

TIRUPATI, INDIA
 .
.
Home » Posts tagged 'osteoarthritis'
Tag Archives: osteoarthritis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
IMRECOXIB
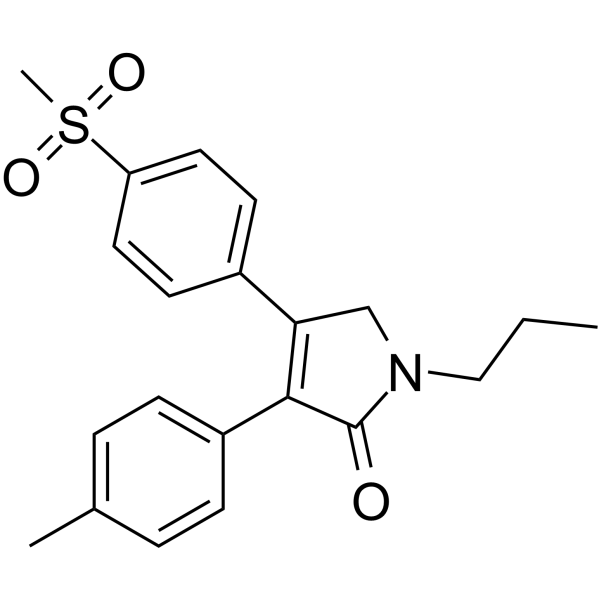
Imrecoxib (Hengyang)
CHINA 2012 osteoarthritis2H-Pyrrol-2-one, 1,5-dihydro-3-(4-methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-
3-(4-Methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-1,5-dihydro-2H-pyrrol-2-one395683-14-4[RN]
Imrecoxib was approved by China Food and Drug Administration (CFDA) on May 20, 2011. It was developed and marketed as 恒扬® by HengRui Pharmaceuticals.
Imrecoxib is a selective COX-2 inhibitor indicated for treatment of osteoarthritis.
恒扬® is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
Common name: Imrecoxib; BAP-909; BAP 909; BAP909
Trademarks: Hengyang
Molecular Formula: C21H23NO3S
CAS Registry Number: 395683-14-4
IUPAC Name: 4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one
Molecular Weight: 369.48
SMILES: O=C1N(CCC)CC(C2=CC=C(S(=O)(C)=O)C=C2)=C1C3=CC=C(C)C=C3
Mechanism: COX-2 Inhibitor; Cyclooxygenase-2 Inhibitor
Activity: Treatment of Osteoarthritis; Analgesic; Antipyritic; Antiinflammatory Drug
Status: Launched 2011 (China)
Originator: HengRuiDrug Name:ImrecoxibResearch Code:BAP-909Trade Name:恒扬®MOA:Selective cyclooxygenase-2 (COX-2) inhibitorIndication:Osteoarthritis (OA)Status:ApprovedCompany:HengRui (Originator)Sales:ATC Code:Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-05-20 | Marketing approval | 恒扬 | Osteoarthritis (OA) | Tablet, Film coated | 100 mg | HengRui |
Reference:1. US7112605B2.Route 2
Reference:1. CN102206178A.
2. Chinese Chem. Lett. 2001, 12, 775-778.Route 3
Reference:1. CN104193664A.
Imrecoxib | NSAID | Treatment of Osteoarthritis | COX-2 Inhibitor
Imrecoxib [4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one] is a novel and moderately selective cyclooxygenase-2 (COX-2) inhibitor that possesses anti-inflammatory effect by inhibition of COX-2 mRNA expression. It belongs to the family of non-steroid anti-inflammtory drugs (NSAIDs). Imrecoxib was found to inhibit COX-1 and COX-2 with IC50 value of 115 ± 28 nM and 18 ± 4 nM, respectively [1].
| Imrecoxib: 2D and 3D Structure |
Imrecoxib effectively inhibited carrageenan-induced acute inflammation at the doses of 5, 10, and 20 mg-kg-1 ig and adjuvant-induced chronic inflammation at the doses of 10 and 20 mg-kg -1·d-1 ig.
NSAIDs and Imrecoxib:
Non-steroidal anti-inflammatory drugs (NSAIDs) are used extensively for the treatment of inflammatory conditions, including pain-releasing, anti-pyretic and rheumatoid arthritis. These functions are believed to inhibit the enzyme cyclooxygenase (COX) that is involved in the biosynthesis of prostaglandins G and H from arachidonic acid. So far two isozymes of COX are known: COX-1 and COX-2. COX-1 is constitutively produced in a variety of tissues and appears to be important to the maintenance of normal physiological functions, including gastric and renal cytoprotection. The COX-2 is an inducible isozyme, which is produced in cells under the stimulation of endotoxins, cytokines, and hormones and catalyzes the production of prostaglandins which cause inflammation.
The currently therapeutic use of NSAIDs has been associated with the inhibition of both COX-1 and COX-2 and causes well-known side effects at the gastrointestinal and renal level. Therefore, the selective COX-2 inhibitors could provide anti-inflammatory agents devoid of the undesirable effects associated with classical, nonselective NSAIDs. In addition, COX-2 is over-expressed in colon cancer tissue. COX-2 inhibitors possess potential prophylactic and therapeutic application to colon cancer.
Imrecoxib is designed in a manner such that it has “moderate selectivity” for COX-2 over COX-1. This balanced inhibition to both COX-1 and COX-2 was pursued to maintain the homeostasis of the two enzymes in the body,which is presumably critical to normal functions of the cardiovascular system.
Imrecoxib was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in May 2011. Hengyang is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
SYN
Imrecoxib Synthesis
Chin Chem Lett 2001, 12, 775-778 (also Ref 2. This route is quoted as industrial method in various texts)
CN104193664A (an improvement here as Br is replaced with Cl)

US7112605B2 (primary reference for synthesis routes)

Identification:

| 1H NMR (Estimated) for Imrecoxib |
Experimental: 1H-NMR (CDCl3, TMS, 400MHz) 1.008 (3H, t, J = 7.2Hz), 1.701-1.756 (2H, m), 2.376 (3H, s), 3.078 (3H, s), 3.575 (2H, t, J = 7.2Hz), 4.317 (2H, s), 7. 175 (2H, d, J = 8.0Hz), 7.294 (2H, d, J = 8.0Hz), 7.505 (2H, t, J = 6.8Hz), 7.870 (2H, t, J = 6.8Hz)
Sideeffects:
Being a mild COX-2 inhibitor, it is expected not to cause any serious cardiovascular risks. Similarly, it should not have any serious gastrointestinal problems too, as it not a good inhibitor of COX-1. None of the reports though have listed any serious adverse event reported by patients in the clinical trials.
References:
- Cheng, G. F.;et. al. Imrecoxib: A novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004, 25(7), 927-931.
- Zhang, F.;et. al.Method for preparing imrecoxib. CN102206178A
- Chao, W.;et. al. Synthesis method of imrecoxib. CN104193664A
- Bai, A. P.;et. al. Synthesis and in vitro Evaluation of a New Class of Novel Cyclooxygenase-2 Inhibitors: 3, 4-diaryl-3-pyrrolin-2 ones.Chin Chem Lett 2001, 12, 775-7785.
- Guo, Z. Discovery of imrecoxib. Chin J New Drugs2012, 21, 223.
- Guo, Z.;et. al. Sulfonyl-containing 3,4-diaryl-3-pyrrolin-2-ones, preparation method, and medical use thereof. US7112605B2
![MS 2 spectrum of the [M þ H] þ ion (m/z 370) of imrecoxib (inset, full-scan mass spectrum).](https://www.researchgate.net/profile/Dafang_Zhong/publication/6930659/figure/fig4/AS:394580018122760@1471086616006/MS-2-spectrum-of-the-M-th-H-th-ion-m-z-370-of-imrecoxib-inset-full-scan-mass.png)
SYN
Imrecoxib (Hengyang)
Imrecoxib, a new non-steroid anti-inflammtory drug (NSAID), was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in 2012. It was originally designed and synthesized by Guo and co-workers at the Institute of Materia Medica (IMM) of the Chinese Academy of Medical Sciences in collaboration with Hengrui Pharmaceuticals.88 Imrecoxib, which is a moderately selective COX-2 inhibitor (with IC50 values against COX-1 and COX-2 being 115 ± 28 and 18 ± 4 nM, respectively),89 is the subject of twwo synthetic routes reported across several publications.90–93
The most likely process-scale route to this drug is described in Scheme 15, 93 which began with 2-bromo-40 -(methylsulfonyl)-acetophenone (84) and p-tolylacetic acid (85) as starting materials. In the presence of base, a-bromoketone 84 was treated with acid 85 which resulted in lactone 86 in 72% yield across the two-step sequence. Exposure of lactone 86 with propylamine triggered a ring-opening-ring closing reaction, which resulted in imrecoxib (XIII) directly in 85% yield.93

