
Home » Posts tagged 'Janus kinase inhibitor'
Tag Archives: Janus kinase inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cenacitinib



Cenacitinib
CAS 2641636-52-2
MF C19H19F2N7O3 MW431.4
Urea, N-[(1R,2S)-2-fluorocyclopropyl]-N′-[5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl]-
N-{5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl}-N′-[(1R,2S)-2-fluorocyclopropyl]urea
Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
VTX958 for the Treatment of Moderately to Severely Active Crohn’s Disease
CTID: NCT05688852
Phase: Phase 2
Status: Terminated
Date: 2025-07-03
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750705&_cid=P22-MKEUDK-45432-1
Example 4: Synthesis of 1-(5-((7-fluoro-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl)-3-((1R,2S)-2-fluorocyclopropyl)urea (5)


| Step 1: To a solution of 1E (100 g, 288 mmol) and 2E (57 g, 345 mmol) in dry 1,4-dioxane (3000 mL) under N 2 atmosphere was added Cs 2CO 3 (141 g, 432 mmol), Pd(OAc) 2 (5.2 g, 23.3 mmol) and BINAP (28.6 g, 46.6 mmol). After stirring at 115° C. overnight, the reaction mixture was cooled to rt. and diluted with hexane (3000 mL). The solid was collected by filtration and washed with 2×1500 mL (50% hexane in DCM). The solid was suspended into 5000 mL water and stirred for 1 h. The solid was collected by filtration and dried under vacuum to afford compound 2 (90 g, 65%) as a brown solid. |
PAT
Publication Number: US-2021139486-A1
Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: EP-4054581-A1Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: US-2023348478-A1Priority Date: 2019-11-08
- Substituted pyrazolo[1,5-a]pyrimidines as TYK2 pseudokinase ligandsPublication Number: US-11753411-B2Priority Date: 2019-11-08Grant Date: 2023-09-12
- TYK2 pseudokinase ligandsPublication Number: CN-114929226-BPriority Date: 2019-11-08Grant Date: 2024-09-27
- TYK2 pseudokinase ligandPublication Number: CN-114929226-APriority Date: 2019-11-08
- Preparation of a tyk2 inhibitorPublication Number: WO-2024151992-A1Priority Date: 2023-01-13
- Crystalline forms of a tyk2 inhibitorPublication Number: US-2024010654-A1Priority Date: 2022-07-06
- Crystalline forms of TYK2 inhibitorsPublication Number: CN-119816502-APriority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: EP-4551576-A1Priority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: WO-2024011136-A1Priority Date: 2022-07-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//////cenacitinib, cenacitinib, Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Zemprocitinib


Zemprocitinib
CAS 2417414-44-7
MF C16H19N5O2S MW 345.4 g/mol
N-[3-(3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-3-yl)-1-bicyclo[1.1.1]pentanyl]propane-1-sulfonamide
N-[3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl]propane-1-sulfonamide
Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Zemprocitinib (also known as LNK01001) is a selective Janus kinase (JAK) 1 inhibitor, a type of small molecule drug being developed for inflammatory and autoimmune conditions like rheumatoid arthritis, atopic dermatitis, and ankylosing spondylitis. It works by blocking the JAK1 enzyme, reducing the inflammatory signals that cause these diseases, and has shown promising results in clinical trials, with development reaching Phase 3.
Key Aspects:
- Drug Class: JAK1 Inhibitor.
- Mechanism: Blocks Janus Kinase 1, a key enzyme in inflammatory pathways.
- Developer: Initially Lynk Pharmaceuticals.
- Potential Uses: Rheumatoid Arthritis, Atopic Dermatitis, Ankylosing Spondylitis, Psoriasis, Alopecia Areata.
- Development Stage: Reached Phase 3 clinical trials for several indications.
- Chemical Info: CAS: 2417414-44-7; Formula: C16H19N5O2S.
