
Home » Posts tagged 'organic chemistry' (Page 8)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bristol-Myers Squibb files NDA in Japan for all-oral hepatitis C treatment
Bristol-Myers Squibb has filed a new drug application (NDA) to Japan’s Pharmaceutical and Medical Devices Agency for the approval of an interferon-free and ribavirin-free treatment regimen for patients with chronic hepatitis C (HCV).
click on title
Bristol-Myers Squibb files NDA in Japan for all-oral hepatitis C treatment
Phase III data show Boehringer Ingelheim’s faldaprevir was highly effective in a broad range of patients with genotype-1 hepatitis C

faldaprevir , 801283-95-4 cas no, BI-201335
(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acid
Molecular Formula: C40H49BrN6O9S
Molecular Weight: 869.82 g.mol-1
2 nd nov 2013
Boehringer Ingelheim today announced new data from its Phase III clinical trial programme, STARTVerso™, which evaluates faldaprevir* in combination with pegylated interferon and ribavirin (PegIFN/RBV). Patients with genotype-1 (GT-1) hepatitis C (HCV) who have not received previous treatment (treatment-naïve: STARTVerso™1&2),1 treatment-experienced patients (STARTVerso™3),2 and HIV co-infected patients (STARTVerso™4)3 participated in this study programme. The results from these and additional studies will be presented at the 64th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD), also known as The Liver Meeting®, taking place 1-5 November in Washington, D.C.
Faldaprevir (formerly BI 201335) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byBoehringer-Ingelheim and is currently in Phase III trials.[1]
Faldaprevir is a hepatitis C virus protease inhibitor.
Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.
Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.
- Efficacy and Safety of BI 201335 (Faldaprevir) in Combination With Pegylated Interferon-alpha and Ribavirin in Treatment-naïve Genotype 1 Hepatitis C Infected Patients (STARTverso 1). Cliicaltrials.gov. March 6, 2013.
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- Bioorganic & Medicinal Chemistry Letters, Volume 23, Issue 14, 15 July 2013, Pages 4267–4271
Synthesis and optimization of a novel series of HCV NS3 protease inhibitors: 4-Arylproline analogs
The following Compound 1):

(1)
wherein B is

; L° is MeO-; L1 is Br; and R2 is and having the chemical name: l-{ [4-[8-Bromo-2-(2-isopropylcarbamoyl-thiazol-4-yl)-7- methoxy-quinolin-4-yloxy]-l-(R)-(2-cyclopentyloxycarbonyl amino-3,3-(S)-dimethyl- butyryl)-pyrrolidine-(S)-2-carbonyl]-amino}-2-(S)-vinyl-cyclopropane-(R)-carboxylic acid, is known as a selective and potent inhibitor of the HCV NS3 serine protease and useful in the treatment of HCV infection. Compound (1) falls within the scope of the acyclic peptide series of HCV inhibitors disclosed in U.S. Patents RE 40,525, 7,514,557 and 7,585,845. Compound (1) is disclosed specifically as Compound # 1055 in U.S. Patent 7,585,845, and as Compound # 1008 in U.S. Patent 7,514,557. Compound (1), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (1) include the crystalline forms, in particular the crystalline sodium salt form, which can be prepared as described in U.S. Patent Application Publication No. 2010/0093792, also incorporated herein by reference. Data demonstrating the activity of Compound (1) as an inhibitor of the HCV NS3 serine protease and its corresponding demonstrated utility in the treatment of HCV infection in mono-infected patients, can be found in U.S. Patent 7,585,845, as well as in numerous publications presenting the preclinical characterization or clinical trial results with Compound (1). See, e.g., Sulkowski MS, et al, Hepatol (2009), Vol. 50, pg. 2A, Abtract LB3; Sulkowski MS, et al., J Hepatol (2010) Vol. 52, Supp. 1, pgs. S462-S463, Abstract 1190; Berg et al., Hepatol (2010), Vol. 52, Supp. SI, Abstract 804; and White PW, et al., Antimicrob Agents Chemother (2010) 54(11):4611-4618.
Combination therapy regimens directed to administering Compound (1) with an interferon- alpha and ribavirin for the treatment of HCV infection are described in U.S. Patent Application Publication Nos. 2010/0068182 and 2011/0268700.
HIV/HCV coinfected persons tend to have higher HCV viral loads and are less likely to clear the HCV spontaneously. The urgency for treatment of persons who are coinfected is greater than it is for those with HCV infection alone. The course of liver disease is more rapid in HIV/HCV-coinfected persons, including an approximately 2-fold increased risk of cirrhosis, more rapid progression to decompensated liver disease and increased risk for hepatocellular carcinoma (Graham CS, et al., Clin Infect Dis (2001 );33:562-569) .
Treatment of HCV might improve the tolerability of highly active antiretroviral therapy (HAART) because HCV infection increases the risk of mitochondrial toxicity and hepatotoxicity from HAART (Sulkowski MS, et al., JAMA (2000);283:74-80; Lafeuil!ade A, et al., Lancet (2001);357:280-281 ). Although there is much less published information on treatment outcomes in those who are HIV/HCV-coinfected than in HCV mono-infected patients, all accumulated data demonstrate that sustained virological response (SVR) and cure from HCV infection with pegylated interferon alpha and ribavirin is achieved in a substantially lower proportion of HIV/HCV coinfected patients when compared to HCV mono-infected patients. Factors associated with a poor treatment response (e.g., a high baseline HCV viral load, cirrhosis, and African American race) are present in a higher proportion of HIV/HCV coinfected populations, when compared to HCV monoinfected populations. It is not clear to what extent HIV infection itself diminishes the SVR rate, and to what extent advanced immunosuppression (e.g., CD4+ T lymphocyte count <200/mm3) further reduces response to HCV treatment (Toriani FJ, et al., N Engl J Med (2004);351(5): 438 -50; Nunez M, et al., ARHR (2007); 23(8):972-982).
Thus, there is a continuing high unmet need in the art for therapies that are effective against HCV in patients that are co-infected with HIV.
FDA approves Gazyva for chronic lymphocytic leukemia
Drug is first with breakthrough therapy designation to receive FDA approval
The U.S. Food and Drug Administration today approved Gazyva (obinutuzumab) for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
read all at
http://www.pharmalive.com/fda-approves-roche-s-gazyva
my old article cut paste
Roche’s new leukaemia drug, Obinutuzumab, superior to Rituxan in clinical trial
JULY 25, 2013 12:52 AM / 6 COMMENTS / EDIT
 July 24 2013 | By Márcio Barra
July 24 2013 | By Márcio Barra
Roche has announced that its experimental leukemia drug GA101, or obinutuzumab, used in combination with chemotherapy, was better than Rituxan at helping people with chronic lymphocytic leukemia live longer without their disease worsening, according to the results from the second phase of the clinical trial. Both drugs were tested and compared in combination with chlorambucil.
Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
- GA101 is the first glycoengineered, type II anti-CD20 mAb.

Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
Obinutuzumab (GA101)
| FORMULA | C6512H10060N1712O2020S44 |
|---|
GA101 is the first glycoengineered, type II anti-CD20 monoclonal antibody (mAb) that has been designed for increased antibody-dependent cellular cytotoxicity (ADCC) and Direct CellDeath.1 This agent is being investigated in collaboration with Biogen Idec.
Swiss pharmaceutical company Roche has announced that its early Phase III trial of Leukemia drug obinutuzumab (GA101) demonstrated significantly improved progression-free survival in people with chronic lymphocytic leukemia (CLL).
The positive results yield from stage 1 of a three-arm study called CLL11, designed to investigate the efficacy and safety profile of obinutuzumab (GA101) plus chlorambucil, a chemotherapy, compared with chlorambucil alone in people with previously untreated chronic lymphocytic leukemia (CLL).
This phase of the study met its primary endpoint and an improvement in progression-free survival was achieved; obinutuzumab plus chlorambucil significantly reduced the risk of disease worsening or death compared to chlorambucil alone.
Roche chief medical officer and global product development head Hal Barron said; “the improvement in progression-free survival seen with GA101 is encouraging for people with CLL, a chronic illness of older people for which new treatment options are needed.”
“GA101 demonstrates our ongoing commitment to the research and development of new medicines for this disease.”
Obinutuzumab is Roche’s most advanced drug in development for the treatment of hematological malignancies.
It has been specifically designed as the first glycoengineered, type 2 anti-CD20 monoclonal antibody in development for B cell malignancies.
Afutuzumab is a monoclonal antibody being developed by Hoffmann-La Roche Inc. for the treatment of lymphoma.[1] It acts as an immunomodulator.[2][3] It was renamed obinutuzumab in 2009.[4]
References
- Robak, T (2009). “GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies”. Current opinion in investigational drugs (London, England : 2000) 10 (6): 588–96. PMID 19513948.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Afutuzumab,American Medical Association.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
-
OBINUTUZUMAB ISMONOCLONAL ANTIBODY TYPE Whole antibody SOURCE Humanized (from mouse) TARGET CD20
GADODIAMIDE, OMNISCAN Drug Patent Expiration, 1 st oct 2013

GADODIAMIDE
GE HEALTHCARE, OMNISCAN
Drug Patent Expiration
1 st oct 2013, US5560903, CAS 122795-43-1
| GADODIAMIDE | INJECTABLE; INJECTION | 287MG/ML | RX | NDA 020123 |
Gadodiamide is a gadolinium-based MRI contrast agent, used in MR imaging procedures to assist in the visualization of blood vessels. It is commonly marketed under the trade name Omniscan.
For intravenous use in MRI to visualize lesions with abnormal vascularity (or those thought to cause abnormalities in the blood-brain barrier) in the brain (intracranial lesions), spine, and associated tissues.

