
Home » Posts tagged 'PEPTIDES'
Tag Archives: PEPTIDES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pepinemab, VX 15
(Heavy chain)
QVQLVQSGAE VKKPGSSVKV SCKASGYSFS DYYMHWVRQA PGQGLEWMGQ INPTTGGASY
NQKFKGKATI TVDKSTSTAY MELSSLRSED TAVYYCARYY YGRHFDVWGQ GTTVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFLGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
DIVMTQSPDS LAVSLGERAT INCKASQSVD YDGDSYMNWY QQKPGQPPKL LIYAASNLES
GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI KRTVAAPSVF
IFPPSDEQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS GNSQESVTEQ DSKDSTYSLS
STLTLSKADY EKHKVYACEV THQGLSSPVT KSFNRGEC
(Disulfide bridge: H22-H96, H132-L218, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’218, H’145-H’201, H’259-H’319, H’365-H’423, L23-L92, L138-L198, L’23-L’92, L’138-L’198)
Pepinemab
VX15/2503
Antineoplastic, Anti-human semaphorin 4D antibody
Monoclonal antibody
Treatment of solid tumors, multiple sclerosis and Huntington’s disease
| Formula | C6442H9910N1702O2052S48 |
|---|---|
| MOL WGT | 145481.0022 |
- Moab VX15/2503
- Pepinemab
- UNII-BPZ4A29SYE
- VX-15
- VX15
- VX15/2503
| Product name | Pepinemab Biosimilar – Anti-SEMA4D mAb – Research Grade |
|---|---|
| Source | CAS 2097151-87-4 |
| Species | Chimeric,Humanized |
| Expression system | Mammalian cells |
- OriginatorVaccinex
- DeveloperBristol-Myers Squibb; Children’s Oncology Group; Emory University; Merck KGaA; National Cancer Institute (USA); Teva Pharmaceutical Industries; UCLAs Jonsson Comprehensive Cancer Center; Vaccinex
- ClassAntibodies; Antidementias; Antineoplastics; Immunotherapies; Monoclonal antibodies
- Mechanism of ActionCD100 antigen inhibitors
- Orphan Drug StatusYes – Huntington’s disease
- New Molecular EntityYes
- Phase IIHuntington’s disease
- Phase I/IIAlzheimer’s disease; Non-small cell lung cancer; Osteosarcoma; Solid tumours; Squamous cell cancer
- Phase IColorectal cancer; Malignant melanoma; Pancreatic cancer
- No development reportedMultiple sclerosis
- 22 May 2021Pepinemab is still in phase I trials for Colorectal cancer and Pancreatic cancer in USA (NCT03373188)
- 17 May 2021Phase-I/II clinical trials in Squamous cell cancer (Combination therapy, Late-stage disease, Metastatic disease, Recurrent, Second-line therapy or greater) in USA (IV) (NCT04815720)
- 17 May 2021Vaccinex plans a phase I/II trial for Alzheimer’s disease (In volunteers), in H2 2021
Semaphorin 4D (SEMA4D) plays a role in multiple cellular processes that contribute to the pathophysiology of neuroinflammatory/neurodegenerative diseases. SEMA4D is, therefore, a uniquely promising target for therapeutic development.
Pepinemab is a novel monoclonal antibody that blocks the activity of SEMA4D, and preclinical testing has demonstrated the beneficial effects of anti-SEMA4D treatment in a variety of neurodegenerative disease models. Vaccinex is committed to the development of this potentially important antibody that has the potential to help people with different neurodegenerative disorders that share common mechanisms of pathology.
Note: Pepinemab (VX15/2503) is an investigational drug currently in clinical studies. It has not been demonstrated to be safe and effective for any disease indication. There is no guarantee that pepinemab (VX15/2503) will be approved for the treatment of any disease by the U.S. Food and Drug Administration or by any other health authority worldwide.
////////////////////Pepinemab, VX15/2503, vx 15, Antineoplastic, Anti-human semaphorin 4D antibody, Monoclonal antibody, solid tumors, multiple sclerosis, Huntington’s disease, PEPTIDES
NEW DRUG APPROVALS
ONE TIME
$10.00
TROFINETIDE

Trofinetide
- Molecular FormulaC13H21N3O6
- Average mass315.322 Da
Tofinetide , NNZ-256610076853400-76-7[RN]
glycyl-2-methyl-L-prolyl-L-glutamic acid
H-Gly-PMe-Glu-OHL-Glutamic acid, glycyl-2-methyl-L-prolyl-UNII-Z2ME8F52QLZ2ME8F52QLтрофинетид [Russian] [INN]تروفينيتيد [Arabic] [INN]曲非奈肽 [Chinese] [INN]
| IUPAC Condensed | H-Gly-aMePro-Glu-OH |
|---|---|
| Sequence | GXE |
| HELM | PEPTIDE1{G.[*C(=O)[C@@]1(CCCN1*)C |$_R2;;;;;;;;_R1;$|].E}$$$$ |
| IUPAC | glycyl-alpha-methyl-L-prolyl-L-glutamic acid |
An (1-3) IGF-1 analog with neuroprotective activity.
OPTICAL ROT; -52.4 ° Conc: 0.19 g/100mL; water ; 589.3 nm; Temp: 20 °C; Len: 1.0 dm…Tetrahedron 2005, V61(42), P10018-10035
EU Customs Code CN, 29339980
Harmonized Tariff Code, 293399
- L-Glutamic acid, glycyl-2-methyl-L-prolyl-
- glycyl-2-methyl-L-prolyl-L-glutamic acid
- Glycyl-L-2-methylprolyl-L-glutamic acid
FDA APPROVED 2023/3/10, Daybue
853400-76-7 CAS
トロフィネチド;
Trofinetide (NNZ-2566) is a drug developed by Neuren Pharmaceuticals that acts as an analogue of the neuropeptide (1-3) IGF-1, which is a simple tripeptide with sequence Gly–Pro–Glu formed by enzymatic cleavage of the growth factor IGF-1 within the brain. Trofinetide has anti-inflammatory properties and was originally developed as a potential treatment for stroke,[1][2] but has subsequently been developed for other applications and is now in Phase II clinical trials against Fragile X syndrome and Rett syndrome.[3][4][5]
Trofinetide (NNZ-2566), a neuroprotective analogue of glypromate, is a novel molecule that has a profile suitable for both intravenous infusion and chronic oral delivery. It is currently in development to treat traumatic brain injury.
In February 2021, Neuren is developing trofinetide (NNZ-2566, phase 2 clinical ), a small-molecule analog of the naturally occurring neuroprotectant and N-terminus IGF-1 tripeptide Glypromate (glycine-proline-glutamate), for intravenous infusion treatment of various neurological conditions, including moderate to severe traumatic brain injury (TBI), stroke, chronic neurodegenerative disorders and peripheral neuropathies. At the same time, Neuren is also investigating an oral formulation of trofinetide (phase 3 clinical) for similar neurological indications, including mild TBI.
Autism Spectrum Disorders and neurodevelopment disorders (NDDs) are becoming increasingly diagnosed. According to the fourth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-4), Autism spectrum disorders (ASD) are a collection of linked developmental disorders, characterized by abnormalities in social interaction and communication, restricted interests and repetitive behaviours. Current classification of ASD according to the DSM-4 recognises five distinct forms: classical autism or Autistic Disorder, Asperger syndrome, Rett syndrome, childhood disintegrative disorder and pervasive developmental disorder not otherwise specified (PDD-NOS). A sixth syndrome, pathological demand avoidance (PDA), is a further specific pervasive developmental disorder.
More recently, the fifth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-5) recognizes recognises Asperger syndrome, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDD-NOS) as ASDs.
This invention applies to treatment of disorders, regardless of their classification as either DSM-4 or DSM-5.
Neurodevelopment Disorders (NDDs) include Fragile X Syndrome (FXS), Angelman Syndrome, Tuberous Sclerosis Complex, Phelan McDermid Syndrome, Rett Syndrome, CDKL5 mutations (which also are associated with Rett Syndrome and X-Linked Infantile Spasm Disorder) and others. Many but not all NDDs are caused by genetic mutations and, as such, are sometimes referred to as monogenic disorders. Some patients with NDDs exhibit behaviors and symptoms of autism.
As an example of a NDD, Fragile X Syndrome is an X-linked genetic disorder in which affected individuals are intellectually handicapped to varying degrees and display a variety of associated psychiatric symptoms. Clinically, Fragile X Syndrome is characterized by intellectual handicap, hyperactivity and attentional problems, autism spectrum symptoms, emotional lability and epilepsy (Hagerman, 1997a). The epilepsy seen in Fragile X Syndrome is most commonly present in childhood, but then gradually remits towards adulthood. Hyperactivity is present in approximately 80 percent of affected males (Hagerman, 1997b). Physical features such as prominent ears and jaw and hyper-extensibility of joints are frequently present but are not diagnostic. Intellectual handicap is the most common feature defining the phenotype. Generally, males are more severely affected than females. Early impressions that females are unaffected have been replaced by an understanding of the presence of specific learning difficulties and other neuropsychiatric features in females. The learning disability present in males becomes more defined with age, although this longitudinal effect is more likely a reflection of a flattening of developmental trajectories rather than an explicit neurodegenerative process.
The compromise of brain function seen in Fragile X Syndrome is paralleled by changes in brain structure in humans. MRI scanning studies reveal that Fragile X Syndrome is associated with larger brain volumes than would be expected in matched controls and that this change correlates with trinucleotide expansion in the FMRP promoter region (Jakala et al, 1997). At the microscopic level, humans with Fragile X Syndrome show abnormalities of neuronal dendritic structure, in particular, an abnormally high number of immature dendritic spines (Irwin et al, , 2000).
Currently available treatments for NDDs are symptomatic – focusing on the management of symptoms – and supportive, requiring a multidisciplinary approach. Educational and social skills training and therapies are implemented early to address core issues of learning delay and social impairments. Special academic, social, vocational, and support services are often required. Medication, psychotherapy or behavioral therapy may be used for management of co-occurring anxiety, ADHD, depression, maladaptive behaviors (such as aggression) and sleep issues, Antiepileptic drugs may be used to control seizures.
Patent
WO 2014085480,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014085480

EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro-Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
WO95/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS to increase TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses certain GPE analogs having amino acid substitutions and certain other modification that are capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including injury or disease in the CNS.
EXAMPLES
The following examples are intended to illustrate embodiments of this invention, and are not intended to limit the scope to these specific examples. Persons of ordinary skill in the art can apply the disclosures and teachings presented herein to develop other embodiments without undue experimentation and with a likelihood of success. All such embodiments are considered part of this invention.
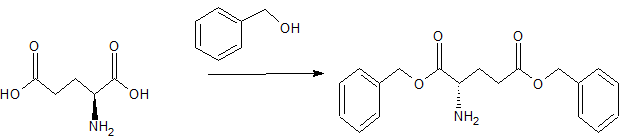
Example 1: Synthesis of N,N-Dimethylglycyl-L-prolyl)-L-glutamic acid
The following non-limiting example illustrates the synthesis of a compound of the invention, N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
All starting materials and other reagents were purchased from Aldrich; BOC=tert-butoxycarbonyl; Bn=benzyl.
BOC-L-proline-(P-benzyl)-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem. Soc: 79, 6810, 1994] (10 mmol) in dichloromethane (50 mi), cooled to 0°C, was added triethylamine (1 .39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl-L-glutamate (10 mmol) was then added and the mixture stirred at 0° C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol 1-1) then dried (MgSO4) and concentrated at reduced pressure to give BOC-L-proline-L-glutamic acid dibenzyl ester (5.0 g, 95%).
L-proline-L-glutamic acid dibenzyl ester
A solution of BOC-L-glutamyl-L-proline dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 h. at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give L-proline-L-glutamic acid dibenzyl ester.
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of L-proline-L-glutamic acid dibenzyl ester (10 mmol), N,N-dimethylglycine (10 mmol) and triethylamine ( 10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0°C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallised from ethyl acetate to yield the tripeptide derivative.
It can be appreciated that following the method of the Examples, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Eample 2: Synthesis of Glycyl-L-2-Methyl-L-Prolyl-L-Glutamate
L-2-Methylproline and L-glutamic acid dibenzyl ester p-toluenesulphonate were purchased from Bachem, N-benzyloxycarbonyl-glycine from Acros Organics and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BoPCl, 97%) from Aldrich Chem. Co.
Methyl L-2-methylprolinate hydrochloride 2
Thionyl chloride (5.84 cm3, 80.1 mmol) was cautiously added dropwise to a stirred solution of (L)-2-methylproline 1 (0.43 g, 3.33 mmol) in anhydrous methanol (30 cm3) at -5 °C under an atmosphere of nitrogen. The reaction mixture was heated under reflux for 24 h, and the resultant pale yellow-coloured solution was. concentrated to dryness in vacuo. The residue was dissolved in a 1 : 1 mixture of methanol and toluene (30 cm3) then concentrated to dryness to remove residual thionyl chloride. This procedure was repeated twice more, yielding hydrochloride 2 (0.62 g, 104%) as an hygroscopic, spectroscopically pure, off-white solid: mp 127- 131 °C; [α]D -59.8 (c 0.24 in CH2Cl2); vmax (film)/cm-1 3579, 3398 br, 2885, 2717, 2681 , 2623, 2507, 1743, 1584, 1447, 1432, 1374, 1317, 1294, 1237, 1212, 1172, 1123, 981 , 894, 861 and 764; δH (300 MHz; CDCl3; Me4Si) 1.88 (3H, s, Proα-CH3), 1 .70-2.30 (3H, br m, Proβ-HAΗΒ and Proγ-H2), 2.30-2.60 (1H, br m, Proβ-HAΗΒ), 3.40-3.84 (2H, br m, Proδ-H2), 3.87 (3H, s, CO2CH3), 9.43 (1H, br s, NH) and 10.49 ( 1H, br s, HCl); δC (75 MHz; CDCl3) 21.1 (CH3, Proα-CH3), 22.4 (CH2, Proγ-C), 35.6 (CH2, Proβ-C), 45.2 (CH2, Proδ-C), 53.7 (CH3, CO2CH3), 68.4 (quat., Proα-C) and 170.7 (quat, CO); m/z (FAB+) 323.1745 [M2.H35Cl.H+: (C7H13NO2)2. H35Cl.H requires 323.1738] and 325.1718 [M2.H37Cl.H+: (C7H13NOz)2. H37Cl.H requires 325.1708],
N-Benxyloxycarbonyl-glycyl-L-2-methylproline 5
Anhydrous triethylamine (0.45 cm3, 3.23 mmol) was added dropwise to a mixture of methyl L-2-methylprolinate hydrochloride 2 (0.42 g, 2.34 mmol) and N-benzyloxycarbonyl-glycine (98.5%) 3 (0.52 g, 2.45 mmol) in methylene chloride (16 cm3), at 0 °C, under an atmosphere of nitrogen. The resultant solution was stirred for 20 min and a solution of 1 ,3-dicyclohexylcarbodiimide (0.56 g, 2.71 mmol) in methylene chloride (8 cm3) at 0 °C was added dropwise and the reaction mixture was warmed to room temperature and stirred for a further 20 h. The resultant white mixture was filtered through a Celite™ pad to partially remove 1 ,3-dicyclohexylurea, and the pad was washed with methylene chloride (50 cm3). The filtrate was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and concentrated to dryness in vacuo. Further purification of the residue by flash column chromatography (35 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) afforded tentatively methyl N-benzyloxycarbonyl-glycyl-L-2-methylprolinate 4 (0.56 g), containing 1 ,3-dicyclohexylurea, as a white semi-solid: Rf 0.65 (EtOAc); m/z (ΕI+) 334.1534 (M+. C17H22N2O5 requires 334.1529) and 224 ( 1 ,3-dicyclohexylurea).
To a solution of impure prolinate 4 (0.56 g, ca. 1.67 mmol) in 1,4-dioxane (33 cm3) was added dropwise 1 M aqueous sodium hydroxide (10 cm3, 10 mmol) and the mixture was stirred for 19 h at room temperature. Methylene chloride ( 100 cm3) was then added and the organic layer extracted with saturated aqueous sodium hydrogen carbonate (2 x 100 cm3). The combined aqueous layers were carefully acidified with hydrochloric acid (32%), extracted with methylene chloride (2 x 100 cm3), and the combined organic layers dried (MgSO4), filtered, and
concentrated to dryness in vacuo. Purification of the ensuing residue (0.47 g) by flash column chromatography ( 17 g SiO2; 50% ethyl acetate – hexane to 30% methanol – dichloromethane; gradient elution) gave N-protected dipeptide 5 (0.45 g, 60%) as a white foam in two steps from hydrochloride 2. Dipeptide 5 was shown to be exclusively the frafw-orientated conformer by NMR analysis: Rf 0.50 (20% MeOH – CH2Cl2); [α]D -62.3 (c 0.20 in CH2Cl2); vmax (film)/cm-1 3583, 3324 br, 2980, 2942, 1722, 1649, 1529, 1454, 1432, 1373, 1337, 1251 , 1219, 1179, 1053, 1027, 965, 912, 735 and 698; δH (300 MHz; CDCl3; Me4Si) 1.59 (3H, s, Proα-CH3), 1 .89 (1H, 6 lines, J 18.8, 6.2 and 6.2, Proβ-HAHB), 2.01 (2H, dtt, J 18.7, 6.2 and 6.2, Proγ-H2), 2.25-2.40 (1H, m, Proβ-HAΗΒ), 3.54 (2H, t, J 6.6, Proδ-H2), 3.89 (1H, dd, J 17.1 and 3.9, Glyα-HAHB), 4.04 (1H, dd, J 17.2 and 5.3, Glyα-HAΗΒ), 5.11 (2H, s, OCH2Ph), 5.84 (I H, br t, J 4.2, N-H), 7.22-7.43 (5H, m, Ph) and 7.89 (1 H, br s, -COOH); δC (75 MHz; CDCl3) 21.3 (CH3, Proα-CH3), 23.8 (CH2, Proγ-C), 38.2 (CH2, Proβ-C), 43.6 (CH2, Glyα-C), 47.2 (CH2, Proδ-C), 66.7 (quat, Proα-C), 66.8 (CH2, OCH2Ph), 127.9 (CH, Ph), 127.9 (CH, Ph), 128.4, (CH, Ph), 136.4 (quat., Ph), 156.4 (quat., NCO2), 167.5 (quat., Gly-CON) and 176.7 (quat., CO); m/z (EI+) 320.1368 (M+. C16Η20Ν2Ο5 requires 320.1372).
Dibenzyl N-benzyloxycarbonyl-glycyl-L-2-methylprolyl-L-glutamate 7
Triethylamine (0.50 cm3, 3.59 mmol) was added dropwise to a solution of dipeptide 5 (0.36 g, 1.12 mmol) and L-glutamic acid dibenzyl ester /Moluenesulphonate 6 (0.73 g, 1.46 mmol) in methylene chloride (60 cm3) under nitrogen at room temperature, and the reaction mixture stirred for 10 min. Bis(2-oxo-3-oxazoIidinyl)phosphinic chloride (BoPCl, 97%) (0.37 g, 1.41 mmol) was added and the colourless solution stirred for 17 h. The methylene chloride solution was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and evaporated to dryness in vacuo. Purification of the resultant residue by repeated (2x) flash column chromatography (24 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) yielded ƒully protected tripeptide 7 (0.63 g, 89%) as a colourless oil. Tripeptide 7 was shown to be exclusively the trans-orientated conformer by NMR analysis: Rf 0.55 (EtOAc); [α]D -41.9 (c 0.29 in CH2Cl2); vmax (film)/cm-1 3583, 3353 br, 2950, 1734, 1660, 1521, 1499, 1454, 1429, 1257, 1214, 1188, 1166, 1051, 911, 737 and 697; δH (400 MHz; CDCl3; Me4Si) 1.64 (3H, s, Proot-CH3), 1.72 (1H, dt, J 12.8, 7.6 and 7.6, Proβ-HAHB), 1.92 (2H, 5 lines, J 6.7, Proγ-H2), 2.04 (1H, 6 lines, J 7.3 Gluβ-HAHB), 2.17-2.27 (1H, m, Gluβ-HAΗΒ), 2.35-2.51 (3H, m, Proβ-HAΗΒ and Gluγ-H2), 3.37-3.57 (2H, m, Proδ-H2), 3.90 (1 H, dd, J 17.0 and 3.6, Glyα-HAHB), 4.00 (1H, dd, J 17.1 and 5.1, Glyα-HAΗΒ), 4.56 (1H, td, J 7.7 and 4.9, Glyα-H), 5.05-5.20 (6H, m, 3 x OCH2Ph), 5.66-5.72 (1H, br m, Gly-NH), 7.26-7.37 (15H, m, 3 x Ph) and 7.44 (1H, d, J 7.2, Glu-NH); δC (100 MHz; CDCl3) 21.9 (CH3, Proα-CH3), 23.4 (CH2, Proγ-C), 26.6 (CH2, Gluβ-C), 30.1 (CH2, Gluγ-C), 38.3 (CH2, Proβ-C),
43.9 (CH2, Glyα-C), 47.6 (CH2, Proδ-C), 52.2 (CH, Glua-C), 66.4 (CH2, OCH2Ph), 66.8 (CH2, OCH2Ph), 67.1 (CH2, OCH2Ph), 68.2 (quat, Proα-C), 127.9 (CH, Ph), 128.0 (CH, Ph), 128.1, (CH, Ph), 128.2, (CH, Ph), 128.2, (CH, Ph), 128.3, (CH, Ph), 128.4, (CH, Ph), 128.5, (CH, Ph), 128.5, (CH, Ph), 135.2 (quat., Ph), 135.7 (quat., Ph), 136.4 (quat, Ph), 156.1 (quat, NCO2), 167.3 (quat., Gly-CO), 171.4 (quat., CO), 172.9 (quat., CO) and 173.4 (quat., CO); m/z (FAB+) 630.2809 (MH+. C35H40N3O8 requires 630.2815).
Glycyl-L-2-methylprolyl-L-glutamic acid (G-2-MePE)
A mixture of the protected tripeptide 7 (0.63 g, 1.00 mmol) and 10 wt % palladium on activated carbon (0.32 g, 0.30 mmol) in 91 :9 methanol – water (22 cm3) was stirred under an atmosphere of hydrogen at room temperature, protected from light, for 23 h. The reaction mixture was filtered through a Celite™ pad and the pad washed with 75 :25 methanol – water (200 cm3). The filtrate was concentrated to dryness under reduced pressure and the residue triturated with anhydrous diethyl ether to afford a 38: 1 mixture of G-2-MePE and tentatively methylamine 8 (0.27 g, 86%) as an extremely hygroscopic white solid. Analytical reverse-phase HPLC studies on the mixture [Altech Econosphere C 18 Si column, 150 x 4.6 mm, 5 ☐m; 5 min flush with H2O (0.05% TFA) then steady gradient over 25 min to MeCN as eluent at flow rate of 1 ml/min; detection using diode array] indicated it was a 38: 1 mixture of two eluting peaks with retention times of 13.64 and 14.44 min at 207 and 197 nm, respectively. G-2-MePE was shown to be a 73 :27 trans:cis mixture of conformers by 1H NMR analysis (the ratio was estimated from the relative intensities of the double doublet and triplet at δ 4.18 and 3.71 , assigned to the Gluα-H protons of the major and minor conformers, respectively):
mp 144 °Cɸ;
[ α]D -52.4 (c 0.19 in H2O);
δα (300 MHz; D2O; internal MeOH) 1.52 (3H, s, Proα-CH3), 1.81-2.21 (6H, m, Proβ-H2, Proγ-H, and Gluβ-H2), 2.34 (1.46H, t, J 7.2, Gluy-H2), 2.42* (0.54H, t, 77.3, Gluγ-H2), 3.50-3.66 (2H, m, Pro6-H2), 3.71 * (0.27H, t, J 6.2, Gluoc-H), 3.85 (1H, d, J 16.6, Glyα-HAHB), 3.92 (1H, d, J 16.6, Glyα-HAΗΒ) and 4.18 (0.73H, dd, J 8.4 and 4.7, Glua-H);
δC (75 MHz; D2O; internal MeOH) 21.8 (CH3, Proα-CH3), 25.0 (CH2, Proγ-C), 27.8* (CH2: Gluβ-C), 28.8 (CH2, Gluβ-C), 32.9 (CH2, Gluγ-C), 40.8 (CH2, Proβ-C), 42.7 (CH2, Glyα-C), 49.5 (CH2, Proδ-C), 56.0* (CH, Gluα-C), 56.4 (CH, Gluα-C), 69.8 (quat, Proα-C), 166.5 (quat., Gly-CO), 177.3 (quat., Pro-CON), 179.2 (quat., Gluα-CO), 180.2* (quat., Gluγ-CO) and 180.6 (quat., Gluγ-CO);
m/z (FAB+) 3 16.1508 (MH+. C13H22N3O6 requires 316.1509).
PATENT
WO02094856
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Testing; Material and Methods
The following experimental protocol followed guidelines approved by the
University of Auckland animal ethics committee.
Preparation of cortical astrocyte cultures for harvest of metabolised cell culture supernatant
One cortical hemisphere from a postnatal day 1 rat was used and collected into
4ml of DMEM. Trituration was done with a 5ml glass pipette and subsequently through an 18 gauge needle. Afterwards, the cell suspension was sieved through a lOOμm cell strainer and washed in 50ml DMEM (centrifugation for 5min at 250g). The sediment was resuspended into 20ml DMEM+10% fetal calf serum. 10 Milliliters of suspension was added into each of two 25cm3 flasks and cultivated at 37°C in the presence of 10% C02, with a medium change twice weekly. After cells reached confluence, they were washed three times with PBS and adjusted to Neurobasal/B27 and incubated for another 3 days. This supernatant was frozen for transient storage until usage at -80°C.
Preparation of striatal and cortical tissue from rat E18/E19 embryos
A dam was sacrificed by C02-treatment in a chamber for up to 4 minutes and was prepared then for cesarean section. After surgery, the embryos were removed from their amniotic sacs, decapitated and the heads put on ice in DMEM/F12 medium for striatum and PBS + 0.65% D(+)-glucose for cortex.
Striatal tissue extraction procedure and preparation of cells
Whole brain was removed from the skull with the ventral side facing upside in DMEM/F12 medium. The striatum was dissected out from both hemispheres under a stereomicroscope and the striatal tissue was placed into the Falcon tube on ice.
The collected striatal tissue was triturated by using a PI 000 pipettor in 1ml of volume. The tissue was triturated by gently pipetting the solution up and down into the pipette tip about 15 times, using shearing force on alternate outflows. The tissue pieces settled to the bottom of the Falcon tube within 30 seconds, subsequently the supernatant was transferred to a new sterile Falcon tube on ice. The supernatant contained a suspension of dissociated single cells. The tissue pieces underwent a second trituration to avoid excessively damaging cells already dissociated by over triturating them. 1 Milliliter of ice-cold DMEM/F12 medium was added to the tissue pieces in the first tube and triturated as before. The tissue pieces were allowed to settle and the supernatant was removed to a new sterile Falcon tube on ice. The cells were centrifuged at 250g for 5 minutes at 4°C. The resuspended cell pellet was ready for cell counting.
Plating and cultivation of striatal cells
Striatal cells were plated into Poly-L-Lysine (O.lmg/ml) coated 96-well plates (the inner 60 wells only) at a density of 200,000 cells /cm2 in Neurobasal/B27 medium (Invitrogen). The cells were cultivated in the presence of 5% C02 at 37°C under 100% humidity. Complete medium was changed on days 1, 3 and 6.
Cortical tissue extraction procedure and preparation of cells
The two cortical hemispheres were carefully removed by a spatula from the whole brain with the ventral side facing upside into a PBS +0.65% D(+)-glucose containing petri dish. Forcips were put into the rostral part (near B. olfactorius) of the cortex for fixing the tissue and two lateral – sagittal oriented cuttings were done to remove the paraform and entorhinal cortices. The next cut involved a frontal oriented cut at the posterior end to remove the hippocampal formation. A final frontal cut was done a few millimeters away from the last cut in order to get hold of area 17/18 of the visual cortex.
The collected cortices on ice in PBS+0.65% D(+)-glucose were centrifuged at 350g for 5min. The supernatant was removed and trypsin/EDTA (0.05%/0.53mM) was added for 8min at 37°C. The reaction was stopped by adding an equal amount of DMEM+10%) fetal calf serum. The supernatant was removed by centrifugation followed by two subsequent washes in Neurobasal/B27 medium.
