
Home » Posts tagged 'KYORIN'
Tag Archives: KYORIN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imidafenacin, イミダフェナシン

Imidafenacin
イミダフェナシン
Cas 170105-16-5
C20H21N3O, 319.408
APPROVED JAPAN 2015-07-29
|
4-(2-methyl-1H-imidazol-1-yl)-2,2-diphenylbutanamide
|
Imidafenacin

C20H21N3O : 319.4
[170105-16-5]
Imidafenacin (INN) is a urinary antispasmodic of the anticholinergic class. It’s molecular weight is 319.40 g/mol
| Imidafenacin (INN) is a urinary antispasmodic of the anticholinergic class. |
Kyorin and Ono have developed and launched imidafenacin, an oral M1 and M3 muscarinic receptor antagonist. Family members of the product case, WO9515951, expire in the US in 2019
Imidafenacin was approved by Pharmaceuticals Medical Devices Agency of Japan (PMDA) on Apr 18, 2007. It was marketed as Uritos® by Kyorin, and marketed as Staybla® by Ono.
Imidafenacin is a potent M1 and M3-subtype antagonist indicated for the treatment of urinary urgency, frequent urination and urgency urinary incontinence due to overactive bladder.
Uritos® is available as tablet for oral use, containing 0.1 mg of free Imidafenacin. The recommended dose is 0.1 mg twice daily, and it can be increased to 0.2 mg twice daily, if the efficacy was not enough.
Uritos® / Staybla®


MOA:Muscarinic acetylcholine receptor antagonist
Indication:Urinary incontinence; Urinary urgency and frequency

PAPER
Imidafenacin, the compound of formula (I), is an antimuscarinic agent marketed in Japan under the brand name Uritos® used to treat overactive bladder, a disease defined by the presence of urinary urgency, usually accompanied by frequency and nocturia, with or without urge incontinence. Overactive bladder dysfunction has a considerable impact on patient quality of life, although it does not affect survival.

(I)
Synthesis of 4-(2-methyl-1 -imidazolyl)-2,2-diphenylbutanamide is first disclosed in Japanese patent JP3294961 B2 as shown in Scheme 1 . 4-bromo-2,2-diphenylbutanenitrile (II) is reacted with three equivalents of 2-methylimidazol, in dimethylformamide and in the presence of triethylamine as a base, to afford 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile, compound of formula (III), which is purified by column chromatography and, further, converted into its hydrochloride salt and recrystallized. Then, compound (III) is hydrolyzed with an excess of 70% sulfuric acid at 140-150 °C, followed by basification and recrystallization to provide imidafenacin (I), in an overall yield of only 25% (as calculated by data provided in docume

(III) (I)
Scheme 1
This route of document JP3294961 B2 implies several drawbacks. Firstly, purification of intermediate (III) is carried out by means of chromatographic methods, which are generally expensive, environmentally unfriendly and time consuming. Secondly, the hydrolysis of the nitrile group is carried out under strong acidic conditions and high temperature not convenient for industrial application.
Japanese document JP2003-201281 discloses a process for preparing imidafenacin as shown in Scheme 2. 4-bromo-2,2-diphenylbutanenitrile (II) is reacted, with five equivalents of 2-methylimidazol, which acts also as a base, in dimethylsufoxide to provide intermediate (III), which after an isolation step is further reacted with phosphoric acid in ethanol to provide the phosphate salt of 4-(2-methyl-1 H-imidazol-1-yl)-2,2-diphenylbutanenitrile. Hydrolysis with potassium hydroxide, followed by purification with a synthetic adsorbent provides imidafenacin (I) in a moderate overall yield

(II) (I)
Scheme 2
The use of a synthetic adsorbent is associated with problems with operativities and purification efficiencies from the viewpoint of industrial production, therefore, the process disclosed in document JP2003-201281 is not suitable for industrial application.
EP1845091 A1 discloses a process for preparing imidafenacin, according to previous document JP2003-201281 , however the purification step is carried out by either preparing the hydrochloride or the phosphate salt of imidafenacin followed by neutralization as shown in Scheme 3. Purified imidafenacin is provided in low yield, overall yield of about 31 % (as calculated by data provided in document EP1845091 A1 ). This process has several disadvantages. Firstly, EP1845091 A1 states that the penultimate intermediate, the 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate is hygroscopic, which implies handling problems. Secondly, the additional steps carried out for purification increases the cost of the final imidafenacin process and the pharmaceutical compositions containing it, which already resulted in expensive medications.

(II) (I)
HCI or
H3PO4

purified (I) HCI or Ή3ΡΟ4
Scheme 3
The intermediate phosphate salt of 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile obtained and used in prior art processes is a solid form having needle-shaped crystals, which are difficult to filtrate. Moreover, said needle-shaped crystals are very hygroscopic and unstable and transform over time to other solid forms. In addition, the water absorbed by this solid form described in the prior art may react with the intermediate to generate further impurities.
Therefore, there is still a need to develop an improved industrially feasible process for the manufacture of imidafenacin in good purity and good yield, involving the use of stable intermediates having also improved handling characteristics.
Example 1 :
Preparation of 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate in solid Form I
4-bromo-2,2-diphenylbutanenitrile (II, 1.000 Kg, 3.33 mol) and 2-methylimidazol (1 .368 Kg, 16.66 mol) were heated in DMSO (0.8 L) at 100-105 °C for 7 hours. The solution was then cooled to 20-25 °C and toluene (2 L) and water (4 L) were added and stirred for 30 minutes. After phase separation, the aqueous layer was extracted with toluene (1 L). Organic layers were combined and washed twice with water (2 x 1 L). Distillation of toluene provided 4-(2-methyl-1 H-imidazol-1-yl)-2,2-diphenylbutanenitrile as a brown oil (0.915 Kg), which was, then, dissolved in dry acetone (3 L) and water (0.1 L), heated to 40-45°C and seeded with 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate. A solution of orthophosphoric acid (0.391 Kg, 3.39 mol) in acetone (2 L) was then added dropwise, maintaining temperature at 40-45 °C. Once the addition was finished, the reaction mixture was maintained 1 hour at 40-45 °C, cooled to 20-25 °C and stirred for 1 hour. The solid was filtered, washed with acetone (1 L), suspended in 2-propanol (10 L), heated at 80 °C and 2 L of solvent were distilled. The obtained suspension was then seeded with 4-(2-methyl-1 H-imidazol-1 -yl)-2,2-diphenylbutanenitrile phosphate solid Form I and maintained at 80 °C for 5 hours. The suspension was cooled down to 20-25°C, filtered off, washed with 2-propanol (1 L) and, finally, dried (45 °C, 0.5 torr, 12 hours).
Yield: 0.967 Kg (73%)
HPLC: 99.5 %
KF: 0.2 %
Optical microscopy: plate-shaped crystal habit as substantially in accordance to Figure 2.
PSD: D90 of 105 m
PXRD: Crystalline solid form as substantially in accordance to Figure 3.
DSC (10 °C/min): Endothermic peak with onset at 177 °C (-1 18 J/g), as substantially in accordance to Figure 4.
TGA (10 °C/min): Decomposition starting at 180 °C.
DVS: No significant weight gain up to 90% of relative humidity. At this humidity, a total increase of only 0.45% in weight was observed.
SCXRD: Crystal structure substantially in accordance to Figure 5. There are not water or solvent molecules in the crystal structure.
PATENT
https://www.google.com/patents/CN103351344A?cl=en
Overactive Bladder (symptomatic overactive bladder, 0AB) is a common chronic lower urinary tract dysfunction. Its incidence, United States and Europe over 75 year-old male incidence up to 42%, slightly lower incidence of women 31%; the incidence of domestic in Beijing 50 years of age for men was 16.4% for women over the age of 18 mixed The overall incidence of urinary incontinence and urge incontinence was 40.4 percent, seriously affecting the physical and mental health of the patient, reduced quality of life. Common antimuscarinic drugs in vivo and in vivo M receptor in some or all of binding with different affinities to improve the symptoms of OAB, but will also cause many side effects, such as dry mouth, constipation, cognitive impairment , tachycardia, blurred vision and so on. Imidafenacin have diphenylbutanoic amide structure, is a new high anticholinergic drugs, which selectively acts on the M3 and Ml receptors, blocking the contraction of the detrusor choline, so detrusor relaxation, reduce side effects of drugs. Meanwhile imidafenacin inhibit smooth muscle of the bladder and inhibiting acetylcholine free dual role, and selectivity for the bladder stronger than the salivary glands.
imidafenacin is a new diphenylbutanoic amides from Japan Ono Pharmaceutical Co., Ltd. jointly developed with Kyorin Pharmaceutical anticholinergics, structure (I) as follows:

The goods listed in June 2007 in Japan under the trade name: STAYBLA, chemical name: 4- (2-methyl-1-imidazolyl) _2,2- diphenylbutyric amide.
At present the preparation imidafenacin few reports, can be summed up as the following ways:
China Patent CN10699098 reported to bromoethyl diphenyl acetonitrile and 2-methylimidazole as a raw material, at 150 ° C condition, after the reaction DMF / triethylamine system, sulfuric acid hydrolysis reuse imidafenacin. The reaction equation is as follows:

BACKGROUND OF THE INVENTION This two-step method was 24% overall yield is too low, and the second step of the reaction is difficult to control. And the reaction product was purified by column chromatography required to obtain a purified product, is not conducive to industrial production.
Chinese patent CN101362721A referred to as the hydrolysis conditions for the preparation of sulfuric acid and organic acid mixed use imidafenacin yield have mentioned the smell.

Although this method increases the yield, but still more by-product of the reaction, the product is not easy purification.
Japanese Patent No. JP2005 / 023216 proposes hydrolysis under alkaline environment, and the use of products and solutions of salts hydrochloride salt and then purified product.

This method improves the yield of the second step of the hydrolysis reaction and simplified purification methods. But the need to use this method to purify salt activated carbon, and filtration devices require more stringent; and a need to be re-crystallized salt solution salt after the operation, a total of four steps of unit operations. Process more cumbersome and more stringent requirements for equipment, it is not conducive to industrial scale production. In addition, the product is dried for a long time, still remaining after solvent treatment product obtained, the purity of the product is still low.

DETAILED DESCRIPTION
The following typical examples are intended to illustrate the present invention, simple replacement of skill in the art of the present invention or improvement made in all part of the present invention within the protection of technical solutions.
Example 1
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butanamide hydrobromide. The 16.5 g (52 mmol) 4- (2- methyl-1-imidazolyl) -2,2-diphenyl butyramide crude into 100 mL of isopropanol, stirring was added 8.0 mL hydrobromic acid and isopropyl alcohol mixed solution (volume ratio of 1: 1), the solid gradually dissolved, was nearly colorless and transparent liquid. After maintaining the reaction mixture was stirred for half an hour, the reaction mixture was added to 100 mL of ethyl acetate, stirred for I hour at room temperature, solid precipitated. Filtration, and the cake was rinsed with an appropriate amount of ethyl acetate. The solid was collected, 40 ° C drying oven and dried to constant weight to give 19.5 g white 4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide hydrobromide, yield 98.9%. ?] \ 1 .228.4-229.00C0MS (m / z): 320 [M + 1] +. 1H-NMR (DMS0-1 / 6, 400 MHz) δ: 2.25 (3H, s), 2.73-2.74 (2H, m), 3.68-3.91 (2H, m), 6.81 (1H, s), 7.28-7.35 (I OH, m), 7.39 (1H, s), 7.49 (1H, d, /=2.4 Hz), 7.55 (1H, d, J = 2.2 Hz), 14.39 (1¾ br s).
Example 2
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide. -2,2-Diphenyl butyric acid amide acetate was dissolved in 900 mL of water to 19.5 g (0.051mmol) obtained in Example 1 4- (2-methyl-1-imidazolyl) embodiment. Extracted with 900mL diethyl ether solution, collecting the inorganic layer. Was added to an aqueous solution of 200 mL of ethanol, was added to the system with stirring in an aqueous solution of KOH 2mol / L, there is a solid precipitated. The reaction was stirred I h after filtration. Cake was washed with 40% ethanol solution rinse, rinsed with water several times. Collect the cake, put 40 ° C drying oven dried to constant weight to give 14.8 g white 4- (2-methyl-1-imidazolyl) -2,2-diphenyl methylbutanamide, yield 91.0% (total yield 90% two steps). Μ.p.192.3-193.00C (CN101076521A 191-193O). MS (m / z): 320 [M + l] +. 1H-NMR (DMSO-J6, 400MHz) δ: 2.11 (3Η, s), 2.69-2.73 (2H, m), 3.61-3.65 (2H, m), 6.75 (1H, d, J = L OMHz), 7.01 (1H, br s), 7.04 (1H, d, J = L 0 MHz), 7.34-7.49 (11H, m).
Example 3
4- (2-methyl-1-imidazolyl) -2,2-diphenyl butyramide. The 14.5 g (0.045mmol) obtained in Example 4- (2-methyl-1-imidazolyl) -2,2-diphenyl butanamide 2 was added 116 mL of ethyl acetate was slowly heated to reflux reflux for 30 min, cooled to room temperature for crystallization 5 h. Suction filtered, the filter cake was rinsed with a small amount of ethanol, collected cake was put 40 ° C drying oven and dried to constant weight to give 13.4 g white 4- (2-methyl-1-imidazolyl) -2,2- diphenyl methylbutanamide refined products, yield 92.4% (three-step total yield 83.1%). Mp192.5-193 (TC (CN101076521A 191_193 ° C) .MS (m / z):.. 320 [M + 1] + 1H-NMR (DMSO-J6, 400 MHz) δ
2.11 (3H, 7.01 (1H,
s), 2.69-2.73 (2H, br s), 7.04 (1H, d,
m), 3.61-3.65 (2H, m), 6.75 (1H, J = L 0 MHz), 7.34-7.49 (11H, m).

PATENT
CN103772286A.
imidafenacin (Imidafenacin) is a new diphenylbutanoic amides from Japan Ono Pharmaceutical Co., Ltd. jointly developed with Kyorin Pharmaceutical anticholinergic drugs, bladder is highly selective for the treatment of overactive bladder, in 2007 in June in Japan. Its chemical name is 4- (2-methyl -1H- imidazol-1-yl) -2,2-diphenyl butyramide chemical structure shown by the following formula I:

Reported in U.S. Patent No. US5932607 imidafenacin preparation method, the method is based on 4-bromo-2 ‘2 ~ phenyl butyronitrile, 2-methylimidazole, triethylamine as raw materials, with DMF as a solvent at 150 ° C reaction 30h, to give the intermediate 4- (2-methyl-imidazol-1-yl) -2,2-diphenyl-butyronitrile, 77% yield, then body 140 ~ 150 ° C with 70% sulfuric acid The resulting intermediate hydrolyzed to the amide, after completion of the reaction required excess soda and sulfuric acid, the reaction is as follows:

Which preclude the use of the dilute sulfuric acid hydrolysis, although succeeded in getting the product, but the yield is very low, only 32%, greatly increasing the production cost, mainly due to 70% sulfuric acid, the reaction is difficult to control amide phase, the product will continue to acid hydrolysis byproducts, resulting in decreased yield.
European Patent No. EP1845091 reports imidafenacin Another preparation method, the method using potassium hydroxide and isopropyl alcohol 4- (2-methyl-imidazol-1-yl) diphenyl _2,2- Hydrolysis of nitrile to amide phosphates, and the crude product was converted to the hydrochloride or phosphate, and recrystallized to remove impurities and then basified imidafenacin obtained, which reaction is as follows:

This method uses a lot of bases, product purification is too much trouble, and the total yield of 45%.
Chinese Patent Publication No. CN102746235 also disclosed imidafenacin preparation method of 4- (2-methyl-1-yl) -2,2-diphenyl phosphate or nitrile salt in methanol / ethanol, dimethyl sulfoxide, and the presence of a base, with hydrogen peroxide in 40 ~ 60 ° C under through improved Radziszewski the target compound, the reaction is as follows:

The method used in the hydrogen peroxide solution, but a solution of hydrogen peroxide has strong oxidizing, and has a certain corrosive, inhalation of the vapor or mist respiratory irritation strong, direct eye contact with the liquid may cause irreversible damage and even blindness, security It is not high on the human body and environmentally unfriendly. Alkaline environment, easily decomposed hydrogen peroxide, as the temperature increases, the decomposition reaction increased, and therefore reaction requires a large excess of hydrogen peroxide solution.

The method comprises the steps of: (1) 4-Bromo-2,2-diphenyl-butyronitrile is hydrolyzed to the amide under basic conditions; (2) The obtained 4-bromo-2,2-diphenylbutyric amide is reacted with 2-methylimidazole to give the desired product.
Example 1
2L reaction flask was added 400mL of dry tetrahydrofuran, under a nitrogen atmosphere was added 60% sodium hydride (82.8g, 2.06mol), stirred to obtain a gray turbid solution A. With 400mL dry tetrahydrofuran was sufficiently dissolved diphenyl acetonitrile (200g, 1.04mol), I, 2- dibromoethane (204.2g, 1.08mol), to give a colorless clear liquid B; 5 ~ 15 ° C, a solution of turbid solution B dropwise to solution A, 10 ~ 15 ° C the reaction was incubated 6h, TLC until the reaction was complete, to the reaction system a small amount of water was added dropwise until no bubbles. After addition of 800mL water, 400mL ethyl acetate and stirred, liquid separation, the organic layer was washed with water, saturated sodium chloride solution, respectively, and the organic layer was dried over anhydrous sodium sulfate, suction filtered, concentrated under reduced pressure to give a yellow liquid 310g.
[0018] The resulting yellow liquid with 800mL 90% ethanol and stirred to dissolve at 40 ° C, then cooling and crystallization, filtration, 45 ° C and concentrated under reduced pressure to give a white solid 232.8g, 75% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0019] Example 2
3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (15 (^, 0.511101), 7501 ^ 6mol / L KOH solution, 750mL dimethylsulfoxide and heated to 100 ~ 120 ° C under stirring The reaction, the reaction lh, until the reaction was complete by TLC after cooling to 40 V, add 2000mL water, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous The organic layer was dried over sodium sulphate, filtration, concentrated under reduced pressure to give brown oily liquid 161.92g, 96% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0020] Example 3
3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (150g, 0.5mol), 666mL 6mol / L NaOH solution, 750mL dimethylsulfoxide, the reaction mixture was stirred and heated to 100 ~ 120 ° C under The reaction lh, until the reaction was complete by TLC after cooling to 40 ° C, add water 2000mL, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous sulfate sodium organic layer was dried, filtration, concentrated under reduced pressure to give brown oily liquid 146.73g, 87% yield.
Preparation of bromo-2,2-diphenyl 4_ butanamide: [0021] Example 4
The reaction was stirred 3L reaction flask was added 4-bromo-2,2-diphenyl-butyronitrile (15 (^, 0.511101), 8331 ^ 36% Na2CO3 solution, 750mL dimethylsulfoxide and heated to 100 ~ 120 ° C under The reaction lh, until the reaction was complete by TLC after cooling to 40 ° C, add water 2000mL, 2000mL of methylene chloride was stirred, liquid separation, the organic layer was washed with water, washed with saturated sodium bicarbonate and sodium chloride solution, separated, dried over anhydrous The organic layer was dried over sodium sulphate, filtration, concentrated under reduced pressure to give brown oily liquid 153.48g, yield 91%.
`[0022] Example 5: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2 2-diphenyl butyric amide (160g, L 5mol), 2- methyl imidazole (123g,
1.5mol), triethylamine (50.6g, 0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, to be After cooling, water was added 3000mL system stirred 0.5h, filtration, washed with water until the filtrate is neutral, concentrated under reduced pressure and dried to give a brown solid 146.14g, a yield of 91%.
[0023] Example 6: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2, 2- diphenyl butyramide (160g, 0.5mol), 2- methyl imidazole (82.1g,
1.011101), triethylamine (50.68,0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, the system was cooled until After adding 3000mL water, stirring 0.5h, filtration, washed with water until the filtrate is neutral, concentrated under reduced pressure and dried to give a brown solid 120.45g, 80% yield.
[0024] Example 7: 4- (2-methyl-imidazol _1_ -1H- yl) butyramide _2,2_ diphenyl (imidafenacin) Preparation 5L reaction flask was added 4-bromo-2, 2- diphenyl butyramide (160g, 0.5mol), 2_ methylimidazole (164.2g,
2.011101), triethylamine (50.68,0.5mol), potassium iodide (5g, 0.03mol), fully dissolved with 1000mL DMF solution was heated to 120 ° C at a reaction 5h, until completion of the reaction by TLC, heating was stopped, the system was cooled until After adding water, stirring 3000mL
0.5h, suction filtered, washed with water until the filtrate was neutral, and concentrated under reduced pressure, and dried to give a brown solid 141.33g, yield 88%.
[0025] Example 8: 4- (2-methyl imidazole -1H- _1_ group) _2,2_ diphenylbutanoic amide (imidafenacin) refining up to 80g microphone said that new crude added 300mL of absolute ethanol, the system was warmed to reflux, refluxed
0.5h, after cooling the ethanol was distilled off to IOOmL about 500mL of ethyl acetate was added to precipitate a white solid, a small amount of ethyl acetate and wash the filter cake, 45 ° C and dried in vacuo to give 74.6g of white crystals, yield 93%.

CLIP
| EP 0733621; US 5932607; US 6103747; WO 9515951 |


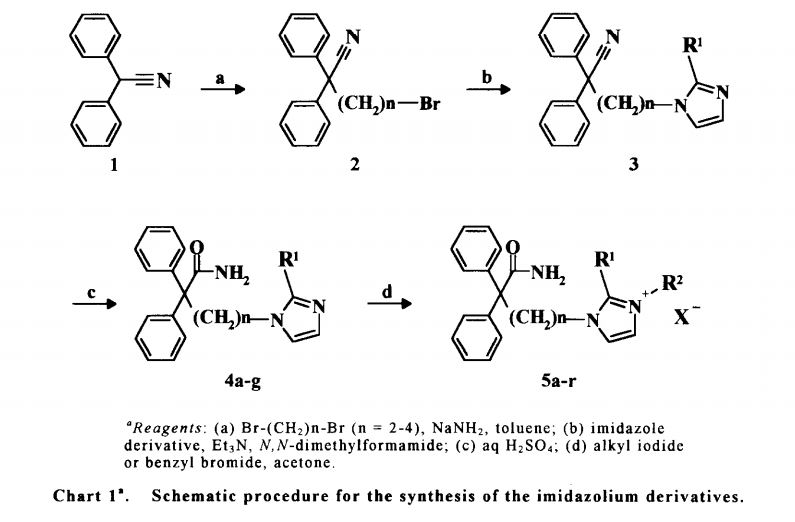
Alkylation of diphenylacetonitrile (I) with dibromoethane provided bromide (II). This was condensed with 2-methylimidazole (III) in the presence of Et3N in DMF to afford the substituted imidazole (IV). Finally, hydrolysis of the cyano group of (IV) with 70% sulfuric acid produced the target amide.

Treatment of acetonitrile derivative (I) with dibromoethane (II) in toluene in the presence of NaNH2 affords bromo compound (III), which is then condensed with imidazole derivative (IV) by means of Et3N in DMF to provide compound (V). Hydrolysis of the cyano group of (V) with aqueous H2SO4 yields amide derivative (VI), which is finally subjected to alkyl quaternization by reaction with bromobenzyl bromide (VI) in acetone to furnish the desired product.
Paper
Bioorganic & Medicinal Chemistry Letters 9 (1999) 3003-3008

PAPER
Bioorganic & Medicinal Chemistry 7 (1999) 1151±1161
4-(2-methyl-1-imidazolyl)- 2,2-diphenylbutyramide (2.02 g, 24%) as a colorless needle:
mp 189.0±190.0 C (from ethyl acetate:ethanol);
High MS (EI+) m/z calcd for C20H21N3O 319.1685, found 319.1671;
1 H NMR (400 MHz, CDCl3) d 2.23 (3H, s), 2.69±2.74 (2H, m), 3.77±3.82 (2H, m), 5.33 (1H, s), 5.49 (1H, s), 6.73 (1H, s), 6.85 (1H, s), 7.31±7.42 (10H, m).
PATENT
CN103880751A.
imidafenacin chemical name 4- (2-methyl–1H–1-yl) -2,2-diphenyl methylbutanamide (I).

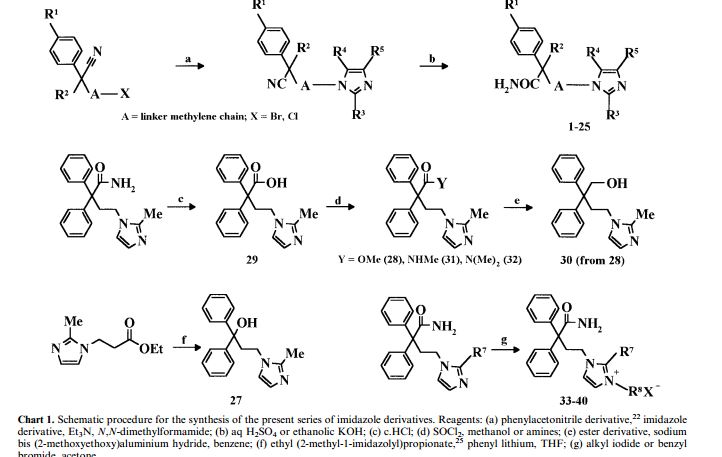
In Patent JP93-341467, JP94-319355 and literature Bioorganic & Medicinal ChemistryLetters, 1999, vol.9,3003 – 3008 reported in the chemical synthesis routes to diphenyl acetonitrile (4) as the starting material,
Condensation and hydrolysis reaction step to give imidafenacin (1).

The new method is simple, mild reaction conditions, easy to control, good high yield and purity of the product, do not pollute the environment, suitable for industrial production.
[0012] The first method from 2-methylimidazole and I, 2- dibromoethane under phase transfer catalyst is tetrabutylammonium bromide (TBAB) and inorganic base catalyzed generate 1- (2-bromoethyl) – methyl -1H- imidazole (5), and diphenyl acetonitrile (4) a phase transfer catalyst and an inorganic base catalyzed condensation of 4- (2-methyl–1H- imidazol-1-yl) -2,2 – diphenylbutyronitrile hydrochloride (2), and then hydrolyzed to imidafenacin (I)


FIG. 1 imidafenacin IH-NMR spectrum
FIG. 2 imidafenacin 13C-NMR spectra

Examples I
1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2C03 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask and stirred and heated to 50 ° C reaction 7h. Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was stirred resolved crystal dissolved, to give the product 5.lg, yield 88.5%, mp.79_80 ° C.
Preparation of 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred for 0.5h in the 40 ° C. 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 20 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL water, extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) of a white solid 7.lg, yield 77.8%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
The preparation imidafenacin (I),
4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 90 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.0g, yield 84.5%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
[0030] 13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 2
[0032] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0033] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium chloride (0.43g) and Na2CO3 (2.8g), NaOH (3.3g) followed by adding 100mL three-necked flask, stirred and heated to 40 ° C reaction 5h.Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.9g, yield 85.1%, mp.79-80 ° C.
Preparation of [0034] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0035] A two phenylethyl chest (5.8g, 30mmol) and 50% aqueous NaOH (15ml), dimethylethylene Bitterness (DMSO) (100ml), tetrabutylammonium chloride (0.8g) was added to a toluene 50ml The reaction flask, stirred 0.5h in the 40 ° C. Join
1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Solution of hydrogen chloride in ether solution with analytical crystal, crystals were filtered with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,
2-phenyl-butyronitrile hydrochloride (2) as a white solid 7.0g, yield 76.8%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
Preparation imidafenacin (I),
[0037] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 110 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.2g, yield 86.8%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H), 6.828 ( s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 3
[0041] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0042] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), benzyltriethylammonium chloride (TEBA) (0.35g) and Na2CO3 (2.8g), Na0H (3.3g) were added sequentially 100mL three-necked flask, stirred and heated to 45 ° C reaction 4h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 5.0g, yield 86.8%, mp.79-80. . .
Preparation of [0043] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), benzyltriethylammonium chloride (TEBA) (0.66g) 50ml Toluene was added to the reaction flask and stirred at 40 ° C under
0.5h0 was added 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 7.0g, yield 76.8%, mp: 156.5-158. . . 1H-NmrgoomHzADCI3), δ (ppm): 7.35-7.42 (10H, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 (2H, m) , 2.25 (3H, s).
Preparation imidafenacin (I),
[0046] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 100 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 7.lg, yield 85.5%, mp: 188.0-190. (TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0047] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 41- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0051] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2C03 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask, stirred and heated to 60 ° C reaction 4h.Cooling to room temperature, the reaction solution was filtered, and the filtrate was washed with saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.5g, yield 78.1%, mp.79_80 ° C.
Preparation of [0052] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0053] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred for 0.5h in the 40 ° C. Plus Λ 1- (2- bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 100 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 6.7g, yield 73.4%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
The preparation imidafenacin (I),
4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 150 ° C at the end of the reaction was monitored TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.2g, yield 74.8%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0056] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2 -), 3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
Example 5
1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), tetrabutylammonium bromide (TBAB) (0.5g) and K2CO3 (3.6g), K0H ( 4.6g) were added sequentially 100mL three-necked flask, stirred and heated to 20 ° C reaction 10h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.1g, yield 71.2%, mp.79-80. . .
Preparation of [0061] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0062] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100mL), tetrabutylammonium bromide (TBAB) (0.9g) in toluene 50ml was added to the reaction flask and stirred at 20 ° C in Ih. Join
1- (2-bromoethyl) -2-methyl -1H- imidazole (4) (5.lg, 27mmol), was heated to 60 ° C, the reaction was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate 240ml water phase. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Solution of hydrogen chloride in ether solution with analytical crystal, crystals were filtered with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,
2-phenyl-butyronitrile hydrochloride (2) as a white solid 6.5g, yield 71.2%, mp: 156.5-158 ° C. 1H-NMR (400MHz, CDCl3), δ (ppm): 7.35-7.42 (IOH, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 ( 2H, m), 2.25 (3H, s).
[0063] Preparation of imidafenacin (I), [0064] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2) (8.78g, 26mmol ) and 70% of concentrated sulfuric acid (25ml) was added to the reaction flask, and stirred in at 50 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.6g, yield 79.7%, mp: 188.0-190 (. TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0065] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H).
[0066] 13C-NMR (CDC13,400MHz) δ: 12.17 (-CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).
[0067] Example 6
[0068] 1- (2-bromoethyl) -1H- -2_ methyl-imidazole (5) Preparation of
[0069] The 1,2_ dibromoethane (50ml), 2- methylimidazole (2.5g, 30.5mmol), benzyltriethylammonium chloride (TEBA) (0.35g) and Na2CO3 (2.8g), Na0H (3.3g) were added sequentially 100mL three-necked flask and stirred and heated to 40 ° C reaction 8h. Cooling to room temperature, the reaction solution was filtered, washed with a saturated aqueous sodium bicarbonate paint filtrate was dried over anhydrous sodium sulfate. Concentrated, added to a mixed solvent of isopropyl ether and ethyl acetate (3: 1) was dissolved with stirring parsing crystal give the product 4.4g, yield 76.4%, mp.79-80. . .
Preparation of [0070] 4- (2-methyl-1-imidazolyl) -2,2-diphenyl-butyronitrile hydrochloride (2)
[0071] The diphenyl acetonitrile (5.8g, 30mmol) and 50% aqueous KOH (15ml), dimethyl sulfoxide (DMSO) (100ml), benzyltriethylammonium chloride (TEBA) (0.66g) 50ml Toluene was added to the reaction flask and stirred 0.5h0 1- (2-bromoethyl) -2-methyl -1H- imidazole (4) at 40 ° C (5.lg, 27mmol), was heated to 50 ° C, the reaction mixture was stirred, TLC tracking and monitoring the reaction was complete, the mixture was poured into 100mL of water and extracted three times with ethyl acetate. The aqueous phase was 240ml. Washed three times with 300ml of water The organic phase was dried over anhydrous sodium sulfate, the organic phase was concentrated. Analytical crystal solution with hydrogen chloride ether solution, filtered crystals with a mixed solvent of isopropyl ether and recrystallized from ethyl acetate to give the condensation product of 4- (2-methyl-1-imidazolyl) -2,2-diphenylbutyric carbonitrile hydrochloride (2) as a white solid 6.8g, yield 74.6%, mp: 156.5-158. . . 1H-NmrgoomHzADCI3), δ (ppm): 7.35-7.42 (10H, m), 6.90 (1H, s), 6.77 (1H, s), 3.90-3.94 (2H, m), 2.75-2.79 (2H, m) , 2.25 (3H, s).
[0072] Preparation imidafenacin (I),
[0073] 4- (2-methyl-1-imidazolyl) -2,2_ diphenyl butyronitrile hydrochloride (2) (8.78g, 26mmol) in 70% concentrated sulfuric acid (25ml) was added to the reaction bottle, the reaction was stirred at 80 ° C, the end of the reaction was monitored by TLC tracking. The reaction solution was poured into 120ml of water, solid sodium carbonate was added to adjust PH to weakly alkaline, sufficiently stirred. With 180ml of dichloromethane and 35ml of ethanol mixed solvent was extracted three times, the organic phase was washed with water, dried over anhydrous sodium sulfate, the organic phase was concentrated. The residue was mixed with a solvent of ethyl acetate and recrystallized from ethanol to give 4- (2-methyl-1-imidazolyl) 2,2-diphenyl butyramide 6.Sg, yield 81.8%, mp: 188.0-190. (TC.1H-NMR and 13C-NMR data are as follows (see Figure 1-2 spectra):
[0074] 1H-NMR (CDC13,400ΜΗζ) δ: 2.209 (s, 3H, -CH3), 2.666-2.707 (t, 2H, -CH2-CH2-),
3.747-3.788 (t, 2H, -CH2-CH2 -), 5.341 (s, 1H, -NH -), 5.757 (s, 1H, -NH -), 6.699 (s, 1H, Ar-H)
, 6.828 (s, 1H, Ar-H), 7.287-7.390 (m, I OH, Ar-H). [0075] 13C-NMR (CDC13,400MHz) δ: 12.17 (_CH3), 41.00 (-CH2 -), 43.74 (-CH2-), 59.44 (quaternary carbon, coupled with strong electron-withdrawing group), 119.08 (-C = C -), 126.95 (aromatic carbon), 127.88 (aromatic carbon), 128.52 (aromatic carbon), 129.10 (aromatic carbon), 142.61 (= CN), 144.54 (-C = N), 176.21 (carbonyl carbon).

PATENT
CN 105399678
https://www.google.com/patents/CN105399678A?cl=en
CLIP
http://dmd.aspetjournals.org/content/35/9/1624/T3.expansion.html
TABLE 3
Chemical shifts of protons and carbons in 1H NMR and 13C NMR spectra of major (M-11b) and minor (M-11a) constituents of reference products obtained from imidafenacin
|
Position of Proton |
1H NMR Data (in D2O) |
||
|---|---|---|---|
| Major Constituent (M-11b)
|
Minor Constituent (M-11a)
|
||
| 1 | 2.18a (3Hb, sc) | 2.11a (3Hb, sc) | |
| 2 | 2.82 (2H, m) | 2.79 (2H, m) | |
| 3 | 3.45 (2H, m) | 3.41 (2H, m) | |
| 5 | 5.26 (1H, s) | 5.43-5.47d (1H, d, J = 8.1e) | |
| 6 | 5.33 (1H, s) | 5.43-5.47d (1H, d, J = 8.1e) | |
| 8, 9, and 10 | 7.39-7.49 (10H, m) | 7.40-7.48 (10H, m) | |
| 13
|
8.45 (1.3H, s)
|
8.45 (2H, s)
|
|
| Position of Carbon
|
13C-NMR Data (in D2O)xc
|
||
|---|---|---|---|
| Major Constituent (M-11b)
|
Minor Constituent (M-11a)
|
||
| 1 | 14.61a | 14.48a | |
| 2 | 39.04 | 38.49 | |
| 3 | 43.49 | 42.90 | |
| 4 | 61.95-61.99f | 61.95-61.99f | |
| 5 | 87.61 | 80.22 or 85.78f | |
| 6 | 93.10 | 80.22 or 85.78f | |
| 7 | 144.2-144.4f | 144.2-144.4f | |
| 8, 9, and 10 | 130.7-131.8f | 130.7-131.8f | |
| 11 | 170.8 | 169.5 | |
| 12 | 181.9-182.2f | 181.9-182.2f | |
| 13
|
173.8
|
173.8
|
|
-
↵ a Chemical shifts are reported in parts per million.
-
↵ b Intensities are represented as number of protons.
-
↵ c Multiplicity: s, singlet; d, doublet; m, multiplet.
-
↵ d These proton signals could not be distinguished.
-
↵ e Coupling constants (J) are given in Hertz.
-
↵ f These carbon signals could not be distinguished.

FIG. 1.
Chemical structures of [14C]imidafenacin and postulated metabolites, and their fragment ions. *, 14C labeled position; broken line, precursor and product ions obtained by collision-induced dissociation in LC/MS/MS.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101076521A * | Dec 13, 2005 | Nov 21, 2007 | 杏林制药株式会社 | Process for producing muscarine receptor antagonist and intermediate therefor |
| CN102746235A * | Jul 20, 2012 | Oct 24, 2012 | 北京科莱博医药开发有限责任公司 | Improved method for preparing imidafenacin |
| CN103275007A * | May 27, 2013 | Sep 4, 2013 | 朱雪琦 | Pyrazole derivatives and preparation method thereof |
| US7351429 | 2008-04-01 | Oral solid preparation |
| US2008004247 | 2008-01-03 | Combinations of Statins with Bronchodilators |
| US2007270436 | 2007-11-22 | NOVEL AMINO- AND IMINO-ALKYLPIPERAZINES |
| US2007219237 | 2007-09-20 | Chromane Derivatives |
| US2007185129 | 2007-08-09 | ACID ADDITION SALTS OF THIENOPYRANCARBOXAMIDE DERIVATIVES |
| US2007092566 | 2007-04-26 | Oral sustained-release tablet |
| US2006188554 | 2006-08-24 | Transdermal absorption preparation |
| EP0733621 | 2002-05-15 | NOVEL IMIDAZOLE DERIVATIVE AND PROCESS FOR PRODUCING THE SAME |
| US6103747 | 2000-08-15 | Imidazole derivatives and process for preparing the same |
| US5932607 | 1999-08-03 | Imidazole derivatives and process for preparing the same |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015064232 | 2015-03-05 | TRANSDERMAL ABSORPTION PREPARATION |
| US8729056 | 2014-05-20 | Preventive and/or therapeutic agent of hand-foot syndrome |
| US8722133 | 2014-05-13 | Method for production of orally rapidly disintegrating tablet comprising imidafenacin as active ingredient |
| US2013211352 | 2013-08-15 | PERCUTANEOUSLY ABSORBED PREPARATION |
| US2013211353 | 2013-08-15 | PERCUTANEOUS ABSORPTION TYPE FORMULATION |
| US8343544 | 2013-01-01 | Oral sustained-release tablet |
| US2012289563 | 2012-11-15 | COMBINATIONS OF IMIDAFENACIN AND SALIVARY STIMULANTS FOR THE TREATMENT OF OVERACTIVE BLADDER |
| US8247415 | 2012-08-21 | Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists |
| US8158152 | 2012-04-17 | Lyophilization process and products obtained thereby |
| US8124633 | 2012-02-28 | HYDROXYMETHYL ETHER HYDROISOINDOLINE TACHYKININ RECEPTOR ANTAGONISTS |
- Kobayashi F, Yageta Y, Segawa M, Matsuzawa S: Effects of imidafenacin (KRP-197/ONO-8025), a new anti-cholinergic agent, on muscarinic acetylcholine receptors. High affinities for M3 and M1 receptor subtypes and selectivity for urinary bladder over salivary gland. Arzneimittelforschung. 2007;57(2):92-100. [PubMed:17396619 ]
- Miyachi H, Kiyota H, Uchiki H, Segawa M: Synthesis and antimuscarinic activity of a series of 4-(1-Imidazolyl)-2,2-diphenylbutyramides: discovery of potent and subtype-selective antimuscarinic agents. Bioorg Med Chem. 1999 Jun;7(6):1151-61. [PubMed:10428387 ]
Reference
- Kobayashi F, Yageta Y, Segawa M, Matsuzawa S (2007). “Effects of imidafenacin (KRP-197/ONO-8025), a new anti-cholinergic agent, on muscarinic acetylcholine receptors. High affinities for M3 and M1 receptor subtypes and selectivity for urinary bladder over salivary gland”. Arzneimittelforschung. 57 (2): 92–100. doi:10.1055/s-0031-1296589. PMID 17396619.
- Miyachi H, Kiyota H, Uchiki H, Segawa M (June 1999). “Synthesis and antimuscarinic activity of a series of 4-(1-Imidazolyl)-2,2-diphenylbutyramides: discovery of potent and subtype-selective antimuscarinic agents”. Bioorg. Med. Chem. 7 (6): 1151–61.doi:10.1016/S0968-0896(99)00003-6. PMID 10428387.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(2-methyl-1H-imidazol-1-yl)-2,2-diphenylbutanamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 170105-16-5 |
| ATC code | none |
| PubChem | CID 6433090 |
| ChemSpider | 4938278 |
| UNII | XJR8Y07LJO |
| ChEMBL | CHEMBL53366 |
| Chemical data | |
| Formula | C20H21N3O |
| Molar mass | 319.40 g/mol |
//////////イミダフェナシン , D06273, KRP-197, KRP 197, ONO-8025, ONO 8025, UNII:XJR8Y07LJO, Kyorin, Ono ,Imidafenacin, 170105-16-5, JAPAN 2015, Uritos® , Staybla®
Lascufloxacin, KRP-AM1977, by Kyorin

Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
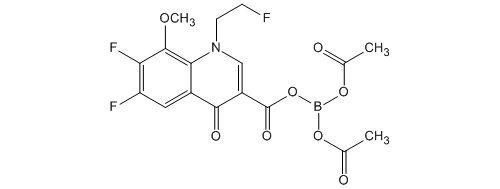
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
KEY INTERMEDIATE
CAS 848498-67-9
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
KEY INTERMEDIATE
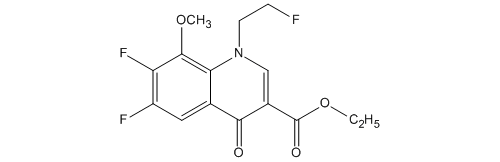
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()




Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line



| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.

Ibudilast

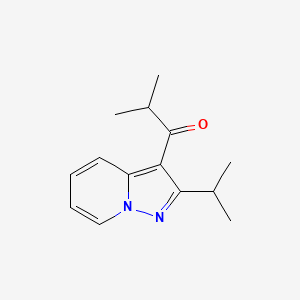
IBUDILAST, MN 166
AV-411
KC-404
MN-166
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one
KYORIN Kyorin Seiyaku Kk……….INNOVATOR

Ibudilast is an anti-inflammatory and neuroprotective oral agent which shows an excellent safety profile at 60 mg/day and provides significantly prolonged time-to-first relapse and attenuated brain volume shrinkage in patients with relapsing-remitting (RR) and/or secondary progressive (SP) multiple sclerosis (MS). Ibudilast is currently in development in the U.S. (codes: AV-411 or MN-166), but is approved for use as an antiinflammatory in Japan.



Ibudilast (development codes: AV-411 or MN-166) is an antiinflammatory drug used mainly in Japan, which acts as aphosphodiesterase inhibitor, inhibiting the PDE-4 subtype to the greatest extent,[1] but also showing significant inhibition of other PDE subtypes.[2][3]
Ibudilast has bronchodilator, vasodilator [4] and neuroprotective effects,[5][6] and is mainly used in the treatment of asthma andstroke.[7] It inhibits platelet aggregation,[8] and may also be useful in the treatment of multiple sclerosis.[9]
Ibudilast crosses the blood–brain barrier and suppresses glial cell activation. This activity has been shown to make ibudilast useful in the treatment of neuropathic pain and it not only enhances analgesia produced by opioid drugs, but also reduces the development oftolerance.[10]
It may have some use reducing methamphetamine[11] and alcohol[12] addiction.
It may have some use reducing methamphetamine addiction.[11]
Avigen has identified the potential of ibudilast (AV-411) for the treatment of neuropathic pain and other neurological indications, including opiate withdrawal. As an inhibitor of glial cells, ibudilast can deactivate these cells which produce various chemicals, including proinflammatory cytokines, in response to nerve damage or viral infection to amplify and maintain pain. Preclinical evaluation to date indicates that it reverses the painful sensory abnormality allodynia in chemotherapy- and trauma-induced neuropathic pain models.
Originator Kyorin and Banyu Pharmaceutical (now MSD KK following the merger of Banyu and Schering-Plough KK in 2010) have been developing ibudilast under a collaborative agreement. MediciNova obtained exclusive, worldwide rights outside of Japan, China, Taiwan and South Korea from Kyorin in October 2004 to develop and commercialize the compound for MS. In 2012, a codevelopment agreement was signed between MediciNova and the University of Colorado for the treatment of post-traumatic brain injury.

Sixteenth revised Japanese Pharmacopoeia chemicals, etc. IBUDILAST Ibudilast C14H18N2O: 230.31 [ 50847-11-5 ] that this product was dried when to quantify, including ibudilast (C14H18N2O) 98.5 ~ 101.0%.


http://www.google.co.in/patents/US3850941PATENT
EXAMPLE 1 Synthesis of 2-isopropyl-3-is0butyrylpyrazolo[1,5-a] pyridine (KC404) A mixture of 1-amino-Z-methylpyridinium iodide g.), isobutyric anhydride (500 g.) and K CO (81 g.) was refluxed for 8 hr. After cooling, the precipitated crystals were filtered off and water was added to the filtrate, The solution was made basic to pH 11 with K CO’ and extracted with ethyl acetate (1000 ml.). The extract’was washed with water (400 ml.), dried over Na SO and concentrated under reduced pressure. The residue was distilled to give 58 g. of colorless crystalline product, hp, 110- 175 (7.5 mm. Hg). Recrystallization from hexane gave colorless prisms, melting point 53.554.
Analysis- Calcd.: C, 73.01; H, 7.88; N, 12.17 Found: C, 72.86; H, 7.94; N, 12.09
CLIP

http://www.customsynthesis.com/ibud.html
PATENT
http://www.google.com/patents/US8119657
FIG. 6 is a synthetic reaction scheme illustrating one approach for preparing (S)-AV1013; the approach employs chiral chromatography of an N-protected form of the racemate as described in detail in Example 1.
FIG. 7 demonstrates additional reaction schemes for synthesizing (S)-AV1013.


Example 1Synthesis of (S)-2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride
(S)-2-Amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride (also referred to herein as S-AV1013.HCl) was prepared on a preparative scale using two different routes to obtain the intermediate isopropylpyrazolo[1,5-a]pyridine (IPPP). In the first approach (method 1), ibudilast was employed as the starting material to obtain IPPP; an alternate synthetic approach (method 2) employed ibudilast acid as the starting material.
Step 1Method 1Preparation of Isopropylpyrazolo[1,5-a]pyridine (IPPP) from ibudilast

A 5 L 3-neck round-bottom flask was equipped with a mechanical stirrer, thermocouple, heating mantle and a Y-adapter with a nitrogen inlet. The flask was charged with water (350 mL, USP), concentrated sulfuric acid (350 mL) and ibudilast (3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine) (140 g, 0.608 mol). The flask was purged with nitrogen, and the mixture was stirred while it was heated to 135° C. An aliquot was removed for HPLC analysis, which showed that all starting material was consumed after 5 hours at 135° C., so the mixture was allowed to cool to room temperature overnight. The mixture was cooled in an ice bath, and water (1400 mL, USP) was added over 10 min, with the temperature maintained below 25° C. With continuous cooling in an ice bath, the mixture was neutralized by adding sodium hydroxide (50% w/w aq., 1150 mL) dropwise, with the temperature maintained below 25° C. Ethyl acetate (250 mL) was added, and the layers were separated. The aqueous layer was washed with ethyl acetate (2×300 mL). The combined ethyl acetate extracts were washed sequentially with 250 mL portions of saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, then dried over anhydrous sodium sulfate for 30 minutes. Activated carbon (20 g) and silica (60 g) were added and stirred before filtering over a pad of Celite. The filtrate was concentrated under reduced pressure to obtain 96.5 g of IPPP (2-isopropyl-pyrazolo[1,5-a]pyridine, 99% crude yield, 99.6 area % pure by HPLC) as an amber oil.
1H-NMR (CDCl3) δ 1.4 (d, 6H), 3.2 (m, 1H), 6.3 (s, 1H), 6.6 (t, 1H), 7.0 (m, 1H), 7.4 (d, 1H), 8.4 (d, 1H). HPLC: RT=9.1 min (99.6 area %).
CLIP
Ibudilast (3-isobutyryl-2-isopropylpyrazolo[l,5-α]pyridine) is a small molecule drug that has been used for many years in Japan and Korea for the treatment of bronchial asthma as well as for treatment of cerebrovascular disorders such as post-stroke dizziness. It is sold in these countries under the tradename, Ketas®. Marketed indications for ibudilast in Japan include its use as a vasodilator, for treating allergy, eye tissue regeneration, ocular disease, and treatment of allergic ophthalmic disease (Thompson Current Drug Reports). Its use in the treatment of both chronic brain infarction (ClinicalTrials.gov) and multiple sclerosis (News.Medical.Net; Pharmaceutical News, 2 Aug 2005) is currently being explored in separate, ongoing clinical trials.
The mechanisms of action of ibudilast have been widely explored. Its role as a non-selective inhibitor of cyclic nucleotide phosphodiesterase (PDE) has been described
(Fujimoto, T., et al., J. of Neuroimmunology, 95 (1999) 35-92). Additionally, ibudilast has been reported to act as an LTD4 antagonist, an anti-inflammatory, a PAF antagonist, and a vasodilatator agent (Thompson Current Drug Reports). Ibudilast is also thought to exert a neuroprotective role in the central nervous system of mammals, presumably via suppression of the activation of glial cells (Mizuno et al. (2004) Neuropharmacology 46: 404-411). New uses for ibudilast continue to be explored.http://www.google.com/patents/WO2007146087A2?cl=en
PATENT
http://www.google.com/patents/WO2007142924A1?cl=en
IBUDILAST
Ibudilast is a small molecule drug (molecular weight of 230.3) having the structure shown below.

Ibudilast is also found under ChemBank ID 3227, CAS # 50847-1 1-5, and Beilstein Handbook Reference No. 5-24-03-00396. Its molecular formula corresponds to [Ci4HIgN2O]. Ibudilast is also known by various chemical names which include 2- methyl-l-(2-(l-methylethyI)pyrazolo(l,5-a)pyridin-3-yl)l-propanone; 3-isobutyryl-2- isopropylpyrazolo(l,5-a)pyridine]; and l-(2-isopropyl-pyrazolo[l,5-a]pyridin-3-yl)-2- methyl-propan-1-one. Other synonyms for ibudilast include Ibudilastum (Latin), BRN 0656579, KC-404, and the brand name Ketas®. Ibudilast, as referred to herein, is meant to include any and all pharmaceutically acceptable salt forms thereof, prodrug forms (e.g., the corresponding ketal), and the like, as appropriate for use in its intended formulation for administration.
Ibudilast is a non-selective nucleotide phosphodiesterase (PDE) inhibitor (most active against PDE-3 and PDE-4), and has also been reported to have LTD4 and PAF antagonistic activities. Its profile appears effectively anti-inflammatory and unique in comparison to other PDE inhibitors and anti-inflammatory agents. PDEs catalyze the hydrolysis of the phosphoester bond on the 3 ‘-carbon to yield the corresponding 5′- nucleotide monophosphate. Thus, they regulate the cellular concentrations of cyclic nucleotides. Since extracellular receptors for many hormones and neurotransmitters utilize cyclic nucleotides as second messengers, the PDEs also regulate cellular responses to these extracellular signals. There are at least eight classes of PDEs: Ca2+/calmodul in-dependent PDEs (PDEl); cGMP-stimulated PDEs (PDE2); cGMP- inhibited PDEs (PDE3); cAMP-specific PDEs (PDE4); cGMP-binding PDEs (PDE5); photoreceptor PDEs (PDE6); high affinity, cAMP-specific PDEs (PDE7); and high affinity cGMP-specific PDEs (PDE9).
SYNTHESIS

DE 2315801; FR 2182914; JP 7714799, WO 0196278
By condensation of 1-amino-2-methylpyridinium iodide (I) with isobutyric anhydride (II) by means of K2CO3 at reflux temperature.
Patent
PATENT
2-methyl -l- [2- (l- methylethyl) – pyrazolo [l, 5_a] pyrimidine _3_ yl] _1_ acetone (ibudilast, generic drug name: IBUDILAST ) is an anti-allergic asthma drugs, anti-leukotrienes can twist and platelet-activating factor, promote the secretion of mucus in the respiratory tract, respiratory cilia function, enhance the role of prostacyclin, increase cerebral blood flow, improve brain metabolism. For the treatment of bronchial asthma, sequelae of cerebral embolism, cerebral arteriosclerosis.
ibudilast preparation methods are mainly the following two:
Method a: (The Jourrtal of Organic Chemistry, 1968, 33, 3766 ~3770) Synthesis Road
Lines are as follows:

The route to 2-picoline as starting material to give amino-2-methyl-pyridine iodide I-, after pyrimidine, the role of isobutyryl chloride to give the title compound. The final product obtained by this route need be purified by column chromatography, thereby increasing the difficulty of the operation, in addition to column chromatography, eluent used larger benzene toxicity, is not suitable for industrial production.
Method II: (Journal of the American Chemical Society, 2005,127, 751-760) co
A route is as follows:

The route to 2-picoline as starting material to obtain the sulfamic acid, potassium iodide I- amino-2-picoline under the action of potassium carbonate, then with isobutyric anhydride to give the title compound effect. This route of the first-stage reaction process locked, the yield is low, is not suitable for industrial production.
so there ibudilast conventional method for preparing the operational difficulties or low yield, making it impossible to achieve industrial production problems.
DETAILED DESCRIPTION IX: with a specific embodiment of the present embodiment is one of one to eight different points: in the second step of the recrystallization specific operation is as follows: First, the collected fractions was cooled to 10 ° c~25 ° C, to give a pale yellow solid, and then n-hexane was added to the pale yellow solid, and the temperature was raised to 50 ° C~68 ° C, at a temperature of 50 ° C~68 ° C incubation 5min~IOmin, then cooled to 10 ° C~ 25 ° C, and at a temperature of 10 ° C~25 ° C incubated O. 5h~Ih, and finally filtered to obtain ibudilast; the volume of the pale yellow solid quality and hexane ratio of Ig: (ImL~2mL), to obtain ibudilast.
PAPER
Bioorganic & medicinal chemistry letters (2011), 21(11), 3307-12.
Ibudilast [1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one] is a nonselective phosphodiesterase inhibitor used clinically to treat asthma. Efforts to selectively develop the PDE3- and PDE4-inhibitory activity of ibudilast led to replacement of the isopropyl ketone by a pyridazinone heterocycle. Structure–activity relationship exploration in the resulting 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3(2H)-ones revealed that the pyridazinone lactam functionality is a critical determinant for PDE3-inhibitory activity, with the nitrogen preferably unsubstituted. PDE4 inhibition is strongly promoted by introduction of a hydrophobic substituent at the pyridazinone N(2) centre and a methoxy group at C-7′ in the pyrazolopyridine. Migration of the pyridazinone ring connection from the pyrazolopyridine 3′-centre to C-4′ strongly enhances PDE4 inhibition. These studies establish a basis for development of potent PDE4-selective and dual PDE3/4-selective inhibitors derived from ibudilast.

UPDATE AS ON JAN 2016
…………..MediciNova’s ibudilast gets FDA rare paediatric disease status to treat Krabbe disease
MediciNova has received rare paediatric disease status from the US Food and Drug Administration (FDA) for its MN-166 (ibudilast) to treat Type 1 Early Infantile Krabbe disease.
![]()
References
- Huang Z, Liu S, Zhang L, Salem M, Greig GM, Chan CC, Natsumeda Y, Noguchi K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sciences. 2006 May 1;78(23):2663-8.
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Research. 1999 Aug 7;837(1-2):203-12.
- Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A, Mackenzie FL, Nagasawa M, Stevens PA, Mackenzie SJ. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. European Journal of Pharmacology. 2006 May 24;538(1-3):39-42.
- Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovascular Drug Reviews. 2001 Fall;19(3):215-25.
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004 Mar;46(3):404-11.
- Yoshioka M, Suda N, Mori K, Ueno K, Itoh Y, Togashi H, Matsumoto M. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacological Research. 2002 Apr;45(4):305-11.
- Wakita H, Tomimoto H, Akiguchi I, Lin JX, Ihara M, Ohtani R, Shibata M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Research. 2003 Nov 28;992(1):53-9.
- Rile G, Yatomi Y, Qi R, Satoh K, Ozaki Y. Potentiation of ibudilast inhibition of platelet aggregation in the presence of endothelial cells. Thrombosis Research. 2001 May 1;102(3):239-46.
- Feng J, Misu T, Fujihara K, Sakoda S, Nakatsuji Y, Fukaura H, Kikuchi S, Tashiro K, Suzumura A, Ishii N, Sugamura K, Nakashima I, Itoyama Y. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Multiple Sclerosis. 2004 Oct;10(5):494-8.
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opinion on Investigational Drugs. 2007 Jul;16(7):935-50.
- http://www.huffingtonpost.com/2013/04/03/meth-addiction-cure-ucla-ibudilast_n_2863126.html?utm_hp_ref=mostpopular#slide=more268305
- http://onlinelibrary.wiley.com/doi/10.1111/adb.12106/abstrac
SEE……Synthesis technology of ibudilast
Shandong Huagong (2014), 43, (8), 29-30. Publisher: (Shandong Huagong Bianjibu, ) CODEN:SHHUA4 ISSN:1008-021X.
Literature References:
Leukotriene D4 antagonist. Prepn: T. Irikura et al., DE 2315801; eidem, US 3850941 (1973, 1974 both to Kyorin).
Pharmacology and antiallergic activity: K. Nishino et al., Jpn. J. Pharmacol. 33, 267 (1983); H. Nagai et al., ibid. 1215.
In vitro cerebral vasodilating activity: M. Ohashi et al., Arch. Int. Pharmacodyn. 280, 216 (1986);
in vivo activity: W. M. Armstead et al., J. Pharmacol. Exp. Ther. 244, 138 (1988).
Bronchodilating activity in animals: S. Mue et al., Arch. Int. Pharmacodyn. 283,153 (1986).
Antiplatelet activity in animals: M. Ohashi et al., ibid. 321; M. Ohashi et al., Gen. Pharmacol. 17, 385 (1986).

| Patent | Submitted | Granted |
|---|---|---|
| Method for treating neuropathic pain and associated syndromes [US7534806] | 2006-07-20 | 2009-05-19 |
| Synergistic combination [US2006205806] | 2006-09-14 | |
| Pharmaceutical composition of a pde4 or pde 3/4 inhibitor and histamine receptor antagonist [US2005112069] | 2005-05-26 | |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005119160] | 2005-06-02 | |
| Methods of inducing ovulation using a non-polypeptide camp level modulator [US7507707] | 2005-07-07 | 2009-03-24 |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005192261] | 2005-09-01 | |
| Synergistic combination [US7056936] | 2004-02-19 | 2006-06-06 |
| Methods for the treatment of respiratory diseases and conditions with a selective iNOS inhibitor and a PDE inhibitor and compositions therefor [US2004087653] | 2004-05-06 | |
| Remedies for multiple sclerosis [US6395747] | 2002-05-28 | |
| Combination [US2005014762] | 2005-01-20 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4097483 | Aug 31, 1976 | Jun 27, 1978 | Kyorin Pharmaceutical Co., Ltd. | Pyrazolo 1,5-a!pyridines |
| US7585875 * | Jun 6, 2007 | Sep 8, 2009 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20070015924 | Jun 15, 2006 | Jan 18, 2007 | Cardiome Pharma Corp. | stereoselective preparation of aminocyclohexyl ether compounds such as trans-(1R,2R)-aminocyclohexyl ether compounds and/or trans-(1S,2S)-aminocyclohexyl ether compounds; useful in treating arrhythmias |
| US20080070912 | Jun 6, 2007 | Mar 20, 2008 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20090062330 | Jul 8, 2008 | Mar 5, 2009 | Medicinova, Inc. | Treatment of progressive neurodegenerative disease with ibudilast |
| US20090318437 * | Jun 10, 2009 | Dec 24, 2009 | Gaeta Federico C A | SUBSTITUTED PYRAZOLO[1,5-a] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2007142924A1 | May 29, 2007 | Dec 13, 2007 | Avigen Inc | Ibudilast for inhibiting macrophage migration inhibitory factor (mif) activity |
| WO2007146087A2 * | Jun 6, 2007 | Dec 21, 2007 | Avigen Inc | SUBSTITUTED PYRAZOLO [1,5-α] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2010151551A1 * | Jun 22, 2010 | Dec 29, 2010 | Medicinova, Inc. | ENANTIOMERIC COMPOSITIONS OF 2-AMINO-1-(2-ISOPROPYLPYRAZOLO[1,5-a]PYRIDIN-3-YL)PROPAN-1-ONE AND RELATED METHODS |
| US8119657 | Jun 22, 2010 | Feb 21, 2012 | Medicinova, Inc. | Enantiomeric compositions of 2-amino-1-(2-isopropylpyrazolo[1,5-α]pyridin-3-yl)propan-1-one and related methods |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003104178A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Napththalene derivatives which inhibit the cytokine or biological activity of macrophage migration inhibitory factor (mif) |
| WO2003104203A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Therapeutic molecules and methods-1 |
| WO2004058713A1 * | Dec 18, 2003 | Jul 15, 2004 | Jason Chyba | Differential tumor cytotoxocity compounds and compositions |
| WO2005058304A1 * | Dec 17, 2004 | Jun 30, 2005 | Cortical Pty Ltd | Implantable device containing inhibitor of macrophage migration inhibitory factor |
| WO2006045505A1 * | Oct 19, 2005 | May 4, 2006 | Novartis Ag | Mif-inhibitors |
| WO2006108671A1 * | Apr 13, 2006 | Oct 19, 2006 | Novartis Ag | 3,4-dihydro-benzo[e][1,3]oxazin-2-ones |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| CAS Number | 50847-11-5 |
| ATC code | R03DC04 |
| PubChem | CID: 3671 |
| IUPHAR/BPS | 7399 |
| DrugBank | DB05266 |
| ChemSpider | 3543 |
| UNII | M0TTH61XC5 |
| KEGG | D01385 |
| ChEMBL | CHEMBL19449 |
| Chemical data | |
| Formula | C14H18N2O |
| Molecular mass | 230.31 g/mol |
Keywords: Antiallergic; Antiasthmatic (Nonbronchodilator); Leukotriene Antagonist; Vasodilator (Cerebral).

SEE………http://apisynthesisint.blogspot.in/2016/01/ibudilast.html
////////////////////
CC(C)C1=NN2C=CC=CC2=C1C(=O)C(C)C
Vibegron ビベグロン
, click to zoom.")
Vibegron, MK-4618, KRP 114V
update FDA APPROVED 12/23/2020, GEMTESA, To treat overactive bladder
Target-based Actions Beta 3 adrenoceptor agonist
Indications Overactive bladder; Urinary incontinence
UPDATE 2018/9/21 pmda Beova JAPAN 2018Kyorin Pharmaceutical, under license from Merck, is developing vibegron (phase II, September 2014) for the treating of overactive bladder. In July 2014, Merck has granted to Kyorin an exclusive license to develop, manufacture and commercialize vibegron in Japan.
MK-4618 is being developed in phase II clinical trials at Merck & Co. for the treatment of overactive bladder. The company had been developing the compound for the treatment of endocrine disorders and hypertension; however, recent progress reports are not available at present.
In 2014, Merck licensed the product to Kyorin for development and commercialization in Japan.
The function of the lower urinary tract is to store and periodically release urine. This requires the orchestration of storage and micturition reflexes which involve a variety of afferent and efferent neural pathways, leading to modulation of central and peripheral neuroeffector mechanisms, and resultant coordinated regulation of sympathetic and parasympathetic components of the autonomic nervous system as well as somatic motor pathways. These proximally regulate the contractile state of bladder (detrusor) and urethral smooth muscle, and urethral sphincter striated muscle.
β Adrenergic receptors (βAR) are present in detrusor smooth muscle of various species, including human, rat, guinea pig, rabbit, ferret, dog, cat, pig and non-human primate. However, pharmacological studies indicate there are marked species differences in the receptor subtypes mediating relaxation of the isolated detrusor; β1AR predominate in cats and guinea pig, β2AR predominate in rabbit, and β3AR contribute or predominate in dog, rat, ferret, pig, cynomolgus and human detrusor. Expression of βAR subtypes in the human and rat detrusor has been examined by a variety of techniques, and the presence of β3AR was confirmed using in situ hybridization and/or reverse transcription-polymerase chain reaction (RT-PCR). Real time quantitative PCR analyses of β1AR, β2AR and β3AR mRNAs in bladder tissue from patients undergoing radical cystectomy revealed a preponderance of β3AR mRNA (97%, cf 1.5% for β1AR mRNA and 1.4% for β2AR mRNA). Moreover, β3AR mRNA expression was equivalent in control and obstructed human bladders. These data suggest that bladder outlet obstruction does not result in downregulation of β3AR, or in alteration of β3AR-mediated detrusor relaxation. β3AR responsiveness also has been compared in bladder strips obtained during cystectomy or enterocystoplasty from patients judged to have normal bladder function, and from patients with detrusor hyporeflexia or hyperreflexia. No differences in the extent or potency of β3AR agonist mediated relaxation were observed, consistent with the concept that the β3AR activation is an effective way of relaxing the detrusor in normal and pathogenic states.
Functional evidence in support of an important role for the β3AR in urine storage emanates from studies in vivo. Following intravenous administration to rats, the rodent selective β3AR agonist CL316243 reduces bladder pressure and in cystomeric studies increases bladder capacity leading to prolongation of micturition interval without increasing residual urine volume.
Overactive bladder is characterized by the symptoms of urinary urgency, with or without urgency urinary incontinence, usually associated with frequency and nocturia. The prevalence of OAB in the United States and Europe has been estimated at 16 to 17% in both women and men over the age of 18 years. Overactive bladder is most often classified as idiopathic, but can also be secondary to neurological condition, bladder outlet obstruction, and other causes. From a pathophysiologic perspective, the overactive bladder symptom complex, especially when associated with urge incontinence, is suggestive of detrusor overactivity. Urgency with or without incontinence has been shown to negatively impact both social and medical well-being, and represents a significant burden in terms of annual direct and indirect healthcare expenditures. Importantly, current medical therapy for urgency (with or without incontinence) is suboptimal, as many patients either do not demonstrate an adequate response to current treatments, and/or are unable to tolerate current treatments (for example, dry mouth associated with anticholinergic therapy). Therefore, there is need for new, well-tolerated therapies that effectively treat urinary frequency, urgency and incontinence, either as monotherapy or in combination with available therapies. Agents that relax bladder smooth muscle, such as β3AR agonists, are expected to be effective for treating such urinary disorders.
PATENT
http://www.google.com/patents/WO2013062881A1?cl=en

EXAMPLE 3

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine i-11 (10.0 g), sodium salt i-12 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 min to adjust pH = 3.3- 3.5, maintaining the batch temperature below 35 °C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 min. The reaction mixture was aged at RT for additional 0.5 – 1 h, aqueous ammonia (14%) was added dropwise to pH ~8.6. The batch was seeded and aged for additional 1 h to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 h. The resulting thick slurry was aged 2-3 h before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound of Formula (I)-
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
HPLC method – For monitoring conversion
Column: XBridge C18 cm 15 cm x 4.6 mm, 3.5 μιη particle size;
Column Temp. : 35 °C; Flow rate: 1.5 mL/min; Detection: 220 nm;
Mobile phase: A. 5 mM Na2B407.10 H20 B: Acetonitrile
Gradient:

HPLC method – For level of amide epimer detection
Column: Chiralpak AD-H 5 μηι, 250 mm x 4.6 mm.
Column Temp: 35 °C; Flow rate: 1.0 mL/min; Detection: 250 nm;
Mobile phase: Isocratic 30% Ethanol in hexanes + 0.1% isobutylamine
PATENT
WO 2009124167
http://www.google.com/patents/WO2009124167A1?cl=en
EXAMPLE 103
(6y)-N-r4-({(‘25′. 5R)-5-r(‘R)-hvdroxy(‘phenvnmethyl1pyrrolidin-2-yl}methvnphenyl1-4-oxo- 4,6J,8-tetrahydropyiτolori,2-α1pyrimidine-6-carboxamide

ter?-butyl(2R. 55f)-2-rCR)-hvdroxy(‘phenvnmethyl1-5-r4-(‘{r(‘65f)-4-oxo-4.6.7.8-
tetrahydropyrrolof 1.2-alpyrimidin-6- yl]carbonyl} amino)benzyl]pyrrolidine- 1 – carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at O0C was added [(65)-4-oxo-4,6,7,8-tetrahydropyrrolo[l,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1 -hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3- dimethylaminopropyl)-Nl-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N- diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from O0C to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml x 2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, IH), 7.93 (d, J = 6.6 Hz, IH), 7.49 (d, J = 8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J = 8.5 Hz, 2H), 6.40 (d, J = 6.7 Hz, IH), 5.36 (d, J = 8.6 Hz, IH), 4.38 (m, IH), 4.12-4.04 (m, 2H), 3.46 (m,lH), 3.15-3.06 (m, 2H), 2.91 (dd, J = 13.1, 9.0 Hz, IH), 2.55 (m, IH), 2.38 (m, IH), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
(6S)-N-\4-( U2S. 5R)-5-r(R)-hvdroxy(phenyl)methyl1pyrrolidin-2-
yl}methyl)phenyl1-4-oxo-4,6J,8-tetrahvdropyrrolori,2-α1pyrimidine-6- carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCCh was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23g, 60%). 1H NMR (DMSO-Cl6): δ 10.40 (s, IH), 7.91 (d, J = 6.7 Hz, IH), 7.49 (d, J = 8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, IH), 7.15 (d, J = 8.4 Hz, 2H), 6.23 (d, J = 6.7 Hz, IH), 5.11 (dd, J = 9.6, 2.9 Hz, IH), 5.10 (br, IH), 4.21 (d, J = 7.1 Hz, IH), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, IH), 1.57 (m, IH), 1.38 (m, IH), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+l).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
CHECK STRUCTURE…………….CAUTION
http://www.google.com/patents/US8247415


CAUTION…………….
Example 103(6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

Step A: tert-butyl(2R,5S)-2-[(R)-hydroxy(phenyl)methyl]-5-[4-({[(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidin-6-yl]carbonyl}amino)benzyl]pyrrolidine-1-carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at 0° C. was added [(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1-hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N-diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from 0° C. to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml×2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, 1H), 7.93 (d, J=6.6 Hz, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J=8.5 Hz, 2H), 6.40 (d, J=6.7 Hz, 1H), 5.36 (d, J=8.6 Hz, 1H), 4.38 (m, 1H), 4.12-4.04 (m, 2H), 3.46 (m, 1H), 3.15-3.06 (m, 2H), 2.91 (dd, J=13.1, 9.0 Hz, 1H), 2.55 (m, 1H), 2.38 (m, 1H), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
Step B: (6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCO3 was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23 g, 60%). 1H NMR (DMSO-d6): δ 10.40 (s, 1H), 7.91 (d, J=6.7 Hz, 1H), 7.49 (d, J=8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, 1H), 7.15 (d, J=8.4 Hz, 2H), 6.23 (d, J=6.7 Hz, 1H), 5.11 (dd, J=9.6, 2.9 Hz, 1H), 5.10 (br, 1H), 4.21 (d, J=7.1 Hz, 1H), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, 1H), 1.57 (m, 1H), 1.38 (m, 1H), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+1).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
WO2014150639
Step 6. Preparation of Compound 1-7 from Compound 1-6 and Compound A-2

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine hemihydrate 1-6 (10.3 g), sodium salt A-2 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 minutes to adjust pH = 3.3-3.5, maintaining the batch temperature below 35°C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 minutes. The reaction mixture was aged at RT for additional 0.5 – 1 hour, aqueous ammonia (14%) was added dropwise to pH -8.6. The batch was seeded and aged for additional 1 hour to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 hours. The resulting thick slurry was aged 2-3 hours before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound 1-7.
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
The crystalline freebase anhydrous form I of Compound 1-7 can be characterized by XRPD by

PATENT
WO-2014150633
Merck Sharp & Dohme Corp
Process for preparing stable immobilized ketoreductase comprises bonding of recombinant ketoreductase to the resin in a solvent. Useful for synthesis of vibegron intermediates. For a concurrent filling see WO2014150639, claiming the method for immobilization of ketoreductase. Picks up from WO2013062881, claiming the non enzymatic synthesis of vibegron and intermediates.
PAPER
Discovery of Vibegron: A Potent and Selective β3 Adrenergic Receptor Agonist for the Treatment of Overactive Bladder
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01372
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01372/suppl_file/jm5b01372_si_001.pdf

The discovery of vibegron, a potent and selective human β3-AR agonist for the treatment of overactive bladder (OAB), is described. An early-generation clinical β3-AR agonist MK-0634 (3) exhibited efficacy in humans for the treatment of OAB, but development was discontinued due to unacceptable structure-based toxicity in preclinical species. Optimization of a series of second-generation pyrrolidine-derived β3-AR agonists included reducing the risk for phospholipidosis, the risk of formation of disproportionate human metabolites, and the risk of formation of high levels of circulating metabolites in preclinical species. These efforts resulted in the discovery of vibegron, which possesses improved druglike properties and an overall superior preclinical profile compared to MK-0634. Structure–activity relationships leading to the discovery of vibegron and a summary of its preclinical profile are described.



| Reference | ||
|---|---|---|
| 1 | H.P. Kaiser, et al., “Catalytic Hydrogenation of Pyrroles at Atmospheric Pressure“, J. Org. Chem., vol. 49, No. 22, p. 4203-4209 (1984). | |
ClinicalTrials.gov Web Site 2011, April 28
| WO2011043942A1 * | Sep 27, 2010 | Apr 14, 2011 | Merck Sharp & Dohme Corp. | Combination therapy using a beta 3 adrenergic receptor agonist and an antimuscarinic agent |
| US20090253705 * | Apr 2, 2009 | Oct 8, 2009 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20110028481 * | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8642661 | Aug 2, 2011 | Feb 4, 2014 | Altherx, Inc. | Pharmaceutical combinations of beta-3 adrenergic receptor agonists and muscarinic receptor antagonists |
| US8653260 | Jun 20, 2012 | Feb 18, 2014 | Merck Sharp & Dohme Corp. | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20120202819 * | Sep 27, 2010 | Aug 9, 2012 | Merck Sharp & Dohme Corporation | Combination therapy using a beta 3 adrenergic receptor agonists and an antimuscarinic agent |
| US20020028835 | Jul 12, 2001 | Mar 7, 2002 | Baihua Hu | Cyclic amine phenyl beta-3 adrenergic receptor agonists |
| US20070185136 | Feb 2, 2007 | Aug 9, 2007 | Sanofi-Aventis | Sulphonamide derivatives, their preparation and their therapeutic application |
| US20110028481 | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| WO2003072572A1 | Feb 17, 2003 | Sep 4, 2003 | Jennifer Anne Lafontaine | Beta3-adrenergic receptor agonists |
|
8-22-2012
|
Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists
|
////////////C1CC(NC1CC2=CC=C(C=C2)NC(=O)C3CCC4=NC=CC(=O)N34)C(C5=CC=CC=C5)O
Japan, Kyorin, Pentasa, mesalazine suppositories launched
PENTASA (mesalamine) for oral administration is a controlled-release formulation of mesalamine, an amino-salicylate anti-inflammatory agent for gastrointestinal use. Chemically, mesalamine is 5-amino-2-hydroxybenzoic acid. It has a molecular weight of 153.14.
The structural formula is:

