
Home » Posts tagged 'neuropathic pain'
Tag Archives: neuropathic pain
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Mazisotine



Mazisotine
CAS 1638588-92-7
MF C16H23N3O2 MW 289.37 g/mol
(1S,5R)-N-[2-methyl-1-[(3-methyl-2-pyridinyl)oxy]propan-2-yl]-3-azabicyclo[3.1.0]hexane-6-carboxamide
rac-(1R,5S,6R)-N-{2-methyl-1-[(3-methylpyridin-2-yl)oxy]propan-2-yl}-3-azabicyclo[3.1.0]hexane-6-carboxamide
(1R,5S,6r)-N-{2-methyl-1-[(3-methylpyridin-2-yl)oxy]propan-2-yl}-3-azabicyclo[3.1.0]hexane-6-carboxamide
somatostatin receptor receptor agonist, LY3556050, CNTX-0290, LY 3556050, CNTX 0290, D3M32WP3MH
Mazisotine (also known as LY3556050 or CNTX-0290) is an experimental, non-opioid chemical compound designed to treat chronic and neuropathic pain. It functions as a selective somatostatin receptor 4 (SSTR4) agonist, meaning it activates specific peripheral nerve pathways to block pain signals without activating the central nervous system’s opioid receptors.
While it showed early promise in animal models, Eli Lilly and Company removed mazisotine from its clinical development pipeline after disappointing results in Phase II clinical trials. It currently remains in use strictly as a chemical tool for laboratory pain research.
Key Facts and Clinical History
- Mechanism of Action: It binds to and activates SSTR4. This triggers a cellular response that suppresses pain and inflammation in peripheral sensory neurons.
- Intended Indications: It was being evaluated to treat diabetic peripheral neuropathic pain, osteoarthritis pain, and chronic low back pain.
- Development Partners: The compound was originally licensed by Eli Lilly from Centrexion Therapeutics in 2019 for an upfront fee of $47.5 million.
- Discontinuation: In mid-2025, Eli Lilly officially dropped the drug. Phase II clinical trials revealed that its efficacy did not meet the high success thresholds required to continue human testing.
- Side Effects: In clinical studies, reported treatment-emergent adverse effects were generally mild to moderate. They included constipation, nausea, dizziness, fatigue, and headaches.
Mazisotine (LY3556050, CNTX-0290) is a chemical compound which acts as an agonist at somatostatin receptor 4. It has analgesic effects and has been researched for the treatment of pain associated with arthritis and neuropathic pain. It was not pursued for human medical use following disappointing results in Phase II clinical trials, but continues to be used in research into the role of SST4 receptors in pain perception.[1][2]
- A Study of LY3556050 in Adult Participants With Diabetic Peripheral Neuropathic PainCTID: NCT06074562Phase: Phase 2Status: CompletedDate: 2025-07-03
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With OsteoarthritisCTID: NCT04627038Phase: Phase 2Status: CompletedDate: 2023-11-02
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04707157Phase: Phase 2Status: TerminatedDate: 2023-11-02
- Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Chronic Low Back PainCTID: NCT04874636Phase: Phase 2Status: CompletedDate: 2023-11-02
- A Study of Effect of LY3556050 on Metformin in Healthy ParticipantsCTID: NCT05615467Phase: Phase 1Status: CompletedDate: 2023-01-18
- A Study of Single and Repeated Doses of LY3556050 in Healthy ParticipantsCTID: NCT05341102Phase: Phase 1Status: CompletedDate: 2022-04-22
- A Study of LY3556050 in Healthy ParticipantsCTID: NCT04156750Phase: Phase 1Status: CompletedDate: 2020-08-19
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025096300&_cid=P12-MQYLW5-94119-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US214328185&_cid=P12-MQYLW3-94084-1
PAT

reparation 1
[0081] Methyl (E)-4-((4-methylphenyl)sulfonamido)but-2-enoate

[0082] Methyl 4-bromobut-2-enoate (36.29g, 202.7mmol) was dissolved in MeCN (500 mL) at 15-25 ºC. tert-Butyl tosylcarbamate (50.00 g, 184.3 mmol) was added at 15-25 ºC. K2CO3 (30.57 g, 221.2mmol) and KI (3.06 g, 202.7 mmol) were added to the solution at 15-25 ºC, and warmed under nitrogen at 30 ºC for 20 hrs. The solution was cooled to 20 ºC and the mixture filtered. The filtered residue was washed with MeCN (100 mL) to give methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate. TFA (101.03 g, 886.06 mmol) was added to the methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate in MeCN solution (483.72 g, 143 mmol) and heated to 55-60 ºC for 16 hrs. The reaction solution was concentrated in vacuo to ~50 mL and solvent exchanged with toluene (2 x 250 mL). Toluene (500 mL) was added followed by EtOAc (50 mL) at 15-25 °C, and heated to 60 ºC for 1 hr., then cooled to 0 ºC for 12 hrs. The solution was filtered, and the wet cake was rinsed with n-heptane (50 mL). The cake was dried in vacuum at 50 ºC to give the title compound (37.85 g, 74.4%) as a white solid.1H NMR (CDCl3) δ 7.68 (d, J = 8.0Hz, 2H) 7.25 (d, J = 8.0Hz, 2H) 6.71 (dt, J = 15.6, 5.2Hz, 1H) 5.88 (dt, J = 15.6, 1.6Hz, 1H) 4.55 (t, J = 6.4Hz, 1H) 3.71 – 3.67 (m, 2H) 3.65 (s, 3H) 2.37 (s, 3H); HRMS (ESI+) Calculated for [C12H15NO4S+H] +: 270.0795, Found: 270.0788 (M+H).
Preparation 2
[0083] (1R,5S,6r)-3-Tosyl-3-azabicyclo[3.1.0]hexane-6-carboxylic acid

0084] Methyl (E)-4-((4- 2-enoate (51.90 g) was dissolved in 2-MeTHF (600mL) at 0 ºC . Added (2-bromoethyl)diphenylsulfonium triflate (36.50 g), KF (6.47 g), KOH (18.75 g) to the solution at 0 ºC. Warmed the solution to 15 ºC for 22 hrs. then 30 ºC for 3 hrs. Added water (100 mL) and MeOH (100 mL) into the solution. Added LiOH.H2O (4.77 g) and stirred at 30 ºC for 16 hrs. Cooled to 15-25oC and added n-heptane (100 mL). Stirred at 15-25oC for 10 min. Separated and collected the aqueous phase and washed the aqueous phase with n-heptane/2-MeTHF (50 mL/200 mL × 2). Concentrated the aqueous phase in vacuo to ~50 mL and added 3M aq. HCl
dropwise to adjust pH to 1~2. Stirred the mixture at 20-30 ºC for 2 hrs. Filtered the solution and rinsed through with EtOH/H2O (15 mL1:4). Dried the wet cake at 45 ºC for 8-10 hrs. to give the title compound as white solid (20.37 g, 65%) 1H NMR (CDCl3) δ 7.67 (d, J = 8.2 Hz, 2 H) 7.34 (d, J = 8.2 Hz, 2 H) 3.63 (d, J = 9.4 Hz, 2 H) 3.12 (d, J = 9.4 Hz, 2 H) 2.46 – 2.40 (m, 4 H) 2.07 – 2.01 (m, 2 H); HRMS (ESI+) Calcd. for [C13H15NO4S+H] +: 282.0795, Found: 282.0795 (M+H).
Preparation 3
[0085] 2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-amine

0086] 2-Amino-2-methylpropan- mmol) was dissolved in toluene (250 mL) at 15-25 ºC. Added potassium tert-butoxide (60.59 g, 534.6 mmol) to the solution and warmed to 30-40 ºC. Added 2-fluoro-3-methylpyridine (50.00 g, 450.0 mmol) to the solution and warmed to 80 ºC. Stirred the mixture for 18 hrs. at 80 ºC. Cooled the mixture to 15-25 ºC, added water (100 mL) and stirred for 30 min. Separated the aqueous layer and washed the organics with 10% aq. NaCl (300 mL x 3). Added toluene (250 mL) to the organics and concentrated in vacuo to give the title compound as a liquid (107.50 g, 98%). 1H NMR (CDCl3) δ 7.92 – 7.82 (m, 1H) 7.32 – 7.22 (m, 1H) 6.74 – 6.66 (m, 1H) 3.97 (s, 3H) 2.14 (s, 3H) 1.15 (s, 6H).
Preparation 4
[0087] (1R,5S,6r)-N-(2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-yl)-3-tosyl-3-azabicyclo[3.1.0]hexane-6-carboxamide

[0088] Dissolved
6-carboxylic acid (32.33 g, 88.2% purity) in toluene (320 mL) at 15~25 ºC. Added DMF (741.0 mg) and (COCl)2 (19.20 g) to the solution and heated to 45-55 ºC for 3~4 hrs. Concentrated the mixture in vacuo and exchanged with THF (100 mL × 2). Added THF (320 mL) and cooled to 0-10 ºC. Added 2-methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-amine (23.60 g), TEA (30.80 g), DMAP (620.0 mg) at 0-10 ºC then warmed to 15-25 ºC for 2~4 hrs.
Concentrated the mixture in vacuo and solvent exchanged with EtOH (140 mL × 2). Concentrated in vacuo to 140 mL and heated to 50 ºC until the solid was dissolved. Added water (210 mL) dropwise into the solution at 40 ºC. Cooled the solution to 10 ºC for 14 hrs. Filtered and rinsed with EtOH/H2O (75 mL, 1:1.5). Dried the wet cake at 45 ºC for 20 hrs. to give the title compound as a solid (41.87 g, 87.4%).1H NMR (CDCl3) δ 7.94 – 7.87 (m, 1 H) 7.60 (d, J=8.0 Hz, 2 H) 7.37 (d, J=6.6 Hz, 1 H) 7.26 (d, J=8.0 Hz, 2 H) 6.78 (dd, J=6.6, 5.0 Hz, 1 H) 6.48 (s, 1 H) 4.25 (s, 2 H) 3.52 (d, J=9.4 Hz, 2 H) 2.95 (d, J=9.4 Hz, 2 H) 2.39 – 2.33 (m, 3 H) 2.17 (s, 3 H) 1.84 (s, 2 H) 1.39 (s, 6 H); HRMS (ESI+) Calcd. for [C23H29N3O4S+H] +: 444.1952, Found: 444.2089 (M+H).
Preparation 5
[0089] (1R,5S,6r)-N-(2-Methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-yl)-3-azabicyclo[3.1.0]hexane-6-carboxamide