In Summary:
Zemprocitinib is an investigational drug targeting inflammation by inhibiting JAK1, with potential to treat various autoimmune disorders, showing strong efficacy in early clinical trials for conditions like rheumatoid arthritis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US347660217&_cid=P21-MJDP3D-82397-1
Example 1



Step 1. 4-Chloro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1b)
Step 2. 4-Chloro-5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1c)
Step 3. Tert-butyl 3-((5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (Id)
Step 4. Tert-butyl 3-((5-amino-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (le)
Step 5. Tert-butyl 3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentane-1-carboxylate (1f)
Step 6. 3-(6-Tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentane-1-carboxylic acid (1g)
Step 7. Tert-butyl (3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1h)
Step 8. Tert-butyl (3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1i)
Step 9. 3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-amine 2,2,2-trifluoroacetate (1j)
Step 10. N-(3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)propane-1-sulfonamide (1)
PAT
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: ES-2993867-T3Priority Date: 2018-11-01Grant Date: 2025-01-10
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: JP-2024147699-APriority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: EP-3856742-B1Priority Date: 2018-11-01Grant Date: 2024-10-02
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2022009927-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023357247-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023339950-A1Priority Date: 2018-11-01
- Tricyclic Janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: AU-2019372677-B2Priority Date: 2018-11-01Grant Date: 2024-05-30
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: TW-202432555-APriority Date: 2018-11-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Zemprocitinib, Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
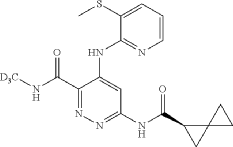
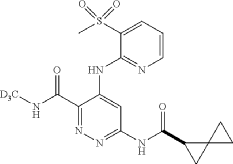
Lomedeucitinib
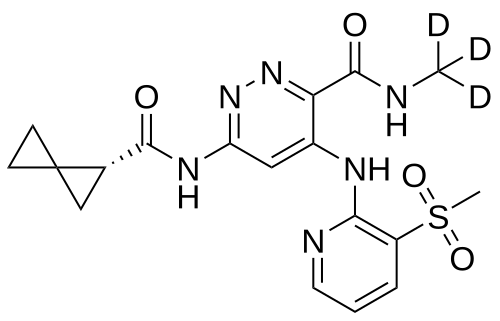
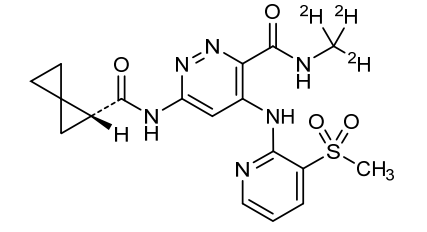
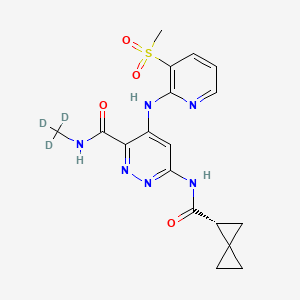
Lomedeucitinib
CAS 2328068-29-5
MF C18H172H3N6O4S
MW 419.5 g/mol

4-{[3-(methanesulfonyl)pyridin-2-yl]amino}-N-(2H3)methyl-6-[(1R)-spiro[2.2]pentane-1-carboxamido]pyridazine-3-carboxamide
4-[(3-methylsulfonyl-2-pyridinyl)amino]-6-[[(2R)-spiro[2.2]pentane-2-carbonyl]amino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Lomedeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis and psoriatic arthritis. It is a tyrosine kinase 2 (TYK2) inhibitor.[1]
- A Study to Evaluate Effectiveness and Safety of BMS-986322 in Participants With Moderate-to-Severe PsoriasisCTID: NCT05730725Phase: Phase 2Status: CompletedDate: 2024-09-19
- A Study to Evaluate the Drug Levels, Metabolism, and Removal of BMS-986322 in Healthy Adult Male ParticipantsCTID: NCT06088264Phase: Phase 1Status: CompletedDate: 2024-03-29
- A Study Investigating Interactions Between BMS-986322 and Rosuvastatin, Metformin and Methotrexate in Healthy ParticipantsCTID: NCT05615012Phase: Phase 1Status: CompletedDate: 2024-03-27
- A Study to Investigate the Interaction of BMS-986322 and a Combined Oral Hormonal Contraceptive (Ethinyl Estradiol [EE]/Norethindrone [NET]) in Healthy Female ParticipantsCTID: NCT05579574Phase: Phase 1Status: CompletedDate: 2023-08-18
- A Study to Assess the Safety and Tolerability of BMS-986322 in Healthy Participants of Japanese DescentCTID: NCT05546151Phase: Phase 1Status: CompletedDate: 2023-06-22
