
Home » Posts tagged 'obesity'
Tag Archives: obesity
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ORZILOBEN
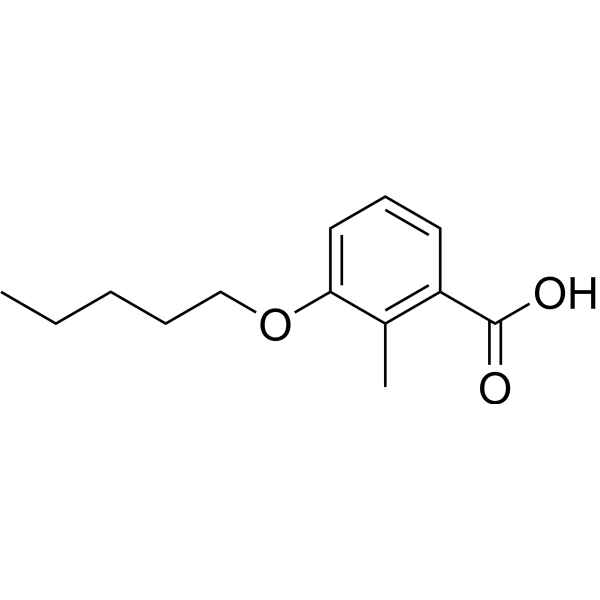

ORZILOBEN,
CAS 1555822-28-0
2-methyl-3-(pentyloxy)benzoic acid
| Molecular Weight | 222.28 |
|---|---|
| Formula | C13H18O3 |
Orziloben is a medium chain fatty acid (MCFA) analogue[1]
Phase IILiver disorders
OriginatorPronova BioPharma
DeveloperNorthSea Therapeutics
ClassAntifibrotics; Hepatoprotectants; Omega 3 fatty acids; Unsaturated fatty acids
Mechanism of ActionUndefined mechanism.
28 Jan 2025No recent reports of development identified for phase-I development in Liver-disorders(In volunteers) in United Kingdom (PO)- 24 Dec 2024Chemical structure information added.
- 23 Aug 2024NorthSea Therapeutics completes a phase I trial in healthy volunteers in the United Kingdom (PO) (ISRCTN12367117)
Patent
WO2020074964
The rising global epidemic of obesity and its comorbidities, e.g., type 2 diabetes mellitus and hyperlipidemia, is placing an enormous burden both on public health (mortality and morbidity) and on the available public health resources required to treat these conditions.
Current drugs that treat hyperlipidemia (e.g., statins, omega-3 fatty acids, fibrates) have mostly neutral effects on glycemic control, whilst drugs targeting glycemic control e.g., insulin, thiazolidinediones (TZDs), have adverse effects upon bodyweight and (for TZDs) other unwanted side-effects restricting their use.
In addition to hyperlipidemia and type 2 diabetes, a marked increase in the prevalence of non-alcoholic fatty liver disease (NAFLD) has occurred. NAFLD has become the most common chronic liver condition in Western populations in relation to the obesity and type 2 diabetes epidemics. The prevalence of non-alcoholic steatohepatitis (NASH), a form of NAFLD that is associated with hepatic inflammation and ballooning of hepatocytes, is expected to increase by 63% between 2015 and 2030 in the United States (Estes, Hepatology, 2018; 67(1): 123-133), where NASH is expected to become the leading cause of liver transplantation by 2020. As liver fibrosis, but not inflammation, is associated with mortality and morbidity in NASH patients, drugs which prevent progression/induce regression of fibrosis are also a focus of biomedical research.
The development of novel compounds that simultaneously target both hyperlipidemia and glycemic control, without the adverse side-effects (e.g., weight gain) typically associated with insulin sensitising drugs is thus a desirable goal. Such compounds would be even more attractive if they could additionally prevent the progression/reverse hepatic fibrosis and reduce hepatic steatosis. The present invention addresses these needs for new treatment methods, compounds, and pharmaceutical compositions.



AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
/////////ORZILOBEN, 1555822-28-0, OBESITY, NST-6179, SEFA-6179, NST 6179, SEFA 6179
O=C(C1=C(C)C(OCCCCC)=CC=C1)O
SIBUTRAMINE
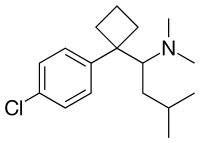
SIBUTRAMINE
- Molecular FormulaC17H26ClN
- Average mass279.848 Da
1-(4-Chlorophenyl)-N,N-dimethyl-a-(2-methylpropyl)cyclobutane methanamine
1-[1-(4-Chlorophenyl)cyclobutyl]-N,N,3-trimethyl-1-butanamine
106650-56-0[RN]
106650-56-0 (Sibutramine );
125494-59-9 (Sibutramine HCl Monohydrate);
84485-00-7 (Sibutramine HCl);
6124
UNII:WV5EC51866, WV5EC51866
сибутрамин[Russian]
سيبوترامين[Arabic]
西布曲明[Chinese]
Drug Name:Sibutramine Hydrochloride Hydrate
Research Code:BTS-54524
Trade Name:Meridia®
MOA:Serotonin-norepinephrine reuptake inhibitor
Indication:Obesity
Status:Withdrawn
Company:Abbott (Originator)
Sibutramine hydrochloride monohydrate, KES-524, BTS-54524, Meridia, Reductil
Sibutramine, formerly sold under the brand name Meridia among others, is an appetite suppressant which has been discontinued in many countries. Until 2010, it was widely marketed and prescribed as an adjunct in the treatment of obesity along with diet and exercise. It has been associated with increased cardiovascular events and strokes and has been withdrawn from the market in several countries and regions including Australia,[1] Canada,[2] China,[3] the European Union,[4] Hong Kong,[5] India,[6] Mexico, New Zealand,[7] the Philippines,[8] Thailand,[9] the United Kingdom,[10] and the United States.[11] However, the drug remains available in some countries.[12]
Sibutramine was originally developed in 1988 by Boots in Nottingham, UK,[13] and marketed by Knoll Pharmaceuticals after BASF/Knoll AG purchased the Boots Research Division in 1995, and was most recently manufactured and marketed by Abbott Laboratories before its withdrawal from most markets. It has been sold under a variety of brand names including Reductil, Meridia, Siredia, and Sibutrex. It is classified as a Schedule IV controlled substance in the United States.
Sibutramine hydrochloride hydrate was approved by the U.S. Food and Drug Administration (FDA) on Nov 16, 1997. It was developed and marketed as Meridia® by Abbott in the US.
Sibutramine hydrochloride hydrate is a serotonin-norepinephrine reuptake inhibitor, it produces its therapeutic effects by norepinephrine, serotonin and dopamine reuptake inhibition. Meridia® is indicated for the management of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a reduced calorie diet.
Meridia® is available as capsule for oral use, containing 5, 10 or 15 mg of Sibutramine hydrochloride hydrate. The recommended dose is initiated at 10 mg once daily with or without food and may increase to 15 mg once daily.
Sibutramine has been withdrawn from the market in several countries and regions since 2010, owning to its side effect that associated with increased cardiovascular events and strokes.Route 1
Reference:1. US4746680A / US4806570A.
2. US4929629A.
SYN

SYN

SYN
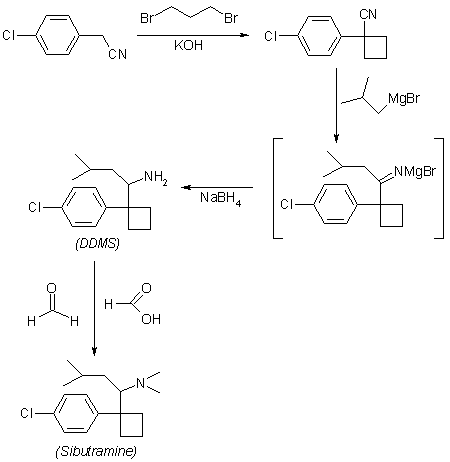
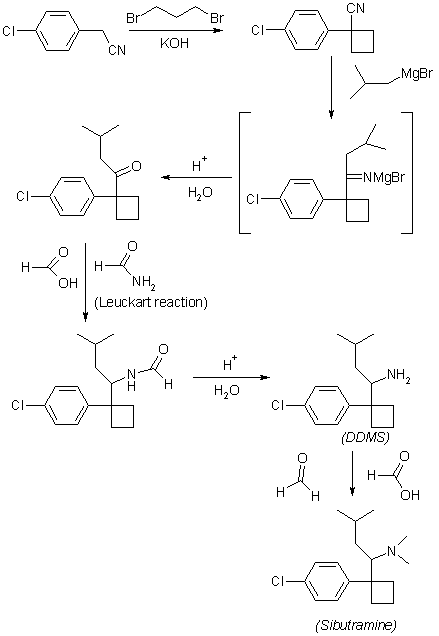
PAT
https://patents.google.com/patent/KR20060019351A/en
Sibutramine hydrochloride (Sibutramine HCl: C 17 H 26 CIN HCl) is a chemical name {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine hydrochloride, and has the structure of Formula 1 .

Sibutramine was originally developed as a drug for the treatment of depression and was found to give weight to patients taking this drug. It was developed as an anti-obesity drug. Let your appetite decrease.
Korean Patent Publication No. 1990-274 (corresponding patent DE3212682 (Boots), filed Oct. 21, 1982; priority GB 1981.4.6.) For the preparation of 1- (1-arylcyclobutyl) alkylamine derivative comprising sibutramine It is described. The method for synthesizing sibutramine described in this document proceeds in a total of five steps as follows.
Step A

1- (4-chlorophenyl) -1-cyclobutyl cyanide is obtained from 4-Chlorobenzyl cyanide. Examples of the actual synthesis method described in this document are as follows.
After dissolving 25 g of 4-chlorobenzyl cyanide and 15 ml of 1,3-dibromopropane in 150 ml of DMSO, the solution was dissolved in nitrogen at room temperature (20-35 ° C.) and 7.5 g of NaH dispersed in mineral oil. And 200 ml of DMSO was added dropwise. The mixture was stirred at room temperature for 2 hours, 8 ml of IPA was added dropwise, and 110 ml of water was added dropwise. The mixture is filtered through CELITE ™ and the solid residue is washed with ether. The ether layer is separated, washed with water, dried, evaporated and vacuum distilled at high temperature to separate the desired 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The total reaction time of this step is 5 hours and the yield from the starting material is 78%.
Step B

1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one is obtained from 1- (4-chlorophenyl) -1-cyclobutyl cyanide. Specific synthesis examples are as follows. 35.2 g of 1- (4-chlorophenyl) -1-cyclobutyl cyanide are dissolved in 100 ml of ether and this solution is added to the product prepared by the reaction of 32 g of propylbromide and 6.36 g of magnesium. The ether is replaced with toluene and the mixture is heated under reflux for 1 hour. After adding water, concentrated hydrochloric acid is added, and the mixture is heated under reflux for 1 hour. The mixture obtained in the same manner as in the previous step was treated with ether, water, dried and evaporated and then vacuum distilled to give the desired 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one. To separate. The reaction time of this step is a total of 22 hours, the yield is 81%. The target product is bp 100-120 ° C / 0.2 mm / Hg.
Step C

N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl from 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one } Formamide is obtained. Specific synthesis examples are as follows. To 23.5 ml of formamide, 37 g (0.14 mol) of 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one and 9 ml of HCOOH were added dropwise at 160-170 ° C. The temperature is maintained at 175 ° C. to 180 ° C. for 24 hours. The mixture is extracted with ether and concentrated to afford an oil, which crystallizes the desired N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide from petroleum ether. The reaction time of this step is a total of 24 hours, the yield is 39%. The target is mp 110-112 ° C.
Step D

1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride from N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide Get Specific synthesis examples are as follows. 4 g of N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide, 25 ml of 2-methoxyethyl ether, 10 ml of water and 22 ml of concentrated hydrochloric acid were refluxed for 18 hours. Stir under. Dilute with water, wash with ether, and add 35 ml of 5M aqueous NaOH solution. After completion of the process by treatment with ether, water and brine, treated with magnesium sulfate, filtered and concentrated. The concentrated crude product is saturated with hydrochloric acid dissolved in 20 ml of ether. The resulting solid is filtered, concentrated and crystallized with petroleum ether to give the desired product 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride. The reaction time of this step is a total of 20 hours, the yield is 96% oil, 46% hydrochloride. The target product is mp 163-165 ° C.
Step E

The final target from 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethyl Amine hydrochloride, ie sibutramine hydrochloride, is obtained. Specific synthesis examples are as follows. 3.3 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride, 2.99 g of HCOOH and 1 ml of water are mixed while cooling. 3.93 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess hydrochloric acid is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. This oil is dissolved in a mixture of 4 mL IPA, 20 mL ether and 2 mL hydrochloric acid is added dropwise. Concentrate, repeatedly dissolve in ethanol and concentrate again. Polishing with petroleum ether gives a yellow solid and recrystallizes with acetone to give the final target sibutramine hydrochloride. The reaction time of this step is a total of 18 hours, the yield is 80%. The target product is mp 195-197 ° C. The yield in 5 steps (A to E) is 18.9%.
As described above, the conventional sibutramine synthesis method has a total of five steps, which is complicated and takes a long time, and requires high temperature vacuum distillation (step A). Since the reaction proceeds at the high temperature of the raw material there was a problem that the yield is reduced. In fact, the synthesis was performed by applying the conventional sibutramine synthesis method, the total yield was very low as 18.9%.
First step
4-Chlorobenzyl cyanide is reacted with 1,3-dibromopropane to give 1- (4-chlorophenyl) -1-cyclobutyl cyanide.

In a flask at room temperature (20-35 ° C.), 14.1 g (352 mmol) of NaH dispersed in mineral oil (200%) and 200 ml of DMSO were added. 25 g (160 mmol) of 4-chlorobenzyl cyanide and 1,3- A solution of 36 g (176 mmol) of dibromopropane dissolved in 200 ml of DMSO was added dropwise. The mixture is stirred at room temperature for 2 hours, 10 ml of IPA is added dropwise and 200 ml of water is added dropwise. The mixture is filtered through a CELITE ™ filter and the solid residue is washed with ether. The ether layer is separated, washed with water, filtered, concentrated and dried to give 34.72 g of crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The yield of crude product is 109.8%.
2nd step
1- (4-chlorophenyl) -1-cyclobutyl cyanide isobutyl magnesium bromide is reacted to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine.

10 g (52 mmol) of 1- (4-chlorophenyl) -1-cyclobutyl cyanide was dissolved in 25 ml of toluene at room temperature, followed by addition of a 2.0 M solution of isobutyl magnesium bromide dissolved in 40 ml of diethyl ether. The mixture is heated at reflux at a temperature of at least 105 ° C. for 2 hours. After completion of the reaction at 0 ° C. with methanol, 2.4 g of NaBH 4 was added to the mixture at 0-25 ° C. and stirred for 1 hour or more. Concentrate, treat with ether, water, concentrate again, and vacuum dry. The yield of crude product is 91.6%.
3rd step
The amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine was dimethylated to obtain {1- [1- (4-chlorophenyl) -cyclobutyl]-which is the final object of the present invention. 3-methylbutyl} -dimethylamine hydrochloride, ie sibutramine hydrochloride, is obtained.