[0090] Dissolved 2-yl)oxy)propan-2-yl)-3-tosyl-3-azabicyclo[3.1.0]hexane-6-carboxamide (5.00 g) in MTBE (50 mL) under nitrogen. Cooled to -70-60 ºC and added dropwise 1M Ph2PK in THF (56 mL, 56 mmol) into the solution. Stirred the mixture for 6-8 hrs. at -70-60 ºC. Added 2M aq. HCl (50 mL) to the solution allowing the temperature to rise to 15-25 ºC. Separated and collected the aqueous phase and washed with 2-MeTHF (50 mL × 3). Added 2-MeTHF (50 mL) and adjusted the pH to 8~9 with K2CO3 powder. Separated and extracted the aqueous phase with 2-MeTHF (50 mL). Combined the organics and concentrated in vacuo to give the title compound as a yellow-brown solid (3.05 g, 89.4%) 1H NMR (CDCl3) δ 8.01 – 7.92 (m, 1 H) 7.45 – 7.36 (m, 1 H) 6.81 (dd, J=7.0, 5.0 Hz, 1 H) 6.39 (s, 1 H) 4.31 (s, 2 H) 3.09 – 2.89 (m, 4 H) 2.21 (s, 3 H) 1.94 – 1.86 (m, 2 H) 1.69 (s, 1 H) 1.47 (s, 6 H) 1.12 (t, J=2.8 Hz, 1 H); HRMS (ESI+) Calcd. for [C16H23N3O2+H] +: 290.1863, Found:
290.1917 (M+H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
References
- Stevens R, Corradini L, Doods H (April 2019). “Preclinical Evaluation of Human Somatostatin Receptor 4 (hSSTR4) Agonist CNTX-0290 for Mixed Pain Conditions”. The Journal of Pain. 20 (4): S73. doi:10.1016/j.jpain.2019.02.092.
- Nguyen TH, Saito T, Chang W, Navarro A, Davies HM (March 2026). “Diastereoselective Cyclopropanation with Secondary Diazoacetamides to Access endo-Azabicyclo[3.1.0]hexane-6-carboxamides”. Organic Letters. 28 (9): 3063–3067. doi:10.1021/acs.orglett.6c00392. PMC 12973298. PMID 41729728.
 | |
| Clinical data | |
|---|---|
| Other names | LY3556050, CNTX-0290 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1638588-92-7 |
| PubChem CID | 86294067 |
| ChemSpider | 77005706 |
| UNII | D3M32WP3MH |
| ChEMBL | ChEMBL5874446 |
| Chemical and physical data | |
| Formula | C16H23N3O2 |
| Molar mass | 289.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
PAT
- Novel methods for the preparation of 3-azabicylco[3.1,0]hexane-6-carboxamide derivativesPublication Number: WO-2025096300-A1Priority Date: 2023-10-30
- Methods for the preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: WO-2024191686-A1Priority Date: 2023-03-10
- Methods for the preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: US-2024307349-A1Priority Date: 2023-03-10
- Methods for preparation and dose regimens for use of sstr4 agonists and salts thereofPublication Number: JP-2024170613-APriority Date: 2023-03-10
- Methods for the preparation of SSTR4 agonists and their salts and dosage regimens for usePublication Number: JP-7569953-B2Priority Date: 2023-03-10Grant Date: 2024-10-18
- Sstr4 agonist saltsPublication Number: US-2024067628-A1Priority Date: 2021-09-14
- SSTR4 agonist saltsPublication Number: US-11834435-B2Priority Date: 2021-09-14Grant Date: 2023-12-05
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: EP-2997021-B1Priority Date: 2013-05-17Grant Date: 2017-12-20
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-9371282-B2Priority Date: 2013-05-17Grant Date: 2016-06-21
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-10166214-B2Priority Date: 2013-05-17Grant Date: 2019-01-01
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2019269650-A1Priority Date: 2013-05-17
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: WO-2014184275-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-10675268-B2Priority Date: 2013-05-17Grant Date: 2020-06-09
- New Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-2014343065-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (SSTR4) agonistsPublication Number: US-9789082-B2Priority Date: 2013-05-17Grant Date: 2017-10-17
- New somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2017014381-A1Priority Date: 2013-05-17
- Somatostatin receptor subtype 4 (sstr4) agonistsPublication Number: US-2018092880-A1Priority Date: 2013-05-17
////////mazisotine, ANAX LABS, somatostatin receptor receptor agonist, LY3556050, CNTX-0290, LY 3556050, CNTX 0290, D3M32WP3MH, PAIN, NEUROPATHIC PAIN
Nispomeben
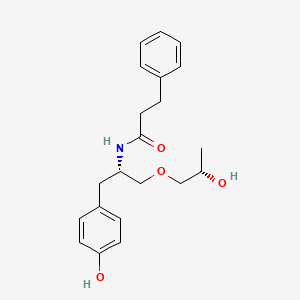
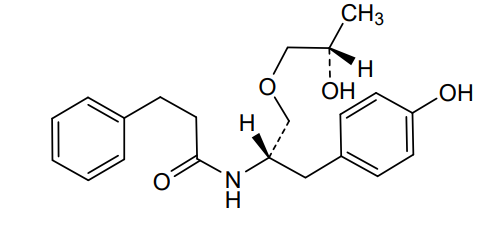
Nispomeben
CAS 1443133-41-2
MF C21H27NO4 MW357.4 g/mol
N-[(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl]-3-phenylpropanamide
N-{(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl}-3-phenylpropanamide
non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1
Nispomeben is a small molecule drug. Nispomeben has a monoisotopic molecular weight of 357.19 Da.
- OriginatorNovaremed
- ClassAlcohols; Amides; Anti-inflammatories; Benzene derivatives; Non-opioid analgesics; Phenols; Small molecules
- Mechanism of ActionLyn protein-tyrosine kinase modulators
- Phase IINeuropathic pain
- 02 Sep 2025Updated adverse events data from a phase II trial in Neuropathic pain released by Novaremed
- 07 May 2025Novaremed completes enrolment in a phase-II clinical trial in Neuropathic pain in USA (PO) (NCT05480228)
- 16 Sep 2022Phase-II clinical trials in Neuropathic pain in USA (PO) (NCT05480228)
PAT
WO 2013/084238
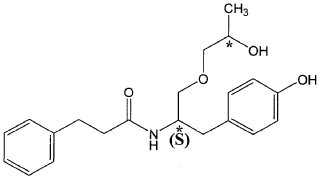
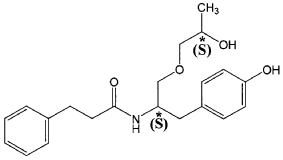
The present invention is based in part on the surprising discovery that the substantially pure enantiomers (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide (also known as the (S,S) enantiomer or El) and (S)2-N(3-0-((R)propan 2-ol)-l -propyl -4-hydroxybenzene)-3-phenylpropyl amide (also known as the (S,R) enantiomer or E2) modulate the activity of specific tyrosine kinases in an opposite manner. It was unexpectedly found that while the (S,S) enantiomer activated protein tyrosine kinases LynA and BLK, the (S,R) enantiomer inhibited their activity. It was further unexpectedly shown that the (S,S) enantiomer was effective as a pain analgesic in animal models of pain, while the (S,R) enantiomer was shown to be ineffective or less effective in these models. Furthermore, the analgesic effect of the (S,S) enantiomer was long acting as it was efficacious for more than 24 hours post administration, in comparison to the commonly used analgesic agent gabapentin which was effective for no longer than 5 hours post administration.
The isolated enantiomers according to some embodiments of the invention may be synthesized as a racemate by known in the art methods described for example in US 7,754,771, US 7,642,290, US 7,674,829 or US 2011/0086910. The racemate may be further separated by known in the art methods for the separation of chiral compounds. According to an exemplary embodiment, the enantiomers may be synthesized as a racemate (comprising (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and (S)2-N(3-0-((R)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and be further separated by a supercritical fluid chromatography (SFC) in combination with chiral stationary phases. Specifically, the (S,S) and (S,R) compounds may be separated on RegisPack™ column a polysaccharide coated chiral column (with a tris-(3,5-dimethylphenyl) carbamoyl cellulose selector) generally used for enantiomeric separations of a wide range of racemate classes (Figure 7A-C).
According to some embodiments, the enantiomers may be synthesized directly using for example, the process described in scheme 1 for the preparation of the (S,S) enantiomer.
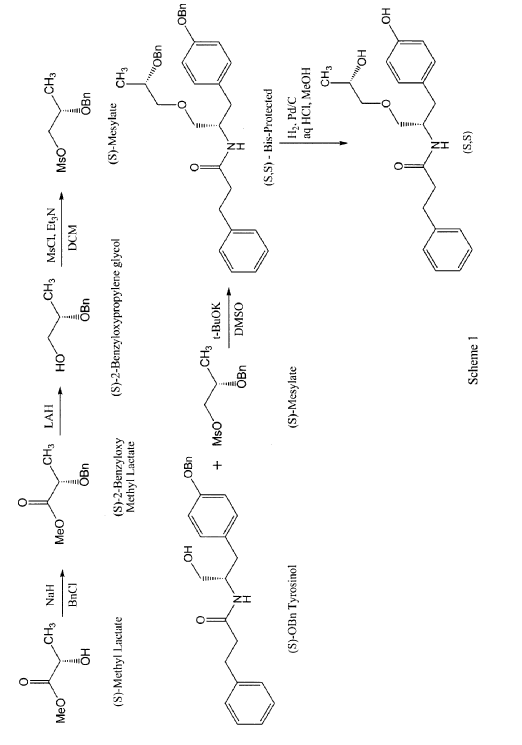
PAT
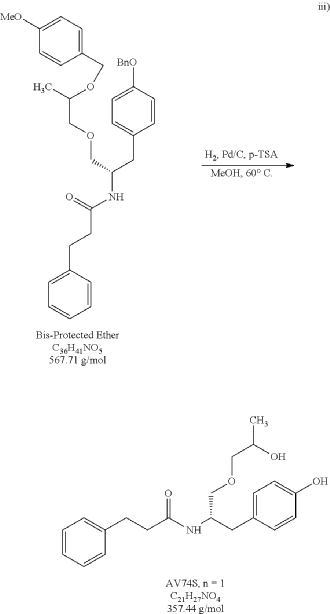
The bis-protected ether (15.7 g) was exposed to one-pot hydrogenation-debenzylation conditions (10% loading of 10% Pd/C and 0.25 eq of p-toluenesulfonic acid) in methanol. After 2 hours at 60° C. under a hydrogen atmosphere, HPLC analysis indicated that the hydrogenation of the benzyl and the debenzylation of PMB ring was complete. The reaction mixture was filtered over Celite and concentrated under reduced pressure. The residue was dissolve in ethyl acetate and a saturated aqueous sodium bicarbonate treatment was conducted to effectively remove p-toluenesulfonic acid, then DURP to provide 12.13 g of an oil (PR030-120-4). Desired product was isolated from an EA/Heptane recrystallization to provide 8.83 g of a white solid (PR030-120-6, 89.4% yield). The purity of PR030-120-6 was 99.3% via HPLC analysis. 1H NMR and Mass spec analysis supported the assigned structure for desired product.
PAT
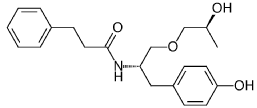
((S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide), including its enantiomers and diastereomers may be prepared as described in WO 2013/084238,
Example 1 – Preparation of -2-N(3-Q-(propan-2-ol)-1-propyl-4-hvdroxybenzene)-3-
phenylpropylamide
(S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 201 1/0086910.
In a first step, 2 g of methyl lactate was reacted with excess of benzyl bromide to get 880 mg of (S)-benzyloxymethyl lactate. The reaction was performed by slurring sodium hydride in THF and cooling down to approximately -15°C. The reaction mixture was then allowed to warm slowly to room temperature and stirred for approximately 1 to 2 hours. The reaction was quenched with saturated ammonium chloride solution and extracted with MTBE twice followed by the removal of solvent on a rotary evaporator to obtain a crude oil. The crude product was purified by column chromatography to yield pure (S)-2-benzyloxymethyl lactate. The (R)-2-benzyloxymethyl lactate isomer was present at 0.93% only. The yield of this step may be increased by avoiding the presence of moisture in the reaction solution.
In a second step, 880 mg (S)-2-benzyloxymethyl lactate obtained in step 1 were reduced using lithium aluminum hydride to obtain (S)-2-benzyloxypropylene glycol in 83.8% yield with 98.7% purity. A solution of pure (S)-2-benzyloxymethyl lactate in methylene chloride was stirred and a solution of lithium aluminum hydride was slowly added thereto at approximately 5°C. The reaction was monitored by TLC and quenched by USP-PW water very carefully. No racemization occurred in this step.
In a third step, the (S)-2-benzyloxypropylene glycol was then reacted with methane sulfonyl chloride in methylene chloride in the presence of triethyl amine to yield the mesylate in 88% yield. A solution of step 2 was stirred in methylene chloride and methane sulfonyl chloride was added to it dropwise at <5°C. After the addition was complete, the progress of the reaction was monitored by TLC. The reaction was quenched with USP-PW water. After the layers were separated, the aqueous layer was back extracted with methylene chloride. The methylene chloride layers were then combined and washed with USP-PW water 3 times to remove most of the methane sulfonic acid. No racemization occurred in this step.
In a fourth step, the mesylate (of step 3) was coupled with S-O-benzyl tyrosinol to form the bis-protected product in 22.7% yield, with a purity of 97.4%. The reaction was carried out at room temperature using a combination of DMF as the solvent and sodium hydride as the base. The reaction went to completion after stirring for at least 12 hours at room temperature.
In a fifth step, 340 mg of the product of step 4 were reduced by hydrogenation in the presence of 10% palladium on carbon catalyst and hydrochloric acid using methylene chloride as a solvent at 50°C. The reaction went to completion in approximately 4 hours with no racemization to yield the desired product in 84.3% yield and 98.9% purity. More specifically, the catalyst was removed by filtration and the filtrate was then concentrated at 33°C. The resulting mixture of solid and oil was mixed with ethyl acetate. The resulting slurry was filtered and the solids washed with ethyl acetate and dried under vacuum at 40 to 45°C to obtain the desired product.
PAT
Example 1—Preparation of (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide
| (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 2011/0086910. |
PAT
- Method of treating or preventing painPublication Number: US-2016317479-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-8802734-B2Priority Date: 2009-09-09Grant Date: 2014-08-12
- Method of Treating or Preventing PainPublication Number: US-2014350099-A1Priority Date: 2009-09-09
- N-substituted benzenepropanamide or benzenepropenamide for use in the treatment of pain and inflammationPublication Number: WO-2011030205-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-2011086910-A1Priority Date: 2009-09-09
- Isolated stereoisomeric forms of (S)2-N(3-O-(propan 2-Ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamidePublication Number: US-9381173-B2Priority Date: 2011-12-08Grant Date: 2016-07-05
- Isolated Stereoisomeric Forms Of (S)2-N(3-O-(Propan 2-Ol)-1-Propyl-4-Hydroxybenzene)-3-PhenylpropylamidePublication Number: US-2014275270-A1Priority Date: 2011-12-08
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: US-9133103-B2Priority Date: 2011-09-21Grant Date: 2015-09-15
- N-Substituted Benzenepropanamide and Benzenepropenamide For Use in the Prevention or the Treatment of Affective DisordersPublication Number: US-2014275273-A1Priority Date: 2011-09-21
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: EP-2758046-B1Priority Date: 2011-09-21Grant Date: 2015-10-21
- Compounds for treatment or prevention of an infection resulting from a coronavirus and/or a coronavirus-induced diseasePublication Number: EP-3939578-A1Priority Date: 2020-07-13
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: EP-3860582-B1Priority Date: 2019-01-23Grant Date: 2022-05-04
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: WO-2020152226-A1Priority Date: 2019-01-23
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: US-2022002228-A1Priority Date: 2019-01-23
- Pharmaceutical composition comprising stereoisomers of n-(1-(4-hydroxyphenyl)-3-(2-hydroxypropoxy)propan-2-yl)-3-phenylpropanamide for the prevention and treatment of type ii diabetesPublication Number: WO-2015173813-A1Priority Date: 2014-05-14

