








Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
Home » Posts tagged 'JAPAN' (Page 2)
Tag Archives: JAPAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan

MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan
KENILWORTH, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) has approved MARIZEV® (omarigliptin) 25 mg and 12.5 mg tablets, an oral, once-weekly DPP-4 inhibitor indicated for the treatment of adults with type 2 diabetes. Japan is the first country to have approved omarigliptin……….http://www.mercknewsroom.com/news-release/prescription-medicine-news/marizev-omarigliptin-mercks-once-weekly-dpp-4-inhibitor-type
syn…….https://newdrugapprovals.org/2014/04/18/omarigliptin-mk-3102-in-phase-3-for-type-2-diabetes/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Linkedin
Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
/////////////MARIZEV, (Omarigliptin), Merck’s, Once-Weekly, DPP-4 Inhibitor, Type 2 Diabetes, Approved, Japan
TAK 272, For Hypertension, Takeda’s Next Sartan
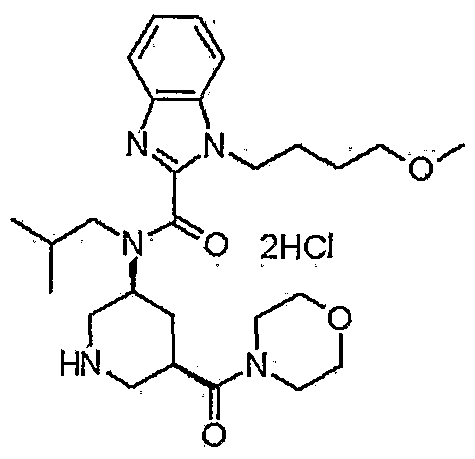
TAK 272
C27 H41 N5 O4 . Cl H, 536.106
CAS.1202269-24-6. MonoHCl
1202265-90-4 DIHCL
Base cas…1202265-63-1
Metanesulfonate…1202266-34-9
Takeda Pharmaceutical Company Limited, INNOVATOR
see……….http://www.allfordrugs.com/2015/10/21/tak-272-for-hypertension-takedas-next-sartan/
1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-ylcarbonyl)-piperidin-3-yl]-1H-benzimidazole-2-carboxamide
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
N-Isobutyl-1-(4-methoxybutyl)-N-[5(R)-(morpholin-4-ylcarbonyl)piperidin-3(S)-yl]-1H-benzimidazole-2-carboxamide hydrochloride
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidine-3 – yl] -1H- benzimidazole-2-carboxamide hydrochloride,
![]()
The compound is used as renin inhibitor for treating diabetic nephropathy and hypertension
Takeda’s TAK-272, was reported to be in phase II in October 2015), an oral renin inhibitor, for treating diabetic nephropathy and hypertension
- 01 Apr 2015Takeda completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 18 Feb 2015Takeda plans a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 13 Feb 2015Takeda plans a phase I pharmacokinetics trial in Renal or Hepatic impairment patients in Japan (NCT02367872)
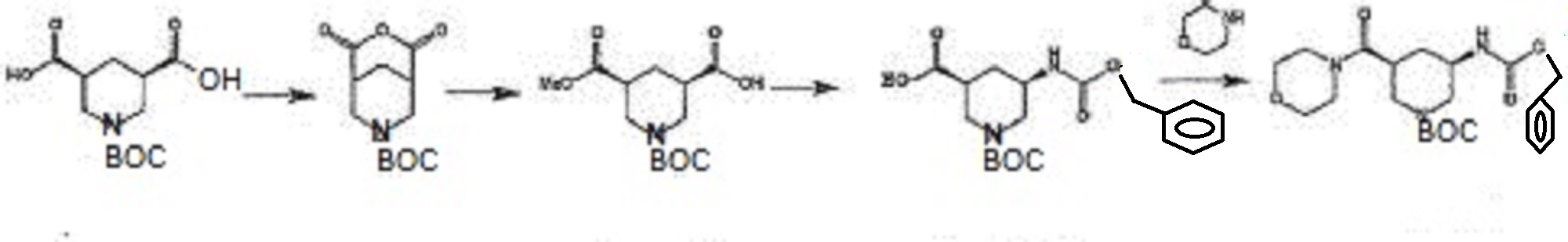
in Patent Document 1, a method for producing a synthetic intermediate of the above heterocyclic compound, the following methods are disclosed.

In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.
(Wherein each symbol is as described in Patent Document 2.)
TABLE In the above method, the acid anhydride of the formula (VI), in the presence of a chiral amine with the formula (VIIa) or (VIIb) is to produce a chiral dicarboxylic acid monoester compound, then reacted with an amine (R1-NH-R2) is subjected to amidation to, to produce a heterocyclic amide compound of the formula (VIII).
Patent literature
Patent Document 1: Patent No. 4,800,445 Patent
Patent Document 2: International Publication No. 2007/077005
Patent Document 2: International Publication No. 2007/077005
SYNTHESIS…click on image to get clear view
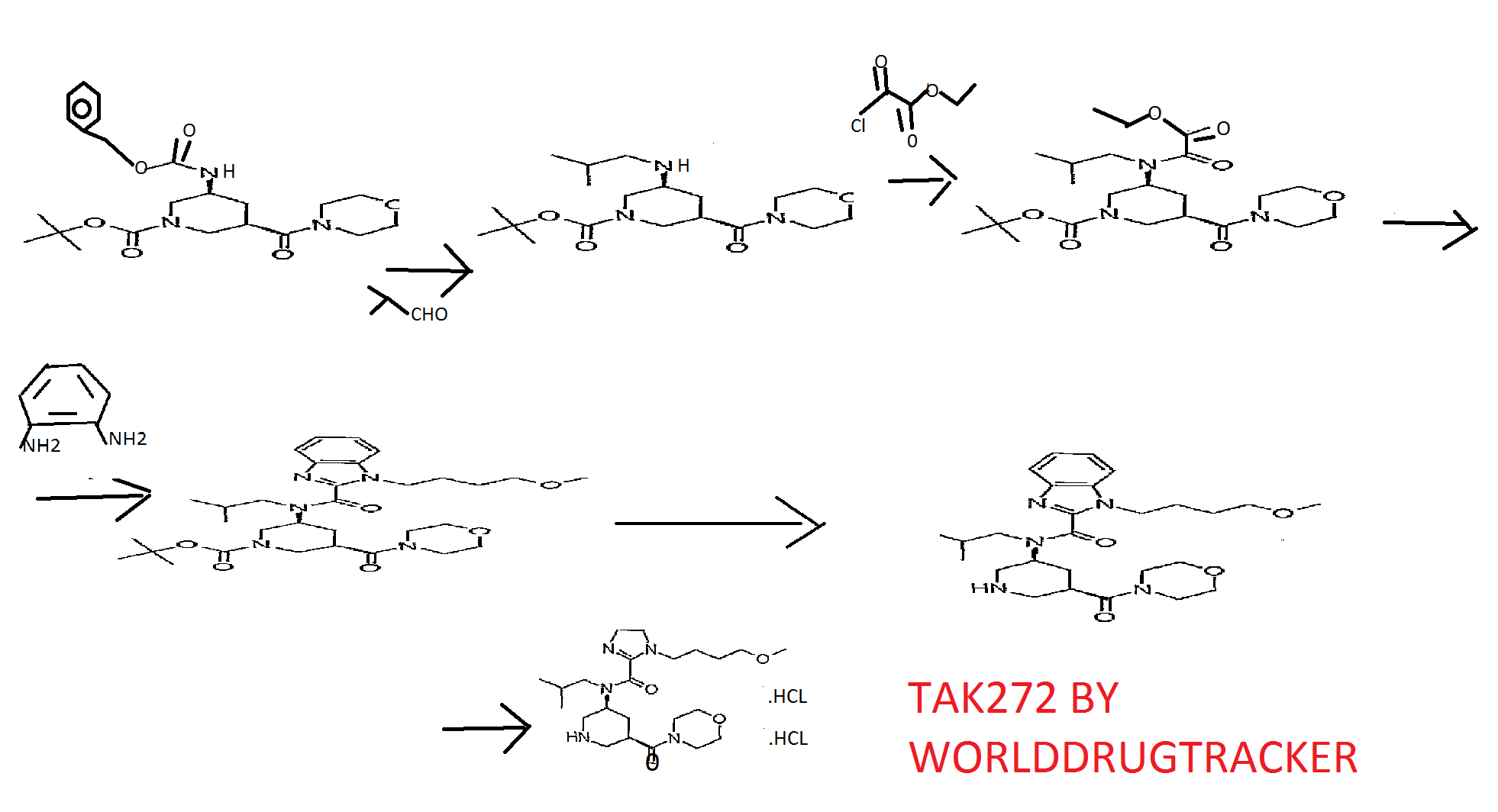
PATENT
WO2009154300
https://www.google.co.in/patents/WO2009154300A2?cl=en
INTERMEDIATES FOR CONSTRUCTION

USE THIS ONE




Reference Example 31 tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate and 1- (4-methoxybutyl) -N-
(2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4- ylcarbonyl)piperidin-3-yl]-lH-benzimidazole-2-carboxamide
tert-Butyl (3S, 5R) -3-{ [ ( {2- [ (4- methoxybutyl) amino] phenyl}amino) (oxo) acetyl] (2- methylpropyl) amino} -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.11 g) was dissolved in acetic acid (50 ml), and the mixture was stirred at 😯0C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give tert- butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl } (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (5.85 g) , and a fraction eluted with ethyl acetate-methanol (85:15) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2- methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin- 3-yl] -lH-benzimidazole-2-carboxamide (580 mg) . [0424] tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl ) piperidine-1-carboxylate 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3. 46 ( 6H, m) , 3.49-3. 91 (1OH, m) , 3. 95-4 . 47 (5H, m) , 7 . 18-7 . 51 (3H, m) , 7. 56-7 . 84 ( IH, m) .
MS (ESI+, m/e) 600 (M+l )
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl)piperidin-3-yl] -lH-benzimidazole-2-carboxamide BASE
1H-NMR (CDCl3) δ 0.64-0.74 (2H, m) , 0.95-1.07 (4H, m) , 1.43-
1.74 (3H, m) , 1.84-2.41 (4H, m) , 2.48-2.67 (IH, m) , 2.67-3.01
(3H, m), 3.03-3.44 (8H, m) , 3.47-3.78 (9H, m) , 4.06-4.46 (3H, m) , 7.28-7.47 (3H, m) , 7.62-7.81 (IH, m) . MS (ESI+, m/e) 500 (M+l)
Example 10
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-
4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
tert-Butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5-
(morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, and the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide (4.40 g) . The obtained 1- (4-methoxybutyl) -N- (2-methylpropyl) – N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -IH- benzimidazole-2-carboxamide (2.20 g) was dissolved in ethyl acetate (20 ml) , 4M hydrogen chloride-ethyl acetate (5 ml) and methanol (20 ml) were added, and the mixture was stirred at room temperature for 5 min. The reaction mixture was concentrated under reduced pressure to give the object product (2.52 g).
dihydrochloride
1H-NMR (DMSO-d6) δ 0.63-0.76 (2H, m) , 0.85-1.00 (4H, m) , 1.40-
1.60 (2H, m) , 1.68-1.89 (2H, m) , 1.93-2.17 (2H, m) , 2.20-2.44
(2H, m) , 2.81-3.81 (2OH, m) , 4.19-4.39 (3H, m) , 7.23-7.46 (2H, m) , 7.57-7.81 (2H, m) , 8.38-9.77 (2H, m) .
MS (ESI+, m/e) 500 (M+l)
Example 252
1- ( 4-methoxybutyl ) -N- ( 2-methylpropyl ) -N- [ ( 3S 1. 5R) -5- (morpholin- 4-ylcarbonyl ) piperidin-3-yl ] -lH-benzimidazole-2-carboxamide methanesulfonate

l-(4-Methoxybutyl) -N- (2-methylpropyl) -N- [ (3S,5R)-5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide (208 mg) was dissolved in ethyl acetate (2 ml) , a solution of methanesulfonic acid (40 μl) in ethyl acetate (1 ml) was added at 75°C, hexane (1 ml) was added, and the mixture was heated under reflux and stood at room temperature overnight. The precipitated crystals were collected by filtration, and dried at 7O0C for 3 hr to give the object product (158 mg) . MS (ESI+, m/e) 500 (M+l) melting point : 144.40C
EXTRAS IF REQD .………….
Example 32
methyl (3R, 5S)-5-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] piperidine-3-carboxylate dihydrochloride [0675]

MS (ESI+, m/e) 445 (M+l)
Example 33
(3R, 5S) -5- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yljcarbonyl} (2-methylpropyl) amino] piperidine-3-carboxylic acid dihydrochloride