5.02 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine and 10 ml of HCOOH are mixed with cooling. 6 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess 2M HCl is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. After dissolving in a small amount of ether, ether saturated with HCl gas is slowly added dropwise at 0 ° C. The solid obtained was filtered and dried in vacuo to give 5.12 g of sibutramine hydrochloride as the final target. Yield of the product is 92%, mp 193.5-194.8 ° C. The total yield of the first to third stages is 52.7%. The H 1 NMR results of the final product are as follows: H 1 NMR (CDCl 3 ) 1.058 (6H, dd), 1.400 (2H, m), 1.508 (2H, m), 2.193 (3H, d), 2.316 (2H , m), 2.784 (2H, m), 2.910 (3H, d), 2.967 (1H, m), 3.568 (1H, m), 7.386 (2H, d), 7.638 (2H, d), 10.771 (1H, s)
The present invention is to shorten the process that was conventionally carried out in five steps to three steps to greatly shorten the process as well as to eliminate the difficult and time-consuming high-temperature vacuum distillation process to enable mass production In addition, it is possible to greatly reduce production time and production costs by improving the process step by step, and to reduce the production cost by showing a yield improvement effect nearly three times that of the conventional synthesis method in terms of overall yield.
Claims (3)
Hide Dependent
- In the method for synthesizing {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine from 4-chlorobenzyl cyanide,(a) reacting 4-chlorobenzyl cyanide with 1,3-dibromopropane to obtain 1- (4-chlorophenyl) -1-cyclobutyl cyanide;(b) To isobutyl magnesium bromide dissolved in diethyl ether is added to 1- (4-chlorophenyl) -1-cyclobutyl cyanide dissolved in toluene, and the mixture is refluxed at a temperature of 105 ° C. or higher at 1-3 ° C. Heating and cooling for a period of time, followed by addition of NaBH 4 at 0-25 ° C., followed by stirring for at least 1 hour to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine;(c) Dimethylating the amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine to yield {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methyl Improved synthesis method of sibutramine consisting of a three-step reaction comprising the step of obtaining butyl} -dimethylamine.
- The method according to claim 1,In step (a), the solution of 4-chlorobenzyl cyanide and 1,3-dibromopropane dissolved in DMSO is added dropwise to the mixture of NaH and DMSO dispersed in mineral oil, followed by filtration. , Washing, concentrating and drying to obtain a crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide, and proceeding to the next step (b) as it is without distillation at high temperature. Improved Synthesis of Sibutramine.
- The method according to claim 1,In step (c), 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine is mixed with formaldehyde in a free base state, and 37% aqueous formaldehyde is added thereto, and 85 An improved method for synthesizing sibutramine, characterized in that sibutramine hydrochloride is obtained by heating at -95 ° C for 15-22 hours followed by addition of hydrochloric acid.
SYN
DE 3212682; GB 2098602; US 4806570
4-Chlorobenzyl cyanide (I) is cycloalkylated with 1,3-dibromopropane to yield 1-(4-chlorophenyl)cyclobutyl cyanide (II). The cyclobutyl cyanide (II) is treated with 2-methylpropyl magnesium bromide te give the imine salt (III), which may be either hydrolyzed to the ketone (IV), which is then formylaminated with formamide and formic acid and subsequently hydrolyzed, or reduced with sodium borohydride in ethanol to give 1-[1-(4-chlorophenyl)cyclobutyl]-3-methylbutylamine (V). Eschweiler-Clarke methylation and hydrochloride formation yield N-[1-[1-(4-chlorophenyl)cyclo butyl]-3-methylbutyl]-N,N-dimethylamine hydrochloride monohydrate
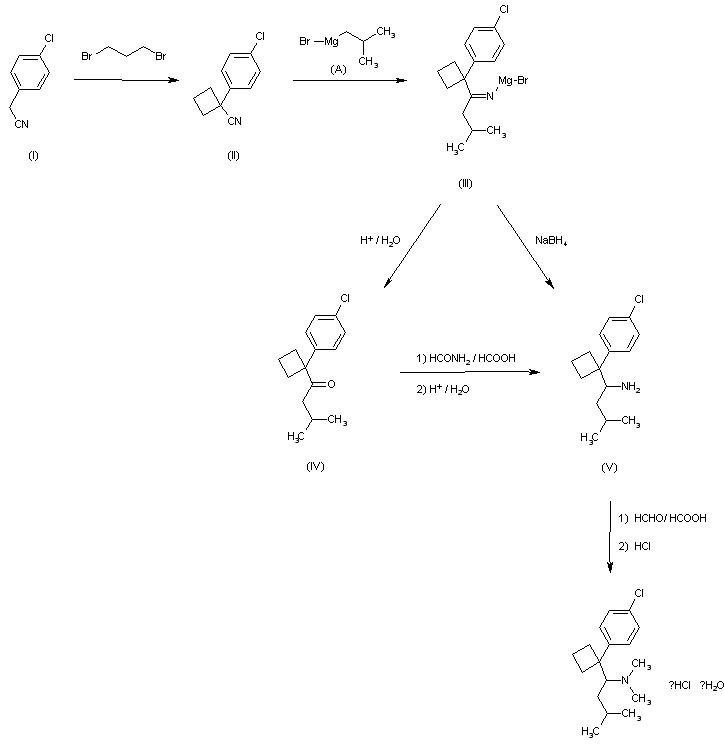
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Sibutramine has been used to produce appetite suppression for the purpose of attaining weight loss in the treatment of patients with obesity.
Contraindications
Sibutramine is contraindicated in patients with:
- Psychiatric conditions as bulimia nervosa, anorexia nervosa, serious depression or pre-existing mania
- Patients with a history of or a predisposition to drug or alcohol abuse
- Hypersensitivity to the drug or any of the inactive ingredients
- Patients below 18 and above 65 years of age[14]
- Concomitant treatment with a MAO inhibitor, antidepressant or other centrally active drugs, particularly other anoretics
- History of peripheral arterial disease
- Hypertension that is not sufficiently controlled (e.g., >145/90 mmHg), caution in controlled hypertension
- Existing pulmonary hypertension
- Existing damage on heart valves, coronary heart disease, congestive heart failure, serious arrhythmias, previous myocardial infarction
- A history of coronary artery disease (e.g., angina, history of myocardial infarction), congestive heart failure, tachycardia, peripheral arterial occlusive disease, arrhythmia or cerebrovascular disease (stroke or transient ischemic attack (TIA))[14]
- Stroke or transient ischemic attack (TIA)
- Hyperthyroidism (overactive thyroid gland)
- Closed angle glaucoma
- Seizure disorders
- Enlargement of the prostate gland with urinary retention (relative contraindication)
- Pheochromocytoma
- Pregnant and lactating women (relative contraindication)
Side effects
A higher number of cardiovascular events has been observed in people taking sibutramine versus control (11.4% vs. 10.0%).[15] In 2010 the FDA noted the concerns that sibutramine increases the risk of heart attacks and strokes in patients with a history of cardiovascular disease.[15]
Frequently encountered side effects are: dry mouth, paradoxically increased appetite, nausea, strange taste in the mouth, upset stomach, constipation, trouble sleeping, dizziness, drowsiness, menstrual cramps/pain, headache, flushing, or joint/muscle pain.
In a 2016 Cochrane review sibutramine was found to substantially increase blood pressure and heart rate in some patients, in the updated review in 2021 sibutramine was not included since the drug had been withdrawn from the market.[16] When used, regular blood pressure monitoring needed to be performed.
The following side effects are infrequent but serious and require immediate medical attention: cardiac arrhythmias, paresthesia, mental/mood changes (e.g., excitement, restlessness, confusion, depression, rare thoughts of suicide).
Symptoms that require urgent medical attention are seizures, problems urinating, abnormal bruising or bleeding, melena, hematemesis, jaundice, fever and rigors, chest pain, hemiplegia, abnormal vision, dyspnea and edema.
Currently, no case of pulmonary hypertension has been noted. (Fenfluramine, of the 1990s “Fen-Phen” combo, forced excess release of neurotransmitters—a different action. Phentermine was uninvolved in the rare—but clinically significant—heart issues of fenfluramine.)
Interactions
Sibutramine has a number of clinically significant interactions. The concomitant use of sibutramine and monoamine oxidase inhibitors (MAOIs, such as selegiline) is not indicated, as it may increase the risk of serotonin syndrome, a somewhat rare but serious adverse drug reaction.[17] Sibutramine should not be taken within two weeks of stopping or starting an MAOI. Taking both sibutramine and certain medications used in the treatment of migraines—such as ergolines and triptans—as well as opioids, may also increase the risk for serotonin syndrome, as may the use of more than one serotonin reuptake inhibitor at the same time.[17]
The concomitant use of sibutramine and drugs which inhibit CYP3A4, such as ketoconazole and erythromycin, may increase plasma levels of sibutramine.[18] Sibutramine does not affect the efficacy of hormonal contraception.[17]
Pharmacology
Pharmacodynamics
| Compound | SERT | NET | DAT |
|---|---|---|---|
| Sibutramine | 298–2,800 | 350–5,451 | 943–1,200 |
| Desmethylsibutramine | 15 | 20 | 49 |
| (R)-Desmethylsibutramine | 44 | 4 | 12 |
| (S)-Desmethylsibutramine | 9,200 | 870 | 180 |
| Didesmethylsibutramine | 20 | 15 | 45 |
| (R)-Didesmethylsibutramine | 140 | 13 | 8.9 |
| (S)-Didesmethylsibutramine | 4,300 | 62 | 12 |
| Values are Ki (nM). |
Sibutramine is a serotonin–norepinephrine reuptake inhibitor (SNRI) that, in humans, reduces the reuptake of norepinephrine (by ~73%), serotonin (by ~54%), and dopamine (by ~16%),[21] thereby increasing the levels of these substances in synaptic clefts and helping enhance satiety; the serotonergic action, in particular, is thought to influence appetite. Older anorectic agents such as amphetamine and fenfluramine force the release of these neurotransmitters rather than affecting their reuptake.[22]
Despite having a mechanism of action similar to tricyclic antidepressants, sibutramine has failed to demonstrate antidepressant properties in animal studies. It was approved by the U.S. Food and Drug Administration (FDA) in November 1997[23] for the treatment of obesity.
Sibutramine is reported to be a prodrug to two active metabolites, desmethylsibutramine (M1; BTS-54354) and didesmethylsibutramine (M2; BTS-54505), with much greater potency as MRIs.[24][25]
Unlike other serotonergic appetite suppressants like fenfluramine, sibutramine and its metabolites have only low and likely inconsequential affinity for the 5-HT2B receptor.[21]
Pharmacokinetics
Sibutramine is well absorbed from the gastrointestinal tract (77%), but undergoes considerable first-pass metabolism, reducing its bioavailability. The drug itself reaches its peak plasma level after 1 hour and has also a half-life of 1 hour. Sibutramine is metabolized by cytochrome P450 isozyme CYP3A4 into two pharmacologically-active primary and secondary amines (called active metabolites 1 and 2) with half-lives of 14 and 16 hours, respectively. Peak plasma concentrations of active metabolites 1 and 2 are reached after three to four hours. The following metabolic pathway mainly results in two inactive conjugated and hydroxylated metabolites (called metabolites 5 and 6). Metabolites 5 and 6 are mainly excreted in the urine.
Chemistry
Sibutramine has usually been used in the form of the hydrochloride monohydrate salt.
Detection in body fluids
Sibutramine and its two active N-demethylated metabolites may be measured in biofluids by liquid chromatography–mass spectrometry. Plasma levels of these three species are usually in the 1–10 μg/L range in persons undergoing therapy with the drug. The parent compound and norsibutramine are often not detectable in urine, but dinorsibutramine is generally present at concentrations of >200 μg/L.[26][27][28]
Society and culture
Regulatory approval
Studies are ongoing into reports of sudden death, heart failure, renal failure and gastrointestinal problems. Despite a 2002 petition by Ralph Nader-founded NGO Public Citizen,[29] the FDA made no attempts to withdraw the drug, but was part of a Senate hearing in 2005.[30] Similarly, David Graham, FDA “whistleblower”, testified before a Senate Finance Committee hearing that sibutramine may be more dangerous than the conditions it is used for.[31]
Between January 2003 and November 2005, a large randomized-controlled “Sibutramine Cardiovascular OUTcomes” (SCOUT) study with 10,742 patients examined whether or not sibutramine administered within a weight management program reduces the risk for cardiovascular complications in people at high risk for heart disease and concluded that use of silbutramine had a RR 1.16 for the primary outcome (composit of nonfatal MI, nonfatal CVA, cardiac arrest, and CV death).[32]
In a dissenting article, “Sibutramine: gone, but not forgotten”, David Haslam (chairman of the National Obesity Forum) says that the SCOUT study is flawed as it only covered high-risk patients and did not consider obese patients who do not have cardiovascular complications or similar contraindications [33]
On January 21, 2010, the European Medicines Agency recommended suspension of marketing authorizations for sibutramine based on the SCOUT study results.[34]
In August 2010 the FDA added a new contraindication for patients over 65 years of age due to the fact that clinical studies of sibutramine did not include sufficient numbers of such patients.[14]
Abbott Laboratories announced on October 8, 2010 that it is withdrawing sibutramine from the US market under pressure from the FDA, citing concerns over minimal efficacy coupled with increased risk of adverse cardiovascular events.[35]
Counterfeit weight-loss products
On December 22, 2008, the United States Food and Drug Administration issued an alert to consumers naming 27 different products marketed as “dietary supplements” for weight loss, that illegally contain undisclosed amounts of sibutramine.[36][37] In March 2009, Dieter Müller et al. published a study of sibutramine poisoning cases from similar Chinese “herbal supplements” sold in Europe, containing as much as twice the dosage of the legally licensed drug.[38]
An additional 34 products were recalled by the FDA on April 22, 2009, further underscoring the risks associated with unregulated “herbal supplements” to unsuspecting persons. This concern is especially relevant to those with underlying medical conditions incompatible with undeclared pharmaceutical adulterants.[39] In January 2010, a similar alert was issued for counterfeit versions of the over-the-counter weight loss drug Alli sold over the Internet. Instead of the active ingredient orlistat, the counterfeit drugs contain sibutramine, and at concentrations at least twice the amount recommended for weight loss.[40]
In March 2010 Health Canada advised the public that illegal “Herbal Diet Natural” had been found on the market, containing sibutramine, which is a prescription drug in Canada, without listing sibutramine as an ingredient.[41] In October 2010 FDA notified consumers that “Slimming Beauty Bitter Orange Slimming Capsules contain the active pharmaceutical ingredient sibutramine, a prescription-only drug which is a stimulant. Sibutramine is not listed on the product label.”[42]
In October 2010 the MHRA in the UK issued a warning regarding “Payouji tea” and “Pai You Guo Slim Capsules” which were found to contain undeclared quantities of sibutramine.[43]
On December 30, 2010 the FDA released a warning regarding “Fruta Planta” dietary products, which were found to contain undeclared amounts of sibutramine. The recall stated that “there is NO SAFE formula on the US market and that all versions of Fruta Planta contain sibutramine. All versions of the formula are UNSAFE and should not be purchased from any source.”[44]
Some illegal weight loss products imported into Ireland have been found to contain sibutramine.[45][46] Similar concerns have been raised in Australia, where illegal imported supplements have been found to contain sibutramine, resulting in public alerts from Australia’s Therapeutic Goods Administration.[47]
In October 2011, the FDA warned that 20 brands of dietary supplements were tainted with sibutramine.[48] In a 2018 study FDA has found synthetic additives including sibutramine in over 700 diet supplements marketed as “natural”, “traditional” or “herbal remedies”.[49]
References
- ^ “Sibutramine (Reductil) – withdrawal in Australia”. Therapeutic Goods Administration (Tga). Therapeutic Goods Administration, Department of Health, Australian Government. 2010. Retrieved 2014-10-06.
- ^ Health Canada Endorsed Important Safety Information on MERIDIA (Sibutramine Hydrochloride Monohydrate): Subject: Voluntary withdrawal of Meridia (sibutramine) capsules from the Canadian market.
- ^ “Notification of Termination of Production, Sale, and Usage of Sibutramine Preparations and Their Active Pharmaceutical Ingredient”. sda.gov in People’s Republic of China. October 30, 2010. Retrieved 2011-05-21.
- ^ (in German) Sibutramin-Vertrieb in der Europäischen Union ausgesetzt [1]. Abbott Laboratories in Germany. Press Release 2010-01-21. Retrieved 2010-01-27
- ^ “De-registration of pharmaceutical products containing sibutramine” (Press release). info.gov in Hong Kong. November 2, 2010. Retrieved 2010-11-08.
- ^ “Banned Medicines” (Press release). Ministry of Health and Family Welfare. February 10, 2011. Retrieved 2011-03-15.
- ^ “Withdrawal of Sibutramine (Reductil) in New Zealand” (Press release). MedSafe in New Zealand. October 11, 2010. Retrieved 2012-11-06.
- ^ “FDA warns online sellers of banned slimming pills”. January 12, 2014. Retrieved February 20, 2014.
- ^ “Thai FDA reveals voluntary withdrawal of sibutramine from the Thai market” (PDF) (Press release). Food and Drug Administration of Thailand. October 20, 2010. Retrieved 2010-12-22.
- ^ “Top obesity drug sibutramine being suspended”. BBC News. 2010-01-22. Retrieved 2010-01-22.
- ^ Rockoff JD, Dooren JC (October 8, 2010). “Abbott Pulls Diet Drug Meridia Off US Shelves”. The Wall Street Journal. Retrieved 8 October 2010.
- ^ “Sibutramine – Drugs.com”. drugs.com.
- ^ Buckett WR, Thomas PC, Luscombe GP (1988). “The pharmacology of sibutramine hydrochloride (BTS 54 524), a new antidepressant which induces rapid noradrenergic down-regulation”. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 12 (5): 575–84. doi:10.1016/0278-5846(88)90003-6. PMID 2851857. S2CID 24787523.
- ^ Jump up to:a b c “The FDA August 2010 drug safety update”. fda.gov.
- ^ Jump up to:a b “Early Communication about an Ongoing Safety Review of Meridia (sibutramine hydrochloride)”. United States Food and Drug Administration. 1 February 2010. Archived from the original on 6 January 2012.
- ^ Siebenhofer, Andrea; Winterholer, Sebastian; Jeitler, Klaus; Horvath, Karl; Berghold, Andrea; Krenn, Cornelia; Semlitsch, Thomas (2021-01-17). “Long-term effects of weight-reducing drugs in people with hypertension”. The Cochrane Database of Systematic Reviews. 1: CD007654. doi:10.1002/14651858.CD007654.pub5. ISSN 1469-493X. PMC 8094237. PMID 33454957.
- ^ Jump up to:a b c “Meridia Side Effects, and Drug Interactions”. RxList.com. 2007. Retrieved 2007-04-29.
- ^ (in Portuguese) Cloridrato de sibutramina monoidratado. Bula. [Sibutramine hydrochloride monohydrate—label information]. Medley (2007).
- ^ Nisoli E, Carruba MO (October 2000). “An assessment of the safety and efficacy of sibutramine, an anti-obesity drug with a novel mechanism of action”. Obesity Reviews. 1 (2): 127–39. doi:10.1046/j.1467-789x.2000.00020.x. PMID 12119986. S2CID 20553857.
- ^ Rothman RB, Baumann MH (May 2009). “Serotonergic drugs and valvular heart disease”. Expert Opinion on Drug Safety. 8 (3): 317–29. doi:10.1517/14740330902931524. PMC 2695569. PMID 19505264.
- ^ Jump up to:a b “Meridia (sibutramine hydrochloride monohydrate) Capsules CIV. Full Prescribing Information” (PDF). Abbott Laboratories, North Chicago, IL 60064, U.S.A. Retrieved 6 February 2016.
- ^ Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, Cheetham SC (August 1998). “Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine”. International Journal of Obesity and Related Metabolic Disorders. 22 Suppl 1: S18–28, discussion S29. PMID 9758240.
- ^ “FDA APPROVES SIBUTRAMINE TO TREAT OBESITY” (Press release). U.S. Food and Drug Administration. November 24, 1997. Retrieved 2007-04-29.
- ^ Kim KA, Song WK, Park JY (November 2009). “Association of CYP2B6, CYP3A5, and CYP2C19 genetic polymorphisms with sibutramine pharmacokinetics in healthy Korean subjects”. Clinical Pharmacology and Therapeutics. 86 (5): 511–8. doi:10.1038/clpt.2009.145. PMID 19693007. S2CID 24789264.
- ^ Hofbauer K (2004). Pharmacotherapy of obesity : options and alternatives. Boca Raton, Fla: CRC Press. ISBN 978-0-415-30321-7.
- ^ Jain DS, Subbaiah G, Sanyal M, et al. Liquid chromatography/electrospray ionization tandem mass spectrometry validated method for the simultaneous quantification of sibutramine and its primary and secondary amine metabolites in human plasma and its application to a bioequivalence study. Rapid Comm. Mass Spec. 20: 3509-3521, 2006.
- ^ Thevis M, Sigmund G, Schiffer AK, Schänzer W. Determination of N-desmethyl- and N-bisdesmethyl metabolites of Sibutramine in doping control analysis using liquid chromatography-tandem mass spectrometry. Eur. J. Mass Spec. 12: 129-136, 2006.
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1426–1427.
- ^ Wolfe SM, Sasich LD, Barbehenn E (March 19, 2002). “Petition to FDA to ban the diet drug sibutramine (MERIDIA) (HRG Publication #1613)”. Public Citizen. Retrieved 2007-04-29.
- ^ Japsen B (13 March 2005). “FDA weighs decision on Meridia; Health advisory likely for Abbott obesity drug”. Chicago Tribune. Chicago, Illinois. p. 1.
- ^ Hearing of 17 November 2004. Related CBS news item 19 November 2004.
- ^ James WP, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, Torp-Pedersen C, Sharma AM, Shepherd GM, Rode RA, Renz CL (September 2010). “Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects” (PDF). The New England Journal of Medicine. 363 (10): 905–17. doi:10.1056/NEJMoa1003114. hdl:2437/111825. PMID 20818901.
- ^ Haslam D (April 2010). “Sibutramine: gone, but not forgotten” (PDF). Pract Diab Int. 27 (3): 96–97. doi:10.1002/pdi.1453. Archived from the original (PDF) on 26 July 2015.
- ^ “European Medicines Agency recommends suspension of marketing authorisations for sibutramine” (PDF). European Medicines Agency. January 21, 2010. Archived from the original (PDF) on 2010-04-01.
- ^ Pollack A (October 8, 2010). “Abbott Labs Withdraws Meridia From Market”. The New York Times.
- ^ “FDA warns consumers about tainted weight loss pills” (Press release). U.S. Food and Drug Administration. 22 December 2008.
- ^ “Consumer directed questions and answers about FDA’s initiative against contaminated weight loss products”. U.S. Food and Drug Administration Center for Drug Evaluation and Research. 22 December 2008.
- ^ Müller D, Weinmann W, Hermanns-Clausen M (March 2009). “Chinese slimming capsules containing sibutramine sold over the Internet: a case series”. Deutsches Ärzteblatt International. 106 (13): 218–22. doi:10.3238/arztebl.2009.0218. PMC 2680571. PMID 19471631.
- ^ 34 weight loss products recalled, WebMD, 22 April 2009.
- ^ “Fake Alli diet pills can pose health risks”. CNN.com. January 23, 2010. Retrieved 2010-01-24.
- ^ “Herbal diet product poses heart risk”. CBC News. March 26, 2010.
- ^ “FDA Alert: Slimming Beauty Bitter Orange Slimming Capsules: Undeclared Drug Ingredient”. drugs.com.
- ^ “Press release: Warning over unlicensed herbal Payouji tea and Pai You Guo Slim Capsules”. United Kingdom Medicines & Healthcare Products Regulatory Agency. 20 October 2010. Archived from the original on 9 February 2012.
- ^ “PRock Marketing, LLC Issues a Voluntary Nationwide Recall of All weight loss formulas and variation of formulas of Reduce Weight Fruta Planta/Reduce Weight Dietary Supplement”. United States Food and Drug Administration. Archived from the original on 23 March 2012.
- ^ Pope C. “Seizures of illegal medicines rise”. The Irish Times.
- ^ “FDA Alert: Slim Xtreme Herbal Slimming Capsule: Undeclared Drug Ingredient”. drugs.com.
- ^ “Majestic slimming capsules: Safety advisory”. Therapeutic Goods Administration. Australian Government. 9 November 2012.
- ^ Carroll L (19 October 2011). “‘Natural’ diet pills tainted with banned prescription drug”. MSNBC. Archived from the original on 11 January 2012.
- ^ Cohen, Ronnie (12 October 2018). “No Wonder It Works So Well: There May Be Viagra In That Herbal Supplement”. NPR.org. Retrieved 2018-10-14.
External links
- Abbott Press Release on Meridia Withdrawal
- Sibutramine drug information from Merck Manual. Includes dosage information and a comprehensive list of international brand names
- U.S. Food and Drug Administration (FDA) product safety website
/////////////SIBUTRAMINE, UNII:WV5EC51866, WV5EC51866, сибутрамин , سيبوترامين , 西布曲明 , ABOTT, OBESITY