The cells were triturated once with a glass Pasteur pipette in 1 ml of
Neurobasal/B27 medium and subsequently twice by using a 1ml insulin syringe with a 22 gauge needle. The cell suspension was passed through a lOOμm cell strainer and subsequently rinsed by 1ml of Neurobasal B27 medium. Cells were counted and adjusted to 50,000 cells per 60μl.
Plating and cultivation of cortical cells
96-well plates were coated with 0.2mg/ml Poly-L-Lysine and subsequently coated with 2μg/ml laminin in PBS, after which 60μl of cortical astrocyte-conditioned medium was added to each well. Subsequently, 60μl of cortical cell suspension was added. The cells were cultivated in the presence of 10% C02 at 37°C under 100%) humidity. At day 1, there was a complete medium change (1:1- Neurobasal/B27 and astrocyte-conditioned medium) with addition of lμM cytosine-β-D-arabino-furanoside (mitosis inhibitor). On the second day, 2/3 of medium was changed. On day 5, 2/3 of the medium was changed again.
Cerebellar microexplants from P8 animals: preparation, cultivation and fixation
The laminated cerebellar cortices of the two hemispheres were explanted from a P8 rat, cut into small pieces in PBS + 0.65% D(+)glucose solution and triturated by a 23gauge needle and subsequently pressed through a 125 μm pore size sieve. The microexplants that were obtained were centrifuged (60 g) twice (media exchange) into serum-free BSA-supplemented START V-medium (Biochrom). Finally, the
microexplants were reconstituted in 1500 μl STARTV-medium (Biochrom). For cultivation, 40μl of cell suspension was adhered for 3 hours on a Poly-D-Lysine
(O.lmg/ml) coated cover slip placed in 35mm sized 6-well plates in the presence of 5% C02 under 100% humidity at 34°C. Subsequently, 1ml of STARTV-medium was added together with the toxins and drugs. The cultures were monitored (evaluated) after 2-3 days of cultivation in the presence of 5% C02 under 100% humidity. For cell counting analysis, the cultures were fixed in rising concentrations of paraformaldehyde (0.4%, 1.2%, 3% and 4% for 3min each) followed by a wash in PBS.
Toxin and drug administration for cerebellar, cortical and striatal cells: analysis
All toxin and drug administration experiments were designed that 1/100 parts of okadaic acid (30nM and lOOnM concentration and 0.5mM 3-nitropropionic acid for cerebellar microexplants only), GPE (InM -ImM) and G-2Methyl-PE (InM-lmM) were used respectively at 8DIV for cortical cultures and 9DIV for striatal cultures. The incubation time was 24hrs. The survival rate was determined by a colorimetric end-point MTT-assay at 595nm in a multi-well plate reader. For the cerebellar microexplants four windows (field of 0.65 mm2) with highest cell density were chosen and cells displaying neurite outgrowth were counted.
Results
The GPE analogue G-2Methyl-PE exhibited comparable neuroprotective capabilities within all three tested in vitro systems (Figures 12-15).
The cortical cultures responded to higher concentrations of GPE (Figure 12) /or
G-2Methyl-PE (lOμM, Figure 13) with 64% and 59% neuroprotection, respectively.
Whereas the other 2 types of cultures demonstrated neuroprotection at lower doses of G-2Methyl-PE (Figures 14 and 15). The striatal cells demonstrated
neuroprotection within the range of InM to ImM of G-2Methyl-PE (Figure 15) while the postnatal cerebellar microexplants demonstrated neuroprotection with G-2Methyl-PE in the dose range between InM and lOOnM (Figure 14).
While this invention has been described in terms of certain preferred embodiments, it will be apparent to a person of ordinary skill in the art having regard to that knowledge and this disclosure that equivalents of the compounds of this invention may be prepared and administered for the conditions described in this application, and all such equivalents are intended to be included within the claims of this application.
PATENT
WO-2021026066
Composition and kits comprising trofinetide and other related substances. Also claims a process for preparing trofinetide and the dosage form comprising the same. Disclosed to be useful in treating neurodegenerative conditions, autism spectrum disorders and neurodevelopmental disorders.
Trofinetide is a synthetic compound, having a similar core structure to Glycyl-Prolyl-Glutamic acid (or “GPE”). Trofinetide has been found to be useful in treating neurodegenerative conditions and recently has been found to be effective in treating Autism Spectrum disorders and Neurodevelopmental disorders.
Formula (Ila),
Example 1: Trofinetide Manufacturing Process
In general, trofinetide and related compounds can be manufactured from a precursor peptide or amino acid reacted with a silylating or persilylating agent at one or more steps. In the present invention, one can use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group.
Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
Step 1: Preparation of Z-Gly-OSu
Several alternative procedures can be used for this step.
Procedure 1A
One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 27 eq of iPrOH and 4 eq of CH2Cl2 at 21 °C. The mixture was cooled and when the temperature reached -4 °C, 1.1 eq of EDC.HCl was added gradually, keeping the temperature below 10 °C. During the reaction a dense solid appeared. After addition of EDC.HCl, the mixture was allowed to warm to 20 °C. The suspension was cooled to 11 °C and filtered. The cake was washed with 4.9 eq of cold iPrOH and 11 eq of IPE before drying at 34 °C (Z-Gly-OSu dried product -Purity: 99.5%; NMR assay: 96%; Yield: 84%).
Procedure 1B
This Procedure is for a variant of Procedure 1A, and differs by replacing iPrOH with ACN. One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 22 eq of ACN at 35 °C. The mixture was cooled in an ice bath. When the temperature reached 1 °C, 0.9 eq of DCC in 5.5 eq of ACN was added gradually to keep the temperature below 5 °C. The coupling reaction took about 20 hrs. During the reaction, DCU precipitated and was removed by filtration at the end of the coupling. After filtration, DCU was washed with ACN to recover the product. The mixture of Z-Gly-OSu was then concentrated to reach 60% by weight. iPrOH (17 eq) was added to initiate the crystallization. Quickly after iPrOH addition a dense solid appeared. An additional 17 eq of iPrOH was needed to liquify the suspension. The suspension was cooled in an ice bath and filtered. The solid was washed with 9 eq of iPrOH before drying at 45 °C (Z-Gly-OSu dried product – Purity: 99.2%; HPLC assay: 99.6%; Yield: 71%).
Step 2: Preparation of Z-Gly-MePro-OH
Several alternative procedures can be used for this step.
Procedure 2A
One (1) eq of MePro.HCl was partially solubilized in 29 eq of CH2Cl2 at 35 °C with 1.04 eq of TEA and 1.6 eq of TMA. The mixture was heated at 35 °C for 2 hrs to perform the silylation. Then 1.02 eq of Z-Gly-OSu was added to the mixture. The mixture was kept at 35 °C for 3 hrs and then 0.075 eq of butylamine was added to quench the reaction. The mixture was allowed to return to room temperature and mixed for at least 15 min. The Z-Gly-MePro-OH was extracted once with 5% w/w NaHCO3 in 186 eq of water, then three times successively with 5% w/w NaHCO3 in 62 eq of water. The aqueous layers were pooled and the pH was brought to 2.2 by addition of 34 eq of HCl as 12N HCl at room temperature. At this pH, Z-Gly-MePro-OH formed a sticky solid that was solubilized at 45 °C with approximately 33 eq of EtOAc and 2.3 eq of iButOH. Z-Gly-MePro-OH was extracted into the organic layer and washed with 62 eq of demineralized water. The organic layer was then dried by azeotropic distillation with 11.5 eq of EtOAc until the peptide began to precipitate. Cyclohexane (12 eq) was added to the mixture to complete the precipitation. The suspension was cooled at 5 °C for 2 hrs and filtered. The solid was washed with 10 eq of cyclohexane before drying at 45 °C (Z-Gly-MePro-OH dried product – Purity: 100%; HPLC assay: 100%; Yield 79%).
Procedure 2B
This Procedure is for a variant of Procedure 2A. One (1) eq of MePro.HCl was partially solubilized in 36.6 eq of CH2Cl2 at 34 °C with 1.01 eq of TEA and 0.1 eq of TMA. Then 1.05 eq of Z-Gly-OSu was added to the mixture, followed by 1.0 eq of TEA. The mixture was maintained at 35 °C for approximately 1 hr, cooled to 25 to 30 °C and 0.075 eq of DMAPA was added to stop the reaction. One hundred (100) eq of water, 8.6 eq of HCl as 12N HCl and 0.3 eq of KHSO4 were added to the mixture (no precipitation was observed, pH=1.7). Z-Gly-MePro-OH was extracted into the organic layer and washed twice with 97 eq of demineralized water with 0.3 eq of KHSO4, then 100 eq of demineralized water, respectively. EtOAc (23 eq) was added to the mixture and CH2Cl2 was removed by distillation until the peptide began to precipitate. Cyclohexane (25 eq) was added to the mixture to complete the precipitation. The suspension was cooled at -2 °C overnight and filtered. The solid was washed with 21 eq of cyclohexane before drying at 39 °C (Z-Gly-MePro-OH dried product – Purity: 98.7%; NMR assay: 98%; Yield 86%).
Procedure 2C
In reactor 1, MePro.HCl (1 eq) was suspended in EtOAc (about 7 eq). DIPEA (1 eq) and TMA (2 eq) were added, and the mixture heated to dissolve solids. After dissolution, the solution was cooled to 0 °C. In reactor 2, Z-Gly-OH (1 eq) was suspended in EtOAc (about 15 eq). DIPEA (1 eq), and pyridine (1 eq) were added. After mixing, a solution was obtained, and cooled to -5 °C. Piv-Cl (1 eq) was added to reactor 2, and the contents of reactor 1 added to reactor 2. Upon completed addition, the contents of reactor 2 were taken to room temperature. The conversion from Z-Gly-OH to Z-Gly-MePro-OH was monitored by HPLC. When the reaction was complete, the reaction mixture was quenched with DMAPA (0.1 eq), and washed with an aqueous solution comprised of KHSO4, (about 2.5 wt%), NaCl (about 4 wt%), and conc. HCl (about 6 wt%) in 100 eq H2O. The aqueous layer was re-extracted with EtOAc, and the combined organic layers washed with an aqueous solution comprised of KHSO4 (about 2.5 wt%) and NaCl (about 2.5 wt%) in 100 eq H2O, and then with water (100 eq). Residual water was removed from the organic solution of Z-Gly-MePro-OH by vacuum distillation with EtOAc. The resulting suspension was diluted with heptane (about 15 eq) and cooled to 0 °C. The product was isolated by filtration, washed with cold heptane (about 7 eq), and dried under vacuum at 45 °C. Z-Gly-MePro-OH (85% yield) was obtained.
Step 3: Preparation of Z-Gly-MePro-Glu-OH
Several alternative procedures can be used in this step.
Procedure 3A
H-Glu-OH (1.05 eq) was silylated in 2 eq of CH2Cl2 with 3.5 eq of TMA at 65 °C. Silylation was completed after 2 hrs. While the silylation was ongoing, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 24 eq of CH2Cl2 and 1.0 eq of DMA at room temperature in another reactor. EDC.HCl (1.0 eq.) was added. The activation rate reached 97% after 15 min. The activated Oxyma Pure solution, was then added to silylated H-Glu-OH at 40 °C and cooled at room temperature. Coupling duration was approximately 15 min, with a coupling rate of 97%. Addition of 8.2% w/w NaHCO3 in 156 eq of water to the mixture at room temperature (with the emission of CO2) was performed to reach pH 8. Z-Gly-MePro-Glu-OH was extracted in water. The aqueous layer was washed twice with 29 eq of CH2Cl2. Residual CH2Cl2 was removed by concentration. The pH was brought to 2.5 with 2.5N HCl, followed by 1.4 eq of solid KHSO4 to precipitate Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 3 x 52 eq of water. The filtered solid was added to 311 eq of demineralized water and heated to 55-60 °C. iPrOH (29 eq) was added gradually until total solubilization of the product. The mixture was slowly cooled to 10 °C under moderate mixing during 40 min to initiate the crystallization. The peptide was filtered and washed with 2 x 52 eq of water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.5%; NMR assay: 96%; Yield 74%).
Procedure 3B
One (1) eq of Z-Gly-MePro-OH and 1.05 eq of Suc-OH were solubilized in 40 eq of ACN and 30 eq of CH2Cl2 at room temperature. The mixture was cooled in an ice bath, and when the temperature was near 0 °C, 1.05 eq of DCC dissolved in 8 eq of ACN was added gradually, keeping the temperature below 5 °C. After addition of DCC, the mixture was progressively heated from 0 °C to 5 °C over 1 hr, then to 20 °C between 1 to 2 hrs and then to 45 °C between 2 to 5 hrs. After 5 hrs, the mixture was cooled to 5 °C and maintained overnight. The activation rate reached 98% after approximately 24 hrs. DCU was removed by filtration and washed with 13.5 eq of ACN. During the activation step, 1.1 eq of H-Glu-OH was silylated in 30 eq of ACN with 2.64 eq of TMA at 65 °C. Silylation was completed after 2 hrs. Z-Gly-MePro-OSu was then added gradually to the silylated H-Glu-OH at room temperature, with 0.4 eq of TMA added to maintain the solubility of the H-Glu-OH. The mixture was heated to 45 °C and 0.7 eq of TMA was added if precipitation occurred. The coupling duration was about 24 hrs to achieve a coupling rate of approximately 91%. The reaction was quenched by addition of 0.15 eq of butylamine and 2.0 eq of TEA. Water (233 eq) was added and the mixture concentrated until gelation occurred. Z-Gly-MePro-Glu-OH was extracted in water by addition of 5% w/w NaHCO3 in 233 eq of water and 132 eq of CH2Cl2. The aqueous layer was washed twice with 44 eq of CH2Cl2. Residual CH2Cl2 was removed by distillation. The pH was brought to 2.0 with 24 eq of HCl as 12N HCl followed by 75 eq of HCl as 4N HCl. At this pH, Z-Gly-MePro-Glu-OH precipitated. The mixture was cooled in an ice bath over 1 hr and filtered. The solid was washed with 186 eq of cold water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – HPLC Purity: 98.4%; NMR assay: 100%; Yield 55%).
Procedure 3C
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq) was silylated in 3.7 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after approximately 1.5 to 2 hrs, as evidenced by solubilization. During the silylation step, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 31.5 eq of CH2Cl2 at 22 °C. One (1.06) eq of EDC.HCl was added to complete the activation. The silylated H-Glu-OH was then added to the activated Oxyma Pure solution. The temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 153 eq of water and 9 eq of iPrOH to reach a pH of 1.65. Residual CH2Cl2 was removed by concentration. The mixture was cooled to 12 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 90 eq of water before drying at 36 °C.
Procedure 3D
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq.) was silylated in 3.9 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after 2 hrs, as evidenced by Solubilization. During the silylation step, 1 eq of Z-Gly-MePro-OH and 1 eq of Oxyma Pure were solubilized in 25 eq of CH2Cl2 at 23 °C. One (1) eq of EDC.HCl was added. To complete the activation, an additional 0.07 eq of EDC. HCl was added. Silylated H-Glu-OH was then added to the activated Oxyma Pure solution. Temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 160 eq of water and 9.6 eq of iPrOH to reach pH 1.63.