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US333829535&_cid=P10-MHIXWK-98212-1
General Scheme for Examples 252 and 253:
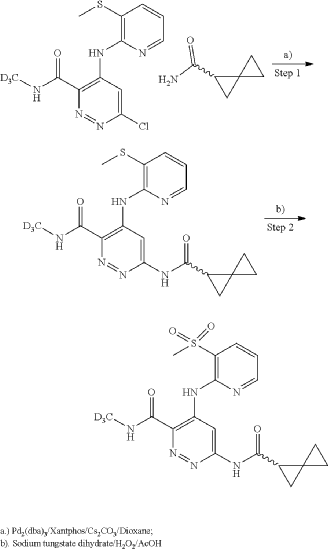
Example 252
Step 1
| A mixture of cesium carbonate (149 mg, 0.457 mmol), Xantphos (14.43 mg, 0.025 mmol), Pd 2(dba) 3 (11.42 mg, 0.012 mmol), 6-chloro-N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)pyridazine-3-carboxamide (65 mg, 0.208 mmol), and (R)-spiro[2.2]pentane-1-carboxamide (50.8 mg, 0.457 mmol) in dioxane (3 mL) was degassed using a vacuum/N2 fill cycle three times. The reaction was heated at 110° C. for 16 hours. The reaction was diluted with water and DCM. The DCM layer was separated and washed two more times with water and then dried (Na 2SO 4), filtered and concentrated. Purification via automated flash chromatography, eluting with methanol in DCM from 0 to 10%, gave the title compound (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (54 mg, 67% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 12.15 (br s, 1H), 9.88 (s, 1H), 8.68 (br s, 1H), 8.36 (br d, J=3.5 Hz, 1H), 8.25 (br s, 1H), 7.72 (br d, J=7.4 Hz, 1H), 6.97 (br dd, J=7.0, 5.1 Hz, 1H), 2.51 (s, 3H), 2.21-2.09 (m, 1H), 1.58-1.10 (m, 6H), 1.08-0.93 (m, 5H). |
| LCMS (ESI) m/e 388.1 [(M+H) +, calc’d C 18H 18D 3N 6O 2S 1, 388.1]; LC/MS retention time (method D): t R=0.80 min. |
Step 2
To a suspension of hydrogen peroxide (30% solution in water, 0.258 mL, 2.52 mmol) and (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (0.0489 g, 0.126 mmol) in AcOH (1 mL) was added sodium tungstate dihydrate (0.042 g, 0.126 mmol) at room temperature. After stirring at room temperature for 1 hour, the reaction was diluted with water, basified with Na 2CO 3 powder and extracted three times with DCM. The DCM layers were combined, washed with Na 2S 2O 3 (5% solution), dried (Na 2SO 4), filtered and concentrated. The crude product was purified using reverse phase prepHPLC to give the title compound (R)—N-(methyl-d3)-4-((3-(methylsulfonyl)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (16.2 mg, 31%) as a colorless solid. 1H NMR (500 MHz, DMSO-d 6) δ 12.07 (s, 1H), 11.22 (s, 1H), 9.49 (s, 1H), 9.16 (s, 1H), 8.63 (dd, J=4.6, 1.5 Hz, 1H), 8.29 (dd, 0.1=7.8, 1.4 Hz, 1H), 7.34 (dd, 0.1=7.8, 4.7 Hz, 1H), 2.48-2.43 (m, 1H), 1.46-1.41 (m, 1H), 1.42-1.36 (m, 1H), 0.95-0.82 (m, 3H), 0.80-0.73 (m, 1H). (3H methyl sulfone was buried under DMSO peak). LCMS (ESI) m/e 420.0 [(M+H) +, calc’d C 18H 18D 3N 6O 4S, 420.1]; LC/MS retention time (method E): t R=1.38 min; OR: −205.39 (20° C.).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US242383764&_cid=P10-MHIXVD-97150-1
PAT
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11787779-B2Priority Date: 2017-11-21Grant Date: 2023-10-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2024002364-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-102702228-B1Priority Date: 2017-11-21Grant Date: 2024-09-02
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: NZ-805343-APriority Date: 2017-11-21
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-2023098942-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2023255024-A1Priority Date: 2017-11-21
- Heteroaryl compounds substituted with sulfone pyridinylalkylamidesPublication Number: CN-111315737-BPriority Date: 2017-11-21Grant Date: 2024-06-18
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B2Priority Date: 2017-11-21
- Sulfonepyridine alkylamide substituted heteroaryl compoundsPublication Number: JP-7490107-B2Priority Date: 2017-11-21Grant Date: 2024-05-24
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: TW-I776994-BPriority Date: 2017-11-21Grant Date: 2022-09-11