88. Guo, Z. R. Chin. J. New Drugs 2012, 21, 223.
89. Chen, X. H.; Bai, J. Y.; Shen, F.; Bai, A. P.; Guo, Z. R.; Cheng, G. F. Acta Pharmacol. Sin. 2004, 25, 927.
90. Bai, A. P.; Guo, Z. R.; Hu, W. H.; Shen, F.; Cheng, G. F. Chin. Chem. Lett. 2001, 12, 775.
91. Guo, Z.; Cheng, G.; Chu, F.; Yang, G.; Xu, B. CN Patent 1134413 C, 2001.
92. Guo, Z.; Cheng, G.; Chu, F. US Patent 2004/0029951 A1, 2004.
93. Zhang, F. Y.; Shen, X. M.; Sun, P. Y. CN Patent 102206178 A, 2011
Patent
CN 111747879
PATENT
CN 111747874
CN 111747873
CN 110386891
CN 109553564
CN 109553563
CN 108997188
CN 108947884
CN 108912030
CN 108864003
CN 108707100
CN 107586268
CN 104193664
CN 102206178
CN 101774958
US 20040029951
PATENT
CN 109678775
https://patents.google.com/patent/CN102206178A/en
Ai Rui former times cloth (N-n-propyl-3-p-methylphenyl-4-is to methylsulfonyl phenyl-3-pyrrolidin-2-one) is the nonsteroidal anti-inflammatory drug that a kind of appropriateness suppresses COX-2; put down in writing the synthetic method of Ai Rui former times cloth in the prior art (US20040029951), may further comprise the steps:
1) is raw material with the methylsulfonyl methyl phenyl ketone, makes alpha-brominated methylsulfonyl methyl phenyl ketone through bromo;
2) sodium borohydride reduction of alpha-brominated methylsulfonyl methyl phenyl ketone obtains the Styrene oxide 98min. derivative;
3) reaction of Styrene oxide 98min. derivative and Tri N-Propyl Amine generates N-n-propyl-capable biology of beta-hydroxyphenyl ethamine;
4) tolyl-acetic acid and the reaction of excessive thionyl chloride are generated the methylbenzene Acetyl Chloride 98Min.;
5) methylbenzene Acetyl Chloride 98Min. and the capable biological respinse of N-n-propyl-beta-hydroxyphenyl ethamine are generated N-n-propyl-N-[2-hydroxyl-2-to the methylsulfonyl styroyl] phenylacetamide;
6) Jone ‘ s reagent or pyridine chromium trioxide oxidation N-n-propyl-N-[2-hydroxyl-2-are to the methylsulfonyl styroyl] phenylacetamide obtains the capable biology of oxo phenylacetamide;
7) the above-mentioned oxo phenylacetamide of condensation makes end product Ai Rui former times cloth under the alkaline medium effect.
Because existing preparation method’s route is longer, and relate to reduction, oxidation, steps such as acid amides coupling, solvent load is big, the cost height, particularly to use oxygenants such as Jone ‘ s reagent or pyridine chromium trioxide in the oxidation step, low and the product of this oxidation step productive rate is difficult for separation and purification, and is difficult to control because chromium metal residual quantity control criterion in bulk drug is extremely strict, thereby makes this preparation method be difficult to be applicable to scale operation.
Synthetic route 1
Step 1), preparation alpha-brominated methylsulfonyl methyl phenyl ketone (III)
51.0g 4-methylsulfonyl methyl phenyl ketone and 760mL acetic acid is added to has magnetic agitation, in three mouthfuls of glass flask of the 1000mL of thermometer and constant pressure funnel.Be heated to 40 ℃, beginning slowly drips 41.1g liquid bromine, after dripping, continues to stir 30 minutes at 40 ℃.Reaction solution 50 ℃ concentrate after, add entry, stir, filter, washing, oven dry obtains the thick product of 70.5g, adds ethyl acetate/normal hexane mixed solvent, reflux 1 hour, slowly be cooled to 25 ℃, filtering drying gets the alpha-brominated methylsulfonyl methyl phenyl ketone of 56.5g off-white color solid (III), yield 80.0%.
1H-NMR(CDCl 3,TMS,400MHz):3.120(3H,s),4.485(2H,s),8.101(2H,dd,J=2.0Hz),8.191(2H,dd,J=2.0Hz)
MS(M+1):279.05
Step 2), prepare 4-(4-methylsulfonyl phenyl)-3-(4-aminomethyl phenyl)-2,5-dihydrofuran-2-ketone (II)
Experiment condition A
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 44.3g (III), 24.0g 4-methylphenyl acetic acid and 600mL acetonitrile are added to and have magnetic agitation, in the 500mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 24.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 1 hour.Add the 36.0mL triethylamine again, reaction solution is heated to 75 ℃, stirring reaction 18 hours.Cool to 25 ℃, concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 28.0g light yellow solid compound (II), yield 53.4%.
1H-NMR(CDCl 3,TMS)2.398(3H,s),3.091(3H,s),5.192(2H,s),7.216(2H,d,J=8.0Hz),7.292(2H,d,J=8.0Hz),7.543(2H,d,J=8.0Hz),7.933(2H,d,J=8.0Hz)
MS(M+1):329.02
Similarly, compound (II) can prepare under experiment condition B, C, D.
Experiment condition B
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 5.0g (III), 2.7g 4-methylphenyl acetic acid and 70mL acetonitrile are added to and have magnetic agitation, in the 100mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 2.3mL tetramethyl guanidine by constant pressure funnel, temperature is controlled at 20 ℃, after adding, continues to stir 1.5 hours.Add the 4.6mL tetramethyl guanidine again, 20 ℃ of stirring reactions 2 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 2.5g light yellow solid compound (II), yield 42.0%.
Experiment condition C
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.85g (III), 1.0g 4-methylphenyl acetic acid and 20mL ethanol are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 1.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 3 hours.Add the 2.0mL triethylamine again, 80 ℃ of stirring reactions 18 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.83g light yellow solid compound (II), yield 38.1%.
Experiment condition D
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.0g (III), 0.54g 4-methylphenyl acetic acid and 12mL acetonitrile are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Add 1.0g salt of wormwood, 25 ℃ were reacted 2 hours.50 ℃ of stirring reactions are 5 hours then.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.13g light yellow solid compound (II), yield 11%.
Step 3), preparation N-n-propyl-3-p-methylphenyl-4-are to methylsulfonyl phenyl-3-pyrrolidin-2-one (Ai Rui former times cloth (I))
Experiment condition A
With the 25.0mL Tri N-Propyl Amine, be added drop-wise in the 17.5mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 10.0g compound (II).Under the nitrogen protection, be heated to 160 ℃, stirring reaction 8 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 8.2g white solid product compound (I), yield 72.8%.
1H-NMR(CDCl 3,TMS,400MHz)1.008(3H,t,J=7.2Hz),1.701-1.756(2H,m),2.376(3H,s),3.078(3H,s),3.575(2H,t,J=7.2Hz),4.317(2H,s),7.175(2H,d,J=8.0Hz),7.294(2H,d,J=8.0Hz),7.505(2H,t,J=6.8Hz),7.870(2H,t,J=6.8Hz)
MS(M+1):370.17
Similarly, compound (I) can prepare under experiment condition B, C, D.
Experiment condition B
2.9g Tri N-Propyl Amine hydrochloride and 1.0g compound (II) are mixed, under the nitrogen protection, be heated to 170 ℃, stirring reaction 2 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 0.9g white solid product compound (I), yield 80.0%.
Experiment condition C
Digest compound (II) with 2.0,3 milliliters of Tri N-Propyl Amines, 1.75 gram Tri N-Propyl Amine hydrochlorides add in the tube sealing of nitrogen protections, are heated to 140 ℃, react 20 hours.Be cooled to room temperature, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 1.8g white solid product compound (I), yield 80.0%.
Experiment condition D
With the 0.5mL Tri N-Propyl Amine, be added drop-wise in the 0.35mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 0.5g compound (II).Under the nitrogen protection, be heated to 120 ℃, stirring reaction 4 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Obtain 0.14g compound (I) after organic phase is concentrated and purified, yield 24.2%.Publication numberPriority datePublication dateAssigneeTitleCN104072467A *2014-07-072014-10-01太仓博亿化工有限公司Synthesis method of 5-chloro-2-benzofuranyl-p-chlorophenyl-oneCN104193664A *2014-08-222014-12-10山东铂源药业有限公司Synthesis method of imrecoxibCN107586268A *2016-07-072018-01-16江苏恒瑞医药股份有限公司A kind of preparation method of imrecoxib and its intermediateCN108864003A *2018-06-152018-11-23江苏美迪克化学品有限公司A kind of preparation method of imrecoxib intermediate and imrecoxibCN108947884A *2018-06-292018-12-07江苏美迪克化学品有限公司A kind of Preparation Method And Their Intermediate of imrecoxibCN109553564A *2017-09-252019-04-02江苏恒瑞医药股份有限公司A kind of purification process of imrecoxibCN109678775A *2017-10-182019-04-26江苏恒瑞医药股份有限公司A kind of crystal form and preparation method thereof of COX-2 selective depressantCN107586268B *2016-07-072021-01-19江苏恒瑞医药股份有限公司Preparation method of dapoxib and intermediate thereofPublication numberPriority datePublication dateAssigneeTitleUS5489693A *1992-04-281996-02-06Linz; GuenterCyclic imino derivatives, pharmaceutical compositions containing these compounds and processes preparing themCN101386590A *2007-09-132009-03-18中国医学科学院药物研究所Pyrrolidone containing hydroxymethyl and carboxyl, preparation method and medicament composition and use thereofCN101497580A *2009-01-092009-08-05华南理工大学HIV-1 inhibitor 2-pyrrolidinone derivative, as well as synthesizing method and use thereof
PAPER
Chinese Chemical Letters (2001), 12(9), 775-778.
PATENT
CN 110386891,

CLIP
For Chinese drugmaker Hengrui, R&D plans pan out
Ambitious program to launch innovative drugs starts to pay off for generics producerby Jean-François TremblayJULY 17, 2017 | APPEARED IN VOLUME 95, ISSUE 29

Credit: Jean-François Tremblay/C&ENHengrui recently invested in a custom-made phage-display library screening system for its Shanghai lab.
Launching their own innovative pharmaceuticals is a common goal for managers of generic drug firms. But it remains a dream for many. Jiangsu Hengrui Medicine, one of China’s largest generic drug makers, has advanced further than most. It has already launched two of its own drugs in China and licensed rights to another to a U.S. firm.
JIANGSU HENGRUI MEDICINE AT A GLANCE
▸ Headquarters: Lianyungang, Jiangsu, China
▸ 2016 sales: $1.6 billion
▸ 2016 profits: $390 million
▸ Employees: More than 13,000, 2,000 of whom work for a Shanghai-based unit developing and commercializing innovative drugs
▸ Innovative drug R&D staff: 800
Obtaining these results required substantial resources, though. Back in 2004, Hengrui built a large R&D lab in Shanghai, hired world-class researchers to lead it, equipped the facility with the latest instruments, and staffed it with hundreds of scientists.
Initially, the project looked like a money pit. In Chinese industry circles, many doubted that it would amount to anything. But revenues from the company’s innovative drugs are starting to pour in, and R&D at Hengrui is well on its way to financial sustainability.
Over the past 10 years, China has made great strides in growing an innovative drug industry. For all the talk, cynics say, China has yet to foster a blockbuster with $1 billion or more in annual sales. But as Hengrui and other Chinese firms launch their own drugs at home and license the foreign rights to others, it is becoming clear that an innovative drug industry is taking root.
“Producing generic drugs funds our R&D,” says Weikang Tao, a Hengrui vice president who doubles as chief executive officer of Shanghai Hengrui, the company’s innovative drug subsidiary.
Overall, Hengrui invests more than 10% of its sales in R&D, “which is big by Chinese standards,” Tao says. The drug giant Pfizer by comparison spent about 15% of its sales on R&D in 2016. With sales of $1.6 billion last year, Hengrui does most of its business in China. But it also exports finished drugs to the U.S., making it one of the few Chinese firms to have the U.S. Food & Drug Administration’s okay to do so.
Hengrui was formed in 1970 as a state-owned company. It began investing in its own R&D in 2004 and has since cultivated an innovative drug subsidiary that employs 2,000 people, including more than 800 at a Shanghai lab and about 20 at a subsidiary in Princeton, N.J. Other staffers work in the usual functions found in an innovative drug firm: clinical trial management, regulatory affairs, marketing and sales, and so on.
The Shanghai subsidiary recruits in China and internationally. Tao, who joined Hengrui in 2014, is a Chinese-trained physician who earned a Ph.D. in molecular and cell biology at the University of Medicine & Dentistry of New Jersey. He focused on tumor cell biology during a postdoc at Princeton University and worked in research at Merck & Co. for 10 years. Hengrui is constantly hiring, he notes.
Hengrui’s research facilities appear to be well equipped. Earlier this year the firm opened a biologics drug lab and a pilot plant for process development in Shanghai. “We spent nearly $7 million just on equipment for the biologics lab,” Tao says.

The lab is equipped with a custom-made automated phage-display library screening system that speeds up the process of discovering antibody drugs. “The machine can do automatically in a few hours what would otherwise take days for several scientists,” says Jiakang Sun, group leader of in vivo pharmacology at Shanghai Hengrui Pharmaceutical. With the phage-display system, Sun adds, a library displaying millions of human antibodies can be screened in vitro to find antibodies that bind to a specific antigen.
However well-staffed and well-equipped, Hengrui’s labs are still smaller than those of Merck or other major drug firms. But Hengrui has made notable strides recently. In 2015, it became the first Chinese firm to license a drug candidate to a U.S. firm. Incyte agreed to pay $25 million up front, and several hundred million dollars more once certain milestones are met, for the rights outside China and Taiwan to camrelizumab, a cancer treatment in Phase III human clinical trials in China.