Residual CH2Cl2 was removed by concentration. The mixture was cooled to 20 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 192 eq of water before drying at about 25 °C for 2.5 days. The solid was then solubilized at 64 °C by addition of 55 eq of water and 31 eq of iPrOH. After solubilization, the mixture was diluted with 275 eq of water and cooled to 10 °C for crystallization. The mixture was filtered and the solid was washed with 60 eq of water before drying at 27 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.6%; NMR assay: 98%; Yield 74%).
Procedure 3E
In reactor 1, H-Glu-OH (1.05 eq) was suspended in ACN (about 2.2 eq). TMA (about 3.5 eq) added, and the mixture was heated to dissolve solids. After dissolution, the solution was cooled to room temperature. In reactor 2, Z-Gly-MePro-OH (1 eq) was suspended in ACN (14 eq). Oxyma Pure (1 eq) and EDC.HCl (1 eq) were added. The mixture was stirred at room temperature until the solids dissolved. The contents of reactor 2 were added to reactor 1. The conversion from Z-Gly-MePro-OH to Z-Gly-MePro-Glu-OH was monitored by HPLC. Upon completion the reaction mixture was added to an aqueous solution comprised of KHSO4 (about 2.5 wt%) dissolved in about 100 eq H2O. ACN was removed from the aqueous suspension of Z-Gly-MePro-Glu-OH by vacuum distillation with H2O. After stirring at room temperature, the product in the resulting suspension was isolated by filtration and washed with water. The solid obtained was dissolved in an aqueous solution comprised of NaHCO3 (about 5 wt%) in 110 eq H2O, and recrystallized by addition of an aqueous solution comprised of KHSO4 (about 10 wt%) in 90 eq H2O. The product was isolated by filtration, washed with water, and dried under vacuum at 45 °C. Z-Gly-MePro-Glu-OH (75% yield) was obtained.
Step 4: Deprotection and Isolation of Trofinetide
Several alternative procedures can be used in this step.
Procedure 4A
Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 25 eq) and EtOAc (about 15 eq). Pd/C (0.025 eq by weight and containing 10% Pd by weight) was added, and the reaction mixture hydrogenated by bubbling hydrogen through the reaction mixture at room temperature. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration, and the layers separated. Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained. Alternatively, deprotection can be accomplished using MeOH only, or a combination of iPrOH and MeOH, or by use of ethyl acetate in water.
Procedure 4B
This Procedure is for a variant of Procedure 4A, excluding EtOAc. Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 50 eq). Pd/C (0.05 eq, 5% Pd by weight) was added, and the reaction mixture hydrogenated at room temperature with a pressure of 5 bar. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon
reaction completion the catalyst was removed by filtration, and the aqueous layer washed with EtOAc (about 5 eq). Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained.
Procedure 4C
This Procedure is for a variant of Procedure 4A, replacing EtOAc with MeOH. Z-Gly-MePro-Glu-OH (1 eq) was suspended in MeOH (100 eq) and water (12 eq). Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration and the layers were washed with MeOH and iPrOH. The solvents were concentrated under vacuum at 45 °C, and trofinetide precipitated. The precipitate was filtered and dried at 45 °C to provide trofinetide.
Procedure 4D
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 105 eq of MeOH and 12 eq of water. Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate approximately 99% after 1 hr), the catalyst was filtered off and washed with 20-30 eq of MeOH. iPrOH (93 eq) was added and MeOH was replaced by iPrOH by concentration at 45 °C under vacuum. The peptide was concentrated until it began to precipitate. The peptide was filtered and dried at 45 °C (H-Gly-MePro-Glu-OH dried product: Purity: 98.1%; NMR assay: 90%; Yield 81%).
Procedure 4E
This Procedure is for a variant of Procedure 4A, removing H2O and replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 44 eq of MeOH. Pd/Si type 340 (0.02 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 99.9%, after 3-3.5 hrs), the catalyst was filtered off and washed with 8 eq of MeOH. Deprotected peptide was then precipitated in 56 eq of iPrOH. After 30 min at 5 °C, the peptide was filtered and washed with three times with 11 eq of iPrOH before drying at 25 °C (H-Gly-MePro-Glu-OH dried product: Purity: 99.4%; HPLC assay: ~98%; Yield: 81%).
Procedure 4F
This Procedure is for a variant of Procedure 4A. One (1) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 14 eq of EtOAc and 25 eq of water. Pd/C (0.01 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 100%, after about 3.5 hrs), the catalyst was filtered off and washed with a mixture of 3.5 eq of EtOAc and 6 eq of water. The aqueous layer was then ready for spray-drying (Aqueous H-Gly-MePro-Glu-OH peptide solution: Purity: 98.6%; Yield: ~95%).
Procedure 4G
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si, EtOAc with MeOH, and removing H2O. Pd/Si type 340 (0.02 eq by weight) was added to 2.9 vols of MeOH for pre-reduction during 30 min. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 34 eq of MeOH. The reduced palladium was then transferred to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. Pd/C type 39 (0.007 eq by weight) was added to the mixture to increase reaction kinetics. At the end of the deprotection, the catalyst was filtered off and washed with 13.6 eq of MeOH. The deprotected peptide was then precipitated in 71 eq of iPrOH. After about 40 min, the peptide was filtered and washed with 35 eq of iPrOH. The peptide was dried below 20 °C and was then ready for solubilization in water and spray-drying.
Procedure 4H
This Procedure is for a variant of Procedure 4A. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 24.8 eq of water and 13.6 eq of EtOAc. Pd/C type 39 (0.025 eq by weight) was added to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (19 hrs), the catalyst was removed by filtration and washed with 5.3 eq of water and 2.9 eq of EtOAc. The biphasic mixture was then decanted to remove the upper organic layer. The aqueous layer was diluted with water to reach an H-Gly-MePro-Glu-OH concentration suitable for spray-drying the solution.
Example 2: Alternative Trofinetide Manufacturing Process
An alternative method for synthesis of Trofinetide is based on U.S. Patent No.
8,546,530 adapted for a tripeptide as follows.
The persilylated compounds used to synthesis Formula (Ia) (trofinetide) are obtained by silylating a corresponding peptide or amino acid by reaction with a silylating agent, optionally in an organic solvent. The persilylated peptide or amino acid can be isolated and purified if desired. One can use the persilylated peptide or amino acid in situ, e.g. by combining a solution containing persilylated peptide or amino acid with a solution containing, optionally activated, peptide or amino acid.
In step 2, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-MePro-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid can be isolated and purified if desired. One can use the persilylated amino acid in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated, amino acid (for example, Z-Gly-OH).
In step 3, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-Glu-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid or peptide can be isolated and purified if desired. It is however useful to use the persilylated amino acid or peptide in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated (for example, by using EDC.HCl and Oxyma Pure), peptide (for example, Z-Gly-MePro-OH).
In the present invention, it is useful to use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group. Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
The reaction of step 2 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 40 °C, and optionally from 15 °C to 30 °C.
The reaction of step 3 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 60 °C, optionally from 15 °C to 50 °C.
In the reaction of step 2, generally 0.5 to 5 equivalents, optionally 1 to 3 equivalents, optionally about 1.5 to 2.5 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2 to 4 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible. “Functional groups to be silylated” means particular groups having an active hydrogen atom that can react with the silylating agent such as amino, hydroxyl, mercapto or carboxyl groups.
In the reaction of step 3, generally 0.5 to 5 equivalents, optionally 2 to 4.5 equivalents, optionally about 3 to 4 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2.5 to 4.5 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible.
It is understood that “persilylated” means an amino acid or peptide or amino acid analogue or peptide analogue in which the groups having an active hydrogen atom that can react with the silylating agent are sufficiently silylated to ensure that a homogeneous reaction medium for a coupling step is obtained.
In the process according to the invention, the reaction between the amino acid or peptide and the persilylated amino acid or peptide is often carried out in the presence of a carboxyl group activating agent. In that case the carboxylic activating reagent is suitably selected from carbodiimides, acyl halides, phosphonium salts and uronium or guanidinium salts. More optionally, the carboxylic activating agent is an acyl halide, such as isobutyl chloroformate or pivaloyl chloride or a carbodiimide, such as EDC.HC1 or DCC.
Good results are often obtained when using additional carboxylic activating reagents which reduce side reactions and/or increase reaction efficiency. For example, phosphonium and uronium salts can, in the presence of a tertiary base, for example, N,N-diisopropylethylamine (DIPEA) and triethylamine (TEA), convert protected amino acids into activated species. Other reagents help prevent racemization by providing a protecting reagent. These reagents include carbodiimides (for example, DCC) with an added auxiliary nucleophile (for example, 1-hydroxy-benzo triazole (HOBt), 1-hydroxy-azabenzotriazole (HOAt), or Suc-OH) or derivatives thereof. Another reagent that can be utilized is TBTU. The mixed anhydride method, using isobutyl chloroformate, with or without an added auxiliary nucleophile, is also used, as is the azide method, due to the low racemization associated with it. These types of compounds can also increase the rate of carbodiimide-mediated couplings. Typical additional reagents include also bases such as N,N-diisopropylethylamine (DIPEA), triethylamine (TEA) or N-methylmorpholine (NMM).
When the silylation is carried out in the presence of a solvent, said solvent is optionally a polar organic solvent, more optionally a polar aprotic organic solvent. An amide type solvent such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAC)
can be used. In the present invention for step 2, one can use an alkyl acetate solvent, in particular ethyl acetate is more particularly optional.
In the present invention for step 3, one can use a chlorinated hydrocarbon solvent or alkyl cyanide solvent, in particular dichloromethane or acetonitrile are more particularly optional.
In another embodiment, silylation is carried out in a liquid silylation medium consisting essentially of silylating agent and amino acid or peptide.
In the present invention, amino acid or peptide is understood to denote in particular an amino acid or peptide or amino acid analogue or peptide analogue which is bonded at its N-terminus or optionally another position, to a carboxylic group of an amino protected amino acid or peptide.
Example 3: Specifications for Compositions Containing Compounds of Formula (I)
1 ICH guideline Q3C on impurities: guideline for residual solvents
Example 4: Alternative Manufacturing of Trofinetide Example 1, Step 4, Procedure 4B
This Procedure is for a variant of Step 4, Procedure 4B. Z-Gly-MePro-Glu-OH (1 eq) was added in portions to Pd/C (0.027 eq by weight and containing 5% Pd by weight) in about 50 eq of water. The reaction mixture was hydrogenated at 20 °C at a pressure of 5 bar for at least 4 cycles of 4 hrs each. Pd/C (0.0027 eq by weight) was charged between cycles, as needed, to speed up the reaction. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon reaction completion the catalyst was removed by filtration, washed with water (12.5 eq) and the aqueous layer washed with EtOAc (about 14 eq). After phase separation, residual EtOAc was removed from the aqueous solution containing
trofinetide by sparging with nitrogen under vacuum at 20 °C for about 3 hrs. The aqueous solution was filtered. The final concentration of trofinetide was about 25 wt% and the solution was then ready for spray-drying to isolate the product.
Example 5: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (I)
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (I) on an anhydrous basis.
Example 6: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (Ia) on an anhydrous basis.
Example 7: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, wherein the product comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101
wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 8: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, and additionally comprising one or more compounds selected from the group consisting of Formula (III), Formula (IIIa), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), and Formula (IX), wherein the composition comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101 wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 9: Analysis of Products and Compositions
The products and compositions disclosed herein may be analyzed by liquid chromatography, a suitable chromatographic method using UPLC, e.g. using materials and conditions such as Waters Acquity CSH C18, 1.7 µm, 150 x 2.1 mm column, water with 0.1 % TFA (mobile phase A), and water/ACN 70/30 + 0.1 % TFA (mobile phase B), ranging from (4% phase A/6% phase B to 100% phase B and flushed with 4% phase A/6% phase B).
Flow rate: 0.35 ml/min, Column temperature: 40 °C, autosampler temperature: 4 °C, injection volume: 4 ml (e.g. prepared by weighing about 10 mg of powder in a 10 ml volumetric flask and diluted to volume with water). Examples of detectors are UV (ultraviolet, UV 220 nm) and MS (mass spectrometry).
INDUSTRIAL APPLICABILITY
This invention finds use in the pharmaceutical, medical, and other health care fields.
PATENT
WO2014085480 ,
claiming use of trofinetide for treating autism spectrum disorders including autism, Fragile X Syndrome or Rett Syndrome.
EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro- Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
W095/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS for increasing TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses GPE analogs capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including but not limited to, injury or disease in the CNS.
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
PAPER
Tetrahedron (2005), 61(42), 10018-10035. (CLICK HERE)
The synthesis of ten proline-modified analogues of the neuroprotective tripeptide GPE is described. Five of the analogues incorporate a proline residue with a hydrophobic group at C-2 and two further analogues have this side chain locked into a spirolactam ring system. The pyrrolidine ring was also modified by replacing the γ-CH2 group with sulfur and/or incorporation of two methyl groups at C-5.
Graphical Abstract

PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(9), 2279-2283
A series of GPE analogues, including modifications at the Pro and/or Glu residues, was prepared and evaluated for their NMDA binding and neuroprotective effects. Main results suggest that the pyrrolidine ring puckering of the Pro residue plays a key role in the biological responses, while the preference for cis or trans rotamers around the Gly-Pro peptide bond is not important.
Graphical abstract
A series of Pro and/or Glu modified GPE analogues is described. Compounds incorporating PMe and dmP showed higher affinity for glutamate receptors than GPE and neuroprotective effects similar to those of this endogenous tripeptide in culture hippocampal neurons exposed to NMDA.

PATENT
US 20060251649
WO 2006127702
US 20070004641
US 20080145335
WO 2012102832
WO 2014085480
US 20140147491
References
- ^ Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. (March 2009). “NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke”. Journal of the Neurological Sciences. 278 (1–2): 85–90. doi:10.1016/j.jns.2008.12.003. PMID 19157421. S2CID 7789415.
- ^ Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, Schmid KE (September 2013). “Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3”. Neuromolecular Medicine. 15 (3): 504–14. doi:10.1007/s12017-013-8236-z. PMID 23765588. S2CID 12522580.
- ^ Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, Cogram P (March 2015). “NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome”. Neuromolecular Medicine. 17 (1): 71–82. doi:10.1007/s12017-015-8341-2. PMID 25613838. S2CID 11964380.
- ^ Study Details – Rett Syndrome Study
- ^ Neuren’s trofinetide successful in Phase 2 clinical trial in Fragile X
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Rett’s Syndrome | 1 |
| 3 | Recruiting | Treatment | Rett’s Syndrome | 1 |
| 2 | Completed | Supportive Care | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Fragile X Syndrome (FXS) | 1 |
| 2 | Completed | Treatment | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Rett’s Syndrome | 2 |
| 2 | Terminated | Treatment | Concussions | 1 |
| 1 | Completed | Treatment | Brain Injuries,Traumatic | 2 |
| Legal status | |
|---|---|
| Legal status | US: Investigational New Drug |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 853400-76-7 |
| PubChem CID | 11318905 |
| ChemSpider | 9493869 |
| UNII | Z2ME8F52QL |
| Chemical and physical data | |
| Formula | C13H21N3O6 |
| Molar mass | 315.322 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@]1(CCCN1C(=O)CN)C(=O)N[C@@H](CCC(=O)O)C(=O)O | |
| InChI[hide]InChI=1S/C13H21N3O6/c1-13(5-2-6-16(13)9(17)7-14)12(22)15-8(11(20)21)3-4-10(18)19/h8H,2-7,14H2,1H3,(H,15,22)(H,18,19)(H,20,21)/t8-,13-/m0/s1Key:BUSXWGRAOZQTEY-SDBXPKJASA-N |
////////////Tofinetide , NNZ 2566, PHASE 2, PHASE 3. NEUREN, Amino Acids, Peptides, Proteins,
CC1(CCCN1C(=O)CN)C(=O)NC(CCC(=O)O)C(=O)O
J. Med. Chem. 2025, 68, 2147−2182
Trofinetide (Daybue). Trofinetide (8) was developed by Neuren Pharmaceuticals and Acadia Pharmaceuticals for the treatment of rare childhood neurodevelopmental disorders and
was approved by the USFDA in March 2023 for adults and pediatric patients two years of age or older with Rett syndrome. 59 In most cases, Rett syndrome is caused by loss of-function mutations in the X-linked gene that encodes methyl CpG-binding protein 2 (MeCP2). 60,61 MeCP2 is a critical transcriptional regulator required for normal neurological development. In the past, treatment of Rett syndrome has
been limited to symptom management based on knowledge from treating other conditions, 62
but new research has focused on targeting the underlying genetic cause and finding agents to
restore MeCP2 function. Trofinetide is an orally available synthetic analog of glycine-proline-glutamate (GPE), the Nterminal tripeptide metabolite of insulin like growth factor-1 (IGF-1). GPE has been shown to partially reverse Rett-like symptoms in MeCP2 deficient mouse models and trofenitide was developed to have an improved pharmakokinetic profile to GPE. 64 Its use has shown significant improvement over placebo in clinical trials.
Anumberofaccountsrelated to the preparation of trofinetide have been reported with various protecting group strategies, but they are moreamenabletosmall-scaleproductionduetoreagent selection and challenging isolations requiring column chromatography.65−67 A commercially viable synthesis of the drug has been described by researchers at Neuren Pharmaceuticals and is depicted in Scheme 12.68
This synthesis takes advantage of in situ silyl protection/deprotection during its amidation steps to
avoid lengthy protecting group manipulations. Activation of Cbz protected glycine 8.1 with hydroxysuccinimide 8.2 using EDCI provided 8.3 in 84% yield as a direct drop crystallization
(isolated by crystallization directly from the reaction mixture). This activated ester was then coupled with commercially available methyl proline 8.4 69 by first silylation of the carboxylic acid of the proline analog in situ with 8.5, then adding 8.3 to couple with the amine. Deprotection of the silyl ester during the
workup provided amide 8.6 in 79% yield. In a similar sequence, 8.6 was activated with Oxyma Pure and EDCI while in a second vessel 8.7 was silyl protected using 8.5. Subsequent combination of these streams followed by workup and crystallization gave amide 8.8 in good yield. Finally, Cbz deprotection was
accomplished using Pd/C and hydrogen. Trofinetide (8) was extracted into the aqueous layer and isolated by spray drying in 90% yield.
(60) Kyle, S. M.; Vashi, N.; Justice, M. J. Rett syndrome: a
neurological disorder with metabolic components. Open Biol. 2018,
8, No. 170216.
(61) Collins, B. E.; Neul, J. L. Rett syndrome and MECP2 duplication
syndrome: disorders of MeCP2 dosage. Neuropsychiatr. Dis. Treat.
2022, 18, 2813−2835.
(62) Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt,
R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus
guidelines on managing Rett syndrome across the lifespan. BMJ
Paediatr. Open 2020, 4, No. e000717.
(63) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D.G.;Marsh,E.D.;Lin,T.;Stankovic,S.;Bishop,K. M.;Youakim,J.M.
Trofinetide for the treatment of Rett syndrome: a randomized phase 3
study. Nat. Med. 2023, 29, 1468−1475.
(64) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D. G.; Peters, S. U.; Jones, N. E.; Youakim, J. M. Design and outcome
measures of LAVENDER, a phase 3 study of trofinetide for Rett
syndrome. Contemp. Clin. Trials 2022, 114, No. 106704.
(65) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl-l-2-methylprolyl-l-glutamic acid. US
20140147491, 2014.
(66) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl L2012102832, 2012.-2-methylprolyl
L-glutamic acid. WO 2012102832, 2012
(67) Brimble, M.A.;Harris, P. W.R.;Sieg, F. Preparation of analogsof
glycyl-prolyl-glutamate as neuroprotective agents. US 20080145335,
2008
(68) Blower, C.; Peterson, M.; Shaw, J. M.; Bonnar, J. A.; Moniotte, E.
D. F. P.; Bousmanne, M. B. C.; Betti, C.; Decroos, K. W. L.; Ayoub, M.
Compositions of trofinetide. WO 2021026066, 2021.
(69) Beck, A. K.; Blank, S.; Job, K.; Seebach, D.; Sommerfeld, T.
Synthesis of (S)-2-methylproline: a general method for the preparation
of α-branched amino acids (L.-proline, 2-methyl-). Org. Synth. 1995, 72, 62−73