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-7258903-B2Priority Date: 2017-11-21Grant Date: 2023-04-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-B2Priority Date: 2017-11-21Grant Date: 2023-08-03
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2019152948-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: CA-3083122-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-20200089706-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11021462-B2Priority Date: 2017-11-21Grant Date: 2021-06-01
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2021253554-A1Priority Date: 2017-11-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986322 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2328068-29-5 |
| PubChem CID | 138620496 |
| IUPHAR/BPS | 13210 |
| UNII | EYQ7KA55XA |
| KEGG | D12725 |
| ChEMBL | ChEMBL5314608 |
| Chemical and physical data | |
| Formula | C18H17D3N6O4S |
| Molar mass | 419.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ahsan S, Degener R, Schlamp M (2024). “Non-Invasive Treatments Invade the Psoriasis Pipeline”. Drugs in Context. 13: 2024–5–6. doi:10.7573/dic.2024-5-6. PMC 11313207. PMID 39131603.
////////lomedeucitinib, Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Girocitinib
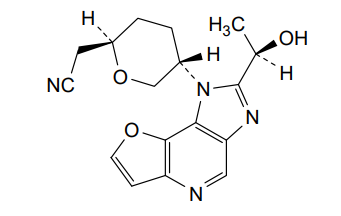
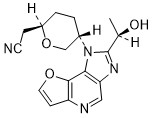
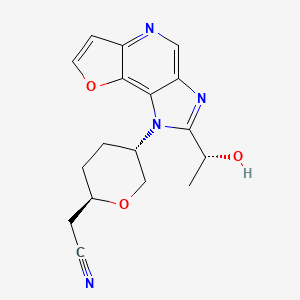
Girocitinib
CAS 2222137-79-1
MFC17H18N4O3 MW 326.36
2-[(2R,5S)-5-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile
[(2R,5S)-5-{2-[(1R)-1-hydroxyethyl]-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl}oxan-2-yl]acetonitrile
2-((2R,5S)-5-(2-((R)-1-hydroxyethyl)-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl)tetrahydro-2H-pyran-2-yl)acetonitrile
Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
In an era where targeted therapies are redefining the landscape of medical treatment, Girocitinib emerges as a beacon of hope for many. This innovative drug, developed by leading pharmaceutical research institutions, primarily targets specific proteins involved in disease progression. Classified as a tyrosine kinase inhibitor (TKI), Girocitinib has shown significant promise in the treatment of various cancers, particularly non-small cell lung cancer (NSCLC). The drug is currently in the advanced stages of clinical trials, with researchers optimistic about its potential to provide a more effective and less toxic treatment option compared to conventional therapies.
Girocitinib is designed to interfere with the signaling pathways that promote cancer cell growth and survival. It does this by inhibiting the activity of tyrosine kinases, enzymes that play a key role in the activation of many proteins by signaling pathways within the cell. Tyrosine kinases are often overactive in cancer cells, leading to unchecked proliferation and survival. By targeting these enzymes, Girocitinib effectively disrupts these malign processes, thereby slowing down or even halting the progression of the disease.
The primary indication for Girocitinib is non-small cell lung cancer (NSCLC), which accounts for approximately 85% of all lung cancer cases. NSCLC is notoriously difficult to treat, especially in its advanced stages, and current treatments often come with significant side effects. Clinical trials have shown that Girocitinib can significantly improve progression-free survival in patients with specific genetic mutations that make them more responsive to TKI therapy. These mutations can be identified through genetic testing, allowing for a more personalized treatment approach that increases the likelihood of success.
