
Home » Posts tagged 'NRD135S'
Tag Archives: NRD135S
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nispomeben
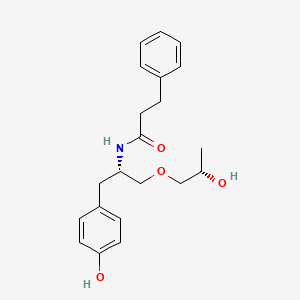
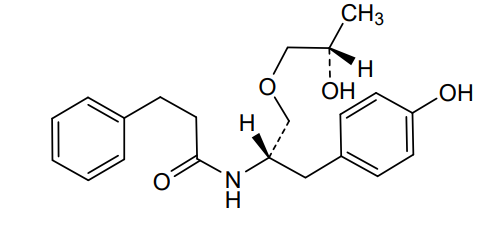
Nispomeben
CAS 1443133-41-2
MF C21H27NO4 MW357.4 g/mol
N-[(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl]-3-phenylpropanamide
N-{(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl}-3-phenylpropanamide
non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1
Nispomeben is a small molecule drug. Nispomeben has a monoisotopic molecular weight of 357.19 Da.
- OriginatorNovaremed
- ClassAlcohols; Amides; Anti-inflammatories; Benzene derivatives; Non-opioid analgesics; Phenols; Small molecules
- Mechanism of ActionLyn protein-tyrosine kinase modulators
- Phase IINeuropathic pain
- 02 Sep 2025Updated adverse events data from a phase II trial in Neuropathic pain released by Novaremed
- 07 May 2025Novaremed completes enrolment in a phase-II clinical trial in Neuropathic pain in USA (PO) (NCT05480228)
- 16 Sep 2022Phase-II clinical trials in Neuropathic pain in USA (PO) (NCT05480228)
PAT
WO 2013/084238
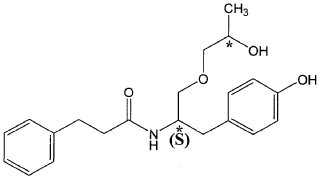
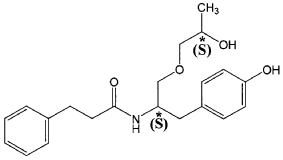
The present invention is based in part on the surprising discovery that the substantially pure enantiomers (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide (also known as the (S,S) enantiomer or El) and (S)2-N(3-0-((R)propan 2-ol)-l -propyl -4-hydroxybenzene)-3-phenylpropyl amide (also known as the (S,R) enantiomer or E2) modulate the activity of specific tyrosine kinases in an opposite manner. It was unexpectedly found that while the (S,S) enantiomer activated protein tyrosine kinases LynA and BLK, the (S,R) enantiomer inhibited their activity. It was further unexpectedly shown that the (S,S) enantiomer was effective as a pain analgesic in animal models of pain, while the (S,R) enantiomer was shown to be ineffective or less effective in these models. Furthermore, the analgesic effect of the (S,S) enantiomer was long acting as it was efficacious for more than 24 hours post administration, in comparison to the commonly used analgesic agent gabapentin which was effective for no longer than 5 hours post administration.
The isolated enantiomers according to some embodiments of the invention may be synthesized as a racemate by known in the art methods described for example in US 7,754,771, US 7,642,290, US 7,674,829 or US 2011/0086910. The racemate may be further separated by known in the art methods for the separation of chiral compounds. According to an exemplary embodiment, the enantiomers may be synthesized as a racemate (comprising (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and (S)2-N(3-0-((R)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and be further separated by a supercritical fluid chromatography (SFC) in combination with chiral stationary phases. Specifically, the (S,S) and (S,R) compounds may be separated on RegisPack™ column a polysaccharide coated chiral column (with a tris-(3,5-dimethylphenyl) carbamoyl cellulose selector) generally used for enantiomeric separations of a wide range of racemate classes (Figure 7A-C).
