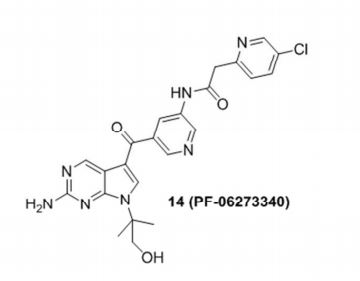
PF-06273340
N-(5-(2-amino-7-(1-hydroxy-2-methylpropan-2-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonyl)pyridin-3-yl)-2-(5-chloropyridin-2-yl)acetamide
CAS 1402438-74-7
Chemical Formula: C23H22ClN7O3
Molecular Weight: 479.925
- Originator Pfizer
- ClassAnalgesics; Small molecules
- Mechanism of ActionUndefined mechanism
- 06 Oct 2014 Pfizer plans a phase I trial in Pain (In volunteers) in the Netherlands (NCT02260947)
- 07 Aug 2014 Discontinued – Phase-I for Pain (In volunteers) in Belgium (PO)
- 07 Aug 2014 Discontinued – Phase-I for Pain (In volunteers) in Singapore (PO)
PF-06273340 is a Potent, Selective, and Peripherally Restricted Pan-Trk Inhibitor with an excellent LipE profile (IC50 value: Trk-A = 6 nM; Trk-B = 4 nM; Trk-C = 3 nM). PF-06273340 has low metabolic turnover in HLM and hHep is a good substrate for efflux transporters P-gp (ER = 35.7) and BCRP (ER = 4.0) and has moderate passive permeability (RRCK Papp = 5.4 × 10−6 cm s−1). PF-06273340 is well-tolerated was selected as a candidate for clinical development.
![ChemSpider 2D Image | N-(5-{[2-Amino-7-(1-hydroxy-2-methyl-2-propanyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]carbonyl}-3-pyridinyl)-2-(5-chloro-2-pyridinyl)acetamide | C23H22ClN7O3](http://www.chemspider.com/ImagesHandler.ashx?id=48244961&w=250&h=250)
Tropomyosin-related kinases (Trks) are a family of receptor tyrosine kinases activated by neurotrophins. Trks play important roles in pain sensation as well as tumour cell growth and survival signaling. Thus, inhibitors of Trk receptor kinases might provide targeted treatments for conditions such as pain and cancer. Recent developments in this field have been reviewed by Wang et al in Expert Opin. Ther.
Patents (2009) 19(3): 305-319 and an extract is reproduced below.
“1.1 Trk receptors
As one of the largest family of proteins encoded by the human genome, protein kinases are the central regulators of signal transduction as well as control of various complex cell processes. Receptor tyrosine kinases (RTKs) are a subfamily of protein kinases (up to 100 members) bound to the cell membrane that specifically act on the tyrosine residues of proteins. One small group within this subfamily is the Trk kinases, with three highly homologous isoforms: TrkA, TrkB, and TrkC. All three isofonns are activated by high affinity growth factors named neurotrophins (NT): i) nerve growth factor (NGF), which activates TrkA; ii) brain-derived neurotrophic factor (BDNF) and NT-4/5, which activate TrkB; and iii) NT-3, which activates TrkC. The binding of neurotrophins to the extracellular domain of Trks causes the Trk kinase to autophosphorylate at several intracellular tyrosine sites and triggers downstream signal transduction pathways. Trks and neurotrophins are well known for their effects on neuronal growth and survival.
1.2 Trks and cancer