In addition to NSCLC, researchers are exploring the potential of Girocitinib to treat other types of cancer, including colorectal cancer and certain forms of leukemia. Early-stage trials have shown encouraging results, suggesting that Girocitinib could become a versatile tool in the oncology arsenal. Its ability to target specific molecular pathways makes it a promising candidate for combination therapies, which aim to enhance treatment efficacy while minimizing resistance and adverse effects.
The development of Girocitinib is a testament to the power of modern science and technology in addressing some of the most challenging health issues of our time. The drug’s journey from the laboratory to clinical trials has been marked by rigorous research and collaboration among scientists, healthcare professionals, and patients. As we await the results of ongoing studies, there is a palpable sense of anticipation in the medical community, as Girocitinib holds the promise of transforming cancer treatment for many patients.
In conclusion, Girocitinib represents a significant advancement in the field of targeted cancer therapy. Its mechanism of action, which involves the inhibition of tyrosine kinases, offers a more precise and potentially less harmful treatment option for patients with NSCLC and possibly other cancers. As research progresses, Girocitinib may well become a cornerstone in the fight against cancer, providing hope and improved outcomes for countless individuals around the world.
PDT PAT
WO2018067422
SYN
https://patents.google.com/patent/US10738060B2/en?oq=US10738060
Example 4: Synthesis of 2-[(2R,5S)-5-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl] acetonitrile (4)
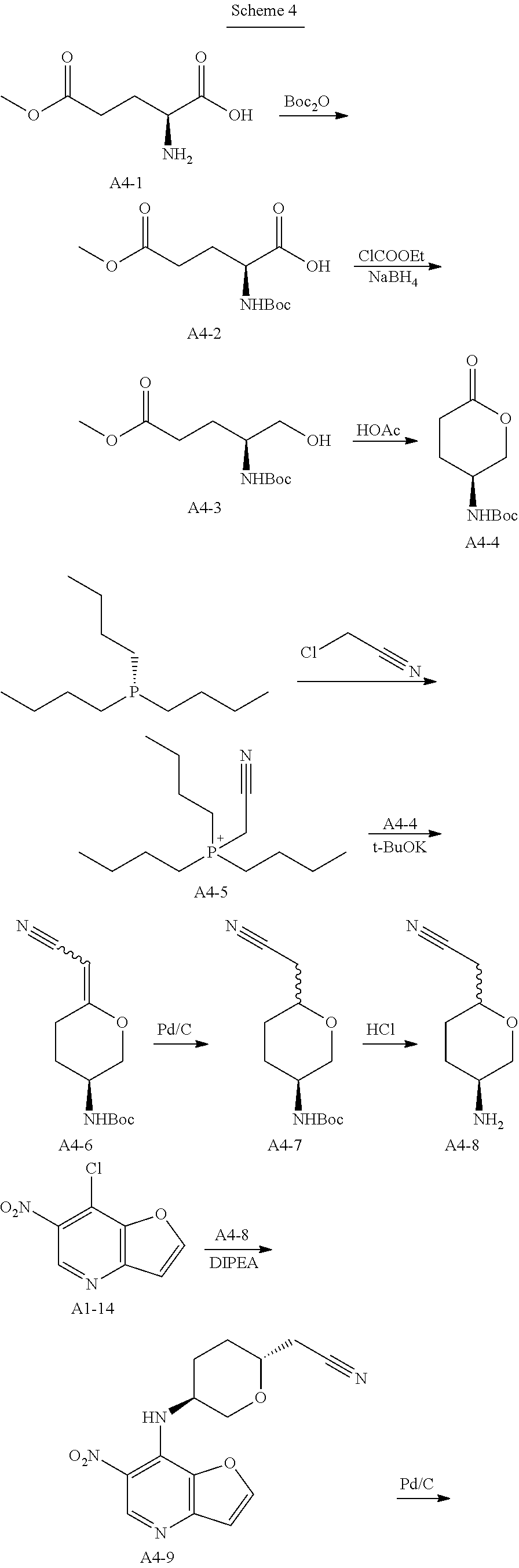
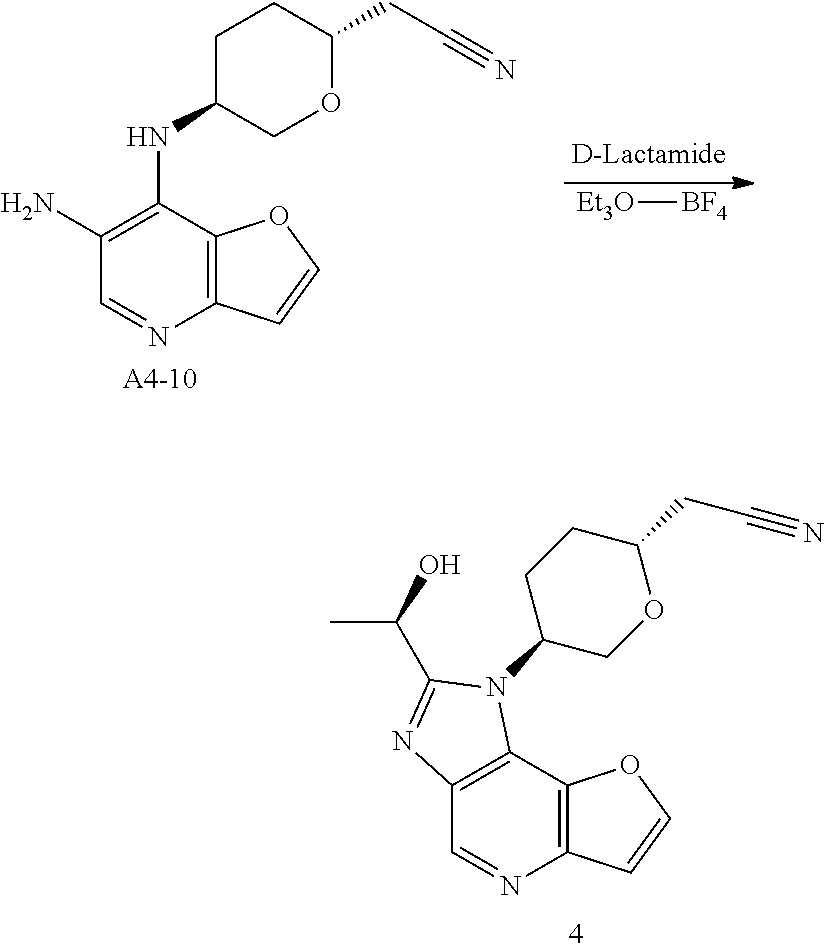
Step 1. In a round bottom flask, triethylamine (188 g, 1.86 mol, 1.0 eq) was added dropwise to a stirred solution of di-tert-butyl dicarbonate (162 g, 0.744 mol, 1.2 eq) and compound A4-1 (100 g, 0.62 mol, 1.0 eq) in water (500 mL) and 1,4-dioxane (500 mL). After stirring for 18 hrs at room temperature, the solution was