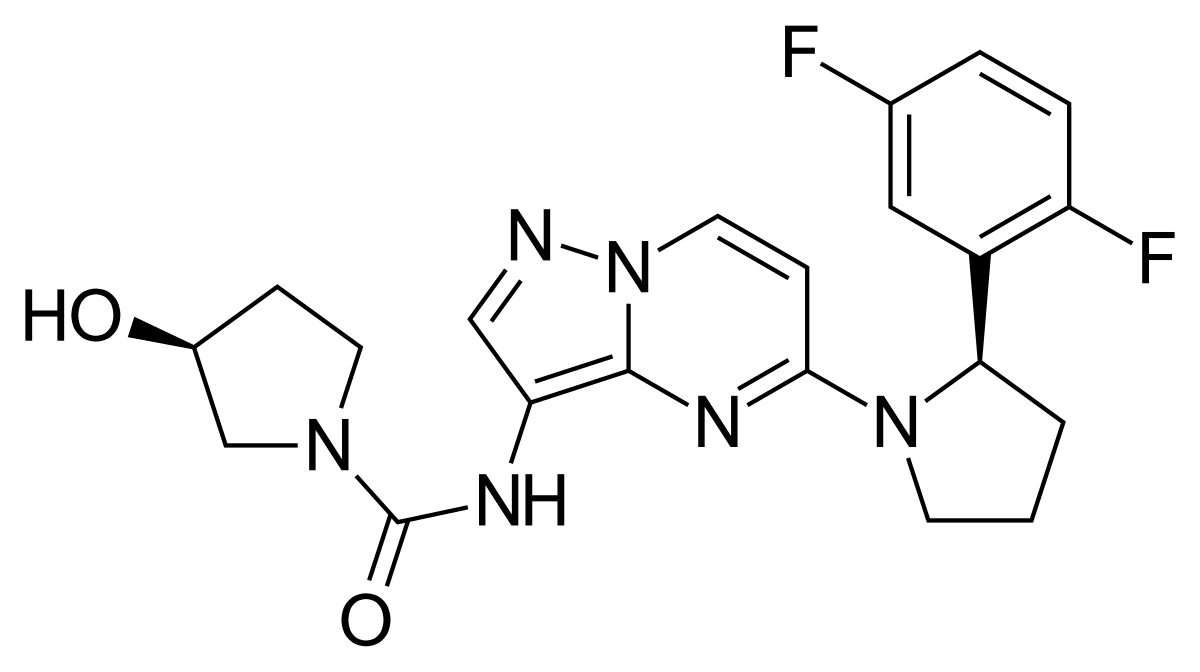
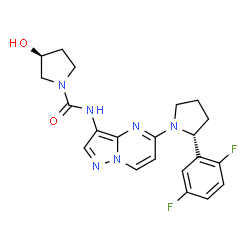
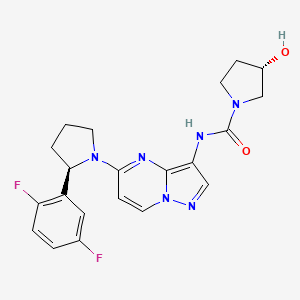

Larotrectinib
ARRY-470, LOXO-101, PF9462I9HX
| Molecular Formula: |
C21H22F2N6O2 |
| Molecular Weight: |
428.444 g/mol |
(3S)-N-{5-[(2R)-2-(2,5-Difluorphenyl)-1-pyrrolidinyl]pyrazolo[1,5-a]pyrimidin-3-yl}-3-hydroxy-1-pyrrolidincarboxamid
(S)-N-{5-[(R)-2-(2,5-Difluorophenyl)pyrrolidin-1-yl]pyrazolo[1,5-a]pyrimidin-3-yl}-3-hydroxypyrrolidine-1-carboxamide
10360
UNII:PF9462I9HX
ларотректиниб [Russian] [INN]
(3S)-N-[5-[(2R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl]pyrazolo[1,5-a]pyrimidin-3-yl]-3-hydroxypyrrolidine-1-carboxamide
NTRK-fusion solid tumours
TRK inhibitor
orphan drug designation in the U.S
In 2013, Array Biopharma licensed the product to Loxo Oncology for development and commercialization in the U.S. In 2016, breakthrough therapy designation was received in the U.S. for the treatment of unresectable or metastatic solid tumors with NTRK-fusion proteins in adult and pediatric patients who require systemic therapy and who have either progressed following prior treatment or who have no acceptable alternative treatments. In 2017, Bayer acquired global co-development and commercialization rights from Loxo Oncology.
- Originator Array BioPharma
- Developer Array BioPharma; Loxo Oncology; National Cancer Institute (USA)
- Class Antineoplastics; Pyrazoles; Pyrimidines; Pyrrolidines; Small molecules
- Mechanism of Action Tropomyosin-related kinase antagonists
- Orphan Drug Status Yes – Solid tumours; Soft tissue sarcoma
Highest Development Phases
- Preregistration Solid tumours
- Phase II Histiocytosis; Non-Hodgkin’s lymphoma
- Phase I/II CNS cancer
- Preclinical Precursor cell lymphoblastic leukaemia-lymphoma
Most Recent Events
- 29 May 2018 FDA assigns PDUFA action date of 26/11/2018 for larotrectinib for Solid tumors
- 29 May 2018 Larotrectinib receives priority review status for Solid tumors in the US
- 29 May 2018 The US FDA accepts NDA for larotrectinib for Solid tumours for review