MS (ESI+, m/e) 431 (M+l)
Reference Example 29
{ [ ( 3S , 5R) -1- (tert-butoxycarbonyl ) -5- (morpholin-4- ylcarbonyl ) piperidin-3~yl ] ( 2-itιethylpropyl ) amino } (oxo ) acetic acid
To a solution of tert-butyl (3S,5R)~3-{ [ethoxy (oxo) acetyl] (2-methylpropyl) amino}-5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate (10.3 g) in ethanol (40 ml) was added 2M aqueous sodium hydroxide solution (22 ml) , and the mixture was stirred at room temperature for 6 hr. The reaction mixture was adjusted to pH 7 with IM hydrochloric acid, and extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give the object product (10.3 g) .
1H-NMR (CDCl3) δ 0.78-0.99 (6H, m) , 1.37-1.52 (9H, m) , 1.79- 2.16 (3H, m) , 2.38-3.86 (14H, m) , 3.93-4.43 (2H, m) . MS (ESI+, m/e) 442 (M+l)
Reference Example 28
tert-butyl (3S, 5R) -3-{ [ethoxy (oxo) acetyl] (2- methylpropyl ) amino } -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate
To a solution of tert-butyl (3S, 5R) -3- [ (2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.24 g) and diisopropylethylamine (10.5 ml) in DMA (100 ml) was added dropwise ethyl chloroglyoxylate (3.4 ml) at 0°C. The reaction mixture was stirred at room temperature for 15 hr, and the reaction mixture was concentrated. An aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (10.3 g) . 1H-NMR (CDCl3) δ 0.84-1.00 (6H, m) , 1.37 (3H, q) , 1.42-1.53 (9H, m) , 1.80-2.19 (3H, m) , 2.26-2.42 (IH, m) , 2.59-2.96 (IH, in) , 2.97-3.30 (3H, m) , 3.37-3.92 (9H, m) , 4.01-4.26 (2H, m) , 4.26- 4.40 (2H, m) . MS (ESI4-, m/e) 470 (M+l) “
Reference Example 22 tert-butyl (3S, 5R) -3- [ (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate
[0369] tert-Butyl (3S,5R)-3-{ [ (benzyloxy) carbonyl] aminoJ-5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (58 g) and palladium (II) hydroxide-carbon (5 g) were suspended in methanol (400 ml) and the mixture was stirred under a hydrogen atmosphere (1 atom) at room temperature for 16 hr. The palladium catalyst was filtered off, and the filtrate was concentrated under reduced pressure. The obtained residue and acetic acid (8.8 ml) were dissolved in methanol (400 ml), 2- methylpropanal (14.0 ml) was added, and the mixture was stirred at room temperature for 1 hr. Sodium triacetoxyborohydride (40.4 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the concentrate was basified with 3.5M aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:5) – ethyl acetate-hexane (1:1) was concentrated under reduced pressure to give the object product (33.3 g) .
1H-NMR (CDCl3) δ: 0.90 (6H, d) , 1.46 (9H, s) , 1.54 (IH, d) , 1.69 (IH, dt), 1.96-2.12 (2H, m) , 2.23-2.37 (IH, m) , 2.47 (3H, d) , 2.66 (IH, d) , 3.61 (IH, br s) , 3.55 (2H, d) , 3.69 (5H, ddd) , 4.01-4.46 (2H, m) .
Example 6 1-tert-butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate [0318]

(3S, 5R) -1- (tert-Butoxycarbonyl) -5-(methoxycarbonyl)piperidine-3-carboxylic acid (2.83 g) was suspended in toluene (36 ml), diphenylphosphoryl azide (2.60 ml) and triethylamine (1.70 ml) were added, and the mixture was stirred at 100°C for 1 hr. The reaction mixture was cooled to room temperature, benzyl alcohol (1.53 ml) and triethylamine (7.00 ml) were added and the mixture was stirred at 80°C for 3 hr. The reaction mixture was concentrated, the residue was dissolved in ethyl acetate, and the solution was washed with water, 0.5M hydrochloric acid, saturated aqueous sodium hydrogen carbonate and saturated brine in this order, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:3 – 3:1) was concentrated under reduced pressure. The obtained residue was dissolved in methanol (60 ml), 10% palladium carbon (50% in water) (150 mg) was added and the mixture was stirred under a hydrogen pressurization (5 atom) at ambient temperature and normal pressure for 5 hr. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the object product (1.83 g) as an oil.
1H-NMR (CDCl3) δ 1.22-1.43 (4H, m) , 1.46 (9H, s), 2.27-2.79 (4H, m) , 3.70 (3H, s) , 4.13 (2H, br s) [0320] In the same manner as in the method shown in Reference Example 6, the following compound (Reference Example 7) was obtained.
Reference Example 8
1-tert-butyl 3-methyl (3R, 5S) -5- [ (2- methylpropyl) amino] piperidine-1, 3-dicarboxylate [0325]

1-tert-Butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate (1.83 g) , isobutyraldehyde (0.78 ml) and acetic acid (0.49 ml) were dissolved in methanol (50 ml), and the mixture was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (3.80 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 7 hr. The reaction mixture was concentrated under reduced pressure, the concentrate was basified with aqueous sodium bicarbonate, and extracted with ethyl acetate. The extract was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:1) – ethyl acetate 100% – ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (1.42 g) as an oil.
1H-NMR (CDCl3) δ 0.90 (6H, d) , 1.22-1.38 (3H, m) , 1.46 (9H, s) , 1.69 (IH, dt), 2.23-2.39 (2H, m) , 2.44-2.59 (IH, m) , 2.47 (2H, d) , 2.74 (IH, br s) , 3.69 (3H, s) , 4.18-4.34 (2H, m)
Reference Example 27
N- (4-methoxybutyl) benzene-1, 2-diamine

To a solution of phenylenediamine (10.8 g) and 4- methoxybutyl methanesulfonate (9.11 g) in acetonitrile (100 ml) was added potassium carbonate (20.7 g) , and the mixture was stirred heated under reflux for 15 hr. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (35:65) was concentrated under reduced pressure to give the object product (5.44 g) . 1H-NMR (CDCl3) δ 1.67-1.82 (4H, m) , 3.13 (2H, t) , 3.24-3.39 (6H, m) , 3 . 38 -3 . 50 ( 2H, m) , 6 . 62 – 6 . 74 ( 3H, m) , 6 . 81 ( IH, in) . MS ( ESI+ , m/e ) 195 (M+l )
Reference Example 146 tert-butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl]carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate

A solution of tert-butyl (3S, 5R) -3- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (200 mg) , 4-itιethoxybutyl methanesulfonate (107 mg) and cesium carbonate (254 mg) in N,N-dimethylacetamide (5 ml) was stirred at 60°C for 15 hr. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (10 ml*2) . The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (5:95 – 3:7) was concentrated under reduced pressure to give the object product (190 mg) . 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3.46 (6H, m) , 3.49-3.91 (1OH, m) , 3.95-4.47 (5H, m) , 7.18-7.51 (3H, m) , 7.56-7.84 (IH, m) . MS (ESI+, m/e) 600 (M+l)
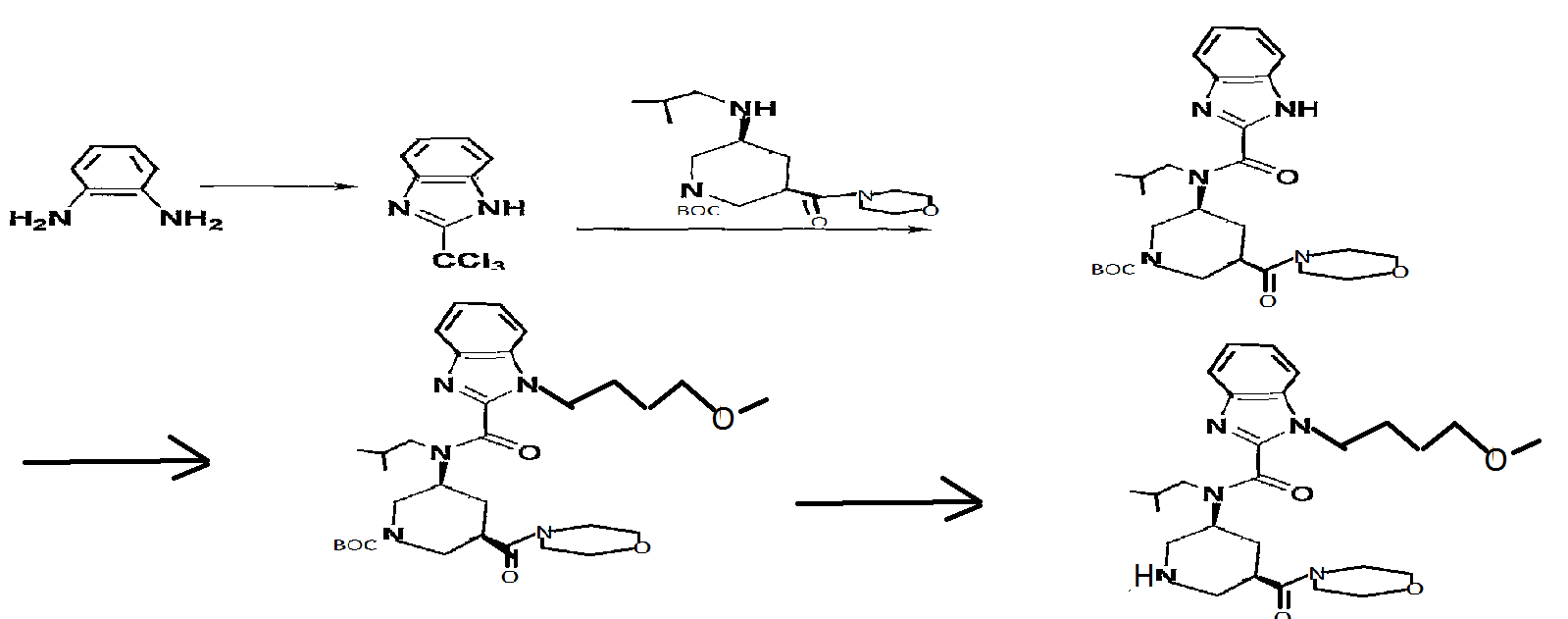
ALTERNATE METHOD IN THIS PATENT



Reference Example 61
2- (trichloromethyl) -lH-benzimidazole

O-Phenylenediamine (25 g) was dissolved in acetic acid (750 ml), and methyl 2, 2, 2-trichloroacetimidate (28.5 ml) was added dropwise over 15 min. After stirring at room temperature for 1 hr, the reaction mixture was concentrated to about 150 ml, and poured into water (1500 ml) . The precipitated crystals were collected by filtration, washed with water (1000 ml) and suspended in toluene (500 ml) . The solvent was evaporated under reduced pressure. The residue was again suspended in toluene (500 ml) and the solvent was evaporated under reduced pressure. The residue was dried under reduced pressure to give the object product (51.8 g) . 1H-NMR (CDCl3) δ 7.31-7.45 (2H, m) , 7.49-7.55 (IH, m) , 7.89 (IH, d) , 9 . 74 ( IH, br s )
Reference Example 64
1-tert-butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate

2- (Trichloromethyl) -lH-benzimidazole (19 g) and 1-tert- butyl 3-methyl (3R, 5S) -5- [ (2-methylpropyl) amino] piperidine- 1,3-dicarboxylate (25 g) were dissolved in THF (1200 ml), sodium hydrogen carbonate (67 g) and water (600 ml) were added, and the mixture was stirred at room temperature for 1 hr and at 5O0C for 1 hr. After evaporation of the solvent, the residue was extracted 3 times with ethyl acetate (700 ml) . The extract was washed successively with 10%-aqueous citric acid solution (500 ml) and brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure.
The residue was dissolved in ethyl acetate (1000 ml), subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (30.6 g) .
1H-NMR (CDCl3) δ 0.78-1.09 (6 H, m) , 1.17-1.55 (9 H, m) , 1.77-2.95 (5 H, m) , 3.11-3.79 (6 H, m) , 3.99-4.73 (4 H, m) , 7.24- 7.41 (2 H, m) , 7.45-7.59 (1 H, m) , 7.72-7.88 (1 H, m) , 10.66-10.98 (1 H, m)MS (ESI+, m/e) 459 (M+l)
Reference Example 69
1-tert-butyl 3-methyl (3R, 5S) -5- [ { [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] piperidine-1 , 3-dicarboxylate

1-tert-Butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate (30 g) and 4-methoxybutyl methanesulfonate (12.5 g) were dissolved in DMA (600 ml), cesium carbonate (32 g) was added, and the mixture was stirred at 70°C for 12 hr. The reaction mixture was poured into ice water (1000 ml), and the mixture was extracted twice with ethyl acetate (1000 ml) . The extract was washed with brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:4 – 1:1) was concentrated under reduced pressure to give the object product (28.7 g) .
1H-NMR (CDCl3) δ 0.76 (4H, d) , 1.01 (2H, d) , 1.30-1.52 (9H, m) , 1.58-2.07 (4H, m) , 2.10-2.93 (4H, m) , 3.27-3.75 (12H, m) , 4.06-4.57 (5H, m) , 7.26-7.48 (3H, m) , 7.79 (IH, d) MS (ESI+, m/e) 545 (M+l)
Example 71
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide

tert-Butyl (3S, 5R) -3- [{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, the residue was diluted with aqueous sodium bicarbonate,…and, the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (4.40 g) . MS (ESI+, m/e) 500 (M+l)
Example 101
1- (5-methoxypentyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide dihydrochloride