extracted with MTBE (500 mL*2) and the aqueous phase was cooled on ice and carefully acidified to pH 3 by slow addition of 10% citric acid solution. The urethane was then extracted twice with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, and concentrated to give compound A4-2 as clear viscous oil (180 g, yield 100%). MS-ESI:[M+1]+: 262.1
Step 2. A solution of compound A4-2 (40 g, 0.153 mmol, 1.0 eq) in THF (600 mL) was treated with 4-methylmorpholine (17 g, 0.168, 1.1 eq) at room temperature. The resulting mixture was cooled to 0° C. before being treated with isobutyl chloroformate (22.7 g, 0.166 mmol, 1.08 eq) dropwise. The resulting reaction mixture was stirred at 0° C. for an addition 20 mins before being filtered and washed with THF. Then the clear filtrate solution was cooed to 0° C., and treated with a solution of NaBH4 (11.2 g, 0.295 mol, 1.93 eq) in water (100 mL). The resulting mixture was stirred overnight at room temperature, and then quenched with an aqueous HCl solution (1.0 mol/L,200 mL) dropwise, The mixture was extracted with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-3 as a yellow oil (25 g, yield 66%). MS-ESI:[M+1]+: 248.1
Step 3. A solution of compound of A4-3 (25 g, 0.1 mol, 1.0 eq) in toluene (300 mL) and acetic acid (150 mL) was heated to reflux for 5 hrs and then cooled, concentrated under vacuum. The residual was added saturated sodium bicarbonate solution to pH 7-8 in ice-bath. Then the mixture was extracted three times with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized by ethyl acetate and PE to give compound A4-4 as a white powder (8.0 g, yield 37.2%). GC-MS: 215
Step 4. A solution of tributyl phosphine (72.9 g, 0.36 mol, 1.0 eq) in nitromethane (500 mL), was added dropwise chloroacetonitrile (27.2 g, 0.36 mol, 1.0 eq) in nitrogen atmosphere. The resulting reaction mixture was stirred for 16 hrs at room temperature, then concentrated. The residual oil solidified when a small amount of ethyl acetate was added. The solid was recrystallized by ethyl acetate and DCM to afford compound A4-5 as a white powder (95 g, yield 95%).