Originally isolated from neuronal tissues, Trks were thought to mainly affect the maintenance and survival of neuronal cells. However, in the past 20 years, increasing evidence has suggested that Trks play key roles in malignant transformation, chemotaxis, metastasis, and survival signaling in human tumors. The association between Trks and cancer focused on prostate cancer in earlier years and the topic has been reviewed. For example, it was reported that malignant prostate epithelial cells secrete a series of neurotrophins and at least one Trks. In pancreatic cancer, it was proposed that paracrine and/or autocrine neurotrophin-Trk interactions may influence the invasive behavior of the cancer. TrkB was also reported to be overexpressed in metastatic human pancreatic cancer cells. Recently, there have been a number of new findings in other cancer settings. For example, a translocation leads to expression of a fusion protein derived from the W-terminus of the ETV9 transcription factor and the C-terminal kinase domain of TrkC. The resulting ETV6-TrkC fusions are oncogenic in vitro and appear causative in secretory breast carcinoma and some acute myelogenous leukemias (AML). Constitutively active TrkA fusions occurred in a subset of papillary thyroid cancers and colon carcinomas. In neuroblastoma, TrkB expression was reported to be a strong predictor of aggressive tumor growth and poor prognosis, and TrkB overexpression was also associated with increased resistance to chemotherapy in neuroblastoma tumor cells in vitro. One report showed that a novel splice variant of TrkA called TrkAIII signaled in the absence of neurotrophins through the inositol phosphate-AKT pathway in a subset of neuroblastoma. Also, mutational analysis of the tyrosine kinome revealed that Trk mutations occurred in colorectal and lung cancers. In summary, Trks have been linked to a variety of human cancers, and discovering a Trk inhibitor and testing it clinically might provide further insight to the biological and medical hypothesis of treating cancer with targeted therapies.
1.3 Trks and pain
Besides the newly developed association with cancer, Trks are also being recognized as an important mediator of pain sensation. Congenital insensitivity to pain with anhidrosis (CIPA) is a disorder of the peripheral nerves (and normally innervated sweat glands) that prevents the patient from either being able to adequately perceive painful stimuli or to sweat. TrkA defects have been shown to cause CIPA in various ethnic groups.
Currently, non-steroidal anti-inflammatory drugs (NSAIDs) and opiates have low efficacy and/or side effects (e.g., gastrointestinal/renal and psychotropic side effects, respectively) against neuropathic pain and therefore development of novel pain treatments is highly desired. It has been recognized that NGF levels are elevated in response to chronic pain, injury and inflammation and the administration of exogenous NGF increases pain hypersensitivity. In addition, inhibition of NGF function with either anti- NGF antibodies or non-selective small molecule Trk inhibitors has been shown to have effects on pain in animal models. It appears that a selective Trk inhibitor (inhibiting at least NGF’s target, the TrkA receptor) might provide clinical benefit for the treatment of pain. Excellent earlier reviews have covered targeting NGF/BDNF for the treatment of pain so this review will only focus on small molecule Trk kinase inhibitors claimed against cancer and pain. However, it is notable that the NGF antibody tanezumab was very recently reported to show good efficacy in a Phase II trial against osteoarthritic knee pain.”