[1144] tert-Butyl (3S, 5R) -3- [ { [1- (5-methoxypentyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (123 mg) was dissolved in 4M hydrogen chloride-ethyl acetate (5 ml) , and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated, and the residue was subjected to reversed-phase preparative HPLC and the eluted fraction was concentrated under reduced pressure. The residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. 4M Hydrogen chloride-ethyl acetate (1 ml) was added and the mixture was stirred for 5 min. The solvent was evaporated under reduced pressure to give the object product (76 mg) . MS (ESI+, m/e) 514 (M+l)
PATENT
WO2013122260
http://www.google.co.in/patents/WO2013122260A1?cl=en
PATENT
WO 2011158880
http://www.google.co.in/patents/WO2011158880A1?cl=en
Reference Example 1
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -1H- benzimidazole -2 – carboxamide hydrochloride (A-type crystal)
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) was suspended dissolved piperidine-1-carboxylate The (300g) in 3N- hydrochloric acid water (1200mL) and Ethyl acetate (60mL), and stirred over 3 h at 25 ~ 35 ℃. After completion of the reaction, it was added ethyl acetate (2400mL) in the same temperature. After the addition, it was added 25% aqueous ammonia (600mL) with cooling. After the addition stirring and extracted the organic layer of 5% aqueous ammonia (600mL) was added and stirred. After stirring, the resulting organic layer it was concentrated until the solvent no longer distilled off. After concentrated, dissolved with ethyl acetate (1500mL), and transferred to solution to the crystallizer vessel, and washed with ethyl acetate (750mL). After washing, it was raised in stirring under 45 ~ 55 ℃. After raising the temperature, at the same temperature 4N- hydrogen chloride – it was dropped ethyl acetate (131.3mL). After dropping, it was to dissolve the precipitate at the same temperature. After dissolution confirmation, it was added heptane (750mL) at 40 ~ 50 ℃, after the addition, then cooled to 25 ~ 35 ℃. After cooling, the addition of A-type crystals of the seed crystals (300mg) which was obtained according to the method described in Example 265 of WO2009 / 154300, and stirred for 30 minutes or more. After stirring, the temperature was raised to 40 ~ 45 ℃, it was dropped heptane (1500mL). After the completion of the dropping, it was stirred at the same temperature. Then gradually cooled to 5 ℃ below, followed by stirring at the same temperature for 1 hour. After stirring, ethyl acetate and filtered crystals – heptane: washed with (1 1,600mL), to obtain a wet crystal. The obtained wet crystals dried under reduced pressure at 50 ℃, 1- (4- methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-yl carbonyl) piperidin-3-yl] -1H- obtained a crystalline powder of benzimidazole-2-carboxamide hydrochloride (A-type crystal, 198.82g, 74.1% yield). FINAL PRODUCT
TERT BUTYL DERIVATIVE, N-1
Reference Example 4
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate 1)
o- nitro aniline (50.0g, 0.362mol), tetrabutylammonium bromide (58.3g, 0.181mol), potassium bromide (43.1g, 0.362mol) in toluene (500mL ) and it was added. At a temperature of 20 ~ 30 ℃ 1- chloro-4-methoxy-butane (66.6g, 0.543mol) and, I was added to 50w / v% sodium hydroxide solution (145mL, 1.81mol). The reaction was heated to a temperature 85 ~ 95 ℃, and stirred for 6 hours. After cooling to a temperature 20 ~ 30 ℃, the reaction mixture water (250mL), 1N- aqueous hydrochloric acid (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), it was washed successively with water (250mL). After concentration under reduced pressure the organic layer to Contents (250mL), was added toluene (100mL), was obtained
N- (4- methoxy-butyl) -2-nitroaniline in toluene (350mL, 100% yield).
1 H-NMR (300MHz, CDCl 3) δ 1.64-1.89 (m, 4H), 3.25-3.39 (m, 2H), 3.35 (s, 3H), 3.44 (t, J = 6.1 Hz, 2H), 6.63 ( ddd, J = 8.5, 6.9, 1.2 Hz, 1H), 6.86 (dd, J = 8.5, 1.2 Hz, 1H), 7.43 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 8.07 (br s, 1H ), 8.17 (dd, J = 8.5, 1.5 Hz, 1H).
2) N- (4-methoxy-butyl) -2-10 percent in nitroaniline of toluene solution (350mL) Pd / C (K-type, 50% water-containing product) (10.0g) and toluene (100mL) it was added. Hydrogen pressure of 0.1MPa, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. A stream of nitrogen, the catalyst was filtered, I was washed with toluene (100mL). After the water in the filtrate was separated off and adding magnesium sulfate (25.0g) at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered over magnesium sulfate, washed with toluene (100mL), was obtained N- (4- methoxybutyl) -o- toluene solution of phenylenediamine (100% yield).
1 H NMR (500 MHz, CDCl 3) δ1.67-1.78 (m, 4H), 3.12-3.14 (m, 2H), 3.32 (br, 3H), 3.35 (s, 3H), 3.41-3.47 (m, 2H), 6.63-6.69 (m, 2H), 6.69-6.74 (m, 1H), 6.82 (td, J = 7.57, 1.58 Hz, 1H).
3) N- (4- methoxy-butyl) -o- After the toluene solution of phenylenediamine cooled to a temperature 0 ~ 10 ℃, acetic acid (65.2g, 1.09mol) and 2,2,2 trichloroacetimide acid methyl ( 70.3g, 0.398mol) and I were added. After stirring for 30 minutes at a temperature 0 ~ 10 ℃, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. The reaction was 5w / v% saline (250mL), 2N- aqueous hydrochloric acid / 5w / v% sodium chloride solution: a mixture of (1 1) (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), 5w / v It was washed successively with% saline solution (250mL). A stream of nitrogen, was added magnesium sulfate (25.0g) to the organic layer at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered magnesium sulfate, and washed with toluene (100mL). The filtrate was concentrated under reduced pressure and the amount of contents (150mL). Stir the concentrated solution at a temperature 20 ~ 30 ℃, was allowed to precipitate crystals, was added dropwise heptane (750mL). The crystals bleeding is heated to a temperature 40 ~ 50 ℃, after stirring for 30 min, cooled to a temperature 0 ~ 10 ℃, and the mixture was stirred at the same temperature for 2 hours.The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,150 mL). And dried under reduced pressure at 40 ℃, it was obtained 1- (4-methoxy-butyl) -2-fine brown crystals of trichloromethyl -1H- benzimidazole (96.5g, 82.9% yield from o- nitroaniline).
1 H-NMR (300MHz, CDCl 3) δ: 1.68-1.85 (m, 2H), 1.99-2.17 (m, 2H), 3.37 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 4.50 -4.65 (m, 2H), 7.27-7.49 (m, 4H), 7.82-7.93 (m, 1H).
. Anal Calcd for C 13 H 15 Cl 3 N 2 O:. C, 48.55; H, 4.70; N, 8.71; Cl, 33.07 Found: C, 48.30; H, 4.61; N, 8.74; Cl, 33.30.
4) pyridine-3,5-dicarboxylic acid (110g, 0.66mol), it was dropped methanol (660 mL) mixture of concentrated sulfuric acid at a temperature of 50 ℃ or less of (226.0g, 2.30mol). Thereafter, the mixture was stirred and heated to a temperature 55 ~ 65 ℃ 7 hours. The reaction was the temperature 40 ~ 50 ℃, was added water (220mL). And further dropping temperature 40-50 5% aqueous ammonia at ℃ (about 1.10L) was adjusted to pH8.0 ~ 8.5. After stirring at a temperature 40 ~ 50 ℃ 30 minutes and stirred for 1 hour and cooled to a temperature 0 ~ 10 ℃. Was collected by filtration precipitated crystals, methanol – water (1: 3,165mL), and washed successively with water (440mL). To obtain a white crystalline powder pyridine-3,5-dicarboxylic acid dimethyl and dried under reduced pressure at 50 ℃ (105.0g, 82.0% yield).
1 H-NMR (300 MHz, CDCl 3) δ 4.00 (s, 6H), 8.87 (s, 1H), 9.37 (s, 2H).
. Anal Calcd for C 9 H 9 NO 4:. C, 55.39; H, 4.65; N, 7.18; O, 32.79 Found: C, 55.42; H, 4.65; N, 7.16.
5) 1 L autoclave pyridine-3,5-dicarboxylic acid dimethyl (100g, 0.51mol) and was charged with dimethylacetamide (400mL), temperature 30 ℃ below with trifluoroacetic acid (59.2mL, after dropping the 0.77mol), 10% Pd-C (PE-type) the (20.0g) it was added. Hydrogen pressure of 0.5 ~ 0.7MPa, it was stirred for 12 hours at a temperature of 55 ~ 65 ℃. The catalyst was filtered off, it was washed with dimethylacetamide (50mL × 2). Triethylamine and the combined filtrates at a temperature 20 ~ 30 ℃ (77.8g, 0.77mol) was added dropwise, and adjusted to pH9.0 ~ 10.0. Temperature 30 ~ 40 ℃ by di -tert- butyl (134g, 0.614mol) was added dropwise and stirred at the same temperature for 2 hours. After the reaction mixture as a 20 ~ 30 ℃, it was added ethyl acetate (600mL), washed with water (900mL). The aqueous layer it was re-extracted with ethyl acetate (400mL). The combined organic layers 5w / v% citric acid -10w / v% sodium chloride solution (600mL), 3% aqueous sodium bicarbonate (600mL), and washed successively with water (600mL). Contents The organic layer (200mL) until it was concentrated under reduced pressure, methanol (250mL) was added to the concentrated solution, and then concentrated under reduced pressure until Contents (200mL). The addition of methanol (250mL) again concentrate, After concentration under reduced pressure until Contents (200mL), was added methanol (2.40L). The solution in water (18.5g, 1.03mol), cesium carbonate (417g, 1.28mol) was added and stirred for about 24 hours at a temperature 55 ~ 65 ℃. The reaction solution was the temperature 20 ~ 30 ℃, concentrated to Contents (700mL), it was added tetrahydrofuran (500mL). The solution temperature at 15 ~ 35 ℃ 2N- hydrochloric acid solution (1.28L, 2.56mol) was added dropwise and adjusted to pH3.0 ~ 3.5, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Extracted with ethyl acetate (750mL × 2), and the organic layer was washed with 10w / v% aqueous sodium chloride solution (500mL × 3). Contents The organic layer (300mL) until it was concentrated under reduced pressure, to obtain a weight content by adding ethyl acetate (650mL).Heating the concentrate to a temperature of 55 ~ 65 ℃, it was added dropwise heptane (500mL). It cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. The precipitated crystals were collected by filtration, ethyl acetate – heptane: was washed with (1 1,120mL). Dried under reduced pressure at 50 ℃ 1- (tert- butoxycarbonyl) to give a white crystalline powder of piperidine-3,5-dicarboxylic acid (113.3g, 80.9% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.40 (s, 9H), 1.44-1.61 (m, 1H), 2.21-2.26 (m, 1H), 2.31-2.41 (m, 2H), 4.10- 4.12 (m, 2H).
. Anal Calcd for C 12 H 19 NO 6:. C, 52.74; H, 7.01; N, 5.13; O, 35.13 Found: C, 52.96; H, 6.99; N, 5.39.
6) Under a nitrogen stream, 1- (tert- butoxycarbonyl) piperidine-3,5-dicarboxylic acid (5.00g, 18.3mmol) was suspended in tetrahydrofuran (10.0mL), trifluoroacetic acid anhydride at a temperature 20 ~ 30 ℃ It was dropping things (3.80mL, 27.5mmol). After the completion of the dropping, it was stirred for 1 hour at a temperature of 20 ~ 30 ℃. It was added dropwise heptane (20.0mL) at a temperature 20 ~ 30 ℃ the reaction solution, and stirred for 3 hours then cooled to a temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with heptane (3.00mL). Dried under reduced pressure at 40 ℃ 2,4- dioxo-3-oxa-7-azabicyclo [3,3,1] white crystalline powder of nonane-7-carboxylic acid tert- butyl was obtained (4.03g, yield 86.1%).
1 H-NMR (300 MHz, CDCl 3) δ 1.43 (s, 9H), 1.93-1.99 (m, 1H), 2.40-2.46 (m, 1H), 3.06-3.11 (m, 4H), 4.50-4.54 ( m, 2H).
. Anal Calcd for C 12 H 17 NO 5:. C, 56.46; H, 6.71; N, 5.49; O, 31.34 Found: C, 56.51; H, 6.63; N, 5.69.
7) Under a nitrogen stream, quinidine (69.9g, 0.215mol) and was charged with tetrahydrofuran (200mL), and cooled to a temperature -5 ~ 5 ℃. At the same temperature 2,4-dioxo-3-oxa-7-azabicyclo [3,3,1] nonane-7-carboxylic acid tert- butyl (50.0g, 0.196mol) was added and washed with tetrahydrofuran (50.0mL) crowded. Temperature -5 ~ 5 methanol at ℃ (9.41g, 0.29 4mol) was added dropwise, and the mixture was stirred for 2 hours at a temperature -5 ~ 5 ℃. Ethyl acetate (350mL) to the reaction mixture, was by adding minute solution 20w / v% citric acid aqueous solution (250mL). The aqueous layer it was re-extracted with ethyl acetate (125mL × 2). The organic layers were combined 20w / v% aqueous solution of citric acid (250mL), I was washed successively with water (250mL × 2). The organic layer it was concentrated under reduced pressure. To the residue ethanol (100mL) was added ethyl acetate (450mL) was heated to a temperature 60 ~ 70 ℃, (R) – was added phenethylamine (23.7g, 0.196mol). Temperature 50-60 for one hour at ℃, 1 hour at a temperature of 20 ~ 30 ℃, it was stirred for 1 hour at a temperature of -5 ~ 5 ℃. The precipitated crystals were collected by filtration, ethanol – ethyl acetate: and washed with (2 9,100mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidin-3 to give a white crystalline powder of the carboxylic acid (1R) -1- phenylethylamine salt It was (55.7g, 69.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.42 (s, 9H), 1.43-1.51 (m, 3H), 2.06-2.14 (m, 1H), 2.21-2.26 (m, 1H), 2.39- 2.44 (m, 1H), 2.52-2.53 (m, 1H), 2.57 (br s, 2H), 3.64 (s, 3H), 4.12 (br s, 2H), 4.19-4.26 (m, 1H), 7.30- 7.40 (m, 3H), 7.45-7.48 (m, 2H).
. Anal Calcd for C 21 H 32 N 2 O 6:. C, 61.75; H, 7.90; N, 6.86; O, 23.50 Found: C, 61.54; H, 7.77; N, 6.86.
8) (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (1R) -1- phenylethylamine salt (20.0g, 49.0mmol), methanol (20mL) and it was charged with water (80mL). Temperature 20-30 citric acid at ℃ (11.3g, 58.8mmol) was added dropwise a solution prepared by dissolving in water (20.0mL), and the mixture was stirred 1.5 hours at the same temperature. The precipitated crystals were collected by filtration and washed with water (60mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- give a white crystalline powder (methoxycarbonyl) piperidine-3-carboxylic acid (13.5g, 96.1% yield ).
1 H-NMR (300 MHz, CDCl 3) δ 1.40 (s, 9H), 1.46-1.59 (m, 1H), 2.22-2.27 (m, 1H), 2.37-2.45 (m, 2H), 2.63-2.73 ( m, 2H), 3.63 (s, 3H), 4.14 (br s, 2H), 12.51 (br s, 1H).
. Anal Calcd for C 13 H 21 NO 6:. C, 54.35; H, 7.37; N, 4.88; O, 33.41 Found: C, 54.14; H, 7.28; N, 4.85.
9) Under a nitrogen stream, (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 104mmol), triethylamine (31.7g, 313mmol) and toluene ( It was charged with 180mL). Diphenylphosphorylazide at a temperature of 15 ~ 35 ℃ (28.7g, 313mmol) I was dropped a toluene (30.0mL) solution. After stirring at a temperature 30 ± 5 ℃ 30 minutes, and the mixture was stirred and heated to a temperature 65 ~ 75 ℃ 30 minutes. Temperature 60 ~ 70 ℃ in the benzyl alcohol (12.4g, 115mmol) it was dropped. To a temperature 80 ~ 90 ℃ was stirred and heated for 3 hours. The reaction mixture was cooled to a temperature 20 ~ 30 ℃, sodium nitrite (7.20g, 104mmol) and after stirring was added a solution prepared by dissolving in water (150mL) 1 hour, the aqueous layer was separated. The organic layer 5w / v% aqueous sodium bicarbonate solution (150mL), 20w / v% aqueous citric acid solution (150mL), washed successively with 5w / v% aqueous sodium chloride solution (150mL), the organic layer was concentrated under reduced pressure. The residue methanol (60.0mL) was added and concentrated under reduced pressure to. The more we went once in the same manner.To the residue was added methanol and the content amount of the (90.0g). Temperature 15 ~ 35 ℃ 2N- aqueous sodium hydroxide (62.6mL, 125mmol) was added and stirred for 1 hour at a temperature 30 ± 5 ℃. Temperature 20 ~ 30 ℃ in methanol (120mL), was added to 20w / v% aqueous citric acid solution (300mL), it was a pH3.0 ~ 3.5. After stirring for 30 minutes at a temperature 50 ~ 60 ℃, cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. It was stirred for 1 hour at the temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with water (90.0mL). And dried under reduced pressure at 50 ℃ (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) to yield a white crystalline powder piperidine-3-carboxylic acid (35.0 g, 88.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 2.11 (d, J = 12.4 Hz, 1H), 2.40-2.48 (m, 4H), 2.62 (br s, 1H), 4.08 (t, J = 14.4 Hz, 2H), 5.04 (s, 2H), 7.31-7.41 (m, 5H), 12.53 (br s, 1H).
. Anal Calcd for C 19 H 26 N 2 O 6:. C, 60.30; H, 6.93; N, 7.40; O, 25.37 Found: C, 60.03; H, 6.99; N, 7.41.
10) Under a nitrogen stream, (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 79.3mmol), morpholine (7.60 g, 87.2mmol), 1- hydroxybenzotriazole monohydrate (2.43g, it was charged with 15.9mmol) and dimethylacetamide (90.0mL). Hydrochloride 1-ethyl at a temperature 20 ~ 30 ℃ -3- (3- dimethylaminopropyl) carbodiimide (16.7g, 87.1mmol) after addition and stirred for 1 hour at a temperature 45 ~ 55 ℃. Temperature 45 ~ 55 ℃ with tetrahydrofuran (90.0mL), sequentially dropwise addition of water (210mL), and stirred for 1 hour. After stirring for 1 hour and cooled to a temperature 20 ~ 30 ℃, were collected by filtration the precipitated crystals, tetrahydrofuran – water: washing with (1 3,120mL). And dried under reduced pressure at 50 ℃ tert- butyl piperidine -1- (3S, 5R) -3 – a white crystalline powder of {[(benzyloxy) carbonyl] amino} -5 (morpholin-4-yl-carbonyl) carboxylate It was obtained (32.7g, 92.3% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 1.49-1.57 (m, 1H), 1.87 (d, J = 12.3 Hz, 1H), 2.43 (br s, 1H), 2.63-2.71 (m, 1H), 2.79-2.83 (m, 1H), 3.37-3.54 (m, 9H), 3.89 (d, J = 11.5 Hz, 1H), 4.06 (br s, 1H), 5.03 (s , 2H), 7.30-7.38 (m, 5H).
. Anal Calcd for C 23 H 33 N 3 O 6:. C, 61.73; H, 7.43; N, 9.39; O, 21.45 Found: C, 61.59; H, 7.50; N, 9.43.
11) tert- Butyl piperidin -1- (3S, 5R) -3 – {[(benzyloxy) carbonyl] amino} -5- (morpholin-4-ylcarbonyl) carboxylate (30.0g, 67.0mmol), isobutyraldehyde (7.25g, 101mmol), it was charged with 10% Pd-C (PE type) (1.50g) and methanol (240mL).Hydrogen pressure of 0.2 ~ 0.3MPa, it was stirred for 4 hours at a temperature of 20 ~ 30 ℃. The catalyst is filtered off and washed with methanol (60.0mL). The filtrate was concentrated under reduced pressure, ethyl acetate was added (60.0mL), and concentrated under reduced pressure again. The residue ethyl acetate was added, followed by the amount of contents (360mL). Temperature 45-55 succinate by heating to ℃ (7.90g, 67.0mmol) was added. After stirring for 1 hour at a temperature 45 ~ 55 ℃, cooled to a temperature 20 ~ 30 ℃, and stirred for 1 hour. The precipitated crystals were collected by filtration, and washed with ethyl acetate (90.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [(2- methyl-propyl) amino] -5- (morpholin-4-yl-carbonyl) piperidine – 1-carboxylate white crystals of alert succinate got sex powder (30.2g, 92.5% yield).
1 H-NMR (300 MHz, D 2 O) δ 1.02 (s, 3H), 1.04 (s, 3H), 1.47 (s, 9H), 1.97-2.09 (m, 2H), 2.26-2.30 (m, 1H ), 2.55 (s, 4H), 2.99 (d, J = 7.0 Hz, 2H), 3.23 (br s, 1H), 3.39-3.45 (m, 2H), 3.53-3.80 (m, 10H), 3.82-3.93 (br s, 1H).
. Anal Calcd for C 23 H 41 N 3 O 8:. C, 56.66; H, 8.48; N, 8.62; O, 26.25 Found: C, 56.48; H, 8.46; N, 8.39.
12) tert- Butyl (3S, 5R) -3 – [(2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine – 1 – carboxylate succinate (30.3g, 62.2mmol), acetonitrile (60.0mL) and, it was charged with water (40.0mL). Then after stirring was added potassium carbonate (34.4g, 0.249mmol) 10 minutes, 1- (4-methoxybutyl) -2-trichloromethyl -1H- benzimidazole (20.0g, 62.2mmol) was added. After stirring for 2 hours at a temperature of 70 ~ 80 ℃, it was added dimethyl sulfoxide (15.0mL), and the mixture was stirred for 6 hours at a temperature 70 ~ 80 ℃. After cooling the reaction mixture to a temperature 20 ~ 30 ℃, water (120mL), it was separated and by adding toluene (240mL). The organic layer 10w / v% sodium chloride solution (100mL), 10w / v% aqueous solution of citric acid (100mL), it was washed sequentially with 10w / v% sodium chloride solution (100mL). The organic layer of activated carbon Shirasagi A a (1.0g) was added, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Activated carbon was filtered, washed with toluene (40.0mL), and concentrated under reduced pressure of the filtrate to 110 mL. By heating to a temperature 35 ~ 45 ℃ was added dropwise heptane (280mL). At a temperature 35 ~ 45 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5 – and the mixture was stirred for 1 hour at (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was added to the same temperature the crystals (10mg) of the acrylate. Heptane (140mL) was stirred and added dropwise to 30 minutes at a temperature 35 ~ 45 ℃. It was cooled to a temperature 20 ~ 30 ℃ and stirred for 2 hours. The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,40.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] – 5- (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was obtained a pale yellowish crystalline powder of alert (27.7g, 74.2% yield).
1 H-NMR (300 MHz, CDCl 3) δ 0.68-0.80 (m, 3H), 0.96-1.08 (m, 3H), 1.31 (br s, 5H), 1.49 (s, 4H), 1.61-1.71 (m , 2H), 1.71 (br s, 0.5H), 1.92-2.05 (m, 3H), 2.05-2.24 (m, 2H), 2.45 (br s, 1H), 2.60 (br s, 1H), 2.72-2.96 (m, 2H), 3.26-3.35 (m, 3H), 3.35-3.47 (m, 2H), 3.47-3.73 (m, 10H), 4.02-4.26 (m, 2H), 4.26-4.34 (m, 1H) , 4.34-4.47 (m, 0.5H), 7.25-7.29 (m, 1H), 7.29-7.41 (m, 1H), 7.41-7.53 (m, 1H), 7.64 (br s, 0.5H), 7.79 (d , J = 8.2 Hz, 0.5H).
. Anal Calcd for C 32 H 49 N 5 O 6:. C, 64.08; H, 8.23; N, 11.68; O, 16.01 Found: C, 63.82; H, 8.12; N, 11.64.
PATENT
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
Provided is a method for producing a synthetic intermediate of a heterocyclic compound having a renin inhibitory activity and effective as a prophylactic or therapeutic drug against diabetic renal disease, hypertension, and the like. A method for producing a compound represented by formula (III-1a), (III-1b), (III-1c), and/or (III-1d) [where the symbols in the formulas are as defined in the description], or a salt thereof, said method characterized in that a compound represented by formula (Ia) or (Ib) [where the symbols in the formulas are as defined in the description] or a salt thereof is reacted with a compound represented by formula (II) [where the symbols in the formula are as defined in the description] or a salt thereof in the presence of an aluminum compound and a chiral amine compound.
in Patent Document 1, a method for producing a synthetic intermediate of the above heterocyclic compound, the following methods are disclosed.
Formula 2]
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.[Formula 3]