Gadodiamide is a contrast medium for cranial and spinal magnetic resonance imaging (MRI) and for general MRI of the body after intravenous administration. The product provides contrast enhancement and facilitates visualisation of abnormal structures or lesions in various parts of the body including the central nervous system (CNS). It does not cross an intactblood brain barrier but might give enhancement in pathological conditions.
Based on the behavior of protons when placed in a strong magnetic field, which is interpreted and transformed into images by magnetic resonance (MR) instruments. Paramagnetic agents have unpaired electrons that generate a magnetic field about 700 times larger than the proton’s field, thus disturbing the proton’s local magnetic field. When the local magnetic field around a proton is disturbed, its relaxation process is altered. MR images are based on proton density and proton relaxation dynamics. MR instruments can record 2 different relaxation processes, the T1 (spin-lattice or longitudinal relaxation time) and the T2 (spin-spin or transverse relaxation time). In magnetic resonance imaging (MRI), visualization of normal and pathological brain tissue depends in part on variations in the radiofrequency signal intensity that occur with changes in proton density, alteration of the T1, and variation in the T2. When placed in a magnetic field, gadodiamide shortens both the T1 and the T2 relaxation times in tissues where it accumulates. At clinical doses, gadodiamide primarily affects the T1 relaxation time, thus producing an increase in signal intensity. Gadodiamide does not cross the intact blood-brain barrier; therefore, it does not accumulate in normal brain tissue or in central nervous system (CNS) lesions that have not caused an abnormal blood-brain barrier (e.g., cysts, mature post-operative scars). Abnormal vascularity or disruption of the blood-brain barrier allows accumulation of gadodiamide in lesions such as neoplasms, abscesses, and subacute infarcts.

1.Schenker MP, Solomon JA, Roberts DA. (2001). Gadolinium Arteriography Complicated by Acute Pancreatitis and Acute Renal Failure, Journal of vascular and interventional radiology 12(3):393.[1]
2 Unal O, Arslan H. (1999). Cardiac arrest caused by IV gadopentetate dimeglumine. AJR Am J Roentgenol 172:1141.[2]
3 Cacheris WP, Quay SC, Rocklage SM. (1990). The relationship between thermodynamics and the toxicity of gadolinium complexes, Magn Reson Imaging 8(6):467-81. doi:10.1016/0730-725X(90)90055-7
4 Canavese, C; Mereu, MC; Aime, S; Lazzarich, E; Fenoglio, R; Quaglia, M; Stratta, P (2008). “Gadolinium-associated nephrogenic systemic fibrosis: the need for nephrologists’ awareness”. Journal of nephrology 21 (3): 324–36. PMID 18587720.
COUNTRY PATENT APPROVED, EXPIRY
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5362475 | 1994-11-08 | 2011-11-08 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
Gadolinium contrast agents are used as contrast media to enhance magnetic resonance imaging as they are paramagnetic. This compound has a low incidence of adverse side effects, although there is a rare association with nephrogenic systemic fibrosis (NSF) when given to people with severe renal impairment (ie, GFRglomerular filtration rate <30mL/min/1·73m2).It seems to be related to the liberation of free gadolinium ions, and UK CHM advice is against using the least stable of the agents – Omniscan (gadodiamide) – in patients with severe renal impairment, and carefully considering whether to use others where renal function is impaired.
OMNISCAN (gadodiamide) Injection is the formulation of the gadolinium complex of diethylenetriamine pentaacetic acid bismethylamide, and is an injectable, nonionic extracellular enhancing agent for magnetic resonance imaging. OMNISCAN is administered by intravenous injection. OMNISCAN is provided as a sterile, clear, colorless to slightly yellow, aqueous solution. Each 1 mL contains 287 mg gadodiamide and 12 mg caldiamide sodium in Water for Injection.
The pH is adjusted between 5.5 and 7.0 with hydrochloric acid and/or sodium hydroxide. OMNISCAN contains no antimicrobial preservative. OMNISCAN is a 0.5 mol/L solution of aqua[5,8-bis(carboxymethyl)11-[2-(methylamino)-2-oxoethyl]-3-oxo-2,5,8,11-tetraazatridecan-13-oato (3-)-N5, N8, N11, O3, O5, O8, O11, O13] gadolinium hydrate, with a molecular weight of 573.66 (anhydrous), an empirical formula of C16H28GdN5O9•xH2O, and the following structural formula:
 |
Pertinent physicochemical data for OMNISCAN are noted below:
PARAMETER
| Osmolality (mOsmol/kg water) | @ 37°C | 789 |
| Viscosity (cP) | @ 20°C | 2 |
| @ 37°C | 1.4 | |
| Density (g/mL) | @ 25°C | 1.14 |
| Specific gravity | @ 25°C | 1.15 |
OMNISCAN has an osmolality approximately 2.8 times that of plasma at 37°C and is hypertonic under conditions of use.
gadodiamide, chemical name: [5,8 _ bis (carboxymethyl) -11 – [2_ (methylamino)-2_ ethyl] -3 – O 2 ,5,8, 11 – tetraazacyclododecane-decane -13 – oxo-(3 -)] gadolinium trihydrate. Its structure is shown in formula one.
[0003] Structural Formula:
[0004]