International Patent Application publication number WO2009/012283 refers to various fluorophenyl compounds as Trk inhibitors; International Patent Application publication numbers WO2009/152087, WO2008/080015 and WO2008/08001 and WO2009/152083 refer to various fused pyrroles as kinase modulators; International Patent Application publication numbers WO2009/143024 and WO2009/143018 refer to various pyrrolo[2,3-d]pyrimidines substituted as Trk inhibitors; International Patent Application publication numbers WO2004/056830 and WO2005/1 16035 describe various 4-amino-pyrrolo[2,3- d]pyrimidines as Trk inhibitors. International Patent Application publication number WO201 1/133637 describes various pyrrolo[2,3-d]pyrimidines and pyrrolo[2,3-b]pyridines as inhibitors of various kinases.
US provisional application US61/471758 was filed 5th April 2012 and the whole contents of that application in it’s entirety are herewith included by reference thereto.
Thus Trk inhibitors have a wide variety of potential medical uses. There is a need to provide new Trk inhibitors that are good drug candidates. In particular, compounds should preferably bind potently to the Trk receptors in a selective manner compared to other receptors, whilst showing little affinity for other receptors, including other kinase and / or GPC receptors, and show functional activity as Trk receptor antagonists. They should be non-toxic and demonstrate few side-effects. Furthermore, the ideal drug candidate will exist in a physical form that is stable, non-hygroscopic and easily formulated. They should preferably be e.g. well absorbed from the gastrointestinal tract, and / or be injectable directly into the bloodstream, muscle, or subcutaneously, and / or be metabolically stable and possess favourable pharmacokinetic properties.
Among the aims of this invention are to provide orally-active, efficacious, compounds and salts which can be used as active drug substances, particularly Trk antagonists, i.e. that block the intracellular kinase activity of the Trk, e.g. TrkA (NGF) receptor. Other desirable features include good HLM/hepatocyte stability, oral bioavailability, metabolic stability, absorption, selectivity over other types of kinase, dofetilide selectivity. Preferable compounds and salts will show a lack of CYP inhibition/induction, and be CNS- sparing.
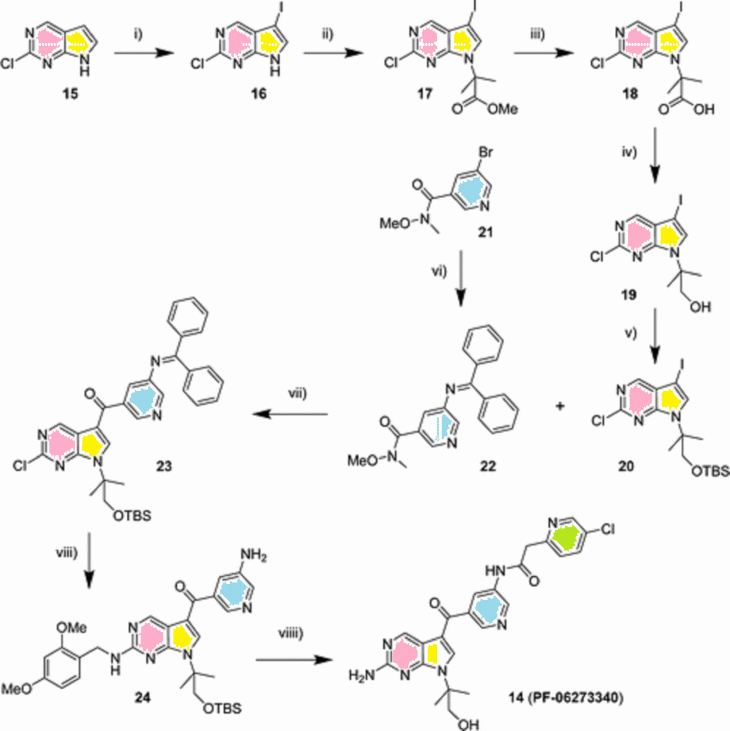
aReagents and conditions:
(i) NIS, MeCN, 12 °C to rt, 1 h, 82%;
(ii) BrMe2CO2Me, KI, Cs2CO3, DMF, 60°C, 19 h, 92%;
(iii) LiOH, THF/H2O, 60 °C, 3 h, 90%;
(iv) DIBAL-H, THF, 0 °C, 1.5 h, 56%;
(v) TBMS-Cl, imidazole, DMF, 0 °C to rt, 16 h, 96%;
(vi) benzophenone imine, Pd2(dba)3, K3PO4, DME, 50 °C, 17 h, 51%;
(vii) 20, i-PrMgCl, THF, 0 °C, then 22, THF, 0 °C to rt, 16 h, 66%;
(viii) 2,4-dimethoxybenzylamine, DMAP, 1,4-dioxane, reflux, 2 d, then citric acid, THF, rt, 5 h, 78%;
(viiii) 2-(5-chloropyridin-2-yl)acetic acid, T3P, Et3N, THF, rt, 2 h, then TFA, 50°C, 3 h, then K2CO3, MeOH, rt, 16 h, 48%.
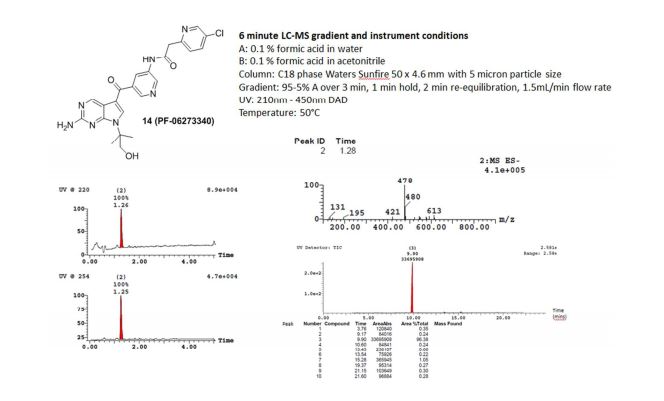
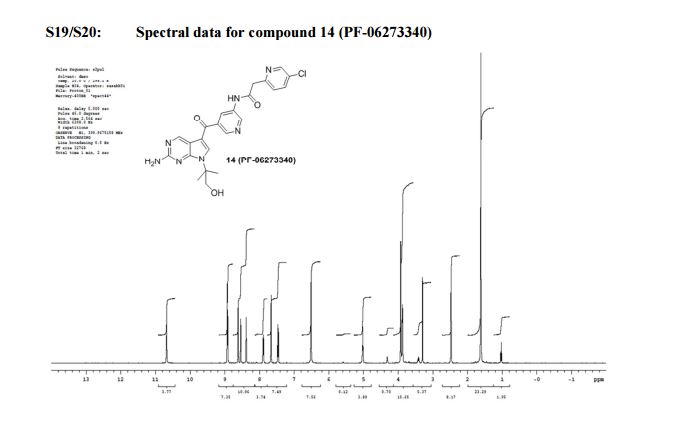
1H NMR (400 MHz, DMSO-d6) δ: 1.64 (s, 6H), 3.90 (d, J = 5.5, 2H), 3.95 (s, 2H), 5.05 (dd, J = 5.7, 5.5, 1H), 6.54 (br s, 2H), 7.49 (d, J = 8.4, 1H), 7.69 (s, 1H), 7.92 (dd, J = 8.3, 2.4, 1H), 8.40 (m, 1H), 8.56 (d, J = 2.5, 1H), 8.64 (d, J = 1.8, 1H), 8.94 (d, J = 2.2, 1H), 8.96 (s, 1H), 10.71 (s, 1H). HPLC (6 min, acid) Rt 1.26 min; UV 220 nM 100% purity; LC-MS (ES–) m/z 478 (M – H+); HRMS (ES+) m/z 480.15468 (M + H+).
SYNTHESIS
WO-2012137089-A1
https://www.google.com/patents/WO2012137089A1?cl=enhttps://www.google.com/patents/WO2012137089A1?cl=en