(Wherein each symbol is as described in Patent Document 2.)
TABLE In the above method, the acid anhydride of the formula (VI), in the presence of a chiral amine with the formula (VIIa) or (VIIb) is to produce a chiral dicarboxylic acid monoester compound, then reacted with an amine (R1-NH-R2) is subjected to amidation to, to produce a heterocyclic amide compound of the formula (VIII).
Prior art documents
Patent literaturePatent Document 1: Patent No. 4,800,445 Patent
Patent Document 2: International Publication No. 2007/077005
Reference Example 1
3-oxabicyclo [3.3.1] nonane-2,4-dione
reaction vessel (1R, 3S) – was added to cyclohexane-1,3-dicarboxylic acid (10g) and THF (20mL), 5 It was cooled to ℃. It was added dropwise trifluoroacetic anhydride (8.19mL), and the mixture was stirred for about 1 hour. The reaction mixture was allowed to warm to room temperature, heptane (20mL) was added, up to 5 ℃ was cooled and stirred for about 30 minutes. The precipitate was filtered off, washed with heptane to give the title compound. Yield (6.7g)
reaction vessel (1R, 3S) – was added to cyclohexane-1,3-dicarboxylic acid (10g) and THF (20mL), 5 It was cooled to ℃. It was added dropwise trifluoroacetic anhydride (8.19mL), and the mixture was stirred for about 1 hour. The reaction mixture was allowed to warm to room temperature, heptane (20mL) was added, up to 5 ℃ was cooled and stirred for about 30 minutes. The precipitate was filtered off, washed with heptane to give the title compound. Yield (6.7g)
Reference Example 2
(3S, 5R) – tert – butyl 3- (isobutyl-amino) -5- (morpholine-4-carbonyl) piperidine-1-carboxylic acid ester succinate
reactor in THF (240ml), (3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholine-4-carbonyl) piperidine-3-carboxylic acid (20.0g), triethylamine (12.2mL) and diphenylphosphoryl azide (15.1mL) They were charged and allowed to react for 1 hour at 60 ℃, cooled to 25 ℃. After cooling the THF (60ml) and sodium trimethyl silanolate (19.7g) to charged 0 ℃ separately reaction vessel, was added dropwise to this was allowed to react before the reaction solution over about 1 hour, 0 at 0 ℃. 5 hours it was allowed to react. 0 slowly added dropwise acetic acid (40mL) at ℃, After stirring for 10 minutes, was added ethanol (60ml) and isobutyraldehyde (5.3mL) at 25 ℃, and stirred for 10 minutes. Then added sodium borohydride (1.88g), and the mixture was stirred for 30 minutes, and further addition of sodium borohydride (1.88g) at 25 ℃, and the mixture was stirred for 30 minutes. After completion of the reaction, water (100mL) was added and stirred for 10 minutes at room temperature. The organic layer was concentrated, then added dropwise slowly toluene (140ml) and 5N aqueous sodium hydroxide solution (120ml), the layers were separated. After washing and addition of aqueous 1N sodium hydroxide (100ml) the organic layer was washed 1N aqueous sodium hydroxide (100ml) was added again organic layer. The aqueous layers were combined and extracted by addition of toluene (100ml). The organic layers were combined, washed with 10w / v% aqueous sodium chloride solution (100ml), and the organic layer was concentrated. It was added ethanol (100ml), after it was concentrated under reduced pressure until about 60ml, warmed to 60 ℃ by the addition of ethyl acetate (40ml). Was added succinic acid (6.9g), After stirring for 30 minutes, it was added dropwise ethyl acetate (200ml) at 60 ℃, and stirred for 30 minutes. After stirring for 1 hour at room temperature, and the mixture was stirred for 1 hour at 0 ℃. The crystals were collected by filtration and washed with a mixture of ethyl acetate / n-heptane (6/1) (60mL). The obtained crystals at an external temperature of 50 ℃ to constant weight and then dried under reduced pressure to give the title compound as almost white crystals. Yield (22.8g)
reactor in THF (240ml), (3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholine-4-carbonyl) piperidine-3-carboxylic acid (20.0g), triethylamine (12.2mL) and diphenylphosphoryl azide (15.1mL) They were charged and allowed to react for 1 hour at 60 ℃, cooled to 25 ℃. After cooling the THF (60ml) and sodium trimethyl silanolate (19.7g) to charged 0 ℃ separately reaction vessel, was added dropwise to this was allowed to react before the reaction solution over about 1 hour, 0 at 0 ℃. 5 hours it was allowed to react. 0 slowly added dropwise acetic acid (40mL) at ℃, After stirring for 10 minutes, was added ethanol (60ml) and isobutyraldehyde (5.3mL) at 25 ℃, and stirred for 10 minutes. Then added sodium borohydride (1.88g), and the mixture was stirred for 30 minutes, and further addition of sodium borohydride (1.88g) at 25 ℃, and the mixture was stirred for 30 minutes. After completion of the reaction, water (100mL) was added and stirred for 10 minutes at room temperature. The organic layer was concentrated, then added dropwise slowly toluene (140ml) and 5N aqueous sodium hydroxide solution (120ml), the layers were separated. After washing and addition of aqueous 1N sodium hydroxide (100ml) the organic layer was washed 1N aqueous sodium hydroxide (100ml) was added again organic layer. The aqueous layers were combined and extracted by addition of toluene (100ml). The organic layers were combined, washed with 10w / v% aqueous sodium chloride solution (100ml), and the organic layer was concentrated. It was added ethanol (100ml), after it was concentrated under reduced pressure until about 60ml, warmed to 60 ℃ by the addition of ethyl acetate (40ml). Was added succinic acid (6.9g), After stirring for 30 minutes, it was added dropwise ethyl acetate (200ml) at 60 ℃, and stirred for 30 minutes. After stirring for 1 hour at room temperature, and the mixture was stirred for 1 hour at 0 ℃. The crystals were collected by filtration and washed with a mixture of ethyl acetate / n-heptane (6/1) (60mL). The obtained crystals at an external temperature of 50 ℃ to constant weight and then dried under reduced pressure to give the title compound as almost white crystals. Yield (22.8g)
Example 1
(3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholin-4-ylcarbonyl) piperidine-3-carboxylic acid
the reaction vessel in chlorobenzene (7.5mL) and quinine (0.70g ) is added and stirred, it was added dropwise DIBAL1.0M hexane solution (2.16mL). The reaction mixture was cooled to -40 ℃, tert – butyl 2,4-dioxo-3-oxa-7-azabicyclo [3.3.1] was added nonane-7-carboxylic acid ester (0.50g), about 1 hour stirring. Was added chlorobenzene to another reaction vessel (2.5mL) and morpholine (0.17mL), the resulting solution was cooled to -40 ℃ was added dropwise to the previous reaction solution. After completion of the reaction, the mixture was separated with ethyl acetate and 10w / w% aqueous citric acid solution, and the resulting aqueous layer was re-extracted with ethyl acetate. The organic layers were combined, washed with 10w / w% saline, and concentrated to give the title compound. 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.41 (s, 9 H), 1.47 – 1.72 (M, 1 H), 1.89 – 2.10 (M, 1 H), 2.36 – 2.49 (M, 1 H ), 2.55 – 2.83 (m, 3 H), 3.40 – 3.50 (m, 2 H), 3.51 -.. 3.57 (m, 4 H), 3.59 (br s, 2 H), 3.83 – 4.04 (m, 1 H), 4.05 – 4.29 (m, 1 H), 12.52 (s, 1 H) optical purity of 94.3% EE <HPLC analytical conditions> column: CHIRALPAK IC (Co., Ltd. Daicel) column temperature: constant around 15 ℃ Temperature Mobile phase: A solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50 : 50 gradient program
the reaction vessel in chlorobenzene (7.5mL) and quinine (0.70g ) is added and stirred, it was added dropwise DIBAL1.0M hexane solution (2.16mL). The reaction mixture was cooled to -40 ℃, tert – butyl 2,4-dioxo-3-oxa-7-azabicyclo [3.3.1] was added nonane-7-carboxylic acid ester (0.50g), about 1 hour stirring. Was added chlorobenzene to another reaction vessel (2.5mL) and morpholine (0.17mL), the resulting solution was cooled to -40 ℃ was added dropwise to the previous reaction solution. After completion of the reaction, the mixture was separated with ethyl acetate and 10w / w% aqueous citric acid solution, and the resulting aqueous layer was re-extracted with ethyl acetate. The organic layers were combined, washed with 10w / w% saline, and concentrated to give the title compound. 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.41 (s, 9 H), 1.47 – 1.72 (M, 1 H), 1.89 – 2.10 (M, 1 H), 2.36 – 2.49 (M, 1 H ), 2.55 – 2.83 (m, 3 H), 3.40 – 3.50 (m, 2 H), 3.51 -.. 3.57 (m, 4 H), 3.59 (br s, 2 H), 3.83 – 4.04 (m, 1 H), 4.05 – 4.29 (m, 1 H), 12.52 (s, 1 H) optical purity of 94.3% EE <HPLC analytical conditions> column: CHIRALPAK IC (Co., Ltd. Daicel) column temperature: constant around 15 ℃ Temperature Mobile phase: A solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50 : 50 gradient program
Example 30 (1R, 3S) -3- (morpholin-4-ylcarbonyl) cyclopentanecarboxylic acid
(anhydride: 3-oxabicyclo [3.2.1] octane-2,4-dione; Amine: Morpholine ) 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.72 – 1.91 (M, 5 H), 2.04 (dt, J = 12.69, 7.84 Hz, 1 H), 2.65 – 2.74 (M, 1 H), 2.99 – 3.07 (m, 1 H), 3.42 – 3.51 (m, 4 H), 3.51 – 3.58 (m, 4 H), 11.96 – 12.17 (m, 1 H) optical purity of 52.3% EE <HPLC analysis conditions > column: CHIRALPAK IF (Co., Ltd. Daicel) column temperature: 15 ℃ constant temperature in the vicinity ofmobile phase: A solution) 0.02 mol / LKH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / LKH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50: 50 gradient Program
| WO2010150840A1 | 24 Jun 2010 | 29 Dec 2010 | Dainippon Sumitomo Pharma Co., Ltd. | N-substituted-cyclic amino derivative |
| WO2011158880A1 | 15 Jun 2011 | 22 Dec 2011 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| WO2012062687A1 * | 7 Nov 2011 | 18 May 2012 | F. Hoffmann-La Roche Ag | Triazole derivatives and their use for neurological disorders |
| WO2013122260A1 | 14 Feb 2013 | 22 Aug 2013 | Takeda Pharmaceutical Company Limited | Tablet |
| CN103221402B * | 7 Nov 2011 | 17 Jun 2015 | 霍夫曼-拉罗奇有限公司 | 三唑衍生物及其用于神经障碍的用途 |
| US8329691 | 14 Oct 2008 | 11 Dec 2012 | Takeda Pharmaceutical Company Limited | Amide compounds and use of the same |
| US8389511 | 19 Dec 2008 | 5 Mar 2013 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic heterocyclic derivative |
| US8658639 | 24 Jun 2010 | 25 Feb 2014 | Dainippon Sumitomo Pharma Co., Ltd | N-substituted-cyclic amino derivative |
| US8742097 | 2 Nov 2011 | 3 Jun 2014 | Hoffmann-La Roche Inc. | Triazole compounds I |
| US9018374 | 15 Jun 2011 | 28 Apr 2015 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| US9090601 | 28 Jan 2010 | 28 Jul 2015 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
///////////TAK 272, Hypertension
Avatrombopag

Avatrombopag
AVATROMBOPAG; UNII-3H8GSZ4SQL; AKR-501; E5501; 570406-98-3; AS 1670542
| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
Elemental Analysis: C, 53.61; H, 5.28; Cl, 10.91; N, 12.94; O, 7.39; S, 9.87
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1-piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

Avatrombopag maleate; UNII-GDW7M2P1IS; E5501 MALEATE; 677007-74-8; YM 477, AKR 501
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid
INTRODUCTION
Avatrombopag, also known as AKR-501, YM477, AS 1670542 or E5501, is a novel orally-active thrombopoietin (TPO) receptor agonist. AKR-501 specifically targeted the TPO receptor and stimulated megakaryocytopoiesis throughout the development and maturation of megakaryocytes just as rhTPO did. Daily oral administration of AKR-501 dose-dependently increased the number of human platelets in these mice, with significance achieved at doses of 1 mg/kg and above. The peak unbound plasma concentrations of AKR-501 after administration at 1 mg/kg in NOD/SCID mice were similar to those observed following administration of an active oral dose in human subjects. AKR-501 may be useful in the treatment of patients with thrombocytopenia. (source: Eur J Haematol. 2009 Apr;82(4):247-54).

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Patent literature 1 discloses a hydrochloride of compound X as example 16 (hereinafter referred to as “compound X hydrochloride”).
Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
The crystals of compound X maleic acid salt disclosed in patent literature 2 (hereinafter referred to as “compound X maleic acid salt A type crystals”) cannot be isolated as compound X maleic acid salt A type crystals when scaled up for mass production using the method disclosed in example 1 of patent literature 2, and therefore must be isolated in a different crystal form. (This other crystal form is referred to as “compound X maleic acid salt B type crystals”). Therefore, the compound X maleic acid salt A type crystals have a possibility that the crystal form will morph depending on the scale of production, and is clearly inappropriate as a drug substance for medicines which require constant quality and constant effects.
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….

REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
UPDATE MAY 2018
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
Lusutrombopag….Oral thrombopoietin (TPO) mimetic
Lusutrombopag
Update…..FDA approved july2018
(E)-3-[2,6-dichloro-4-[[4-[3-[(1S)-1-hexoxyethyl]-2-methoxyphenyl]-1,3-thiazol-2-yl]carbamoyl]phenyl]-2-methylprop-2-enoic acid
(S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid
(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylacrylic acid
UNII 6LL5JFU42F, CAS 1110766-97-6,
D10476, MW591.546 , [US2010267783], MF C29H32Cl2N2O5S, S-888711
Shionogi & Co., Ltd., 塩野義製薬株式会社 INNOVATOR
Optically active compound (C-3B) Melting point: 142-145°C…………….EP2184279B1
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)………….EP2184279B1
Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C), NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs)………EP2184279B1
Thrombopoietin receptor agonist, Oral thrombopoietin (TPO) mimetic
- 24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
- 31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
- 08 Nov 2013 Phase II development is ongoing in the US and the Europe
Process for preparing intermediates of an optically active 1,3-thiazole containing thrombopoietin receptor agonist Also claims crystalline forms of lusutrombopag intermediates and a process for preparing lusutrombopag. Shionogi is developing lusutrombopag, a small-molecule thrombopoietin mimetic, as an oral tablet formulation for treating thrombocytopenia.
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.

The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, “(E) -3- [2,6-Dichloro-4- [4- [3 – [(S) -1-hexyloxyethyl] – 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid “. lusutrombopag is represented by the following chemical structural formula.