In China, Hengrui’s priority market, the firm launched the osteoarthritis treatment imrecoxib in 2011 and the gastric cancer drug apatinib in 2014. The two will eventually achieve combined annual sales of $160 million, Hengrui expects.
Together with the licensing deal with Incyte, this will allow the firm to nearly recoup its R&D investment. Launching a few more compounds, particularly in the U.S., would make innovative R&D at Hengrui solidly profitable. The company is making good progress in that direction. A neutropenia treatment awaits final market approval in China, and five others have reached Phase III trials. Hengrui also has drugs in Phase I clinical trials in the U.S.
“I wouldn’t say that our lab is more productive than a lab operated by a multinational drug firm,” Tao says. Merck and other major players operate excellent facilities staffed by top people, he says. “But I would say that our researchers work very hard, and our decision-making at the top is very quick.”
Unlike biotech start-ups that tend to be built around groundbreaking technology, promising drug leads, or star researchers, Hengrui at first approached innovative drug development with a conservative strategy designed to reduce the risk of failure.
Relying on developmental compounds licensed from other organizations, the company initially aimed to develop drugs with the same mechanisms of action as others already on the market. Imrecoxib, for example, is part of the well-known family of COX-2 inhibitor anti-inflammatory drugs.
Later, it sought to invent compounds offering slight improvements over existing ones. Today, Hengrui is aiming to launch pharmaceuticals that are clearly superior to the competition. The company’s ultimate goal, Tao says, is to develop groundbreaking pharmaceuticals.
“We went from me-too to me-better to now best in class, and then we will do first in class,” he says.
And as Hengrui’s research strategy has become more ambitious, its scientists have broadened the range of diseases and drugs that they work on. Six or seven years ago, Tao says, Hengrui limited itself to the development of small-molecule drugs that treat cancer. Today the company is looking at small molecules, peptides, antibodies, antibody-drug conjugates, and other drug types to treat diseases as diverse as psoriasis and diabetes. “We have expanded our focus,” Tao says.
Most research is conducted in-house, Tao says. This includes medicinal chemistry, process chemistry, biology, drug metabolism, and pharmacokinetics. But the company leans on contract research firms for certain specific tasks, such as developing animal models. “They help accelerate our R&D,” Tao says.
Although Hengrui is a pioneer in launching new drugs in China, several other Chinese firms have made progress in advancing their own drug development. For instance, in the southern city of Dongguan, the generic drug producer HEC Pharma is conducting Phase II trials of the hepatitis C drug yimitasvir.
In Beijing, the biotech firm BeiGene just sold the U.S. company Celgene rights in much of the world to one of its immuno-oncology compounds for $263 million. Celgene also agreed to inject $150 million into BeiGene.
Chinese companies increasingly have the resources required to sustain innovative drug discovery and development, China watchers say. “Hengrui has the financial resources and the commitment to become a world-class innovative drugmaker,” says George Baeder, a former pharmaceutical industry executive who is now a director of China Global Insight, a California-based think tank.
But developing drugs and selling them are two different things, Baeder warns. “It is easy to underestimate the complexity a firm faces when moving into the arena of innovative medicines,” he says. “Chinese companies typically lack the capabilities in medical affairs, marketing, and sales needed to build a successful franchise.”
For the time being, Hengrui’s innovative drug subsidiary will stay focused on developing new drugs and not worry about the fine points of marketing them. Tao expects the former will keep his firm busy. “Don’t be surprised if several of our drugs begin clinical trials in the U.S., Europe, and Australia in the next year or two,” he says.
Rich pipeline
Hengrui boasts a diverse portfolio of drugs in late-stage development.
| China approval stage | Name | Application | Mechanism or target |
|---|---|---|---|
| Phase II clinical trials | Hetrombopaga | Idiopathic thrombocytopenia | Thrombopoietin receptor agonist |
| HR7056 | Anesthesia | na | |
| Pyrotiniba | Non-small cell lung cancer | EGFR/HER2 | |
| SHR3680b | Prostate cancer | Androgen receptor | |
| Phase III | Apatinib | Liver and non-small cell lung cancer | VEGFR-2 |
| Camrelizumabc | Cancer | PD-1 blocker | |
| Pyrotinib | HER2-positive breast cancer | EGFR/HER2 | |
| Retagliptin | Type 2 diabetes | Dipeptidyl peptidase 4 | |
| SHR3824 | Type 2 diabetes | SGLT2 inhibitor | |
| New drug application (China) | Mecapegfilgrastim | Neutropenia | PEG G-CSF |
| Launched | Apatinib | Gastric cancer | VEGFR-2 |
| Imrecoxib | Osteoarthritis | COX-2 inhibitor |
a In Phase I in U.S. b In Phase I in Australia. c The U.S. firm Incyte has acquired the rights to this drug outside China. na = not available. Source: Hengrui
////////////////Imrecoxib, Hengyang, CHINA 2012, osteoarthritis
Novel Autotaxin Inhibitors for the Treatment of Osteoarthritis Pain from Lilly Research Laboratories
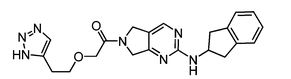
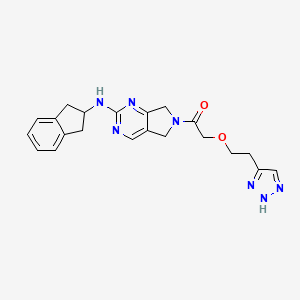
2-(2-(1H-1,2,3-triazol-5-yl)ethoxy)-1-(2-((2,3-dihydro-1H-inden-2-yl)amino)-5,7-dihydro-6Hpyrrolo[3,4-d]pyrimidin-6-yl)ethan-1-one
l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
CAS 1619971-30-0
| 1-[2-(2,3-dihydro-1H-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidin-6-yl]-2-[2-(1H-1,2,3-triazol-4-yl)ethoxy]ethanone; | |
| Molecular Formula: | C21H23N7O2 |
|---|---|
| Molecular Weight: | 405.45302 g/mol |
Scheme A


Scheme B

Scheme C

VI


Scheme E

Autotaxin is an enzyme reported to be the source of lysophosphatidic acid (LPA) which up-regulates pain-related proteins through one if its cognate receptors, LPAi. LPA is an intracellular lipid mediator which influences a multiplicity of biological and biochemical processes. Targeted inhibition of autotaxin-mediated LPA biosynthesis may provide a novel mechanism to prevent nerve injury-induced neuropathic pain.
Compounds that inhibit autotaxin are desired to offer a potential treatment option for patients in need of treatment for pain.
Pain associated with osteoarthritis (OA) is reported to be the primary symptom leading to lower extremity disability in OA patients. Over 20 million Americans have been diagnosed with OA, the most common of the arthropathies. The currently approved treatments for OA pain may be invasive, lose efficacy with long term use, and may not be appropriate for treating all patients. Additional treatment options for patients suffering from pain associated with OA are desired. Compounds that inhibit autotaxin represent another possible treatment option for patients with pain associated with OA.
U.S. Patent 7,524,852 (‘852) discloses substituted bicyclic pyrimidine derivatives as anti-inflammatory agents.
PCT/US2011/048477 discloses indole compounds as autotoxin inhibitors.
There is a need for novel compounds that provide autotaxin inhibition. The present invention provides novel compounds which are autotaxin inhibitors. The present invention provides certain novel compounds that inhibit the production of LPA.
Autotaxin inhibitor compounds are desired to provide treatments for autotaxin mediated conditions, such as pain and pain associated with OA.
PAPER

In an effort to develop a novel therapeutic agent aimed at addressing the unmet need of patients with osteoarthritis pain, we set out to develop an inhibitor for autotaxin with excellent potency and physical properties to allow for the clinical investigation of autotaxin-induced nociceptive and neuropathic pain. An initial hit identification campaign led to an aminopyrimidine series with an autotaxin IC50 of 500 nM. X-ray crystallography enabled the optimization to a lead compound that demonstrated favorable potency (IC50 = 2 nM), PK properties, and a robust PK/PD relationship.


Novel Autotaxin Inhibitors for the Treatment of Osteoarthritis Pain: Lead Optimization via Structure-Based Drug Design
Lilly Research Laboratories, A Division of Eli Lilly and Company, Indianapolis, Indiana 46285, United States
ACS Med. Chem. Lett., 2016, 7 (9), pp 857–861
DOI: 10.1021/acsmedchemlett.6b00207
*E-mail: jonessp@lilly.com. Tel: +1-317-277-5543.
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00207

Spencer Jones
Senior Research Scientist at Eli Lilly and Company
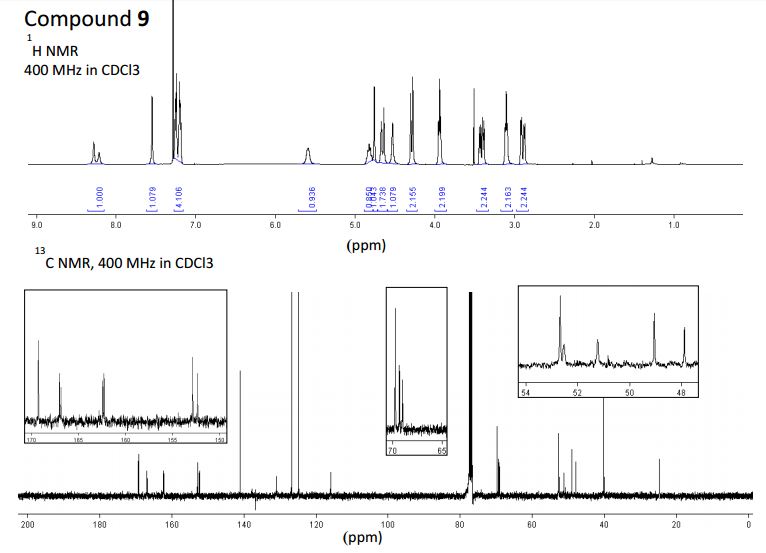
2-(2-(1H-1,2,3-triazol-5-yl)ethoxy)-1-(2-((2,3-dihydro-1H-inden-2-yl)amino)-5,7-dihydro-6Hpyrrolo[3,4-d]pyrimidin-6-yl)ethan-1-one (9)
………… Purified the resulting residue by silica gel chromatography (gradient elution: 0-9% methanol in ethyl acetate ) to give the title compound……..
1H NMR (400 MHz, CDCl3): 60:40 mixure of rotamers * indicates minor rotamer δ 8.18 (bs, 0.6H), *8.13 (bs, 0.4H), 7.49 (s, 1H), 7.21-7.09 (m, 4 H), 5.70-5.50 (m, 1H), 4.87-4.78 (m, 1H), 4.75 (s, 1.2H), *4.67 (s, 0.8H), 4.64 (s, 1.2H) *4.53 (s, 0.8H), *4.30 (s, 0.8H), 4.28 (s, 1.2H), 3.93 (t, J = 5.6 Hz, 2H), 3.43 (dd, J = 16.2, 7.1 Hz, 2H), 3.10 (t, J = 5.6 Hz, 2H), 2.89 (dd, J = 16.2, 4.9 Hz, 2H).
13C NMR (400 MHz, CDCl3): * indicates minor δ *169.3, 16 169.2, 167.0, *166.8, *162.4, 162.2, 152.8, *152.3, 141.1, 137.8, 130.9, 126.7, 124.9, 115.9, 69.8, 69.3, *69.0, 52.7, *52.5, 51.2, 49.0, *47.9, 40.1, 24.7.
LC/MS (ESI+ ): (m/z) 406 (C21H24N7O2 = (M+1)+ ).
PATENT
Example 2
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.

Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
PATENT
US-20140200231-A1
https://www.google.com/patents/US20140200231
Scheme E

Preparation 7
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid.

Pressurize 1 atmosphere of hydrogen (g) to a flask containing [2-(l-benzyl-lH- l,2,3-triazol-5-yl)ethoxy]acetic acid (10.1 g; 1.00 equiv; 38.66 mmoles) and palladium (II) chloride (3 g; 16.92 mmoles; 3.00 g) in isopropyl alcohol (300 mL) and water (60 mL). Maintain the flask under a hydrogen atmosphere for 3 h, then filter through Celite™ and concentrate. Add toluene (2×50 mL) and concentrate to afford the title compound (7.96 g, 100%). ]H NMR (d6-DMSO): 2.86 (t, / = 7 Hz, 2 H), 3.65 (t, / = 7 Hz, 2 H), 3.98 (s, 2 H), 7,77 (s, 1 H), 13.4 – 13.6 (br s, 2 H).
Example 1
Synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin- 6(5H)-yl]-2-[2-(lH-l,2,3-triazol-4- l)ethoxy]ethanone.