MARK ANDREWS

Sharanjeet Kaur Bagal,

Karl Gibson

Kiyoyuki OMOTO

Thomas Ryckmans,
Example 46: N-(5-{[2-Amino-7-(2-hydroxy-1 ,1-dimethylethyl)-7H-pyrrolo[2,3-d]pyrimidin-5- yl]carbonyl}pyridin-3-yl)-2-(5-chloropyridin-2-yl)acetamide
(5-Chloropyridin-2-yl)acetic acid (26.1 g, 152 mmol) (see Preparation 90) was added to (5-aminopyridin- 3-yl){7-(2-{[ferf-butyl(dimethyl)silyl]oxy}-1 , 1-dimethylethyl)-2-[(2,4-dimethoxybenzyl)ami
d]pyrimidin-5-yl}methanone (75.0 g, 130 mmol ) (see Preparation 51 ), 1-propylphosphonic acid cyclic anhydride (187 mL, 317 mmol, 50% solution in EtOAc) and triethylamine (61.9 mL, 444 mmol ) in THF (423 mL). The mixture was stirred at 25°C for 2 hours then saturated aqueous sodium bicarbonate (400 mL) was added and the organic layer was separated. The aqueous phase was extracted with EtOAc (400 mL) and all organic phases were combined and dried over sodium sulfate then evaporated in vacuo. The residue brown solid was dissolved in trifluoroacetic acid (300 mL) and the solution was stirred at 50°C for 3 hours then evaporated in vacuo. Methanol (1800 mL) was added to the residue and the mixture was filtered. The filtrate was evaporated in vacuo and azeotroped with ethanol (3 x 200 mL).
Potassium carbonate (87.7 g, mmol) was added to the crude trifluoroacetamide in methanol (300 mL) and the mixture was stirred at room temperature for 16 hours. The mixture was poured into water (2000 mL) and filtered. The solid was washed with water (200 mL) then triturated with ethanol (2 x 200 mL at room temperature then 380 mL at 50°C) to afford the title compound as a yellow solid in 48% yield, 29.9 g. H NMR (400 MHz, DMSO-c/6) δ: 1.64 (s, 6H), 3.90 (d, 2H), 3.95 (s, 2H), 5.05 (t, 1 H), 6.54 (br s, 2H), 7.49 (d, 1 H), 7.69 (s, 1 H), 7.92 (dd, 1 H), 8.40 (m, 1 H), 8.56 (m, 1 H), 8.64 (d, 1 H), 8.94 (d, 1 H), 8.96 (s, 1 H), 10.71 (s, 1 H); LCMS (System 3): Rt = 9.92 min; m/z 480 [M+H]+.


