
Home » Posts tagged 'NEW DRUGS' (Page 10)
Tag Archives: NEW DRUGS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Novavax announces positive preclinical data for vaccine against influenza

Novavax announces positive preclinical data for vaccine against influenza
Novavax has announced positive preclinical results for its virus-like particle (VLP) vaccine candidate against A (H7N9) influenza.
The study examined the immunogenicity, the ability to provoke an immune response, and efficacy of two doses of its A(H7N9) VLP vaccine candidate against a lethal wild-type challenge mouse model.

There were three control groups, including Novavax’ non-homologous A(H7N3) VLP vaccine candidate, its A(H5N1) VLP vaccine candidate, and a placebo. All vaccine candidates were administered with or without Iscomatrix, a saponin-based adjuvant.
read all at
http://www.pharmaceutical-technology.com/news/newsnovavax-announces-positive-preclinical-data-for-vaccine-against-influenza?WT.mc_id=DN_News



………….
………….
Animal testing failures put drug trial volunteers in danger

© Shutterstock
Poor pre-clinical data and an absence of negative results are pushing candidate molecules into the clinic too soon
The reporting of animal studies is biased, inflating the efficacy of drug candidates and pushing them into the clinic before they are ready. This is the verdict of new research, which finds that more treatments go from pre-clinical to human trials than ought to, wasting valuable resources and potentially putting trial participants in danger.1 It adds to a growing body of evidence pointing to problems in the way animal testing is reported and managed.
READ AT
http://www.rsc.org/chemistryworld/2013/07/failures-animal-testing-put-drug-trial-volunteers-danger
………..
Gene Therapy for Melanoma: Progress and Perspectives

FIGURE 1.
Schematic representation of the wild type counterpart of the typically used recombinant viral vectors. (A) Gammaretroviruses and (B) lentiviruses share similar structures, but differ greatly in their genomes and their impact on cellular function. Gag, pro, pol and env genes encode structural proteins of the capsid, protease, reverse transcriptase and envelope proteins, respectively. The additional lentiviral genes perform regulatory functions as well as alter cellular function. (C) The serotype 5 adenovirus has a protein capsid (non-enveloped) and a large, complex genome that encodes critical genes for viral replication (E1a, E1b) as well as structural and functional genes that regulate both viral and cellular activities.
Introduction
Gene therapy, the therapeutic transfer of genetic information to a target cell, continues to be a promising alternative in the fight against cancer. In the case of melanoma, the use of an experimental treatment is justified since this disease is incurable in its advanced stages. Is gene therapy a viable option for the treatment of melanoma patients? In this chapter, we will attempt to answer this question by exploring the intersection between the technology of gene therapy and the biology of melanoma, a point at which opportunities for intervention are revealed.
Gene Therapy for Melanoma: Progress and Perspectives
[1] Cancer Institute of Sao Paulo, University of Sao Paulo School of Medicine, Brazil
[2] University of Sao Paulo, Biomedical Sciences Institute, Brazil

……………….
Analysis Of Vical’s Allovectin-7: Best Results Ever In A Melanoma Phase 3 Trial
check this video
…………….


Vical’s (VICL) Allovectin-7 is a pure immune therapy.(1) Which means it does not directly kill cancer cells, but activates the immune system to do so. Vical will soon announce A-7 phase 3 results in Melanoma, but the mechanism of action is not specific to Melanoma, and can be used in any solid tumor cancer.(2) For this reason, I expect that Allovectin-7 will become one of the best selling cancer drugs of all time.
Allovectin-7 is a substance that is being studied as a gene therapy agent in the treatment of cancer, such as malignant melanoma. It is a plasmid/lipid complex containing the DNA sequences encoding HLA-B7 and ß2 microglobulin – two components of major histocompatibility complex (MHC, class I). It increases the ability of the immune system to recognize cancer cells and kill them.
In 1999, FDA granted Allovectin-7 orphan drug designation for the treatment of invasive and metastatic melanoma.
- Allovectin-7 entry in the public domain NCI Dictionary of Cancer Terms
![]() This article incorporates public domain material from the U.S. National Cancer Institute document “Dictionary of Cancer Terms”.
This article incorporates public domain material from the U.S. National Cancer Institute document “Dictionary of Cancer Terms”.
Vical’s Allovectin-7
Allovectin is a first-class DNA-based immunotherapeutic designed to stimulate both innate and adaptive immune responses in local tumors and distal metastases. The goal is to become a first-line treatment for Stage III and IV melanoma, where it is intended to provide improved efficacy, a better safety profile, and simple outpatient administration.
As last reported, the company is approaching completion of a Phase III registration trial versus chemotherapy in patients with metastatic melanoma. The reporting of end results has had numerous delays, but the results are now expected by Q3.
Outside of Allovectin, Vical has ten clinical trials ongoing, three of those independent and the rest in collaboration. Clearly, Vical is not totally dependent on this immunotherapy though it is the most advanced independent program in the company’s pipeline.
Vical has a market cap of $257M, so clearly a homerun therapy could send the stock soaring.
Indian Pharma Market Needs Strong Regulatory Set-up: Kiran Mazumdar-Shaw

Kiran Mazumdar-Shaw, MD, BIOCON
Indian Pharma Market Needs Strong Regulatory Set-up: Kiran Mazumdar-Shaw, MD, BIOCON
Biocon is looking at gaining market share and improving its margins with a greater focus on its product mixes and organizational efficiencies. Biocon Chairman and Managing Director Kiran Mazumdar-Shaw tells Financial Express that the company has outpaced the market, despite various challenges and that the pharmaceutical market needs to have a more robust regulatory set-up. Edited excerpts:
READ ALL AT
http://kiranmazumdarshaw.blogspot.in/2013/07/indian-pharma-market-needs-strong.html
………………
Serum Institute of India acquires rights to TB vaccine
Serum Institute of India acquires rights to TB vaccine
Serum Institute of India, a Pune-based manufacturer of vaccines, is planning on taking a promising vaccine – originally developed in Germany – and introducing it into the clinic. Studies have shown that the new vaccine is more effective and better tolerated than currently available options.
By signing a contract with Hannover-based Vakzine Projekt Management (VPM), Serum has secured the licence to the various patents and technologies related to the…
…..
UK regulator approves access to Revolade drug after three-year process
25 July 2013
The National Institute for Health and Care Excellence (NICE) in the UK has recommended immune disorder drug Revolade for use on the NHS after a process of three and a half years.
The GSK once-daily oral treatment is now available to adult patients in England and Wales living with chronic immune (idiopathic) thrombocytopenic purpura (cITP), an immune disorder associated with low-blood platelet counts.
In patients with cITP, the immune system prematurely destroys platelets or impairs their production so that platelets are lost from the circulation faster than they can be replaced from the bone marrow, where they are made.
This results in patients developing mild bruising or serious bleeding, which affects their quality of life and, in some instances, may be fatal.
It is estimated that cITP currently affects 50 in 100,000 people in the UK.
The only other licensed TPO-RA recommended by NICE is romiplostim, which is given in the form of a weekly injection.
The Royal London Hospital’s clinical director for pathology Prof Adrian Newland said: “I was very pleased to see that NICE has recognised the clinical value and cost-effectiveness of eltrombopag in their guidance.
“We now have an important addition to the treatment options for patients with severe or refractory disease.”
Revolade is an oral thrombopoietin receptor agonist (TPO-RA) that stimulates the growth and maturation of cells in the bone marrow (megakaryocytes) that produce platelets, increasing platelet production.
When added to conventional immunosuppressive therapy, Revolade, also known as eltrombopag, increases response rates compared with placebo and in some patients.
GlaxoSmithKline UK general manager Erik Van Snippenberg said: “This has been a lengthy three and a half year long appraisal process. We are pleased that NICE has recommended eltrombopag and that the small number of cITP patients in England and Wales are granted access to an alternative treatment option offering the benefit of oral convenience.
“With eltrombopag, we hope to ultimately make a meaningful difference in the quality of life of cITP patients and contribute to potential savings for the NHS.”
![]()
Compound Suffocates Tumors
Scientists have discovered a new molecule that prevents cancer cells from responding and surviving when starved of oxygen and which could be developed into new treatments for the disease, according to new research published in the Journal of the American Chemical Society.
Cancer Research UK scientists at the University of Southampton found that this molecule targets the master switch—HIF-1—that cancer cells use to adapt to low oxygen levels, a common feature in the disease.
read all at
Find out more:
- Cancer Sciences Unit
- Is it your ambition to help cure cancer? If so, visit: www.southampton.ac.uk/medicine/undergraduate/index.page
Cabergoline therapy for cushing disease throughout pregnancy

CABERGOLINE
Obstet Gynecol. 2013 Aug;122(2 Pt 2):485-7. doi: 10.1097/AOG.0b013e31829e398a.
http://www.ncbi.nlm.nih.gov/pubmed/23884269
.
Woo I, Ehsanipoor RM.
Source
Department of Gynecology and Obstetrics and Division of Maternal-Fetal Medicine, Department of Gynecology and Obstetrics, Johns Hopkins University School of Medicine, Baltimore, Maryland.
Abstract
BACKGROUND:
Cushing disease during pregnancy is rare and is associated with significant maternal and fetal morbidity and mortality. Transsphenoidal pituitary surgery is the first-line therapy; however, in cases of failed surgery or in patients who are not surgical candidates, medical therapy has been used to control symptoms.
CASE:
A 29-year-old woman with Cushing disease and a noncurative transsphenoidal pituitary surgery was successfully treated with cabergoline, a dopamine agonist. After approximately 1 year of therapy, she became pregnant. She was maintained on high-dose cabergoline throughout her pregnancy and had an uncomplicated antenatal course. She went into spontaneous labor at 38 weeks of gestation and delivered a healthy female neonate.
CONCLUSION:
Cabergoline can be used to manage Cushing disease successfully during pregnancy with an opportunity for a favorable outcome.
Cabergoline (brand names Dostinex and Cabaser), an ergot derivative, is a potent dopamine receptor agonist on D2 receptors. In vitro, rat studies show cabergoline has a direct inhibitory effect on pituitary lactotroph (prolactin) cells.[1] It is frequently used as a first-line agent in the management of prolactinomas due to higher affinity for D2 receptor sites, less severe side effects, and more convenient dosing schedule than the older bromocriptine.
History
Cabergoline was invented by scientists working for the Italian drug company Farmitalia-Carlo Erba SpA in Milan in 1981/82,[2] who were experimenting with semisynthetic derivatives of the ergot alkaloids. Farmitalia-Carlo Erba was acquired by Pharmacia in 1992, which in turn was acquired by Pfizer in 2002. The drug was approved by the FDA on December 23, 1996. It went generic in late 2005 following US patent expiration.
Intellectual property
Farmitalia filed a patent application for Cabergoline in 1982, and U.S. Patent 4,526,892 issued in July 1985.
Pharmacology
Although cabergoline is commonly described principally as a dopamine D2 receptor agonist, it also possesses significant affinity for the D3, D4, 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C, α2B– receptors, and moderate/low affinity for the D1 and 5-HT7 receptors. Cabergoline functions as an agonist at all of these receptors except for 5-HT7 and α2B–, where it acts as an antagonist.[3]
Following a single oral dose, resorption of cabergoline from the gastrointestinal (GI) tract is highly variable, typically occurring within 0.5 to 4 hours. Ingestion with food does not alter its absorption rate. Human bioavailability has not been determined since the drug is intended for oral use only. In mice and rats the absolute bioavailability has been determined to be 30 and 63 percent, respectively. Cabergoline is rapidly and extensively metabolized in the liver and excreted in bile and to a lesser extent in urine. All metabolites are less active than the parental drug or inactive altogether. The human elimination half-life is estimated to be 63 to 68 hours in patients with Parkinson’s disease and 79 to 115 hours in patients with pituitary tumors. Average elimination half-life is 80 hours.
The therapeutic effect in treatment of hyperprolactinemia will typically persist for at least 4 weeks after cessation of treatment.
Mechanism of action
Cabergoline is a long-acting dopamine D2 receptor agonist and in vitro rat studies show a direct inhibitory effect on the prolactin secretion in the pituitary’s lactotroph cells. Cabergoline decreased serum prolactin levels in reserpinized rats.
Receptor binding studies indicate a low affinity for dopamine D1 receptors, α1-adrenergic receptors, and α2-adrenergic receptors.[1]
- 1 Dostinex at www.rxlist.com”. Retrieved 2007-04-27.
- 2 US Patent 4526892 – Dimethylaminoalkyl-3-(ergoline-8′.beta.carbonyl)-ureas
- 3 Sharif NA, McLaughlin MA, Kelly CR, Katoli P, Drace C, Husain S, Crosson C, Toris C, Zhan GL, Camras C (March 2009). “Cabergoline: Pharmacology, ocular hypotensive studies in multiple species, and aqueous humor dynamic modulation in the Cynomolgus monkey eyes”. Experimental Eye Research 88 (3): 386–97. doi:10.1016/j.exer.2008.10.003. PMID 18992242.
- 4 National Institute ofMental Health. PDSD Ki Database (Internet) [cited 2013 Jul 24]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: http://pdsp.med.unc.edu/pdsp.php
- 5 Sayyah-Melli, M; Tehrani-Gadim, S; Dastranj-Tabrizi, A; Gatrehsamani, F; Morteza, G; Ouladesahebmadarek, E; Farzadi, L; Kazemi-Shishvan, M (2009). “Comparison of the effect of gonadotropin-releasing hormone agonist and dopamine receptor agonist on uterine myoma growth. Histologic, sonographic, and intra-operative changes”. Saudi medical journal 30 (8): 1024–33. PMID 19668882. edit
6 Sankaran, S.; Manyonda, I. (2008). “Medical management of fibroids”. Best Practice & Research Clinical Obstetrics & Gynaecology 22 (4): 655. doi:10.1016/j.bpobgyn.2008.03.001. PMID 18468953. edit http://www.britishfibroidtrust.org.uk/journals/bft_Sankaran.pdf
CUSHING DISEASE VIDEO
LUMACAFTOR an Orphan drug in clinical trial for oral the treatment of cystic fibrosis

Lumacaftor
3-[6-[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropylcarboxamido]-3-methylpyridin-2-yl]benzoic acid
3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
VRT-826809
VX-809
US patents: US8124781, US8461342
Indication:Cystic fibrosis
Developmental status:Phase III (US, UK, EU)
Developer:Vertex
| Vertex Pharmaceuticals |
| Company | Vertex Pharmaceuticals Inc. |
| Description | Small molecule cystic fibrosis transmembrane conductance regulator (CFTR) corrector |
| Molecular Target | Cystic fibrosis transmembrane conductance regulator (CFTR) |
| Mechanism of Action | CFTR stabilizer |
| Latest Stage of Development | Phase III |
| Indication | Cystic fibrosis (CF) |
| cas | 936727-05-8 |
http://www.ama-assn.org/resources/doc/usan/lumacaftor.pdf for all data
see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.html
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients in Europe,[1] and in about 90% of persons with some mutation in the CFTR gene.
A corrector molecule, one of two new classes of ion channel modulators. The corrector modulators enhance the number of channels of the CFTR protein at the cell surface. in combination with ivacaftor in homozygous F508del pts
Results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis—homozygous F508del—had a mean increase of 7.4% in lung function (FEV1) on a combination of lumacaftor and ivacaftor.[2]
VX-809 is an investigational corrector compound in a phase II clinical trial for oral the treatment of cystic fibrosis. The trial will evaluate single and multiple doses of VX-809 in healthy volunteers. This compound has resulted from a collaboration with the Cystic Fibrosis Foundation Therapeutics, Inc. (CFFT) . In 2010, orphan drug designation was assigned in the E.U. and the U.S. for the treatment of CF.
VX-809 may act to restore the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, the defective cell membrane protein responsible for the progression of CF. VX-809 and other corrector compounds were designed to increase the amount of DF508-CFTR on the surface of cells lining the airway, which may result in an increase in chloride transport across the cell surface in patients with the DF508-CFTR mutation.
On January 11, 2013, the combination regimen of Lumacaftor (VX-809) and Kalydeco (Ivacaftor) was awarded by U.S. FDA with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease.Breakthrough Therapy Designation for the combination regimen of VX-809 with ivacaftor was based on the Phase II combination data announced in 2012. Vertex Pharmaceuticals will report results from two Phase III trials (NCT01807949 (TRANSPORT) and NCT01807923 (TRAFFIC)) of the combination of Kalydeco + VX-809 in the middle of 2014. Positive data from TRAFFIC and TRANSPORT could open up a market with peak sales of approximately $6 billion, estimate analysts.

- 1 Merk; Schubert-Zsilavecz. Pharmazeutische Zeitung (in German) 156 (37): 24–27.
- 2 Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115

see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm
…………………………
PATENT
http://www.google.com/patents/EP2639222A1?cl=en
-
CFTR correctors useful in the treatment of cystic fibrosis. Such compounds include 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (hereinafter “Compound 1”) which has the structure below:

-
Compound 1 and pharmaceutically acceptable compositions thereof are useful for treating or lessening the severity of a variety of CFTR mediated diseases.

-
Scheme 1. Synthesis of the acid chloride moiety.


Scheme 2. Synthesis of the amine moiety.

Scheme 3. Formation of an acid salt of 3-(6-(1-(2,2-difluorobcnzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

-
Synthesis of 3-(6-(1-(2,2-difluorobenzord[d][1,3]dioxol-5-yl) cyclopropancearboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl.
-
[0238]Acid Chloride Moiety
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-methanol (Compound 18).

-
Commercially available 2,2-difluoro-1,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude Compound 18 that is used directly in the next step.
-
Synthesis of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (Compound 19).

-
Compound 18 (1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and SOCl2 (1.2 eq) is added via addition funnel. The SOCl2 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO4), filtered, and concentrated to afford crude Compound 19 that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 20).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 20 (95%) that is used directly in the next step.
-
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 21).

-
A mixture of compound 20 (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) is heated at 70 °C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford compound 21.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 22).

-
Compound 21 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE, 1 M HCl was added and the layers are separated. The MTBE layer was then treated with dicyclohexylamine (0.97 equiv). The slurry is cooled to 0 °C, filtered and washed with heptane to give the corresponding DCHA salt. The salt is taken into MTBE and 10% citric acid and stirred until all solids dissolve. The layers are separated and the MTBE layer was washed with water and brine. Solvent swap to heptane followed by filtration gives compound 22 after drying in a vacuum oven at 50 °C overnight.
-
Synthesis of 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 7).

-
Compound 22 (1.2 cq) is slurried in toluene (2.5 vol) and the mixture heated to 60 °C. SOCl2 (1.4 eq) is added via addition funnel. The toluene and SOCl2 are distilled from the reaction mixture after 30 minutes. Additional toluene (2.5 vol) is added and distilled again.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (compound 23).

-
A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of Na14 CN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 23 that is purified by chromatography.
-
Synthesis of 14C-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile (compound 24).

-
A mixture of compound 23 (1.0 eq) and 1,2-dibromoethane (1.8 eq) in THF (3 vol) is cooled to -10 °C via external chiller. 1 M LHMDS in THF (2.5 eq) is added via an addition funnel and at a rate to maintain the temperature in the reactor below 10 °C. One hour after addition is complete, 20% w/v aq. citric acid (13 vol) is added via addition funnel maintaining the temperature in the reactor below 20 C. The external chiller is turned off and after stirring for 30 min the layers are separated. The organic layer is filtered and concentrated to afford crude compound 24 that is purified by chromatography.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid (compound 25).

-
Compound 24 is hydrolyzed using 6 M NaOH (8 equiv) in ethanol (5 vol) at 80 °C overnight. The mixture is cooled to room temperature and ethanol is evaporated under vacuum. The residue is taken into water and MTBE. 1 M HCl is added to the mixture and the organic layer is filtered and concentrated to afford compound 25.
-
Synthesis of 14C-1-(2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonyl chloride (compound 26).

-
A mixture of Compound 25, 4-dimethylaminopyridine, and thionyl chloride (SOCl2) in CH2Cl2 is stirred to produce compound 26, which may be further reacted with compound 6 without isolation.
-
Amine Moiety
-
Synthesis of tert-butyl-3-(3-methylpyridin-2-yl)benzoate (compound 4).

-
2-Bromo-3-methylpyridine (1.0 eq) is dissolved in toluene (12 vol). K2CO3 (4.8 eq) is added followed by water (3.5 vol) and the mixture heated to 65 °C under a stream of N2 for 1 hour. 3-(t-Butoxycarbonyl)phenylboronic acid (1.05 eq) and Pd(dppf)Cl2-CH2Cl2 (0.015 eq) are then added and the mixture is heated to 80 °C. After 2 hours, the heat is turned off, water is added (3.5 vol) and the layers are allowed to separate. The organic phase is then washed with water (3.5 vol) and extracted with 10% aqueous methanesulfonic acid (2 eq MsOH, 7.7 vol). The aqueous phase is made basic with 50% aqueous NaOH (2 eq) and extracted with EtOAc (8 vol). The organic layer is concentrated to afford crude compound 4 (82%) that is used directly in the next step.
-
Synthesis of 2-(3-(tert-butoxycarbonyl)phenyl)-3-methylpyridine-1-oxide (compound 5).

-
Compound 4 (1.0 eq) is dissolved in EtOAc (6 vol). Water (0. 3 vol) is added followed by urea-hydrogen peroxide (3 cq). The phthalic anhydride (3 cq) is added portion-wise as a solid to maintain the temperature in the reactor below 45 °C. After completion of phthalic anhydride addition, the mixture is heated to 45 °C. After stirring for an additional 4 hours, the heat is turned off. 10% w/w aqueous Na2SO3 (1.5 eq) is added via addition funnel. After completion of Na2SO3 addition, the mixture is stirred for an additional 30 minutes and the layers separated. The organic layer is stirred and 10% w/w aq. Na2CO3 (2 eq) is added. After stirring for 30 minutes, the layers are allowed to separate. The organic phase is washed 13% w/v aq NaCl. The organic phase is then filtered and concentrated to afford crude compound 5 (95%) that is used directly in the next step.
-
Synthesis of tert-butyl-3-(6-amino-3-methylpyridin-2-yl)benzoate (compound 6).

-
A solution of compound 5 (1 eq) and pyridine (4 eq) in MeCN (8 vol) is heated to 70 °C. A solution of methanesulfonic anhydride (1.5 eq) in MeCN (2 vol) is added over 50 min via addition funnel maintaining the temperature at less than 75 °C. The mixture is stirred for an additional 0.5 hours after complete addition. The mixture is then allowed to cool to ambient. Ethanolamine (10 eq) is added via addition funnel. After stirring for 2 hours, water (6 vol) is added and the mixture is cooled to 10 °C. After stirring for NLT 3 hours, the solid is collected by filtration and washed with water (3 vol), 2:1 MeCN/water (3 vol), and MeCN (2×1.5 vol). The solid is dried to constant weight (<1% difference) in a vacuum oven at 50 °C with a slight N2 bleed to afford compound 6 as a red-yellow solid (53% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (compound 8).

-
Compound 7 is dissolved in toluene (2.5 vol based on acid chloride) and added via addition funnel to a mixture of compound 6 (1 eq), dimethylaminopyridine (DMAP, 0.02 eq), and triethylamine (3.0 cq) in toluene (4 vol based on compound 6). After 2 hours, water (4 vol based on compound 6) is added to the reaction mixture. After stirring for 30 minutes, the layers are separated. The organic phase is then filtered and concentrated to afford a thick oil of compound 8 (quantitative crude yield). MeCN (3 vol based on crude product) is added and distilled until crystallization occurs. Water (2 vol based on crude product) is added and the mixture stirred for 2 h. The solid is collected by filtration, washed with 1:1 (by volume) MeCN/water (2 x 1 vol based on crude product), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate as a brown solid.
-
Syntheisis of Syntheisis of 3-(6-(1-(2,2-difluorobenzo[d] [1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt (compound 9).

-
To a slurry of compound 8 (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 °C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford compound 9 as an off-white solid.















-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1).

-
A slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) using water and base.


-
To a slurry of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 °C or 90 °C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.

-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.



-
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
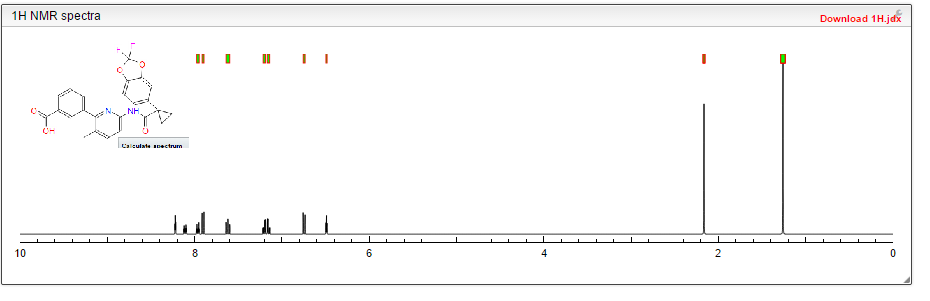
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)



| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |
| see……http://orgspectroscopyint.blogspot.in/2015/03/lumacaftor.htm |
References
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8124781 B2 ;Also published as CA2707494A1, CN101910134A, EP2231606A2, EP2231606B1, EP2639222A1, EP2639223A1, EP2639224A1, US8592602, US20090176989, US20120190856, WO2009076142A2, WO2009076142A3;Filing date:Dec 4, 2008;Original Assignee:Vertex Pharmaceuticals Incorporated
David Andrew Siesel;Processes for producing cycloalkylcarboxamido-pyridine benzoic acids,US patent number US8461342 B2 ;Also published as US20100036130, US20120203006, US20130274477, WO2010138484A2, WO2010138484A3;Original Assignee:Vertex Pharmaceuticals Incorporated
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis;PCT Int. Appl., WO2011133956
Van Goor, Fredrick F. et al;Pharmaceutical compositions in the treatment of CFTR-mediated diseases such as cystic fibrosis.PCT Int. Appl., WO2011133951
Van Goor, Fredrick F. et al;Pharmaceutical compositions for treatment of CFTR-mediated diseases;PCT Int. Appl., WO2011133953
Verwijs, Marinus Jacobus et al;Preparation and pharmaceutical compositions of Lumacaftor for the treatment of cystic fibrosis and other diseases associated with CFTR mutations;PCT Int. Appl., WO2011127241
Keshavarz-Shokri, Ali et al;Preparation of Lumacaftor for therapeutical use;PCT Int. Appl., WO2011127290
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;U.S. Pat. Appl. Publ., US20100036130
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2010138484
Young, Christopher;Dosage units of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid;PCT Int. Appl., WO2010037066
Hadida-Ruah, Sara et al;paration of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;U.S. Pat. Appl. Publ., 20080019915
Siesel, David;A process for the preparation of solid forms of (((difluorobenzodioxolyl)cyclopropanecarboxamido)methylpyridinyl)benzoic acid;PCT Int. Appl., WO2009076142
Hadida Ruah, Sara et al;Preparation of N-pyridinyl carboxamide derivatives as modulators of ATP-binding cassette transporters;PCT Int. Appl., WO2007056341
video on cystic fibrosis
second video
Update on 26 mar 2015
LUMACAFTOR
VX 809
| 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic Acid | |
| CAS No.: | 936727-05-8 |
|---|---|
| Synonyms: |
|
| Formula: | C24H18F2N2O5 |
| Exact Mass: | 452.11800 |
SMILLES…. Cc1ccc(nc1c2cccc(c2)C(=O)O)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F
NMR…………….http://file.selleckchem.com/downloads/nmr/S156503-VX-809-HNMR-Selleck.pdf
-
Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1) directly from benzoate.
-
A solution of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 °C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 °C and the mixture stirred. The mixture is then heated to 70 ± 10 °C until the level of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 °C with a slight N2 bleed to afford Compound 1 as an off-white solid.
-
1HNMR spectra of Compound 1 are shown in Figures 9-11 (Figures 9 and 10 depict Compound 1 in Form I in a 50 mg/mL, 0.5 methyl cellulose-polysorbate 80 suspension, and Figure 11 depicts Compound 1 as an HCl salt).
-
Table 3 below recites additional analytical data for Compound 1.
-
Table 3.
Cmpd. No. LC/MS M+1 LC/RTmin NMR 1 453.3 1.93 H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)

1H NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-1h.png)
13C NMR PREDICT
![3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid NMR spectra analysis, Chemical CAS NO. 936727-05-8 NMR spectral analysis, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-29/001/530/195/936727-05-8-13c.png) CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT
CAS NO. 936727-05-8, 3-[6-[[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino]-3-methylpyridin-2-yl]benzoic acid C-NMR spectral analysisCOSY PREDICT


13C NMR PREDICT




| WO2002096421A1 * | May 22, 2002 | Dec 5, 2002 | Neurogen Corp | 5-substituted-2-arylpyridines as crf1 modulators |
| WO2004072038A1 * | Feb 10, 2004 | Aug 26, 2004 | Vertex Pharma | Processes for the preparation of n-heteroaryl-n-aryl-amines by reacting an n-aryl carbamic acid ester with a halo-heteroaryl and analogous processes |
| WO2007056341A1 | Nov 8, 2006 | May 18, 2007 | Vertex Pharma | Heterocyclic modulators of atp-binding cassette transporters |

http://www.google.co.in/patents/US8124781



Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1)

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Using Water and Base

Synthesis of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic Acid (Compound 1) Directly from Benzoate

Compound 1
Compound 1 is used as the starting point for the other solid state forms and can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

1. NaCN
2. H20

socio

Scheme 1 depicts the preparation of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride, which is used in Scheme 3 to make the amide linkage of Compound 1.
The starting material, 2,2-difluorobenzo[d][l,3]dioxole-5-carboxylic acid, is commercially available from Saltigo (an affiliate of the Lanxess Corporation). Reduction of the carboxylc acid moiety in 2,2-difluorobenzo[d][l ,3]dioxole-5-carboxylic acid to the primary alcohol, followed by conversion to the corresponding chloride using thionyl chloride (SOCl2), provides 5-(chloromethyl)-2,2-difluorobenzo[d][l,3]dioxole, which is subsequently converted to 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile using sodium cyanide. Treatment of 2-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile with base and l-bromo-2-chloroethane provides 1- (2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile. The nitrile moiety in l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarbonitrile is converted to a carboxylic acid using base to give l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarboxylic acid, which is converted to the desired acid chloride using thionyl chloride.
Scheme 2. Alternative synthesis of the acid chloride moiety.

Touene, H20, 70 °C3 N HC1,
DMSO,
75 °C

Scheme 2 depicts an alternative synthesis of the requisite acid chloride. 5- bromomethyl-2,2-difluoro-l,3-benzodioxole is coupled with ethyl cyanoacetate in the presence of a palladium catalyst to form the corresponding alpha cyano ethyl ester. Saponification of the ester moiety to the carboxylic acid gives the cyanoethyl compound. Alkylation of the cyanoethyl compound with l-bromo-2-chloro ethane in the presence of base gives the cyanocyclopropyl compound. Treatment of the cyanocyclopropyl compound with base gives the carboxylate salt, which is converted to the carboxylic acid by treatment with acid. Conversion of the carboxylic acid to the acid chloride is then accomplished using a chlorinating agent such as thionyl chloride or the like.
Scheme 3. Synthesis of the amine moiety.

ptBu urea-hydrogen peroxide hthalic anhydride EtOAc, water

Scheme 3 depicts the preparation of the requisite tert-butyl 3-(6-amino-3- methylpyridin-2-yl)benzoate, which is coupled with l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride in Scheme 3 to give Compound 1. Palladium-catalyzed coupling of 2-bromo-3-methylpyridine with 3-(tert-butoxycarbonyl)phenylboronic acid gives tert-butyl 3-(3-methylpyridin-2-yl)benzoate, which is subsequently converted to the desired compound. Scheme 4. Formation of an acid salt of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid.

Scheme 4 depicts the coupling of l-(2,2-difluorobenzo[d][l,3]dioxol-5- yl)cyclopropanecarbonyl chloride with tert-butyl 3-(6-amino-3-methylpyridin-2-yl)benzoate using triethyl amine and 4-dimethylaminopyridine to initially provide the tert-butyl ester of Compound 1.
……………………..
WO2010037066
http://www.google.im/patents/WO2010037066A2?cl=en
Syntheisis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCL salt.

HCl
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in MeCN (3.0 vol) is added water (0.83 vol) followed by concentrated aqueous HCl (0.83 vol). The mixture is heated to 45 ± 5 0C. After stirring for 24 to 48 hours the reaction is complete and the mixture is allowed to cool to ambient. Water (1.33 vol) is added and the mixture stirred. The solid is collected by filtration, washed with water (2 x 0.3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford 3-(6-(l-(2,2- difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2- yl)benzoic acid • HCl as an off-white solid.
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I).

HCl

Compound 1 in Form I
A slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) is stirred at ambient temperature. A sample is taken after stirring for 24 hours. The sample is filtered and the solid washed with water (2 x). The solid sample is submitted for DSC analysis. When DSC analysis indicates complete conversion to Compound 1, the solid is collected by filtration, washed with water (2 x 1.0 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (98% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) using water and base.


Compound 1 in Form I
To a slurry of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid • HCl (1 eq) in water (10 vol) stirred at ambient temperature is added 50% w/w aq. NaOH (2.5 eq). The mixture is stirred for NLT 15 min or until a homogeneous solution. Concentrated HCl (4 eq) is added to crystallize Compound 1. The mixture is heated to 60 0C or 90 0C if needed to reduce the level of the t-butylbenzoate ester. The mixture is heated until HPLC analysis indicates NMT 0.8% (AUC) t-butylbenzoate ester. The mixture is then cooled to ambient and the solid is collected by filtration, washed with water (3 x 3.4 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 as an off-white solid (97% yield).
Synthesis of 3-(6-(l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid (Compound 1 in Form I) directly from benzoate.


Compound 1 in Form I
A solution of 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate (1.0 eq) in formic acid (3.0 vol) is heated to 70 ± 10 0C. The reaction is continued until the reaction is complete (NMT 1.0% AUC 3-(6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)-t-butylbenzoate) or heating for NMT 8 h. The mixture is allowed to cool to ambient. The solution is added to water (6 vol) heated at 50 0C and the mixture stirred. The mixture is then heated to 70 ± 10 0C until the level of 3- (6-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin- 2-yl)-t-butylbenzoate is NMT 0.8% (AUC). The solid is collected by filtration, washed with water (2 x 3 vol), and partially dried on the filter under vacuum. The solid is dried to constant weight (<1% difference) in a vacuum oven at 60 0C with a slight N2 bleed to afford Compound 1 in Form I as an off-white solid.