[0005] Magnetic resonance contrast agent gadodiamide resonance than ionic contrast agents safer generation of products, it is non-ionic structure significantly reduces the number of particles in solution, osmotic balance of body fluids is very small.Meanwhile, gadodiamide relatively low viscosity to bring the convenience of nursing staff, making it easier to bolus. In addition, gadodiamide pioneered the use of amide-substituted carboxyl part, not only reduces the toxicity of carboxyl groups and ensure the non-ionic nature of the product solution.
[0006] reported in the literature and their intermediates gadodiamide synthetic route is as follows:
[0007] 1. Compound III synthetic routes for its preparation in U.S. Patent No. US5508388 described as: In the synthesis process, the inventors using acetonitrile as solvent, acetic anhydride as dehydrating agent, pyridine as acid-binding agent, at 55 ~ 60 ° C, the reaction 18h. Anti-
See the reaction should be a process. The disadvantage of this synthesis are acetonitrile toxicity, not widely used.
[0008]

[0009] Reaction a
[0010] (2) Synthesis of Compound III in many articles are reported in the patent and its implementation method similar to the patent US5508388.
[0011] In US3660388, the diethylenetriamine pentaacetic acid (Compound II), pyridine, acetic anhydride, the mixture was reacted at 65 ° C or 20h at 125 ° C the reaction 5min, to give compound III.
[0012] In US4822594, the compounds II, pyridine, acetic anhydride mixture was reacted at 65 ° C 20h, to give compound III.
[0013] In US4698263, the compounds II, pyridine, acetic anhydride heated in a nitrogen or argon atmosphere under reflux for 18h, to give compound III. [0014] In the EPO183760B1, the compounds II, pyridine, acetic anhydride mixture was reacted at 55 ° C 24h, to give compound III.
[0015] In CN1894223A, the compounds II, pyridine, acetic anhydride, the mixture above 65 ° C the reaction mixture, and the pyridine of DTPA feed ratio is: 1: (0.5 to 3).
[0016] The above patents do not provide for the compound III is post-processing method.
[0017] 3 Synthesis of Compound IV.
[0018] In U.S. Patent US4859451, the diethylenetriamine pentaacetic acid dianhydride (compound III) and ammonia, methanol and the reaction of compounds IV, see Reaction Scheme II.
[0019]

[0020] Reaction two
[0021] In the patent US5087439, the compound III with methylamine in aqueous solution for several hours, or overnight reactions, see reaction formula III.
[0022]

[0023] Reactive three
[0024] These two patents using ammonia and methylamine, which can form explosive mixtures with air, in case of fire or high pressure can cause an explosion in the production process of great insecurity. Although raw material prices are lower, but higher production conditions (such as requiring sealed, low temperature, etc.). Compared to this synthesis process,
[0025] 4, gadodiamide (Compound I) synthesis.
[0026] In the patent US4859451, the use of gadolinium chloride with the compound IV is carried out under acidic conditions, complexing. Finally, tune
Section PH neutral, see reaction IV.
[0027]

[0028] Reaction formula tetrakis [0029] in the patent US5087439, the chlorides are used as reactants, and details of the post-processing method of Compound I.
[0030] In the patent US5508388, the use of gadolinium oxide with compound IV in acetonitrile, water with stirring, the resulting compound I.
[0032] The synthetic route is as follows:
[0033]

[0034] 1) Compound II (diethylenetriamine pentaacetic acid) in pyridine, acetic anhydride in the presence of a dehydration reaction into the acid anhydride, and the product was stirred with cold DMF, leaving the solid filtered, washed with ether reagents, drying , to obtain a white powdery solid compound III (diethylenetriamine pentaacetic acid anhydride);
[0035] 2) Compound III in DMF with methylamine hydrochloride, the reaction of the compound IV (5,8 _ bis carboxymethyl methyl-11 – [2 – (dimethylamino) -2 – oxoethyl] – 3 – oxo -2,5,8,11 – tetraazacyclododecane _13_ tridecyl acid); and the control compound III: MeNH2 · HCl molar ratio = 1: (1 to 4), control the temperature between 20 ~ 80 ° C, the reaction time is 4 ~ 6h, after the treatment, the method of distillation under reduced pressure to remove DMF, the product is dissolved in a polar solvent, methanol, and then adding a solvent polarity modulation, so that the target Compound IV from system completely precipitated;
[0036] 3) Compound IV with gadolinium oxide formed in the presence of hydrochloric acid of the complex, after the reaction, filtration and drying, to obtain a white powdery compound I, i.e. gadodiamide.
[0037] Existing gadodiamide Synthesis basically from the synthesis of Compound IV as a starting material, the present invention is first introduced to the compound II as a starting material to synthesize gadodiamide. Synthesis of the conventional method of gadodiamide, the present invention has the advantage of inexpensive starting materials, convenient and easy to get. In addition, the synthetic pathway intermediates are involved in the post-processing is simple, enabling continuous reaction, saving time and cost savings, the reaction becomes controlled step by step, and try to avoid the use of toxic reagents, reducing the possibility of operator injury , while also greatly reducing damage to the environment.
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

Biosimilars-in-India
http://www.ibef.org/download/Biosimilars-in-India-30312.pdf
-
Biosimilars – India Brand Equity Foundation
www.ibef.org/download/Biosimilars-in-India-30312.pdfpatented/registered biotech products, but are manufactured by new companies after the patent expiry of the originator product. The global. Biosimilars market is …
Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder

cyclic pyranopterin monophosphate (cPMP, ALXN1101)
Alexion Pharma International Sàrl has received a breakthrough therapy designation from the US Food and Drug Administration (FDA) for its cyclic pyranopterin monophosphate (cPMP, ALXN1101), an enzyme co-factor replacement therapy to treat patients with molybdenum cofactor deficiency (MoCD) type A.
Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder
read all at
Cyclic pyranopterin monophosphate (cPMP) is an experimental treatment formolybdenum cofactor deficiency type A, which was developed by José Santamaría-Araujo and Schwarz at the German universities TU Braunschweig and the University of Cologne.[1][2]
cPMP is a precursor to molybdenum cofactor, which is required for the enyzme activity ofsulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
- Guenter Schwarz Laboratory, Institute for Biochemistry – University of Cologne (English, German)
- Günter Schwarz, José Angel Santamaria-Araujo, Stefan Wolf, Heon-Jin Lee, Ibrahim M. Adham, Hermann-Josef Gröne, Herbert Schwegler, Jörn Oliver Sass, Tanja Otte, Petra Hänzelmann, Ralf R. Mendel, Wolfgang Engel and Jochen Reiss (2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli“. Human Molecular Genetics 13 (12): 1249–1255. doi:10.1093/hmg/ddh136.PMID 15115759.
- Doctors risk untried drug to stop baby’s brain dissolving, TimesOnline, November 5, 2009
- José Angel Santamaria-Araujo, Berthold Fischer, Tanja Otte, Manfred Nimtz, Ralf R. Mendel, Victor Wray and Günter Schwarz (2004). “The Tetrahydropyranopterin Structure of the Sulfur-free and Metal-free Molybdenum Cofactor Precursor”. The Journal of Biological Chemistry 279 (16): 15994–15999.doi:10.1074/jbc.M311815200. PMID 14761975.
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclicpyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, unbeatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis {see U.S. Patent No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
WO 2012112922 A1
In this synthesis, the deprotection may involve, for example, either sequential or one-pot deprotection of certain amino and hydroxyl protecting groups on a compound of formula (VII) to furnish the compound of formula (I). Suitable reagents and conditions for the deprotection of a compound of formula (VII) can be readily determined by those of ordinary skill in the art. For example, compound (I) may be formed upon treatment of a compound of formula (VII) under conditions so that hydroxyl protecting groups, such as acetate, isopropylidine, and benzylidine protecting groups, are removed from the formula (VII) structure. The acetate group can be cleaved, for example, under Zemplen conditions using catalytic NaOMe as a base in methanol. The benzylidene and isopropylidene groups can be cleaved by hydrogenation or using acidic hydrolysis as reported by R.M. Harm et ah, J. Am. Chem. Soc, 72, 561 (1950). In yet another example, the deprotection can be performed so that amino protecting groups, such as 9- fluorenylmethyl carbamate (Fmoc), t-butyl carbamate (Boc), and carboxybenzyl carbamate (cbz) protecting groups are cleaved from the compound of formula (VII). 9-fluorenylmethyl carbamate (Fmoc) can be removed under mild conditions with an amine base (e.g. , piperidine) to afford the free amine and dibenzofulvene, as described by E. Atherton et al, “The
Fluorenylmethoxycarbonyl Amino Protecting Group,” in The Peptides, S. Udenfriend and J. Meienhofer, Academic Press, New York, 1987, p. 1. t-butyl carbamate (Boc) can be removed, as reported by G.L. Stahl et al., J. Org. Chem., 43, 2285 (1978), under acidic conditions (e.g., 3 M HC1 in EtOAc). Hydrogenation can be used to cleave the carboxybenzyl carbamate (cbz) protecting group as described by J. Meienhofer et al., Tetrahedron Lett., 29, 2983 (1988).
To prevent oxidation of formula (I) during the reaction, the deprotection may be performed under anaerobic conditions. The deprotection may also be performed at ambient temperature or at temperatures of from about 20 – 60 °C (e.g. , 25, 30, 35, 40, 45, 50, or 55 °C).
The compound of formula (I) may be isolated in the form of a pharmaceutically acceptable salt. For example, the compound of formula (I) may be crystallized in the presence of HC1 to form the HC1 salt form of the compound. In some embodiments, the compound of formula (I) may be crystallized as the HBr salt form of the compound. The compound of formula (I) may also be isolated, e.g., by precipitation as a sodium salt by treating with NaOH. The compound of formula (I) is labile under certain reaction and storage conditions. In some embodiments, the final solution comprising the compound of formula (I) may be acidified by methods known in the art. For example, the compound of formula (I), if stored in solution, can be stored in an acidic solution.
In some embodiments, the compound of formula (I) may be prepared, for example, by: reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV- A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-A):

wherein:
Rj is a protecting group, as defined above;
phosphorylating the compound of formula (V-A) to prepare a compound of formula (VI- A):

oxidizing the compound of formula (VI-A) to prepare a compound of formula (VII- A):

; and deprotecting the compound of formula (VII-A) to prepare the compound of formula (I). For example, a compound of formula (I) can be prepared as shown in Scheme 3.
Scheme 3.

5 R = Fraoc

In another embodiment, the compound of formula (I) is prepared by:
reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV-A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-B):

wherein:
each Ri is independently a protecting group, as defined above;
phosphorylating the compound of formula (V-B) to prepare a compound of formula (VI-B):

oxidizing the compound of formula (VI-B) to prepare a compound of formula (VII-B):

; and deprotecting the compound of formula (VII-B) to prepare the compound of formula (I), example, a compound of formula (I) can be prepared as shown in Scheme 4.
Scheme 4.

Alternatively, a compound of formula (I) can be formed as shown in Scheme 5. A diaminopyrimidinone compound of formula (II) can be coupled with a phosphorylated hexose sugar of formula (VIII), to give a compound of formula (IX). The piperizine ring nitrogen atoms can be protected to give a compound of formula (X) which can be oxidized to give a diol of formula (XI). The diol of formula (XI) can then be deprotected using appropriate conditions and converted to the compound of formula (I).
Scheme 5

In this embodiment, the phosphate may be introduced at the beginning of the synthesis to avoid undesirable equilibrium between the pyrano and furano isomers during subsequent steps of the synthesis. For example, a compound of formula (I) can be prepared as shown in Scheme 6.
Scheme 6.
ridine

A compound of formula (I) can also be formed as shown in Scheme 7. A diaminopyrimidinone compound of formula (II) can be coupled to a compound of formula (III) to afford the piperizine derivative of formula (IV). The piperizine ring nitrogen atoms of the compound of formula (IV) can be protected under standard conditions to give a derivative of formula (V). The formula (V) structure can be oxidized to afford compounds of formula (XII). Phosphorylation of a compound of formula (XII) gives a compound of formula (VII). Global deprotection of the compound of formula (VII) can afford the compound of formula (I).
Scheme 7
Piperizine ring protection
sphorylation
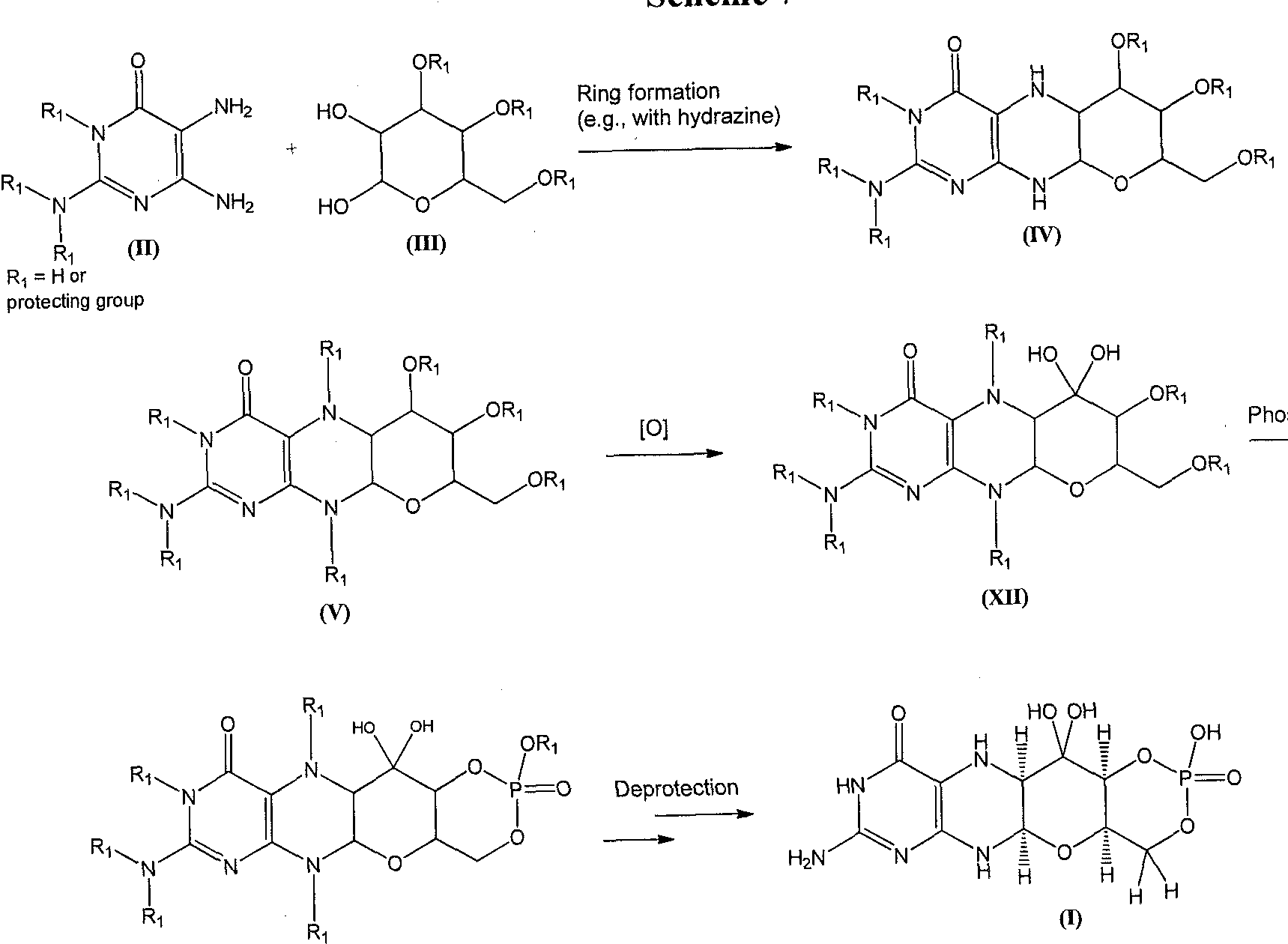
(VII)
For example, a compound of formula (I) can be prepared as shown in Scheme 8.
Scheme 8.

Founder of leading Indian pharmaceutical company, Sun Pharmaceuticals has been named India’s third richest man

This is the first time Mr Dilip Shanghvi, founder of Sun Pharmaceutical has been named in the top three with a 66 percent surge in his wealth –
25 oct 2013
Singapore: For the first time, founder of leading Indian drugmaker Sun Pharmaceuticals, Mr Dilip Shanghvi has been named in the top three of India’s richest men. Mr Shanghvi has made an appearance in the list prepared by China based ‘Hurun India Rich List’ as the third richest Indian man with a 66 percent surge in his wealth. Energy tycoon and Reliance Industries chairman Mr Mukesh Ambani has topped the list as India’s richest man with personal assets of $18.9 billion, news reports mentioned. The report pointed out that Mr Ambani has retained the top position for the second year even after a wealth decrease of two percent. London-based steel baron Mr Lakshmi Narayan Mittal has been named the second richest, with assets of $15.9 billion. – See more at: http://www.biospectrumasia.com/biospectrum/news/199210/sun-pharma-s-founder-named-india-s-3rd-richest#.Umzu4fmnqls
Read more at: http://www.biospectrumasia.com/biospectrum/news/199210/sun-pharma-s-founder-named-india-s-3rd-richest#.Umzu4fmnql
s
Potential of US pipeline continues to be under appreciated
SUNP’s US pipeline is shaping up well, with an interesting mix of complex products, branded generics and me-too products. Motilal Oswal believe that SUNP’s strong pipeline in the US is well placed to deliver revenue CAGR of 25% to USD1.8b. While a meaningful contribution to this growth is being led by Doxil and recently acquired URL Pharma, we estimate that SUNP’s own pipeline is set to witness revenue CAGR of 40% to USD6 20m. They build flat sales growth for Taro as they expect incremental competition to impact the market share for key Taro products. If competition is delayed, their estimates may have room for positive surprise.
Even after a 50-plus per cent return in one year, Sun Pharma’s stock continues to hold promise. While domestic revenues are growing well, US sales growth has been phenomenal, driven by a strong product pipeline and the acquisitions of Taro, DUSA, URL generics and Caraco.
-
[PDF]
NCE & NDDS Development Pipeline – Sun Pharma
www.sunpharma.com/pressdownload.php?download_file=329.pdfNCE & NDDS Development Pipeline / 2. Disclaimer. Except for the historical information contained herein, statements in this presentation and the subsequent …
-
[PDF]
Pipeline of Potential Ocular Therapies to Help Millions … – Sun Pharma
www.sunpharma.com/pressdownload.php?…Press%20Release%20SunPh…Oct 1, 2013 – 1. For Immediate Release. Sun Pharma and Intrexon Form Joint Venture to Develop. New Class of Therapeutics for Ocular Diseases. Pipeline …

pipeline
Sun Pharma Advanced Research (SPARC, US$345
mn market cap), a proprietary product research
company, detailed its NDDS and NCE product pipelines
in its latest filing with the SEBI, pursuant to its proposed
rights issue. Overall, there has been modest
clinical/regulatory progression in the NDDS pipeline
since the last disclosure six-seven months back. See
Exhibits 1 and 4 inside.
Key highlights: Big picture – New Drug Delivery
System (NDDS) – Overall SPARC is developing a
diverse collection of seven technology platforms for oral,
injectable and topical delivery systems (including nano
particulate, bio-degradable depot, DPI, SMM, GFR).
SPARC has 19 NDDS products under development.
New Chemical Entity (NCE) – The company has a
pipeline of five compounds that are pro-drugs of existing
molecules (such as baclofen, gabapentin etc). The NCE
pipeline appears to be in its early stages, with market
launch a few years away.
Update on key products (NDDS) – Three lead drugs:
1) levetiracetam XR 1000/1500 mg (505(b)(2) filing with
USFDA is now targeted for F1Q13), 2) baclofen GRS
(Special Protocol Assessment, SPA, agreement has
been received from FDA and patient enrollment for
Phase 3 studies is targeted in F4Q12) and 3) BAK-free
latanoprost (Phase III patient enrollment over in US).
These timelines imply that the first product launch in the
US is likely in 2H13. SPARC continues to validate its
underlying technology platforms by launching drugs in
the domestic market (six proprietary launches, seven
generic launches so far).
Other milestones for CY12: IND filing with USFDA for
two drugs – Octreotide Depot Inj (1 month) and Dry
Powder Inhaler (Salmeterol and Fluticasone
combination); paclitaxel (with carboplatin) Phase I
results in US and clinical progress of second WRAP
based drug, cardiovascular agent (and its combination)NDDS
LATANOPROST
LATANOPROST+TIMOLOLOL
BACLOFEN VENLAFAXINE
LEVETIRACETAM
PICN, PACLITAXEL
NCE
SUN597,
SUN L731
SUN K706
OTHERS
DRY POWDER INHALER
DOCETAXEL NANODISPERSION
OCTEOTIDE

MAY 2013
mn market cap), a proprietary product research
company, detailed its NDDS and NCE product pipelines
in its latest filing with the SEBI, pursuant to its proposed
rights issue. Overall, there has been modest
clinical/regulatory progression in the NDDS pipeline
since the last disclosure six-seven months back. See
Exhibits 1 and 4 inside.
Key highlights: Big picture – New Drug Delivery
System (NDDS) – Overall SPARC is developing a
diverse collection of seven technology platforms for oral,
injectable and topical delivery systems (including nano
particulate, bio-degradable depot, DPI, SMM, GFR).
SPARC has 19 NDDS products under development.
New Chemical Entity (NCE) – The company has a
pipeline of five compounds that are pro-drugs of existing
molecules (such as baclofen, gabapentin etc). The NCE
pipeline appears to be in its early stages, with market
launch a few years away.
Update on key products (NDDS) – Three lead drugs:
1) levetiracetam XR 1000/1500 mg (505(b)(2) filing with
USFDA is now targeted for F1Q13), 2) baclofen GRS
(Special Protocol Assessment, SPA, agreement has
been received from FDA and patient enrollment for
Phase 3 studies is targeted in F4Q12) and 3) BAK-free
latanoprost (Phase III patient enrollment over in US).
These timelines imply that the first product launch in the
US is likely in 2H13. SPARC continues to validate its
underlying technology platforms by launching drugs in
the domestic market (six proprietary launches, seven
generic launches so far).
Other milestones for CY12: IND filing with USFDA for
two drugs – Octreotide Depot Inj (1 month) and Dry
Powder Inhaler (Salmeterol and Fluticasone
combination); paclitaxel (with carboplatin) Phase I
results in US and clinical progress of second WRAP
based drug, cardiovascular agent (and its combination)

{kind=link}