.
TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 ,


TERIPARATIDE
テリパラチド;
- PTH 1-34
- LY 333334 / LY-333334 / LY333334 / ZT-034
|
Ser Val Ser Glu Ile Gln Leu Met His Asn Leu Gly Lys His Leu Asn
Ser Met Glu Arg Val Glu Trp Leu Arg Lys Lys Leu Gln Asp Val His Asn Phe-OH |
|
| Type |
Peptide
|
|---|
| Formula |
C181H291N55O51S2
|
|---|---|
| CAS |
52232-67-4
99294-94-7 (acetate)
|
| Mol weight |
4117.7151
|
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-hydroxypropanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-methylpentanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]-4-methylsulfanylbutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-oxobutanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]hexanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-amino-1-[[(1S)-1-carboxy-2-phenylethyl]amino]-1,4-dioxobutan-2-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-oxopentanoic acid
| SVG Image |
 |
|---|---|
| IUPAC Condensed | H-Ser-Val-Ser-Glu-Ile-Gln-Leu-Met-His-Asn-Leu-Gly-Lys-His-Leu-Asn-Ser-Met-Glu-Arg-Val-Glu-Trp-Leu-Arg-Lys-Lys-Leu-Gln-Asp-Val-His-Asn-Phe-OH |
| Sequence | SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF |
| PLN | H-SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF-OH |
| HELM | PEPTIDE1{S.V.S.E.I.Q.L.M.H.N.L.G.K.H.L.N.S.M.E.R.V.E.W.L.R.K.K.L.Q.D.V.H.N.F}$$$$ |
| IUPAC | L-seryl-L-valyl-L-seryl-L-alpha-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparagyl-L-leucyl-glycyl-L-lysyl-L-histidyl-L-leucyl-L-asparagyl-L-seryl-L-methionyl-L-alpha-glutamyl-L-arginyl-L-valyl-L-alpha-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-alpha-aspartyl-L-valyl-L-histidyl-L-asparagyl-L-phenylalanine |
Other Names
- L-Seryl-L-valyl-L-seryl-L-α-glutamyl-L-isoleucyl-L-glutaminyl-L-leucyl-L-methionyl-L-histidyl-L-asparaginyl-L-leucylglycyl-L-lysyl-L-histidyl-L-leucyl-L-asparaginyl-L-seryl-L-methionyl-L-α-glutamyl-L-arginyl-L-valyl-L-α-glutamyl-L-tryptophyl-L-leucyl-L-arginyl-L-lysyl-L-lysyl-L-leucyl-L-glutaminyl-L-α-aspartyl-L-valyl-L-histidyl-L-asparaginyl-L-phenylalanine
- (1-34)-Human parathormone
- (1-34)-Human parathyroid hormone
- 1-34-Human PTH
- 1-34-Parathormone (human)
- 11: PN: WO0039278 SEQID: 17 unclaimed protein
- 14: PN: WO0181415 SEQID: 16 claimed protein
- 15: PN: WO0123521 SEQID: 19 claimed protein
- 1: PN: EP2905289 SEQID: 1 claimed protein
- 1: PN: WO0198348 SEQID: 13 claimed protein
- 1: PN: WO2011071480 SEQID: 14 claimed protein
- 225: PN: US20090175821 SEQID: 272 claimed protein
- 22: PN: US6110892 SEQID: 22 unclaimed protein
- 2: PN: US20100261199 SEQID: 4 claimed protein
- 31: PN: US20070099831 PAGE: 7 claimed protein
- 32: PN: WO2008068487 SEQID: 32 claimed protein
- 5: PN: WO2008033473 SEQID: 4 claimed protein
- 692: PN: WO2004005342 PAGE: 46 claimed protein
- 69: PN: US20050009742 PAGE: 20 claimed sequence
- 7: PN: WO0031137 SEQID: 8 unclaimed protein
- 7: PN: WO0040611 PAGE: 1 claimed protein
- 93: PN: WO0069900 SEQID: 272 unclaimed protein
- Forsteo
- Forteo
- HPTH-(1-34)
- Human PTH(1-34)
- Human parathormone(1-34)
- Human parathyroid hormone-(1-34)
- LY 333334
- Osteotide
- Parathar
- Parathormone (human)
- Teriparatide
- ZT 034
Product Ingredients
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Teriparatide acetate | 9959P4V12N | 99294-94-7 |
Teriparatide is a form of parathyroid hormone consisting of the first (N-terminus) 34 amino acids, which is the bioactive portion of the hormone. It is an effective anabolic (promoting bone formation) agent[2] used in the treatment of some forms of osteoporosis.[3] It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone.
Recombinant teriparatide is sold by Eli Lilly and Company under the brand name Forteo/Forsteo. A synthetic teriparatide from Teva Generics has been authorised for marketing in European territories[4]. Biosimilar product from Gedeon Richter plc has been authorised in Europe[5]. On October 4, 2019 the US FDA approved a recombinant teriparatide product, PF708, from Pfenex Inc. PF708 is the first FDA approved proposed therapeutic equivalent candidate to Forteo.
Teriparatide (recombinant human parathyroid hormone) is a potent anabolic agent used in the treatment of osteoporosis. It is manufactured and marketed by Eli Lilly and Company.
Teriparatide is a recombinant form of parathyroid hormone. It is an effective anabolic (i.e., bone growing) agent used in the treatment of some forms of osteoporosis. It is also occasionally used off-label to speed fracture healing. Teriparatide is identical to a portion of human parathyroid hormone (PTH) and intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone. Teriparatide is sold by Eli Lilly and Company under the brand name Forteo.
Indication
For the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. Also used to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Associated Conditions
Pharmacodynamics
Clinical trials indicate that teriparatide increases predominantly trabecular bone in the lumbar spine and femoral neck; it has less significant effects at cortical sites. The combination of teriparatide with antiresorptive agents is not more effective than teriparatide monotherapy. The most common adverse effects associated with teriparatide include injection-site pain, nausea, headaches, leg cramps, and dizziness. After a maximum of two years of teriparatide therapy, the drug should be discontinued and antiresorptive therapy begun to maintain bone mineral density.
Mechanism of action
Teriparatide is the portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34 of the complete molecule which contains amino acid sequence 1 to 84. Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. Daily injections of teriparatide stimulates new bone formation leading to increased bone mineral density.
Medical uses
Teriparatide has been FDA-approved since 2002.[6] It is effective in growing bone (e.g., 8% increase in bone density in the spine after one year)[7] and reducing the risk of fragility fractures.[6][8] When studied, teriparatide only showed bone mineral density (BMD) improvement during the first 18 months of use. Teriparatide should only be used for a period of 2 years maximum. After 2 years, another agent such a bisphosphonate or denosumab should be used in cases of osteoporosis. [9]
Teriparatide cuts the risk of hip fracture by more than half but does not reduce the risk of arm or wrist fracture.[10]
Other
Teriparatide can be used off-label to speed fracture repair and treat fracture nonunions.[11] It has been reported to have been successfully used to heal fracture nonunions.[12] Generally, due to HIPAA regulations, it is not publicized when American athletes receive this treatment to improve fracture recovery.[11] But an Italian football player, Francesco Totti, was given teriparatide after a tibia/fibula fracture, and he unexpectedly recovered in time for the 2006 World Cup.[11] It has been reported that Mark Mulder used it to recover from a hip fracture Oakland A’s for the 2003 MLB playoffs[13] and Terrell Owens to recover from an ankle fracture before the 2005 Super Bowl.[13]
Administration
Teriparatide is administered by injection once a day in the thigh or abdomen.
Contraindications
Teriparatide should not be prescribed for people who are at increased risks for osteosarcoma. This includes those with Paget’s Diseaseof bone or unexplained elevations of serum alkaline phosphate, open epiphysis, or prior radiation therapy involving the skeleton. In the animal studies and in one human case report, it was found to potentially be associated with developing osteosarcoma in test subjects after over 2 years of use. [14]
Patients should not start teriparatide until any vitamin D deficiency is corrected. [15]
Adverse effects
Adverse effects of teriparatide include headache, nausea, dizziness, and limb pain.[6] Teriparatide has a theoretical risk of osteosarcoma, which was found in rat studies but not confirmed in humans.[2] This may be because unlike humans, rat bones grow for their entire life.[2] The tumors found in the rat studies were located on the end of the bones which grew after the injections began.[15]After nine years on the market, there were only two cases of osteosarcoma reported.[7] This risk was considered by the FDA as “extremely rare” (1 in 100,000 people)[6] and is only slightly more than the incidence in the population over 60 years old (0.4 in 100,000).[6]
Mechanism of action
Teriparatide is a portion of human parathyroid hormone (PTH), amino acid sequence 1 through 34, of the complete molecule (containing 84 amino acids). Endogenous PTH is the primary regulator of calcium and phosphate metabolism in bone and kidney. PTH increases serum calcium, partially accomplishing this by increasing bone resorption. Thus, chronically elevated PTH will deplete bone stores. However, intermittent exposure to PTH will activate osteoblasts more than osteoclasts. Thus, once-daily injections of teriparatide have a net effect of stimulating new bone formation leading to increased bone mineral density.[16][17][18]
Teriparatide is the first FDA approved agent for the treatment of osteoporosis that stimulates new bone formation.[19]
FDA approval
Teriparatide was approved by the Food and Drug Administration (FDA) on 26 November 2002, for the treatment of osteoporosis in men and postmenopausal women who are at high risk for having a fracture. The drug is also approved to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture.
Combined teriparatide and denosumab
Combined teriparatide and denosumab increased BMD more than either agent alone and more than has been reported with approved therapies. Combination treatment might, therefore, be useful to treat patients at high risk of fracture by increasing BMD. However, there is no evidence of fracture rate reduction in patients taking a teriparatide and denosumab combination. Moreover, the combination therapy group showed a significant decrease in their bone formation marker, indicating that denosumab, an antiresorptive agent, might actually counteract the effect of teriparatide, a bone formation anabolic agent, in bone formation. [20]
PATENT
KR 2011291
WO 2019077432
CN 109897099
CN 109879955
CN 109879954
CN 108373499
PATENT
WO-2020000555
Process for preparing teriparatide as parathyroid hormone receptor agonist, useful for treating osteoporosis in menopausal women. Appears to be the first filing from the assignee and the inventors on this compound, however, this invention was previously seen as a Chinese national filing published in 12/2013. Daiichi Sankyo , through its subsidiary Asubio Pharma , was developing SUN-E-3001 , a nasally administered recombinant human parathyroid hormone, for the treatment of osteoporosis.
Teriparatide is a 1-34 fragment of human parathyroid hormone, which has the same biological activity as human parathyroid hormone. Hypogonadous osteoporosis and osteoporosis in menopausal women have great market prospects.
In patent CN201410262511, a pseudoproline dipeptide Fmoc-Asn (Trt) -Ser (ψ Me, Me Pro) -OH is used instead of the two amino acids at the original 16-17 positions for coupling one by one, and the final cleavage yields teriparatide. This method adopts the method of feeding pseudoproline dipeptide to avoid the generation of oxidative impurities, but it cannot avoid a variety of missing peptides due to the excessively long peptide chain. At the same time, the pseudoproline dipeptide is expensive and difficult to obtain.
References
- ^ http://www.minsa.gob.pa/sites/default/files/alertas/nota_seguridad_teriparatida.pdf
- ^ Jump up to:a b c Riek AE and Towler DA (2011). “The pharmacological management of osteoporosis”. Missouri Medicine. 108 (2): 118–23. PMC 3597219. PMID 21568234.
- ^ Saag KG, Shane E, Boonen S, et al. (November 2007). “Teriparatide or alendronate in glucocorticoid-induced osteoporosis”. The New England Journal of Medicine. 357 (20): 2028–39. doi:10.1056/NEJMoa071408. PMID 18003959.
- ^ BfArM (2017-05-08). “PUBLIC ASSESSMENT REPORT – Decentralised Procedure – Teriparatid-ratiopharm 20 µg / 80ml, Solution for injection” (PDF).
- ^ “Summary of the European public assessment report (EPAR) for Terrosa”. Retrieved 2019-08-14.
- ^ Jump up to:a b c d e Rizzoli, R.; Reginster, J. Y.; Boonen, S.; Bréart, G. R.; Diez-Perez, A.; Felsenberg, D.; Kaufman, J. M.; Kanis, J. A.; Cooper, C. (2011). “Adverse Reactions and Drug–Drug Interactions in the Management of Women with Postmenopausal Osteoporosis”. Calcified Tissue International. 89 (2): 91–104. doi:10.1007/s00223-011-9499-8. PMC 3135835. PMID 21637997.
- ^ Jump up to:a b Kawai, M.; Mödder, U. I.; Khosla, S.; Rosen, C. J. (2011). “Emerging therapeutic opportunities for skeletal restoration”. Nature Reviews Drug Discovery. 10 (2): 141–156. doi:10.1038/nrd3299. PMC 3135105. PMID 21283108.
- ^ Murad, M. H.; Drake, M. T.; Mullan, R. J.; Mauck, K. F.; Stuart, L. M.; Lane, M. A.; Abu Elnour, N. O.; Erwin, P. J.; Hazem, A.; Puhan, M. A.; Li, T.; Montori, V. M. (2012). “Comparative Effectiveness of Drug Treatments to Prevent Fragility Fractures: A Systematic Review and Network Meta-Analysis”. Journal of Clinical Endocrinology & Metabolism. 97(6): 1871–1880. doi:10.1210/jc.2011-3060. PMID 22466336.
- ^ O’Connor KM. Evaluation and Treatment of Osteoporosis. Med Clin N Am. 2016; 100:807-26
- ^ Díez-Pérez A, Marin F, Eriksen EF, Kendler DL, Krege JH, Delgado-Rodríguez M (September 2018). “Effects of teriparatide on hip and upper limb fractures in patients with osteoporosis: A systematic review and meta-analysis”. Bone. 120: 1–8. doi:10.1016/j.bone.2018.09.020. PMID 30268814.
- ^ Jump up to:a b c Bruce Jancin (2011-12-12). “Accelerating Fracture Healing With Teriparatide”. Internal Medicine News Digital Network. Retrieved 2013-09-20.
- ^ Giannotti, S.; Bottai, V.; Dell’Osso, G.; Pini, E.; De Paola, G.; Bugelli, G.; Guido, G. (2013). “Current medical treatment strategies concerning fracture healing”. Clinical Cases in Mineral and Bone Metabolism. 10 (2): 116–120. PMC 3796998. PMID 24133528.
- ^ Jump up to:a b William L. Carroll (2005). “Chapter 1: Defining the Issue”. The Juice: The Real Story of Baseball’s Drug Problems. ISBN 1-56663-668-X. Retrieved 2013-09-23.
- ^ Harper KD, Krege JH, Marcus R, et al. Osteosarcoma and teriparatide? J Bone Miner Res 2007;22(2):334
- ^ Jump up to:a b https://www.drugs.com/pro/forteo.html
- ^ Bauer, E; Aub, JC; Albright, F (1929). “Studies of calcium and phosphorus metabolism: V. Study of the bone trabeculae as a readily available reserve supply of calcium”. J Exp Med. 49 (1): 145–162. doi:10.1084/jem.49.1.145. PMC 2131520. PMID 19869533.
- ^ Selye, H (1932). “On the stimulation of new bone formation with parathyroid extract and irradiated ergosterol”. Endocrinology. 16 (5): 547–558. doi:10.1210/endo-16-5-547.
- ^ Dempster, D. W.; Cosman, F.; Parisien, M.; Shen, V.; Lindsay, R. (1993). “Anabolic actions of parathyroid hormone on bone”. Endocrine Reviews. 14 (6): 690–709. doi:10.1210/edrv-14-6-690. PMID 8119233.
- ^ Fortéo: teriparatide (rDNA origin) injection Archived 2009-12-27 at the Wayback Machine
- ^ Tsai, Joy N; Uihlein, Alexander V; Lee, Hang; Kumbhani, Ruchit; Siwila-Sackman, Erica; McKay, Elizabeth A; Burnett-Bowie, Sherri-Ann M; Neer, Robert M; Leder, Benjamin Z (2013). “Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: The DATA study randomised trial”. The Lancet. 382 (9886): 1694–1700. doi:10.1016/S0140-6736(13)60856-9. PMC 4010689. PMID 24517156.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Forteo/Forsteo, Teribone[1] |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Metabolism | Hepatic (nonspecific proteolysis) |
| Elimination half-life | Subcutaneous: 1 hour |
| Excretion | Renal (metabolites) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.168.733 |
| Chemical and physical data | |
| Formula | C181H291N55O51S2 |
| Molar mass | 4117.72 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| CA2314313 | No | 2005-02-08 | 2018-12-08 |  |
| CA2325371 | No | 2004-08-17 | 2019-08-19 | |
| US6770623 | No | 2004-08-03 | 2018-12-08 |  |
| US6977077 | No | 2005-12-20 | 2019-08-19 | |
| US7163684 | No | 2007-01-16 | 2019-08-19 | |
| US7351414 | No | 2008-04-01 | 2019-08-19 | |
| US7550434 | No | 2009-06-23 | 2018-12-08 | |
| US7144861 | No | 2006-12-05 | 2018-12-08 | |
| US7517334 | No | 2009-04-14 | 2025-03-25 | |
FORTEO (teriparatide [rDNA origin] injection) contains recombinant human parathyroid hormone (1- 34), and is also called rhPTH (1-34). It has an identical sequence to the 34 N-terminal amino acids(the biologically active region) of the 84-amino acid human parathyroid hormone.
Teriparatide has a molecular weight of 4117.8 daltons and its amino acid sequence is shown below:
 |
Teriparatide (rDNA origin) is manufactured using a strain of Escherichia coli modified by recombinant DNA technology. FORTEO is supplied as a sterile, colorless, clear, isotonic solution in a glass cartridge which is pre-assembled into a disposable delivery device (pen) for subcutaneous injection. Each prefilled delivery device is filled with 2.7 mL to deliver 2.4 mL. Each mL contains 250 mcg teriparatide (corrected for acetate, chloride, and water content), 0.41 mg glacial acetic acid, 0.1 mg sodium acetate (anhydrous), 45.4 mg mannitol, 3 mg Metacresol, and Water for Injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4.
Each cartridge, pre-assembled into a delivery device, delivers 20 mcg of teriparatide per dose each day for up to 28 days.
REFERENCES
1: Lindsay R, Krege JH, Marin F, Jin L, Stepan JJ. Teriparatide for osteoporosis: importance of the full course. Osteoporos Int. 2016 Feb 22. [Epub ahead of print] Review. PubMed PMID: 26902094.
2: Im GI, Lee SH. Effect of Teriparatide on Healing of Atypical Femoral Fractures: A Systemic Review. J Bone Metab. 2015 Nov;22(4):183-9. doi: 10.11005/jbm.2015.22.4.183. Epub 2015 Nov 30. Review. PubMed PMID: 26713309; PubMed Central PMCID: PMC4691592.
3: Babu S, Sandiford NA, Vrahas M. Use of Teriparatide to improve fracture healing: What is the evidence? World J Orthop. 2015 Jul 18;6(6):457-61. doi: 10.5312/wjo.v6.i6.457. eCollection 2015 Jul 18. Review. PubMed PMID: 26191492; PubMed Central PMCID: PMC4501931.
4: Lecoultre J, Stoll D, Chevalley F, Lamy O. [Improvement of fracture healing with teriparatide: series of 22 cases and review of the literature]. Rev Med Suisse. 2015 Mar 18;11(466):663-7. Review. French. PubMed PMID: 25962228.
5: Sugiyama T, Torio T, Sato T, Matsumoto M, Kim YT, Oda H. Improvement of skeletal fragility by teriparatide in adult osteoporosis patients: a novel mechanostat-based hypothesis for bone quality. Front Endocrinol (Lausanne). 2015 Jan 30;6:6. doi: 10.3389/fendo.2015.00006. eCollection 2015. Review. PubMed PMID: 25688232; PubMed Central PMCID: PMC4311704.
6: Wheeler AL, Tien PC, Grunfeld C, Schafer AL. Teriparatide treatment of osteoporosis in an HIV-infected man: a case report and literature review. AIDS. 2015 Jan 14;29(2):245-6. doi: 10.1097/QAD.0000000000000529. Review. PubMed PMID: 25532609; PubMed Central PMCID: PMC4438749.
7: Campbell EJ, Campbell GM, Hanley DA. The effect of parathyroid hormone and teriparatide on fracture healing. Expert Opin Biol Ther. 2015 Jan;15(1):119-29. doi: 10.1517/14712598.2015.977249. Epub 2014 Nov 3. Review. PubMed PMID: 25363308.
8: Yamamoto M, Sugimoto T. [Glucocorticoid and Bone. Beneficial effect of teriparatide on fracture risk as well as bone mineral density in patients with glucocorticoid-induced osteoporosis]. Clin Calcium. 2014 Sep;24(9):1379-85. doi: CliCa140913791385. Review. Japanese. PubMed PMID: 25177011.
9: Chen JF, Yang KH, Zhang ZL, Chang HC, Chen Y, Sowa H, Gürbüz S. A systematic review on the use of daily subcutaneous administration of teriparatide for treatment of patients with osteoporosis at high risk for fracture in Asia. Osteoporos Int. 2015 Jan;26(1):11-28. doi: 10.1007/s00198-014-2838-7. Epub 2014 Aug 20. Review. PubMed PMID: 25138261.
10: Eriksen EF, Keaveny TM, Gallagher ER, Krege JH. Literature review: The effects of teriparatide therapy at the hip in patients with osteoporosis. Bone. 2014 Oct;67:246-56. doi: 10.1016/j.bone.2014.07.014. Epub 2014 Jul 15. Review. PubMed PMID: 25053463.
11: Meier C, Lamy O, Krieg MA, Mellinghoff HU, Felder M, Ferrari S, Rizzoli R. The role of teriparatide in sequential and combination therapy of osteoporosis. Swiss Med Wkly. 2014 Jun 4;144:w13952. doi: 10.4414/smw.2014.13952. eCollection 2014. Review. PubMed PMID: 24896070.
12: Krege JH, Lane NE, Harris JM, Miller PD. PINP as a biological response marker during teriparatide treatment for osteoporosis. Osteoporos Int. 2014 Sep;25(9):2159-71. doi: 10.1007/s00198-014-2646-0. Epub 2014 Mar 6. Review. PubMed PMID: 24599274; PubMed Central PMCID: PMC4134485.
13: Nakano T. [Once-weekly teriparatide treatment on osteoporosis]. Clin Calcium. 2014 Jan;24(1):100-5. doi: CliCa1401100105. Review. Japanese. PubMed PMID: 24369286.
14: Yano S, Sugimoto T. [Daily subcutaneous injection of teriparatide : the progress and current issues]. Clin Calcium. 2014 Jan;24(1):35-43. doi: CliCa14013543. Review. Japanese. PubMed PMID: 24369278.
15: Lewiecki EM, Miller PD, Harris ST, Bauer DC, Davison KS, Dian L, Hanley DA, McClung MR, Yuen CK, Kendler DL. Understanding and communicating the benefits and risks of denosumab, raloxifene, and teriparatide for the treatment of osteoporosis. J Clin Densitom. 2014 Oct-Dec;17(4):490-5. doi: 10.1016/j.jocd.2013.09.018. Epub 2013 Oct 25. Review. PubMed PMID: 24206867.
16: Delivanis DA, Bhargava A, Luthra P. Subungual exostosis in an osteoporotic patient treated with teriparatide. Endocr Pract. 2013 Sep-Oct;19(5):e115-7. doi: 10.4158/EP13040.CR. Review. PubMed PMID: 23757619.
17: Borges JL, Freitas A, Bilezikian JP. Accelerated fracture healing with teriparatide. Arq Bras Endocrinol Metabol. 2013 Mar;57(2):153-6. Review. PubMed PMID: 23525295.
18: Thumbigere-Math V, Gopalakrishnan R, Michalowicz BS. Teriparatide therapy for bisphosphonate-related osteonecrosis of the jaw: a case report and narrative review. Northwest Dent. 2013 Jan-Feb;92(1):12-8. Review. PubMed PMID: 23516715.
19: Lamy O. [Bone anabolic treatment with Teriparatide]. Ther Umsch. 2012 Mar;69(3):187-91. doi: 10.1024/0040-5930/a000272. Review. German. PubMed PMID: 22403112.
20: Narváez J, Narváez JA, Gómez-Vaquero C, Nolla JM. Lack of response to teriparatide therapy for bisphosphonate-associated osteonecrosis of the jaw. Osteoporos Int. 2013 Feb;24(2):731-3. doi: 10.1007/s00198-012-1918-9. Epub 2012 Mar 8. Review. PubMed PMID: 22398853.
/////TERIPARATIDE, テリパラチド , терипаратид , تيريباراتيد , 特立帕肽 , PTH 1-34, LY 333334, LY-333334, LY333334, ZT-034, 52232-67-4, PEPTIDES
Difelikefalin


