
Home » Posts tagged 'JAPAN' (Page 4)
Tag Archives: JAPAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Biota Reports That Laninamivir Octanoate is Approved for the Prevention of Influenza in Japan

Laninamivir
(4S,5R,6R)-5-acetamido-4-carbamimidamido-6-[(1R,2R)-3-hydroxy-2-methoxypropyl]-5,6-dihydro-4H-pyran-2-carboxylic acid
| Formula | C13H22N4O7 |
|---|---|
| Mol. mass | 346.33638 g/mol |
cas 203120-17-6,
Laninamivir (L174000) prodrug; a novel long-acting neuraminidase inhibitor.
laninamivir octanoate
 472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
Daiichi Sankyo (Originator)
R-118958 is a potent, long-acting neuraminidase inhibitor (LANI) approved and launched in 2010 in Japan as an inhalable formulation for the treatment of influenza A and influenza B in adults and pediatric patients. In 2013 the product was approved in Japan for the prevention of influenza A and influenza B.
| 5-(Acetylamino)-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-7-O-methyl-D-glycero-D-galacto-non-2-enonic Acid 9-Octanoate |
| (2R,3R,4S)-3-Acetamido-4-guanidino-2-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-3,4-dihydro-2H-pyran-6-carboxylic Acid |
| (4S,5R,6R)-5-Acetamido-4-guanidino-6-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-5,6-dihydro-4H-pyran-2-carboxylic Acid |
| CS 8958 |
ATLANTA, Dec. 20, 2013 (GLOBE NEWSWIRE) — Biota Pharmaceuticals, Inc.
(Nasdaq:BOTA) (“Biota” or the “Company”) today reported that Daiichi Sankyo Company, Limited (“Daiichi Sankyo”) has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan

Laninamivir (CS-8958) is a neuraminidase inhibitor which is being researched for the treatment and prophylaxis of Influenzavirus A and Influenzavirus B.[1] It is currently in Phase III clinical trials. [2]
Laninamivir was approved for influenza treatment in Japan in 2010 and is currently marketed under the name “Inavir” by Daiichi Sankyo. Biota Pharmaceuticals [3] and Daiichi Sankyo co-own Laninamivir. On 1st April 2011, BARDA awarded up to an estimated U$231m to Biota Pharmaceuticals (Formerly Biota Holdings Ltd) wholly owned subsidiary, Biota Scientific Management Pty Ltd, for the advanced development of Laninamivir in the US. [4]
patent
|
8-13-2010
|
DRUG FOR TREATMENT OF INFLUENZA
|
The recent flu scares – first H5N1 bird flu and then H1N1 swine flu – transformed Roche’s neuraminidase inhibitor Tamiflu (oseltamivir) into a household name, along with GSK’s Relenza (zanamivir). Both of these require twice-daily dosing, and the orally available oseltamivir is the first choice, but resistance is starting to appear.
A new neuraminidase inhibitor, laninamivir, is being developed by Daiichi Sankyo.5 When administered as the octanoate prodrug form, it appears that a single dose might be sufficient to treat influenza, weekly doses could be preventative, and it is active against extremely pathogenic H5N1 strains.
Laninamivir octanoateIn a double blind, randomised, placebo-controlled Phase I study in 76 healthy male volunteers, subjects were given inhaled single doses of 5, 10, 20, 40, 80 or 120mg of the prodrug, or twice-daily doses of 20 or 40mg for three days.6 No adverse events were observed, and while the prodrug disappeared from the plasma with a half-life of about two hours, the laninamivir itself was much more slowly eliminated, with a half-life of the order of three days, suggesting the potential for giving long-lasting activity against influenza.
In another Phase I trial, a total of 20 healthy subjects with renal function ranging from normal to severely impaired were given single inhaled 20mg doses of the prodrug.7 The degree of renal impairment did not affect the maximum concentration or the time to achieve it, but the half-life increased as renal function reduced. This indicates that the rate-limiting step for elimination is drug release rate to plasma from tissues rather than renal excretion. It was well tolerated, but systemic exposure increased with increasing renal impairment.
It has also been compared with oseltamivir in patients with influenza. A total of 186 children under 10 who had had febrile influenza symptoms for no longer than 36 hours were randomised to receive 20 or 40mg of laninamivir octanoate as a single inhalation or 2mg/kg oseltamivir orally twice a day for five days.8
The new drug gave a significant reduction, of 61 hours for the 40mg group and 66 for the 20mg group, in median time to illness alleviation compared with oseltamivir in those with oseltamivir-resistant H1N1 influenza A. However, there was no significant difference in the time to alleviation of illness with H3N2 influenza A, or influenza B.
The most common side-effects were gastrointestinal problems.
In a Phase III trial, a total of 1,003 adult patients with febrile influenza symptoms for no more than 36 hours were given similar doses to those in the trial in children.9 Median time to alleviation of illness was 73h for 40mg, 86h for 20mg, and 74h for oseltamivir, and the proportion of patients shedding virus at day 3 was significantly lower in the 40mg group than for those given oseltamivir.
- Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (January 2009).“CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity”. Antimicrobial Agents and Chemotherapy53 (1): 186–92.doi:10.1128/AAC.00333-08. PMC2612152. PMID18955520.
- Hayden F (January 2009). “Developing new antiviral agents for influenza treatment: what does the future hold?”. Clinical Infectious Diseases. 48. Suppl 1 (S1): S3–13.doi:10.1086/591851. PMID19067613.
- http://www.biotapharma.com
- http://www.biotapharma.com/?page=1021001&subpage=1021019
5. T. Honda et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en, Bioorg Med Chem Lett 2009, 19(11): 2938
6. H. Ishizuka et al. J. Clin. Pharmacol. 2010, 50, 1319
7. H. Ishizuka et al. J. Clin. Pharmacol. 2010, epub ahead of print, doi 10.1177/0091270010361914
8. N. Sugaya and Y. Ohashi, Antimicrob. Ag. Chemother. 2010, 54, 2575
9 A. Watanabe et al. Clin. Inf. Dis. 2010, 51, 1167
A new route toward 2-acetamido-4-O-methyl-2-deoxy-D-mannopyranose from a Ferrier derivative of tri-O-acetyl-D-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958)
Tetrahedron 2013, 39(37): 7931

