimuli with U-46619 [TXA2 stable analogue (7 μM)], arachidonic acid [AA (1 mM)], collagen (3 μg/mL) and adenosine diphosphate [ADP (5 μM and 20 μM)] with and without in vitro EV-077. EV-077 completely inhibited U-46619-stimulated platelet aggregation (p = 0.005 for both aspirin and clopidogrel) and also showed a significant reduction of collagen-induced aggregation (aspirin p = 0.008; clopidogrel p = 0.005). EV-077 significantly reduced AA-induced platelet aggregation in clopidogrel (p = 0.009), but not aspirin (p = 0.667) treated patients. Ultimately, EV-077 significantly reduced ADP-mediated platelet aggregation in both aspirin (ADP 5 μM p = 0.012; ADP 20 μM p = 0.032) and clopidogrel (ADP 5 μM p = 0.007; ADP 20 μM p = 0.008) treated patients. In conclusion, in DM patients with CAD on aspirin or clopidogrel monotherapy, in vitro EV-077 exerts potent platelet inhibitory effects on multiple platelet signaling pathways. These data support that EV-077 has only additive platelet inhibiting effects on top of standard antiplatelet therapies. These findings warrant further investigation in ex vivo settings.
Description
EV-077 is a small compound being developed for the treatment of complications of diabetes. In Phase 2. Outlicensed to Serodus in 2013.
Situation Overview
Diabetes and its complications are major global health care problems. Based on estimates by the International Diabetes Federation (IDF), there were 366 million diabetics worldwide in 2011, a number which is expected to increase to 552 million by 2030. IDF estimates the number of deaths in 2011 at 4.6 million and total spending on diabetic health care at USD 465 billion.
EV-077 is an oral, small molecule compound, belonging to a new structural class. EV-077 is being developed for the reduction of vascular inflammation by inhibiting the activity of prostanoids and isoprostanes ��� in particular in diabetes. Towards the end of 2011, the Russian Patent Office granted patent protection for EV-077 in the treatment of complications of diabetes for a term extending to 2026. Additional patent applications are pending in all major territories. Evolva has outlicenced EV-077 to Serodus in 2013.
Mechanism of Action
Preclinical and early clinical studies indicate EV-077 has potential in reducing vascular inflammation by inhibiting the activity of prostanoids and isoprostanes in particular in diabetes. The mechanism of action of EV-077 means that it can potentially ameliorate or prevent a range of diabetic complications (including loss of kidney function, reduced peripheral blood flow and increased risk of thrombosis) that derive from the following chain of events:
- Diabetic patients have a reduced sensitivity to insulin which increases overall glucose levels in the body;
- This increase in glucose increases oxidative stress;
- The oxidative stress generates a high level of isoprostanes and prostanoids;
- The isoprostanes and prostanoids chronically activate thromboxane prostanoid receptors, that are located on the walls of blood vessels (endothelial cells and smooth muscle cells) and the surface of platelets;
- Activation of the thromboxane prostanoid receptors causes vascular inflammation and increased platelet reactivity;
- An increased number of vascular events and a progressive deterioration of circulatory and renal function.
Clinical Trials
In November 2011, Evolva received regulatory clearance to progress EV-077 into Phase IIa clinical studies for the treatment of complications of diabetes. It is a single-centre study, conducted in Germany. The study was a randomized, double-blind, and placebo-controlled, and investigated the efficacy and safety of EV-077 in type 2 diabetics with a heightened risk of diabetic vascular complications. Measurements included blood flow and platelet reactivity, biomarkers for oxidative stress and vascular inflammation as well as markers of the function of organs that are often impaired in diabetes (e.g. kidney).
In May 2012, the study was terminated. Interim results for the first 32 patients enrolled in the Phase IIa study show promising efficacy data, indicating that 300mg EV-077 given orally twice daily to patients with type 2 diabetes provided anti-platelet activity, reduced exercise-induced proteinuria and increased forearm blood flow. This was achieved with only a slight increase in bleeding time. The analysis also indicated that EV-077 was generally well tolerated, with adverse events mostly limited to increases in liver enzymes, which were transient or resolved after discontinuation.
In parallel with the Phase IIa study, Evolva is conducting epidemiological studies to identify high risk diabetic patient subgroups that can potentially derive particular benefit from the administration of EV-077. Given success, this is expected to expedite both further clinical development (by reducing the size and duration of late stage clinical trials) and the eventual approval process.
Partners by Region
Evolva has outlicensed EV-077 to Serodus in 2013. Serodus aims to bring EV-077 further through clinical development and at a future time point decide whether Serodus or a partner will conduct the final clinical trials.
WO 2014011273
http://www.google.com/patents/WO2014011273A2?cl=en
Journal of Thrombosis and Haemostasis (2011), 9(10), 2109-2111
Thrombosis Research (2012), 130(5), 746-752
European Journal of Clinical Pharmacology (2013), 69(3), 459-465
Biochemical and Biophysical Research Communications (2013), 441(2), 393-398
Journal of Thrombosis and Thrombolysis (2014), 37(2), 131-138
http://www.google.co.in/patents/WO2008089461A1?cl=en
(Z)-6-((2S,4S,5R)-2-(2-chlorophenyl)-4-(2-hydroxyphenyl)-1 ,3-dioxan-5-yl)hex-4-enoic acid has the 3 groups all up, which has a dramatic effect on its biological activities:

see
WO 2011057262
/////////////// SEE……..http://drugsynthesisint.blogspot.in/2015/11/ev-077.html
ZYD 1/ZYDPLA 1 From Zydus Cadila, a New NCE in Gliptin class of Antidiabetic agents.

GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..
![]()
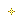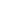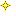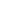


ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………


Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

Scheme 2:

Scheme 3:

Scheme 4A:


Scheme 4B.
] Scheme 5 A:

Scheme 5B:

Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.


http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Session: LBSU 1074-1087-Diabetes & Obesity
Translational
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors