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN THE BLOG SUBSCRIPTION
$10.00
HM04 or H0900
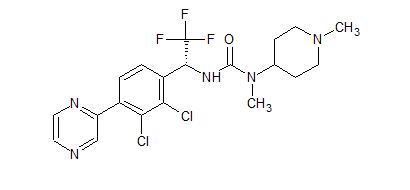
![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)
HM04 or H0900
Cas 1808913-24-7
(R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea
The compound was disclosed in WO2015134839 . Helsinn under license from Novo Nordisk , is investigating ghrelin antagonists for treating obesity, Prader-Willi syndrome and other metabolic disorders; in May 2015, the program was listed as being in preclinical development
Helps reducing ghrelin signaling activity and treating disorder associated with an increase in ghrelin level (eg food abuse, alcohol addiction, and Prader-Willi syndrome).
Ghrelin, a growth hormone-releasing peptide produced by ghrelinergic cells in the gastrointestinal tract, is understood to function as a neuropeptide that regulates energy metabolism by stimulating appetite. The modulation, for example inhibition, of ghrelin signaling, through the ghrelin/growth hormone secretagogue receptor (GHS-Rla), is an attractive target for pharmacological treatment of disorders associated with high ghrelin level. Potential disorders for treatment using ghrelin modulators include food abuse (such as binge eating, obesity, hyperphagia (or uncontrollable appetite), post-dieting body weight rebound (including post-dieting hyperphagia), alcohol addiction, and genetic diseases associated with increased ghrelin level (e.g., Prader-Willi syndrome (PWS)).
PATENT
US 20150252021

PATENT
WO2015134839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015134839
Example 1
nthesis of Intermediate lk
Intermediate k
Step 1:
To a solution of la (100 g, 0.62 mol) in DMF (1.2 L) was added N-bromosuccinimide (110 g, 0.62 mol) at 0 °C. The mixture was stirred at room temperature for 4 h, then water (800 mL) was added and the resulting mixture was extracted with EtOAc (3 x 500 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was triturated with petroleum ether to provide lb (133.7 g, 89% yield) as a brown solid. !H-NMR (CDC13, 300 MHz): δ= 7.30 (d, 1 H), 6.59 (d, 1 H), 4.22 (br, 2 H). LC-MS: 241 [M+l]+.
Step 2:
To a solution of lb (133.7 g, 0.55 mol) in dry CH2C12 (1.5 L) was added acetic anhydride (110 g, 0.62 mol) dropwise over a period of 20 minutes at room temperature. The mixture was stirred at room temperature overnight, then diluted with CH2C12 (300 mL) and washed with water (150 mL) and brine (200 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was triturated with petroleum ether (300 mL) to provide compound lc (143.0 g, 91% yield) as a white solid. ¾-NMR (CDC13, 400 MHz): δ= 8.26 (d, 1 H), 7.63 (br, 1 H), 7.54 (d, 1 H), 2.26 (s, 3 H). LC-MS: 280 [M-l]“.
Step 3:
A mixture of compound lc (50.0 g, 0.18 mol), butyl vinyl ether (Id, 89.0 g, 0.89 mol), bis(l,3-diphenylphosphino)propane (DPPP, 22.0 g, 0.053 mol), TEA (100 mL, 0.71 mol) and Pd(OAc)2 (6.4 g, 0.027 mol) in DMSO (1.2 L) was heated at 130 °C under N2 overnight. After the reaction was completed, the mixture was cooled to 0 °C and 2N HC1 (480 mL) was added dropwise over a period of 30 minutes. Then, the mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous a2S04 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide le (19.5 g, 45% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 8.46 (d, 1 H), 7.82 (br, 1 H), 7.51 (d, 1 H), 2.63 (s, 3 H), 2.29 (s, 3 H). LC-MS: 244 [M-l]“.
Step 4:
To a solution of le (21.9 g, 89.4 mmol) in MeOH (350 mL) was added 2N NaOH solution (350 mL) at room temperature. The mixture was heated at 50 °C overnight, then cooled and concentrated under reduced pressure. The resulting solid was triturated with water (100 mL) for 30 min and filtered to provide If (18.0 g, 98% yield) as a brown solid. ¾-NMR (CDC13, 400 MHz): 3= 7.48 (d, 1 H), 6.68 (d, 1 H), 4.56 (br, 2 H), 2.62 (s, 3 H). LC-MS: 202[M-1]\
Step 5:
To a mixture of compound If (18.0 g, 89.2 mmol) and ice (360 g) in cone. HC1 (180 mL) was added a solution of NaN02 (9.2 g, 133.7 mmol) in water (20 mL) dropwise over a period of 30 minutes, and the resulting mixture stirred in an ice bath for 30 min. A solution of KI (74.0 g, 446 mmol) in water (360 mL) was added dropwise over 45 min at 0 °C. The mixture was stirred for 30 min and then extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 :40) to provide lg (23.9 g, 86% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 7.6 (d, 1 H), 7.06 (d, 1 H), 2.62 (s, 3 H).
Step 6:
To a solution of lg (23.9 g, 76.1 mmol) in MeOH (100 mL)/THF (100 mL) was slowly added NaB¾ (2.9 g, 76.1 mmol) at 0 °C. The mixture was stirred at room temperature for 5 min, and then quenched with water (100 mL). The mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide lh (22.4 g, 93% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.26 (d, 1 H), 5.23 (q, 1 H), 2.17 (br, 1 H), 1.47 (d, 3 H).
Step 7:
To a mixture of lh (22.4 g, 70.9 mmol), phthalimide (12.5 g, 85.0 mmol) and PPh3 (22.3 g, 85.0 mmol) in dry THF (450 mL) was added DIAD (21.5 g, 106.3 mmol) at room temperature under N2 protection. The mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 15) to provide li (18.5 g, 58% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.78-7.84 (m, 3 H), 7.70-7.73 (m, 2 H), 7.41-7.43 (d, 1 H), 5.76-5.81 (q, 1 H), 1.84 (d, 3 H).
Step 8:
A solution of li (7.2 g, 16.2 mmol) and hydrazine hydrate (98%, 4.0 g, 80.9 mmol) in MeOH (150 mL) was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with CH2C12 (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure to give lj (3.8 g, 75% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.25 (d, 1 H), 4.55 (q, 1 H), 1.36-1.38 (d, 3 H). LC-MS: 316 [M+l]+.
Step 9:
To a solution of lj (41. Og, 0.13 mol) in methyl tert-butyl ether (750 mL) was added slowly a solution of D-mandelic acid (7.8 g, 0.052 mol) in methyl tert-butyl ether (1 10 mL) at 45°C. The mixture was stirred at this temperature for 30 min then cooled and filtered. White solid obtained was partitioned between 5% NaOH solution (300 mL) and methyl tert-butyl ether (300 mL). The bi -phases were separated and the aqueous phase was extracted with methyl tert-butyl ether (300 mL). The combined organic layer was concentrated to provide Intermediate lk (12 g, 58.5% yield) as a white solid (ee%=98.0%, Chiralpak AD-H, 5 μπι, 4.6*250mm, mobile phase: Hex: EtOH : DEA=80 : 20 : 0.2), retention time = 6.408 min).
Example 2
Synthesis of Compoun
A suspension of N-methyl-4-piperidone 2a (13.3 g, 58.6 mmol), NH2Me (30% in MeOH, 100 mL) and Pd/C (0.66 g) in MeOH (200 mL) was heated at 60 °C under H2 atmosphere (50 psi) overnight, then cooled and filtered. The filtrate was concentrated under reduced pressure and the residue was dissolved in HC1 in dioxane (3N, 100 mL) and stirred for 30 min. The precipitate was filtered and washed with EtOAc (50 mL) to provide 2b (7.7g, 54% yield) as white powder. 1H-NMR (DMSO, 400 MHz): δ= 9.50 (br, 2 H), 3.48 (d, 2 H), 3.15-3.16 (m, 1 H), 2.96-3.01 (m, 2 H), 2.70 (s, 3 H), 2.51 (s, 3 H), 2.22-2.28 (m, 2 H), 1.94-2.02 (m, 2 H), LC-MS: 129 [M+l]+ .
Example 20
Synthesis of H0900
Step 1:
To a mixture of 16d (32 g, 120 mmol) in dry CH2CI2 (800 mL) was added Dess-Martin peroxide reagent (76 g, 180 mmol) portion- wise at 0 °C. The mixture was stirred at room temperature for 1 h, then diluted with DCM (800 mL), washed with aqueous NaHC03 solution (300 mL) and brine (300 mL). The organic phase was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude 18a (31.4 g) which was used directly in the next step without further purification.
Step 2:
To a solution of 18a (12 g, 40 mmol) and 3b (22.2 g, 60 mmol) in DME (560 mL) were added Pd(PPh3)4 (9.25 g, 8 mmol) and Cul (1.52 g, 8 mmol) at room temperature. The mixture was stirred at 90 °C overnight, then concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE = 1 :5) to provide 18b (8.0 g, 79.3%) as a white solid. LC-MS: 253 [M+l]+.
Step 3:
To a solution of 18b (7 g, 27.7 mmol) and (¾)-tert-butylsulfinamide (7.27 g, 30.56 mmol) in dry THF (200 mL) was added Ti(i-OPr)4 (15.7 g, 55.4 mmol) dropwise at room temperature. The mixture was stirred at 80 °C overnight, and then cooled. Ethyl acetate (40 mL) was added, the resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA:PE =1 :5) to provide 18c (6.8 g, 69%) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 9.10 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.64 (d, 1H),8.12 (d, 1H), 7.59 (d, 1H), 1.30 (s, 9H).LC-MS: 356 [M+l]+.
Step 4:
To a stirred solution of 18c (6.8 g, 19 mmol) and Tetrabutylammonium difluorotnphenylsilicate (15.8 g, 29 mmol) in dry THF (250 mL) was added a solution of TMSCF3 (11 g, 77 mmol) in anhydrous THF (50 mL) at -65 °C. The mixture was then stirred at -65 °C for 2 h, and at that point aqueous NH4CI solution (250 mL) was added. The mixture was diluted with ethyl acetate (250 mL), washed with brine (250 mL), dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE=1 :2) to provide 18d (4.3 g, 52%) as a yellow solid. LC-MS: 426 [M+l]+.
Step 5:
To a stirred solution of 18d (4.3 g, 10.1 mmol) in MeOH (40 mL) was added a solution of HCl/MeOH (4N, 40 mL) at room temperature. The mixture was stirred for 1 h, then concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 mL) to afford crude 18e (4.3g) which was directly in the next step without further purification. LC-MS: 322 [M+l]+.
Step 6:
To a solution of 18e (2.7 g, 7.1 mmol), 2b (3.4 g, 21.3 mmol) and TEA (80 mL) in DCM (220 mL) was added thiphosgene (3.15 g, 10.6 mmol) in DCM (40 mL) dropwise at 0 °C. The solution was warmed to ambient temperature and stirred for 1 h, then diluted with DCM ( 100 mL) and washed with aqueous Na2C03 solution (100 mL) and brine (100 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated. The residue was purified with silica gel column chromatography (silica, DCM : CH3OH=10 : 1) to provide crude H0900 (2.13 g, ee%=92.5%) which was further purified through chiral separation to afford H0900 (1.6 g, 49% yield) as a white solid. (ee%=98.5%, Chiralpak IC 5um, 4.6*250mm, Phase: Hex: EtOH:
DEA=90: 10:0.2), retention tine =12.829 min. 1H-NMR (CDC13, 400 MHz): δ= 8.86 (d, 1H), 8.63 (dd, 1H), 8.55 (d, 1H), 7.47 (d, 1H), 7.40 (d, 1H), 6.28 (m, 1H), 5.18 (d, 1H), 4.12 (m, 1H), 2.88 (t, 2H), 2.77 (s, 3H), 2.22 (s, 3H), 2.05 (m, 2H), 2.48 (m, 2H), 1.52 (m, 2H), 1.73-1.49 (m, 4H). LC-MS: 476 [M+l]+.
PATENT
WO-2019118298
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118298&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline fumarate salt forms of (R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea (also referred to as HM04 or H0900; designated as Forms 1-4), process for their preparation and compositions comprising them are claimed.
PWS occurs in approximately 1 in 10,000 births and is associated with deletion or lack of expression of region 15ql 1.2 of the paternal chromosome 15.
Characteristics of PWS include short stature, low muscle tone, and hyperphagia. Growth hormone replacement is frequently used to treat growth deficiencies and hypotonia. However, treatment for the insatiable appetite is lacking and PWS children can mature into adults suffering from obesity and type 2 diabetes. Levels of ghrelin are generally elevated in PWS; however, the relationship with ghrelin signaling and food intake in PWS remains unclear. See Purtell L., et ah, In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-l levels. Neuropeptides. 201 l;45(4):30l-7; Cummings D.E., et ah, Elevated plasma ghrelin levels in Prader Willi syndrome. Nature Medicine . 2002;8(7):643-4; DelParigi A., et ah, High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(l2):546l-4.
[005] Accordingly, it is desirable to find treatments that effectively inhibit GHSRla, that are tolerable to the patient, and that do not interfere with other functions of the growth hormones. GHSRla modulators, including inhibitors such as (R)-3-(l-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2, 2, 2-trifluoroethyl)-l -methyl- l-(l-m ethylpiperidin-4-yl) urea (HM04, H0900) depicted below, are reported in LT.S. Patent No. 9,546,157.
Step 1 : Synthesis of compound 2A
[00106] 2,2,6,6-tetramethylpiperidine (7.20 kg, 51.1 mol, 3.0 eq.,
KF=0.30%) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer and mixed at RT under nitrogen protection. THF (50 L) was added into the reactor and stirred. The vessel was purged with nitrogen three times and cooled to 0 °C. n-BuLi (20.4 L, 3.0 eq.; 2.5 M hexane solution) was added to the mixture dropwise while keeping the temperature at about 0 °C to about 5 °C for over one hour. The color of the solution turned yellow. The mixture was stirred at about 0 °C to about 5 °C for 30 minutes. The mixture was cooled to about -78 °C to about -70 °C to form Solution A.
[00107] Compound 1 (3.25 kg, 17.0 mol. 1.0 eq., KF=0.03%) was dissolved in 15 L of THF to form Solution B.
[00108] Solution B was added to solution A dropwise at a temperature of about -70 °C to about -78 °C over one hour and then stirred for 30 minutes to form solution C. Tri-isopropyl borate ((i-PrO)3B) (3.52 kg, 18.7 mol., 1.1 eq.) was added dropwise into solution C over 10 minutes. The reaction mixture was stirred at a
temperature of about -70 °C to about -78 °C for one hour. HC1 (40 L, 3M, 7.0 eq.) was added over 30 minutes to quench the reaction. A 10 degree rise in temperature was noted.
[00109] The resulting aqueous layer was separated and extracted with EtOAc (40 L). The aqueous layer was separated and extracted twice again with EtOAc (35 L, 30 L). The organic layers were combined resulting in about 160 L of liquid. The combined organic layer was washed twice with 50 L of a 1M aqueous HC1 solution saturated with NaCl. The organic layer was concentrated to about 5 L in a 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 8 hours.
[00110] The residual EtOAc was swapped with DME for 3 times (10 L x 3). The organic layer was concentrated in the 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 6 hours. Each time about 5 L of residual remained. DME (20 L) was added to the residual to obtain a deep brown solution of 14.2% compound 2A (3.55 kg in 25 kg of solution; 88.8% yield; 97.4% purity (AETC by HPLC, retention time = 1.6 minutes); 0.24% residual ethyl acetate). 1H-NMR (400 MHz, DMSO): 5=8.55 (s, 2H), 7.36 (d, 1H), 7.69 (d, 1H). A second batch of compound 2A was prepared by the same method to produce 3.29 kg (95.4% purity, 82.3% yield, 0.11% residual ethyl acetate).
[00111] Step 2: Synthesis of Compound 3A
C! , N
M
K2CO3 (I .O equiv)
2A OH
DME/H20 3:1 (20 vol), 50 e C 3A N
[00112] Compound 2 A (2.91 kg in 20.5 kg solution) was added into a 100
L reactor at room temperature under nitrogen. DME (45 mL), 2-chloropyrazin (1.42 kg,
12.4 mol., 1.0 eq.), and Pd(dppf)Cl2 (10% w/w, 291 g) were added sequentially, and each
mixed at room temperature under nitrogen. Nitrogen was bubbled into the mixture for 20
minutes and the resulting mixture was purged and filled with nitrogen (3 times). The
mixture was heated to 48-52°C over 60 minutes. K2CO3 (2.57 kg, 18.6 mol, 1.5 eq.) was
added to 22 L of water in another reactor at room temperature and then added dropwise to
the compound 2 A mixture over 10 minutes. The mixture was stirred at 48-52°C for 16
hours and then cooled to room temperature. This procedure was repeated twice and all
three batches were combined.
[00113] An aqueous solution of K2CO3 (1.0 kg) was dissolved in 22 L of
water and added to the combined mixture to adjust the pH to 9. TBME (50 L) was added
into the mixture and filtered (PET filter, 3-5 pm, 205g/m2) to remove about 50 g of
sticky, brown solid material (catalyst analog). The aqueous layer was twice separated and
extracted with TBME (40 L, 40L).
[00114] The aqueous layer was combined with the aqueous layer of a
fourth batch prepared according to the above method. The pH of the combined aqueous layers was adjusted to pH<3 with HC1 (2N, 48 L). The solid precipitated out slowly as
the mixture was stirred at room temperature for 1 hour. The mixture was filtered (PET
filter, 3-5 pm, 205g/m2) over 30 minutes to obtain 20 kg of wet product. ACN (40 L) was
added into a 100 L reactor equipped with an overhead stirrer at room temperature. The 20
kg of wet product was added into the reactor and the reaction mixture heated to reflux
and stirred at reflux for 4 hours. The reaction mixture was cooled to room temperature
over 3 hours (around 15 °C/hour) and filtered to obtain 8.5 kg of wet solid. The wet solid
was dried under vacuum (20-30 mmHg) at 50-55 °C for 15 hours to obtain compound 3 A
as a pale white solid (6.1 kg; 97.4% purity (AUC by HPLC, retention time = 3.7
minutes); 83.8% yield). 1H-NMR (400 MHz, DMSO): 5=7.67 (d, 1H), 7.82 (d, 1H), 8.75
(d, 1H), 8.82 (t, 1H), 8.98 (d, 1H), 13.89 (bs, 1H).
[00115] Step 3: Synthesis of compound 6A
3A 6A N
N
[00116] Compound 3 A (6.1 kg, 22.7 mol, 1.0 eq.) was added into a 100 L
reactor equipped with a temperature probe, overhead stirrer, and condenser. Methanol
(92 L) was added into the reactor at room temperature. The mixture was cooled to
0-10 °C and added with SOCk (5.4 kg, 45.3 mol, 2.0 eq.) dropwise at 0-10 °C over 30
minutes. The reaction mixture was heated to reflux (65 °C) and stirred at reflux for 15
hours. A suspension was formed. Most of the solvent and SOCk was removed under
vacuum distillation until about 30 L remained. The mixture was concentrated under
vacuum (30-40 mmHg) at 50-55 °C for about 6 hours. Water (10 L) was added to the residual at -5 to 15 °C. The pH was adjusted to 8-9 with an aqueous solution of K2CO3 (200 g, dissolved in 2L of water) at -5 to 15 °C. The resulting aqueous layer was extracted twice with isopropyl acetate (25 L, 25 L). The combination of organic layers (about 50 kg) was washed with 20 L of NaHCCb aqueous layer. The organic layer was separated and washed with 10 L of of an aqueous solution of NaHCCb. All the aqueous layers were combined (55.8 kg). The organic layer was filtered through a silica pad (30 cm) and the pad washed with extra isopropyl acetate until the compound 6 A was filtered from the silica gel (about 3 hours). The organic layer was concentrated to about 5 L. THF (10 L) was added to the residual and concentrated to about 5 L (3 times) under vacuum (30-40 mmHg) at 50-55 °C for about 3 hours. Another 10 L of THF was added to the residual concentrate, giving a concentrated solution of compound 6A (15.8 kg; 32.83%,
5.19 kg compound 6A in solution; 97.9% purity (AUC by HPLC, retention time = 8.5 min); 80.8% yield). 1H-NMR (400 MHz, DMSO): 5=3.98 (s, 3H), 7.54 (d, 1H), 7.78 (d, 1H), 8.63 (d, 1H), 8.72 (t, 1H), 8.94 (d, 1H).
[00117] Step 4: Synthesis of compound 6B
[00118] THF (26 L) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer under nitrogen. DIBAL-H (26 kg, 46 mol, 5.0 eq.) was added and the system purged and filled with nitrogen three times. The mixture was cooled to -78 to -70 °C to form solution A. A room temperature solution of compound 6A (2.6 kg, 9.2 mol, 1.0 eq.) in 52 L of THF was added dropwise at -78 to -70 °C over 30 minutes under nitrogen. The mixture was warmed to -30 °C over about 5-6 hours. The reaction mixture was stirred at -40 to -30 °C for 30 minutes. The mixture was slowly added to 42 L of 2N HCL over 1 hour reaching a maximum temperature of 35 °C. The mixture was extracted with 26 L of isopropyl acetate. The organic layer was separated and washed with 30 L of brine. This procedure was repeated and both batches of organic layer were combined and concentrated from about 100 L to about 5-10 L under vacuum.
A solid slowly formed during concentration. The mixture was cooled to 5-15 °C and stirred for 1 hour. The mixture was filtered (30-50 pm) over 30 minutes. The solid was dried under vacuum at 50 °C for 6 hours to obtain compound 6B as a brown solid (2.1 kg; 97.5% purity (AUC by HPLC, retention time = 8.6 min); 45.7% yield). 1H-NMR (400 MHz, DMSO): d = 4.65 (d, 2H), 5.68 (t, 1H), 7.62 (d, 1H), 7.68 (d, 1H), 8.72 (d, 1H),
8.80 (t, 1H), 8.94 (d, 1H).
[00119] Step 5: Synthesis of compound 7
[00120] DMSO (10 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 6B (2.05 kg, 8.04 mol, 1.0 eq.) was added under nitrogen at room temperature. Et3N (8 L) was added under nitrogen at RT and the mixture was then cooled to 15-20 °C.
SCb. pyridine (5.1 kg, 32.08 mol, 4.0 eq.) was dissolved into 10 L of DMSO at 5-15 °C in a separate flask and added to the mixture dropwise over 3.5 hours at about 20 °C. The reaction mixture was transferred to 70 L of ice-water. The suspension mixture was stirred at 0-10 °C for 1 hour and filtered (PET, 3-5 pm, 205 g/m2) by centrifuge over 1.5 hours to obtain compound 7 as a brown solid. The solid was dissolved in 35 L of DCM at room temperature. The resulting DCM layer was washed with 5 L of brine. The organic layer was separated and concentrated under vacuum at 40-45 °C to dryness to obtain compound 7 as a brown solid (2.33 kg; 96.3% purity (AEiC by HPLC, retention time = 9.2 minutes); 93.5% yield). 1H-NMR (400 MHz, DMSO): d = 7.67 (d, 1H), 7.99 (d, 1H), 8.67 (d, 1H), 8.75 (s, 1H), 8.99 (d, 1H), 10.56 (s, 1H).