Infection of poultry with H5N1 avian influenza virus has been expanding since 2003 in wide areas including Asia, Europe and Africa. Humans infected with this virus have been found not only in Asia but also in Middle East and Africa. If a new type of H5N1 influenza virus has appeared and its infection has started, it is believed that the infection will rapidly expand to cause a worldwide spread (i.e., influenza pandemic) because most people do not possess immunity against that virus and influenza viruses spread via droplet infection and airborne infection. More than half of human patients infected with H5N1 influenza virus have died so far. Thus, the virus is highly pathogenic. It is known that three influenza pandemics, the Spanish Flu, the Asian Flu and the Hong Kong Flu, occurred in the 20th century. In the Spanish Flu which caused the largest number of patients, it is estimated that about 20-40 million people died in the world and about 0.5 million people in Japan.
According to a report from Japanese Ministry of Health, Labour and Welfare made in November, 2005, if a new type influenza virus has spread, the number of patients who will consult medical doctors in Japan as a result of infection with that virus is estimated about 18-25 million. Further, when the pathogenicity of that new type influenza virus is severe, the number of inpatients is estimated about 0.2 million while the number of dead is estimated about 0.64 million. Therefore, not only health hazard but also significant influences upon social activities are feared.
Thus, a new type influenza can cause a highly severe disease. Early development of effective therapeutics is demanded.
Although it is reported that zanamivir (in particular, zanamivir hydrate) and oseltamivir (in particular, oseltamivir phosphate or oseltamivir carboxylate) which are influenza therapeutics with neuraminidase inhibitory activity show an inhibitory activity against H5N1 influenza virus, compounds with more excellent activity are desired (Non-Patent Document 1 or 2). Further, H5N1 influenza virus strains against which oseltamivir does not show any inhibitory activity (i.e., oseltamivir resistant virus strains) have been reported. Compounds which possess an inhibitory activity against these oseltamivir resistant H5N1 influenza virus strains are desired (Non-Patent Document 1 or 2).
Compounds represented by formula (I) are known to be useful as influenza therapeutics with neuraminidase inhibitory activity (Patent Documents 1 to 3). However, it has not been reported that these compounds have an inhibitory activity against H5N1 influenza virus. Further, the structures of the compounds represented by formula (I) resemble the structure of zanamivir but are completely different from the structure of oseltamivir.
Non-Patent Document 1: Nature, 2005, vol. 437, p. 1108
Non-Patent Document 2: N. Engl. J. Med., 2005, vol. 353, (25):2667-72
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)

………………………
Preparation Example 1 5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

(1) Diphenylmethyl 5-acetamido-4-(N,N-bis-t-butyloxycarbonyl)guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoate (3.46 g, 4.1 mmol) disclosed in Example 35 (i) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was dissolved in methylene chloride (27 ml) and trifluoroacetic acid (14 ml). The resultant solution was stirred at room temperature overnight. The reaction solution was concentrated to dryness under reduced pressure, followed by three cycles of azeotropic distillation to dryness with toluene (5 ml). The resultant oily material was dissolved in ethyl acetate (10 ml). The solution was poured into a saturated aqueous solution of sodium hydrogencarbonate (50 ml). The pH of the resultant solution was adjusted to 8.5 by addition of 20% aqueous solution of sodium carbonate. Then, the solution was stirred at room temperature for 3 hr and its pH was adjusted to 5.0 with hydrochloric acid (3 ml), followed by stirring at room temperature for another 1 hr. The solution was further stirred for 1 hr while ice-cooling. Subsequently, precipitating crystals were suction filtered and vacuum dried for 10 hr at an external temperature of 50° C. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (0.97 g; yield 51%).
Infrared Absorption Spectrum (KBr) ν max cm−1: 3412, 2929, 2856, 1676, 1401, 1320, 1285, 1205, 1137, 722.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.88 (1H, d, J=2.5 Hz), 4.45 (3H, m), 4.27 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.15 (1H, m), 3.47 (21-1, m), 3.42 (3H, s), 2.37 (2H, t, J=7.4 Hz), 2.10 (3H, s), 1.31 (2H, m), 1.20-1.40 (8H, m), 0.85-0.95 (3H, m).
13C Nuclear Magnetic Resonance Spectrum (100 MHz, CD3OD) δ ppm: 176.5, 173.7, 164.7, 158.9, 146.7, 108.7, 80.1, 78.0, 69.3, 66.8, 61.4, 52.4, 35.1, 32.8, 30.2, 30.1, 26.0, 23.7, 22.8, 14.4.
(2) The subject compound was also obtained by the method described below.
5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 35 (ii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was subjected to reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque), 100 g] and eluted with methanol/water (0/1-1/1-2/1). Fractions containing the compound of interest were vacuum concentrated. The precipitating crystals were suction filtered and vacuum dried. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (1.2 g; yield 49%). The property data of the resultant compound were consistent with those of the compound obtained in (1) above.
Preparation Example 2 5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 28 (viii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was purified in an ion-exchange resin column [Dowex-50X; (i) water and (ii) 5% aqueous ammonium solution] and further purified by reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque); water]. Fractions containing the compound of interest were vacuum concentrated. The resultant solid was washed with methanol, filtered and dried to thereby yield the subject compound (1.43 g) as a colorless solid.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.64 (1H, d, J=2.0 Hz), 4.43 (2H, m), 4.22 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.00 (1H, m), 3.85 (1H, dd, J=10.0 Hz, 2.4 Hz), 3.68 (1H, dd, J=10.0 Hz, 5.5 Hz), 3.58 (1H, m), 3.43 (3H, s).
………………………….


…………………………..
Process W is known as a method for manufacturing a compound represented by the formula (Ia), which is embraced in a compound represented by the formula (I) or a pharmacologically acceptable salt thereof, (hereinafter also referred to as “compound (Ia)”; the same shall be applied with respect to other (Patent Document 1). In Process W, n-Hep represents a 1-heptyl group.


Process X is known as a method for manufacturing compound (Ib), which is embraced in compound (I) or a pharmacologically acceptable salt thereof (Patent Document 2). Compound (IVk) is a synthetic intermediate in Process W. In Process X, n-Hep represents a 1-heptyl group.

Process Y is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 1). The procedures from compound (IVc) to compound (IVe) and from compound (IVf) to compound (IVh) in Process Y are the same as in Process W.


Process Z is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 2). In Process Z, the procedure from compound (IVf) to compound (IVh) is the same as in Process W, and the procedure from compound (IVh) to compound (IIIa) is the same as in Process Y.
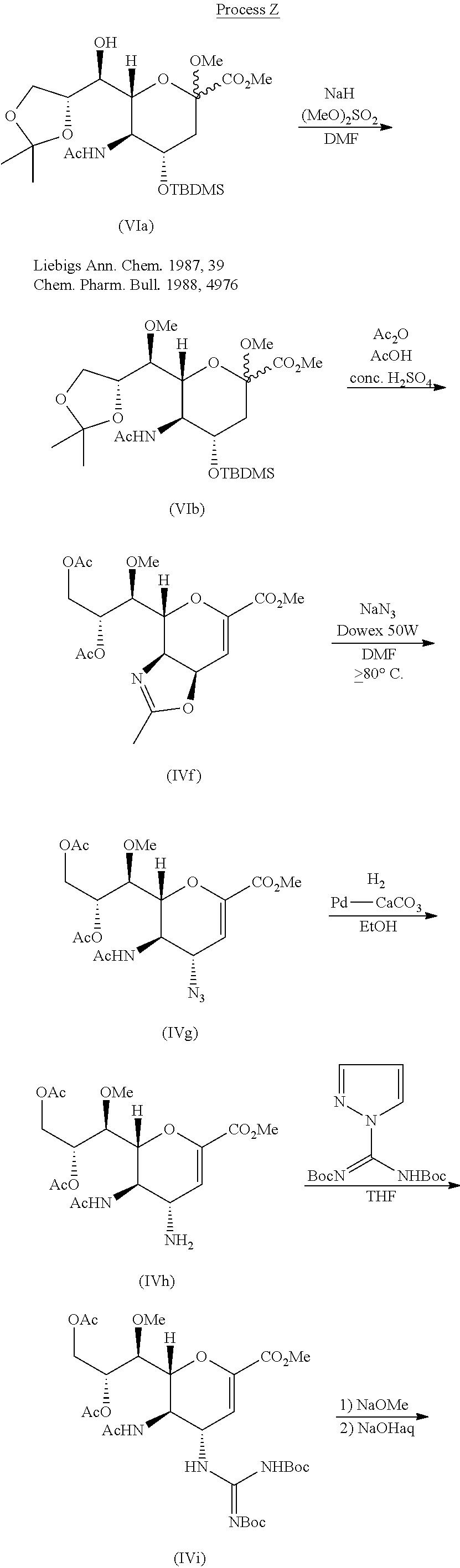

From the viewpoint of industrial production, the aforementioned Process W, Process Y, or Process Z could be improved in points such as the following:
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
update…………

:ACIE 10.1002/anie.201408138
Scheme zanamivir and Lanimiwei is based on N- acetylneuraminic acid as a starting material, the price is more expensive (ca.13000RMB / kg). Ma recently from Shanghai Institute of Organic Chemistry greatly researcher on ACIE published zanamivir, Lanimiwei and CS-8958 is more simple synthetic route. References: ACIE 10.1002 / anie.201408138
EYLEA® (aflibercept) Injection Approved For The Treatment of Macular Edema Following Central Retinal Vein Occlusion In Japan

TARRYTOWN, N.Y., Nov. 22, 2013 /PRNewswire/ — Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that EYLEA® (aflibercept) Injection has received approval for the treatment of Macular Edema Following Central Retinal Vein Occlusion (CRVO) from the Japanese Ministry of Health, Labour and Welfare.http://www.pharmalive.com/japan-approves-eylea
In November 2011 the United States Food and Drug Administration approved aflibercept for the treatment of wet macular degeneration.
On August 3, 2012 the United States Food and Drug Administration approved Zaltrap (ziv-aflibercept) for use in combination with 5-fluorouracil, leucovorin and irinotecan to treat adults with metastatic colorectal cancer that is resistant to or has progressed following an oxaliplatin‑containing regimen.
In November 2012 the European Medicines Agency (EMA) approved aflibercept for the treatment of wet macular degeneration.
On February 1, 2013 the European Commission granted a marketing authorisation valid throughout the European Union for treatment of adults with metastatic colorectal cancer for whom treatment based on oxaliplatin has not worked or the cancer got worse.

Bristol-Myers Squibb files NDA in Japan for all-oral hepatitis C treatment
Bristol-Myers Squibb has filed a new drug application (NDA) to Japan’s Pharmaceutical and Medical Devices Agency for the approval of an interferon-free and ribavirin-free treatment regimen for patients with chronic hepatitis C (HCV).
click on title
Bristol-Myers Squibb files NDA in Japan for all-oral hepatitis C treatment
Simeprevir has been approved in Japan for the treatment of genotype 1 chronic hepatitis C infection

simeprevir
| CAS number | 923604-59-5 | ||
| Formula | C38H47N5O7S2 | ||
| Weight | 749.93908 |
Stockholm, Sweden — Medivir AB (OMX: MVIR) today reports that Janssen Pharmaceutical R&D Ireland (Janssen) has been informed by the Japanese Ministry of Health, Labour and Welfare (MHLW) that simeprevir has been approved for the treatment of genotype 1 chronic hepatitis C virus (HCV) infection.
read all at
http://www.pharmalive.com/japan-approves-simeprevir
Hepatitis C virus (HCV) infections affect approximately 3 percent of the worldwide population and often lead to cirrhosis and hepatocellular carcinoma. The standard therapy of pegylated- interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferonare currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. Numerous direct acting agents (DAAs) have been or are being developed for treatment of HCV, such as telaprevir and boceprevir (both received MA approved in 2011 for use with interferon and ribavirin based therapy), however direct acting agents are linked to increased toxicity of treatment, the emergence of resistance, and to date do not provide a standard of care which is interferon free. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byMedivir and Johnson & Johnson‘s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Food and Drug Administration (FDA) has granted Priority Review to the New Drug Application (NDA) for simeprevir (TMC435). Simeprevir is an investigational NS3/4A protease inhibitor taken orally (150 mg capsule) once a day along with pegylated interferon and ribavirin for genotype 1 chronic hepatitis C virus (HCV) infection in adult patients with compensated liver disease (meaning the liver is heavily scarred but still functional).
“Hepatitis C is a complex disease and Janssen is committed to working with the HCV community, caregivers, and health care systems to address this global epidemic,” said Gaston Picchio, Hepatitis Disease Area Leader, Janssen Research & Development. “We are pleased that the FDA has granted simeprevir Priority Review, as it is a significant step forward in making this therapy available to physicians and their hepatitis C patients.”
The FDA grants Priority Review to medicines that may offer major advances in care or provide a treatment option where no adequate therapy exists. Under the Prescription Drug User Fee Act, FDA review will begin approximately 60 days after receipt of the application and will aim to be completed within six months from when the review period begins.
The regulatory submission for simeprevir is supported in part by data from three pivotal Phase 3 studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. Janssen also recently submitted simeprevir for marketing authorization to regulatory authorities in Japan and Europe.

- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
|displayauthors=suggested (help) - “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
IUPAC standard name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 – (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 – (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
Aliases
TMC435
TMC435350

,,,,,,,,,,,,,
NS3/4A protease inhibitors
Ciluprevir (BILN 2061) Boehringer Ingelheim
Boceprevir (SCH503034) Merck
Telaprevir (VX-950) Vertex
Danoprevir (RG7227) Roche
simeprevir /TMC435 Tibotec / Medivir
Vaniprevir (MK-7009) Merck
Bl 201335 Boehringer Ingelheim
BMS-650032 Bristol-Myers Squibb
GS-9256 Gilead
ABT-450 Abbott / Enanta
Narlaprevir (SCH900518) Merck
PHX1766 Phenomix
ACH-1625 Achillion
IDX320 Idenix
MK-5172 Merck
VX-985 Vertex Drug name Company
GS-9451 Gilead
Telaprevir
Accordin to http://en.wikipedia.Org/wiki/File:Telaprevir.svg, Teaprevir has the structure

Systematic lUPAC Name: (1 S,3aR,6aS)-2-[(2S)-2-[[(2S)-2-Cyclohexyl-2-(pyrazine-2- carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-/\/-[(3S)-1-(cyclopropylamino)-1 ,2- dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1/-/-cyclopenta[c]pyrrole-1-carboxamide
Telaprevir may be administered in a unit dose of, for example between about 250 and about l OOOmg, such as about 750mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Boceprevir
Accordin to http://en.wikipedia.0rg/wiki/File:B0ceprevir.svg, Boceprevir has the structure:

Systematic lUPAC Name: (1 R,2S,5S)-N-[(2≡)-4-amino-1-cyclobutyl-3,4-dioxobutan-2-yl)]- 3-{(2S)-2-[(tert-butylcarbamoyl)amino]-3,3-dimethylbutanoyl}- 6,6-dimethyl-3- azabicyclo[3.1.0]hexane-2-carboxamide
Boceprevir may be administered in a unit dose of, for example between about 250 and about 1000mg, such as about 800mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Compound 1: miR-122 inhibitor
As reported in Young et al., JACS 2010, 132, 7976-7981) (hereby incorporated by reference), it is possible to assay for small molecule inhibitors of miR122 and small molecule are known, such as those illustrated below:



» {7.02 ± 1.40) If (4. S3 * 0.45)

The numerical values refer to luciferase expression due to miR-122 deprepression, and values greater than 1 indicate miR-122 inhibition.
First biosimilar filgrastims launched in Japan


International nonproprietary name: Filgrastim
Chemical name: N-L- Methionyl colony-stimulating factor (human genetically engineered); non-glycated protein consisted of 175 amino acids.
Chemical name: N-L- Methionyl colony-stimulating factor (human genetically engineered); non-glycated protein consisted of 175 amino acids.
Filgrastim is a granulocyte colony-stimulating factor (G-CSF) analog used to stimulate the proliferation and differentiation of granulocytes.[1] It is produced by recombinant DNA technology. The gene for human granulocyte colony-stimulating factor is inserted into the genetic material of Escherichia coli. The G-CSF then produced by E. coli is different from G-CSF naturally made in humans.

Hematopoietic growth factor. Interacting with receptors on the surface of hematopoietic cells it regulates production and release of neutrophils from the bone marrow to the peripheral blood. Dose dependant number growth of neutrophils with normal or increased functional activity is passing for 24 hours.
Filgrastim is marketed under several brand names, including Neupogen (Amgen), Imumax(Abbott Laboratories), Grafeel (Dr. Reddy’s Laboratories), Neukine (Intas Biopharmaceuticals), Emgrast (Emcure Pharmaceuticals), Religrast (Reliance Life Sciences), Zarzio (Sandoz), Nufil (Biocon) and others.
Apricus Biosciences is currently developing and testing a product under the brand nameNupen which can deliver filgrastim through the skin to improve post-chemotherapy recovery of neutrophil counts.
Filgrastim is also used to increase the number of hematopoietic stem cells in the blood before collection by leukapheresis for use in hematopoietic stem cell transplantation.Filgrastim is used to treat neutropenia,[2] stimulating the bone marrow to increase production of neutrophils. Causes of neutropenia include chemotherapy and bone marrow transplantation.
Filgrastim should not be used in patients with known hypersensitivity to E. coli-derived proteins.
The most commonly observed adverse effect is mild-to-moderate bone pain after repeated administration and local skin reactions at the site of injection.[3] Other observed adverse effects include serious allergic reactions (including a rash over the whole body, shortness of breath, wheezing, dizziness, swelling around the mouth or eyes, fast pulse, and sweating), ruptured spleen (sometimes resulting in death), alveolar hemorrhage, acute respiratory distress syndrome, and hemoptysis.[3] Severe sickle cell crises, in some cases resulting in death, have been associated with the use of filgrastim in patients with sickle cell disorders.[4]
Drug interactions between filgrastim and other drugs have not been fully evaluated. Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes; this should be considered when interpreting bone-imaging results.[5]
Filgrastim has not been studied in pregnant women and its effects on unborn babies is unknown. If taking filgrastim while pregnant, it is possible that traces of the drug could be found in the baby’s blood. It is not known if the drug can get into human breast milk.
- Beveridge, R. A.; Miller, J. A.; Kales, A. N.; Binder, R. A.; Robert, N. J.; Harvey, J. H.; Windsor, K.; Gore, I. et al. (1998). “A Comparison of Efficacy of Sargramostim (Yeast-Derived RhuGM-CSF) and Filgrastim (Bacteria-Derived RhuG-CSF) in the Therapeutic Setting of Chemotherapy-Induced Myelosuppression”. Cancer Investigation 16 (6): 366–373. doi:10.3109/07357909809115775.PMID 9679526. edit
- Crawford, J.; Glaspy, J. A.; Stoller, R. G.; Tomita, D. K.; Vincent, M. E.; McGuire, B. W.; Ozer, H. (2005). “Final Results of a Placebo-Controlled Study of Filgrastim in Small-Cell Lung Cancer: Exploration of Risk Factors for Febrile Neutropenia”. Supportive Cancer Therapy 3 (1): 36–46. doi:10.3816/SCT.2005.n.023. PMID 18632435. edit
- Neupogen “Neupogen: Patient Information Leaflet”. Amgen. Retrieved 24 June 2013.
- “NEUPOGEN® Patient Guide”. Amgen. Retrieved 24 June 2013.
- “Neupogen”. RxList. 4 June 2012. Retrieved 23 June 2013.
- Budiono Santoso; Chris J. van Boxtel; Boxtel, Christoffel Jos van (2001). Drug benefits and risks: international textbook of clinical pharmacology. New York: Wiley. ISBN 0-471-89927-5.
- “Neupogen information”. Retrieved 20 October 2005.
Kyowa Hakko Kirin seeks MHLW Approval for Additional Indication for ATL, PTCL and CTCL of Mogamulizumab

Kyowa Hakko Kirin Co., Ltd. has been filed an application to Japan’s Ministry of Health, Labour and Welfare (“MHLW”) seeking approval for additional indication for untreated CCR4-positive adult T-cell leukemia-lymphoma (ATL), relapsed CCR4-positive peripheral T-cell lymphoma (PTCL) and cutaneous T-cell lymphoma (CTCL) of Mogamulizumab (brand name: POTELIGEO® Injection 20 mg).
read at…………
Mogamulizumab (USAN; trade name Poteligeo) is a humanized monoclonal antibodytargeting CC chemokine receptor 4 (CCR4). It has been approved in Japan for the treatment of relapsed or refractory adult T-cell leukemia/lymphoma.[1]
Mogamulizumab was developed by Kyowa Hakko Kirin Co., Ltd.[2] It has also been licensed to Amgen for development as a therapy for Asthma.[3]
- Subramaniam, J; Whiteside G, McKeage K, Croxtall J (18). “Mogamulizumab: First Global Approval”. Drugs 72 (9): 1293–1298. doi:10.2165/11631090-000000000-00000. Retrieved 10 September 2012.
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Mogamulizumab”. American Medical Association.
- “Kyowa Hakko Kirin R&D Pipeline”. Kyowa Hakko Kirin. Retrieved 10 September 2012.

Poteligeo(mogamulizumab)-单克隆抗体
| 单克隆抗体Poteligeo(mogamulizumab)获得日本厚生劳动省批准治疗白血病-淋巴瘤 日本厚生劳动省批准Kyowa Hakko Kirin公司的Poteligeo治疗复发或难治性CC趋化因子受体4(CCR4,CD194)阳性的T细胞性白血病-淋巴瘤。厚生劳动省还批准了Kyowa公司这一抗体的两个诊断方法,用于测试IHC和FCM,从而确定最有可能对治疗有应答的患者亚群。Amgen公司拥有Poteligeo在除日本、韩国、中国大陆和台湾以外地区的所有非癌症适应症的开发和商业化独占权。Amgen公司正在进行本品用于治疗哮喘的Ⅰ期临床研究。 |

Enbrel (etanercept), Biosimilar innovator drug companies scrambling to copy

Enbrel (etanercept)

http://www.biosimilarnews.com/enbrel-patent-in-the-us
Biosimilars are protein products that are sufficiently similar to a biopharmaceutical already approved by a regulatory agency. Several biotechnology companies and generic drug manufacturers in Asia and Europe are developing biosimilars of tumor necrosis factor inhibitors and rituximab. A biosimilar etanercept is already being marketed in Colombia and China. In the US, several natural source products and recombinant proteins have been approved as generic drugs under Section 505(b)(2) of the Food, Drug, and Cosmetic Act. However, because the complexity of large biopharmaceuticals makes it difficult to demonstrate that a biosimilar is structurally identical to an already approved biopharmaceutical, this Act does not apply to biosimilars of large biopharmaceuticals. Section 7002 of the Patient Protection and Affordable Care Act of 2010, which is referred to as the Biologics Price Competition and Innovation Act of 2009, amends Section 351 of the Public Health Service Act to create an abbreviated pathway that permits a biosimilar to be evaluated by comparing it with only a single reference biological product.
Amgen announced the issuance of U.S. Patent No. 8,063,182 related to Enbrel (etanercept).owned by Hoffmann-la roche and licensed to Amgen (exp2028) VIA immunex
A biosimilar etanercept, manufactured in China by CP Guojian Pharmaceutical Co., Ltd. (Shanghai), is already being marketed in China as Yisaipu [3] and in Colombia as Etanar [4]. Several biotechnology companies in Asia are also developing biosimilar versions of tumor necrosis factor inhibitors. Protalix Biotherapeutics, Inc. (Carmiel, Israel) is developing a biosimilar etanercept that is expressed in plant cells [5]. Mycenax Biotech (Taiwan) has completed early-phase clinical trials of a biosimilar etanercept in Southeast Asia: a phase I trial among 24 healthy subjects in South Korea and a phase I/II trial that enrolled 18 patients with rheumatoid arthritis in Taiwan [6]. Avesthagen (Bangalore, India) has received a patent from the Indian patent office for a biosimilar etanercept [7]. In South Korea, both Celltrion (Yeonsu-gu Incheon City) and Aprogen (Daejeon) are developing a biosimilar of infliximab [8] and LG Life Sciences (Seoul) is developing biosimilars of both etanercept and infliximab to treat rheumatoid arthritis and other inflammatory diseases [9].

Drug developers:
- Avesthagen: Avent™ in clinical studies
![]()
read this doc
http://www.avesthagen.com/docs/020910pr.pdf
…………………………………………………………………………………..
- BioXpress Therapeutics: Biosimilar in active development

http://www.bioxpress.com/pipeline/
………………………………………………………………………………………
- Cipla:Etacept, Launches biosimilar in India on April 17, at a price of Rs. 6,150 ($113.43), 30% less than the innovator product.

- read this
………………………………………………………………………………….
- Hanwha Chemical: HD203 “scheduled for launch,” company states on its website without including a date, following submission for marketing approval to South Korea’s Korea Ministry of Food and Drug Safety following completion of Phase I and Phase III trials. Hanwha has said it will seek a partner to commercialize HD203 and a biosimilar for Herceptin (trastuzumab).
- http://www.thepharmaletter.com/file/105028/merck-co-links-with-koreas-hanwha-on-biosimilar-of-enbrel.html
………………………………………………………………………………
- LG Life Sciences: LBEC0101 completed Phase I trial in South Korea
![]()
http://www.lgls.co.kr/rd/pipeline.jsp
………………………………………………………………………………
- Mycenax Biotech: TuNEX in Phase III clinical trials in Japan and South Korea
…………………………………………………………………………..
- Protalix Biotherapeutics: PRX-106 in preclinical studies
http://www.protalix.com/product-development/prx-106.asp
![]()
…………………………………………………………………………………
- Shanghai CP Goujian Pharmaceutical: Etanar®, marketed in Colombia; Yisaipu, marketed in China
……………………………………………………………………………………………..
Recently discontinued effort: Merck & Co. and Hanwha Chemical: Hanwha disclosed December 18, 2012, that Merck terminated agreement to develop and manufacture the biosimilar MK-8953, now called HD203, as well as market it in all countries except South Korea and Turkey, an up to $720 million deal signed June 2011.1
Nature and indication: Tumor necrosis factor (TNF) blocker for rheumatoid arthritis, polyarticular Juvenile Idiopathic Arthritis (JIA) in patients aged two years or older; psoriatic arthritis; ankylosing spondylitis; and plaque psoriasis
2012 sales: $7.963 billion (includes $4.236 billion Amgen + $3.737 billion Pfizer). Amgen markets Enbrel in U.S. and Canada under an agreement with Pfizer set to expire October 31, 2013
Patent status: Patents set to expire in EU in 2015; in U.S., 2019, 2023, 2028, and 2029
Etanercept is a fusion protein produced by recombinant DNA, which fuses a soluble human TNF receptor with an IgG1 antibody. This modified protein works by blocking TNF activity, thereby reducing their ability to cause an inflammatory response as well as severe, chronic pain and discomfort to patients. The fusion protein is protected by five different molecule Key patent families (Fig 2) and are all considered to be a constraint to generic entry until expiry. Although the patent families are owned by different patentees, Amgen have entered into licensing agreements with all parties allowing them sole distributing and marketing rights of Enbrel®.
- Public Health Service Act Sec. 262. Regulation of biological products.http://www.fda.gov/RegulatoryInformation/Legislation/ucm149278.htm
- Woodcock J, Griffin J, Behrman R, Cherney B, Crescenzi T, Fraser B, Hixon D, Joneckis C, Kozlowski S, Rosenberg A, Schrager L, Shacter E, Temple R, Webber K, Winkle H. The FDA’s assessment of follow-on protein products: a historical perspective. Nat Rev Drug Discov. 2007;6:437–442. doi: 10.1038/nrd2307. [PubMed] [Cross Ref]
- Yisaipu. http://www.cpgj-pharm.com/en/product_patient.asp?proid=22&action=intro
- Rondon F, Bautista A, Salazar JC, Casas N, Santos P, Vargas F, Marquez J. Etanar therapy in real-life patients with rheumatoid arthritis [abstract]Arthritis Rheum. 2010;62(Suppl 10):1811.
- Pipeline products. http://www.protalix.com/pipeline_products.html
- Biosimilar TuNEX® completes Phase I/II clinical trial in Taiwan, Phase I in Korea. http://www.mycenax.com.tw/webe/html/02news/news_show.aspx?page=1
- Singh K. Avesthagen gets patent for Enbrel biosimilar. Economic Times. 2010.
- Korea’s Celltrion and Aprogen in race to sell biosimilars in Japan.http://sis.windhover.com/buy/abstract.php?id=28101102003
- LG Life Sciences’ Pipeline Overview. http://thinklgls.com/rnd/pipeline
- Dr. Reddy’s Marketed Pharmaceutical Products.http://www.drreddys.com/products/bio_mproducts.html#
- TL011 in severe, active rheumatoid arthritis patients.http://clinicaltrials.gov/ct2/show/NCT01123070
- GP2013 in the treatment of RA patients refractory to or intolerant of standard therapy. http://www.clinicaltrials.gov/ct2/show/NCT01274182
- View Opportunities. http://www.sourcegenerics.com/viewAllListing.asp
- Rudick RA, Simonian NA, Alam JA, Campion M, Scaramucci JO, Jones W, Coats ME, Goodkin DE, Weinstock-Guttman B, Herndon RM, Mass MK, Richert JR, Salazar AM, Munschauer FE, Cookfair DL, Simon JH, Jacobs LD. Incidence and significance of neutralizing antibodies to interferon beta-1a in multiple sclerosis. Multiple Sclerosis Collaborative Research Group (MSCRG)Neurology. 1998;50:1266–1272. [PubMed]
- Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. doi: 10.1056/NEJMoa011931. [PubMed] [Cross Ref]
- Schellekens H, Jiskoot W. Eprex-associated pure red cell aplasia and leachates. Nat Biotechnol. 2006;24:613–614. doi: 10.1038/nbt0606-613.[PubMed] [Cross Ref]
- Bennett CL, Luminari S, Nissenson AR, Tallman MS, Klinge SA, McWilliams N, McKoy JM, Kim B, Lyons EA, Trifilio SM, Raisch DW, Evens AM, Kuzel TM, Schumock GT, Belknap SM, Locatelli F, Rossert J, Casadevall N. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351:1403–1408. doi: 10.1056/NEJMoa040528. [PubMed] [Cross Ref]
- Federal Food, Drug, and Cosmetic Act (FD&C Act) SEC. 505. [21 USC §355] New Drugs.http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/ucm108125.htm
- Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products. London: European Medicines Agency; 2005.
- Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London: European Medicines Agency; 2006.
- European Medicines Agency. Work plan for the biosimilar medicinal products working party (BMWP) 2011. London: European Medicines Agency; 2010.
- H. R. 3590–686. Patient Protection and Affordable Care Act. Title VII– Improving Access to Innovative Medical Therapies. Subtitle A–Biologics Price Competition and Innovation. Sec. 7002. Approval Pathway for Biosimilar Biological Products.http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf
- Implementation of the Biologics Price Competition and Innovation Act of 2009.http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/ucm215089.htm
see details of etanercept

Etanercept
ATC (Anatomical Therapeutic Chemical Classification)
L04AA11,L04AB01
CAS registry number (Chemical Abstracts Service)
0185243-69-0
Chemical Formula
C2224-H3472-N618-O701-S36
Molecular Weight
51238
Therapeutic Categories
Immunosuppressant
Disease-modifying antirheumatic drug, DMARD
Biological response modifier, BRM
Anti-inflammatory agent
Tumor necrosis factor alpha (TNF-α) inhibitor
Chemical Name
Dimeric fusion protein consisting of the extracellular ligand-binding portion of the human 75 kilodalton (p75) tumor necrosis factor receptor (TNFR) linked to the Fc portion of human IgG1
is made from the combination of two naturally occurring soluble human 75-kilodalton TNF receptors linked to an Fc portion of an IgG1. The effect is an artificially engineered dimeric fusion protein.
• Sandoz continues to advance biosimilar pipeline with seven Phase III trials across five molecules
• Global program underscores Sandoz’s leadership in biosimilarsHolzkirchen, Germany, June 24, 2013 – Sandoz, the global leader in biosimilars, announced it has initiated a major Phase III clinical trial with its biosimilar version of etanercept (Amgen’s Enbrel®).
Read more at
http://www.drugs.com/news/novartis-begins-enbrel-phase-iii-trial-45414.html
| Etanercept (trade name Enbrel) is a biopharmaceutical that treats autoimmune diseases by interfering with tumor necrosis factor (TNF; a soluble inflammatory cytokine) by acting as a TNF inhibitor. It has U.S. F.D.A. approval to treat rheumatoid, juvenile rheumatoid andpsoriatic arthritis, plaque psoriasis and ankylosing spondylitis. TNF-alpha is the “master regulator” of the inflammatory (immune) response in many organ systems. Autoimmune diseases are caused by an overactive immune response. Etanercept has the potential to treat these diseases by inhibiting TNF-alpha. Etanercept is a fusion protein produced by recombinant DNA. It fuses the TNF receptor to the constant end of the IgG1 antibody. First, the developers isolated the DNA sequence that codes the human gene for soluble TNF receptor 2, which is a receptor that binds to tumor necrosis factor-alpha. Second, they isolated the DNA sequence that codes the human gene for the Fc end of immunoglobulin G1 (IgG1). Third, they linked the DNA for TNF receptor 2 to the DNA for IgG1 Fc. Finally, they expressed the linked DNA to produce a protein that links the protein for TNF receptor 2 to the protein for IgG1 Fc.The prototypic fusion protein was first synthesized and shown to be highly active and unusually stable as a modality for blockade of TNF in vivo in the early 1990s by Bruce A. Beutler, an academic researcher then at the University of Texas Southwestern Medical Center at Dallas, and his colleagues.[2][3][4] These investigators also patented the protein, selling all rights to its use to Immunex, a biotechnology company that was acquired by Amgen in 2002.It is a large molecule, with a molecular weight of 150 kDa., that binds to TNFα and decreases its role in disorders involving excess inflammation in humans and other animals, including autoimmune diseases such as ankylosing spondylitis, juvenile rheumatoid arthritis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and, potentially, in a variety of other disorders mediated by excess TNFα.In North America, etanercept is co-marketed by Amgen and Pfizer under the trade name Enbrel in two separate formulations, one in powder form, the other as a pre-mixed liquid. Wyeth is the sole marketer of Enbrel outside North America excluding Japan whereTakeda Pharmaceuticals markets the drug.Etanercept is an example of a protein-based drug created using the tools of biotechnologyand conceived through an understanding afforded by modern cell biology.  |

Figure 2: Molecule Key Patents landscape
International Market
Patents protecting the various technologies of the Etanercept molecule (Fig. 2) across all five families have now expired in Europe, Canada and Australia. In Europe, SPCs and paediatric extensions were granted based on the EP0418014 (1989-09-05) and EP0939121 (1989-09-12) however the last of the paediatric extensions expired in early August, 2015. Finland has been granted a national patent disclosing the Etanercept sequence in the family with priority US40324189A (1989-09-05), which would constrain generic entry until April, 2020. Cyprus has also received a five year patent extension on a national patent set to expire in mid-2016 and would be a constraint for biosimilars entering the market there.
Although the Etanercept molecule is no longer protected in the European, Canadian or Australian markets, no biosimilar has been approved in these major markets suggesting the difficulty of developing a biosimilar which complies with the stringent regulatory pathways in place. Having said that, Merck and Samsung Bioepis (a joint venture from electronics giant Samsung and biotech firm Biogen Idec) has submitted their Etanercept biosimilar candidate SB4 to the EMA, which is currently awaiting review. If approved, it is expected that they will obtain further approval in other territories where Etanercept is no longer protected. With the regulatory approval pathways differing from country to country, Etanercept biosimilars have been approved in smaller markets including India, China and South Korea.
US Market
In the US, the ‘molecule’ patents protecting active ingredient Etanercept have all expired aside from US8,063,182 (‘182) and US8,163,522 (‘522) members from priority CH331989 (1989-09-12) owned by Roche (exclusively licensed to Amgen), which are set to expire in 2028 and 2029, respectively. These patents members disclose a portion of the Etanercept sequence, so are considered to constrain biosimilar entry until expiry. The members are continuation patents filed from US5,610,279 (another member of the same family) and while they were both filed in May, 1995, were not issued until 2011 (‘182) and 2012 (‘522). Under the 35 U.S. Code § 154, these patents received 17 year patent term from the issuing date. Since these patents were applied for in 1995 during the transitional period of the TRIPS agreement, they were not published by the USPTO until they were issued. This situation often gives rise to the term ‘submarine patents’.
Currently there is no system to link relevant patents to biologic drugs in the US as with small molecule drugs (Orange Book) which makes filing biosimilars in the US a convoluted process. While the FDA are currently working on an equivalent to the Orange Book, the ‘Purple book’, companies wishing to develop biosimilars in the US need to do considerable patent landscape searching in order to avoid infringement of any patents potentially protecting the biologic drug. In the case of US member ‘182 and ‘522, upon inspection these patents are clearly relevant to Enbrel®, however without a registry there is no easy way of making this link. The patents have been flagged in the Key Patent module in Ark due to SPCs and paediatric extensions on the equivalent EP0939121 member and litigation in the US (see below).
Currently, biologic drugs approved in the US receive a 12 year data exclusivity period and in Europe, an 8 year data exclusivity period with additional 2 year market exclusivity, starting from the market authorisation date. Enbrel® was approved in 1998 and 2000, in the US and Europe, respectively and data exclusivity protection has therefore now expired.
Development of biosimilars takes considerably longer than generic medicine making it a costly venture for generic pharmaceutical manufacturers. According to Amgen, Enbrel® was protected by US5395760 (‘760) and US5605690 (‘690) members from priority 1989-09-05 which were set to lose patent protection in 2012 and 2014, respectively. In 2004, Sandoz began developing GP2015 a biosimilar equivalent of Etanercept, investing millions of dollars in the hope that they would be ready to launch by the time all the patent protection for Enbrel® expired. Currently, GP2015 is in Phase III study in the US and European Union for patients with moderate to severe chronic plaque-type psoriasis with respect to PASI 75 response rate at Week 12.
In June 2013, Sandoz filed a suit against Amgen and Roche in the US District Court for the Northern District of California seeking declaratory judgment of non-infringement, invalidity and unenforceability of the ‘182 and ‘522 patents. Sandoz claimed a ‘case of controversy’ regarding the patents, as their research and development was based on the understanding that ‘760 and ‘690 patents members were protecting Enbrel®. With the issuing of ‘182 and ‘522 patents this has essentially delayed the prospect of an Etanercept biosimilar from entering the US market until 2029.
Amgen and Roche sought a dismissal of the proceeding due to lack of subject matter jurisdiction, which was granted. Although Sandoz appealed the decision, the Court of Appeals affirmed the dismissal, since there was no real and immediate controversy as Sandoz had not yet filed an FDA application, and they had based their suit on future events and were not able to establish “real and immediate injury or threat of future injury.”

DRUG DISCOVERY PRESENTATION BY DR ANTHONY CRASTO

Please wait and allow slideshare to load…………..
Oramed Enrolls First Patient in its Phase 2a U.S. Oral Insulin Clinical Trial
![]()

Nadav Kidron
Marks initiation of Oramed’s first FDA trial for its flagship ORMD-0801 oral insulin product
JERUSALEM, July 8, 2013
Oramed Pharmaceuticals Inc. (NASDAQCM: ORMP) (http://www.oramed.com), a developer of oral drug delivery systems, announced today that the first patient has been enrolled in a Phase 2a trial of ORMD-0801, an orally ingestible insulin capsule, on patients with type 2 diabetes. The current trial is to be a randomized, double-blind study designed to assess the safety of ORMD-0801.
Read more at

In addition to ORMD-0801, Oramed is also developing an oral GLP-1 analog, known as exenatide, and a combination therapy of ORMD-0801 and exenatide.
GLP-1, or glucagon-like peptide-1, possesses a number of physiological properties that make it and its analogs the subject of intensive investigation as a potential treatment for diabetes. Among other things, it aids in the balance of blood sugar levels by decreasing glucose levels, especially after a meal; promotes weight loss; and does not cause hypoglycemia.
Let’s Set a Global Drug Quality Benchmark by Kiran M Shaw, Biocon

With Indian-made generics accounting for a US market share of over 25 per cent, it is not surprising that it is gaining significant mindshare of the Food and Drug Administration ( FDA). The spate of quality issues with leading Indian pharmaceutical companies in the past couple of years however should not be viewed in isolation. Big Pharma in the West, too, has been facing increasing flak from the FDA and other regulators over good manufacturing practice ( GMP) violations. High profile names like J& J, Genzyme (Sanofi), GSK, Sandoz, Watson, Teva and many others have encountered their share of quality problems and have been served with ‘warning letters’ from FDA
http://kiranmazumdarshaw.blogspot.in/2013/06/lets-set-global-drug-quality-benchmark.html
READ ALL AT THE LINK ABOVE

{kind=link}
{kind=link}