Difelikefalin, CR-845; MR-13A-9; MR-13A9
4-amino-1- (D-phenylalanyl-D-phenylalanyl-D-leucyl-D-lysyl) piperidine-4-carboxylic acid
Phase III
C36H53N7O6, 679.40573
| Originator | Ferring Pharmaceuticals |
|---|---|
| Developer | Cara Therapeutics |
| Class | Analgesic drugs (peptides) |
| Mechanism Of Action | Opioid kappa receptor agonists |
| Who Atc Codes | D04A-X (Other antipruritics), N02A (Opioids) |
| Ephmra Codes | D4A (Anti-Pruritics, Including Topical Antihistamines, Anaesthetics, etc), N2A (Narcotics) |
| Indication | Pain, Osteoarthritis, Pruritus |
A kappa opioid receptor agonist potentially for treatment of post-operative pain and uremic pruritus.
Difelikefalin, also known CR845, is a novel and potent kappa opioid receptor agonist. CR845 exhibit low P450 CYP inhibition and low penetration into the brain. CR845 may be useful in the prophylaxis and treatment of pain and inflammation associated with a variety of diseases and conditions .
![]()
No. CAS 1024828-77-0
Difelikefalin ( INN ) (Developmental Code Names CR845 , FE-202845 ), Also Known As D -Phe- D -Phe- D -Leu- D -Lys- [Ganma- (4-N-Piperidinyl) Amino Carboxylic Acid] (As The Acetate Salt ), Is An Analgesic Opioid Peptide [2] Acting As A Peripherally-Specific , Highly Selective Agonist Of The kappa-Opioid Receptor (KOR). [1] [3] [4] [5] It Is Under Development By Cara Therapeutics As An Intravenous Agent For The Treatment Of Postoperative Pain . [1] [3] [5] An Oral Formulation Has Also Been Developed. [5] Due To Its Peripheral Selectivity, Difelikefalin Lacks The Central Side Effects Like Sedation , Dysphoria , And Hallucinations Of Previous KOR-Acting Analgesics Such As Pentazocine And Phenazocine . [1] [3] In Addition To Use As An Analgesic, Difelikefalin Is Also Being Investigated For The Treatment Of Pruritus (Itching). [1] [3] [4 ] Difelikefalin Has Completed Phase II Clinical Trials For Postoperative Pain And Has Demonstrated Significant And “Robust” Clinical Efficacy, Along With Being Safe And Well-Tolerated. [3] [5] It Is Also In Phase II Clinical Trials For Uremic Pruritus In Hemodialysis Patients. [4]
Difelikefalin Acts As An Analgesic By Activating KORs On Peripheral Nerve Terminals And KORs Expressed By Certain Immune System Cells . [1] Activation Of KORs On Peripheral Nerve Terminals Results In The Inhibition Of Ion Channels Responsible For Afferent Nerve Activity , Causing Reduced Transmission Of Pain Signals , While Activation Of KORs Expressed By Immune System Cells Results In Reduced Release Of Proinflammatory , Nerve-Sensitizing Mediators (Eg, Prostaglandins ). [1]
PATENT
Patent Document 2: Japanese Unexamined Patent Publication No. 2013-241447
1 (1) Synthesis of Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3)
to the four-necked flask of 2L, α-Boc-Pic- OMe · HCl [α-Boc-4 – aminopiperidine-4-carboxylic acid methyl hydrochloride] were charged (2) 43.7g (148mmol), was suspended in EtOAc 656mL (15v / w). To the suspension of 1-hydroxybenzotriazole (HOBt) 27.2g (178mmol), while cooling with Cbz-D-Lys (Boc) -OH 59.2g (156mmol) was added an ice-bath 1-ethyl -3 – (3-dimethylcarbamoyl amino propyl) was added to the carbodiimide · HCl (EDC · HCl) 34.1g (178mmol). After 20 minutes, stirring was heated 12 hours at room temperature. After completion of the reaction, it was added and the organic layer was 1 N HCl 218 mL of (5.0v / w). NaHCO to the resulting organic layer 3 Aq. 218ML (5.0V / W), Et 3 N 33.0 g of (326Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 218ML 1N (5.0V / W), NaHCO 3 Aq. 218mL (5.0v / w), NaClaq . Was washed successively with 218ML (5.0V / W), Na 2 SO 4 dried addition of 8.74g (0.2w / w). Subjected to vacuum filtration, was concentrated under reduced pressure resulting filtrate by an evaporator, and pump up in the vacuum pump, the Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3) 88.9g as a white solid obtained (96.5% yield, HPLC purity 96.5%).
In An Eggplant-Shaped Flask Of 2L, Cbz-D-Lys (Boc) -Arufa-Boc-Pic-OMe (3) 88.3g (142mmol) were charged, it was added and dissolved 441mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 17.7g (0.2w / w) was added, After three nitrogen substitution reduced pressure Atmosphere, Was Performed Three Times A Hydrogen Substituent. The Reaction Solution Was 18 Hours With Vigorous Stirring At Room Temperature To Remove The Pd / C And After The Completion Of The Reaction Vacuum Filtration. NaHCO The Resulting Filtrate 3 Aq. 441ML And (5.0V / W) Were Added For Liquid Separation, And The Organic Layer Was Extracted By The Addition Of EtOAc 200ML (2.3V / W) In The Aqueous Layer. NaHCO The Combined Organic Layer 3 Aq. 441ML And (5.0V / W) Were Added for liquid separation, and the organic layer was extracted addition of EtOAc 200mL (2.3v / w) in the aqueous layer. NaClaq the combined organic layers. 441mL and (5.0v / w) is added to liquid separation, was extracted by the addition EtOAc 200ML Of (2.3V / W) In The Aqueous Layer. The Combined Organic Layer On The Na 2 SO 4 Dried Addition Of 17.7 g of (0.2W / W), Then The Filtrate Was Concentrated Under Reduced Pressure Obtained Subjected To Vacuum Filtration By an evaporator, and pump up in the vacuum pump, D-Lys (Boc) -α-Boc-Pic- OMe (4) to give 62.7g (90.5% yield, HPLC purity 93.6%).
in the four-necked flask of 2L, D-Lys (Boc) -α-Boc-Pic-OMe (4) was charged 57.7 g (120 mmol), was suspended in EtOAc 576mL (10v / w). HOBt 19.3g (126mmol) to this suspension, was added EDC · HCl 24.2g (126mmol) while cooling in an ice bath added Cbz-D-Leu-OH 33.4g (126mmol). After 20 minutes, after stirring the temperature was raised 5 hours at room temperature, further the EDC · HCl and stirred 1.15 g (6.00 mmol) was added 16 h. After completion of the reaction, it was added liquid separation 1N HCl 576mL (10v / w) . NaHCO to the resulting organic layer 3 Aq. 576ML (10V / W), Et 3 N 24.3 g of (240Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 576ML 1N (10V / W), NaHCO 3 Aq. 576mL (10v / w), NaClaq . Was washed successively with 576ML (10V / W), Na 2 SO 4 dried addition of 11.5g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, the Cbz-D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe (5) 85.8g It was obtained as a white solid (98.7% yield, HPLC purity 96.9%).
in an eggplant-shaped flask of 1L, Cbz-D-Leu- D-Lys (Boc) -α-Boc-Pic -OMe the (5) 91.9g (125mmol) were charged, was added and dissolved 459mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 18.4g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 8 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 200mL (2.2v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 200mL (2.2v / w), NaClaq . It was sequentially added washed 200mL (2.2v / w). To the resulting organic layer Na 2 SO 4 dried added 18.4g (0.2w / w), to the filtrate concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and a pump-up with a vacuum pump. The resulting amorphous solid was dissolved adding EtOAc 200mL (2.2v / w), was crystallized by the addition of heptane 50mL (1.8v / w). Was filtered off precipitated crystals by vacuum filtration, the crystals were washed with a mixed solvent of EtOAc 120mL (1.3v / w), heptane 50mL (0.3v / w). The resulting crystal 46.1g to added to and dissolved EtOAc 480mL (5.2v / w), was crystallized added to the cyclohexane 660mL (7.2v / w). Was filtered off under reduced pressure filtered to precipitate crystals, cyclohexane 120mL (1.3v / w), and washed with a mixed solvent of EtOAc 20mL (0.2v / w), and 30 ° C. vacuum dried, D-Leu- as a white solid D-Lys (Boc) -α- Boc-Pic-OMe (6) to give 36.6 g (48.7% yield, HPLC purity 99.9%).
to the four-necked flask of 1L, D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe with (6) 35.8g (59.6mmol) was charged, it was suspended in EtOAc 358mL (10v / w). To this suspension HOBt 9.59g (62.6mmol), Cbz- D-Phe-OH 18.7g was cooled in an ice bath is added (62.6mmol) while EDC · HCl 12.0g (62.6mmol) It was added. After 20 minutes, a further EDC · HCl After stirring the temperature was raised 16 hours was added 3.09 g (16.1 mmol) to room temperature. After completion of the reaction, it was added and the organic layer was 1N HCl 358mL of (10v / w). NaHCO to the resulting organic layer 3 Aq. 358ML (10V / W), Et 3 N 12.1 g of (119Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 358ML 1N (10V / W), NaHCO 3 Aq. 358mL (10v / w), NaClaq . Was washed successively with 358ML (10V / W), Na 2 SO 4 dried addition of 7.16g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, Cbz-D-Phe-D -Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7) was obtained 52.5g as a white solid (yield quant, HPLC purity 97.6%).
in an eggplant-shaped flask of 2L, Cbz-D-Phe- D-Leu-D-Lys ( Boc) -α-Boc-Pic- OMe (7) the 46.9g (53.3mmol) were charged, the 840ML EtOAc (18V / W), H 2 added to and dissolved O 93.8mL (2.0v / w) It was. The 5% Pd / C to the reaction mixture 9.38g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 10 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 235mL (5.0v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 235mL (5.0v / w), NaClaq . It was added sequentially cleaning 235mL (5.0v / w). To the resulting organic layer Na 2 SO 4 dried addition of 9.38g (0.2w / w), then the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, pump up with a vacuum pump to D-Phe -D-Leu-D-Lys ( Boc) -α-Boc-Pic-OMe (7) was obtained 39.7g (yield quant, HPLC purity 97.3%).
In An Eggplant-Shaped Flask Of 20ML Boc-D-Phe-D -Phe-D- Leu-D- lys (Boc) -α -Boc- Pic-OMe (9) and 2.00gg, IPA 3.3mL (1.65v / w), was suspended by addition of PhMe 10mL (5v / w). It was stirred at room temperature for 19 hours by addition of 6N HCl / IPA 6.7mL (3.35v / w). The precipitated solid was filtered off by vacuum filtration and dried under reduced pressure to a white solid of D-Phe-D-Phe- D- Leu-D-Lys-Pic- OMe 1.59ghydrochloride (1) (yield: 99 .0%, HPLC purity 98.2%) was obtained.
In An Eggplant-Shaped Flask Of 20ML-D-Phe-D- Phe D-Leu -D-Lys- pic-OMe hydrochloride crude crystals (1) were charged 200mg, EtOH: MeCN = 1: after stirring for 1 hour then heated in a mixed solvent 4.0 mL (20v / w) was added 40 ° C. of 5 , further at room temperature for 2 was time stirring slurry. Was filtered off by vacuum filtration, the resulting solid was dried under reduced pressure a white solid ((1) Purification crystals) was obtained 161 mg (80% yield, HPLC purity 99.2% ).
(1)) Of (A) To A Round-Bottomed Flask Of 10ML D-Phe-D-Phe-D- -D-Lys Leu-Pic-OMe Hydrochloride Salt (1) Was Charged With Purified Crystal 38.5Mg (0.0488Mmol), H 2 Was Added And Dissolved O 0.2ML (5.2V / W). 1.5H Was Stirred Dropwise 1N NaOH 197MyuL (0.197mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 48.8μL (0.0488mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys- Pic (A) (yield: quant , HPLC purity 99.7%).
D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe (1) physical properties 1 H NMR (400 MHz, 1M DCl) [delta] ppm by: 0.85-1.02 (yd,. 6 H), 1.34-1.63 ( m, 5 H), 1.65-2.12 ( m, 5 H), 2.23-2.45 (m, 2 H), 2.96-3.12 (m, 4 H), 3.19 (ddt, J = 5.0 & 5.0 & 10.0 Hz), 3.33-3.62 (m, 1 H), 3.68-3.82 (m, 1 H), 3.82-3.95 (m, 4 H), 3.95-4.18 (m, 1 H), 4.25-4.37 (m, 2 H), 4.61-4.77 (M, 2 H), 7.21-7.44 (M, 10 H) 13 C NMR (400MHz, 1M DCl) Deruta Ppm: 21.8, 22.5, 24.8, 27.0, 30.5, 30.8, 31.0, 31.2, 31.7, 37.2 , 37.8, 38.4, 39.0, 39.8, 40.4, 40.6, 41.8, 42.3, 49.8, 50.2, 52.2, 52.6, 54.6, 55.2, 57.7, 57.9, 127.6, 128.4, 129.2, 129.6, 129.7, 129.8 dp 209.5 ℃
(Trifluoroacetic Acid (TFA)
Use) (1) D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe TFA Synthesis Of Salt (1)
TFA 18ML Eggplant Flask Of 50ML (18V / W) , 1- Dodecanethiol 1.6ML (1.6V / W), Triisopropylsilane 0.2ML (0.2V / W), H 2 Sequentially Added Stirring The O 0.2ML (0.2V / W) Did. The Solution To The Boc-D-Phe- D- Phe-D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe the (9) 1.00g (1.01mmol) was added in small portions with a spatula. After completion of the reaction, concentrated under reduced pressure by an evaporator, it was added dropwise the resulting residue in IPE 20mL (20v / w). The precipitated solid was filtered off, the resulting solid was obtained and dried under reduced pressure to D-Phe-D-Phe- D-Leu -D-Lys-Pic-OMe · TFA salt as a white solid (1) (Osamu rate 93.0%, HPLC purity 95.2%).
to a round-bottomed flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe TFA were charged salt (1) 83mg (0.0843mmol), was added and dissolved H2O 431μL (5.2v / w). Was 12h stirring dropwise 1N NaOH 345μL (0.345mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 84.3μL (0.0843mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 95.4%).
3 (HCl / EtOAc
Use) (1) In An Eggplant-Shaped Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OMe (9) 1. It was charged with 00g (1.01mmol ), was added and dissolved EtOAc7.0mL (7.0v / w). 4N HCl / EtOAc 5.0mL (5.0v / w) was added after 24h stirring at room temperature, the precipitated solid was filtered off by vacuum filtration, washed with EtOAc 2mL (2.0v / w). The resulting solid D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe hydrochloride (1) was obtained 781mg of a white solid was dried under reduced pressure (the 96.7% yield, HPLC purity 95.4%).
eggplant flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe hydrochloride were charged salt (1) 90 mg (0.112 mmol), H 2 was added and dissolved O 0.47mL (5.2v / w). Was 12h stirring dropwise 1N NaOH 459μL (0.459mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.112μL (0.112mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 93.1%).
Compound (1) Of The Compound By Hydrolysis Synthesis Of (The A) (Compound (1) Without
Purification) Eggplant Flask 10ML D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe (1) Charged Hydrochloride Were (Without Pre-Step Purification) 114.5Mg (0.142Mmol), H 2 Was Added And Dissolved O 595MyuL (5.2V / W). Was 14H Stirring Dropwise 1N NaOH 586MyuL (0.586Mmol) At Room Temperature. After Completion Of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.15μL (0.150mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) (yield: quant, HPLC purity 95.2 %).
Path Not Via The Compound (1) (Using Whole Guard Boc-D-Phe-D-Phe-D-Leu-D-Lys (Boc) -Alpha-Boc-Pic-OMe
(A)) (1) D–Boc Phe- D-Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OH Synthesis Of
Eggplant Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D- Lys (Boc) -α- Boc-Pic -OMe (9) were charged 1.00g (1.00mmol), was added and dissolved MeOH 5.0mL (5.0v / w). After stirring for four days by the addition of 1N NaOH 1.1 mL (1.10mmol) at room temperature, further MeOH 5.0mL (5.0v / w), 1N NaOH 2.0mL the (2.0mmol) at 35 ℃ in addition 3h and the mixture was stirred. After completion of the reaction, 1 N HCl 6.1 mL was added, After distilling off the solvent was concentrated under reduced pressure was separated and the organic layer was added EtOAc 5.0mL (5.0mL) .NaClaq. 5.0mL (5.0v / w) Wash the organic layer was added, the organic layer as a white solid was concentrated under reduced pressure to Boc-D-Phe-D- Phe-D-Leu-D-Lys (Boc) – α-Boc-Pic-OH 975.1mg (99.3% yield, HPLC purity 80.8% )
to a round-bottomed flask of 20mL Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) It was charged -α-Boc-Pic-OH ( 10) 959mg (0.978mmol), was added and dissolved EtOAc 4.9mL (5.0v / w). And 4h stirring at room temperature was added dropwise 4N HCl / EtOAc 4.9mL (5.0mL) at room temperature. After completion of the reaction, it was filtered under reduced pressure, a white solid as to give D-Phe-D-Phe- D-Leu-D-Lys-Pic the (A) (96.4% yield, HPLC purity 79.2%) .
References
- S. Sinatra Raymond; Jonathan S. Jahr;. J. Michael Watkins-Pitchford (14 October 2010) The Essence Of Analgesia And Analgesics …. Cambridge University Press Pp 490-491 ISBN 978-1-139-49198-3 .
- A Janecka, Perlikowska R, Gach K, Wyrebska A, Fichna J (2010) “Development Of Opioid Peptide Analogs For Pain Relief”.. Curr Pharm Des… 16 (9):. 1126-35 Doi : 10.2174 / 138161210790963869 . PMID 20030621 .
- Apfelbaum Jeffrey (8 September 2014). Ambulatory Anesthesia, An Issue Of Anesthesiology Clinics, . Elsevier Health Sciences. Pp. 190-. ISBN 978-0-323-29934-3 .
- Cowan Alan;. Gil Yosipovitch (10 April 2015) Pharmacology Of Itch …. Springer Pp 307- ISBN 978-3-662-44605-8 .
- Allerton Charlotte (2013). Pain Therapeutics: Current And Future Treatment Paradigms …. Royal Society Of Chemistry Pp 56- ISBN 978-1-84973-645-9 .
REFERENCES
1: Cowan A, Kehner GB, Inan S. Targeting Itch With Ligands Selective For kappa Opioid
. Receptors Handb Exp Pharmacol 2015; 226:.. 291-314 Doi:
.. 10.1007 / 978-3-662-44605-8_16 Review PubMed PMID: 25861786.
 |
|
| Systematic (IUPAC) Name | |
|---|---|
|
Amino–4 1- ( D -Phenylalanyl- D -Phenylalanyl- D -Leucyl- D -Lysyl) Piperidine-4-Carboxylic Acid
|
|
| Clinical data | |
| Of Routes Administration |
Intravenous |
| Pharmacokinetic Data | |
| Bioavailability | Pasento 100 ( IV ) [1] |
| Metabolism | Metabolized Not [1] |
| Biological half-life | Hours 2 [1] |
| Excretion | As Unchanged Excreted Drug Via Bile And Urine [1] |
| Identifiers | |
| CAS Number | 1024828-77-0 |
| ATC code | None |
| ChemSpider | 44208824 |
| Chemical data | |
| Formula | C 36 H 53 N 7 O 6 |
| Molar mass | 679.85 g / mol |
///// Difelikefalin, CR845 , FE-202845, Phase III, PEPTIDES
CC (C) C [C @ H] (C (= O) N [C @ H] (CCCCN) C (= O) N1CCC (CC1) (C (= O) O) N) NC (= O) [ C @@ H] (Cc2ccccc2) NC (= O) [C @@ H] (Cc3ccccc3) N
AUNP-12 from Aurigene Discovery Technologies Limited
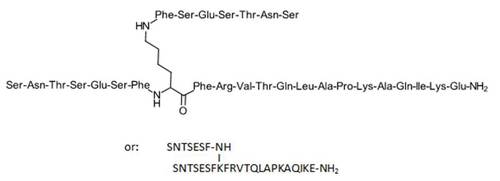
AUNP-12
AUR-012; Aurigene-012; NP-12, Aurigene; PD-1 inhibitor peptide (cancer), Aurigene; PD-1 inhibitor peptide (cancer), Aurigene/ Pierre Fabre; W-014A
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Laboratoires Pierre Fabre S.A. |
Aurigene Discovery Technologies Limited