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////nispomeben, non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1, Neuropathic pain
Pudafensine
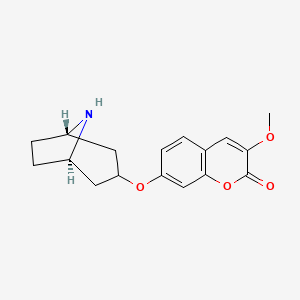
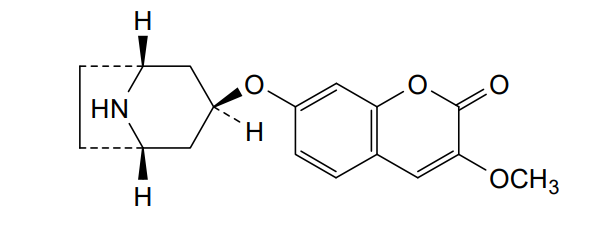
Pudafensine
CAS 1320346-14-2
MFC17H19NO4 MW 301.34 g/mol
7-{[(1R,3s,5S)-8-azabicyclo[3.2.1]octan-3-yl]oxy}-3-methoxy2H-1-benzopyran-2-one
monoamine reuptake inhibitor, erectile dysfunction, neuropathic pain, NS18313, NS 18313, L9NG7US8GE, IP2015, IP 2015
Pudafensine is a monoamine reuptake inhibitor being developed as a potential treatment for erectile dysfunction (ED) and neuropathic pain. As a drug candidate, it works by preferentially inhibiting the reuptake of dopamine and serotonin. It is designed to be a first-line treatment for patients with organic ED who are not adequately served by existing therapies like PDE5 inhibitors.
How it works
- Pudafensine is a monoamine reuptake inhibitor that increases the levels of dopamine and serotonin in the brain by preventing their reabsorption into neurons.
- It has been shown in animal models and human trials to improve erectile function and reduce pain, including neuropathic pain.
Potential uses
- Erectile Dysfunction (ED): Pudafensine is being investigated for its potential to help men with organic ED who do not respond well to or cannot tolerate current treatments. Phase IIb clinical trial results are expected in late 2023.
- Neuropathic Pain: A clinical trial on pain involving pudafensine indicated it reduced allodynia and was well-tolerated with a favorable safety profile compared to pregabalin.
Development status
- Initiator Pharma is developing pudafensine as an oral tablet.
- Phase IIb studies for erectile dysfunction and Phase II studies for neuropathic pain have been completed, with positive results.
- The company is exploring its use in treating patients who are inadequately treated with existing medications.
Erectile dysfunction (ED)
Pudafensine, Initiator’s most advanced drug program has successfully demonstrated efficacy in a Clinicial Phase 2a Proof-of-Concept study and in a Phase 2b study to treat patients who suffer from organic erectile dysfunction (ED) that do not respond or cannot tolerate the currently marketed drugs in the PDE5i class (e.g. Viagra®, Cialis®, Levitra®).
Pudafensine strengthens the natural erection response by having a dual-action, both a central effect initiating erection and a peripheral effect potentiating erection through smooth muscle relaxation. Pudafensine is aimed for treatment of organic erectile dysfunction in patients who have erectile dysfunction (ED) due to abnormalities of the penile arteries and/or veins. Most common risk factors for organic ED are diabetes, overweight, lack of exercise, high cholesterol, high blood pressure, and cigarette smoking. Since Initiator Pharma was founded and pudafensine acquired, all preclinical development of the drug candidate to enable an application for clinical trials (CTA) has been carried out by the company’s auspices. Pudafensine is developed as a tablet that is taken orally on-demand. It is the company’s goal to be able to create a new “First-Line” treatment (recommended treatment) for the large group of men who have organic erectile dysfunction, who are sub-optimally treated with PDE5i products or for whom PDE5i treatment is contraindicated.
In Q4 2023 positive results from the Phase IIb clinical trial with pudafensine (IP2015) was announced. The Phase 2b trial is a randomized, double-blind, placebo-controlled, parallel-dosing group trial studying the efficacy and safety of high and low doses of pudafensine (IP2015) and placebo in otherwise healthy patients suffering from moderate to severe ED. The study comprises 130 patients divided into 3 parallel arms receiving a higher and a lower dose of pudafensine and placebo, respectively, with treatment duration of 4 weeks with frequent assessments of erectile dysfunction, safety and pharmacokinetics. The study has been conducted at the MAC clinical sites in the UK.
The study demonstrated statistically significant efficacy on the primary endpoint (related to improvements in intercourse settings) compared to placebo [p=0.034] and baseline [p=0.046]. Furthermore, the results were consistent throughout the study. Several other clinical endpoints related to improved intercourse activities (obtained from the International Index of Erectile Function Questionnaire, IIEF-15) demonstrated significant effects compared to the baseline. The frequency and type of adverse effects were mild to moderate and comparable to those observed in the placebo group. There was no reporting of critical safety observations.
Neuropathic pain
Pudafensine have shown effects in a human model of pain ie. in a clinical Phase I study in healthy subjects dosed with the drug pudafensine and challenged with a pain-inducing ingredient (capsaicin).
The Phase I study was a randomized, double blind, placebo controlled study in 24 healthy male subjects, investigating the effects on pain measures (hyperalgesia, allodynia, and subjects pain rating) of single doses of pudafensine, pregabalin as an active control, and placebo. The pain was induced by intradermal capsaicin. Pudafensine demonstrated a statistically significant effect on allodynia (p=0.049) and showed a dose-dependent effect on the measured pain parameters. Pregabalin (p=0.083) and IP2015 (p=0.051) tended to reduce hyperalgesia, although the effects on hyperalgesia were not statistically significant compared to placebo-treated subjects.
Syn
US20130040985
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76705962&_cid=P22-MIFE0H-55553-1
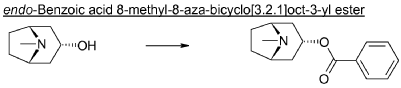
endo-Benzoic acid 8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl ester
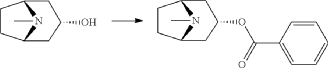
Benzoylchloride (84.3 g, 600 mmol) was added during 30 min at <30° C. to a mixture of tropine (70.6 g, 500 mmol), potassium tert-butoxide (67.3 g, 600 mmol) and THF (500 ml). The mixture was stirred at room temperature for 2 h. Water (1 L) was added followed by extraction with diethylether (2×500 ml). The organic phase was washed twice with water (2×200 ml) followed by a solution of saturated aqueous sodium chloride (200 ml). The ether phase was dried and hydrochloric acid in ethanol (170 ml, 3 M) was added. The precipitated hydrochloride was filtered and washed with diethylether. The free base was obtained by adding an excess of aqueous ammonia followed by extraction with a mixture of ethylacetate and diethylether. Yield 66.8 g (54%).
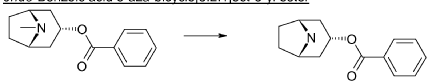
endo-Benzoic acid 8-aza-bicyclo[3.2.1]oct-3-yl ester
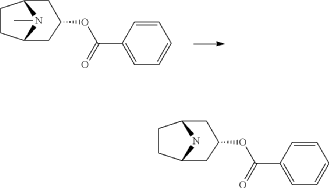
2,2,2-Trichloroethylchloroformate (75.0 ml, 544 mmol) was added dropwise to a mixture of endo-benzoic acid 8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl ester (66.8 g, 272 mmol) and dry toluene (500 ml). The mixture was allowed to stir for 1 h at room temperature, followed by 15 h at 100° C. Water (250 ml) was added followed by stirring 1 h. The phases were separated and the organic phase was washed twice with water (2×200 ml). The mixture of the intermediate 3-benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid trichloromethyl ester, was dried and evaporated. Acetic acid (350 ml) was added followed by addition of zinc (53.4 g, 817 mmol) over 3 h time period. Water (100 ml) was added, cooled by adding ice and made alkaline by adding concentrated aqueous ammonia (ca: 400 ml) and the mixture was extracted with dichloromethane (2×300 ml). Yield 44.5 g (61%).
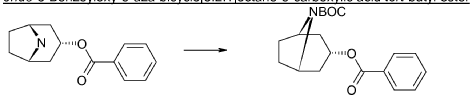
endo-3-Benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester
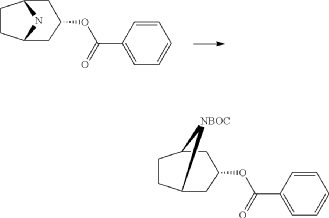
Di-tert-butyl-dicarbonate (39.9 g, 183 mmol) solved in THF (100 ml) was added to a stirred mixture of endo-benzoic acid 8-aza-bicyclo[3.2.1]oct-3-yl ester (44.5 g, 166.4 mmol), triethylamine (67.4 g, 666 mmol) and THF (250 ml) during 0.5 h at room temperature, followed by stirring for 1 h. Water (1 L) was added and the mixture was extracted with diethylether (2×300 ml). The collected ether phase was washed twice with water (2×200 ml), dried and evaporated. Yield 60.1 g (100%).
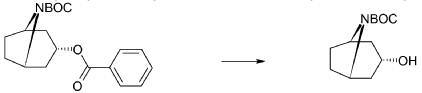
endo-3-Hydroxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester
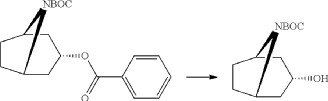
A mixture of endo-3-benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (55.0 g, 166 mmol), potassium hydroxide (11.2 g 199 mmol) and ethanol (99%, 400 ml) was stirred for 3 days at room temperature. Potassium benzoate was separated by filtration and the filtrate was evaporated. Diethylether (200 ml) was added and remaining potassium benzoate was separated by filtration and the filtrate was evaporated. The product was triturated with petroleum. Yield 30.0 g (80%). Mp 139.5-140.8° C.
xample 1
Exo-tert-butyl-3-(3-methoxy-2-oxo-chromen-7-yl)oxy-8-azabicyclo[3.2.1]octane-8-carboxylate (Intermediate)
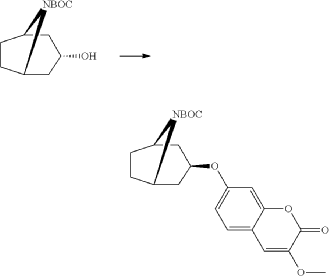
Triphenylphosphine (1.15 g, 4.37 mmol) was solved in toluene (20 ml) and cooled to <20° C. Diethylazodicarboxylate (40% in toluene) (2.0 ml, 4.37 mmol) was added to the mixture below 20° C., followed by stirring for 10 minutes. endo-3-Hydroxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (0.828 g, 3.64 mmol) was added and after 10 minutes 7-hydroxy-3-methoxy-chromen-2-one (0.70 g, 3.64 mmol) (prepared according to J. Med. Chem. 1999, 42, p2662-2672) was added to the mixture. The temperature raised to 25° C. due to an exothermic reaction. The mixture precipitates. The mixture was allowed to stir for 15 h at room temperature. Water (20 ml) and sodium hydroxide (0.5 ml, 4 M) was added followed by stirring. The mixture was cooled on an ice-bath, filtered and washed with water and diethylether. Yield 0.92 g (63%).
Exo-7-[(-8-azabicyclo[3.2.1]octan-3-yl)oxy]-3-methoxy-chromen-2-one hydrochloride (Compound 1.1)
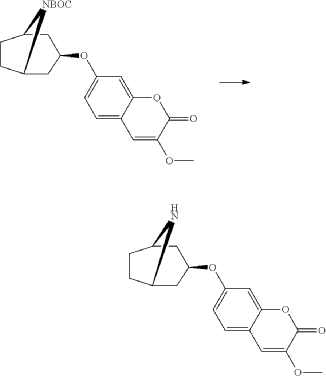
Exo-tert-butyl-3-(3-methoxy-2-oxo-chromen-7-yl)oxy-8-azabicyclo[3.2.1]octane-8-carboxylate (0.92 g, 2.29 mmol) and hydrogen chloride (15 ml, 1 M) in acetic acid was mixed as a solution and stirred at room-temperature and precipitated after a few minutes. The product was filtered and washed with diethylether. Yield 0.48 g (62%). LC-ESI-HRMS of [M+H]+ shows 302.13856 Da. Calc. 302.138689 Da, dev. −0.4 ppm.
Syn
WO2011092061
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011092061&_cid=P22-MIFE80-61015-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024008808&_cid=P22-MIFDSB-50229-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024089247&_cid=P22-MIFDSB-50229-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024146892&_cid=P22-MIFDSB-50229-1
PAT
- Compound for treatment of erectile dysfunctionPublication Number: WO-2024146892-A1Priority Date: 2023-01-03
- Compound for treatment of painPublication Number: WO-2024089247-A1Priority Date: 2022-10-28
- Compound for treatment of female sexual dysfunctionPublication Number: WO-2024008808-A1Priority Date: 2022-07-08
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Pudafensine, monoamine reuptake inhibitor, erectile dysfunction, neuropathic pain, NS18313, NS 18313, L9NG7US8GE, IP2015, IP 2015
Pilavapadin
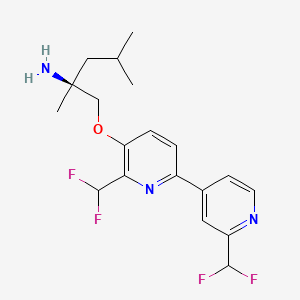

Pilavapadin
CAS1815613-42-3
MFC19H23F4N3O MW 385.4 g/mol
(2S)-1-{[2′,6-bis(difluoromethyl)[2,4′-bipyridin]-5-yl]oxy}-2,4-dimethylpentan-2-amine
(2S)-1-[[2-(difluoromethyl)-6-[2-(difluoromethyl)-4-pyridinyl]-3-pyridinyl]oxy]-2,4-dimethylpentan-2-amine
adaptor protein 2-associated kinase 1 (AAK1) inhibitor, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
Pilavapadin (also known as LX9211 or BMS-986176) is an investigational, orally available small molecule developed by Lexicon Pharmaceuticals for the treatment of neuropathic pain, primarily diabetic peripheral neuropathic pain (DPNP).
Key Information
- Mechanism of Action: Pilavapadin is a selective inhibitor of AAK1 (AP2 associated kinase 1), a novel target identified through Lexicon’s gene science research. It is designed to inhibit the reuptake and recycling of neurotransmitters involved in pain signaling in the central nervous system without affecting opiate pathways.
- Indication: It is being investigated for the management of chronic and debilitating conditions such as diabetic peripheral neuropathic pain (DPNP), chemotherapy-induced peripheral neuropathy (CIPN), and multiple sclerosis (MS) pain.
- Development Stage: Pilavapadin has completed Phase 2 clinical trials for DPNP and is expected to advance to a Phase 3 trial.
- Status/Designation: The U.S. Food and Drug Administration (FDA) has granted Fast Track designation for the development of pilavapadin in DPNP.
Clinical Trial Results
Phase 2 studies (RELIEF-DPN-1 and PROGRESS) demonstrated that pilavapadin can provide meaningful pain reduction in adults with DPNP.
- In a post-hoc analysis of the PROGRESS study, the 10 mg dose was found to be effective, achieving a clinically meaningful, two-point reduction in average daily pain scores from baseline, with an acceptable safety and tolerability profile.
- The data has been presented at several medical meetings, including the European Association for the Study of Diabetes (EASD).
- OriginatorBristol-Myers Squibb; Lexicon Pharmaceuticals
- DeveloperLexicon Pharmaceuticals
- ClassAnalgesics; Small molecules
- Mechanism of ActionAdaptor-associated kinase 1 inhibitors
- Phase IINeuropathic pain; Postherpetic neuralgia
- 20 Jun 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain presented at 85th Annual Scientific Sessions of the American Diabetes Association (ADA-2025)
- 13 May 2025Lexicon Pharmaceuticals plans an End of Phase 2 meeting with FDA for Pilavapadin
- 13 May 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain released by Lexicon Pharmaceuticals
- A Dose-ranging Study in Patients With Diabetic Peripheral Neuropathic Pain (DPNP)CTID: NCT06203002Phase: Phase 2Status: CompletedDate: 2025-08-29
- Efficacy, Safety, and PK of LX9211 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04455633Phase: Phase 2Status: CompletedDate: 2025-06-25
- Efficacy and Safety of LX9211 in Participants With Postherpetic NeuralgiaCTID: NCT04662281Phase: Phase 2Status: CompletedDate: 2023-11-18
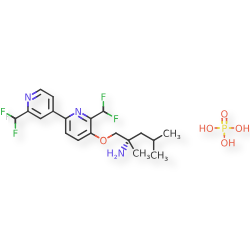
Molecular FormulaC19H23F4N3O.H3O4P
Molecular Weight483.4
CAS 2977251-24-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215884039&_cid=P11-MI4ESM-19570-1
Example 123
(S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine

Part A: (2-(difluoromethyl)pyridin-4-yl)boronic acid

Part B: (S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021216441&_cid=P11-MI4EP8-16561-2
REF
- Discovery and Optimization of Biaryl Alkyl Ethers as a Novel Class of Highly Selective, CNS-Penetrable, and Orally Active Adaptor Protein-2-Associated Kinase 1 (AAK1) Inhibitors for the Potential Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-09PMID: 35261239DOI: 10.1021/acs.jmedchem.1c02132
- Discovery of (S)-1-((2′,6-Bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine (BMS-986176/LX-9211): A Highly Selective, CNS Penetrable, and Orally Active Adaptor Protein-2 Associated Kinase 1 Inhibitor in Clinical Trials for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-08PMID: 35257579DOI: 10.1021/acs.jmedchem.1c02131
- Discovery, Structure–Activity Relationships, and In Vivo Evaluation of Novel Aryl Amides as Brain Penetrant Adaptor Protein 2-Associated Kinase 1 (AAK1) Inhibitors for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-07-16PMID: 34270254DOI: 10.1021/acs.jmedchem.1c00472
PAT
- Biaryl kinase inhibitorsPublication Number: US-2021277001-A1Priority Date: 2014-04-02
- Biaryl kinase inhibitorsPublication Number: KR-102379518-B1Priority Date: 2014-04-02Grant Date: 2022-03-25
- Biaryl kinase inhibitorsPublication Number: US-12065437-B2Priority Date: 2014-04-02Grant Date: 2024-08-20
- Biaryl kinase inhibitorsPublication Number: US-2024360131-A1Priority Date: 2014-04-02
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////////Pilavapadin, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
TRK 700
![1-[4-(Dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=71738795&t=l)
TRK-700
CAS 1463432-16-7C14 H24 N4 O264.371-Propanone, 1-[4-(dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-
1-[4-(dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one
- 1-[4-(Dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-1-propanone
- OriginatorToray Industries
- ClassAnalgesics
- Mechanism of ActionUndefined mechanism
- Phase IIPostherpetic neuralgia
- PreclinicalPeripheral nervous system diseases
- 12 Sep 2018Pharmacodynamics data from a preclinical trial in Peripheral neuropathy presented at the 17th World Congress on Pain (WCP-2018)
- 01 Jul 2017Toray Industries completes a phase II trial for Postherpetic neuralgia (In adults, In the elderly) in Japan (PO) (NCT02701374)
- 21 May 2017Toray Industries completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (PO) (NCT03043248)
developed by Toray for treating neuropathic pain and investigating for fibromyalgia. In August 2021, this drug was reported to be in phase 1 clinical development.
PATENT
WO 2016136944
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016136944

(Reference Example 22) Synthesis of (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate:
[Chemical 56]
1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, Methyl (triphenylphosphoranylidene) acetate (33.4 g, 99.9 mmol) was added to a solution of 90.8 mmol) in dichloromethane (240 mL) at room temperature, and the mixture was stirred for 16 hours and then concentrated under reduced pressure. The residue was washed with a mixed solvent of hexane / dichloromethane = 19/1, and the washing liquid was concentrated. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate as a white solid (11.9 g, 71. 6 mmol, 79%).
1 H-NMR (400 MHz, CDCl 3 ) δ: 3.76 (3H, s), 3.81 (3H, s), 6.82 (1H, d, J = 15.6 Hz), 6.98 (1H, brs), 7.16 (1H, brs), 7.53 (1H, d, J = 15.6Hz).
ESI-MS: m / z = 167 (M + H) + .
(Reference Example 27) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 61]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propane. -1-one (0.179 g, 0.68 mmol, 63%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Comparative Example 1) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one hydrochloride:
[Chemical 66]
1- (4- (Dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (1.50 g, 5.67 mmol) diethyl ether (60) A dioxane solution of hydrogen chloride (4.0 M, 3.69 mL, 14.8 mmol) was added to the (0.0 mL) solution at 0 ° C. The reaction mixture was stirred at the same temperature for 1 hour and then at room temperature for 30 minutes. The precipitated white solid was collected by filtration, washed with diethyl ether (100 mL), dried at room temperature for 36 hours, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-). Imidazole-2-yl) propan-1-one hydrochloride (1.41 g, 4.18 mmol, 74%) (hereinafter, the compound of Comparative Example 1) was obtained as a white solid.
1 1 H-NMR (400 MHz, D 2 O) δ: 1.53-1.80 (2H, m), 2.12-2.23 (2H, m), 2.68-2.80 (1H, m), 2.88 (6H, s), 3.01- 3.08 (2H, m), 3.15-3.26 (3H, m), 3.47-3.58 (1H, m), 3.84 (3H, s), 4.08-4.16 (1H, m), 4.50-4.59 (1H, m), 7.29-7.33 (2H, m).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) as propan-1-one : m / z = 265 (M + H) + .
(Comparative Example 2) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one sulfate monohydrate:
[Chemical 67]
1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (6.72 g, 25.4 mmol) Concentrated sulfuric acid (2.49 g, 25.4 mmol), water (1.83 g, 102 mmol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl) in a DMSO (100 mL) solution. Seed crystals (50 mg, 0.13 mmol) of -1H-imidazol-2-yl) propan-1-one sulfate monohydrate were added at 80 ° C. The reaction was stirred at the same temperature for 2.5 hours, at 50 ° C. for 2.5 hours and at room temperature for 15 hours. The precipitated white solid was collected by filtration, washed successively with DMSO (20 mL) and methyl ethyl ketone (40 mL), dried at room temperature, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl). -1H-imidazol-2-yl) propan-1-one sulfate monohydrate (8.42 g, 22.1 mmol, 87%) (hereinafter, the compound of Comparative Example 2) was obtained as white crystals.
1 1 H-NMR (400 MHz, DMSO-d 6)) δ: 1.36 (1H, m), 1.58 (1H, m), 1.95 (2H, br), 2.44-2.57 (1H, m), 2.65 (6H, s), 2.74-2.88 (4H, m), 3.00 (1H, t, J = 12.0 Hz), 3.22 (1H, m), 3.61 (3H, s), 4.02 (1H, d, J = 14.0 Hz), 4.47 (1H, d, J = 12.8 Hz), 6.87 (1H, d, J = 1.2 Hz), 7.11 (1H, d, J = 1.2 Hz).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-) As 1H-imidazol-2-yl) propan-1-one: m / z = 265 (M + H) + .
NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153744
PATENT
WO-2021153743
Novel crystalline polymorphic form of 1-(4-(dimethylamino) piperidin-1-yl)-3-(1-methyl-1H-imidazol-2-yl)propan-1-one, useful as an analgesic in treating neuropathic pain and/or fibromyalgia.Pain is an experience with unpleasant sensations and emotions that occurs when or may cause tissue damage. Pain is mainly classified into nociceptive pain, neuropathic pain or psychogenic pain according to its cause. In addition, fibromyalgia is known as pain of unknown cause.
Neuropathic pain is pathological pain caused by dysfunction of the peripheral or central nervous system itself, and is caused by direct damage or compression of nervous tissue even though nociceptors are not stimulated. It refers to the pain that occurs. As a therapeutic agent for neuropathic pain, an anticonvulsant, an antidepressant, anxiolytic, or an antiepileptic drug such as gabapentin or pregabalin is used.
Fibromyalgia is a disease in which systemic pain is the main symptom and neuropsychiatric symptoms and autonomic nervous system symptoms are secondary symptoms. Pregabalin approved in the United States and Japan, duloxetine and milnacipran approved in the United States are mainly used as therapeutic agents for fibromyalgia, and non-approved agents for fibromyalgia are not approved. It has also been used for steroidal anti-inflammatory agents, opioid compounds, antidepressants, anticonvulsants and antiepileptic drugs. However, the therapeutic effects of non-steroidal anti-inflammatory drugs and opioid compounds are generally considered to be low (Non-Patent Document 1).
On the other hand, Patent Document 1 discloses that certain substituted piperidins have cardiotonic activity, and Patent Document 2 discloses that an imidazole derivative exhibits an FXa inhibitory effect. Patent Document 3 suggests that the substituted piperidins may have a medicinal effect on overweight or obesity, and Patent Documents 4 to 6 and Non-Patent Document 2 indicate that the imidazole derivative has an analgesic effect. It is disclosed.
In addition, the quality of pharmaceutical products needs to be maintained over a long period of time such as distribution and storage, and the compound as an active ingredient is required to have high chemical and physical stability. Therefore, as the active ingredient of a pharmaceutical product, a crystal that can be expected to have higher stability than an amorphous substance is generally adopted. Further, if crystals are obtained, a purification effect due to recrystallization during production can be expected. Further, it is preferable to have low hygroscopicity from the viewpoint of maintaining stability and handling during manufacturing, storage, formulation and analysis of the drug substance. In addition, since a drug needs to be dissolved in the digestive tract in order to exhibit its medicinal effect, it is preferable that the drug has excellent solubility, which is a physical property contrary to stability.
In order to obtain crystals of a compound that is an active ingredient of a pharmaceutical product, it is necessary to study various conditions for precipitating crystals from the solution. It is common to carry out crystallization under the condition of being dissolved in.
Patent documents
Patent Document 1: French Patent Invention No. 2567885
Patent Document 2: Japanese Patent Application Laid-Open No. 2006-0083664
Patent Document 3: International Publication No. 2003/031432
Patent Document 4: International Publication No. 2013/147160
Patent Document 5: International Publication No. 2015/046403
Patent Document 6: International Publication No. 2016/136944
Non-patent literature
Non-Patent Document 1: Okifuji et al., Pain and Therapy, 2013, Volume 2, p. 87-104
Non-Patent Document 2: Takahashi et al., Toxicological Pathology, 2019, Vol. 47. p. 494-503
Compound (I) was synthesized by the method described in the following reference example. For the compounds used in the synthesis of the reference example compounds for which the synthesis method is not described, commercially available compounds were used.
(Reference Example 4) Synthesis of amorphous compound (I):
[Chemical formula 2] 2 of
crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanol (5.00 g, 27.4 mmol) Aqueous sodium hydroxide solution (1.0N, 30.2 mL, 30.2 mmol) was added to a solution of -propanol (55 mL) at 0 ° C., and the mixture was stirred at room temperature for 12 hours. 2-Propanol (220 mL) was added to the reaction solution at room temperature, and crude 4- (dimethylamino) piperidine (3.17 g, 24.7 mmol) and DMT-MM (8.35 g, 30.2 mmol) were added at room temperature to react. The liquid was stirred at the same temperature for 3 hours. A 10% aqueous sodium chloride solution and a 1.0N aqueous sodium hydroxide solution were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give compound (I) (6.98 g) as an amorphous substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 (5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz) ), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Reference Example 5) Synthesis of crude 4- (dimethylamino) piperidine:
[Chemical
formula 3] 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (20.1 g, 77.0 mmol) in methanol (154.0 mL) Palladium-carbon (10% wet, 2.01 g) was added thereto, and the mixture was stirred at room temperature for 19 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 4- (dimethylamino) piperidine (9.86 g).
(Reference Example 6) Synthesis of crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate:
[Chemical
formula 4] Sodium hydride (55%, 4.36 g, 100 mmol) aqueous solution and tetrahydrofuran (150 mL) To the mixture was added triethylphosphonoacetate (19.1 mL, 95.0 mmol) at 0 ° C. After stirring the reaction solution for 20 minutes, a solution of 1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, 91.0 mmol) in tetrahydrofuran (150 mL) was added at 0 ° C., and then ethanol (30 mL) was added in the same manner. The mixture was added at temperature and stirred at room temperature for 2 hours. A 10% aqueous sodium chloride solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, chloroform / methanol). After adding methanol (310 mL) to the residue, palladium-carbon (10% wet, 1.40 g) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (14.2 g) of ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate.
(Reference Example 7) Synthesis of 1-benzyloxycarbonyl-4- (dimethylamino) piperidine:
[Chemical
formula 5] dichloromethane (55.7 mL) of 1-benzyloxycarbonyl-4-oxopiperidine (13.0 g, 55.7 mmol) ) Solution of dimethylamine in tetrahydrofuran (2.0 M, 34.8 mL, 69.7 mmol), acetic acid (0.32 mL, 5.6 mmol) and sodium triacetoxyborohydride (4.8 g, 22.6 mmol). Added at ° C. After stirring the reaction solution at the same temperature for 30 minutes, sodium triacetoxyborohydride (4.8 g, 22.6 mmol) was added at 0 ° C. The reaction mixture was stirred at the same temperature for 30 minutes, sodium triacetoxyborohydride (8.1 g, 38.2 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 12 hours. The reaction solution was cooled to 0 ° C. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) and then again by flash chromatography (silica gel, chloroform / methanol) to obtain 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (dimethylamino) piperidine. 13.6 g, 51.8 mmol, 93%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.34-1.46 (2H, m), 1.78-1.86 (2H, m), 2.28 (6H, s), 2.29-2.34 (1H, m), 2.75-2.85 (2H, m), 4.14-4.28 ( 2H, m), 5.12 (2H, s), 7.29-7.36 (5H, m).
ESI-MS: m / z = 263 (M + H) + .
(Reference Example 8) Synthesis of 1-benzyloxycarbonyl-4-oxopiperidine:
[Chemical
formula 6] Hydrochloride (130 mL) and water (130 mL) of 4-piperidinone hydrochloride monohydrate (10.0 g, 65.1 mmol) Sodium carbonate (13.8 g, 130.2 mmol) and benzyl chloroformate (8.79 mL, 61.8 mmol) were added to the mixed solution with and at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) to give 1-benzyloxycarbonyl-4-oxopiperidine (13.1 g, 56.2 mmol, 86%) as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 2.42-2.50 (4H, m), 3.78-3.82 (4H, m), 5.18 (2H, s), 7.32-7.38 (5H, m).
(Example 1) Production of A-type crystal of
compound (I): Amorphous compound (6.98 g) of compound (I) prepared in Reference Example 4 is purified and concentrated with chloroform / methanol by silica gel column chromatography. After that, the wall surface of the flask was rubbed with a spartel and mechanical stimulation was applied to obtain A-type crystals of compound (I) as a powder. For the obtained crystals, measurement of powder X-ray diffraction using a powder X-ray diffractometer (Rigaku Co., Ltd .; 2200 / RINT ultima + PC) and TG-DTA using a TG-DTA device (Rigaku Co., Ltd .; TG8120) Was done. The results of these measurements are shown in FIGS. 1 and 2.
Diffraction angle 2θ: 5.9, 16.5, 17.7, 20.8, 26.7 °
Endothermic peak: 55 ° C
PATENT
WO2013147160
Example 1 Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 27]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan- 1-one (0.179 g, 0.68 mmol, 63%) (hereinafter, the compound of Example 1) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016136944-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| JP-WO2013147160-A1 | Cyclic amine derivatives and their pharmaceutical use | 2012-03-29 | |
| TW-201350119-A | Cyclic amine derivatives and their medical uses | 2012-03-29 | |
| WO-2013147160-A1 | Cyclic amine derivative and use thereof for medical purposes | 2012-03-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| RU-2667062-C1 | Dynamic cyclic amine and pharmaceutical application thereof | 2015-02-27 | 2018-09-14 |
| TW-201639826-A | Cyclic amine derivatives and their medical uses | 2015-02-27 | |
| TW-I682927-B | Cyclic amine derivatives and their medical uses | 2015-02-27 | 2020-01-21 |
| US-10173999-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-01-08 |
| US-2018065950-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3263565-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| EP-3263565-B1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-06-26 |
| ES-2744785-T3 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2020-02-26 |
| JP-6569671-B2 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 | 2019-09-04 |
| JP-WO2016136944-A1 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019189781-A1 | Agent for inhibiting rise in intraneuronal calcium concentration | 2018-03-30 | |
| AU-2016224420-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| AU-2016224420-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-08-22 |
| CA-2977614-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| CN-107250128-B | Cyclic amine derivatives and its medical usage | 2015-02-27 | 2019-07-26 |
//////////TRK-700, phase 1, neuropathic pain, fibromyalgia, toray
O=C(CCc1nccn1C)N1CCC(CC1)N(C)C
BMS-741672


BMS-741672
N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide
N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide;
CAS 1004757-96-3
PHASE 2, , Treatment of Type 2 Diabetes, Agents for Neuropathic Pain
Chemokine CCR2 (MCP-1 Receptor) Antagonists

| Molecular Formula: | C25H33F3N6O2 |
|---|---|
| Molecular Weight: | 506.574 g/mol |

| Michael G. Yang, Robert J. Cherney | |
| Original Assignee | Bristol-Myers Squibb Company |
| Michael G. Yang, Robert J. Cherney, Martin G. Eastgate, Jale Muslehiddinoglu, Siva Josyula Prasad, Zili Xiao, | |
| Bristol-Myers Squibb Company |
- Originator Bristol-Myers Squibb
- Class Analgesics; Antihyperglycaemics
- Mechanism of Action CCR2 receptor antagonists
- Discontinued Diabetic neuropathies; Type 2 diabetes mellitus
Most Recent Events
- 10 Apr 2007 Preclinical trials in Inflammation in USA (unspecified route)
BMS-741672, 1 , is a highly selective CCR2 antagonist (IC50 = 1.4 nM) featuring a complex array of four stereocenters. The key synthetic challenge was efficient assembly of the densely functionalized 1,2,4-triaminocyclohexane (TACH) core in a minimum number of linear steps.

N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
Mp 161.3 °C.
1H NMR (400 MHz, CDCl3) δ 9.50–9.20 (1H), 9.04 (s, 1H), 8.68 (s, 1H), 8.41 (d, J = 7.1 Hz, 1H), 7.87 (s, 1H), 5.04 (dt, J = 1.3, 7.3 Hz, 1H), 4.9 (m, 1H), 4.07 (dt, J = 3.7, 12.9 Hz, 1H), 3.53 (dt, J = 1.4, 9.9 Hz, 1H), 3.44–3.30 (m, 2H), 2.39 (dq, J = 13.6, 8.4 Hz, 1H), 2.26 (m, 1H), 2.21 (s, 3H), 2.17 (q, J = 2.9 Hz, 1H), 2.03–1.91 (m, 5H), 1.71–1.54 (m, 5H), 1.04 (s, br., 6H).
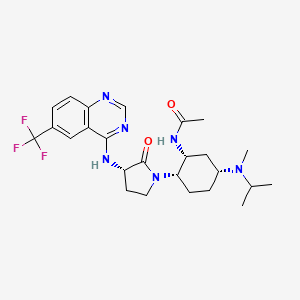
13C NMR (100 MHz, d6-DMSO) δ 171.46, 169.49, 159.62, 156.92, 151.22, 129.28, 128.27 (q, 4JCF = 3 Hz), 125.78 (q, 2JCF = 32 Hz), 124.11 (q, 1JCF = 272 Hz), 121.57 (q, 3JCF = 4 Hz), 114.33, 54.83, 53.54, 52.36, 47.34, 46.94, 43.13, 30.76, 30.24, 26.94, 26.38, 23.28, 20.87, 17.65 (br.), 16.73 (br.).
13C NMR (100 MHz, CDCl3) δ 172.17. 170.73, 159.89, 156.91, 151.16, 128.68, 128.06 (q,4JCF = 3.0 Hz), 127.25 (q, 2JCF = 32 Hz), 123.98 (q, 1JCF = 272 Hz), 121.78 (q, 3JCF = 4 Hz), 115.11, 54.89, 53.21, 52.40, 47.40, 46.98, 43.72, 30.84, 30.70, 29.96, 27.80, 23.55, 19.96, 17.70 (2C).
LCMS (ESI, pos.): 508 (16.8), 507 (66.2), 254 (5.0). HR-ESI(pos)-MS: calcd for C25H34F3N6O2 507.2690 [M + H]+, found 507.2694.
IR (KBr): ν = 3428 (m, br.), 2966 (w), 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870 (w), 845 (w).
[α]20D−187.9 (c 1.0, CHCl3).
Anal. Calcd for C25H33F3N6O2: C, 59.28; H, 6.57; F, 11.25; N, 16.59. Found: C, 59.21; H, 6.43; F, 11.07; N, 16.53.

PATENT
WO 2008014381
http://www.google.ch/patents/WO2008014381A2?cl=en&hl=de
EXAMPLE 1
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide

[00212] Example 1, Step 1: (IR, 2S, 5R)-tert-Butyl 2-benzyloxycarbonylamino- 7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCCh (2 x 0.45 L) and sat. NaCl (I x 0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3 -necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (-1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with Z-N-Cbz- methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and Ν,Ν-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2Cθ3 (2 x 0.9 L) and sat. NaCl (1 x 0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (IR,2S,5R)- tert-butyl 2-((5)-2-(benzyloxycarbonylamino)-4-
(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 406.3; [M+Naf = 528.3. 1H-NMR (400 MHz, d4-Me0H): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, IH), 4.2 (m, IH), 4.0 (m, IH), 2.5 – 2.7 (m, 3H), 2.25 (m, IH), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, IH), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83
(S)].
[00213] Example 1, Step 2: A sample of (1^,25,5^)- tert-butyl 2-((5)-2- (benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza- bicyclo[3.2. l]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M- Me2S+H]+ = 458.4; [M]+ = 520.4. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, IH), 4.28 (m, IH), 3.98 (m, IH), 3.3 – 3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, IH), 2.0 – 2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)]. [00214] Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into -0.22 L portions and worked up as follows: the reaction mixture (-0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3 x 0.5 L) and brine (1 x 0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (\R,2S,5R)- tert-bvXyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6- azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+ = 358.4; [M+Na]+ = 480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, IH), 3.6 (m, IH), 3.3 (m, IH), 2.64 (m, IH), 2.28 – 2.42 (m, 2H), 2.15 (m, IH), 1.7 – 2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
[00215] Example 1, Step 4: A stirring solution of (\R,2S,5R)- tert-butyl 2-((S>3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6- carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20 0C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to -4 through the addition of IN HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2 x 1 L). The solid was dissolved in dichloromethane (1.5 L) and water (~ 1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (\R,2S,5R)-2-((S)-3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+ = 420.2; [M-Boc+H]+ = 376.2; [M+H]+ = 476.2. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, IH), 3.45 – 3.6 (m, 2H), 2.99 (m, IH), 2.41 (m, IH), 2.15 (m, IH), 2.0 (m, 2H), 1.6 – 1.9 (m, 4H), 1.46 (s, 9H).
[00216] Example 1, Step 5: A 3 L round bottom flask was charged with (lR,25′,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDOHCl (33.5 g, 0.175 mol), 1 -hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCθ3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, -100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 375.3; [M+H]+ = 475.4; [M-tBu+H]+ = 419.3. 1H-NMR (400 MHz, Cl4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, IH), 3.6 (m, IH), 3.45 (m, IH), 2.91 (m, IH), 2.38 (m, IH), 2.12 (m, IH), 1.9 – 2.05 (m, 2H), 1.65 – 1.9 (m, 4H), 1.46 (s, 9H).
[00217] Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0 0C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp < 10 0C). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCθ3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40 0C before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75 0C for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2CI2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-bυXyl (lR,3R,45)-3-acetamido-4-((5)-3-(benzyloxycarbonylamino)-2- oxopyrrolidin-l-yl)cyclohexylcarbamate (purity >99.5% by HPLC). LC/MS for primary peak: [M+H]+ = 489.4; [M-tBu+H]+ = 433.3. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, IH), 4.15 (m, IH), 4.04 (m, IH), 3.8 (m, IH), 3.6 (m, 2H), 2.44 (m, IH), 2.12 (m, IH), 1.87 – 2.05 (m, 4H), 1.87 (s, 3H), 1.55 – 1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in Figure 1. [00218] Example 1, Step 7: A stirring solution of tert-butyl (\R,3R,4S)-3- acetamido-4-((5′)-3 -(benzyloxycarbonylamino)-2-oxopyrrolidin- 1 – yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl («S)-l-((l«S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M + H]+ = 389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, IH), 4.15 (m, IH), 4.00 (t, J= 9.3 Hz, IH), 3.81 (t, J= 9.1 Hz, IH), 3.65 (q, J= 8.4 Hz, IH), 3.3 – 3.4 (m, IH), 2.45 (m, IH), 1.95 – 2.24 (m, 5H), 2.00 (s, 3H), 1.6 – 1.8 (m, 2H). [00219] Example 1, Step 8: A stirring solution of benzyl (S)- 1-(( \S,2R,4R)-2- acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (-0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with IN NaOH (1 L). The solids collected in the filtration were dissolved in IN NaOH (IL) at 0 0C and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL IN HCl + 800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-I- ((lS,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3- ylcarbamate as an oil. LC/MS found [M + H]+ = 431.45. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, IH), 4.24 (t, J= 9.4 Hz, IH), 4.11 (m, IH), 3.61 (t, J= 9.1 Hz, IH), 3.52 (q, J= 8.6 Hz, IH), 3.04 (br. s, IH), 2.96 (sep, J= 6.3 Hz, IH), 2.40 (m, IH), 2.15 (m, IH), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.65 (m, IH), 1.12 (app. dd, J= 6.3, 1.1 Hz, 6H).
[00220] Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-I -((lS’,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2- oxopyrrolidin-3-ylcarbamate (-115 mmol) in dichloromethane (600 mL) was cooled to 0 0C and charged sequentially with formaldehyde (18.6 g, 37 wt% solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCθ3 + NaOH (sat. NaHCO3, pH to 11 w/ IN NaOH). The organic layer was extracted with aq. HCl (200 mL IN HCl + 600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl {S)-\-{{\S,2R,AR)-2- acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M + H]+ = 445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, IH), 4.25 (t, J= 9.2 Hz, IH), 4.11 (br s, IH), 3.5 – 3.6 (m, 2H), 2.77 (v br s, 2 H), 2.41 (m, IH), 2.26 (s, 3H), 2.0 – 2.1 (m, 2H), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.10 (app. dd, J = 17, 6.4 Hz, 6H). [00221] Example 1, Step 10: To a solution of benzyl (S)- 1-(( 15″,2R,4R)-2- acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3 -ylcarbamate (-0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((li?,25,5i?)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M + H]+ = 311.47. 1H-NMR (400 MHz, (I4-MeOH): δ 4.39 (br s, IH), 4.00 (m, IH), 3.3 –
3.5 (m, 4H), 2.73 (m, IH), 2.38 (m, IH), 2.25 (s, 3H), 2.0 – 2.2 (m, 3H), 1.94 (s, 3H),
1.6 – 1.75 (m, 4H), 1.07 (app. dd, J= 21, 6.4 Hz, 6H). [00222] Example 1, Step 11: To a solution of N-((lR,25′,5R)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (~35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P.H. Carter et al, PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60 0C for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0 0C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2 x 600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+ = 507.3. 1H-NMR (400 MHz, U4– MeOH): δ 8.82 (s, IH), 8.59 (s, IH), 8.05 (dd, J= 8.8, 1.8 Hz, IH), 7.9 (d, J= 8.7 Hz, IH), 5.28 (t, J= 8.6 Hz, IH), 4.58 (br s, IH), 4.06 (m, IH), 3.52 – 3.68 (m, 2H), 3.43 (m, IH), 2.76 (br s, IH), 2.55 (m, IH), 2.28 (s, 3H), 2.1 – 2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, IH), 1.65 – 1.8 (m, 3H), 1.09 (app. dd, J= 24, 6.4 Hz, 6 H).
Example 1, Alternative Step 9

[00223] Example 1, Alternative step 9a1: To a hydrogenator were charged ethyl (7R,SS)-S-((S)- l-phenyl-ethylamino)-l,4-dioxa-spiro[4.5]decane-7-carboxylate A- toluenesulfonate salt I A (1417 g, 2.8 moles, c.f : WO2004098516, prepared analogous to US Pat.6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40 0C until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60 0C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ~2 L until most ethanol was removed (<0.5%) and residual moisture content was <l,000 ppm. Batch volume was adjusted to -7.5 L by the addition of isopropyl acetate. The mixture was heated to 80 0C until clear, then cooled 65°-70 0C. Seed crystals of 1 (5 g) were added and the batch was cooled to 500C over 2 hours, then further cooled to 20 0C over 4 hours and held for ~10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at -35 0C until volatiles were recduced below -1% (LOD). Ethyl (7R,85′)-8-amino-l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0Hz, 2H), 7.15 (d, J 8.0Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, IH,), 3.20-3.13 (m, IH), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm x 150 mm Ld., 3.5μm particle size, 0.05% NH40H (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).

Example 1, Alternative Step 9a”: Aminoester 1 (63g, 0.16M, leq.; the product of reductive deprotection of a known compound – (See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (50OmL) was added. EDAC (33.1g, 0.17M, l. leq), HOBt-H2O (21.2g, 0.16M, l.Oeq) and N-Cbz-Z-methionine (46.7g, 0.17M, 1.05eq) were then added followed by TEA (48.OmL, 0.35M, 2.2eq). An exotherm to 38 0C was observed. The reaction mass was left to stir at RT. After 30mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5L) and washed with KHCO3 (4x500mL, 20wt% aq. solution) and brine (50OmL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,85)-8- {(2S)-2-benzyloxycarbonylamino- 4-methylsulfanyl-butyr-yl-amino}-l,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2g, 98% yield, 93AP purity). 1H-NMR: (300MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J9.0Hz, IH), 5.66 (d, J 8.0Hz, IH), 5.10 (s, 2H), 4.35- 4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, IH), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0Hz, 7.0Hz, IH), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. lO.Olmin (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [CaIc: C24H35N2O7S 495.2165].

2 3 [00224] Example 1, Alternative Step 9b: Methionine amide 2 (75.Og, 0.15M) was dissolved in MeI (225mL, 3mL/g) – some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200mmHg and some of the MeI removed. The remaining material was slurried in TBME (50OmL), after a 30min stir-out the slurry was filtered, the cake washed with TBME (50OmL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBME (50OmL), filtered, washed with TBME (50OmL) and dried under vacuum to give [(35)-3-benzyloxycarbonylamino-3-{(7R,85′)-7- ethoxycarbonyl-l,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5g, 97%, 99 area% purity). 1H-NMR: (300MHz, CDCl3) δ 7.75 (d, J 9.0Hz, IH), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0Hz, IH), 5.10 (s, 2H), 4.76-4.65 (m, IH), 4.48-4.39 (m, IH), 4.14-3.85 (m, 6H), 3.84-7.73 (m, IH), 3.68-3.55 (m, IH), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, IH), 2.52-1.55 (m, 8H), 1.24 (t, J7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 2.45min (I-), 8.14min (Compound 3, 43.6AP, I“ 54.6AP). HRMS: m/z 509.2341 [CaIc: C25H37N2O7S 509.2321].

[00225] Example 1, Alternative Step 9c: Cs2CO3 (61.5g, 0.19M, 1.5eq) was placed in an round bottom flask and anhydrous DMSO (2.4L) was added. Sulfonium salt 3 (80.Og, 0.13M, 1.Oeq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 2Oh. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0L) and washed with brine (2L), the organic phase was washed with brine (50OmL). The combined aq. layers were then washed EtOAc (50OmL). The combined organic phases were then washed with brine (3x750mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S>3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl}-l,4-dioxa-spiro[4.5]decane-7- carboxylate 4 as a light colored oil (56.5g, 0.13M, -100 area-% purity) pure by NMR analysis. 1H-NMR: (300MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J4.0Hz, IH), 5.11 (s, 2H), 4.27-4.18 (m, IH), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0Hz, IH), 3.23 (q, J5.0Hz, IH), 2.63-2.57 (m, IH), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.99min (Compound 5, produced on column, 4.2AP), 9.48 (Compound 4, 74.3AP). HRMS: m/z 447.2127 [CaIc: C23H31N2O7 447.2131].

4 5
[00226] Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.Og, 0.1 IM) was dissolved in acetone (50OmL) and IN HCl (50OmL) was added. The reaction mass was then heated to 65°C. After 20mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3x3OOmL) to give ethyl (\R,2S)-2-((3S)-3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8g, 88%, 97 area-% purity). 1H-NMR: (300MHz, CDCl3) δ 7.37- 7.32 (m, 5H), 6.65 (br d, J4.0Hz, IH), 5.12 (s, 2H), 4.54-4.47 (m, IH), 4.34-4.26 (m, IH), 4.18 (dq, J 11.0Hz, 7.0Hz, IH), 4.09 (dq, J 11.0Hz, , 7.0Hz, IH), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, IH), 1.81 (quin., J l l.OHz, IH), 1.24 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.95min (Compound 5). HRMS: m/z 403.1864 [CaIc: C2iH27N2O6403.1869].

[00227] Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5g, 0.06M, leq), DMSO (3OmL) and Ti(O-ZPr)4 (33.7mL, 0.1 IM, 2.04eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6mL, 0.1 IM, 2.0eq) was then added in one portion. The mixture was left to stir for 30mins at room temperature before being cooled to <3°C in ice/water. MeOH (3OmL) was then added followed by the portionwise addition OfNaBH4 (4.33g, 0.1 IM, 2.04eq) – temperature kept <8°C. 30mins after the addition was completed the reaction mass was diluted with methylene chloride (30OmL) and then NaOH (IN, 4OmL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (10OmL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (50OmL). This solution was extracted with IN HCl (2x400mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4x250mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6g, 96%, 7: 1 d.r.). This material was then slurried overnight in hexane (67OmL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (lR,25′,5R)-2-((3S)-3-benzyloxycarbonylamino- 2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9g, 81%, 24: 1 d.r.). 1H-NMR: (300MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J4.5, IH), 5.10 (s, 2H), 4.42 (q, J4.5, IH), 4.23-4.12 (m, IH), 4.08 (dq, J 10.5, 7.0, IH), 4.02 (dq, J 10.5, 7.0, IH), 3.84 (t, J9.0, IH), 3.46-3.36 (m, IH), 3.04 (septet, J6.5, IH), 2.86-2.80 (m, IH), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC- Pack Pro C18 5μm 4.6 x 150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15min gradient. 8.23 (Compound 6), 8.88 (5-e/«-Compound 6). HRMS: 460.2798 [CaIc: C25H38N3O5 460.2811].

[00228] Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to -55 0C under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6 – 7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0 0C and stirred for Ih. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (li?,25r,5R)-2-((35′)-3-Benzyloxycarbonylamino-2- oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 500C, CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, IH), 5.19 (app s, 2H), 4.42 (dd, J= 15.6, 7.8 Hz, IH), 4.29-4.23 (m, IH), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, IH), 2.99 (broad s, IH), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, IH), 2.00 (ddd, J= 9.0, 8.6, 3.9 IH), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [CaIc: C23H34N3O5 432.25].
NHCbz



[00229] Example 1, Alternative Step 9g: Amino acid 7 (6.3g, 14.7mmol, l.Oeq) was dissolved in THF (8OmL) under N2 and NaH (584mg, 14.7mmol, l.Oeq, 60wt% dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59wt%). This solid was slurried in toluene (100 mL) under N2and heated to 900C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ~2min. After ~5min all the solids had dissolved, after lOmins precipitation of a white solid was observed. After 30mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into ACOH/AC2O (80/20, 168mL) solution at 900C. After 45mins HPLC still indicated some isocyanate. At 1.15h , the reaction mass was cooled to RT and diluted with toluene (10OmL) and water (10OmL). The organic layer was removed and the toluene washed with IN HCl
(10OmL). The combined aq. phases were then basified with K2Cθ3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 200C. The aq layer was then extracted with methylene chloride (4xl50mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-l-((lS,2R,4R)-2-acetamido-4- (isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 7.20min (Compound 8), 7.85min (urea dimer). HRMS: 445.2809 [CaIc: C24H37N4O4 445.2815].
Alternative Preparation of Example 1

2 3
[00230] Example 1, Alternative Preparation, Step 1: Ethyl (7R,85)-8-amino- l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450. Ig), was combined with l-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3g), 1-hydroxy benzotriazole hydrate (171.9g), N-carbobenzyloxy-Z -methionine (333.4g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5g) below 30 0C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,85)-8-((5)-2- benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(5)-3-benzyloxy-carbonylamino-3-((7R,8«S’)-7-ethoxycarbonyl-l,4- dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, -94% yield) as an off-white solid (HPLC purity 99.9%).

[00231] Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides.
Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20 % brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8«S)-8-((«S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (~0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60 0C until conversion of 4 to ethyl (IR,2S)- 2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo- cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40 0C, cooled to -30 0C, and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10 0C and agitated for ~1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40 0C in a vacuum oven. Cyclohexanone 5 (272g, 70% yield) was obtained (HPLC purity 98.7%).

[00232] Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25 0C) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5 % relative to 5) was added and hydrogenation was performed at -30 psig for at least 6 h, yielding a mixture of ethyl (lR,25′,5R)-2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl- methyl-amino)-cyclohexanecarboxylate 6 and its 5-epz-isomer (-7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. -600 mL. Wet ethyl acetate (-3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to -400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2 x 400 mL). The aqueous layer was warmed to 50° to 60 0C for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH -10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ~6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to -600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (\R,2S,5R)-2- ((5*)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)- cyclohexane carboxylic acid 7 (-148 g) in THF (-4 L). Seed crystals of 8 were added, followed by 25 % solution of sodium methoxide in methanol (81.24 g) below 25 0C. The slurry was held for at least additional 16h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30 0C. Dry (lR,25′,5R)-2-((5)-3-benzyloxycarbonyl- amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139g, -60% yield from 5).

[00233] Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (10Og), diphenyl phosphate (3.86g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80 0C. Upon complete conversion (HPLC), tert- BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (lx800mL, 1×400 mL) and water (40OmL). The organic phase was heated and concentrated in vacuo to approx. 27OmL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80 0C, followed by seeds of 9 (~lg). The slurry was slowly cooled to room temperature and benzyl {(S)-l-[(lS,2R,4R)-2- tert- butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3- yl} -carbamate 9 was isolated by filtration as a white solid (86.76g, 78% yield).

[00234] Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (5Og) was dissolved in Toluene (50OmL) and /-PrOH (15OmL). The resulting solution was then heated to 6O0C. Methanesulfonic acid (19.6mL) was added below 65°C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4mL) added slowly below 25°C. Acetic anhydride was then added below 25°C. After Ih acetic acid (25OmL) was added below 25°C. The toluene phase was discarded and 2-methyl-THF (50OmL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (25OmL). The organic layer was concentrated under reduced pressure and continuously replaced with /-PrOH. The solution was cooled and filtered to provide benzyl {(5′)-l-[(15r,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2- oxo-pyrrolidin-3-yl} -carbamate 10 in /-PrOH solution which was used directly in the hydrogenation.
[00235] Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (~61g) in /-PrOH (-625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25 0C for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in /-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with /-PrOH (2×100 mL) and dried in vacuo to constant weight at -50 0C to give N-[(li?,25r,5R)-2-((5′)-3-amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl- amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).
NH,
CL,
Example 1

[00236] Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl- quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50 0C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give -400 mL of slurry. The mixture was cooled to room temperature and washed with 10 % aqueous K2HPO4 (lxl.O L, 1×0.5 L) to afford 4-chloro-6- trifluoromethyl-quinazoline 13 (-21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8 %). [00237] Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl- N-ethylamine (61 mL) at room temperature was added a solution of 13 (-21.2 g) in MeCN (-450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert- butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH >11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-bvAyl ether (-250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60 0C, followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((lR,25′,5R)-5- (isopropylamino)-2-((5′)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4- ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1
[00238] Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60 0C and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2θ, which provided the salt free from any residual methanol. HPLC purity = 99.8%. 1H ΝMR (500 MHz, D2O) δ ppm 8.75 (1 H, s), 8.66 (1 H, s), 8.25 (1 H, d, J=8.80 Hz), 7.90 (1 H, d, J=8.80 Hz), 7.75 (4 H, d, J=8.25 Hz), 7.43 – 7.57 (6 H, m), 5.42 (1 H, t), 4.33 – 4.44 (1 H, m), 4.09 – 4.19 (1 H, m), 3.83 – 3.91 (1 H, m), 3.74 – 3.83 (2 H, m), 3.61 (1 H, t, J=I 1.55 Hz), 2.75 (3 H, d, J=6.60 Hz), 2.61 – 2.70 (1 H, m), 2.31 – 2.44 (1 H, m), 2.20 – 2.27 (1 H, m), 2.17 (2 H, d, J=12.10 Hz), 1.94 – 2.04 (1 H, m, J=12.65 Hz), 1.90 – 1.95 (3 H, m), 1.72 – 1.91 (2 H, m), 1.37 (3 H, d, J=6.05 Hz), 1.29 (3 H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10 °C/min and revealed a melting / decomposition endotherm with an onset temperature of 297.6 0C and a peak temperature at 299.1 0C. [00239] Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((lR,25r,5R)-5-(isopropyl(methyl)amino)-2-((5′)-2-oxo-3- (6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin- 1 -yl)cyclohexyl)acetamide ( 1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40 0C; a white solid precipitated. The suspension was heated to 90 0C and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.
![]()
PATENT
US 7671062
http://google.com/patents/US7671062

or a pharmaceutically acceptable salt, solvate or prodrug, thereof, having an unexpected combination of desirable pharmacological characteristics. Crystalline forms of the present invention are also provided. Pharmaceutical compositions containing the same and methods of using the same as agents for the treatment of inflammatory diseases, allergic, autoimmune, metabolic, cancer and/or cardiovascular diseases is also an objective of this invention. The present disclosure also provides a process for preparing compounds of Formula (I), including N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide:

wherein R1, R8, R9, R10, and
are as described herein. Compounds that are useful intermediates of the process are also provided herein.
1st embodiment, the disclosure provides a process for preparing a compound of formula IV, or a salt thereof:

Example 1 N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide

Example 1, Step 1: (1R,2S,5R)-tert-Butyl 2-benzyloxycarbonylamino-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCO3 (2×0.45 L) and sat. NaCl (1×0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3-necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (˜1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with L-N-Cbz-methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and N,N-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2CO3 (2×0.9 L) and sat. NaCl (1×0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=406.3; [M+Na]+=528.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, 1H), 4.2 (m, 1H), 4.0 (m, 1H), 2.5-2.7 (m, 3H), 2.25 (m, 1H), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, 1H), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83 (s)].
Example 1, Step 2: A sample of (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M-Me2S+H]+=458.4; [M]+=520.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, 1H), 4.28 (m, 1H), 3.98 (m, 1H), 3.3-3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, 1H), 2.0-2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)].
Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into ˜0.22 L portions and worked up as follows: the reaction mixture (˜0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3×0.5 L) and brine (1×0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+=358.4; [M+Na]+=480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, 1H), 3.6 (m, 1H), 3.3 (m, 1H), 2.64 (m, 1H), 2.28-2.42 (m, 2H), 2.15 (m, 1H), 1.7-2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
Example 1, Step 4: A stirring solution of (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20° C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to ˜4 through the addition of 1N HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2×1 L). The solid was dissolved in dichloromethane (1.5 L) and water (˜1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+=420.2; [M-Boc+H]+=376.2; [M+H]+=476.2. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, 1H), 3.45-3.6 (m, 2H), 2.99 (m, 1H), 2.41 (m, 1H), 2.15 (m, 1H), 2.0 (m, 2H), 1.6-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 5: A 3 L round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDC.HCl (33.5 g, 0.175 mol), 1-hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCO3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, ˜100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=375.3; [M+H]+=475.4; [M-tBu+H]+=419.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, 1H), 3.6 (m, 1H), 3.45 (m, 1H), 2.91 (m, 1H), 2.38 (m, 1H), 2.12 (m, 1H), 1.9-2.05 (m, 2H), 1.65-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0° C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp<10° C.). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCO3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40° C. before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75° C. for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2Cl2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (purity>99.5% by HPLC). LC/MS for primary peak: [M+H]+=489.4; [M-tBu+H]+=433.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, 1H), 4.15 (m, 1H), 4.04 (m, 1H), 3.8 (m, 1H), 3.6 (m, 2H), 2.44 (m, 1H), 2.12 (m, 1H), 1.87-2.05 (m, 4H), 1.87 (s, 3H), 1.55-1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in FIG. 1.
Example 1, Step 7: A stirring solution of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M+H]+=389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, 1H), 4.15 (m, 1H), 4.00 (t, J=9.3 Hz, 1H), 3.81 (t, J=9.1 Hz, 1H), 3.65 (q, J=8.4 Hz, 1H), 3.3-3.4 (m, 1H), 2.45 (m, 1H), 1.95-2.24 (m, 5H), 2.00 (s, 3H), 1.6-1.8 (m, 2H).
Example 1, Step 8: A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with 1N NaOH (1 L). The solids collected in the filtration were dissolved in 1N NaOH (1 L) at 0° C. and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL 1N HCl+800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil. LC/MS found [M+H]+=431.45. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, 1H), 4.24 (t, J=9.4 Hz, 1H), 4.11 (m, 1H), 3.61 (t, J=9.1 Hz, 1H), 3.52 (q, J=8.6 Hz, 1H), 3.04 (br. s, 1H), 2.96 (sep, J=6.3 Hz, 1H), 2.40 (m, 1H), 2.15 (m, 1H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.65 (m, 1H), 1.12 (app. dd, J=6.3, 1.1 Hz, 6H).
Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜115 mmol) in dichloromethane (600 mL) was cooled to 0° C. and charged sequentially with formaldehyde (18.6 g, 37 wt % solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCO3+NaOH (sat. NaHCO3, pH to 11 w/1N NaOH). The organic layer was extracted with aq. HCl (200 mL 1N HCl+600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M+H]+=445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, 1H), 4.25 (t, J=9.2 Hz, 1H), 4.11 (br s, 1H), 3.5-3.6 (m, 2H), 2.77 (v br s, 2H), 2.41 (m, 1H), 2.26 (s, 3H), 2.0-2.1 (m, 2H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.10 (app. dd, J=17, 6.4 Hz, 6H).
Example 1, Step 10: To a solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M+H]+=311.47. 1H-NMR (400 MHz, d4-MeOH): δ 4.39 (br s, 1H), 4.00 (m, 1H), 3.3-3.5 (m, 4H), 2.73 (m, 1H), 2.38 (m, 1H), 2.25 (s, 3H), 2.0-2.2 (m, 3H), 1.94 (s, 3H), 1.6-1.75 (m, 4H), 1.07 (app. dd, J=21, 6.4 Hz, 6H).
Example 1, Step 11: To a solution of N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (˜35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P. H. Carter et al., PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60° C. for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0° C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2×600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+=507.3. 1H-NMR (400 MHz, d4-MeOH): δ 8.82 (s, 1H), 8.59 (s, 1H), 8.05 (dd, J=8.8, 1.8 Hz, 1H), 7.9 (d, J=8.7 Hz, 1H), 5.28 (t, J=8.6 Hz, 1H), 4.58 (br s, 1H), 4.06 (m, 1H), 3.52-3.68 (m, 2H), 3.43 (m, 1H), 2.76 (br s, 1H), 2.55 (m, 1H), 2.28 (s, 3H), 2.1-2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, 1H), 1.65-1.8 (m, 3H), 1.09 (app. dd, J=24, 6.4 Hz, 6 H).
Example 1 Alternative Step 9

Example 1, Alternative step 9ai: To a hydrogenator were charged ethyl (7R,8S)-8-((S)-1-phenyl-ethylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1A (1417 g, 2.8 moles, c.f.: WO2004098516, prepared analogous to U.S. Pat. No. 6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40° C. until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60° C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ˜2 L until most ethanol was removed (<0.5%) and residual moisture content was <1,000 ppm. Batch volume was adjusted to ˜7.5 L by the addition of isopropyl acetate. The mixture was heated to 80° C. until clear, then cooled 65°-70° C. Seed crystals of 1 (5 g) were added and the batch was cooled to 50° C. over 2 hours, then further cooled to 20° C. over 4 hours and held for ˜10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at ˜35° C. until volatiles were reduced below ˜1% (LOD). Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300 MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0 Hz, 2H), 7.15 (d, J 8.0 Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, 1H,), 3.20-3.13 (m, 1H), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0 Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm×150 mm i.d., 3.5 μm particle size, 0.05% NH4OH (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).

Example 1, Alternative Step 9aii: Aminoester 1 (63 g, 0.16M, 1 eq.; the product of reductive deprotection of a known compound—(See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (500 mL) was added. EDAC (33.1 g, 0.17M, 1.1 eq), HOBt.H2O (21.2 g, 0.16M, 1.0 eq) and N-Cbz-L-methionine (46.7 g, 0.17M, 1.05 eq) were then added followed by TEA (48.0 mL, 0.35M, 2.2 eq). An exotherm to 38° C. was observed. The reaction mass was left to stir at RT. After 30 mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5 L) and washed with KHCO3 (4×500 mL, 20 wt % aq. solution) and brine (500 mL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,8S)-8-{(2S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyr-yl-amino}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2 g, 98% yield, 93 AP purity). 1H-NMR: (300 MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J 9.0 Hz, 1H), 5.66 (d, J 8.0 Hz, 1H), 5.10 (s, 2H), 4.35-4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, 1H), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0 Hz, 7.0 Hz, 1H), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 10.01 min (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [Calc: C24H35N2O7S 495.2165].

Example 1, Alternative Step 9b: Methionine amide 2 (75.0 g, 0.15M) was dissolved in MeI (225 mL, 3 mL/g)—some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5 h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200 mmHg and some of the MeI removed. The remaining material was slurried in TBMF (500 mL), after a 30 min stir-out the slurry was filtered, the cake washed with TBMF (500 mL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBMF (500 mL), filtered, washed with TBMF (500 mL) and dried under vacuum to give [(3S)-3-benzyloxycarbonylamino-3-{(7R,8S)-7-ethoxycarbonyl-1,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5 g, 97%, 99 area % purity). 1H-NMR: (300 MHz, CDCl3) δ 7.75 (d, J 9.0 Hz, 1H), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0 Hz, 1H), 5.10 (s, 2H), 4.76-4.65 (m, 1H), 4.48-4.39 (m, 1H), 4.14-3.85 (m, 6H), 3.84-7.73 (m, 1H), 3.68-3.55 (m, 1H), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, 1H), 2.52-1.55 (m, 8H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 2.45 min (I−), 8.14 min (Compound 3, 43.6 AP, I−54.6 AP). HRMS: m/z 509.2341 [Calc: C25H37N2O7S 509.2321].

Example 1, Alternative Step 9c: Cs2CO3 (61.5 g, 0.19M, 1.5 eq) was placed in an round bottom flask and anhydrous DMSO (2.4 L) was added. Sulfonium salt 3 (80.0 g, 0.13M, 1.0 eq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 20 h. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0 L) and washed with brine (2 L), the organic phase was washed with brine (500 mL). The combined aq. layers were then washed EtOAc (500 mL). The combined organic phases were then washed with brine (3×750 mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 as a light colored oil (56.5 g, 0.13M, ˜100 area-% purity) pure by NMR analysis. 1H-NMR: (300 MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J 4.0 Hz, 1H), 5.11 (s, 2H), 4.27-4.18 (m, 1H), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0 Hz, 1H), 3.23 (q, J 5.0 Hz, 1H), 2.63-2.57 (m, 1H), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.99 min (Compound 5, produced on column, 4.2 AP), 9.48 (Compound 4, 74.3 AP). HRMS: m/z 447.2127 [Calc: C23H31N2O7 447.2131].

Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.0 g, 0.11M) was dissolved in acetone (500 mL) and 1N HCl (500 mL) was added. The reaction mass was then heated to 65° C. After 20 mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3×300 mL) to give ethyl (1R,2S)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8 g, 88%, 97 area-% purity). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.32 (m, 5H), 6.65 (br d, J 4.0 Hz, 1H), 5.12 (s, 2H), 4.54-4.47 (m, 1H), 4.34-4.26 (m, 1H), 4.18 (dq, J 11.0 Hz, 7.0 Hz, 1H), 4.09 (dq, J 11.0 Hz, 7.0 Hz, 1H), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, 1H), 1.81 (quin., J 11.0 Hz, 1H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.95 min (Compound 5). HRMS: m/z 403.1864 [Calc: C21H27N2O6403.1869].

Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5 g, 0.06M, 1 eq), DMSO (30 mL) and Ti(O-iPr)4 (33.7 mL, 0.11M, 2.04 eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6 mL, 0.11M, 2.0 eq) was then added in one portion. The mixture was left to stir for 30 mins at room temperature before being cooled to <3° C. in ice/water. MeOH (30 mL) was then added followed by the portionwise addition of NaBH4 (4.33 g, 0.11M, 2.04 eq)—temperature kept <8° C. 30 mins after the addition was completed the reaction mass was diluted with methylene chloride (300 mL) and then NaOH (1N, 40 mL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (100 mL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (500 mL). This solution was extracted with 1N HCl (2×400 mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4×250 mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6 g, 96%, 7:1 d.r.). This material was then slurried overnight in hexane (670 mL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (1R,2S,5R)-2-((3S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9 g, 81%, 24:1 d.r.). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J 4.5, 1H), 5.10 (s, 2H), 4.42 (q, J 4.5, 1H), 4.23-4.12 (m, 1H), 4.08 (dq, J 10.5, 7.0, 1H), 4.02 (dq, J 10.5, 7.0, 1H), 3.84 (t, J 9.0, 1H), 3.46-3.36 (m, 1H), 3.04 (septet, J 6.5, 1H), 2.86-2.80 (m, 1H), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15 min gradient. 8.23 (Compound 6), 8.88 (5-epi-Compound 6). HRMS: 460.2798 [Calc: C25H38N3O5 460.2811].

Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to ˜55° C. under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6-7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0° C. and stirred for 1 h. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (1R,2S,5R)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 50° C., CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, 1H), 5.19 (app s, 2H), 4.42 (dd, J=15.6, 7.8 Hz, 1H), 4.29-4.23 (m, 1H), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, 1H), 2.99 (broad s, 1H), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, 1H), 2.00 (ddd, J=9.0, 8.6, 3.9 1H), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [Calc: C23H34N3O5 432.25].

Example 1, Alternative Step 9g: Amino acid 7 (6.3 g, 14.7 mmol, 1.0 eq) was dissolved in THF (80 mL) under N2 and NaH (584 mg, 14.7 mmol, 1.0 eq, 60 wt % dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59 wt %). This solid was slurried in toluene (100 mL) under N2and heated to 90° C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ˜2 min. After ˜5 min all the solids had dissolved, after 10 mins precipitation of a white solid was observed. After 30 mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into AcOH/Ac2O (80/20, 168 mL) solution at 90° C. After 45 mins HPLC still indicated some isocyanate. At 1.15 h, the reaction mass was cooled to RT and diluted with toluene (100 mL) and water (100 mL). The organic layer was removed and the toluene washed with 1N HCl (100 mL). The combined aq. phases were then basified with K2CO3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 20° C. The aq layer was then extracted with methylene chloride (4×150 mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5 g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 7.20 min (Compound 8), 7.85 min (urea dimer). HRMS: 445.2809 [Calc: C24H37N4O4445.2815].
Alternative Preparation of Example 1

Example 1, Alternative Preparation, Step 1: Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450.1 g), was combined with 1-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3 g), 1-hydroxy benzotriazole hydrate (171.9 g), N-carbobenzyloxy-L-methionine (333.4 g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5 g) below 30° C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,8S)-8-((S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(S)-3-benzyloxy-carbonylamino-3-((7R,8S)-7-ethoxycarbonyl-1,4-dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, ˜94% yield) as an off-white solid (HPLC purity 99.9%).

Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides. Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20% brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8S)-8-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (˜0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60° C. until conversion of 4 to ethyl (1R,2S)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40° C., cooled to ˜30° C., and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10° C. and agitated for ˜1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40° C. in a vacuum oven. Cyclohexanone 5 (272 g, 70% yield) was obtained (HPLC purity 98.7%).

Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25° C.) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5% relative to 5) was added and hydrogenation was performed at ˜30 psig for at least 6 h, yielding a mixture of ethyl (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 and its 5-epi-isomer (˜7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. ˜600 mL. Wet ethyl acetate (˜3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to ˜400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2×400 mL). The aqueous layer was warmed to 50° to 60° C. for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH ˜10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ˜6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to ˜600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane carboxylic acid 7 (˜148 g) in THF (˜4 L). Seed crystals of 8 were added, followed by 25% solution of sodium methoxide in methanol (81.24 g) below 25° C. The slurry was held for at least additional 16 h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30° C. Dry (1R,2S,5R)-2-((S)-3-benzyloxycarbonyl-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139 g, ˜60% yield from 5).

Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (100 g), diphenyl phosphate (3.86 g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4 mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80° C. Upon complete conversion (HPLC), tert-BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (1×800 mL, 1×400 mL) and water (400 mL). The organic phase was heated and concentrated in vacuo to approx. 270 mL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80° C., followed by seeds of 9 (˜1 g). The slurry was slowly cooled to room temperature and benzyl {(S)-1-[(1S,2R,4R)-2-tert-butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 9 was isolated by filtration as a white solid (86.76 g, 78% yield).

Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (50 g) was dissolved in Toluene (500 mL) and i-PrOH (150 mL). The resulting solution was then heated to 60° C. Methanesulfonic acid (19.6 mL) was added below 65° C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4 mL) added slowly below 25° C. Acetic anhydride was then added below 25° C. After 1 h acetic acid (250 mL) was added below 25° C. The toluene phase was discarded and 2-methyl-THF (500 mL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (250 mL). The organic layer was concentrated under reduced pressure and continuously replaced with i-PrOH. The solution was cooled and filtered to provide benzyl {(S)-1-[(1S,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 10 in i-PrOH solution which was used directly in the hydrogenation.
Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (˜61 g) in i-PrOH (˜625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25° C. for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in i-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with i-PrOH (2×100 mL) and dried in vacuo to constant weight at ˜50° C. to give N-[(1R,2S,5R)-2-((S)-3-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).

Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl-quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50° C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give ˜400 mL of slurry. The mixture was cooled to room temperature and washed with 10% aqueous K2HPO4 (1×1.0 L, 1×0.5 L) to afford 4-chloro-6-trifluoromethyl-quinazoline 13 (˜21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8%).
Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl-N-ethylamine (61 mL) at room temperature was added a solution of 13 (˜21.2 g) in MeCN (˜450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert-butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH>11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-butyl ether (˜250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60° C., followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((1R,2S,5R)-5-(isopropylamino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60° C. and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2O, which provided the salt free from any residual methanol. HPLC purity=99.8%. 1H NMR (500 MHz, D2O) δ ppm 8.75 (1H, s), 8.66 (1H, s), 8.25 (1H, d, J=8.80 Hz), 7.90 (1H, d, J=8.80 Hz), 7.75 (4H, d, J=8.25 Hz), 7.43-7.57 (6H, m), 5.42 (1H, t), 4.33-4.44 (1H, m), 4.09-4.19 (1H, m), 3.83-3.91 (1H, m), 3.74-3.83 (2H, m), 3.61 (1H, t, J=11.55 Hz), 2.75 (3H, d, J=6.60 Hz), 2.61-2.70 (1H, m), 2.31-2.44 (1H, m), 2.20-2.27 (1H, m), 2.17 (2H, d, J=12.10 Hz), 1.94-2.04 (1H, m, J=12.65 Hz), 1.90-1.95 (3H, m), 1.72-1.91 (2H, m), 1.37 (3H, d, J=6.05 Hz), 1.29 (3H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10° C./min and revealed a melting/decomposition endotherm with an onset temperature of 297.6° C. and a peak temperature at 299.1° C.
Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide (1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40° C.; a white solid precipitated. The suspension was heated to 90° C. and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.

PAPER

A concise bulk synthesis of stereochemically complex CCR2 antagonist BMS-741672 is reported. A distinct structural feature is the chiral all-cis 1,2,4-triaminocyclohexane (TACH) core, which was assembled through consecutive stereocontrolled heterogeneous hydrogenations: efficient Pt-catalyzed reduction of a β-enaminoester, directed by (S)-α-methylbenzylamine as a low-cost chiral template, and reductive amination of a 3,4-cis-disubstituted cyclohexanone over sulfided Pt/C introduced a tert-amine, setting the third stereocenter in the all-cis cyclohexane core. The heterogeneous catalysts were recycled. Ester hydrolysis produced a γ-amino acid, isolated as its Na salt. A challenging Curtius reaction to introduce the remaining C–N bond at C-2 was strongly influenced by the presence of the basic tert-amine, providing a stereoelectronically highly activated isocyanate. Detailed mechanistic and process knowledge was required to enable clean trapping with an alcohol (t-BuOH) while avoiding formation of side products, particularly an unusual carbamoyl phosphate. Deprotection, N-acetylation, and uncatalyzed SNAr coupling with known 4-chloroquinazoline provided the final product. The resulting 12-step synthesis was used to prepare 50 kg of the target compound in an average yield of 82% per step.
Stereoselective Bulk Synthesis of CCR2 Antagonist BMS-741672: Assembly of an All-cis (S,R,R)-1,2,4-Triaminocyclohexane (TACH) Core via Sequential Heterogeneous Asymmetric Hydrogenations


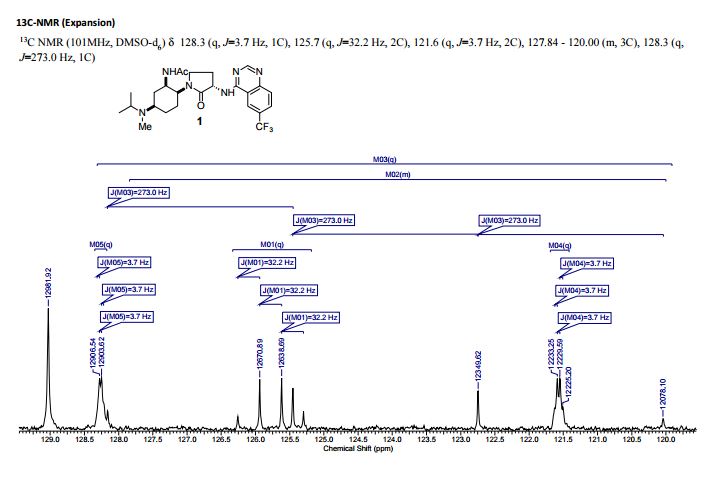

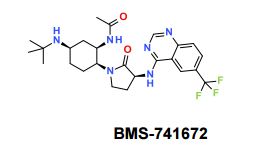
Patents
| Patent ID | Date | Patent Title |
|---|---|---|
| US7687508 | 2010-03-30 | CYCLIC DERIVATIVES AS MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY |
| US7671062 | 2010-03-02 | N-((IR, 2S, 5R)-5-(ISOPROPYL(METHYL)AMINO)-2-((S)-2-0XO-3-(6-TRIFLUOROMETHYL)QUINAZOLIN-4-YLAMINO)PYRROLIDIN-1-YL)CYCLOHEXYL)ACETAMIDE AND OTHER MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY, CRYSTALLINE FORMS AND PROCESS. |
Bristol-Myers Squibb, Paul Biondi Senior Vice President, Head of Business Development
About Bristol-Myers Squibb
Bristol-Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information, please visit www.bms.com or follow us on Twitter at http://twitter.com/bmsnews.
////////// ////////////////BMS-741672, BMS 741672, BRISTOL MEYER SQUIB, PHASE 2, type 2 Diabetes, Neuropathic Pain, Bristol-Myers Squibb
CC(C)N(C)[C@H]1C[C@@H](NC(C)=O)[C@H](CC1)N4CC[C@H](Nc3ncnc2ccc(cc23)C(F)(F)F)C4=O
Glenmark’s TRPA1 antagonist ‘GRC 17536’ shows positive data in a proof of concept study

MUMBAI, India, Sep 17, 2014
- Glenmark's first in class TRPA1 antagonist, GRC 17536, has shown positive data in a Phase 2a proof of concept study in patients with painful diabetic neuropathy
Glenmark Pharmaceuticals today announced that its first in class Transient Receptor Potential Ankyrin 1 (TRPA1) antagonist, GRC 17536 has shown positive data in a Phase 2a double blind, placebo controlled, multi-centre, proof of concept study conducted on 138 patients in Europe and India.
A statistically significant and clinically relevant response was seen in a prospectively-identified, substantial sub-group of patients with moderate to severe pain who had relatively intact sensory responses as detected by a standardized testing methodology. GRC 17536 was well-tolerated with no evidence of CNS or other drug related side effects.
Patrick Keohane, Chief Medical Officer, Glenmark stated “Diabetic neuropathy remains a difficult to manage chronic clinical condition with limited therapeutic options. These initial efficacy and safety data with GRC 17536, a peripherally acting novel therapeutic, are encouraging, and Glenmark intends to be ready to file for a Phase 2b dose range finding study in patients with neuropathic pain before the end of this financial year. This announcement also reaffirms our position globally in the development of novel pain therapies”.
Commenting on this result, Dr. Michael Buschle, Chief Scientific Officer & President – Biologics, Glenmark Pharmaceuticals mentioned, “This is very promising and GRC 17536 may be useful for several indications which we will pursue”.
The Glenmark TRPA1 program includes indications in pain as well as respiratory. Inhaled doses of GRC 17536 are also being tested in a Phase 2A proof of concept study in patients with Chronic Cough.
, Managing Director and CEO, along with Dr. Michael Buschle, President Biologics, Glenmark Pharmaceuticals at a press conference in Mumbai on Monday. Photo: Paul Noronha")
Note on TRPA1
TRPA1 is an ion channel expressed on peripheral and spinal sensory neurons and it mediates pain signal transmission. It functions as a cellular sensor for detecting painful mechanical, biochemical and thermal stimuli that cause sensory nerve hyperactivity during chronic pathologies including chronic pain, inflammation, itch and cough. TRPA1 receptor is shown to induce pain hypersensitivity in animal models of diabetic neuropathic pain and its blockade attenuates pain hypersensitivity as well as later loss of the nerve fibers and their function. GRC 17536 is a potent, selective and first in class antagonist of TRPA1 receptor. Preclinical studies have demonstrated its effectiveness in animal models of neuropathic and inflammatory pain including the peripheral diabetic neuropathic pain, osteoarthritic pain, postoperative pain and chemotherapy induced pain which supports potential utility of TRPA1 blockade in therapeutic pain management.
About Glenmark Pharmaceuticals Ltd
Glenmark Pharmaceuticals Ltd. (GPL) is a research-driven, global, integrated pharmaceutical company and ranked among the top 80 Pharma & Biotech companies of the world in terms of revenues as per SCRIP 100 Rankings. Glenmark is a leading player in the discovery of new molecules both NCEs and NBEs. Glenmark has several molecules in various stages of clinical development and primarily focused in the areas of Inflammation, Pain and Oncology. The company has significant presence in branded formulations across emerging economies including India. Its subsidiary, Glenmark Generics Limited services the requirements of the US and Western Europe markets.

Cebranopadol GRT 6005 セブラノパドール a Potent Analgesic NOP and Opioid Receptor Agonist

- C24H27FN2O
- Average mass: 378.482391 Da

Neuropathic pain
Neuropathic pain is caused when peripheral nerves are damaged by mechanical, metabolic or inflammatory way. The pain occurring images are mainly due to the occurrence of spontaneous pain, hyperalgesia and allodynia (pain is already triggered by non-noxious stimuli) in. As a result, the lesions to increased expression of Na + channels and thus to spontaneous activity in the damaged axons and their Nachbaraxonen (England et al., Neurology, 1996, 47, 272-276).The excitability of the neurons is increased and they react to incoming stimuli with an increased discharge frequency. This results in an increased sensitivity to pain, which contributes to the development of hyperalgesia and spontaneous pain (Baron, Clin J Pain 2000;. 16 (2 Suppl), 12-20). The causes and manifestations, and therefore the treatment needs of neuropathischerm pain are varied. They arise as a result of injury or disease of the brain, spinal cord or peripheral nerves.Causes may be operations, such as phantom pain after amputation, stroke, multiple sclerosis, spinal cord injury, alcohol or drug abuse or other toxins, cancers but also
Metabolic diseases such as diabetes, gout, kidney failure or liver cirrhosis, or infectious diseases such as mononucleosis, ehrlichiosis, typhoid, diphtheria, HIV, syphilis or Lyme disease. The pain experience is very different signs and symptoms that can change over time in number and intensity. Paradoxically, patients with neuropathic pain outline a slowdown or failure of acute pain perception and the simultaneous increase of neuropathic pain. The typical symptoms of neuropathic pain as tingling, burning, shooting or described, or radiating electrifying. Pharmacological basis for treatment of neuropathic pain include tricyclic antidepressants and anticonvulsants, which are used as monotherapy or in combination with opioids. These drugs usually provide only a certain pain relief during a pain-free but is often not achieved. The often-adjusting side effects are dose increases while the drug to achieve adequate pain relief often in the way. In fact, a higher dosage of a μ-opioid is often required as the treatment of acute pain, thereby reducing the side effects get even more important for satisfactory treatment of neuropathic pain. By the occurrence of typical μ-opioid tolerance development and the concomitant need for dose escalation of this problem is exacerbated. In summary it can be stated that neuropathic pain is difficult to treat and today is alleviated by high doses of μ-opioids only partially (Saudi Pharm J. 2002, 10 (3), 73-85). There is therefore an urgent need for medicines for the treatment of chronic pain, the dose should not be increased until the occurrence of intolerable side effects to ensure a satisfactory pain treatment.
……………
http://www.google.com/patents/US7547707
Example 24 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Non-polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (651 mg, 3 mmoles) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 537 mg, 3 mmoles) were initially introduced into abs. MC (20 ml) under argon. Trifluoromethanesulfonic acid trimethylsilyl ester (0.6 ml, 3.1 mmoles) was then added very rapidly. The mixture was stirred at RT for 20 h. For working up, 1 M NaOH (30 ml) was added to the reaction mixture and the mixture was stirred for 30 min. The organic phase was separated, and the aqueous phase which remained was extracted with MC (3×60 ml). The combined organic phases were washed with water (2×30 ml) and dried over sodium sulfate. Methanol (30 ml) was added to the solid residue obtained after the solvent had been distilled off, and the mixture was heated, and stirred for 15 hours. The solid contained in the suspension was filtered off with suction and dried. 955 mg of the more non-polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole were obtained (m.p. 284-292° C.). 850 mg of this were dissolved in hot ethanol (900 ml), and a similarly hot solution of citric acid (1 g, 5.2 mmoles) in ethanol (20 ml) was added. After approx. 15 minutes, crystals precipitated out at the boiling point. After cooling to approx. 5° C., the mixture was left to stand for 2 h. The solid formed was filtered off with suction. 640 mg of the hemicitrate were obtained as a white solid (m.p. 258-282° C.).
Example 25 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (217 mg, 1 mmole) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 179 mg, 1 mmole) were dissolved in conc. acetic acid (4 ml). Phosphoric acid (1 ml, 85 wt. %) was slowly added dropwise to this mixture. The mixture was stirred at RT for 16 h. For working up, the mixture was diluted with water (20 ml), brought to pH 11 with 5 M NaOH and extracted with MC (3×20 ml). The combined organic phases were dried with sodium sulfate and evaporated. The residue (364 mg of white solid) was suspended in hot ethanol (20 ml), and a similarly hot solution of citric acid (185 mg, 0.96 mmole) in ethanol (5 ml) was added. The residue thereby dissolved completely and no longer precipitated out even on cooling to approx. 5° C. Ethanol was removed on a rotary evaporator and the hemicitrate of the more polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole was obtained in this way in a yield of 548 mg as a white solid (m.p. 148-155° C.).
| 24 |
 |
hemicitrate | more non-polar diastereomer |
| 25 |
 |
hemicitrate | more polar diastereomer |
(1 r,4r)-6′-fluoro-N,N- dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1 ,1 ‘-pyrano[3,4-b]indol]-4-amine (free base), has the following structural formula (I):
http://www.google.com/patents/WO2013113690A1?cl=en


One particular drug that is of great interest for use in treating cancer pain (and other acute, visceral, neuropathic and chronic pain pain disorders) is (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine. This drug is depicted below as the compound of formula (I).

The solid forms of (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine that are known so far are not satisfactory in every respect and there is a demand for advantageous solid forms

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol
http://pubs.acs.org/doi/full/10.1021/ml500117c
b]indol]-4-amine, trans-, 2-hydroxy-1,2,3-propanetricarboxylate (2:1)
2.76 (m,6 H); 3.88 (t, 2 H); 6.86 (dt, 1 H); 7.10 (dd, 1 H); 7.30-7.43 (m, 6 H); 10.91 (br
s, 1 H).
overlap); 71.5; 72.2; 102.3 (2JC,F = 23 Hz); 105.6 (3JC,F = 4 Hz); 108.3 (2JC,F = 26 Hz);
156,7 (1JC,F = 231 Hz); 171.3 (2 C), 175.3.HPLC-MS: m/z 378.9 [M + H]+
| US20120034297 * | Aug 4, 2011 | Feb 9, 2012 | Gruenenthal Gmbh | Pharmaceutical dosage forms comprising 6′-fluoro-(N-methyl- or N,N-dimethyl-)-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| WO2004043967A1 | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2008040481A1 | Sep 26, 2007 | Apr 10, 2008 | Gruenenthal Gmbh | MIXED ORL 1/μ AGONISTS FOR TREATING PAIN |
-
CORAL – Cebranopadol Versus Morphine Prolonged-release in Patients With Chronic Moderate to Severe Pain Related to Cancer
Efficacy, Safety, and Tolerability of Oral Cebranopadol Versus Morphine Sulfate PR in Subjects With Chronic Moderate to Severe Pain Related to Cancer.Average amount of daily rescue medication at the end of the maintenance period.
UK Clinical Trials Gateway, 07 October 2013
-
CORAL XT – Open-label Extension Trial of the CORAL Trial
An Open-label, Multi-site Trial to Describe the Safety and Tolerability of Oral Cebranopadol Administered for 26 Weeks in Subjects With Cancer-related Pain Who Have Completed Treatment in the KF6005/07 Trial.Absolute…
UK Clinical Trials Gateway, 12 December 2013
| WO2004043967A1 * | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2005066183A1 * | Dec 21, 2004 | Jul 21, 2005 | Gruenenthal Gmbh | Spirocyclic cyclohexane derivatives with affinity for the orl1-receptor |
| US20050153998 * | Aug 19, 2004 | Jul 14, 2005 | Fumitaka Ito | Tetrahydroisoquinoline or isochroman compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7799931 * | Feb 17, 2009 | Sep 21, 2010 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7951948 * | Apr 19, 2010 | May 31, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7960404 | Aug 21, 2009 | Jun 14, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8034936 | Nov 4, 2010 | Oct 11, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds useful to treat substance dependency |
| US8053576 | Feb 17, 2009 | Nov 8, 2011 | Gruenenthal Gmbh | Treating conditions associated with the nociceptin/ORL1 receptor system, e.g. pain, drug withdrawal, anxiety, muscle relaxants, anxiolytic agents; e.g. 1,1-[3-dimethylamino-3-(pyridin-2-yl)pentamethylene]-3,4-dihydro-1H-2,9-diazafluorene |
| US8288406 | Sep 22, 2010 | Oct 16, 2012 | Gruenenthal Gmbh | Hydroxymethylcyclohexylamines |
| US8288430 | Mar 25, 2009 | Oct 16, 2012 | Grunenthal Gmbh | Spiro(5.5)undecane derivatives |
| US8293758 * | Mar 25, 2009 | Oct 23, 2012 | Grunenthal Gmbh | Substituted spirocyclic cyclohexane derivatives |
| US8357705 | Mar 25, 2009 | Jan 22, 2013 | Gruenenthal Gmbh | Substituted cyclohexyldiamines |
| US8404740 | Aug 21, 2009 | Mar 26, 2013 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8614245 * | Jan 8, 2013 | Dec 24, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8618156 * | Jul 6, 2012 | Dec 31, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
http://www.stumbleupon.com/stumbler/amcrasto
VIETNAM
ICELAND
RUSSIA

========================


{kind=link}