Eltrombopag is represented by the following chemical structural formula.

Avatrombopag is represented by the following chemical structural formula.
Totrombopag choline is represented by the following chemical structural formula.


C 3B IS THE COMPD OF ROT (-) AND S, E FORM

Example 2
Synthesis of (R)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3A) (not included in the present invention) and (S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3B)
According to the same method as in Example 1, an optically active compound (C-3A) and an opticallly active compound (C-3B) were synthesized from (RS)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (B-3) obtained in Reference Example 3.Optically active compound (C-3A)Melting point: 139-141°C UNDESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82 – 0.87 (m, 3H) Optical rotaion +4.5 degrees (DMSO, c = 1.001, 25°C)
Optically active compound (C-3B)Melting point: 142-145°C DESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)
First step: Synthesis of (S)-1-(3-bromo-2-methyloxyphenyl)ethane-1-ol (17)
Using the same method as that of the first step of Example 3, the compound (17) was obtained from the compound (16) at a yield 77%.
-
-
Optical rotation: -23.5 ± 0.6 degrees (CHCl3, c = 1.050, 21°C)
NMR (CDCl3) θ ppm: 1.49 (3H, d, J = 6.6 Hz), 2.33 (1H, brs), 3.88 (3H, s), 5.19 (1H, q, J = 6.4 Hz), 7.01 (1H, t, J = 7.9 Hz), 7.40 (1H, dd, J = 7.7 Hz, J = 1.1 Hz), 7.46 (1H, dd, J = 8.0 Hz, J = 1.4 Hz)
-
Second step: Synthesis of (S)-1-bromo-3-(1-hexyloxyethyl)-2-methyloxybenzene (18)
-
-
Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
-
Third step and fourth step: Synthesis of (S)-4-(3-(1-hexyloxyethyl)-2-methyloxyphenyl)thiazole-2-amine (20)
-
-
Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
-
Compound (19)
-
-
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2-1.4 (6H, m), 1.45 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 3.29 (2H, m), 3.78 (3H, s), 4.73 (2H, m), 4.80 (1H, q, J = 6.4 Hz), 7.24 (1H, t, J = 7.8Hz), 7.52 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.65 (1H, dd, J = 7.7 Hz, J = 1.8 Hz)
-
Compound (20)
-
Optical rotation: -4.2 ± 0.4 degrees (DMSO, c = 1.025, 21°C)
NMR (CDCl3) δ ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, m), 1.35 (3H, d, J = 6.5 Hz), 1.48 (2H, m), 3.25 (2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) -
Fifth step: Synthesis of ethyl (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylate (21)
-
-
Using the same method as that of the fifth step of Example 3, the compound (21) was obtained from the compound (20) at a yield of 94%.
Optical rotation: +4.7 ± 0.4 degrees (CHCl3, c = 1.07, 21°C)
NMR (CDCl3 ) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2 – 1.35 (6H, m), 1.38 (3H, t, J = 7.1
Hz), 1.44 (3H, d, J = 6.4 Hz), 1.57 (2H, m), 1.77 (3H, d, J = 1.4 Hz), 3.30 (2H, m), 3.59 (3H, s), 4.31 (2H, q, J = 7.1 Hz), 4.83 (1H, q, J = 6.4 Hz), 7.17 (1H, t, J = 7.7 Hz), 7.42 (1H, d, J = 1.7 Hz), 7.42 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.51 (1H, s), 7.67 (1H, dd, J = 7.6 Hz, J = 1.7 Hz), 7.89 (2H, s), 10.30 (1H, brs)
-
Sixth step: Synthesis of (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylic acid (C-3B)
-
Using the same method as that of the sixth step of Example 3, the compound (C-3B) was obtained from the compound (21) at a yield of 80%.Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) -
Results of powder X-ray deffraction are shown in Fig. 5.
-
Diffraction angle of main peak: 2θ = 17.8, 21.1, 22.5, 23.3, 24.1, and 24.4 degrees
WO2005014561/EP1655291A1
https://www.google.co.in/patents/EP1655291A1?cl=en
WO2014003155, claiming a composition comprising lusutrombopag, useful for treating thrombocytopenia.
https://www.google.co.in/patents/US20150148385?cl=en
.
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
(Step 1) Synthesis of compound (VII ‘) under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran – heptane – ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ‘) (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI ‘))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ‘) under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ‘)). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ‘)). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ‘) (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ‘)).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II ‘). Next, the above-mentioned compound (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound ‘Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ‘) (1.50kg) and compound (VII’) (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ‘) (2.98kg, 95.2% yield).
1H-NMR (CDCl 3 ) delta: 0.87 (t, J = 6.8 Hz, 3H), 1.20-1.34 (M, 6H), 1.37 (t, J = 7.1 Hz, 3H), 1.44 (D, J = 6.5 Hz , 3H), 1.52-1.59 (m, 2H), 1.77 (d, J = 1.3Hz, 3H), 2.62 (s, 6H), 3.28-3.34 (m, 2H), 3.59 (s, 3H), 4.31 ( q, J = 7.1Hz, 2H), 4.83 (q, J = 6.5Hz, 1H), 7.16 (t, J = 7.7Hz, 1H), 7.40-7.43 (m, 2H), 7.51 (s, 1H), 7.68 (dd, J = 7.7, 1.8Hz, 1H), 7.92 (d, J = 1.3Hz, 2H), 10.58 (s, 1H). The results of the powder X-ray diffraction and I are shown in Figure 9 and Table 7. [Table 7]
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ‘) The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ‘) and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ‘) (1.99kg, 93.9% yield).
NMR (CDCl 3 ) delta ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, M), 1.48 (3H, D, J = 6.4 Hz), 1.52 – 1.64 (2H, M), 1.86 (3H, d, J = 1.4Hz), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) I is shown in Figure 15 the results of powder X-ray diffraction.
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (…. Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
| 101 | Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama, Hajime Yamada, Hiroshi Takemoto, Takeshi Shiota, Yoshikazu Tanaka, Noriko Yamane, Kouji Takahashi, Naoki Oyabu, Kenji Kuwabara, Itsuki Oshima, Kenzo Koizumi, Hiroshi Yoshida, Ayumu Nogami, Tomomi Yamada, Yutaka Yoshida, Takami Murashi, Shinichiro Hara. |
| 101 – Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia
Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). |
update………..
FDA approves lusutrombopag for thrombocytopenia in adults with chronic liver disease
https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm615348.htm
synthesis………..https://newdrugapprovals.org/2015/08/20/lusutrombopag-oral-thrombopoietin-tpo-mimetic/
On July 31, 2018, the Food and Drug Administration approved lusutrombopag (Mulpleta, Shionogi Inc.) for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure.
Approval was based on two randomized, double-blind, placebo-controlled trials (L-PLUS 1 and L-PLUS 2, NCT02389621) involving 312 patients with chronic liver disease and severe thrombocytopenia who were undergoing an invasive procedure and had a platelet count less than 50 x 109/L. Patients were randomized 1:1 to receive 3 mg of lusutrombopag or placebo once daily for up to 7 days.
In L-PLUS 1, 78% of patients (38/49) receiving lusutrombopag required no platelet transfusion prior to the primary invasive procedure, compared with 13% (6/48) who received placebo (95% CI for treatment difference: 49%, 79%; p<0.0001). In L-PLUS 2, 65% (70/108) of patients who received lusutrombopag required no platelet transfusion prior to the primary invasive procedure or rescue therapy for bleeding from randomization through 7 days after the procedure, compared with 29% (31/107) receiving placebo (95% CI for treatment difference: 25%, 49%; p<0.0001).
The most common adverse reaction in ≥ 3% of patients was headache.
The recommended lusutrombopag dosage is 3 mg orally once daily with or without food for 7 days.
View full prescribing information for Mulpleta.
FDA granted this application priority review and fast track designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
Follow the Oncology Center of Excellence on Twitter @FDAOncology.
Check out recent approvals at the OCE’s podcast, Drug Information Soundcast in Clinical Oncology.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
//////
phase 3, shionogi, japan, lusutrombopag, S 888711
CCCCCCOC(C)C1=CC=CC(=C1OC)C2=CSC(=N2)NC(=O)C3=CC(=C(C(=C3)Cl)C=C(C)C(=O)O)Cl
TENELIGLIPTIN

TENELIGLIPTIN
| Teneligliptin; 760937-92-6; UNII-28ZHI4CF9C; Teneligliptin (INN); 28ZHI4CF9C | |
| MF | C22H30N6OS |
|---|---|
| MW | 426.5782 g/mol |
Teneligliptin (INN; trade name Tenelia) is a pharmaceutical drug for the treatment of type 2 diabetes mellitus. It is approved for use in Japan.[1] It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or “gliptins”.[2] {(2S,4S)-4-[4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)-1-piperazinyl]-2-pyrrolidinyl}(1,3-thiazolidin-3-yl)methanone
Teneligliptin was launched in Japan in 2012 by Mitsubishi Pharma and Daiichi Sankyo for the treatment of type 2 diabetes mellitus. In 2013, the indication was partially changed to include it as a combination therapy with existing oral hypoglycemic agents, such as biganides, alpha-glucosidaseinhibitors, rapid-acting insulin secretagogues, and insulin preparations, as well as sulfonylureas and thiazolidines that had been approved for the combination.
In 2014, the product was registered in KR for the treatment of type 2 diabetes mellitus.
In 2013, Mitsubishi Tanabe Pharma filed for approval in Japan for use of the compound as combination therapy for the treatment of diabetes type 2.
| CAS | 760937-92-6 |
|---|
3-{(2S,4S)-4-[4-(3-methyl-l -phenyl- 1 H- pyrazol-5-yl)- l-piperazinyl]-2-pyrrolidinylcarbonyl}-l , 3-thiazolidine is represented structurally by a compound of formula (I):

Teneligliptin (CAS 760937-92-6) is a novel, potent and long-lasting dipeptidyl peptidase-4 inhibitor in treatment of type 2 diabetes. Dipeptidyl-peptidase-4 (DPP- 4) inhibitor has been demonstrated to improve glycemic control, in particular postparandial hyperglycemic control.
Despite of their common mechanism of action, DPP-4 inhibitors show marked structural heterogeneity. DPP-4 inhibitors may be classified into peptidomimetic (i.e. sitagliptin, vildagliptin, saxagliptin, and anagliptin) and non-peptidomimetic (i.e. alogliptin and linagliptin) subtypes.
Teneligliptin, is chemically known as a 3- {((2S,4S)-4-(4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-yl 25 carbonyl}thiazolidine hemipentahydrobromide hydrate and is peptidomimetic with the molecular formula of C22H30N6OS.2½HBr.xH2O and molecular weight of 642.88 g/mol for hemipentahydrobromide. The hydrate can be from mono to dihydrate.
U.S. Patent No. 7,074,794 B2 (the US ‘794) discloses teneligliptin as L-proline derivative and its pharmaceutically acceptable salts which exhibits a Dipeptidyl 5 peptidase IV (DPP-IV) inhibitory activity, which is useful for the treatment or prophylaxis of diabetes, obesity, HIV infection, cancer metastasis, dermopathy, prostatic hyperplasia, periodontitis, autoimmune diseases and the like.
The example-222 of the US ‘794 discloses the process for the preparation of teneligliptin as trihydrochloride salt U.S. Patent No. 8,003,790 B2 (the US ‘790) discloses salts of proline derivative, solvate thereof and production method thereof. In particular, the US ‘790 discloses 2.0 hydrochloride or 2.5 hydrochloride; 2.0 hydrobromide or 2.5 hydrobromide, and hydrates thereof teneligliptin.
The US ‘790 B2 further discloses different salts 15 of teneligliptin which are incorporated herein as reference in their entirety U.S. PG-Pub. No. 2011/0282058 A1 discloses salts of 3-{((2S,4S)-4-(4-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-ylcarbonyl}thiazolidine with mono-, di- and tri-basic acids or a solvate thereof. 20 International (PCT) publication No. WO 2012/165547 A1 discloses a process for preparation of teneligliptin and pharmaceutically acceptable salts thereof.
International (PCT) publication No. WO 2007/127635 A2 (the WO ‘635 A2) discloses a process for the preparation of diketo-piperazine and piperidine 25 derivatives. In particular, the WO ‘635 A2 discloses the process for preparation of 4-oxo-2-(thiazolidine-3-carbonyl)-pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (III)] by reacting piperazine with aryl halide.
International (PCT) publication No. WO 2012/099915 A1 (the WO ‘915 A1) 5 discloses the process for the preparation of deuterated thiazolidine derivatives. The WO ‘915 A1 also discloses the process for the preparation of 1-(3-methyl-1- phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by condensation of 5- chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by deprotection of Boc-protected 1-(3-methyl-1-phenyl-1Hpyrazol-5-yl)piperazine with triflouroacetic acid.
U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine by condensation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine in presence of n-BuLi in tetrahydrofuran. Bioorganic & Medicinal Chemistry, 14(11), 3662-3671 (2006),
Bioorganic & Medicinal Chemistry, 20(16), 5033-5041 (2012) and U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate by reacting (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid with 25 thiazolidine in presence of HOBT and EDC.HCl in dimethylformamide solvent.
Bioorganic & Medicinal Chemistry, 15(2), 641-655 (2007) discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3- carbonyl)pyrrolidine-1-carboxylate by treating (2S,4S)-tert-butyl 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate with tetrabutylammonium fluoride in tetrahydrofuran.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the 5 process for the preparation of herein compound (II) after by reacting 1-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) with (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate in presence of sodium triacetoxyborohydride. There is provided different alternative processes for the preparation of teneligliptin and intermediates thereof.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) also discloses the process for the preparation of 4-[4-(5-methyl-2-phenyl-2H-pyrazol-3-yl)-piperazin- 1-yl]-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (II)] after by reacting 1-(3-methyl-1-phenyl-1H-pyrazol-5- 15 yl)piperazine [herein compound (V)] with (2S,4S)-tert-butyl 4-[[(1,1- dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate in presence of trifluoromethylsulfonic anhydride and diisopropylethylamine. 3 – [[(2S, 4S) -4- [4- (3- methyl-1-phenyl–1H- pyrazol-5-yl) -1-piperazinyl ] -2-pyrrolidinyl] carbamoyl] thiazolidine, having the formula below, is a very novel DPP-4 inhibitor potential.