[00121] Step 6: Synthesis of compound 8
[00122] THF (23 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 7 (2.3 kg, 9.1 mol, 1.0 eq.) and (S)-2-methylpropane-2-sulfmamide (1.21 kg, 10 mol, 1.1 eq.) were added sequentially to the flask under nitrogen. Ti(OEt)4 (6.22 kg, 27.3 mol, 3.0 eq.) was added dropwise to the flask over 1 hour at 30-35 °C under nitrogen. The system was purged with nitrogen three times and then the mixture was stirred at room temperature for 2 hours. Isopropyl acetate (40 L) was added to the reaction mixture. The entire reaction mixture was then charged to 20 L of brine while stirring slowly at RT. A lot of solid was formed and no heat release was observed. The solid (about 18 kg) was filtered using centrifuge, and then the solid was slurried with 20 L of isopropyl acetate again for 20 minutes, and filtered again, resulting is slightly less solid (17.3 kg). The filtrates were then combined and washed with 20 L of brine. The organic layer was separated and concentrated in a rotavapor under vacuum (30-40 mmHg) at 40-50 °C for about 4 hours to remove the solvents and obtain a brown oil (compound 8). The oil was dissolved in DMF to obtain a black solution (7.36 kg; 40.1%; 3.0 kg compound 8 in solution; 92.1% purity (AUC by HPLC, retention time = 9.7 minutes); >100% yield). 1H-NMR (400 MHz, CDCb): d = 1.30 (s, 9H), 7.59 (d, 1H), 8.11 (d, 1H), 8.64 (s, 1H), 8.73 (m, 1H), 8.97 (s, 1H), 9.10 (s, 1H).
[00123] Step 7: Synthesis of compound 11
O
S
10 s C
8
11 N
[00124] DMF (26 L, 10 v/w) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at 15 °C. Compound 8 (7.3 kg of
DMF solution, containing 2.9 kg, 8.1 mol, 1.0 eq.) and TBAA (2.44 kg, 8.1 mol, 1.0 eq.) were added sequentially to the flask under nitrogen. The mixture was cooled to 0-10 °C.
TMSCF3 (2.88 kg, 20.3 mol, 2.5 eq.) was then added to the flask over 60 min at 0-10 °C.
The reaction mixture was stirred at 0-5 °C under nitrogen protection for 3 hours.
Isopropyl acetate (60 L) was added to the mixture, followed by the addition of 45 L of
NaHCCb under stirring at 5-25 °C. The organic layer was separated, washed three times with NaHC03 (30 L x 3), and concentrated from 60 kg to 2.5 kg of brown oil. The oil product was dissolved in 20 L of TBME and filtered through a pad of silica gel (about 40 cm high, 30 cm diameter) over 2 hours to obtain 2.14 kg of compound 1 1 in TBME solution. The solution was concentrated at 45-50 °C to dryness to obtain compound 1 1 as a black oil (1.85 kg; 85.2% purity (AETC by HPLC, retention time = 9.1 minutes, 9.6 minutes for diastereoisomer); 53.6% yield). 1H-NMR (400 MHz, CDCh): d = 1.33 (s, 9H), 3.82-3.85 (d, 1H), 5.61-5.66 (m, 1H), 7.53-7.60 (m, 2H), 8.63-8.64 (d, 1H), 8.71-8.72 (m, 1H), 8.95 (s, 1H).
[00125] Step 8: Synthesis of compound 12 (free base)
[00126] Compound 1 1 (1.8 kg, 4.23 mol, 1.0 eq., crude) was added to a 50 L reactor equipped with a temperature probe and overhead stirrer under nitrogen at 25 °C. Anhydrous MeOH (18 L) was added to dissolve compound 1 1. Then MeOH/HCl (18 L, 1 N) was added dropwise at 25-30 °C over 10 minutes and the mixture was stirred at 25-30 °C for 1 hour. Water (15 L) was added to the reaction and the mixture concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 4 hours to remove the solvent. The pH of the mixture was adjusted to 10 with 5 L of K2CO3 solution. 20 L of EtOAc was then added to the mixture and the organic layer was separated and the aqueous layer extracted twice with EtOAc (15 L x 2). The organic layers were combined and washed with 10 L of brine. The combined organic layers contained 996 g of
compound 12 in 40 kg of EtOAc solution (84% purity (AUC by HPLC, retention time =
2.8 minutes). The organic layers were concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to a 7.5 kg volume of compound 12 in EtOAc solution (83% purity (AETC by HPLC, retention time = 2.7 minutes).
[00127] In a separate 50 L reactor equipped with a temperature probe and overhead stirrer, D-CSA was added (930 g, 4.0 mol, 1.0 eq. to 1.26 kg compound 12) and stirred at room temperature under nitrogen. EtOAc (10 L) and then the EtOAc solution of compound 12 (1.26 kg, 3.9 mol, 1.0 eq.) were each sequentially added to the reactor. The mixture was stirred at room temperature for 1 hour and slowly became a suspension. The mixture was filtered by centrifuge and washed with EtOAc to produce 2.3 kg of compound 12 as an off-white solid (96.0% purity).
[00128] The solid product, 20 L of EtOAc, and 10 L of 10% aqueous K2CO3 were added sequentially to a 50 L flask and stirred at room temperature until no solid remained (pH = 9-10). The organic layer was separated and the aqueous layer extracted twice with EtOAc (10 L x 2). The organic layers were combined (about 32 kg) and washed with 10 L of brine. The organic layer contained 716 g of compound 12 in
31.8 kg of solution.
[00129] The organic layer was concentrated under vacuum at 45-50 °C to about 8 L. Activated carbon (200 g) was added to the organic layer and the mixture stirred at 60-70 °C for 1 hour, cooled to room temperature, and filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to remove the activated carbon. The mixture was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to yield 710 g of compound 12 as a yellow solid (99.4% purity). [00130] D-CSA (410 g, 1.77 mol, 1.0 eq. to 680 g compound 12), 3.4 L iPrOH, and 68 mL of water were added sequentially to a 10 L reactor equipped with a temperature probe and overhead stirrer and stirred at room temperature under nitrogen. The mixture was heated to reflux (84 °C) to form solution A after 1 hour. Compound 12 (680 g) was dissolved in 3.4 L of iPrOH and added into solution A for one partition. A clear solution was formed and the temperature decreased to 65 °C. The mixture was stirred at 65 °C for about 15 minutes after which a solid appeared. The mixture was cooled to 10 °C over 2 hours, stirred at 10 °C for an additional 30 minutes, and filtered through a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to collect the 1.1 kg of white solid.
[00131] EtOAc (10 L), 1.1 kg of white solid product, and 5 L of 10% K2CO3 were added sequentially to a 20 L flask and mixed for 5 minutes. The solid dissolved (pH = 9-10). The EtOAc layer was separated and the aqueous layer extracted twice with EtOAc (5 L each). The organic layers were combined (about 20 L), washed with 5 L of brine, and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-55 °C for about 3 hours to remove most of the solution and until the residue weight reached 1 kg. Heptanes (1 L) was added to the mixture and stirred at room temperature for 30 minutes. The mixture was filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to obtain 419 g of compound 12 base as a white solid (99.7% purity). The filtrate was concentrated to 135 g of compound 12 as a yellow solid (98.7% purity). 1H-NMR (400 MHz, CDCh): d = 1.85 (bs, 2H), 5.17 (m, 1H), 7.56 (d, 1H), 7.68 (d, 1H), 8.62 (d, 1H), 8.70-8.71 (m, 1H), 8.93 (s, 1H). Combined, the products resulted in a 40.7% yield of compound 12.
[00132] Step 9: Synthesis of compound 10
10A 10
[00133] Pd/C (40 g, 5% w/w) was added into a 10 L autoclave reactor at room temperature under nitrogen. THF (2 L), 2 L of methylamine (27%-30% alcoholic solution, 2.1 eq.), and 800 g of compound 10A (7 mol, 1.0 eq.) were sequentially added into the reactor. The system was purged with hydrogen three times. The mixture was stirred at hydrogen pressure (50 psi) at 70-75 °C overnight and was then filtered using a Biichner funnel and filter paper (pore size: 30-50 pm) over 10 minutes to remove the Pd/C. The filtrate was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to obtain 933 g of yellow oil. The mixture was distilled without a column at atmospheric pressure and the 140-170 °C portion was collected to obtain 763 g of compound 10 as a colorless oil (98.6% purity (AUC by HPLC, retention time = 4.8 minutes); 84.2% yield; 8000 ppm residual ethanol). A portion of the oil (563 g) was distilled using a 3 cm column at atmospheric pressure and the 140-170 °C portion was collected to obtain 510 g of compound 10 (75.8% yield; 134 ppm residual ethanol). 1H-NMR (400 MHz, CDCb): d = 0.82 (bs, 1H), 1.10-1.12 (q, 2H), 1.66 (d, 2H), 1.73-1.81 (t, 2H), 2.05 (s, 3H), 2.08-2.19 (m, 1H), 2.22 (s, 3H), 2.60 (d, 2H).
[00134] Step 10: Synthesis of HM04 fumarate salt
[00135] DCM (1L), 200 g CDI (1.23 mol, 2.0 eq.), and 35 g DABCO (0.31 mol, 0.5 eq.) were sequentially added into a 3 L reactor equipped with a temperature probe and overhead stirrer, and stirred at room temperature under nitrogen. The mixture was cooled to -10 to -5 °C. Compound 12 (200 g) was dissolved in 1 L of DCM and added into the mixture dropwise over 1 hour, followed by stirring for 16 hours at -10 to -5 °C. Compound 10 (159 g, 1.24 mol, 2.0 eq.) was added at -10 to 0 °C over 10 minutes. The mixture was then warmed to 0 to 5 °C and held for 2 hours. The mixture was concentrated under vacuum at 40-45 °C to about 1 L. HC1 (1 L of 1 N) was added to the residual and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to remove the DCM. Another 3 L of 1N HC1 was added to the residual and extracted three times with TBME (4 L, 2 L, 2 L). The aqueous layer was slowly adjusted to pH = 9-10 with 20% aqueous K2CO3 (about 1.5 L) and extracted with DCM (2 L x 3). The organic layers were combined (about 4 L) and washed three times with 0.25 N KH2PO4 (1.2 L x 3). The organic layer was washed with 2 L of brine to bring the pH to neutral and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to 450 g (335 mL). MTBE (1.5 L) was added to the residual and distilled until 500 mL of liquid was collected. This step was repeated four times with the addition of 500 mL of TBME and collection of 500 mL of distillate, with the exception that 330 mL of liquid was collected at the final distillation. About 1 to 1.2 L of residual remained in the flask. The residual was slowly cooled to room temperature and stirred at room temperature overnight. The mixture was filtered, washed twice with TBME (400 mL x 2), and dried to obtain 192 g of HM04 free base a light yellow solid (99.3% purity (AUC by HPLC, retention time = 11.0 minutes). The product on the wall was dissolved in DCM and concentrated under vacuum to obtain 22 g of HM04 free base as a brown sticky oil (97.6% purity). The filtrate was concentrated under vacuum to obtain 22.5 g of yellow solid (94.0% purity).
[00136] HM04 free base (187 g, 0.39 mol, 1.0 eq., 99.3% purity) and 1.9 L of ACN were sequentially added to a 3 L flask equipped with a temperature probe and overhead stirrer and stirred at 15 °C under nitrogen to obtain a light-yellow suspension. Fumaric acid (45.6 g, 0.39 mol, 1.0 eq.) was added to the flask and generated a white suspension after 1 minute. The reaction suspension was stirred overnight at room temperature, filtered (15-20 pm, ash<0.l5), washed twice with ACN (50 mL x 2), and dried under vacuum at 50 °C for 6 hours to obtain 207 g of HM04 fumarate salt as a light yellow solid (99.4% purity (AUC by HPLC, retention time = 11.1 minutes); 57.8% yield; 3100 ppm residual ACN). The filtrate was concentrated under vacuum to obtain 20.1 g of HM04 fumarate salt as a light yellow solid (97.3% purity).
[00137] A portion of the product (117 g) was further dried in a vacuum oven (20-40 mmHg) to lower the residual acetonitrile content. After drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours, the residual acetonitrile content was measured as 3100 ppm, 2570 ppm, 1300 ppm, and 256 ppm, respectively. After the drying process, 98 g of HM04 fumarate salt was isolated (99.4% purity (AUC by HPLC, retention time = 11.0 minutes); 1H-NMR (400 MHz, DMSO): d = 1.49-1.58 (m, 2H),
1.81-1.92 (m, 2H), 2.44-2.53 (m, 5H), 2.78 (s, 3H), 3.12 (m, 2H), 4.06-4.13 (m, 1H), 6.36-6.41 (m, 1H), 6.55 (s, 2H), 7.47 (d, 1H), 7.73 (d, 1H), 8.11 (d, 1H), 8.75 (d, 1H),
8.81-8.82 (m, 1H), 8.99 (d, 1H). The yield of 98g of HM04 fumarate salt isolated after drying the partial batch was extrapolated over the whole batch to calculate an
approximate yield of 48% for step 10.
[00138] XRPD analysis of HM04 fumarate salt products obtained after drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours was performed (see Figures 6-9, respectively). The XRPD profile showed that the HM04 fumarate salt product was consistent with Form 1.
Example 6. Streamlined Synthesis of HM04 Fumarate Salt Form 1
[00139] The overall yield of HM04 fumarate salt produced using Step 10 of Example 5 was calculated as approximately 48%. In order to increase the overall yield, a streamlined synthesis was investigated that eliminated the step of isolating HM04 free base. In particular, step 10 of the method of Example 5 shown in Figure 5 was changed. An overview of the streamlined synthesis beginning after step 9 of Example 5 is shown in Figure 10.
[00140] Streamlined HM04 Fumarate Salt Trial 1 : PCM (121.4 g). CPI (20.0 g, 123 mmol, 2 eq.) and DABCO (3.5 g, 31 mmol) were sequentially added into an inertized 1 L reactor. The mixture was cooled to -10 °C. Separately, a solution of DCM (132.5 g) and compound 12 (20.0 g, 62.1 mmol) were charged into a vessel and stirred until a solution was obtained. This solution was dropped into the 1 L reactor over 33 minutes by keeping the internal temperature at -10 to -5 °C. At the end of the addition, the vessel was rinsed with DCM (7.0 g), which was then added to the reaction mixture.
After stirring overnight (19 hours) and positive IPC, compound 10 (15.9 g, 124 mmol, 2 eq.) was added over 15 minutes and the vessel rinsed with DCM (9.0 g). After heating at 0 °C, 1 hour of stirring, positive IPC, and a further 1.5 hours of stirring, the mixture was heated at room temperature and charged with water (200.1 g). The aqueous layer was separated and the organic layer extracted twice with 1 N HC1 (201, 200 g). The combined aqueous layers containing the product were washed with TBME (148 g). After removal of the organic layer, the aqueous layer was charged with DCM (265.0 g) and 50% K2CO3 solution (about 240 ml) until reaching pH 9.61.
[00141] Meanwhile, a solution of KH2PO4 (8.2 g) in water (240 g) was prepared. The organic layer containing the product was charged with the KH2PO4 solution until reaching pH 7.12 (142.2 g). After separation of the aqueous layer, the organic layer was washed with water (200 g). After separation of the aqueous layer, the organic layer was evaporated at 50 °C. ACN (314.4 g) was added and the solvent distilled again at 70-75 °C under vacuum. ACN (235.8 g) was added and the solvent distilled again under vacuum. ACN (141.5 g) was added, the resulting solution polish filtered and the filter washed with ACN (16 g). After heating at 60 °C, fumaric acid (7.2 g, 62 mmol) was added to the solution, causing a white precipitate. After cooling to 20 °C over 1 hour, the suspension was filtered and washed twice with TBME (2 x 30 g). After drying on the filter with nitrogen flow, 70.7 g of wet raw product was obtained. This was slurried with TBME (177.0 g) for 1 hour, filtered, and washed with TBME (70 g). After drying on the filter under nitrogen flow, 33.0 g of wet product was obtained. Heating at 50 °C under vacuum afforded the dry product as a white powder of HM04 fumarate salt (21.1 g;
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9926337 | SUBSTITUTED ASYMMETRIC UREAS AND MEDICAL USES THEREOF | 2016-12-02 | |
| US9546157 | p-Substituted Asymmetric Ureas and Medical Uses Thereof | 2015-03-06 | 2015-09-10 |
////////////HM04, H0900, Helsinn, Novo Nordisk, PRECLINICAL, obesity, Prader-Willi syndrome, ghrelin
CN(C1CCN(C)CC1)C(=O)N[C@H](c3ccc(c2cnccn2)c(Cl)c3Cl)C(F)(F)F
ACLIMOSTAT
Aclimostat
CAS: 2082752-83-6
Chemical Formula: C26H42N2O6
Molecular Weight: 478.63
Elemental Analysis: C, 65.25; H, 8.85; N, 5.85; O, 20.06
ZGN-1061; ZGN1061; ZGN 1061; Aclimostat,
UNII-X150A3JK8R
X150A3JK8R
(3R,4S,5S,6R)-5-Methoxy-4-[(2R,3R)-2-methyl-3-(3- methylbut-2-en-1-yl)oxiran-2-yl]-1-oxaspiro[2.5]octan-6-yl 3-[2-(morpholin-4-yl)ethyl]azetidine-1-carboxylate
1-Azetidinecarboxylic acid, 3-[2-(4-morpholinyl)ethyl]-, (3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-buten-1-yl)-2-oxiranyl]-1-oxaspiro[2.5]oct-6-yl ester
3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1- oxaspiro[2.5]octan-6-yl 3-(2-morpholinoethyl)azetidine-1-carboxylate
ZAFGEN, PHASE 2, DIABETES
Aclimostat, also known as ZGN-1061, is an anti-diabetic, anti-obesity MetAP2 inhibitor.
Over 1.1 billion people worldwide are reported to be overweight. Obesity is estimated to affect over 90 million people in the United States alone. Twenty-five percent of the population in the United States over the age of twenty is considered clinically obese. While being overweight or obese presents problems (for example restriction of mobility, discomfort in tight spaces such as theater or airplane seats, social difficulties, etc.), these conditions, in particular clinical obesity, affect other aspects of health, i.e., diseases and other adverse health conditions associated with, exacerbated by, or precipitated by being overweight or obese. The estimated mortality from obesity-related conditions in the United States is over 300,000 annually (O’Brien et al. Amer J Surgery (2002) 184:4S-8S; and Hill et al. (1998) Science, 280:1371). [0003] There is no curative treatment for being overweight or obese. Traditional pharmacotherapies for treating an overweight or obese subject, such as serotonin and noradrenergic re-uptake inhibitors, noradrenergic re-uptake inhibitors, selective serotonin re- uptake inhibitors, intestinal lipase inhibitors, or surgeries such as stomach stapling or gastric banding, have been shown to provide minimal short-term benefits or significant rates of relapse, and have further shown harmful side-effects to patients. [0004] MetAP2 encodes a protein that functions at least in part by enzymatically removing the amino terminal methionine residue from certain newly translated proteins such as glyceraldehyde-3-phosphate dehydrogenase (Warder et al. (2008) J. Proteome Res.7:4807). Increased expression of the MetAP2 gene has been historically associated with various forms of cancer. Molecules inhibiting the enzymatic activity of MetAP2 have been identified and have been explored for their utility in the treatment of various tumor types (Wang et al. (2003) Cancer Res.63:7861) and infectious diseases such as microsporidiosis, leishmaniasis, and malaria (Zhang et al. (2002) J. Biomed. Sci.9:34). Notably, inhibition of MetAP2 activity in obese and obese-diabetic animals leads to a reduction in body weight in part by increasing the oxidation of fat and in part by reducing the consumption of food (Rupnick et al. (2002) Proc. Natl. Acad. Sci. USA 99:10730).
[0005] Such MetAP2 inhibitors may be useful as well for patients with excess adiposity and conditions related to adiposity including type 2 diabetes, hepatic steatosis, and
cardiovascular disease (via e.g. ameliorating insulin resistance, reducing hepatic lipid content, and reducing cardiac workload). Accordingly, compounds capable of modulating MetAP2 are needed to address the treatment of obesity and related diseases as well as other ailments favorably responsive to MetAP2 modulator treatment.
Synthesis