PAPER
The Discovery of a Potent, Selective, and Peripherally Restricted Pan-Trk Inhibitor (PF-06273340) for the Treatment of Pain
Sarah E. Skerratt*†, Mark Andrews‡, Sharan K. Bagal†, James Bilsland†, David Brown‡, Peter J. Bungay†, Susan Cole‡, Karl R Gibson‡, Russell Jones‡, Inaki Morao‡, Angus Nedderman‡, Kiyoyuki Omoto†, Colin Robinson‡,Thomas Ryckmans‡, Kimberly Skinner‡, Paul Stupple‡, and Gareth Waldron†
†Pfizer Global Research & Development, The Portway Building, Granta Park, Great Abington, Cambridge, CB21 6GS, U.K.
‡Pfizer Global Research & Development, Ramsgate Road, Sandwich CT13 9NJ, U.K.
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.6b00850
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.


Sarah E. Skerratt
MARK ANDREWS
Abstract
The neurotrophin family of growth factors, comprised of nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), neurotrophin 3 (NT3), and neurotrophin 4 (NT4), is implicated in the physiology of chronic pain. Given the clinical efficacy of anti-NGF monoclonal antibody (mAb) therapies, there is significant interest in the development of small molecule modulators of neurotrophin activity. Neurotrophins signal through the tropomyosin related kinase (Trk) family of tyrosine kinase receptors, hence Trk kinase inhibition represents a potentially “druggable” point of intervention. To deliver the safety profile required for chronic, nonlife threatening pain indications, highly kinase-selective Trk inhibitors with minimal brain availability are sought. Herein we describe how the use of SBDD, 2D QSAR models, and matched molecular pair data in compound design enabled the delivery of the highly potent, kinase-selective, and peripherally restricted clinical candidate PF-06273340.
ADDITIONAL INFORMATION
The aqueous solubility of PF-06273340 is 131 μM, much improved over previous analogues, it is highly kinase-selective (Gini score of 0.92) and has no measurable activity at the hERG channel. PF-06273340 was profiled in a series of in vitro safety assays, showing little cytotoxicity in THLE or HepG2 cell lines (IC50 > 42 μM and >300 μM, respectively) and was evaluated for broader pharmacological activity in a panel of receptors, ion channels, and enzymes. In this broad panel, all IC50/Ki values were >10 μM except for COX-1 (IC50 = 2.7 μM) and dopamine transporter assays (Ki = 5.2 μM) and PDEs 4D, 5A, 7B, 8B, and 11 (54−89% inhibition at 10 μM). PF-06273340 was screened in the Invitrogen wide kinase panel of 309 kinases, and all were inhibited by <40% when tested at 1 μM except the following: MUSK (IC50 53 nM), FLT-3 (IC50 395 nM), IRAK1 (IC50 2.5 μM), MKK (90% @ 1 μM), and DDR1 (60% @ 1 μM).
REFERENCES
1: Skerratt SE, Andrews MD, Bagal SK, Bilsland J, Brown D, Bungay PJ, Cole S,
Gibson KR, Jones R, Morao I, Nedderman A, Omoto K, Robinson C, Ryckmans T,
Skinner K, Stupple PA, Waldron G. The Discovery of a Potent, Selective and
Peripherally Restricted Pan-Trk Inhibitor (PF-06273340) for the Treatment of
Pain. J Med Chem. 2016 Oct 21. [Epub ahead of print] PubMed PMID: 27766865.

Gareth Waldron

Paul Stupple
/////////
CC(C)(CO)n1cc(C(=O)c2cncc(NC(=O)Cc3ccc(Cl)cn3)c2)c4cnc(N)nc14

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....