-
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
-
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
-
The PD-1 protein’s structure comprise of—
-
- an extracellular IgV domain followed by
- a transmembrane region and
- an intracellular tail
-
-
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
-
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
-
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
-
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
-
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
-
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
-
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
-
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
-
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
-
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
-
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
-
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
-
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
-
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.


Patent
http://www.google.com/patents/US20110318373
| 8. | SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49) |
|

Example 2 Synthesis of

Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
-
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
-
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
STRUCTURE , READER DISCRETION IS NEEDED
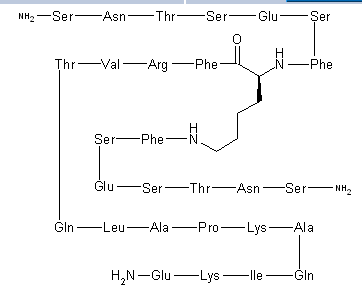
N2,N6-Bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-alpha-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-L-alpha-glutamine
C142 H226 N40 O48, 3261.553
SEE ALSO
US 2015087581
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+;); 816.2[M/4+H]+.
SMILES
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O
NEXT………..
- Molecular Weight, 3261.55
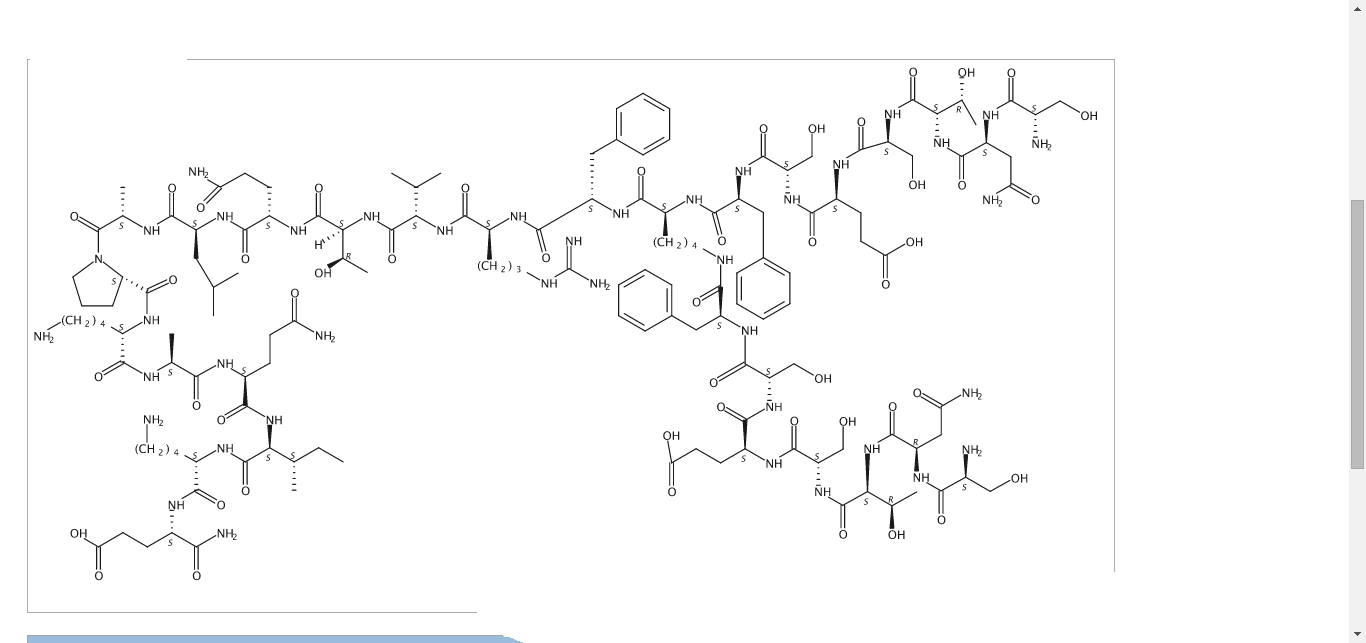
NEXT……….

Clips
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
Feb 13, 2014, 03:14 ET from Aurigene and Pierre Fabre Pharmaceuticals
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapy and targeted therapy as the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12 is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1 It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways. AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway. Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
For more information: http://aurigene.com/
About Pierre Fabre:
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10 000 people worldwide, including 1 300 in R&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
REFERENCES
http://slideplayer.com/slide/5760496/
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
M. Cordingley, “Battle of PD-1 blockade is on”, February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
Mr. CSN Murthy
Chief Executive Officer, Aurigene Discovery Technologies Ltd.

Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click here or Select File and then Print from your browser's menu “Strategic partnerships will boost drug discovery activities in India”Wednesday, May 16, 2007 08:00 IST
What are the factors led to the scores of partnerships in research and drug discovery space? Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects. What do you think is the uniqueness of Aurigene that sets it apart from other companies? It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this? The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market? In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India. How much do you acknowledge science and strategy in Aurigene’s growth? What according to you is the future of the drug discovery sector? |
 The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
FEW MORE COMPDS FROM PATENT
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54, Sequence Length: 29, 22, 7
multichain; modified (modifications unspecified)
SNTSESFK FRVTQ LAPKAQIKE, 1353564-66-5
SNTSESF
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54

NEXT……………………
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF
CAS 1353564-64-3
C142 H226 N40 O48
L-α-Glutamine, L-seryl-D-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
MW 3261.55, Sequence Length: 29, 22, 7
multichain; modified

smiles
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O
NEXT……………..
CAS 1353564-60-9
C142 H226 N40 O48
L-α-Glutamine, D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFKFR VTQLAPKAQI KE
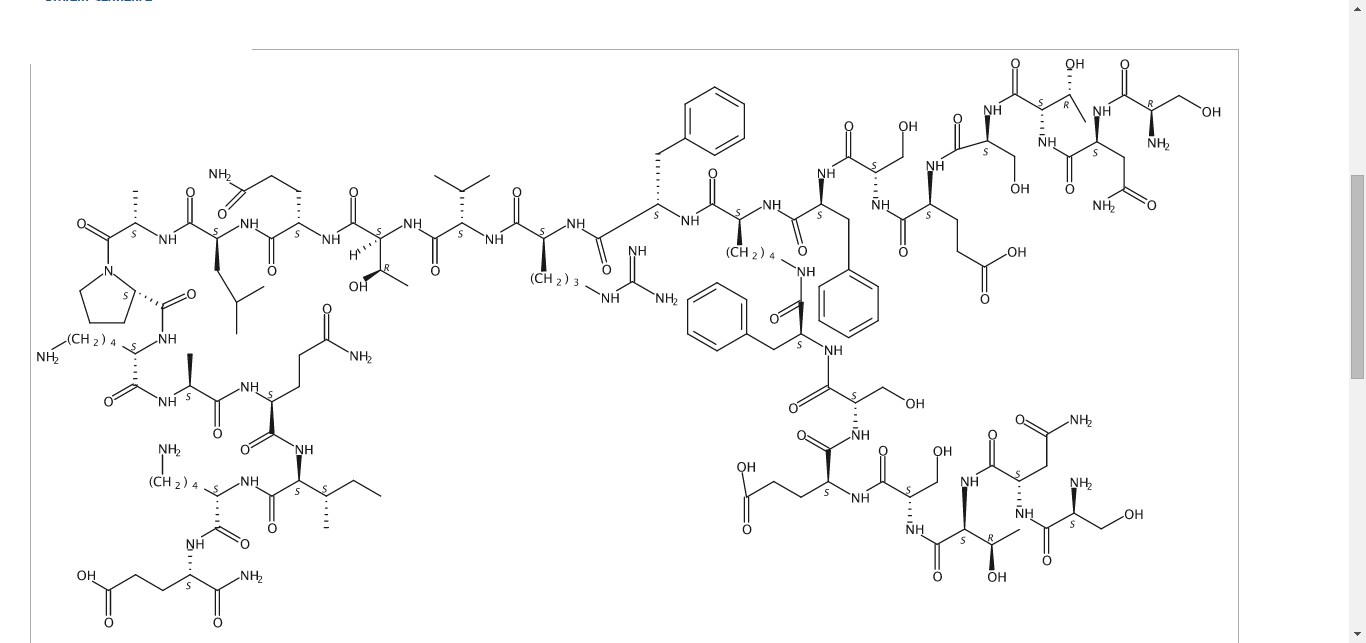
NRXT…………………….
. CAS 1353564-61-0
C142 H226 N40 O48
L-α-Glutamine, N2,N6-bis(D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF

/////////////

{kind=link}