CONTD……………….

contd………………….

Tetrahedron, 73(30), 4371-4379; 2017

WO 2017027684
PATENT
WO 2017027684
https://patents.google.com/patent/WO2017027684A1/en
Example 1
(3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1- oxaspiro[2.5]octan-6-yl 3-(2-morpholinoethyl)azetidine-1-carboxylate
[00312] To a mixture of 4-(2-(azetidin-3-yl)ethyl)morpholine, trifluoroacetate (2.33 g, 3.7 mmol) in CH3CN (150 mL) was added DIPEA (2.9 mL, 17 mmol) drop-wise at 0-5oC. The mixture was then stirred at 0-5oC for 10 min, and carbonate Intermediate 1 (1.3 g, 2.9 mmol) was added to the mixture in portions at 0oC under a N2atmosphere. The reaction mixture was stirred at 25oC for 16 hrs. TLC (PE : EtOAc = 3 : 1) showed that the reaction was complete. The solvent was removed under vacuum below 40oC. The residue was diluted with DCM (60 mL), and the DCM solution was washed with ammonium acetate buffer (pH~4, 15 mL x 2). The combined aqueous layers were back-extracted with DCM (20 mL x 2). The combined organic layers were washed with aq. NaHCO3 solution (15 mL x 2, 5% wt), dried over Na2SO4 and concentrated. Purification by silica gel column chromatography (DCM: MeOH=100: 0~60: 1), followed by preparative HPLC (Method A, H2O (0.1% FA) / CH3CN) gave the title compound (1.15 g) as a light yellow syrup. LC-MS: m/z = 479 [M+H]+; 1H-NMR (400 MHz, CDCl3) δ 5.43 (br, 1H), 5.13 (t, J = 7.6 Hz, 1H), 3.87-4.15 (m, 2H), 3.63-3.65 (m, 4H), 3.52- 3.56 (m, 3H), 3.49 (s, 3H), 2.90 (d, J = 4.4 Hz, 1H), 2.46-2.54 (m, 3H), 2.19-2.36 (m, 7H), 1.97-2.13 (m, 2H), 1.78-1.89 (m, 5H), 1.73 (s, 3H), 1.62 (s, 3H), 1.13 (s, 3H), 0.99 (d, J = 13.6 Hz, 1H).
REFERENCES
1: Malloy J, Zhuang D, Kim T, Inskeep P, Kim D, Taylor K. Single and multiple dose evaluation of a novel MetAP2 inhibitor: Results of a randomized, double-blind, placebo-controlled clinical trial. Diabetes Obes Metab. 2018 Aug;20(8):1878-1884. doi: 10.1111/dom.13305. Epub 2018 Apr 23. PubMed PMID: 29577550; PubMed Central PMCID: PMC6055687.
2: Burkey BF, Hoglen NC, Inskeep P, Wyman M, Hughes TE, Vath JE. Preclinical Efficacy and Safety of the Novel Antidiabetic, Antiobesity MetAP2 Inhibitor ZGN-1061. J Pharmacol Exp Ther. 2018 May;365(2):301-313. doi: 10.1124/jpet.117.246272. Epub 2018 Feb 28. PubMed PMID: 29491038.
//////////////Aclimostat, ZGN-1061, ZAFGEN, PHASE 2, DIABETES
O=C(N1CC(CCN2CCOCC2)C1)O[C@H](CC3)[C@@H](OC)[C@H]([C@@]4(C)O[C@@H]4C/C=C(C)\C)[C@]53CO5
SRT 1720

SRT-1720 diHCl
CAY10559
CAS: 1001645-58-4 (di HCl) , 925434-55-5 (free base) 1001645-58-4 (HCl)
Chemical Formula: C25H25Cl2N7OS
Molecular Weight: 542.483
Elemental Analysis: C, 55.35; H, 4.65; Cl, 13.07; N, 18.07; O, 2.95; S, 5.91
SRT-1720 HCl, SRT-1720 hudrochloride; SRT1720; SRT-1720; SRT 1720; CAY10559; CAY-10559; CAY 10559; SIRT-1933; SIRT 1933; SIRT1933.
N-(2-(3-(piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide dihydrochloride

- Molecular FormulaC25H23N7OS
- Average mass469.561 Da
SRT-1720, also known as CAY10559 and is a drug developed by Sirtris Pharmaceuticals intended as a small-molecule activator of the sirtuin subtype SIRT1. It has similar activity in the body to the known SIRT1 activator resveratrol, but is 1000x more potent. In animal studies it was found to improve insulin sensitivity and lower plasma glucose levels in fat, muscle and liver tissue, and increased mitochondrial and metabolic function. A study of SRT1720 conducted by the National Institute on Aging found that the drug may extend the lifespan of obese mice by 44% .
SRT1720 is an experimental drug that was studied by Sirtris Pharmaceuticals intended as a small-molecule activator of the sirtuinsubtype SIRT1. The compound has been studied in animals, but safety and efficacy in humans have not been established.
Animal research
In animal models of obesity and diabetes SRT1720 was found to improve insulin sensitivity and lower plasma glucose levels in fat, muscle and liver tissue, and increase mitochondrial and metabolic function.[1] In mice rendered obese and diabetic by feeding a high-fat, high-sugar diet, a study performed at the National Institute of Aging found that feeding chow infused with the highest dose of SRT1720 beginning at one year of age increased mean lifespan by 18%, and maximum lifespan by 5%, as compared to other short-lived obese, diabetic mice; however, treated animals still lived substantially shorter lives than normal-weight mice fed normal chow with no drug.[2] In a later study, SRT1720 increased mean lifespan of obese, diabetic mice by 21.7%, similar to the earlier study, but there was no effect on maximum lifespan in this study.[3] In normal-weight mice fed a standard rodent diet, SRT1720 increased mean lifespan by just 8.8%, and again had no effect on maximum lifespan.[3]
Since the discovery of SRT1720, the claim that this compound is a SIRT1 activator has been questioned[4][5][6] and further defended.[7][8]
Although SRT1720 is not currently undergoing clinical development, a related compound, SRT2104, is currently in clinical development for metabolic diseases.[9]
PAPER
Letters in Drug Design & Discovery, 10(9), 793-797; 2013
The Identification of the SIRT1 Activator SRT2104 as a Clinical Candidate
Author(s): Pui Yee Ng, Jean E. Bemis, Jeremy S. Disch, Chi B. Vu, Christopher J. Oalmann, Amy V. Lynch,David P. Carney, Thomas V. Riera, Jeffrey Song, Jesse J. Smith, Siva Lavu, Angela Tornblom, Meghan Duncan, Marie Yeager, Kristina Kriksciukaite, Akanksha Gupta, Vipin Suri, Peter J. Elliot, Jill C. Milne, Joseph J. Nunes, Michael R. Jirousek, George P. Vlasuk, James L. Ellis, Robert B. Perni.
Journal Name: Letters in Drug Design & Discovery
Volume 10 , Issue 9 , 2013