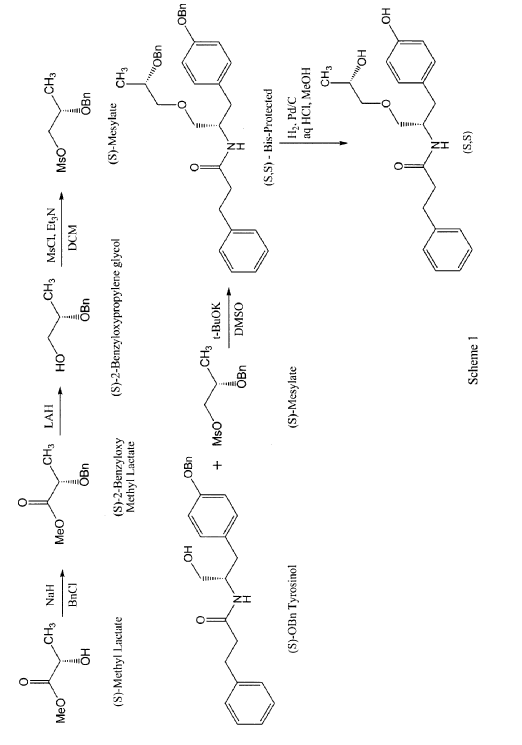
According to some embodiments, the enantiomers may be synthesized directly using for example, the process described in scheme 1 for the preparation of the (S,S) enantiomer.
PAT
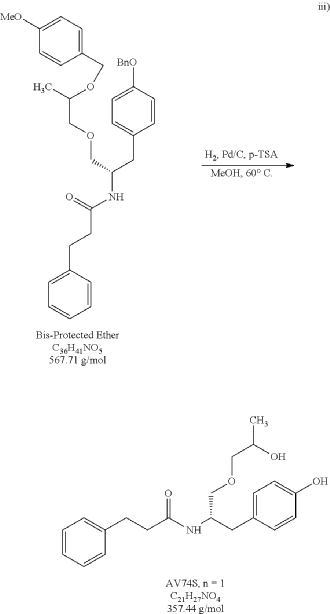
The bis-protected ether (15.7 g) was exposed to one-pot hydrogenation-debenzylation conditions (10% loading of 10% Pd/C and 0.25 eq of p-toluenesulfonic acid) in methanol. After 2 hours at 60° C. under a hydrogen atmosphere, HPLC analysis indicated that the hydrogenation of the benzyl and the debenzylation of PMB ring was complete. The reaction mixture was filtered over Celite and concentrated under reduced pressure. The residue was dissolve in ethyl acetate and a saturated aqueous sodium bicarbonate treatment was conducted to effectively remove p-toluenesulfonic acid, then DURP to provide 12.13 g of an oil (PR030-120-4). Desired product was isolated from an EA/Heptane recrystallization to provide 8.83 g of a white solid (PR030-120-6, 89.4% yield). The purity of PR030-120-6 was 99.3% via HPLC analysis. 1H NMR and Mass spec analysis supported the assigned structure for desired product.
PAT
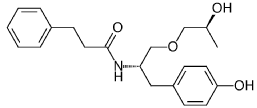
((S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide), including its enantiomers and diastereomers may be prepared as described in WO 2013/084238,
Example 1 – Preparation of -2-N(3-Q-(propan-2-ol)-1-propyl-4-hvdroxybenzene)-3-
phenylpropylamide
(S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 201 1/0086910.
In a first step, 2 g of methyl lactate was reacted with excess of benzyl bromide to get 880 mg of (S)-benzyloxymethyl lactate. The reaction was performed by slurring sodium hydride in THF and cooling down to approximately -15°C. The reaction mixture was then allowed to warm slowly to room temperature and stirred for approximately 1 to 2 hours. The reaction was quenched with saturated ammonium chloride solution and extracted with MTBE twice followed by the removal of solvent on a rotary evaporator to obtain a crude oil. The crude product was purified by column chromatography to yield pure (S)-2-benzyloxymethyl lactate. The (R)-2-benzyloxymethyl lactate isomer was present at 0.93% only. The yield of this step may be increased by avoiding the presence of moisture in the reaction solution.
In a second step, 880 mg (S)-2-benzyloxymethyl lactate obtained in step 1 were reduced using lithium aluminum hydride to obtain (S)-2-benzyloxypropylene glycol in 83.8% yield with 98.7% purity. A solution of pure (S)-2-benzyloxymethyl lactate in methylene chloride was stirred and a solution of lithium aluminum hydride was slowly added thereto at approximately 5°C. The reaction was monitored by TLC and quenched by USP-PW water very carefully. No racemization occurred in this step.
In a third step, the (S)-2-benzyloxypropylene glycol was then reacted with methane sulfonyl chloride in methylene chloride in the presence of triethyl amine to yield the mesylate in 88% yield. A solution of step 2 was stirred in methylene chloride and methane sulfonyl chloride was added to it dropwise at <5°C. After the addition was complete, the progress of the reaction was monitored by TLC. The reaction was quenched with USP-PW water. After the layers were separated, the aqueous layer was back extracted with methylene chloride. The methylene chloride layers were then combined and washed with USP-PW water 3 times to remove most of the methane sulfonic acid. No racemization occurred in this step.
In a fourth step, the mesylate (of step 3) was coupled with S-O-benzyl tyrosinol to form the bis-protected product in 22.7% yield, with a purity of 97.4%. The reaction was carried out at room temperature using a combination of DMF as the solvent and sodium hydride as the base. The reaction went to completion after stirring for at least 12 hours at room temperature.
In a fifth step, 340 mg of the product of step 4 were reduced by hydrogenation in the presence of 10% palladium on carbon catalyst and hydrochloric acid using methylene chloride as a solvent at 50°C. The reaction went to completion in approximately 4 hours with no racemization to yield the desired product in 84.3% yield and 98.9% purity. More specifically, the catalyst was removed by filtration and the filtrate was then concentrated at 33°C. The resulting mixture of solid and oil was mixed with ethyl acetate. The resulting slurry was filtered and the solids washed with ethyl acetate and dried under vacuum at 40 to 45°C to obtain the desired product.
PAT
Example 1—Preparation of (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide
| (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 2011/0086910. |
PAT
- Method of treating or preventing painPublication Number: US-2016317479-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-8802734-B2Priority Date: 2009-09-09Grant Date: 2014-08-12
- Method of Treating or Preventing PainPublication Number: US-2014350099-A1Priority Date: 2009-09-09
- N-substituted benzenepropanamide or benzenepropenamide for use in the treatment of pain and inflammationPublication Number: WO-2011030205-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-2011086910-A1Priority Date: 2009-09-09
- Isolated stereoisomeric forms of (S)2-N(3-O-(propan 2-Ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamidePublication Number: US-9381173-B2Priority Date: 2011-12-08Grant Date: 2016-07-05
- Isolated Stereoisomeric Forms Of (S)2-N(3-O-(Propan 2-Ol)-1-Propyl-4-Hydroxybenzene)-3-PhenylpropylamidePublication Number: US-2014275270-A1Priority Date: 2011-12-08
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: US-9133103-B2Priority Date: 2011-09-21Grant Date: 2015-09-15
- N-Substituted Benzenepropanamide and Benzenepropenamide For Use in the Prevention or the Treatment of Affective DisordersPublication Number: US-2014275273-A1Priority Date: 2011-09-21
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: EP-2758046-B1Priority Date: 2011-09-21Grant Date: 2015-10-21
- Compounds for treatment or prevention of an infection resulting from a coronavirus and/or a coronavirus-induced diseasePublication Number: EP-3939578-A1Priority Date: 2020-07-13
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: EP-3860582-B1Priority Date: 2019-01-23Grant Date: 2022-05-04
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: WO-2020152226-A1Priority Date: 2019-01-23
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: US-2022002228-A1Priority Date: 2019-01-23
- Pharmaceutical composition comprising stereoisomers of n-(1-(4-hydroxyphenyl)-3-(2-hydroxypropoxy)propan-2-yl)-3-phenylpropanamide for the prevention and treatment of type ii diabetesPublication Number: WO-2015173813-A1Priority Date: 2014-05-14



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

////////nispomeben, non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1, Neuropathic pain