World Patent Application No. W02012099915 for Ge Lieting discloses a process for the preparation route is as follows:

Journal B10rganic & Medicinal Chemistry, 2012, 20, 5705-5719 also discloses a preparation method for Ge Lieting, the route is as follows:

[0009] 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine, was prepared for the Ge Lieting key intermediate. Journals B10rganic & Medicinal Chemistry, 2012,20,5705-5719 reported the preparation of the intermediates prepared route is as follows:

[0011] The preparative route after the N-Boc-N- acetoacetyl piperazine phenylhydrazine and methanesulfonic acid in an ethanol solution of the reaction at room temperature 14h, concentrated under reduced pressure after addition of pyridine.Was added phosphorus oxychloride in pyridine, 20h post treatment reaction at room temperature the reaction system. The compound obtained above was then added trifluoroacetic acid was dissolved in methylene chloride after, after treatment at room temperature for 1.5h to give 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine.
The reaction process requires mesylate mesylate flammable, easy-absorbent deliquescence, and has a strong corrosive and irritating, easy to cause the body burns; phosphorus oxychloride, a highly toxic substance, water violent hair in the air smoke, hydrolyzed into phosphoric acid and hydrogen chloride, is very unstable, to operate a lot of trouble; trifluoroacetic acid is highly corrosive and irritant, can cause the body burns; low yield of the reaction (10%). Seeking a simple operation, high reaction yield, low cost and suitable for industrial production production process 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine has a very important role in the field of medicine.
…………………………………….


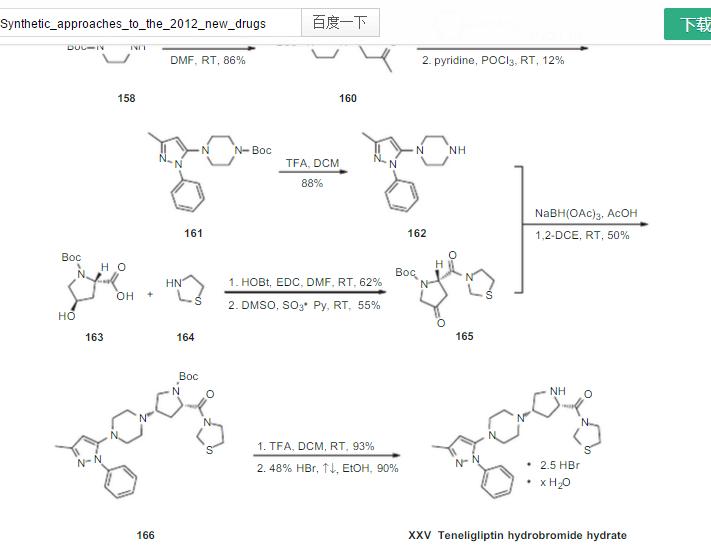
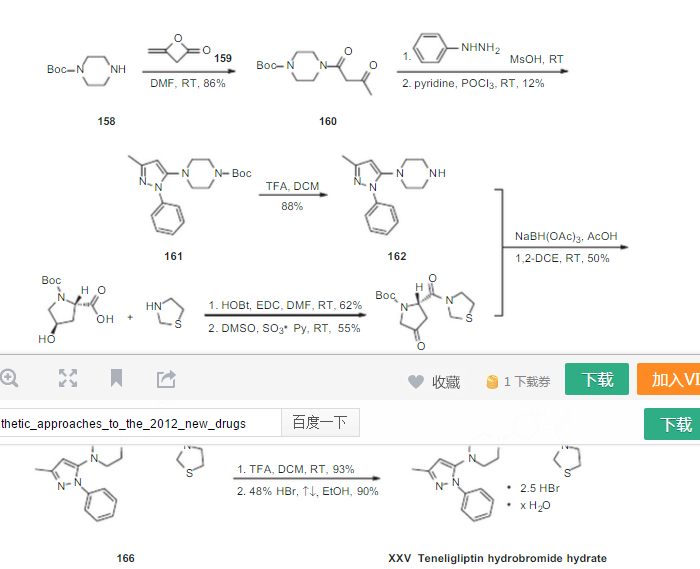
since the capture is staggered, compd 165 is not clear in above pic see below
…………
if above section iis not clear see at ……..http://www.allfordrugs.com/2015/07/03/teneligliptin/
…………………….
reaction scheme in http://www.google.com/patents/CN104177295A?cl=en

Description: LR as Lawesson reagent (Lawesson Reagent), is a sulfur oxygen exchange reagent. The present invention provides a method for preparing key intermediates Ge Lieting method, comprising the steps of: (I) N-Boc-N- acetoacetyl piperazine Lawesson’s reagent in the presence of an organic solvent, with a phenylhydrazine of the formula occurs ⑴ reaction shown:

(2) the step (1) The product was dissolved in an organic solvent, the following formula (II) in concentrated hydrochloric acid to deprotected shown:

格列汀 refers to 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine
……………………………..
Volume 20, Issue 19, 1 October 2012, Pages 5705–5719

…………………………………..

………………………..
http://www.google.co.in/patents/WO2015019238A1?cl=en
Example 5: Preparation of {(2^,.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -vHpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
Activated carbon (10 g) was added to a solution of the residue (obtained in Example 4) in isopropyl alcohol (1000 mL) at 30°C to 35°C. The reaction mixture was filtered through a Hyflo® bed. The filtrate was heated to a temperature of 70°C to 75°C. Hydrobromic acid (48%; 168 g) was slowly added to the filtrate at 70°C to 75°C over a period of 10 minutes to 15 minutes. The reaction mixture was stirred for 2.5 hours at 70°C to 77°C. The progress of the reaction was monitored by HPLC. After completion of the reaction, the reaction mixture was cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes. The reaction mixture was filtered to obtain a solid. The solid obtained was washed with isopropyl alcohol (2 x 200 mL), and dried at 50°C under reduced pressure for 15 hours to obtain crude {(25*,45)-4-[4-(3-methyl-l-phenyl-lH- pyrazol-5 -yl)piperazin- 1 -yl]pyrrolidin-2-yl} ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate.
Yield: 90%
Example 6: Purification of {(2^’.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -yllpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
A reaction mixture containing {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5- yl)piperazin- 1 -yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate (100 g; prepared according to the process of Example 5) in ethanol (700 mL) was heated at 70°C to 75°C to obtain a solution. The solution was filtered at the same temperature. The filtrate was allowed to cool to a temperature of 65 °C to 68°C, and deionized water (10 mL) was added at the same temperature. The solution was cooled to a temperature of 55°C to 60°C, and stirred at the same temperature for 2 hours. The solution was further cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes to obtain a solid. The solid was filtered, washed with ethanol (100 mL), and dried at 45°C to 50°C under reduced pressure for 18 hours to 20 hours to obtain pure {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin-l- yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone hemipentahydrobromide hydrate .
Yield: 90%
HPLC Purity: 99.93%
| WO2012099915A1 * | 18 Jan 2012 | 26 Jul 2012 | Hongwen Zhu | Thiazolidine derivatives and their therapeutic use |
| WO2012165547A1 * | 31 May 2012 | 6 Dec 2012 | Mitsubishi Tanabe Pharma Corporation | Method for manufacturing pyrazole derivative |
| WO2014041560A2 * | 28 Aug 2013 | 20 Mar 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of teneligliptin |
| US7074794 | 10 Aug 2001 | 11 Jul 2006 | Mitsubishi Pharma Corporation | Proline derivatives and the use thereof as drugs |
| US8003790 | 17 Feb 2006 | 23 Aug 2011 | Mitsubishi Tanabe Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| US20050256310 * | 12 May 2005 | 17 Nov 2005 | Pfizer Inc | Therapeutic compounds |
| EP1854795A1 * | 17 Feb 2006 | 14 Nov 2007 | Mitsubishi Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| EP1894567A1 * | 2 Jun 2006 | 5 Mar 2008 | Mitsubishi Tanabe Pharma Corporation | Concomitant pharmaceutical agents and use thereof |
| US20040106655 * | 10 Aug 2001 | 3 Jun 2004 | Hiroshi Kitajima | Proline derivatives and the use thereof as drugs |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015019238A1 * | 28 Jul 2014 | 12 Feb 2015 | Ranbaxy Laboratories Limited | Process for the preparation of n-protected (5s)-5-(1,3-thiazolidin-3-ylcarbonyl)pyrrolidin-3-one |
| Patent | Submitted | Granted |
|---|---|---|
| Proline derivatives and use thereof as drugs [US7060722] | 2005-11-03 | 2006-06-13 |
| Proline derivatives and the use thereof as drugs [US7074794] | 2004-06-03 | 2006-07-11 |
| Proline derivatives and use thereof as drugs [US2006173056] | 2006-08-03 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US8003790] | 2009-08-27 | 2011-08-23 |
| METHOD OF TREATING ABNORMAL LIPID METABOLISM [US2010305139] | 2010-12-02 | |
| COMBINED USE OF DIPEPTIDYL PEPTIDASE 4 INHIBITOR AND SWEETENER [US2010113382] | 2010-05-06 | |
| CONCOMITANT PHARMACEUTICAL AGENTS AND USE THEREOF [US2009082256] | 2009-03-26 | |
| PROPHYLACTIC/THERAPEUTIC AGENT FOR ABNORMALITIES OF SUGAR/LIPID METABOLISM [US2009088442] | 2009-04-02 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US2011282058] | 2011-11-17 |
- Joanne Bronson, Amelia Black, T. G. Murali Dhar, Bruce A. Ellsworth, and J. Robert Merritt. “Teneligliptin (Antidiabetic)”. Annual Reports in Medicinal Chemistry 48: 523–524. doi:10.1016/b978-0-12-417150-3.00028-4.
- Kishimoto, M (2013). “Teneligliptin: A DPP-4 inhibitor for the treatment of type 2 diabetes”. Diabetes, metabolic syndrome and obesity : targets and therapy 6: 187–95. doi:10.2147/DMSO.S35682. PMC 3650886. PMID 23671395.
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
Lascufloxacin, KRP-AM1977, by Kyorin

Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
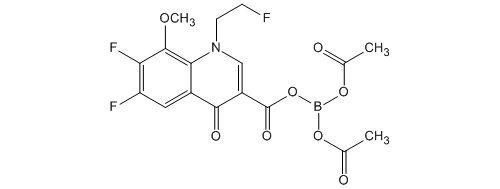
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
KEY INTERMEDIATE
CAS 848498-67-9
Boron, bis(acetato-κO)[6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-(oxo-κO)-3-quinolinecarboxylato-κO]-, (T-4)-
Coordination Compound
ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル)
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
KEY INTERMEDIATE
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
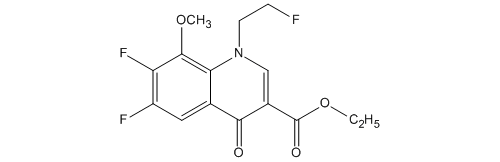 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()


Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line
| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |
Shimotsuga-gun, Tochigi

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.MARKET
Ochanomizu station


Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon

Firategrast, T-0047

 Japan
Japan
Firategrast, 402567-16-2;
Firategrast, MS, Alpha4beta1 integrin
PHASE 2 GSK
Mitsubishi Tanabe Pharma INNOVATOR
Glaxo Group Limited, Mitsubishi Tanabe Pharma Corporation
SB 683699, SB-683699, UNII-OJY3SK9H5F
Firategrast; UNII-OJY3SK9H5F; SB-683699; Firategrast (USAN); 402567-16-2; SB683699; T-0047
Molecular Formula: C27H27F2NO6
Molecular Weight: 499.503186 g/mol
SYSTEMATIC NAME:
1,1′-Biphenyl)-4-propanoic acid, alpha-((2,6-difluorobenzoyl)amino)-4′-(ethoxymethyl)-2′,6′-dimethoxy-, (alphaS)-
N-(2,6-Difluorobenzoyl)-4-[4-(ethoxymethyl)-2,6-dimethoxyphenyl]-L-phenylalanine
N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
2S)-2-((2,6-Difluorobenzoyl)amino)-3-(4′-(ethoxymethyl)-2′,6′-dimethoxybiphenyl-4- yl)propanoic acid
(2S)-2-{[(2,6- difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4- biphenylyl]propanoic acid
(2S)-2-[[2,6-bis(fluoranyl)phenyl]carbonylamino]-3-[4-[4-(ethoxymethyl)-2,6-dimethoxy-phenyl]phenyl]propanoic acid
Pharmacological half-life is 2.5 – 4.5 hours, compared to 11 days for natalizumab, a drug in the same class
Orally bioavailable small molecule α4-integrin antagonist
see
http://www.msdiscovery.org/node/1377#node-biblio-1338
http://multiple-sclerosis-research.blogspot.com/2012/01/research-oral-tysabri-analogue.html
SB683699 is an alpha4 integrin antagonist that had been studied in phase II trials at GlaxoSmithKline under a license from Mitsubishi Tanabe Pharma for the oral treatment of multiple sclerosis (MS) in Europe. GlaxoSmithKline and Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) had been studying the drug candidate for the treatment of asthma, rheumatoid arthritis (RA) and Crohn’s disease
MECHANISMS/EFFECTS
HUMAN:
Similar mechanism of action to natalizumab (α4-integrin blocker), but its faster elimination could improve safety profile

Firategrast
SYNTHESIS
………………….
PATENT
Scheme 1

Scheme 2

In a further aspect the present invention provides for a process for the preparation of compound of formula (II) which comprises coupling the compound of formula (V)

Suitable coupling conditions for the compound of formula (V) and the compound of formula (VI) include those shown in Scheme 2. In a further aspect of the invention there is provided the compound of formula (V):

1H NMR characterisation data for the compound of formula (V) were generated on an isolated and purified batch. 1H-NMR spectra were recorded on a Bruker Avance 400 at 400MHz, using TMS as an internal reference.1H NMR (400 MHz, DMSO-D6) δ ppm 1.17 (t, J=7.09 Hz, 3 H) 2.96 (dd, J=13.82, 9.90 Hz, 1 H) 3.1 1 (dd, J=13.82, 5.26 Hz, 1 H) 4.12 (q, J=7.09 Hz, 2 H) 4.63 (ddd, J=9.78, 7.82, 5.38 Hz, 1 H) 7.15 (t, J=7.95 Hz, 2 H) 7.25 (d, J=8.31 Hz, 2 H) 7.47 – 7.55 (m, 3 H) 9.23 (d, J=7.83 Hz, 1 H).
The present invention provides a process for the preparation of the compound of formula

which process comprises the steps: a) hydrolysis of an ester of formula (I la):

Recrvstallisation of (2S)-2-{r(2,6-difluorophenyl)carbonyllamino)-3-r4′-r(ethyloxy)methyll- 2′,6′-bis(methyloxy)-4-biphenylyllpropanoic acid
(2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)- 4-biphenylyl]propanoic acid (9.38Kg) was charged into a clean reactor, followed by ethyl acetate (46.9L). The solution was heated to 50°C and filtered into the pre-warmed (35°C) crystallizing vessel. A line-wash with ethyl acetate (9.4L) was carried out. The combined ethyl acetate solutions were heated to 50°C, stirred to ensure complete dissolution. Filtered heptane (9.4L) was added maintaining the temperature at 50°C then the solution cooled to 30°C and seeded with (2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4 – [(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4-biphenylyl]propanoic acid (47g) slurried in 1 :9 ethyl acetate:heptane (0.47L). The slurry was aged for 2 hours at 30°C. Filtered heptane (75L) was added over 3 hours. The slurry was then cooled to 0°C over 1 hour. The mixture was aged at 0°C for 1 hour then the solid was filtered off, washed with isopropyl ether (29.6L and dried under vacuum at 50±3°C to give the product (8.55Kg, 91 %). Characterised by having an infrared absorption spectrum with significant absorption bands at about 754, 768, 800, 820, 849, 866, 1006, 1 100, 1 122, 1 157, 1 188, 1225, 1242, 1268, 1292, 1317, 1352, 1417, 1466, 1530, 1580, 1624, 1650, 1662, 171 1 , 1728, 2938, 3302cm“
…………………………………..
PATENT
Example 10: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine ethyl ester.
(1) The product obtained in Example l-(4) (2.1 g) was acylated with 2 , 6-difluorobenzoyl chloride in a similar manner as described in Example 1 -(5) to give N- (2, 6-difluorobenzoyl) – 4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L-phenylalanine ethyl ester (2.75 g) . mp . 70-72 °C; IR (Nujol) 3400, 3263, 1735, 1654, 1624 cm“1; MS (APCI) m/z 500 (M+H) . (2) To a solution of the product obtained above (1.72 g) in DMSO (20 ml) were added Et3N (4.8 ml) and S03«pyridine (5.6 g) successively at room temperature. The whole mixture was stirred at room temperature for 25 minutes. The reaction mixture was poured into ice-water, and then the mixture was extracted with EtOAc. The organic layer was sequentially washed with 5% aqueous HCl, H20 and brine, dried (Na2S04) and then evaporated. The residue was purified by column chromatography (silica gel; eluent: n-hexane/EtOAc 5:1 to 1:1) to yield N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-formylphenyl) -L- phenylalanine ethyl ester (1.54 g) . mp. 114-116°C; IR (Nujol)
3332, 1735, 1695, 1657, 1644, 1623 cm“1; MS (APCI) m/z 498 (M+H) .
(3) The product obtained above (716 mg) was converted into the title compound (428 mg) in a similar manner as described in Example 1- (7) . mp . 87-89°C; IR (Neat+CHC13) 3300, 1739, 1668 cm“ 1; MS (APCI) m/z 528 (M+H) .
Example 11: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl ) -L-phenylalanine methyl ester.
(1) The product obtained in Example 2- (4) (1.00 g) was acylated with 2 , 6-difluorobenzoyl chloride to give N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L- phenylalanine methyl ester (873 mg) in a similar manner as described in Example l-(5). IR (Nujol) 3257, 1743, 1655, 1624 cm“ 1; MS (APCI +Q1MS) m/z 503 (M+NH4) , 486 (M+H) . (2) The product obtained above (860 mg) was converted into the title compound (220 mg) in a similar manner as described in Example 2- (6) and (7).
Example 12: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
…………………….
PATENT
https://www.google.com/patents/WO2003072536A1?cl=en
OUT LINE
phenylalanine derivative of the formula (I) :

wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :

wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :

wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :

wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :

wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :

wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :

Ethyl (ocS) – – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4- hydroxybenzene propionate and ethyl (otS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.

Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Ethyl (αS) -α-amino-4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate
Another name: ethyl (2S) -2-amino-3- [4- (4-ethoxymethyl- 2, 6-dimethoxyphenyl) phenyl]propanoate
Ethyl (αS) – [ [1, 1-dimethylethoxy] carbonyl] amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate
Another name 1: ethyl (2S) -2- [ (t-butoxycarbonyl) – amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) – phenyl]propanoate
Another name 2: Ethyl N- (t-butoxycarbonyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
Ethyl (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionate Another name 1: Ethyl (2S) -2- [ (2, 6- difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6- di ethoxyphenyl) phenyl] propanoate
Another name 2: Ethyl N- [2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
(ocS) – – [ (2, 6-Difluorobenzoyl) amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionic acid
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.
References
5. Firategrast
| WO2002018320A2 | 27 Ago 2001 | 7 Mar 2002 | Tanabe Seiyaku Co | INHIBITORS OF α4 MEDIATED CELL ADHESION |
| WO2003072536A1 | 27 Fev 2003 | 4 Set 2003 | Tanabe Seiyaku Co | A process for preparing a phenylalanine derivative and intermediates thereof |
| WO2003072537A2 | 6 Fev 2003 | 4 Set 2003 | Abbott Lab | Selective protein tyrosine phosphatatase inhibitors |
Mitsubishi Tanabe Pharma Corporation


Mitsubishi Tanabe Pharma Corporation
Pharmacological research building

 |
||
| ■Mitsubishi Tanabe Pharma Corporation Pharmacological research building |

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
K 912, NC 6300, Epirubicin nano



Cancer Research UK

Irish Cancer Society
Macmillan Cancer Support
NHS Evidence
 Epirubicin – Substance Summary
Epirubicin – Substance Summary
PubChem
MedlinePlus
– See more at: http://www.cancerindex.org/Epirubicin#sthash.BBioCC6K.dpuf
PHASE 1 JAPAN SOLID TUMOURS
DNA/RNA Synthesis Inhibitor
WITH Nano Carrier Co.,Ltdhttp://pdf.irpocket.com/C4571/qnwX/eFou/vG1J.pdf
KOWA COMPANY LTD
CAS FREE FORM. 56420-45-2
Smiles
- O=C2c1c(O)c5c(c(O)c1C(=O)c3cccc(OC)c23)C[C@@](O)(C(=O)CO)C[C@@H]5O[C@@H]4O[C@H]([C@H](O)[C@@H](N)C4)C
- NMR
- http://file.selleckchem.com/downloads/nmr/S122302-Epirubicin-Hydrochloride-NMR-Selleck.pdf
- http://www.medkoo.com/Product-Data/Epirubicin/Epirubicin-QC-TZC20130604web.pdf
-
Epirubicin CAS No.: 56420-45-2 Synonyms: - 4′-Epi-DX;
- Epirubicina;
- WP 697;
- Pidorubicin;
- 4′-epiadriamycin;
- Adriblastin;
- epi-dx;
- Epiadriamycin;
- farmorubicin;
- imi28;
- Farmarubicin;
Formula: C27H29NO11 Exact Mass: 543.17400
NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin.
Epirubicin is widely used to treat various human tumors. However, it is difficult to achieve a sufficient antitumor effect because of dosage limitation to prevent cardiotoxicity. We hypothesized that epirubicin-incorporating micelle would reduce cardiotoxicity and improve the antitumor effect. NC-6300 comprises epirubicin covalently bound to PEG polyaspartate block copolymer through an acid-labile hydrazone bond. The conjugate forms a micellar structure of 40-80 nm in diameter in an aqueous milieu. NC-6300 (10, 15 mg/kg) and epirubicin (10 mg/kg) were given i.v. three times to mice bearing s.c. or liver xenograft of human hepatocellular carcinoma Hep3B cells. Cardiotoxicity was evaluated by echocardiography in C57BL/6 mice that were given NC-6300 (10 mg/kg) or epirubicin (10 mg/kg) in nine doses over 12 weeks. NC-6300 showed a significantly potent antitumor effect against Hep3B s.c. tumors compared with epirubicin. Moreover, NC-6300 also produced a significantly longer survival rate than epirubicin against the liver orthotopic tumor of Hep3B. With respect to cardiotoxicity, epirubicin-treated mice showed significant deteriorations in fractional shortening and ejection fraction. In contrast, cardiac functions of NC-6300 treated mice were no less well maintained than in control mice. This study warrants a clinical evaluation of NC-6300 in patients with hepatocellular carcinoma or other cancers.

K-912(NC-6300)の概要 K-912(NC-6300)は、世界的に幅広く使用されているアントラサイクリン系の抗が ん剤の一つであるエピルビシンを内包したミセル化ナノ粒子製剤で、その特性により、 エピルビシンの有する心毒性の軽減が期待できます。さらに、pH 応答性システムを採 用することで、腫瘍細胞内でのエピルビシンの放出量を高め、既存のエピルビシンに比 べより強力な抗腫瘍効果が期待できます。
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US andPharmorubicin or Epirubicin Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound’s cytotoxic effects. Epirubicin also generates free radicalsthat cause cell and DNA damage.
Epirubicin is favoured over doxorubicin, the most popular anthracycline, in some chemotherapy regimens as it appears to cause fewer side-effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar – it has the opposite chirality – which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas.
Development history
The first trial of epirubicin in humans was published in 1980.[1] Upjohn applied for approval by the U.S. Food and Drug Administration(FDA) in node-positive breast cancer in 1984, but was turned down because of lack of data.[2] It appears to have been licensed for use in Europe from around this time however.[3] In 1999 Pharmacia (who had by then merged with Upjohn) received FDA approval for the use of epirubicin as a component of adjuvant therapy in node-positive patients.
Patent protection for epirubicin expired in August 2007.
References
- Bonfante, V; Bonadonna, G; Villani, F; Martini, A (1980). “Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia”. Recent results in cancer research 74: 192–9. PMID 6934564. PM6934564.
- “On Target”.
- According to the proprietary database iddb.com
External links
- http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/Epirubicin.htm
- http://www.pfizerpro.com/page_not_found?rid=/wyeth_html/home/minisites/oncology/ellence/pi/description.html
1H NMR PREDICT

13C NMR PREDICT

COSY
1H NMR

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (8R,10S)-10-((2S,4S,5R,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione | |
| Clinical data | |
| Trade names | Ellence |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603003 |
| Intravenous | |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 77% |
| Metabolism | Hepatic glucuronidationand oxidation |
| Excretion | Biliary and renal |
| Identifiers | |
| 56420-45-2 |
|
| L01DB03 | |
| PubChem | CID 41867 |
| DrugBank | DB00445 |
| ChemSpider | 38201 |
| UNII | 3Z8479ZZ5X |
| KEGG | D07901 |
| ChEBI | CHEBI:47898 |
| ChEMBL | CHEMBL417 |
| Chemical data | |
| Formula | C27H29NO11 |
| 543.519 g/mol | |
KOWA COMPANY LTD


Nano Carrier Co


P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