Paper
Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; Bemis, J.E.; Xie, R.; Disch, J.S.; Ng, P.Y.; Nunes, J.J.; Lynch, A.V.; Yang, H.; Galonek, H.; Israelian, K.; Choy, W.; Iffland, A.; Lavu, S.; Medvedik, O.; Sinclair, D.A.; Olefsky, J.M.; Jirousek, M.R.; Elliott, P.J.; Westphal, C.H.
Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes
Nature 2007, 450(7170): 712
PATENT
WO 2007019417
WO 2007019416
WO 2007019345
WO 2007019344
WO 2007019346
WO 2008115518
PAPER
Vu, Chi B.; Journal of Medicinal Chemistry 2009, VOL 52(5), PG 1275-1283
https://pubs.acs.org/doi/abs/10.1021/jm8012954

A series of imidazo[1,2-b]thiazole derivatives is shown to activate the NAD+-dependent deacetylase SIRT1, a potential new therapeutic target to treat various metabolic disorders. This series of compounds was derived from a high throughput screening hit bearing an oxazolopyridine core. Water-solubilizing groups could be installed conveniently at either the C-2 or C-3 position of the imidazo[1,2-b]thiazole ring. The SIRT1 enzyme activity could be adjusted by modifying the amide portion of these imidazo[1,2-b]thiazole derivatives. The most potent analogue within this series, namely, compound 29, has demonstrated oral antidiabetic activity in the ob/ob mouse model, the diet-induced obesity (DIO) mouse model, and the Zucker fa/fa rat model.
Discovery of Imidazo[1,2-b]thiazole Derivatives as Novel SIRT1 Activators
* To whom correspondence should be addressed. Phone: (617)-252-6920, extension 2129. Fax: (617)-252-6924. E-mail: cvu@sirtrispharma.com., †
Present address: Department of Medicine, Division of Endocrinology and Metabolism, University of California—San Diego, 9500 Gilman Drive, La Jolla, CA 92093.
Preparation of N-(2-(3-(Piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide (29)
References
- ^ Milne JC; Lambert PD; Schenk S; Carney DP; Smith JJ; Gagne DJ; Jin L; Boss O; Perni RB; Vu CB; Bemis JE; Xie R; Disch JS; Ng PY; Nunes JJ; Lynch AV; Yang H; Galonek H; Israelian K; Choy W; Iffland A; Lavu S; Medvedik O; Sinclair DA; Olefsky JM; Jirousek MR; Elliott PJ; Westphal CH (November 2007). “Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes”. Nature. 450(7170): 712–6. doi:10.1038/nature06261. PMC 2753457. PMID 18046409.
- ^ Minor RK; Baur JA; Gomes AP; Ward TM; Csiszar A; Mercken EM; Abdelmohsen K; Shin YK; Canto C; Scheibye-Knudsen M; Krawczyk M; Irusta PM; Martín-Montalvo A; Hubbard BP; Zhang Y; Lehrmann E; White AA; Price NL; Swindell WR; Pearson KJ; Becker KG; Bohr VA; Gorospe M; Egan JM; Talan MI; Auwerx J; Westphal CH; Ellis JL; Ungvari Z; Vlasuk GP; Elliott PJ; Sinclair DA; de Cabo R (Aug 2011). “SRT1720 improves survival and healthspan of obese mice”. Scientific Reports. 1 (70): 70. doi:10.1038/srep00070. PMC 3216557. PMID 22355589. Retrieved 1 March 2014.
- ^ Jump up to:a b Mitchell SJ; Martin-Montalvo A; Mercken EM; et al. (Feb 2014). “The SIRT1 Activator SRT1720 Extends Lifespan and Improves Health of Mice Fed a Standard Diet”. Cell Reports. 6 (4): 836–43. doi:10.1016/j.celrep.2014.01.031. PMC 4010117. PMID 24582957. Retrieved 1 March 2014.
- ^ Pacholec M; Chrunyk BA; Cunningham D; Flynn D; Griffith DA; Griffor M; Loulakis P; Pabst B; Qiu X; Stockman B; Thanabal V; Varghese A; Ward J; Withka J; Ahn K (January 2010). “SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1”. J Biol Chem. 285 (11): 8340–8351. doi:10.1074/jbc.M109.088682. PMC 2832984. PMID 20061378.
- ^ Beher D; Wu J; Cumine S; Kim KW; Lu SC; Atangan L; Wang M (December 2009). “Resveratrol is not a direct activator of SIRT1 enzyme activity”. Chem Biol Drug Des. 74 (6): 619–24. doi:10.1111/j.1747-0285.2009.00901.x. PMID 19843076.
- ^ Zarse, K.; Schmeisser, S.; Birringer, M.; Falk, E.; Schmoll, D.; Ristow, M. (2010). “Differential Effects of Resveratrol and SRT1720 on Lifespan of AdultCaenorhabditis elegans”. Hormone and Metabolic Research. 42 (12): 837–839. doi:10.1055/s-0030-1265225. PMID 20925017.
- ^ Callaway E (2010-08-16). “GlaxoSmithKline strikes back over anti-ageing pills: Drugs do work as thought, says pharmaceutical giant”. Nature. doi:10.1038/news.2010.412.
- ^ Dai H; Kustigian L; Carney D; Case A; Considine T; Hubbard BP; Perni RB; Riera TV; Szczepankiewicz B; Vlasuk GP; Stein RL (August 2010). “SIRT1 activation by small molecules – kinetic and biophysical evidence for direct interaction of enzyme and activator”. J Biol Chem. 285 (43): 32695–32703. doi:10.1074/jbc.M110.133892. PMC 2963390. PMID 20702418.
- ^ “Sirtuin Pipeline”. Sirtris Pharmaceuticals.
 |
|
| Identifiers | |
|---|---|
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C25H23N7OS |
| Molar mass | 469.560 g/mol g·mol−1 |
| 3D model (JSmol) | |
////////////SRT-1720 DI HCl, obesity, diabetes, SRT 1720, Sirtris Pharmaceuticals, CAY10559, CAY 10559, Preclinical
O=C(NC1=CC=CC=C1C2=CN3C(SC=C3CN4CCNCC4)=N2)C5=NC6=CC=CC=C6N=C5.[H]Cl.[H]Cl
Beloranib, 성분명 벨로라닙 ZGN-433….Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe


Beloranib
CAS 251111-30-5 (beloranib),529511-79-3 (beloranib hemioxalate)
(E)-(3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1-oxaspiro[2.5]octan-6-yl 3-(4-(2-(dimethylamino)ethoxy)phenyl)acrylate
6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol
Mechanism of Action:methionine aminopeptidase 2 (MetAP2) inhibitor
Indication:Obesity US Patent : US6063812 Patent Exp Date: May 13, 2019
Originator: Chong Kun Dang (CKD) Pharma (종근당) Chong Kun Dang Pharm Corp
Developer: Zafgen Inc. (자프젠)Zafgen Corporation
Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe The European Commission (EC) has granted orphan drug designation to US-based Zafgen for its beloranib for treating Prader-Willi syndrome. Beloranib is a potent inhibitor of Methionine aminopeptidase-2 that reduces hunger while stimulating the use of stored fat as an energy source (MetAP2). MetAP2 is an enzyme that modulates the activity of key cellular processes that control metabolism. http://www.pharmaceutical-technology.com/news/newszafgens-prader-willi-syndrome-therapy-receives-orphan-drug-designation-in-europe-4316842?WT.mc_id=DN_News
INTRODUCTION Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen. Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication. At such low doses, says Thomas E. Hughes, president and chief executive officer of Zafgen, toxicity concerns tend to evaporate, in part because so little opportunity exists to inhibit off-target proteins.
Zafgen, a small pharmaceutical company in Cambridge, Mass., sees high selectivity and low toxicity with its covalent molecule for treating obesity, beloranib hemioxalate, also known as ZGN-433. “You’re passing a wave of the molecule through the body,” he says. “It hits the different tissues, silences the target enzyme where it finds it, and then it goes away.” Zafgen’s drug candidate inhibits an enzyme called methionine aminopeptidase 2 (MetAP2), which had been of interest in oncology circles until it turned out to be a poor target for treating cancer in mice. However, animals treated with a MetAP2 inhibitor lost weight. Zafgen pursued the enzyme as a target for obesity. Its drug candidate contains a spiroepoxide that bonds with a histidine in the protein’s active site.
ZGN-433 has undergone a Phase I clinical trial, in which obese volunteers lost up to 2 lb per week. It will enter Phase II trials within a year, Hughes says, funded by $33 million the company raised from investors. With dosing of up to 2 mg twice per week, ZGN-433 reaches a maximum concentration in the body of just a few nanomolar for several hours before the body quickly eliminates it, Hughes says. During that time, the drug is much more likely to interact with MetAP2 than with anything else. “You’re flying under the radar of a lot of concerns,” he says. “Drug-drug interactions are not an issue. There’s just not enough inhibitor to go around.
The same is true for off-target inhibition: The chance of off-target toxicity is largely gone.” Proponents of covalent inhibitors are quick to point out that dozens of such drugs are already on the market. They include aspirin, the world’s most widely used medicine; penicillin and related antibiotics; and recently developed blockbusters such as Plavix, Prevacid, and Nexium. The drugs treat a broad range of conditions, and many have minimal side effects, even when taken for years. By one count, of the marketed drugs that inhibit enzymes, more than one-third work by covalent modification (Biochemistry, DOI: 10.1021/bi050247e).
![]()
6-O-(4-dimethylaminoethoxy) cinnamoyl fumagillol hemioxalate
| Beloranib, ZGN-433, CKD-732 | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 251111-30-5 |
| PubChem | 6918502 |
| ChemSpider | 26286923 |
| UNII | FI471K8BU6 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C29H41NO6 |
| Molar mass | 499.64 g mol−1 |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |
Beloranib (previously known as CKD-732; ZGN-433), a methionine aminopeptidase 2 (MetAP2) inhibitor originally designed as an anticancer agent, is being developed by Zafgen as a first-in-class obesity therapy. Beloranib, a twice-daily injection, is discovered by korean company Chong Kun Dang (CKD) Pharmaceuticals and was licensed to Cambridge, MA-based startup Zafgen, Inc. Zafgen holds exclusive worldwide rights (exclusive of Korea) for development and commercialization of beloranib. Beloranib, an analog of the antimicrobial agent fumagillin, is an inhibitor of the enzyme METAP2 involved in fatty acid production. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects.
Zafgen has chosen to develop beloranib not for the folks that need to shed a few pounds, but for severely obese people, and smaller groups of patients with rare and dangerous conditions. In January 2013, beloranib was granted orphan drug designation by the U.S. Food and Drug Administration to treat a rare genetic condition known as Prader-Willi Syndrome (PWS) that causes obesity through compulsive eating. Zafgen plans to seek the same designation for beloranib in craniopharyngioma (a rare benign brain tumor) related obesity as well. By going after these orphan indications, Zafgen can get onto the market quicker and cheaper than if it went straight for the larger obesity market. Zafgen recently completed two Phase 2a clinical trials evaluating beloranib’s ability to reduce body weight and to improve hyperphagia, one in PWS patients and one in severely obese patients. In its Phase 2a clinical trials, Zafgen observed reductions in body weight, body mass and body fat content in both patient populations and reductions in hyperphagia-related behaviors in PWS patients.
On June 19, 2014, Zafgen Inc. raised $96 million in its initial public offering (IPO) on the Nasdaq under the symbol “ZFGN” amid strong demand from investors. With its IPO cash, Zafgen plans to initiate its Phase 3 clinical program, consisting of two Phase 3 clinical trials, of beloranib in PWS patients, with the first Phase 3 trial to start in the second half of 2014, after finalizing the program design based on ongoing conversations with the FDA and certain European regulatory authorities. Zafgen is also planning a phase 2a trial in craniopharyngioma, and a Phase 2b trila in patients with severe obesity, all this year. The composition of matter patent (US6063812) on beloranib will each expire in May 2019. Zafgen owns two issued U.S. patents relating to beloranib polymorph compositions of matter that will expire in 2031 and two issued U.S. patents to methods of treating obesity that will expire in 2029.  Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
………………………………..
http://www.google.com/patents/WO2005082349A1?cl=en
compound O-(4- dimethylaminoethoxycinnamoyl)fumagillol can be used in the form of a salt, e.g., acetate, lactate, benzoate, salicylate, mandelate, oxalate, methanesulfonate, or p- toluenesulfonate. Korean Patent No. 0357542 and its corresponding patents (U.S. Patent No. 6,063,812, Japanese Patent No. 3370985, and European Patent No. 1077964), filed by the present applicant, disclose fumagiUol derivatives, including the compounds used in the present invention. The composition of the present invention can be prepared in combination with pharmaceutically acceptable carriers commonly used in pharmaceutical formulations.
………………………..
http://www.google.com/patents/WO2012064838A1?cl=en
MetAP2 encodes a protein that functions at least in part by enzymatically removing the amino terminal methionine residue from certain newly translated proteins, such as, glyceraldehyde-3- phosphate dehydrogenase (Warder et al. (2008) J Proteome Res 7:4807). Increased expression of the MetAP2 gene has been historically associated with various forms of cancer. Molecules inhibiting the enzymatic activity of MetAP2 have been identified and have been explored for their utility in the treatment of various tumor types (Wang et al. (2003) Cancer Res 63:7861) and infectious diseases, such as, microsporidiosis, leishmaniasis, and malaria (Zhang et al. (2002) J. Biomed Sci. 9:34). Notably, inhibition of MetAP2 activity in obese and obese-diabetic animals leads to a reduction in body weight in part by increasing the oxidation of fat and in part by reducing the consumption of food (Rupnick et al. (2002) Proc Natl Acad Sci USA 99: 10730). [0003] 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is a METAP2 inhibitor and is useful in the treatment of, e.g., obesity. 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is characterized by formula I:

Example 1 [0060] Crystalline, Form A material of 6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol was prepared as follows: [0061] Approximately 423 mg of amorphous gum/oil-like 6-O-(4- dimethylaminoethoxy)cinnamoyl fumagillol free base compound was dissolved in ca. 6 mL of diisopropylether (IPE). The solution was allowed to stir for ca. 24 hours at ambient temperature (18-22°C) during which time solid precipitated. The resulting solid was isolated by filtration and dried under vacuum at ambient for ca. 4 hours (yield 35.8 %).
…………………..

………………….
http://www.google.com/patents/WO1999059986A1?cl=en
Example 14 : 0-(4-dimethylaminocinnamoyl)fumagillol 1) To a solution of 4-dimethylaminocinnamic acid (950 mg) in toluene (20 ml), dipyridyl disulfide (1.64 g) and triphenyl phosphine (1.97 g) were added, and the mixture was stirred for 12 hours. 2) The resultant solution of 1) was added to fumagillol (500 mg) at room temperature. Sodium hydride (142 mg) was added thereto, and the reaction mixture was stirred for 30 minutes. After adding saturated ammonium chloride solution (20 ml), the reaction mixture was extracted with ethyl acetate (100 ml). The organic layer was washed with brine and dried over anhydrous magnesium sulfate. After filtering, the solvent was distilled off under reduced pressure, and the residue was purified by column chromatography (eluent: ethyl acetate/ n-hexane = 1/2) to obtain yellow solid (470 mg). ‘H-NMR (CDCI3) δ : 7.60 (d, IH, J=15.8Hz), 7.41 (d, 2H, J=8.9Hz), 6.67 (d, 2H, J=8.9Hz), 6.27 (d, IH, J=15.8Hz), 5.71 (m, IH), 5.22 (bit, IH), 3.70 (dd, IH, J=2.8, 11.0Hz), 3.45 (s, 3H), 3.02 (s, 6H), 3.01 (d, IH, J=4.3Hz), 2.63 (t, IH, J=6.3Hz), 2.56 (d, IH, J=4.3Hz), 2.41 – 1.81 (m, 6H), 1.75 (s, 3H), 1.67 (s, 3H), 1.22 (s, 3H), 1.15 – 1.06 (m, IH)
………..
Organic Letters, 16(3), 792-795; 2014

An efficient, two-step construction of highly complex alkaloid-like compounds from the natural product fumagillol is described. This approach, which mimics a biosynthetic cyclase/oxidase sequence, allows for rapid and efficient structure elaboration of the basic fumagillol scaffold with a variety of readily available coupling partners. Mechanistic experiments leading to the discovery of an oxygen-directed oxidative Mannich reaction are also described.
References
- “News Release: Zafgen Secures $33 Million Series C Financing”. Zafgen, Inc. July 7, 2011.
- Chun, E; Han, CK; Yoon, JH; Sim, TB; Kim, YK; Lee, KY (2005). “Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model”. International Journal of Cancer. Journal International Du Cancer 114 (1): 124–30. doi:10.1002/ijc.20687. PMID 15523682.
- Kim, EJ; Shin, WH (2005). “General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system”. Biological & Pharmaceutical Bulletin 28 (2): 217–23. doi:10.1248/bpb.28.217. PMID 15684472.
- “Zafgen Announces Positive Topline Phase 1b Data for ZGN-433 in Obesity”. MedNews. Drugs.com. 5 January 2011.
- “Fat-busting pill helps obese to shed two pounds a week – without changing their diets”. UK Daily Mail. 11 January 2011.
MORE REF Grenning, Alexander J. et al.Remodeling of Fumagillol: Discovery of an Oxygen-Directed Oxidative Mannich Reaction.Organic Letters, 16(3), 792-795; 2014
Hughes, T. E.; Kim, D. D.; Marjason, J.; Proietto, J.; Whitehead, J. P.; Vath, J. E. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (2013), 21(9), 1782-1788.
Chung Il Hong, Jung Woo Kim, Sang Joon Lee, Soon Kil Ahn, Nam Song Choi, Ryung Kee Hong, Hyoung Sik Chun, Seung Kee Moon, Cheol Kyu Han. Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis. US patent Number US6063812 A, Also published as CA2331873A1, CA2331873C, CN1301260A, CN100352810C, DE69903279D1, DE69903279T2, EP1077964A1,EP1077964B1,WO1999059986A1, Filing date: May 13, 1999.Original Assignee:Chong Kun Dang Corporation Crawford, Thomas; Reece, Hayley A.Preparation of crystalline forms of 6-O-(4-dimethylaminoethoxy)cinnamoylfumagillol.PCT Int. Appl. (2012), WO2012064838 A1, 20120518
Egorov, Maxim et al. Preparation of fumagillol derivatives useful for the treatment or prevention of bone tumors. PCT Int. Appl., WO2012130906, 04 Oct 2012
Stevenson, Cheri A.; Akullian, Laura C.; Petter, Russell C.; Kane, John J.; Hammond, Charles E.; Yin, Mao; Yurkovetskiy, Aleksandr.Preparation of biocompatible biodegradable fumagillin analog conjugates for the treatment of cancer. PCT Int. Appl. (2009), WO2009073445 A2, 20090611
Lee, Hong Woo et al.Design, synthesis, and antiangiogenic effects of a series of potent novel fumagillin analogues.Chemical & Pharmaceutical Bulletin, 55(7), 1024-1029; 2007
Lee, Hong Woo et al.Selective N-demethylation of tertiary aminofumagillols with selenium dioxide via a non-classical Polonovski type reaction.Heterocycles, 68(5), 915-932; 2006
|
References OTHERS |
1: Yin SQ, Wang JJ, Zhang CM, Liu ZP. The development of MetAP-2 inhibitors in cancer treatment. Curr Med Chem. 2012;19(7):1021-35. Review. PubMed PMID: 22229417.
2: Shin SJ, Ahn JB, Park KS, Lee YJ, Hong YS, Kim TW, Kim HR, Rha SY, Roh JK, Kim DH, Kim C, Chung HC. A Phase Ib pharmacokinetic study of the anti-angiogenic agent CKD-732 used in combination with capecitabine and oxaliplatin (XELOX) in metastatic colorectal cancer patients who progressed on irinotecan-based chemotherapy. Invest New Drugs. 2012 Apr;30(2):672-80. doi: 10.1007/s10637-010-9625-x. Epub 2010 Dec 29. PubMed PMID: 21188464.
3: Shin SJ, Jeung HC, Ahn JB, Rha SY, Roh JK, Park KS, Kim DH, Kim C, Chung HC. A phase I pharmacokinetic and pharmacodynamic study of CKD-732, an antiangiogenic agent, in patients with refractory solid cancer. Invest New Drugs. 2010 Oct;28(5):650-8. doi: 10.1007/s10637-009-9287-8. Epub 2009 Jul 8. PubMed PMID: 19585083.
4: Rhee Y, Park SY, Kim YM, Lee S, Lim SK. Angiogenesis inhibitor attenuates parathyroid hormone-induced anabolic effect. Biomed Pharmacother. 2009 Jan;63(1):63-8. doi: 10.1016/j.biopha.2007.10.013. Epub 2007 Nov 20. PubMed PMID: 18457934.
5: Kim YM, An JJ, Jin YJ, Rhee Y, Cha BS, Lee HC, Lim SK. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007 Apr;38(4):455-65. PubMed PMID: 17446235.
6: Kim EJ, Shin WH. General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system. Biol Pharm Bull. 2005 Feb;28(2):217-23. PubMed PMID: 15684472.
7: Chun E, Han CK, Yoon JH, Sim TB, Kim YK, Lee KY. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005 Mar 10;114(1):124-30. PubMed PMID: 15523682.
8: Lee HS, Choi WK, Son HJ, Lee SS, Kim JK, Ahn SK, Hong CI, Min HK, Kim M, Myung SW. Absorption, distribution, metabolism, and excretion of CKD-732, a novel antiangiogenic fumagillin derivative, in rats, mice, and dogs. Arch Pharm Res. 2004 Feb;27(2):265-72. PubMed PMID: 15029870.
9: Kim JH, Lee SK, Ki MH, Choi WK, Ahn SK, Shin HJ, Hong CI. Development of parenteral formulation for a novel angiogenesis inhibitor, CKD-732 through complexation with hydroxypropyl-beta-cyclodextrin. Int J Pharm. 2004 Mar 19;272(1-2):79-89. PubMed PMID: 15019071.
10: Myung SW, Kim HY, Min HK, Kim DH, Kim M, Cho HW, Lee HS, Kim JK, Hong CI. The identification of in vitro metabolites of CKD-732 by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16(21):2048-53. PubMed PMID: 12391579.
| WO2007072083A1 | Dec 22, 2006 | Jun 28, 2007 | Prosidion Ltd | Treatment of type 2 diabetes with a combination of dpiv inhibitor and metformin or thiazolidinedione |
| WO2011085201A1 * | Jan 7, 2011 | Jul 14, 2011 | Zafgen Corporation | Fumagillol type compounds and methods of making and using same |
| WO2011088055A2 * | Jan 11, 2011 | Jul 21, 2011 | Zafgen Corporation | Methods and compositions for treating cardiovascular disorders |
| WO2012064838A1 | Nov 9, 2011 | May 18, 2012 | Zafgen Corporation | Crystalline solids of a metap-2 inhibitor and methods of making and using same |
| WO2013169727A1 * | May 7, 2013 | Nov 14, 2013 | Zafgen, Inc. | Polymorphic salt of the oxalate salt of 6 – o – ( 4 – dimethylaminoethoxy) cinnarnoyl fumagillol and methods of making and using same |
| WO2013169857A1 * | May 8, 2013 | Nov 14, 2013 | Zafgen, Inc. | Treating hypothalamic obesity with metap2 inhibitors |
| EP2317845A1 * | Jul 17, 2009 | May 11, 2011 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8349891 | Aug 7, 2012 | Jan 8, 2013 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US8367721 | Aug 7, 2012 | Feb 5, 2013 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8642650 | Dec 4, 2009 | Feb 4, 2014 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8735447 | Nov 16, 2012 | May 27, 2014 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US20130018095 * | Jan 7, 2011 | Jan 17, 2013 | Vath James E | Fumigillol Type Compounds and Methods of Making and Using Same |
| WO2003027104A1 * | Jun 11, 2002 | Apr 3, 2003 | Byung-Ha Chang | Fumagillol derivatives and preparing method thereof |
| EP0682020A1 * | Aug 31, 1989 | Nov 15, 1995 | Takeda Chemical Industries, Ltd. | Fumagillol derivatives useful as angiogenesis inhibitors |
| US6040337 * | May 13, 1999 | Mar 21, 2000 | Chong Kun Dang Corporation | 5-demethoxyfumagillol derivatives and processes for preparing the same |
| US6063812 * | May 13, 1999 | May 16, 2000 | Chong Kun Dang Corporation | Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis |
| WO1999059986A1 * | May 11, 1999 | Nov 25, 1999 | Soon Kil Ahn | Fumagillol derivatives and processes for preparing the same |
| WO2005082349A1 | Feb 25, 2005 | Sep 9, 2005 | Chong Kun Dang Pharm Corp | Composition for the treatment of obesity comprising fumagillol derivative |
| WO2010065883A2 | Dec 4, 2009 | Jun 10, 2010 | Zafgen Corporation | Method of treating an overweight or obese subject |
| KIM ET AL. JOURNAL OF MOLECULAR ENDOCRINOLOGY vol. 38, 2007, pages 455 – 465 | ||
| 2 | RUPNICK ET AL. PROC NATL ACAD SCI USA vol. 99, 2002, page 10730 | |
| 3 | WANG ET AL. CANCER RES vol. 63, 2003, page 7861 | |
| 4 | WARDER ET AL. J PROTEOME RES vol. 7, 2008, page 4807 | |
| 5 | * | YOO MEE KIM ET AL: “Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732“, JOURNAL OF MOLECULAR ENDOCRINOLOGY, SOCIETY FOR ENDOCRINOLOGY, GB, vol. 38, no. 4, 1 April 2007 (2007-04-01), pages 455-465, XP002632891, ISSN: 0952-5041, DOI: 10.1677/JME.1.02165 |
| 6 | ZHANG ET AL. J. BIOMED SCI. vol. 9, 2002, page 34 |
………

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


Veterinary-Dirlotapide, drug used to treat obesity in dogs

1-Methyl-N-[(1S)-2-(methyl-(phenylmethyl)amino)-2-oxo-1-phenylethyl]-5-[[oxo-[2-[4-(trifluoromethyl)phenyl]phenyl]methyl]amino]-2-indolecarboxamide
Chemical Formula
C40-H33-F3-N4-O3
Molecular Weight
674
Therapeutic Category, dog
Antiobesity agent
Chemical Names
N-{(1S)-2-[Benzyl(methyl)amino]-2-oxo-1-phenylethyl}-1-methyl-5-[4′-(trifluoromethyl)biphenyl-2-carboxamido]-1H-indol-2-carboxamide (WHO)
1H-Indole-2-carboxamide, 1-methyl-N-[(1S)-2-[methyl(phenylmethyl)amino]-2-oxo-1-phenylethyl]-5-[[[4′-(trifluoromethyl)[1,1′-biphenyl]-2-yl]carbonyl]amino]- (USAN)
1-Methyl-5-[(4′-trifluormethylbiphenyl-2-carbonyl)amino]-1H-indol-2-carbonsäure-[(S)-(benzylmethylcarbamoyl)phenylmethyl]amid (IUPAC)
5-[4′-(Trifluoromethylbiphenyl-2-carbonyl)amino]-1H-indole-2-carboxylic acid benzylmethyl carbamoylamide
| Identifiers | |
|---|---|
| CAS number | 481658-94-0 |
| ATCvet code | QA08AB91 |
Dirlotapide is a drug used to treat obesity in dogs. It is manufactured by Pfizer and marketed as Slentrol.
It works as a selective microsomal triglyceride transfer protein (MTTP) inhibitor. This blocks the assembly and release of lipoproteins into the bloodstream, thereby reducing fat absorption. It also elicits a satiety signal from lipid-filled cells lining the intestine.
It is supplied as an oral solution. It is not intended for use in humans, cats, or parrots.
On January 5 2007, the U.S. Food and Drug Administration (FDA) approved Slentrol, the first time the FDA has approved a drug for obese dogs.[1]
Dirlotapide is used to manage obesity in dogs and helps by reducing appetite. It should be used as part of an overall weight control program that also includes proper diet and exercise, under the supervision of a veterinarian. Side effects may include vomiting, diarrhea, lethargy, drooling, or uncoordination. Allergic reaction to the medication may include, facial swelling, hives, scratching, sudden onset of diarrhea, vomiting, shock, seizures, pale gums, cold limbs, or coma. Contact your veterinarian if you observe any of these signs. The dose of dirlotapide will need to be recalculated each month, based on your dog’s weight.
Canine patient information sheet http://www.drsfostersmith.com/Rx_Info_Sheets/rx_dirlotapide.pdf

- “FDA approves 1st drug for obese dogs”. Yahoo. Archived from the original on January 8, 2007. Retrieved 2007-01-06.
Generic Names
- Dirlotapide (OS: USAN)
- CP-742033 (IS)
Brand Names
- Slentrol (veterinary use)
Pfizer, Poland; Pfizer Animal Health, Belgium; Pfizer Animal Health, Switzerland; Pfizer Animal Health, United Kingdom; Pfizer Animal Health, United States; Pfizer GmbH Tiergesundheit, Germany; Pfizer Limited, Austria; Pfizer Santé Animale, France


{kind=link}
{kind=link}
{kind=link}