Add N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (4.2 g, 15.8 mmol) to a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid (2.7 g, 15.8 mmol), 1-hydroxybenzotriazole (3.20 g, 23.7 mmol), and dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (5.44 g, 28.4 mmol) in dichloromethane (40 mL) at 25 °C. Add triethylamine (4.40 mL, 31.6 mmol) to the reaction mixture and stir for 16 h. Wash with water (2 x 50 mL) and concentrate the organic layer. Purify by silica gel column chromatography, eluting with ethyl acetate/methanol, to give the title compound (4.0 g, 60%) as a solid. MS (m/z): 420 (M + Η). Preparation 8
Synthesis of 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]ethanone.

To N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (11.0 g, 41.3 mmol) and triethylamine (7.48 mL, 53.7 mmol) in dichloromethane (200 mL), add 2- chloroacetyl chloride (3.61 mL, 5.13 g, 45.4 mmol) dropwise over five minutes at 23 °C. Stir for 30 minutes and pour the reaction mixture into 1 : 1 50% saturated aqueous sodium bicarbonate: dichloromethane (75 mL). Separate the organic layer from the aqueous layer and further extract the aqueous layer with dichloromethane (2 x 25 mL). Combine the organic extracts and dry over anhydrous sodium sulfate, filter, and concentrate. Dissolve the residue in chloroform (10 mL) and purify via silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (9.75 g, 69%). ]H NMR (CDC13, * = minor amide rotamer) δ 2.77* (t, 2H), 2.84 (dd, 2H), 2.87 (t, 2H), 3.35 (dd, 2H), 3.76 (t, 2H), 3.85* (t, 2H), 4.12 (s, 2H), 4.52* (s, 2H), 4.57 (s, 2H), 4.72-4.82 (m, IH), 5.48-5.64 (m, IH), 7.12-7.21 (m, 4H), 8.03-8.10 (m, IH).
Preparation 9
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8- dihydropyrido[4,3-d]p rimidin-6(5H)-yl]ethanone.

To sodium hydride (60 wt% in mineral oil, 1.58 g, 39.6 mmol) in tetrahydrofuran (50 mL) at 23 °C, add 3-butyn-l-ol (7.93 g, 8.59 mL, 113.2 mmol) dropwise, then stir at 23 °C for 20 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (9.70 g, 28.3 mmol) in tetrahydrofuran (150 mL) at 23 °C and stir for one hour. Pour the reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether (x 2) and ethyl acetate (x 2). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Purify the resulting crude product by silica gel column chromatography (gradient elution: 20% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (8.16 g, 77%). MS (m/z): 377 (M + 1).
Example la
Alternative synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]-2-[2-(lH- l,2,3-triazol-4- l)ethoxy]ethanone.

Sparge a solution of 2-(but-3-yn- l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (8.15 g, 21.7 mmol) and L-ascorbic acid sodium salt (8.58 g, 43.3 mmol) in dimethylformamide (60 mL) and water (60 mL) with nitrogen for ten minutes, then evacuate and backfill with nitrogen three times. Add copper (II) sulfate pentahydrate (1.08 g, 4.33 mmol) and heat to 90 °C, then add azidotrimethylsilane (23.1 mL, 20.0 g, 173 mmol) dropwise and stir for one hour. Cool reaction mixture to 23 °C and pour into water (50 mL). Extract this mixture with ethyl acetate (4 x 50 mL). Combine the organic extracts and wash with saturated aqueous sodium chloride, dry over anhydrous sodium sulfate, filter, and concentrate.
Purify the resulting crude product by silica gel column chromatography (gradient elution: 0 to 10% methanol in ethyl acetate) to give the title compound (3.60 g, 40%). MS (m/z): 420 (M + 1). Preparation 10
Synthesis of tert-butyl-2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidine-6-carboxylate.

Charge 450 rriL (2.58 mol) of N-ethyl-N-isopropylpropan-2-amine into a 15 °C solution of tert-butyl 2-chloro-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidine-6-carboxylate (220 g, 860.37 mmol) and 2,3-dihydro-lH-inden-2-amine (137.7 g, 1.03 mol) in 1- methylpyrrolidin-2-one (3.6 L). Heat the resulting mixture to 80 °C for 16 h, then cool to 30 °C and transfer the resulting mixture into 5 L of water at 25 °C. Filter the resulting solid and rinse the filter cake with water (2 x 300 rriL). Reslurry the solid in ethyl acetate (350 iriL) for 45 min at 15 °C. Filter the slurry, rinsing with 15 °C ethyl acetate ( 2 x 250 rriL), and dry to give the title compound (226 g, 75%) as an off-white solid. ‘H NMR (d6-DMSO) 1.45 (s, 9 H), 2.87 (dd, /= 7.2, 15.8 Hz, 2 H), 3.24 (dd, /= 7.2, 15.8 Hz, 2 H), 4.36 (d, 10.4 Hz, 2 H), 4.44 (d, /= 12.8 Hz, 2 H), 4.60 (m, 1 H), 7.14 (m, 2 H), 7.20 (m, 2 H), 7.55 (d, /= 6.8 Hz, 1 H), 8.27 (d, /= 7.2 Hz, 1 H).
Preparation 11
Synthesis of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidin-2- amine dihydrochloride hydrate.

Charge 670 rriL of 5 M hydrochloric acid (3.35 mol) to a solution of tert-butyl 2-
(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H pyrrolo[3,4-d]pyrimidine-6- carboxylate (226 g, 641.25 mmol) in tetrahydrofuran (2.0 L) at 17 °C, maintaining the internal temperature below 26 °C during the addition. Heat the resulting solution to 50 °C for 16 h, cool to 25 °C and dilute with 500 rriL of water and 500 mL of tert- butylmethylether. Separate the resulting layers and extract with tert-butylmethylether (3 x 1 L). Concentrate the water phase down to a reaction volume of ca. 200 mL, and filter the resulting slurry. Rinse the cake with tert-butylmethylether (2 x 200 mL) and dry to give the title product (177 g, 80%) as a light brown solid. MS (m/z): 253.2 (M-2HC1- H20+1).
Preparation 12
Syntheis of tert-butyl 2-but-3-ynox acetate.

Stir a mixture of but-3-yn-l-ol (6.00 g; 85.60 mmol), tetrabutylammonium sulfate (2.07 g; 8.54 mmol) and sodium hydroxide (40% wt/wt; 150 mL) in dichloromethane (150 mL) at 0°C. Add tert-butyl bromoacetate (19.34 mL; 128.40 mmol) dropwise and stir the mixture for 2.5 hours at room temperature. Dilute the reaction mixture with dichloromethane (200 mL) and water (100 mL), separate the layers, and further extract the aqueous layer with dichloromethane (2 x 100 mL). Wash the combined organic layers with brine (100 mL), dry over anhydrous sodium sulfate, and concentrate to afford the crude title compound as a brown oil (11.93 g). Purify the oil by silica gel column chromatography, eluting with hexane: ethyl acetate (0% to 10% mixtures) to give the title compound (11.35 g; 72%) as a colorless oil. ]H NMR (CDCI3) δ 1.48 (s, 9H), 2.00 (m, 1H), 2.52 (m, 2H), 3.67 (m, 2H), 4.01 (bs, 2H).
Preparation 13
Synthesis of tert-butyl 2-[2-(lH-triazol-5- l)ethoxy]acetate.

Stir tert-Butyl 2-but-3-ynoxyacetate (11.34 g; 61.55 mmol) and copper(I)iodide (584 mg; 3.07 mmol) in a mixture of dimethylformamide (56.70 mL) and methanol (11.34 mL) at 0°C. Add azido(trimethyl)silane (12.33 mL; 86.47 mmol) dropwise and heat the mixture at 90°C for 18 hours.
In a second batch, stir tert-butyl 2-but-3-ynoxyacetate (4.38 g; 23.77 mmol) and copper(I)iodide (226 mg; 1.19 mmol) in a mixture of dimethylformamide (22 mL) and methanol (6 mL) at 0°C. Add azido(trimethyl)silane (4.8 mL; 33.66 mmol) dropwise and the mixture heated at 90°C for 18 hours.
Upon cooling to room temperature, combine the crude products from both batches and concentrate the mixture to afford a greenish residue. Purify the crude product by filtration through a plug of silica eluting with dichloromethane: ethyl acetate (75% to 100% mixtures) to afford the title compound (14.15 g, 73%) as a colorless oil. MS (m/z): 228.15 (M+l).
Preparation 14
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid.

Stir a mixture of ieri-butyl 2-[2-(lH-triazol-5-yl)ethoxy]acetate (14.15 g; 62.26 mmol) and trifluoroacetic acid (70.75 mL, 935.69 mmol) in dichloromethane (70.75 mL) for 2 hours at room temperature. Concentrate the reaction mixture under reduced pressure to provide the title compound containing additional trifluoroacetic acid (20.22 g, >100%) as a brown solid. MS (m/z): 172.05 (M+l).
Example 2
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
Preparation 15
Synthesis of 2-chloro- l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H- pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.

Stir a suspension of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (14.4 g, 41.9 mmol) and triethylamine (14.3 g, 19.7 mL, 141.4 mmol) in dichloromethane (200 mL) at 23 °C for 10 minutes, then cool to -30 °C. Add 2-chloroacetyl chloride (5.49 g, 3.86 mL, 48.6 mmol) over two minutes and warm to 23 °C over 10 minutes. Add methanol (5 mL) and remove the solvent in vacuo. Slurry the crude reaction mixture in methanol (30 mL), add 50 g silica gel and remove solvent in vacuo. Load the resulting residue onto a loading column and purify via silica gel column chromatography (gradient elution: 50% ethyl acetate in hexanes to ethyl acetate to 10% methanol in ethyl acetate) to give the title compound (11.5 g, 84%). MS (m/z): 329(M+1).
Preparation 16
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- 6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.

To sodium hydride (60 wt% in mineral oil, 2.06 g, 51.4 mmol) in tetrahydrofuran (86 mL) at 0 °C, add 3-butyn-l-ol (4.64 g, 5.03 mL, 64.3 mmol), then stir at 23 °C for 15 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7- dihydro-6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone (8.45 g, 25.7 mmol) in
tetrahydrofuran (86 mL) at 0 °C and stir for five minutes. Pour reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether and ethyl acetate (2 x 50 mL each). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Combine the crude product with the crude product from a second reaction (run reaction under identical conditions and stoichiometry employing 2-chloro- 1- [2-(indan-2-ylamino)-5,7-dihydropyrrolo[3,4-d]pyrimidin-6-yl]ethanone (3.0 g, 9.1 mmol)) and purify by silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (2.90 g, 23%). MS
(m/z): 363(M+1). Example 2a
Alternative synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- pyrrolo[3,4-d]pyrimidin-6-yl]-2-[2-(lH-l,2,3-triazol-4-yl)ethoxy]ethanone.

Add dimethylformamide (27 mL) and water (27 mL) to a flask containing 2-(but- 3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]ethanone (2.90 g, 8.00 mmol). Add copper (II) sulfate pentahydrate (400 mg, 1.60 mmol) and L-ascorbic acid sodium salt (3.17 g, 16.0 mmol). Evacuate flask and backfill with nitrogen (x 2), then add azidotrimethylsilane (7.37 g, 8.53 mL, 64.0 mmol) and heat the reaction to 90 °C for 70 minutes. Cool the reaction mixture to 23 °C and remove all solvent in vacuo. Suspend the residue in methanol/dichloromethane and then add silica gel and remove solvent in vacuo. Load this material onto a loading column and purify via silica gel column chromatography (gradient elution: 0-9% methanol in ethyl acetate) to give the title compound (980 mg, 30%). MS (m/z):
406(M+1).
/////////Autotaxin, LPA, osteoarthritis, tool molecule, lily, Spencer Jones, PRECLINICAL
N1(Cc2cnc(nc2C1)NC3Cc4ccccc4C3)C(=O)COCCc5cnnn5
S-flurbiprofen (TT-063)
Cas 51543-39-6,
MW 244.26,
MF C15 H13 F O2
[1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (αS)-
- [1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (S)-
- (+)-(S)-Flurbiprofen
- (+)-Flurbiprofen
- (2S)-2-(2-Fluoro-1,1′-biphenyl-4-yl)propanoic acid
- (2S)-2-(2-Fluoro-4-biphenyl)propanoic acid
- (S)-Flurbiprofen
- Dexflurbiprofen
- Esflurbiprofen
- S-(+)-Flurbiprofen
- d-Flurbiprofen
On October 20, 2014, Taisho filed for manufacturing and marketing approval for TT-063 from the Ministry of Health, Labour and Welfare as a new drug candidate that will follow the Type 2 diabetes treatment Lusefi®, which was launched in May 2014. TT-063 is a patch formulation that has been co-developed by Taisho and TOKUHON Corporation with the aim of obtaining an indication for osteoarthritis. In Phase 3 clinical trials comparing TT-063 with therapeutic drugs already on the market, TT-063 has been found to be more effective than the control drugs in patients with osteoarthritis of the knee joint (January 16, 2014 announcement ).
Furthermore, Taisho is also preparing to file for approval from the Ministry of Health, Labour and Welfare for CT-064, an oral formulation of the osteoporosis treatment agent Bonviva launched in August 2013. Taisho has confirmed the effectiveness of CT-064 for osteoporosis patients through Phase 3 clinical trials (September 22, 2014 announcement).

In the central nervous system field, TS-091 transitioned from Phase 1 to Phase 2 in Japan in May 2014. Clinical trials of TS-091 have commenced to confirm the effectiveness of this drug in patients with central disorders of hypersomnolence. In addition, Phase 1 clinical trials of TS-091 have commenced overseas. TS-111 and TS-121 are undergoing Phase 1 clinical trials overseas with the aim of obtaining an indication for depression.
Faced with intensifying competition in new drug discovery, we will jointly implement R&D activities with research institutions outside the Taisho Group, and with companies in Japan and overseas, as we work to enhance our drug development pipeline (lineup of drugs in development). Our goal is to discover many more new drugs, primarily in our priority fields.
| Company | Taisho Pharmaceutical Holdings Co. Ltd. |
| Description | Topical anti-inflammatory analgesic patch containing S-flurbiprofen |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Standard Indication | Osteoarthritis |
| Indication Details | Treat osteoarthritis (OA) and scapulohumeral periarthritis |
| Regulatory Designation | |

Scheme 2.
Reagents and conditions: (a) THF, EDC, Et3N; (b) TFA; (c) 0.5 equiv 2,5-dimethoxybenzoquinone, EtOH, 50–80 °C for 3–5 h; (d) 1 equiv naphthoquinone, MeOH, rt, overnight.
http://www.sciencedirect.com/science/article/pii/S0960894X13011773
……………………………………………
2-(6-methoxynaphthalen-2-yl) propanoic acid By way of illustration, chemically, flurbiprofen is 2-(2-fluoro-4-biphenylyl) propionic acid and is described in US Patent No. 3,755,427. NSAIDs, such as flurbiprofen, are usually supplied as a racemate. However, recently there has been renewed interest in the separate enantiomers of flurbiprofen, i.e. S-flurbiprofen and R-flurbiprofen.

R-Flurbιprofen

S-Flurtιprofen
Flurbiprofen is a potent inhibitor of cyclooxygenase (both COX-I and COX-2) in humans and it is understood that the inhibitory effect lies predominantly in the S- enantiomer.
Flurbiprofen is generally produced in the form of a racemic compound. It is known that from the racemic compound, flurbiprofen having a high optical purity can be produced by an optical resolution method using, for example, an optically active amine compound, such as α-phenylethylamine, as an optical resolution agent, as is described in US Patent No. 5,599,969. In addition, whether dealing with racemic, S- or R- 2-aryl propionic acid, there is also a need to make the synthetic process as efficient as possible.
Example 2 – Ibuprofen
Example 2.1 Resolution procedure
Racemic ibuprofen (530g) is dissolved in toluene (1335ml) and methanol (900ml).
The mixture is heated to dissolve the solid. S-1-Phenylethylamine (247g) is dissolved in toluene (200ml) and the solution is added with stirring at 600C over about 3 hours while the temperature is maintained at about 65-700C. The mixture is cooled gradually to 0 to 50C to induce crystallisation and stirred at this temperature for 1 hour. The crystals are filtered off, washed with toluene (600ml) and dried in a Vacuum oven at 550C to form crude S-ibuprofen / S-1-phenylethylamine salt (635g).
Crude S-ibuprofen / S-1-phenylethylamine salt (635g) is stirred with toluene (1930ml) and methanol (800ml) and the mixture is heated to 6O0C to dissolve the solid. The solution is cooled gradually to 0 to 5°C to induce crystallisation. The crystals are filtered off and dried in a vacuum oven at 55°C to form pure S-ibuprofen / S-I- phenylethylamine salt (510g). This recrystallisation of the S-ibuprofen / S-I- phenylethylamine salt may be repeated if necessary to upgrade the enantiomeric purity if required.
Pure S-ibuprofen / S-1-phenylethylamine salt (485g) is mixed with toluene (1700ml) with stirring. Water (300ml) and concentrated hydrochloric acid (17Og) are added and
÷ibe mixture is stirred at 600C. The lower aqueous layer is separated off and the upper organic layer is retained. The hydrochloric acid wash is repeated, then the toluene solution is washed with water. Water (370ml) and 47% sodium hydroxide
(118g) are added and the solution is heated to 600C and allowed to settle. The lower aqueous layer is separated and the upper toluene layer is washed with water. The aqueous phases are combined and heptane (420ml) is added. Hydrochloric acid
(130g) is added and the mixture is heated to 600C, stirred and settled. The organic layer is separated off and washed with water. The solution is cooled to -100C to induce crystallisation and the crystals are separated off by filtration, washed with heptane and dried under vacuum to yield (S)-ibuprofen (28Og) at an enantiomeric purity of over 99%.
Example 2.2 Racemisation procedure
Toluene/methanol mother liquors from the filtration of crude S-ibuprofen / S-I- phenylethylamine salt in the resolution procedure (2400ml, containing an estimated 130g of ibuprofen) is charged into a 3 L 3 necked round bottomed flask and methanol and toluene are distilled out at atmospheric pressure (volume removed approximately 1400 ml). The batch is then cooled to around 60°C and washed twice with hydrochloric acid (20 ml concentrated hydrochloric acid in 200 ml of water), and then twice with water (200 ml). Toluene is charged (80 ml) followed by methanol (200 ml) and caustic soda solution (45Og of 28% w/w solution, 5 molar equivalents). The mixture is heated to reflux for about 6 hours. Solvent is then removed at atmospheric pressure until the vapour temperature reaches approximately 85°C. The mixture is cooled to around 60°C and concentrated hydrochloric acid is charged at about 60 to 70°C until the pH of the mixture is 1 or less. The layers are allowed to separate and the bottom aqueous layer removed. The organic layer is washed with water (200 ml) and then azeotroped to dryness using a Dean and Stark trap. A solution of racemic ibuprofen in toluene remains.
…………………………………………
PATENT
http://www.google.com/patents/CN104478703A?cl=en
Preparation of R – (+) _ flurbiprofen:
The racemic flurbiprofen as a starting material, to obtain an intermediate product of formula I as shown and then the ester prepared as shown in Formula II with 5-isosorbide monobenzyl ether, ester hydrolysis after obtained R – (+) – flurbiprofen;

wherein, in formula I, X is Cl or Br;
(2) by the R – (+) _ flurbiprofen obtained (RS) – flurbiprofen:
The R _ (+) _ flurbiprofen 200mg, potassium hydroxide 150mg, 0. 5mL water into IOmL reaction flask and heated to 120 ° C and held for 2h, then water was added 15mL, cooled to room temperature, the resulting stirring the mixed solution with 10% hydrochloric acid to pH = 0. 5, extracted with ethyl acetate, combined several layers, washed with water until neutral, the organic solvent is recovered, the resulting residue was added at 60~90 ° C under an appropriate amount of petroleum ether by recrystallization, obtained (RS) – flurbiprofen 100mg, 50% yield.
(3) Preparation of (S) -⑴- flurbiprofen:
In 25mL single-necked flask, followed by adding (RS) – flurbiprofen 123mg, Portugal TOA 29. 8mg, isopropanol lmL, the mixture was stirred at reflux until clear, half the amount of the solvent evaporated under reduced pressure except , set the refrigerator overnight. The precipitate was collected by suction filtration as white crystals, after washing a small amount of isopropanol, which was dissolved in water, washed with 10% aqueous sodium hydroxide (10% NaOH mean mass fraction) adjusted pH = 13, the sheet-like precipitate was filtered off Portuguese octylamine white crystals. The resulting filtrate was added dropwise with stirring 10% hydrochloric acid to pH = 1, extracted with ethyl acetate, the organic layer was washed with water to recover the solvent, the resulting residue was purified by an appropriate amount of petroleum ether and recrystallized at 60~90 ° C. The product was collected by filtration, and dried in vacuo to give a white (S) – (+) _ flurbiprofen needle crystal 45. 3mg, 65% yield, mp 102~103 ° C, [α] = + 44 ° (C = 1, methanol), ee value of 92.6% (ee value measurement method: (S) – (+) – flurbiprofen after chiral amine derivatization reagents, by HPLC analysis).
wherein in step (3) is a byproduct eleven R _ (+) _ flurbiprofen, its follow step (1) of racemic reused.
Step (1) of the specific operation is as follows:
(la) 1:. Synthesis of 2,6-sorbitol dehydration -D- -5- benzyl ether: 4: 3
250ml volumetric flask isosorbide 18. 25g (125mmol), lithium hydroxide monohydrate 5. 25g (125mmol) and 60ml of dimethyl sulfoxide (DMSO), heated to 90 ° C, stirred for 30min, constant pressure equalizing dropping funnel was added dropwise benzyl chloride 14. 4ml (125mmol), 90 ° C the reaction 19-20h, reaction mixture was adjusted to pH 1 with 2M hydrochloric acid, extracted with ethyl acetate (50ml * 3), the organic layers combined, washed with water ( 30ml * 2), dried over anhydrous sodium sulfate overnight, filtered and concentrated residue Cheng baby gel column chromatography (petroleum ether: ethyl acetate = 5: 1) to give a cream solid, that is 1: 4: 3: 2,6 Dehydration -D- sorbitol -5- benzyl ether 24. 5g, m.p. 59 ~61 ° C.
(Ib) · 2- (2- fluoro-4-biphenylyl) propionyl chloride Synthesis
50ml vial before racemic flurbiprofen was added 2. 44g (IOmmol), anhydrous toluene 20ml, freshly distilled thionyl chloride was added dropwise 0. 8ml (Ilmmol), N, N- dimethylformamide amide (DMF) 2 dropwise, stirred at room temperature 2h, the solvent was distilled off under reduced pressure to give a pale yellow gum, i.e., 2- (2-fluoro-4-biphenylyl) propionyl chloride, it was used directly in the reaction without isolation.
(lc). R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester synthesis
The (Ib) resulting acid chloride was dissolved in 20ml of dry toluene was added dropwise at room temperature, dimethyl amine 3. 5ml, solid precipitation, stirred for about Ih, ice salt bath, a bath temperature of minus 10-15Ό, stirred at this temperature IOmin so, and then the constant pressure dropping funnel (Ia) 5 isosorbide monobenzyl ether (2. 83g, 12mmol) in toluene, keeping the reaction temperature, stirring 8h. The ice bath was removed and the reaction mixture under reduced pressure to remove the solvent, the residue was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous sodium sulfate overnight, ethyl acetate was removed under reduced pressure, the residue was a white gel, recrystallized from petroleum ether to give a white solid that R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester 3. 65g (7. 88mmol), in order to put the racemic flurbiprofen yield based on 78.8%.
(ld) R – Synthesis of flurbiprofen – (+)
Under ice bath (Ic) obtained R-2- (2- fluoro-4-biphenylyl) propionic acid monobenzyl ether isosorbide 5- ester 2. 3Ig (5mmol) was dissolved in 20ml of acetone / water (1/1) was added Iml hydrochloric acid to adjust pH to 3, stirred for 3-4h, the reaction solution was extracted with ethyl acetate (20ml * 2), sash organic layer was washed with ice (10ml * 2), dried over anhydrous sodium sulfate overnight , filtration, and the filtrate was concentrated, the residue was recrystallized from ether to give white crystals, i.e. L-flurbiprofen 1.02g (4 18mmol.), yield 83.5%, optical purity 93% (HPLC method); input-racemic flurbiprofen dollars, the total yield of 78.8% * 83.5% = 65.8%.
Step (1) reaction of the formula:

FLURBIPROFEN RACEMIC
3-Fluoro-4-phenyl-α-methylphenylacetic acid 1
M.p. 110-113°C (lit.3d 111-113.5°C).
1 H NMR (CDCl3, δ ppm) 7.51-7.55 (m, 2H), 7.49-7.37 (m, 4H), 7.21-7.16 (m, 2H), 3.85-3.78 (q, 1H, J = 7.1 Hz, CH), 1.60-1.57 (d, 3H, J = 7.1 Hz, CH3);
13C NMR (CDCl3 δ ppm) 180.4 (COOH), 161.3 & 158.0 (3-Ar-C), 140.9 & 140.8, 135.4, 130.9 & 130.8 (5-Ar-C), 128.9, 128.4, 128.2 & 128.0 (4-Ar-C), 127.7 (4′-Ar-C), 123.7 & 123.7 (6-Ar-C), 115.5 & 115.2 (2-Ar-C), 44.8 (CH), 18.0 (CH3).
(d) Sagami Chemical Research Center. Jpn. Kokai Tokkyo Koho JP 8216840, 1982 (Chem. Abstr. 1982, 97: 5996s).

 |
| RACEMIC |
|
Flurbiprofen
CAS : 5104-49-4
: 2-Fluoro-a-methyl[1,1¢-biphenyl]-4-acetic acid
Additional Names: 2-(2-fluoro-4-biphenylyl)propionic acid; 3-fluoro-4-phenylhydratropic acid
Manufacturers’ Codes: BTS-18322; U-27182
Trademarks: Adfeed (Lead Chem.); Ansaid (Pfizer); Antadys (Thamex); Cebutid (Boots); Froben (Boots); Flurofen (Boots); Ocufen (Allergan); Stayban (Boots); Zepolas (Mikasa)
Molecular Formula: C15H13FO2
Molecular Weight: 244.26
Percent Composition: C 73.76%, H 5.36%, F 7.78%, O 13.10%
Literature References: Prepn: FR M5737; Adams et al., US 3755427 (1968, 1973 both to Boots Co., Ltd.). Pharmacology: Chalmers et al., Ann. Rheum. Dis. 31, 319 (1972); ibid. 32, 58 (1973); Glenn et al., Agents Actions 3, 210 (1973); Nishizawa et al.,Thromb. Res. 3, 577 (1973). HPLC determn in urine and plasma: J. M. Hutzler et al., J. Chromatogr. B 749, 119 (2000). Symposium on pharmacokinetics and clinical efficacy in pain management: Am. J. Med. 80, Suppl. 3A, 1-157 (1986).
Properties: Crystals from petr ether, mp 110-111°. Slightly sol in water (pH 7.0); readily sol in most polar solvents.
Melting point: mp 110-111°
Therap-Cat: Anti-inflammatory; analgesic.
|
racemic


s form


| Patent | Submitted | Granted |
|---|---|---|
| Methods to accelerate the isolation of novel cell strains from pluripotent stem cells and cells obtained thereby [US2008070303] | 2006-11-21 | 2008-03-20 |
| Herpes Virus-Based Compositions and Methods of Use in the Prenatal and Perinatal Periods [US2008226601] | 2006-06-05 | 2008-09-18 |
| METHOD OF REDUCING ABETA42 AND TREATING DISEASES [US2008021085] | 2007-06-21 | 2008-01-24 |
| METHODS TO ACCELERATE THE ISOLATION OF NOVEL CELL STRAINS FROM PLURIPOTENT STEM CELLS AND CELLS OBTAINED THEREBY [US2010184033] | 2009-07-16 | 2010-07-22 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US2011112064] | 2011-05-12 | |
| PROCESS FOR THE MANUFACTURE OF RACEMIC 2-ARYL-PROPIONIC ACID [US2011172460] |
| Patent | Submitted | Granted |
|---|---|---|
| Nitroxyderivatives having antinflammatory, analgesic and antithrombotic activity [US6613784] | 2003-09-02 | |
| Global method for mapping property spaces [US6675136] | 2004-01-06 | |
| Method of reducing Abeta42 and treating diseases [US2006004086] | 2006-01-05 | |
| 11-Beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia [US7179802] | 2004-06-03 | 2007-02-20 |
| 11-BETA-HYDROXYSTEROID DEHYDROGENASE 1 INHIBITORS USEFUL FOR THE TREATMENT OF DIABETES, OBESITY AND DYSLIPIDEMIA [US6730690] | 2004-03-11 | 2004-05-04 |
| Process for producing optically active flurbiprofen [US7214820] | 2006-06-22 | 2007-05-08 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US7875636] | 2009-11-05 | 2011-01-25 |
| METHOD FOR PRODUCING OPTICALLY ACTIVE ESTER AND METHOD FOR PRODUCING OPTICALLY ACTIVE CARBOXYLIC ACID [US8115008] | 2010-09-16 | 2012-02-14 |
| DRUG SUBSTANCE PREPARATIONS, PHARMACEUTICAL COMPOSITIONS AND DOSAGE FORMS [US2010087538] | 2010-04-08 | |
| (R)-2-(3-Benzoylphenyl)propionic acid salts and pharmaceutical preparations containing them [EP0935961] | 1999-08-18 | 2008-04-02 |
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Taisho Pharmaceutical Co., Ltd. (大正製薬株式会社 Taishō Seiyaku Kabushiki-gaisha?) (TYO: 4535) is a Japanese pharmaceutical company based in Tokyo.

.
////////////
| Tirupati తిరుపతి |
|
|---|---|
| City | |

Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
|
|
|
Tirupati
Location in Andhra Pradesh, India |
|
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| Country | India |
| State | Andhra Pradesh |
| Region | Rayalaseema |
| District | Chittoor |
| Government | |
| • Member of Parliament | Varaprasad Rao Velagapalli |
| Area | |
| • City | 24 km2 (9 sq mi) |
| Elevation | 161 m (528 ft) |
| Population (2011)[1] | |
| • City | 287,035 |
| • Density | 12,000/km2 (31,000/sq mi) |
| • Metro[2] | 459,985 |
| Languages | |
| • Official | Telugu |
| Time zone | IST (UTC+5:30) |
| PIN | 517501 |
| Telephone code | +91–877 |
| Vehicle registration | AP 03 |
| Website | Tirupati Mucnicipal Corporation |


 .
.
 .
.
Kapila Theertham in Tirupati







Food Service During Tirumala Tirupati Devastanam’s ‘Srinivasa Kalyanam Utsavam’ at MARG Swarnabhoomi



Rosa canina for osteoarthritis

Rosiflex contains a unique natural supplement that is good for joint health. If you are looking forward to a natural way to minimize your joint pain and stiffness, then Rosiflex is the ideal choice for you. Rosiflex is for anyone who wants healthy, flexible and mobile joints for a better quality of life. The unique natural ingredient in Rosiflex has been clinically proven to soothe the inflamed joints and improve joint comfort and flexibility.

What is Rosiflex?
Rosiflex is a Unique Dietary Supplement containing 100% Rosehip powder, made from a species of wild rose, Rosa canina. Rosiflex is available in capsule form with each capsule containing 750 mg (of imported) rosehip powder. Rosehip powder has been shown to decrease joint pain, improve joint health and increase mobility and flexibility in arthritic patients, particularly osteoarthritic patients.

The speciality of Rosiflex is as given below:
The speciality of Rosiflex is as given below:
- European supplement now brought to Indian arthritic patients
- Huge success internationally
- Effective within 3 weeks
- Good pain relief
- Reduces the need for regular pain killers
- Very Safe, being a herbal supplement
- Dosage: 2 capsules thrice daily for the initial 3 weeks followed by maintenance dose of 2 capsules twice daily
-
Rosa canina 
Photograph showing Rosa canina flowers. Scientific classification Kingdom: Plantae (unranked): Angiosperms (unranked): Eudicots (unranked): Rosids Order: Rosales Family: Rosaceae Genus: Rosa Species: R. canina Binomial name Rosa canina
L.Synonyms See text

History:
 ROSE HIPS
ROSE HIPS
Rose Hips (also called rose haws) are the pomaceous fruit of the rose plant. Roses are a group of herbaceous shrubs found in temperate regions throughout both hemispheres and grown in sunny areas or light shade and thrive in well-drained, slightly acid soil. Probably cultivated first in ancient Persia and carried to Greece and Rome, there are now hundreds of species of this beautiful flower cultivated throughout the world that occupy a vital place in medicine, as well as cosmetics, perfumes, soaps and foods. The leaves of Rosa canina were once even used as a substitute for tea. The botanical genus, Rosa, is derived from the Greek, roden, meaning “red” and the Latin, ruber, also meaning “ruby” or “red,” as apparently, the Roses of the ancient Mediterranean region were deep crimson, giving birth to the legend that the flowers sprang from the blood of Adonis.
Roses have a long tradition of medicinal use. The ancient Romans used Rosa canina (or Dog Rose) for the bites of rabid dogs, and in the first century A.D., the Roman, Pliny, recorded thirty-two different disorders that responded well to Rose preparations. An oriental species (Rosa laevigata) was mentioned in Chinese medical literature about A.D. 470, and in China, Rose Hips are still used for chronic diarrhea with stomach weakness.
It is typically red to orange but may be dark purple to black in some species. In Ayurvedic medicine, Roses have long been considered “cooling” to the body and a tonic for the mind, and Native Americans used Rose Hips to treat muscle cramps. In 1652, the esteemed British herbalist, Nicholas Culpeper, prescribed them for “consumptive persons,” for “tickling rheums,” to “break the stone” (kidneys) and to help digestion.
Long used for medicinal purposes in Great Britain, Rose Hips remained listed in the official British Pharmacopœia well into the 1930s, and were considered an overall cooling tonic, an astringent, a great help for sore throats and a source of the essential vitamin C. During World War II, there was a shortage of citrus fruit in England, and the British government organized the harvesting of all the Rose Hips in England as a substitute for vitamin C. This illuminated the importance of Rose Hips as a superior source of the vitamin and began its worldwide popularity. Rose Hips have a reported sixty times the amount of vitamin C than citrus fruit, and we now know how absolutely essential vitamin C is to the maintenance of good health and the prevention of many diseases.
Rose Hips contain one of the highest measures of vitamin C (about 1700-2000 mgs. per 100 g. in the dried product) than is known in other herbs. Rose Hips are the fruits of the Rose, the ripe seed receptacles that remain after the petals are removed, and they contain many vitamins and other beneficial supplements, including lycopene, essential fatty acids, beta-carotene, bioflavonoids, pectin, sugar, resin, wax, malates, citrates and other salts, tannin, malic and citrus acids, magnesium, calcium, iron, manganese, sulfur, phosphorus, potassium, selenium, zinc and vitamins A, B-1, B-2, B-3, B-5, C, D, E and K.
Beneficial Uses:
Probably the greatest known use of Rose Hips is as an extraordinary and powerful source of vitamin C, which is most beneficial for the prevention and treatment of infection and a great many common diseases, including the common cold, flu and pneumonia. It is said to prevent ailments before they happen by using a prophylactic dosage on a daily basis. Vitamin C is necessary for every cell in our bodies and without it, we would not be able to sustain life.
Natural vitamin C and bioflavonoids are combined in nature, and for efficacy, it is vital that they be used together. Rose Hips are rich in both, and together they help to strengthen body tissues and build and maintain a healthy vascular system and are said to heal and prevent damage to fragile capillaries. The combination is also thought to enhance the body’s ability to absorb vitamin C in those who have difficulty absorbing it.
Rose Hips, with its abundance of vitamin C, are useful in treating infections of all kinds and have been used for centuries for the relief of diarrhea and dysentery. It is considered to be a cleansing agent and may be helpful for temporary bladder problems, gallbladder dysfunction, kidney health, general debility and exhaustion.
Current research indicates that large doses of vitamin C in Rose Hips could be helpful in enhancing our immune systems, which may be valuable in warding off infectious invaders and serious malignant disease.
Rose Hips are said to have mild laxative and diuretic properties.
Rosa canina, commonly known as the dog-rose,[1] is a variable climbing wild rose species native to Europe, northwest Africa and western Asia.
It is a deciduous shrub normally ranging in height from 1–5 m, though sometimes it can scramble higher into the crowns of taller trees. Its stems are covered with small, sharp, hooked prickles, which aid it in climbing. The leaves are pinnate, with 5-7 leaflets. The flowers are usually pale pink, but can vary between a deep pink and white. They are 4–6 cm diameter with five petals, and mature into an oval 1.5–2 cm red-orange fruit, or hip.
 It’s that time of year again and the hedgerows are heaving with fruit. But with most people intent on collecting juicy blackberries, the vibrantly coloured and perhaps mystifying rose-hip is often overlooked. Maybe it’s because they are a suspicious red colour or maybe it’s because they’re a fruit that’s never seen in supermarkets. Whatever the reason, the conclusion is the same: there’s more to collect for yourself!
It’s that time of year again and the hedgerows are heaving with fruit. But with most people intent on collecting juicy blackberries, the vibrantly coloured and perhaps mystifying rose-hip is often overlooked. Maybe it’s because they are a suspicious red colour or maybe it’s because they’re a fruit that’s never seen in supermarkets. Whatever the reason, the conclusion is the same: there’s more to collect for yourself!
Rose-hips are the fruit of the rose bush and in the summer are found as a swollen green part of the stem just underneath the flower. Every rose left uncut will eventually produce a hip but some will appear in the summer and others later in the autumn depending on species. To my knowledge all rose hips are edible, though some varieties have better flavour than others.
Blessed with a delicate fruity taste and rich in vitamins A, B and C, Rose-hips can be used to make an assortment of products including jellies, syrups, teas, wine and even cosmetics. Both the fruit and the seeds are edible but you should not eat rose-hips whole due to irritating hairs which are found inside the berries. These hairs must be removed either by filtering during the cooking process.
The best variety for making edible products is the hip of the common wild rose, also known as the Dog Rose, Latin name Rosa Canina. It produces small, firm, deep-red hips that are rich in flavour and easy to find and harvest. They are available in the autumn but it’s said the best time to harvest them is directly after a frost. Being that birds favour other foods over these hard seed-laden hips, you can often find them hanging onto bare branches in the darkest days of winter. If you choose to use them to make edible products please know that it’s not necessary to separate the seeds from the red fruit as both have their own nutritious values. But of course beware the hairs mentioned previously and make sure they are excluded from your end product.

Synonyms
From DNA analysis using amplified fragment length polymorphisms of wild-rose samples from a transect across Europe (900 samples from section Caninae, and 200 from other sections), it has been suggested that the following named species are best considered as part of a single Rosa canina species complex, and are therefore synonyms of R. canina:[2]
- R. balsamica Besser
- R. caesia Sm.
- R. corymbifera Borkh.
- R. dumalis Bechst.
- R. montana Chaix
- R. stylosa Desv.
- R. subcanina (Christ) Vuk.
- R. subcollina (Christ) Vuk.
- R. × irregularis Déségl. & Guillon
Cultivation and uses

A botanical illustration showing the various stages of growth by Otto Wilhelm Thomé
The plant is high in certain antioxidants. The fruit is noted for its high vitamin C level and is used to make syrup, tea and marmalade. It has been grown or encouraged in the wild for the production of vitamin C, from its fruit (often as rose-hip syrup), especially during conditions of scarcity or during wartime. The species has also been introduced to other temperate latitudes. During World War II in the United States Rosa canina was planted in victory gardens, and can still be found growing throughout the United States, including roadsides, and in wet, sandy areas up and down coastlines. In Bulgaria, where it grows in abundance, the hips are used to make a sweet wine, as well as tea. In the traditional Austrian medicine Rosa canina fruits have been used internally as tea for treatment of viral infections and disorders of the kidneys and urinary tract.[3]
Forms of this plant are sometimes used as stocks for the grafting or budding of cultivated varieties. The wild plant is planted as a nurse or cover crop, or stabilising plant in land reclamation and specialised landscaping schemes.
Numerous cultivars have been named, though few are common in cultivation. The cultivar Rosa canina ‘Assisiensis’ is the only dog rose without prickles. The hips are used as a flavouring in Cockta, a soft drink made in Slovenia.
Canina meiosis

A tall, climbing Rosa canina shrub

Rose hips

Rose bedeguar gall on a dog rose
The dog roses, the Canina section of the genus Rosa (20-30 species and subspecies, which occur mostly in Northern and Central Europe), have an unusual kind of meiosis that is sometimes called permanent odd polyploidy, although it can occur with even polyploidy (e.g. in tetraploids or hexaploids). Regardless of ploidy level, only seven bivalents are formed leaving the other chromosomes as univalents. Univalents are included in egg cells, but not in pollen.[4][5] Similar processes occur in some other organisms.[6] Dogroses are most commonly pentaploid, i.e. five times the base number of seven chromosomes for the genus Rosa, but may be tetraploid or hexaploid as well.
Names and etymology
The botanical name is derived from the common names ‘dog rose’ or similar in several European languages, including classical Latin and ancient (Hellenistic period) Greek.
It is sometimes considered that the word ‘dog’ has a disparaging meaning in this context, indicating ‘worthless’ (by comparison with cultivated garden roses) (Vedel & Lange 1960). However it also known that it was used in the eighteenth and nineteenth centuries to treat the bite of rabid dogs, hence the name “dog rose” may result from this[7] (though it seems just as plausible that the name gave rise to the treatment).
Other old folk names include dogberry and witches’ briar.[citation needed]
Invasive species
Dog rose is an invasive species in the high country of New Zealand. It was recognised as displacing native vegetation as early as 1895[8] although the Department of Conservation does not consider it to be a conservation threat.[9]
Dog rose in culture
The dog rose was the stylized rose of medieval European heraldry, and is still used today.[citation needed] It is also the county flower of Hampshire.[10] Legend states the Thousand-year Rose or Hildesheim Rose, that climbs against a wall of Hildesheim Cathedral dates back to the establishment of the diocese in 815.[11]
Rose hip, rose hip and seed and rose hip seed, all were negatively monographed by the German Commission E due to insufficient evidence of effects and effectiveness. Therefore a comprehensive review of the literature was conducted to summarize the pharmacological and clinical effects of Rosa canina L. to reevaluate its usefulness in traditional medicine. For various preparations of rose hip and rose hip and seed, antioxidative and antiinflammatory effects have been demonstrated. Lipophilic constituents are involved in those mechanisms of action. The proprietary rose hip and seed powder Litozin has been employed successfully in a number of exploratory studies in patients suffering from osteoarthritis, rheumatoid arthritis and low back pain. However, the sizes of the clinical effects for the different indications need to be determined to assure clinical significance. There is also a rationale behind the use of Litozin as part of a hypocaloric diet based on the rose hip probiotic, stool regulating and smooth muscle-relaxing actions, as well as the rose hip seed lipid-lowering, antiobese and antiulcerogenic effects. Further research is needed to clarify the importance of the reported promising experimental effects in clinical use and to characterize the optimum rose hip seed oil preparation for topical use in the treatment of skin diseases.

Rosiflex Discovery
The Rosiflex™ story began in the early 1990s, when Erik Hansen, a farmer from Langeland, Denmark, discovered, quite by chance that rosehips from the Rosa Canina plant appeared to help soothe his aching joints.
Encouraged by this realisation, he developed the first of his rosehip powders. Made from rosehips grown on his own farm, he sold the powder to friends and neighbours after telling them of his own positive experiences.
The response from these early customers was so positive that Erik, and his son Torbjorn, decided to seek scientific verification of what they had found. They contacted scientists at the local hospital to see if they could find what it was in the rosehip that was producing the positive joint-health benefits being reported.
At first, the scientists were sceptical about the claimed benefits of the rosehip fruit – more commonly associated with teas and marmalades than with potential joint-health benefits. They did however agree to begin some scientific studies.
As the results of the testing began to emerge, the researchers became more and more convinced about the Langeland rosehip powder. Since then, several well designed scientific studies involving a couple of hundred people have been undertaken and published in recognised scientific journals.

Anti-inflammatory action of Rose hip
Rose hip is a typical daily food supplement traditionally used for its vitamin C content and other active principles to treat several discomforts: respiratory disorders, infectious diseases, gastrointestinal and urinary system illnesses and prophylaxis of vitamin C deficiencies. Rose hips have been eaten as jam or drunken as fruit tea for centuries. Therefore the separated Rose hip peels have always been regarded as everyday food.
In the last ten years it was scientifically documented, that the daily use of food containing rose hip fruits was positive to treat inflammatory joint diseases, in particular osteoarthritis. Several human studies with rose hip powder showed pain reducing properties and could also reduce symptoms such stiffness or even the need for additional medication.
However, the daily amount of 5 g over a period of 12 weeks showed moderate beneficial effects and low compliance demonstrating what the limits of a treatment with rose hip powder are.
Rose hip fruit skin powder contains remarkable active principles able to inhibit pro-inflammatory mediators and oxidative substances as well as enzymes responsible for the degradation of the organic matrix of joints and bones. A marked action on the inhibition of different cytokines has been observed as the interleukin 1β (IL-1β), the interleukin 6 (IL-6) and the alpha tumoral necrosis factor (TNF- α).
However herbal drug powders are usually not as stable and uniform as extracts. Using purification techniques and water as extraction solvent Finzelberg get a new extract, which compared with the rose hip drug powder is 7 fold stronger in their anti-inflammatory activity.
References
- ^ “BSBI List 2007” (xls). Botanical Society of Britain and Ireland. Retrieved 2014-10-17.
- ^ De Riek, Jan; De Cock, Katrien; Smulders, Marinus J.M.; Nybom, Hilde (2013). “AFLP-based population structure analysis as a means to validate the complex taxonomy of dogroses (Rosa section Caninae)”. Molecular Phylogenetics and Evolution 67 (3): 547–59. doi:10.1016/j.ympev.2013.02.024. PMID 23499615.
- ^ Vogl, Sylvia; Picker, Paolo; Mihaly-Bison, Judit; Fakhrudin, Nanang; Atanasov, Atanas G.; Heiss, Elke H.; Wawrosch, Christoph; Reznicek, Gottfried; Dirsch, Verena M.; Saukel, Johannes; Kopp, Brigitte (2013). “Ethnopharmacological in vitro studies on Austria’s folk medicine—An unexplored lore in vitro anti-inflammatory activities of 71 Austrian traditional herbal drugs”. Journal of Ethnopharmacology 149 (3): 750–71. doi:10.1016/j.jep.2013.06.007. PMC 3791396. PMID 23770053.
- ^ Täckholm, Gunnar (1922) Zytologische Studien über die Gattung Rosa. Acta Horti Bergiani 7, 97-381.
- ^ Lim, K Y; Werlemark, G; Matyasek, R; Bringloe, J B; Sieber, V; El Mokadem, H; Meynet, J; Hemming, J; Leitch, A R; Roberts, A V (2005). “Evolutionary implications of permanent odd polyploidy in the stable sexual, pentaploid of Rosa canina L”. Heredity 94 (5): 501–6. doi:10.1038/sj.hdy.6800648. PMID 15770234.
- ^ Stock, M.; Ustinova, J.; Betto-Colliard, C.; Schartl, M.; Moritz, C.; Perrin, N. (2011). “Simultaneous Mendelian and clonal genome transmission in a sexually reproducing, all-triploid vertebrate”. Proceedings of the Royal Society B: Biological Sciences 279 (1732): 1293. doi:10.1098/rspb.2011.1738.
- ^ Howard, Michael. Traditional Folk Remedies (Century, 1987); p133
- ^ Kirk, T (1895). “The Displacement of Species in New Zealand”. Transactions of the New Zealand Institute 1895 (Wellington: Royal Society of New Zealand) 28. Retrieved 2009-04-17.
- ^ Owen, S. J. (1997). Ecological weeds on conservation land in New Zealand: a database. Wellington: Department of Conservation.
- ^ “County Flowers | Wild plants”. Plantlife. Retrieved 2012-02-04.
- ^ Lucy Gordan. “Hildesheim’s Medieval Church Treasures at the Met”. Inside the Vatican. Archived from the original on 30 April 2014. Retrieved 30 April 2014.
Further reading
- Flora Europaea: Rosa canina
- Blamey, M. & Grey-Wilson, C. (1989). Flora of Britain and Northern Europe. Hodder & Stoughton. ISBN 0-340-40170-2.
- Vedel, H. & Lange, J. (1960). Trees and bushes. Metheun, London.
- Graham G.S. & Primavesi A.L. (1993). Roses of Great Britain and Ireland. B.S.B.I. Handbook No. 7. Botanical Society of the British Isles, London.
External links
………….
Contraindications:
As a natural diuretic, Rose Hips Herbal Supplement may increase the efficacy of prescription diuretics and should not be used at the same time. Make sure your doctor knows if you are taking a blood thinner, such as Coumadin®.
Disclaimer:
The information presented herein by this post is intended for educational purposes only. These statements have not been evaluated by the FDA and are not intended to diagnose, cure, treat or prevent disease. Individual results may vary, and before using any supplements, it is always advisable to consult with your own health care provider.
Boswellia serrata, -The cure for osteoarthritis in ayurveda, Shallaki,

in Kinnerasani Wildlife Sanctuary,Andhra Pradesh, India.
Boswellia serrata, -The cure for osteoarthritis in ayurveda, Shallaki,
Shallaki-Boswellia serrata
In degenerative and inflammatory pathologies invoving joints, there is no other drug as useful as Guggulu. Many international companies today use shallaki for the manufacture of drugs, ayurvedic and allopathic alike.
Family : Berseraceae
Scientific name : Boswellia serrata
Nomenclature in other languages :
Sanskrit : Shallaki, Susrava, Gajabhakshya
Hindi : Salei
Gujarathi : Dhoopa
Bengali : Salei
Tamil : Olibana
English : Indian Olibanum
Distribution : Gujarat, Rajasthan, Bihar are most commonly the residence of this plant.
Botanical description : It’s a resinous tree that grows to a height of 12m. A tree of moderate height , its bark are grey in colour. Upon time the bark sheds off like scales of a snake. The younger branches and leaflets of this tree are very smooth. The leaves which are compound(pinnate) in nature are 20-37 cm long. The leaflets are 2-5cm long and 1-2.5cm wide. The leaflets are oval shaped. The leaves contains 8 pairs or more of the leaflets . The margins of leaflets are serrated. Flowers are many and the inflorescence is terminal raceme, with it seen in the axilla of the leaf and stem. The petals and sepals are hairy and five in number. The stamen are 10 in number, they are diercted inwards. The fruits are seen in 3-4 numbers and are seen as drupes along with cones. The flowering season in April-May.



C hemical constituents and action
The bark contains carbohydrates, glycosides, beta-sitosterol. The resin contains ditrepene alcohol. This is knownn by the name sitosterol. In addition to that 11-keto-b-boswellic acid also has been extracted from the resin.
Ayurvedic Pharmacoepia
Rasa : kashaya, tikta, madhura
Guna : laghu, rooksha
Veerya : sheeta
Vipaka : katu
Medicinal properties :
Alleiviates vata kapha disorders. Also cures chronic skin lesions of all kinds infective and inflammatory, ulcers, wounds, piles, diseases of mouth, diarhhoea, hepatic disorders etc.
Useful parts : Bark, Resin
Therapeutic uses :
-1gm of resin taken in tablet form daily three times cures rheumatic, neurologic complaints and rheumatic fever.
-for gangrenes in diabetes the resin of this palnt may be applied externally and it taken internally as pills regularly
-the resin of this plant when chewed cures bad odour of mouth and mouth ulcers.
Medical uses
In Ayurvedic medicine Indian frankincense (Boswellia serrata) has been used for hundreds of years for treating arthritis.
Extracts of Boswellia serrata have been clinically studied for osteoarthritis and joint function, particularly for osteoarthritis of the knee, with the research showing a slight improvement of both pain and function compared to a placebo. Positive effects of Boswellia in some chronic inflammatory diseases including rheumatoid arthritis, bronchial asthma, osteoarthritis, ulcerative colitis and Crohn’s disease have been reported. A Boswellia extract marketed under the name Wokvel has undergone human efficacy, comparative, pharmacokinetic studies. Some see Boswellia serrata as a promising alternative to NSAIDs, warranting further investigation in pharmacological studies and clinical trials.

Topical application
Boswellia serrata has been recently developed for topical use in a patent-pending formula in Sano Relief Gel. Boswellia serrata is used in the manufacture of the supposed anti-wrinkle agent “Boswelox”,which has been criticised as being ineffective.
Potential for anti-cancer activity
Boswellic acid, an extract from Boswellia serrata, has been studied for anti-neoplastic activity, especially in experimental primary and secondary brain tumors, indicating potential efficacy from in vitro and limited clinical research. Boswellic acid is also undergoing an early-stage clinical trial at the Cleveland Clinic.
Active constituents
Boswellic acid and other pentacyclic triterpene acids are present. Beta-boswellic acid is the major constituent.
Mechanism of action
Animal studies performed in India show ingestion of a defatted alcoholic extract of Boswellia decreased polymorphonuclear leukocyte infiltration and migration, decreased primary antibody synthesis and almost totally inhibited the classical complement pathway.
Properties
Shallaki has potent analgesic and anti-inflammatory effects that can reduce the pain and inflammation of joints.
Frankincense ‘can ease arthritis’ researches have suggested |
|||
Extracts from Boswellia serrata, a similar species to the variety famous for its role in the Christian nativity, were tested on dozens of patients. Those who received it reported better movement and less pain and stiffness. The herb has been used for thousands of years in Indian Ayurvedic medicine, reports the journal Arthritis Research and Therapy. Current treatments carry a great many adverse effects, and scientists have been hunting for an alternative. The investigation into the properties of Boswellia serrata was led by Dr Siba Raychaudhuri at the University of California, Davis. Eventually they tested an extract of the plant enriched with the chemical – AKBA – thought to be its active ingredient. Some of the 70 patients with severe arthritis in their knees recruited into the trial were given a low-dose capsule, some a higher dose capsule, and the remainder were given a dummy pill with no active ingredients. In as little as seven days, patients taking the frankincense drug reported improvements in their pain and stiffness levels compared with the placebo group, and these continued until the 90-day mark, when the study ended. Alternative therapies Tests of the fluid within affected joints also revealed falls in levels of enzymes linked to the condition. Dr Raychaudhuri said: “We have shown that B. serrata enriched with AKBA can be an effective treatment for osteoarthritis of the knee.” However, UK experts urged caution. Professor Philip Conaghan, from Leeds University, and a spokesman for the Arthritis Research Campaign, said: “Certainly osteoarthritis is in need of new safe analgesics, although many effective therapies that reduce pain such as muscle strengthening exercises, shock-absorbing footwear and weight loss have very few bad side-effects. “This report on treating knee pain with a chemical derivative of B. serrata is interesting but the patient numbers are small, there were some problems with the reported trial design and we need more information on its medium to long-term safety.” |
|||
Boswellia serrata: an overall assessment of in vitro, preclinical, pharmacokinetic and clinical data.
Non-steroidal anti-inflammatory drug (NSAID) intake is associated with high prevalence of gastrointestinal or cardiovascular adverse effects. All efforts to develop NSAIDs that spare the gastrointestinal tract and the cardiovasculature are still far from achieving a breakthrough. In the last two decades, preparations of the gum resin of Boswellia serrata (a traditional ayurvedic medicine) and of other Boswellia species have experienced increasing popularity in Western countries. Animal studies and pilot clinical trials support the potential of B. serrata gum resin extract (BSE) for the treatment of a variety of inflammatory diseases like inflammatory bowel disease, rheumatoid arthritis, osteoarthritis and asthma. Moreover, in 2002 the European Medicines Agency classified BSE as an ‘orphan drug’ for the treatment of peritumoral brain oedema. Compared to NSAIDs, it is expected that the administration of BSE is associated with better tolerability, which needs to be confirmed in further clinical trials. Until recently, the pharmacological effects of BSE were mainly attributed to suppression of leukotriene formation via inhibition of 5-lipoxygenase (5-LO) by two boswellic acids, 11-keto-β-boswellic acid (KBA) and acetyl-11-keto-β-boswellic acid (AKBA). These two boswellic acids have also been chosen in the monograph of Indian frankincense in European Pharmacopoiea 6.0 as markers to ensure the quality of the air-dried gum resin exudate of B. serrata. Furthermore, several dietary supplements advertise the enriched content of KBA and AKBA. However, boswellic acids failed to inhibit leukotriene formation in human whole blood, and pharmacokinetic data revealed very low concentrations of AKBA and KBA in plasma, being far below the effective concentrations for bioactivity in vitro. Moreover, permeability studies suggest poor absorption of AKBA following oral administration. In view of these results, the previously assumed mode of action – that is, 5-LO inhibition – is questionable. On the other hand, 100-fold higher plasma concentrations have been determined for β-boswellic acid, which inhibits microsomal prostaglandin E synthase-1 and the serine protease cathepsin G. Thus, these two enzymes might be reasonable molecular targets related to the anti-inflammatory properties of BSE. In view of the results of clinical trials and the experimental data from in vitro studies of BSE, and the available pharmacokinetic and metabolic data on boswellic acids, this review presents different perspectives and gives a differentiated insight into the possible mechanisms of action of BSE in humans. It underlines BSE as a promising alternative to NSAIDs, which warrants investigation in further pharmacological studies and clinical trials.
Reference :
http://www.ncbi.nlm.nih.gov/pubmed/21553931
http://en.wikipedia.org/wiki/Boswellia_serrata
http://news.bbc.co.uk/2/hi/health/7535733.stm

Grape consumption may offer benefits for symptomatic knee osteoarthritis
New research presented last week at the Experimental Biology conference in San Diego, California, suggests that regular grape consumption may help alleviate pain associated with symptomatic osteoarthritis of the knee, and improve joint flexibility and overall mobility. Researchers attribute these potential benefits to the polyphenols found in grapes.
The sixteen week clinical study, undertaken by Texas Woman’s University, was designed to investigate the benefits of grape consumption on inflammation and osteoarthritis outcomes. 72 men and women with knee osteoarthritis (OA) were assigned to either consume grapes in the form of a whole grape freeze-dried powder, or a placebo powder.
The study results, presented by lead investigator Shanil Juma, Ph.D., showed that both men and women consuming a grape-enriched diet had a significant decrease in self-reported pain related to activity and an overall decrease in total knee symptoms. This beneficial effect was more pronounced in females. Additionally, age-related differences were observed:…
View original post 374 more words

{kind=link}
{kind=link}
{kind=link}