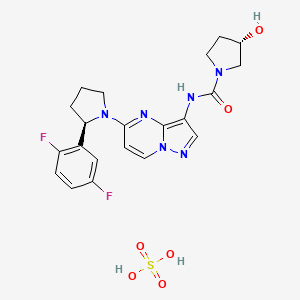
Larotrectinib sulfate
(3S)-N-[5-[(2R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl]pyrazolo[1,5-a]pyrimidin-3-yl]-3-hydroxypyrrolidine-1-carboxamide;sulfuric acid
Larotrectinib (LOXO-101) sulfate is an oral potent and selective ATP-competitive inhibitor of tropomyosin receptor kinases (TRK).
-
Crystalline Form (I-HS) OF
SULFATE SALT REPORTED IN https://patents.google.com/patent/US20170165267
nmr http://file.selleckchem.com/downloads/nmr/s796001-loxo-101-methanol-hnmr-selleck.pdf


| Molecular Weight |
526.51 |
| Formula |
C21H22F2N6O2.H2O4S |
| CAS No. |
1223405-08-0 |
- LOXO-101 sulfate
- Larotrectinib sulfate
- LOXO-101 (sulfate)
- 1223405-08-0
- UNII-RDF76R62ID
- RDF76R62ID
- ARRY-470 sulfate
- LOXO-101(sulfate)
- Larotrectinib sulfate [USAN]
- PXHANKVTFWSDSG-QLOBERJESA-N
- HY-12866A
- s7960
- AKOS030526332
- CS-5314
LOXO-101 is a small molecule that was designed to block the ATP binding site of the TRK family of receptors, with 2 to 20 nM cellular potency against the TRKA, TRKB, and TRKC kinases. IC50 value: 2 – 20 nM Target: TRKA/B/C in vitro: LOXO-101 is an orally administered inhibitor of the TRK kinase and is highly selective only for the TRK family of receptors. LOXO-101 is evaluated for off-target kinase enzyme inhibition against a panel of 226 non-TRK kinases at a compound concentration of 1,000 nM and ATP concentrations near the Km for each enzyme. In the panel, LOXO-101 demonstrates greater than 50% inhibition for only one non-TRK kinase (TNK2 IC50, 576 nM). Measurement of proliferation following treatment with LOXO-101 demonstrates a dose-dependent inhibition of cell proliferation in all three cell lines. The IC50 is less than 100 nM for CUTO-3.29 and less than 10 nM for KM12 and MO-91, consistent with the known potency of this drug for the TRK kinase family. [1] LOXO-101 demonstrates potent and highly-selective inhibition of TRKA, TRKB, and TRKC over other kinase- and non-kinase targets. LOXO-101 is a potent, ATP-competitive TRK inhibitor with IC50s in low nanomolar range for inhibition of all TRK family members in binding and cellular assays, with 100x selectivity over other kinases. [2] in vivo: Athymic nude mice injected with KM12 cells are treated with LOXO-101 orally daily for 2 weeks. Dose-dependent tumor inhibition is observed, demonstrating the ability of this selective compound to inhibit tumor growth in vivo. [1]


DOI
https://doi.org/10.1038/nrd.2018.4
SYNTHESIS
WO 2010048314

Synthesis of larotrectinib
N-Boc-pyrrolidine as starting material The method involves enantioselective deprotonation, transmetalation with ZnCl2, Negishi coupling with 2-bromo-1,4-difluorobenzene,
N-arylation with 5-chloropyrazolo[1,5-a]pyrimidine, nitration, nitro reduction and condensation with CDI and 3(S)-pyrrolidinol.
PRODUCT Patent
WO 2010048314
https://patents.google.com/patent/WO2010048314A1
InventorJulia HaasSteven W. AndrewsYutong JiangGan Zhang
Original AssigneeArray Biopharma Inc.
Priority date 2008-10-22
Example 14

(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-alpyrimidin-3-yl)- 3 -hydroxypyrrolidine- 1 -carboxamide
[00423] To a DCM (0.8 mL) solution of (R)-5-(2-(2,5-difiuorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-amine (Preparation B; 30 mg, 0.095 mmol) was added CDI (31 mg, 0.19 mmol) at ambient temperature in one portion. After stirring two hours, (S)-pyrrolidin-3-ol (17 mg, 0.19 mmol) [purchased from Suven Life Sciences] was added in one portion. The reaction was stirred for 5 minutes before it was concentrated and directly purified by reverse-phase column chromatography, eluting with 0 to 50% acetonitrile/water to yield the final product as a yellowish foamy powder (30 mg, 74% yield). MS (apci) m/z = 429.2 (M+H).
Example 14A

(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolori,5-alpyrimidin-3-yl)- 3 -hydroxypyrrolidine- 1 -carboxamide sulfate
[00424] To a solution of (S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo [ 1 ,5 -a]pyrimidin-3 -yl)-3 -hydroxypyrrolidine- 1 -carboxamide (4.5 mg, 0.011 mmol) in methanol (1 mL) at ambient temperature was added sulfuric acid in MeOH (105 μL, 0.011 mmol). The resulting solution was stirred for 30 minutes then concentrated to provide (S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-yl)-3 -hydroxypyrrolidine- 1 -carboxamide sulfate (5.2 mg, 0.0099 mmol, 94 % yield) as a yellow solid.
PATENT
WO 2017201241
Examples
Preparation of 10:
1)

(R,E)-N-(2,5-difluorobenzylidene)-2-methylpropane-2-sulfinamide (17): Compound 16 and (R)-2-methylpropane-2-sulfinamide (1.05 eq.) were charged to a reactor outfitted with a mechanical stirrer, reflux condensor, J-Kem temperature probe under N2. DCM (3 mL/g of 14) was added (endothermic from 22 °C to about 5 °C) followed by addition of cesium carbonate (0.70 eq.) (exothermic to -50 °C). Once the addition was complete, the reaction mixture was stirred at room temperature for 3 h (slowly cools from about 40 °C). When the reaction was called complete (HPLC) the mixture was filtered through Celite. The Celite pad (0.3 wt eq) was equilibrated with DCM (1 mL/g of 16), and the reaction mixture was poured through the pad. The Celite cake was washed with DCM (2 x 1 mL/g), and the filtrate concentrated partially to leave about 0.5 to 1 mL/g DCM remaining. The orange solution was stored at room temperature (generally overnight) and used directly in the next reaction. (100% yield was assumed).
2)

(R)-N-((R)-l-(2,5-difluorophenyl)-3-(l,3-dioxan-2-yl)propyl)-2-methylpropane-2-sulfinamide (19): To a reactor equipped with overhead stirring, reflux condensor, under
nitrogen, was added magnesium turnings (2.0 eq), and THF (8 mL/g of 17). The mixture was heated to 40 °C. Dibal-H (25% wt in toluene, 0.004 eq) was added to the solution, and the suspension heated at 40 °C for 25 minutes. A solution of 2-(2-bromoethyl)-l,3-dioxane (18) (2 eq) in THF (4.6 mL/g of 17) was added dropwise to the Mg solution via addition funnel. The solution temperature was maintained < 55 °C. The reaction progress was monitored by GC. When the Grignard formation was judged complete, the solution was cooled to -30 °C, and 17 (1.0 eq, in DCM) was added dropwise via addition funnel. The temperature was kept between -30 °C and -20 °C and the reaction was monitored for completion (FIPLC). Once the reaction was called complete, the suspension (IT = -27.7 °C) was vacuum transferred to a prepared and cooled (10 °C) 10% aqueous citric acid solution (11 mL/g of 17). The mixture temperature rose to 20 °C during transfer. The milky solution was allowed to stir at ambient temperature overnight. MTBE (5.8 mL/g) was added to the mixture, and it was transferred to a separatory funnel. The layers were allowed to separate, and the lower aqueous layer was removed. The organic layer was washed with sat. NaHC03 (11 mL/g) and then sat. NaCl (5.4 mL/g). The organic layer was removed and concentrated to minimum volume via vacuum distillation. MTBE (2 mL/g) was added, and the mixture again concentrated to minimum volume. Finally MTBE was added to give 2 mL/g total MTBE (GC ratio of MTBE:THF was about 9: 1), and the MTBE mixture was heated to 50 °C until full dissolution occurred. The MTBE solution was allowed to cool to about 35 °C, and heptane was added portion -wise. The first portion (2 mL/g) is added, and the mixture allowed to stir and form a solid for 1-2 h, and then the remainder of the heptane is added (8 mL/g). The suspension was allowed to stir for >lh. The solids were collected via filtration through polypropylene filter cloth (PPFC) and washed with 10% MTBE in heptane (4 mL/g. The wet solid was placed in trays and dried in a vacuum oven at 55 °C until constant weight (3101 g, 80.5%, dense white solid, 100a% and 100wt%).
3)

(R)-2-(2,5-difluorophenyl)pyrrolidine (R)-2-hydroxysuccinate (10): To a flask containing 4: 1 TFA:water (2.5 mL/g, pre-mixed and cooled to <35 °C before adding 19) was added (R)-N-((R)-l-(2,5-difluorophenyl)-3-(l,3-dioxan-2-yl)propyl)-2-methylpropane-2-sulfinamide (19) (1 eq). The mixture temperature rose from 34 °C to 48 °C and was stirred at ambient temperature for 1 h. Additional TFA (7.5 mL/g) was added, followed by triethylsilane (3 eq) over 5 minutes. The biphasic mixture was stirred vigorously under nitrogen for 21 h until judged complete (by GC, <5% of imine). The mixture was then concentrated under vacuum until -10 kg target mass (observed 10.8 kg after concentration). The resulting concentrate was transferred to a separatory funnel and diluted with MTBE (7.5 mL/g), followed by water (7.5 mL/g). The layers were separated. The MTBE layer was back-extracted with 1M HC1 (3 mL/g). The layers were separated, and the aqueous layers were combined in a round-bottomed flask with DCM (8 mL/g). The mixture was cooled in an ice bath and 40% NaOH was charged to adjust the pH to >12 (about 0.5 mL/g; the temperature went from 24 °C to 27 °C, actual pH was 13), and the layers separated in the separatory funnel. The aqueous layer was back-extracted twice with DCM (2 x 4 mL/g). The organic layers were concentrated to an oil (<0.5 mL/g) under vacuum (rotovap) and EtOH (1 mL/g based on product) was added. The yellow solution was again concentrated to an oil (81% corrected yield, with 3% EtOH, 0.2% imine and Chiral HPLC showed 99.7%ee).
Salt formation: To a solution of (R)-2-(2,5-difluorophenyl)pyrrolidine 10 (1 eq) in EtOH (15 mL/g) was added Z)-(+)-Malic Acid (1 eq). The suspension was heated to 70 °C for 30 minutes (full dissolution had occurred before 70 °C was reached), and then allowed to cool to room temperature slowly (mixture was seeded when the temperature was < 40 °C). The slurry was stirred at room temperature overnight, then cooled to <5 °C the next morning. The suspension was stirred at <5 °C for 2h, filtered (PPFC), washed with cold EtOH (2 x 2 mL/g), and dried (50-55 °C) under vacuum to give the product as a white solid (96% based on 91% potency, product is an EtOH solvate or hemi- solvate).
Preparation of the compound of Formula I:
1)

(R)-5-(2-(2,5-difluorophenyl)pyrrolidin-l-yl)-3-nitropyrazolo[l,5-a]pyrimidine (11):
Compound 5 and 10 (1.05 eq) were charged to a reactor outfitted with a mechanical stirrer, J-Kem temperature probe, under N2. EtOH and THF (4: 1, 10 mL/g of 5) were added and the mixture was cooled to 15-25 °C. Triethylamine (3.5 eq) was added and the internal temp generally rose from 17.3 – 37.8 °C. The reaction was heated to 50 – 60 °C and held at that temperature for 7 h. Once the reaction is judged complete (HPLC), water (12 mL/g of 5) is added maintaining the temperature at 50 – 60 °C. The heat is removed and the suspension was slowly cooled to 21 °C over two h. After stirring at -21 °C for 2 h, the suspension was centrifuged and the cake was washed with water (3 x 3 mL/g of 5). The solid was transferred to drying trays and placed in a vacuum oven at 50 – 55 °C to give 11.
2)

(R)-5-(2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-amine fumarate Pt/C hydrogenation (12 fumarate): To a Parr reactor was charged 11 (1.0 eq), 5% Pt/C ~ 50 wt% water (2 mol% Pt / Johnson Matthey B 103018-5 or Sigma Aldrich 33015-9), and MeOH (8 mL/g). The suspension was stirred under hydrogen at 25-30 psi and the temperature was maintained below 65 °C for ~8 h. When the reaction was called complete (HPLC), the reaction was cooled to 15 – 25 °C and the hydrogen atmosphere was replaced with a nitrogen atmosphere. The reaction mixture was filtered through a 2 micron bag filter and a 0.2 micron line filter in series. The filtrate from the Pt/C hydrogenation was transferred to a reactor under nitrogen with mechanical stirring and then MTBE (8 mL/g) and fumaric acid (1.01 eq) were charged. The mixture was stirred under nitrogen for 1 h and solids formed after -15 min. The mixture was cooled to -10 to -20 °C and stirred for 3 h. The suspension was filtered (PPFC), washed with MTBE (-2.5 mL/g), and the solids was dried under vacuum at 20-25 °C with a nitrogen bleed to yield an off-white solid (83% yield).
3)

Phenyl (5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-3,3a-dihydropyrazolo[l,5-a]pyrimidin-3-yl)carbamate (13): To a 5 to 15°C solution of 12-fumarate (1.0 eq) in 2-MeTHF (15 mL/g) was added a solution of potassium carbonate (2.0 eq.) in water (5 mL/g) followed by phenyl chloroformate (1.22 eq.) (over 22 min, an exotherm from 7 °C to 11 °C occurred). The mixture was stirred for 2 h and then the reaction was called complete (HPLC). The stirring ceased and the aqueous layer was removed. The organic layer was washed with brine (5 mL/g) and concentrated to ca. 5 mL/g of 2-MeTHF under vacuum and with heating to 40 °C. To the 2-MeTHF solution was added heptanes (2.5 mL/g) followed by seeds (20 mg, 0.1 wt%). This mixture was allowed to stir at room temperature for 2 h (until a solid formed), and then the remainder of the heptanes (12.5 mL/g) was added. The mixture was stirred at ambient temperature for 2 h and then the solids were collected via filtration (PPFC), washed with 4: 1 heptanes :MeTHF (2 x 2 mL/g), and dried to give 13 (96%).
4)

(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate: To a flask containing 13 (1.0 eq) was added a solution of (S)-pyrrolidin-3-ol (1.1 eq.) in EtOH (10 mL/g). The mixture was heated at 50 – 60 °C for 5 h, called complete (HPLC), and then cooled to 20-35 °C. Once <35°C, the reaction was polish-filtered (0.2 micron) into a clean reaction vessel and the mixture was cooled to -5 to 5 °C. Sulfuric acid (1.0 eq.) was added over 40 minutes, the temperature rose to 2 °C and the mixture was seeded. A solid formed, and the mixture was allowed to stir at -5 to 5 °C for 6.5 h. Heptanes (10 mL/g) was added, and the mixture stirred for 6.5 h. The
suspension was filtered (PPFC), washed with 1 : 1 EtOH:heptanes (2 x 2 mL/g), and dried (under vacuum at ambient temperature) to give Formula I (92.3%).
Preparation of the hydrogen sulfate salt of the compound of Formula I:
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)-N-(5- ((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (,S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1 : 1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5 :95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5 :95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate.
PATENT
US2017165267
https://patents.google.com/patent/US20170165267
Provided herein is a novel crystalline form of the compound of Formula I:
[0000]

also known as (S)—N-(5-((R)-2-(2, 5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide. In particular, the novel crystalline form comprises the hydrogen sulfate salt of the compound of Formula I in a stable polymorph form, hereinafter referred to as crystalline form (I-HS) and LOXO-101, which can be characterized, for example, by its X-ray diffraction pattern—the crystalline form (I-HS) having the formula:
[0000]

In some embodiments of the above step (c), the base is an alkali metal base, such as an alkali metal carbonate, such as potassium carbonate.

Preparation of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine Step A—Preparation of sodium pyrazolo[1,5-a]pyrimidin-5-olate
A solution of 1H-pyrazol-5-amine and 1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1.05 equiv.) were charged to a round bottom flask outfitted with a mechanical stirrer, a steam pot, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control. Under mechanical stirring the solids were suspended with 4 vol. (4 mL/g) of absolute EtOH under a nitrogen atmosphere, then charged with 2.1 equivalents of NaOEt (21 wt % solution in EtOH), and followed by line-rinse with 1 vol. (1 mL/g) of absolute EtOH. The slurry was warmed to about 75° Celsius and stirred at gentle reflux until less than 1.5 area % of 1H-pyrazol-5-amine was observed by TRK1PM1 HPLC to follow the progression of the reaction using 20 μL of slurry diluted in 4 mL deionized water and 5 μL injection at 220 nm.
After 1 additional hour, the mixture was charged with 2.5 vol. (2.5 mL/g) of heptane and then refluxed at 70° Celsius for 1 hour. The slurry was then cooled to room temperature overnight. The solid was collected by filtration on a tabletop funnel and polypropylene filter cloth. The reactor was rinsed and charged atop the filter cake with 4 vol. (4 mL/g) of heptane with the cake pulled and the solids being transferred to tared drying trays and oven-dried at 45° Celsius under high vacuum until their weight was constant. Pale yellow solid sodium pyrazolo[1,5-a]-pyrimidin-5-olate was obtained in 93-96% yield (corrected) and larger than 99.5 area % observed by HPLC (1 mg/mL dilution in deionized water, TRK1PM1 at 220 nm).
Step B—Preparation of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one
A tared round bottom flask was charged with sodium pyrazolo[1,5-a]pyrimidin-5-olate that was dissolved at 40-45° Celsius in 3.0 vol. (3.0 mL/g) of deionized water, and then concentrated under high vacuum at 65° Celsius in a water-bath on a rotary evaporator until 2.4× weight of starting material was observed (1.4 vol/1.4 mL/g deionized water content). Gas chromatography (GC) for residual EtOH (30 μL of solution dissolved in ˜1 mL MeOH) was performed showing less than 100 ppm with traces of ethyl nitrate fumes being observed below upon later addition of HNO3. In some cases, the original solution was charged with an additional 1.5 vol. (1.5 mL/g) of DI water, then concentrated under high vacuum at 65° Celsius in a water-bath on a rotary evaporator until 2.4× weight of starting material was observed (1.4 vol/1.4 mL/g DI water content). Gas chromatograph for residual EtOH (30 μL of solution dissolved in about 1 mL MeOH) was performed showing <<100 ppm of residual EtOH without observing any ethyl nitrate fumes below upon later addition of HNO3.
A round bottom vessel outfitted with a mechanical stirrer, a steam pot, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control was charged with 3 vol. (3 mL/g, 10 equiv) of >90 wt % HNO3 and cooled to about 10° Celsius under a nitrogen atmosphere using external ice-water cooling bath under a nitrogen atmosphere. Using a pressure equalizing addition funnel, the HNO3solution was charged with the 1.75-1.95 volumes of a deionized water solution of sodium pyrazolo[1,5-a]pyrimidin-5-olate (1.16-1.4 mL DI water/g of sodium pyrazolo[1,5-a]pyrimidin-5-olate) at a rate to maintain 35-40° Celsius internal temperature under cooling. Two azeotropes were observed without any ethyl nitrate fumes. The azeotrope flask, the transfer line (if applicable) and the addition funnel were rinsed with 2×0.1 vol. (2×0.1 mL/g) deionized water added to the reaction mixture. Once the addition was complete, the temperature was gradually increased to about 45-50° Celsius for about 3 hours with HPLC showing >99.5 area % conversion of sodium pyrazolo[1,5-a]pyrimidin-5-olate to 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one.
Step C—Preparation of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine
3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one was charged to a round bottom flask outfitted with a mechanical stirrer, a heating mantle, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2pressure control. Under mechanical stirring the solids were suspended with 8 volumes (8 mL/g) of CH3CN, and then charged with 2,6-lutitine (1.05 equiv) followed by warming the slurry to about 50° Celsius. Using a pressure equalizing addition funnel, the mixture was dropwise charged with 0.33 equivalents of POCl3. This charge yielded a thick, beige slurry of a trimer that was homogenized while stirring until a semi-mobile mass was observed. An additional 1.67 equivalents of POCl3 was charged to the mixture while allowing the temperature to stabilize, followed by warming the reaction mixture to a gentle reflux (78° Celsius). Some puffing was observed upon warming the mixture that later subsided as the thick slurry got thinner.
The reaction mixture was allowed to reflux until complete dissolution to a dark solution and until HPLC (20 μL diluted in 5 mL of CH3CN, TRK1PM1 HPLC, 5 μL injection, 268 nm) confirmed that no more trimer (RRT 0.92) was present with less than 0.5 area % of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one (RRT 0.79) being observed by manually removing any interfering and early eluting peaks related to lutidine from the area integration. On a 1.9 kg scale, 0 area % of the trimer, 0.25 area % of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one, and 99.5 area % of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine was observed after 19 hours of gentle reflux using TRK1PM1 HPLC at 268 [0000]

Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxysuccinate Step A—Preparation of tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate
2-bromo-1,4-difluorobenzene (1.5 eq.) was dissolved in 4 volumes of THF (based on weight of tert-butyl 2-oxopyrrolidine-1-carboxylate) and cooled to about 5° Celsius. A solution of 2.0 M iPrMgCl in THF (1.4 eq.) was added over 2 hours to the mixture while maintaining a reaction temperature below 25° Celsius. The solution was allowed to cool to about 5° Celsius and stirred for 1 hour (GC analysis confirmed Grignard formation). A solution of tert-butyl 2-oxopyrrolidine-1-carboxylate (1.0 eq.) in 1 volume of THF was added over about 30 min while maintaining a reaction temperature below 25° Celsius. The reaction was stirred at about 5° Celsius for 90 min (tert-butyl 2-oxopyrrolidine-1-carboxylate was confirmed to be less than 0.5 area % by HPLC). The reaction was quenched with 5 volumes of 2 M aqueous HCl while maintaining a reaction temperature below 45° Celsius. The reaction was then transferred to a separatory funnel adding 10 volumes of heptane and removing the aqueous layer. The organic layer was washed with 4 volumes of saturated aqueous NaCl followed by addition of 2×1 volume of saturated aqueous NaCl. The organic layer was solvent-switched to heptane (<1% wt THF confirmed by GC) at a distillation temperature of 35-55° Celsius and distillation pressure of 100-200 mm Hg for 2×4 volumes of heptane being added with a minimum distillation volume of about 7 volumes. The mixture was then diluted to 10 volumes with heptane while heating to about 55° Celsius yielded a denser solid with the mixture being allowed to cool to room temperature overnight. The slurry was cooled to less than 5° Celsius and filtered through polypropylene filter cloth. The wet cake was washed with 2×2 volumes of heptane. The solids were dried under vacuum at 55° Celsius until the weight was constant, yielding tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate as a white solid at about 75% to 85% theoretical yield.
Step B—Preparation of 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole
tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate was dissolved in 5 vol. of toluene with 2.2 eq. of 12M HCl being added observing a mild exotherm and gas evolution. The reaction was heated to 65° Celsius for 12-24 hours and monitored by HPLC. Upon completion the reaction was cooled to less than 15° Celsius with an ice/water bath. The pH was adjusted to about 14 with 3 equivalents of 2M aqueous NaOH (4.7 vol.). The reaction was stirred at room temperature for 1-2 hours. The mixture was transferred to a separatory funnel with toluene. The aqueous layer was removed and the organic layer was washed with 3 volumes of saturated aqueous NaCl. The organic layer was concentrated to an oil and redissolved in 1.5 volumes of heptane. The resulting suspension was filtered through a GF/F filter paper and concentrated to a light yellow oil of 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole with a 90% to 100% theoretical yield.
Step C—Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine
Chloro-1,5-cyclooctadiene iridium dimer (0.2 mol %) and (R)-2-(2-(diphenylphosphino)phenyl)-4-isopropyl-4,5-dihydrooxazole (0.4 mol %) were suspended in 5 volumes of MTBE (based on 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole) at room temperature. The mixture was stirred for 1 hour and most of the solids dissolved with the solution turning dark red. The catalyst formation was monitored using an HPLC/PDA detector. The reaction was cooled to less than 5° Celsius and 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole (1.0 eq.) was added using a 0.5 volumes of MTBE rinse. Diphenylsilane (1.5 eq.) was added over about 20 minutes while maintaining a reaction temperature below 10° Celsius. The reaction was stirred for 30 minutes below 10° Celsius and then allowed to warm to room temperature. The reaction was stirred overnight at room temperature. The completion of the reaction was confirmed by HPLC and then cooled to less than 5° Celsius. The reaction was quenched with 5 volumes of 2M aqueous HCl maintaining temperature below 20° Celsius. After 10 minutes the ice/water bath was removed and the reaction temperature was allowed to increase to room temperature while stirring for 2 hours. The mixture was transferred to a separatory funnel with 3 volumes of MTBE. The aqueous layer was washed with 3.5 volumes of MTBE followed by addition of 5 volumes of MTBE to the aqueous layer while adjusting the pH to about 14 by adding 0.75 volumes of aqueous 50% NaOH. The organic layer was washed with 5 volumes of aqueous saturated NaCl, then concentrated to an oil, and diluted with 3 volumes of MTBE. The solution was filtered through a polypropylene filter cloth and rinsed with 1 volume of MTBE. The filtrate was concentrated to an oil of (R)-2-(2,5-difluorophenyl)-pyrrolidine with a 95% to 100% theoretical yield and with 75-85% ee.
Step D—Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate
(R)-2-(2,5-difluorophenyl)-pyrrolidine (1.0 eq.) was transferred to a round bottom flask charged with 15 volumes (corrected for potency) of EtOH (200 prf). D-malic acid (1.05 eq.) was added and the mixture was heated to 65° Celsius. The solids all dissolved at about 64° Celsius. The solution was allowed to cool to RT. At about 55° Celsius the solution was seeded with (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate (about 50 mg, >97% ee) and stirred at room temperature overnight. The suspension was then filtered through a polypropylene filter cloth and washed with 2×1 volumes of EtOH (200 prf). The solids were dried under vacuum at 55° Celsius, yielding (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate with a 75% to 90% theoretical yield and with >96% ee.
Referring to Scheme 1, suitable bases include tertiary amine bases, such as triethylamine, and K2CO3. Suitable solvents include ethanol, heptane and tetrahydrofuran (THF). The reaction is conveniently performed at temperatures between 5° Celsius and 50° Celsius. The reaction progress was generally monitored by HPLC TRK1PM1.
[0247]
Compounds II (5-chloro-3-nitropyrazolo[1,5-a]pyrimidine) and III ((R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxysuccinate, 1.05 eq.) were charged to a round bottom flask outfitted with a mechanical stirrer, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control. A solution of 4:1 EtOH:THF (10 mL/g of compound II) was added and followed by addition of triethylamine (NEt3, 3.50 eq.) via addition funnel with the temperature reaching about 40° Celsius during addition. Once the addition was complete, the reaction mixture was heated to 50° Celsius and stirred for 0.5-3 hours to yield compound IV.
To a round bottom flask equipped with a mechanical stirrer, a J-Kem temperature probe, and an N2 inlet compound IV was added and followed by addition of tetrahydrofuran (10 mL/g of compound IV). The solution was cooled to less than 5° Celsius in an ice bath, and Zn (9-10 eq.) was added. 6M HCl (9-10 eq.) was then added dropwise at such a rate to keep the temperature below 30° Celsius (for 1 kg scale the addition took about 1.5 hours). Once the exotherm subsided, the reaction was allowed to warm to room temperature and was stirred for 30-60 min until compound IV was not detected by HPLC. At this time, a solution of potassium carbonate (K2CO3, 2.0 eq.) in water (5 mL/g of compound IV) was added all at once and followed by rapid dropwise addition of phenyl chloroformate (PhOCOCl, 1.2 eq.). Gas evolution (CO2) was observed during both of the above additions, and the temperature increased to about 30° Celsius after adding phenyl chloroformate. The carbamate formation was stirred at room temperature for 30-90 min. HPLC analysis immediately followed to run to ensure less than 1 area % for the amine being present and high yield of compound VI in the solution.
To the above solution amine VII ((S)-pyrrolidin-3-ol, 1.1 eq. based on theoretical yield for compound VI) and EtOH (10 mL/g of compound VI) was added. Compound VII was added before or at the same time as EtOH to avoid ethyl carbamate impurities from forming. The above EtOH solution was concentrated to a minimum volume (4-5 mL/g) using the batch concentrator under reduced pressure (THF levels should be <5% by GC), and EtOH (10 mL/g of compound VI) was back-added to give a total of 10 mL/g. The reaction was then heated at 50° Celsius for 9-19 hours or until HPLC shows that compound VI is less than 0.5 area %. The reaction was then cooled to room temperature, and sulfuric acid (H2SO4, 1.0 eq. to compound VI) was added via addition funnel to yield compound I-HS with the temperature usually exotherming at about 30° Celsius.
Example 1 Preparation of Crystalline Form (I-HS) (Method 1)
(S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (0.500 g, 1.17 mmol) was dissolved in EtOH (2.5 mL) and cooled to about 5° Celsius. Concentrated sulfuric acid (0.0636 mL, 1.17 mmol) was added to the cooled solution and stirred for about 10 min, while warming to room temperature. Methyl tert-butyl ether (MTBE) (2 mL) was slowly added to the mixture, resulting in the product gumming out. EtOH (2.5 mL) was then added to the mixture and heated to about reflux until all solids were dissolved. Upon cooling to room temperature and stirring for about 1 hour, some solids formed. After cooling to about 5° Celsius, the solids were filtered and washed with MTBE. After filtration and drying at air for about 15 minutes, (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate was isolated as a solid.
Example 2 Preparation of Crystalline Form (I-HS) (Method 2)
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1:1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5:95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5:95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate.
Example 3 Preparation of Amorphous Form AM(HS)
To a solution of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (9.40 g, 21.94 mmol) in MeOH (220 mL) was slowly added sulfuric acid (0.1 M in MeOH, 219.4 mL, 21.94 mmol) at ambient temperature under rapid stirring. After 30 minutes, the reaction was first concentrated by rotary evaporator to near dryness, then on high vacuum for 48 h to provide amorphous form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide sulfate (11.37 g, 21.59 mmol, 98.43% yield). LCMS (apci m/z 429.1, M+H).
PATENT
CN 107987082
PATENT
https://patents.google.com/patent/US20170281632A1/en
WO 2010/048314 discloses in Example 14A a hydrogen sulfate salt of (S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide. WO 2010/048314 does not disclose the particular form of the hydrogen sulfate salt described herein when prepared according to the method of Example 14A in that document. In particular, WO 2010/048314 does not disclose crystalline form (l-HS) as described below.
(S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide having the formula (I):

Example 1 Preparation of Crystalline Form (I-HS) (Method 1)
(S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (0.500 g, 1.17 mmol) was dissolved in EtOH (2.5 mL) and cooled to about 5° Celsius. Concentrated sulfuric acid (0.0636 mL, 1.17 mmol) was added to the cooled solution and stirred for about 10 min, while warming to room temperature. Methyl tert-butyl ether (MTBE) (2 mL) was slowly added to the mixture, resulting in the product gumming out. EtOH (2.5 mL) was then added to the mixture and heated to about reflux until all solids were dissolved. Upon cooling to room temperature and stirring for about 1 hour, some solids formed. After cooling to about 5° Celsius, the solids were filtered and washed with MTBE. After filtration and drying at air for about 15 minutes, (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidi n-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate was isolated as a solid.
Example 2 Preparation of Crystalline Form (I-HS) (Method 2)
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)—N-(5-((R)-2-(2, 5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1, 5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1:1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5:95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5:95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate.
Example 3 Preparation of Amorphous Form AM(HS)
To a solution of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (9.40 g, 21.94 mmol) in MeOH (220 mL) was slowly added sulfuric acid (0.1 M in MeOH, 219.4 mL, 21.94 mmol) at ambient temperature under rapid stirring. After 30 minutes, the reaction was first concentrated by rotary evaporator to near dryness, then on high vacuum for 48 h to provide amorphous form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide sulfate (11.37 g, 21.59 mmol, 98.43% yield). LCMS (apci m/z 429.1, M+H).
References
External links
Larotrectinib
 |
| Identifiers |
|
|
| CAS Number |
|
| ChemSpider |
|
| UNII |
|
| Chemical and physical data |
| 3D model (JSmol) |
|
-
O[C@H]1CCN(C1)C(=O)Nc2cnn3ccc(nc23)N4CCC[C@@H]4c5cc(F)ccc5F
|
-
InChI=1S/C21H22F2N6O2/c22-13-3-4-16(23)15(10-13)18-2-1-7-28(18)19-6-9-29-20(26-19)17(11-24-29)25-21(31)27-8-5-14(30)12-27/h3-4,6,9-11,14,18,30H,1-2,5,7-8,12H2,(H,25,31)/t14-,18+/m0/s1
-
Key:NYNZQNWKBKUAII-KBXCAEBGSA-N
|

FDA Accepts Larotrectinib New Drug Application and Grants Priority Review
https://ir.loxooncology.com/press-releases/fda-accepts-larotrectinib-new-drug-application-and-grants-priority-review
Released on May 29, 2018
– PDUFA date set for November 26, 2018 –
STAMFORD, Conn., May 29, 2018 (GLOBE NEWSWIRE) — Loxo Oncology, Inc. (Nasdaq:LOXO), a biopharmaceutical company innovating the development of highly selective medicines for patients with genetically defined cancers, today announced that the U.S. Food and Drug Administration (FDA) has accepted the company’s New Drug Application (NDA) and granted Priority Review for larotrectinib for the treatment of adult and pediatric patients with locally advanced or metastatic solid tumors harboring an NTRK gene fusion. The FDA has set a target action date of November 26, 2018, under the Prescription Drug User Fee Act (PDUFA).
“We are excited the larotrectinib NDA has been accepted by FDA and granted Priority Review status,” said Josh Bilenker, M.D., chief executive officer of Loxo Oncology. “Larotrectinib marks an important shift towards treating cancer based on the tumor’s genetics rather than its site of origin in the body.”
The FDA grants Priority Review for the applications of medicines that, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. Larotrectinib has also been granted Breakthrough Therapy Designation, Rare Pediatric Disease Designation and Orphan Drug Designation by the FDA.
Loxo Oncology and Bayer are engaged in a collaboration for the development and commercialization of larotrectinib. Bayer plans to submit a Marketing Authorization Application (MAA) in the European Union in 2018.
About Larotrectinib (LOXO-101)
Larotrectinib is an oral and highly selective investigational tropomyosin receptor kinase (TRK) inhibitor in clinical development for the treatment of patients with cancers that harbor a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. Growing research suggests that the NTRK genes, which encode for TRKs, can become abnormally fused to other genes, resulting in growth signals that can lead to cancer in many sites of the body. In clinical trials, larotrectinib demonstrated anti-tumor activity in patients with tumors harboring NTRK gene fusions, regardless of patient age or tumor type. In an analysis of 55 RECIST-evaluable adult and pediatric patients with NTRK gene fusions, larotrectinib demonstrated a 75 percent centrally-assessed confirmed overall response rate (ORR) and an 80 percent investigator-assessed confirmed ORR, across many different types of solid tumors. The majority of all adverse events were grade 1 or 2.
Larotrectinib has been granted Priority Review, Breakthrough Therapy Designation, Rare Pediatric Disease Designation and Orphan Drug Designation by the U.S. FDA.
In November 2017, Loxo Oncology and Bayer entered into an exclusive global collaboration for the development and commercialization of larotrectinib and LOXO-195, a next-generation TRK inhibitor. Bayer and Loxo Oncologywill jointly develop the two products with Loxo Oncology leading the ongoing clinical studies as well as the filing in the U.S., and Bayer leading ex-U.S. regulatory activities and worldwide commercial activities. In the U.S., Loxo Oncology and Bayer will co-promote the products.
For additional information about the larotrectinib clinical trials, please refer to www.clinicaltrials.gov. Interested patients and physicians can contact the Loxo Oncology Physician and Patient Clinical Trial Hotline at 1-855-NTRK-123 or visit www.loxooncologytrials.com/trk-trials.
About TRK Fusion Cancer
TRK fusion cancer occurs when a neurotrophic tyrosine receptor kinase (NTRK) gene fuses with another unrelated gene, producing an altered tropomyosin receptor kinase (TRK) protein. The altered protein, or TRK fusion protein, is constantly active, triggering a permanent signal cascade. These proteins become the primary driver of the spread and growth of tumors in patients with TRK fusion cancer. TRK fusion cancer is not limited to certain types of cells or tissues and can occur in any part of the body. NTRK gene fusions occur in various adult and pediatric solid tumors with varying prevalence, including appendiceal cancer, breast cancer, cholangiocarcinoma, colorectal cancer, GIST, infantile fibrosarcoma, lung cancer, mammary analogue secretory carcinoma of the salivary gland, melanoma, pancreatic cancer, thyroid cancer, and various sarcomas. It may affect greater than 60 percent of both adult and pediatric patients with certain rare tumor types, such as secretory breast, secretory salivary gland and infantile fibrosarcoma. Only sensitive and specific tests can reliably detect TRK fusion cancer. Next-generation sequencing (NGS) can provide a comprehensive view of genomic alterations across a large number of genes. Fluorescence in situ hybridization (FISH) can also be used to test for TRK fusion cancer, and immunohistochemistry (IHC) can be used to detect the presence of TRK protein
About Loxo Oncology
Loxo Oncology is a biopharmaceutical company innovating the development of highly selective medicines for patients with genetically defined cancers. Our pipeline focuses on cancers that are uniquely dependent on single gene abnormalities, such that a single drug has the potential to treat the cancer with dramatic effect. We believe that the most selective, purpose-built medicines have the highest probability of maximally inhibiting the intended target, with the intention of delivering best-in-class disease control and safety. Our management team seeks out experienced industry partners, world-class scientific advisors and innovative clinical-regulatory approaches to deliver new cancer therapies to patients as quickly and efficiently as possible. For more information, please visit the company’s website at www.loxooncology.com.
larotrectinib
larotrectinib
Treatment for Solid Tumors
FDA Accepts Larotrectinib New Drug Application and Grants Priority Review
WHIPPANY, N.J., May 29, 2018 /PRNewswire/ — Bayer announced today that the U.S. Food and Drug Administration (FDA) has accepted the New Drug Application (NDA) submitted by its collaboration partner Loxo Oncology, Inc. (NASDAQ: LOXO), and granted Priority Review for larotrectinib for the treatment of adult and pediatric patients with locally advanced or metastatic solid tumors harboring a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. The FDA has set a target action date of November 26, 2018, under the Prescription Drug User Fee Act (PDUFA).
NTRK gene fusions are genetic alterations that result in production of tropomyosin receptor kinase (TRK) fusion proteins, and lead to the development of tumor growth. Bayer and Loxo Oncology are jointly developing larotrectinib, which is being studied globally for the treatment of patients across a wide range of cancers that harbor an NTRK gene fusion. Bayer plans to submit a Marketing Authorization Application (MAA) in the European Union in 2018.
“TRK fusion cancer is not limited to any organ or site of the body and occurs in both adults and children,” said Scott Fields, MD, senior vice president and head of oncology development at Bayer’s Pharmaceutical Division. “The Priority Review designation for larotrectinib may help bring this treatment option to patients, facing a high unmet medical need, as soon as possible.”
The FDA grants Priority Review for the applications of medicines that, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. Larotrectinib has also been granted Breakthrough Therapy Designation, which is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the medicine may demonstrate substantial improvement over available therapies on a clinically significant endpoint, Rare Pediatric Disease Designation and Orphan Drug Designation by the U.S. FDA.
About Larotrectinib (LOXO-101)
Larotrectinib is an investigational tropomyosin receptor kinase (TRK) inhibitor in clinical development for the treatment of patients with cancers that harbor a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. Growing research suggests that the NTRK genes can become abnormally fused to other genes, producing a TRK fusion protein that can lead to the development of solid tumors across multiple sites of the body.
In November 2017, Bayer and Loxo Oncology entered into an exclusive global collaboration for the development and commercialization of larotrectinib and LOXO-195, a TRK inhibitor in clinical development. Bayer and Loxo Oncology will jointly develop the two products with Loxo Oncology leading the ongoing clinical studies as well as the filing in the U.S., and Bayer leading ex-U.S. regulatory activities and worldwide commercial activities. In the U.S., Bayer and Loxo Oncology will co-promote the products.
For additional information about the larotrectinib clinical trials, please refer to http://www.clinicaltrials.gov or visit http://www.loxooncologytrials.com. Larotrectinib has not been approved by the U.S. Food and Drug Administration, the European Medicines Agency or any other health authority.
About TRK Fusion Cancer
TRK fusion cancer occurs when a neurotrophic tyrosine receptor kinase (NTRK) gene fuses with another unrelated gene, producing an altered tropomyosin receptor kinase (TRK) protein. The altered protein, or TRK fusion protein, becomes active and triggers a signal cascade. These proteins become the primary oncogenic driver of the spread and growth of tumors. NTRK gene fusion has been identified in various adult and pediatric solid tumors with varying frequencies.
About Oncology at Bayer
Bayer is committed to delivering science for a better life by advancing a portfolio of innovative treatments. The oncology franchise at Bayer now includes four oncology products and several other compounds in various stages of clinical development. Together, these products reflect the company’s approach to research, which prioritizes targets and pathways with the potential to impact the way that cancer is treated.
About Bayer
Bayer is a global enterprise with core competencies in the Life Science fields of health care and agriculture. Its products and services are designed to benefit people and improve their quality of life. At the same time, the Group aims to create value through innovation, growth and high earning power. Bayer is committed to the principles of sustainable development and to its social and ethical responsibilities as a corporate citizen. In fiscal 2017, the Group employed around 99,800 people and had sales of EUR 35.0 billion. Capital expenditures amounted to EUR 2.4 billion, R&D expenses to EUR 4.5 billion. For more information, go to http://www.bayer.us.
///////////Larotrectinib, UNII:PF9462I9HX, ларотректиниб , 拉罗替尼 , ARRY-470, LOXO-101, PF9462I9HX, phase 3, Array BioPharma, Loxo Oncology, National Cancer Institute, BAYER, orphan drug designation, breakthrough therapy designation
C1CC(N(C1)C2=NC3=C(C=NN3C=C2)NC(=O)N4CCC(C4)O)C5=C(C=CC(=C5)F)F.OS(=O)(=O)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





















 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye… OSAKA
OSAKA
 OSAKA
OSAKA


![ChemSpider 2D Image | 8,8'-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) | C51H40N6O23S6](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fwww.chemspider.com%2FImagesHandler.ashx%3Fid%3D5168%26w%3D200%26h%3D200&container=blogger&gadget=a&rewriteMime=image%2F*) suramin
suramin