Step 5. To a solution of dry compound A4-5 (8.3 g, 30 mmol, 3.0 eq) in N,N-dimethylacetamide (30 mL) in nitrogen atmosphere, was added solid Potassium tert-butoxide (3.1 g, 28 mmol, 2.8 eq) in portions at 0° C. The resulting mixture was gradually warmed to 30° C. and stirred for 2 hrs. The resulting ylide solution was then treated with compound A4-4 (2.15 g, 10 mmol, 1.0 eq), and stirred overnight at 70° C. After cooled to room temperature, the resulting slurry was poured into the mixture of ice-water (100 mL) and saturated sodium bicarbonate solution (100 mL). The mixture was extracted twice with ethyl acetate, and the combined extracts was washed three times with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-6 as yellow oil without purification (7.5 g, yield 100%). MS-ESI:[M+1]+: 239.1
Step 6. To a solution of compound A4-6 (7.5 g, 10 mmol, 1.0 eq) in methanol (200 mL), was added 10% Pd/C (0.5 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and purified by silica gel column chromatography to give compound A4-7 as off-white powder (1.6 g, yield 66.7%). MS-ESI:[M+1]+: 241.1
Step 7. To a solution of compound A4-7 (1.6 g, 6.67 mmol, 1.0 eq) in DCM (20 mL), was added TFA (10 g, 88.5 mmol, 13.2 eq). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A4-8 as light-brown oil (950 mg, yield 100%). MS-ESI:[M+1]+: 141.1
Step 8. To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (600 mg, 3.0 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A4-8 (950 mg, 6.7 mmol, 2.26 eq) and DIPEA (1.36 g, 10.5 mmol, 3.5 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A4-9 (2R,5S) as light-yellow powder (254 mg, yield 28.0%).MS-ESI: [M+1]+: 303.1.
1H NMR (300 MHz, d6-DMSO): 9.063 (s, 1H), 8.503 (d, 1H), 9.326 (d, 1H), 7.176 (d, 1H), 4.431-4.513 (m, 1H), 4.128-4.156 (m, 1H), 3.633-3.659 (m, 1H), 3.448-3.518 (m, 1H), 2.775-2.841 (m, 2H), 2.205-2.312 (m, 1H), 1.829-1.859 (m, 2H), 1.501-1.521 (m, 1H).
Step 9. To a solution of compound A4-9 (254 g, 0.84 mmol, 1.0 eq) in methanol (20 mL), was added 10% Pd/C (0.15 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and compound A4-10 was obtained as yellow oil (230 mg, yield 100%). MS-ESI:[M+1]+: 273.1
Step 10. A solution of D-Lactamide (388 mg, 4.2 mmol, 5.0 eq) and Et3O—BF4 (1.3 g, 6.72 mmol, 8.0 eq) in THF (10 mL) was stirred for 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (230 mg, 0.84 mmol, 1.0 eq) in ethanol (10 mL). After stirring for 3 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added water and extracted four times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution to pH 8, extracted twice with ethyl acetate. The second organic phases was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as light-yellow powder (120 mg, yield 43.8%). MS-ESI: [M+1]+: 327.6,
1H NMR (300 MHz, CDCl3): 9.039 (s, 1H), 7.939 (d, 1H), 7.196 (d, 1H), 5.235-5.336 (m, 1H), 4.806-4.973 (m, 1H), 4.403-4.483 (t, 1H), 4.096-6.116 (m, 2H), 2.700-2.807 (m, 4H), 2.105-2.312 (m, 2H), 1.830-1.852 (d, 3H).
SYN
US2022227777
https://patents.google.com/patent/US20220227777A1
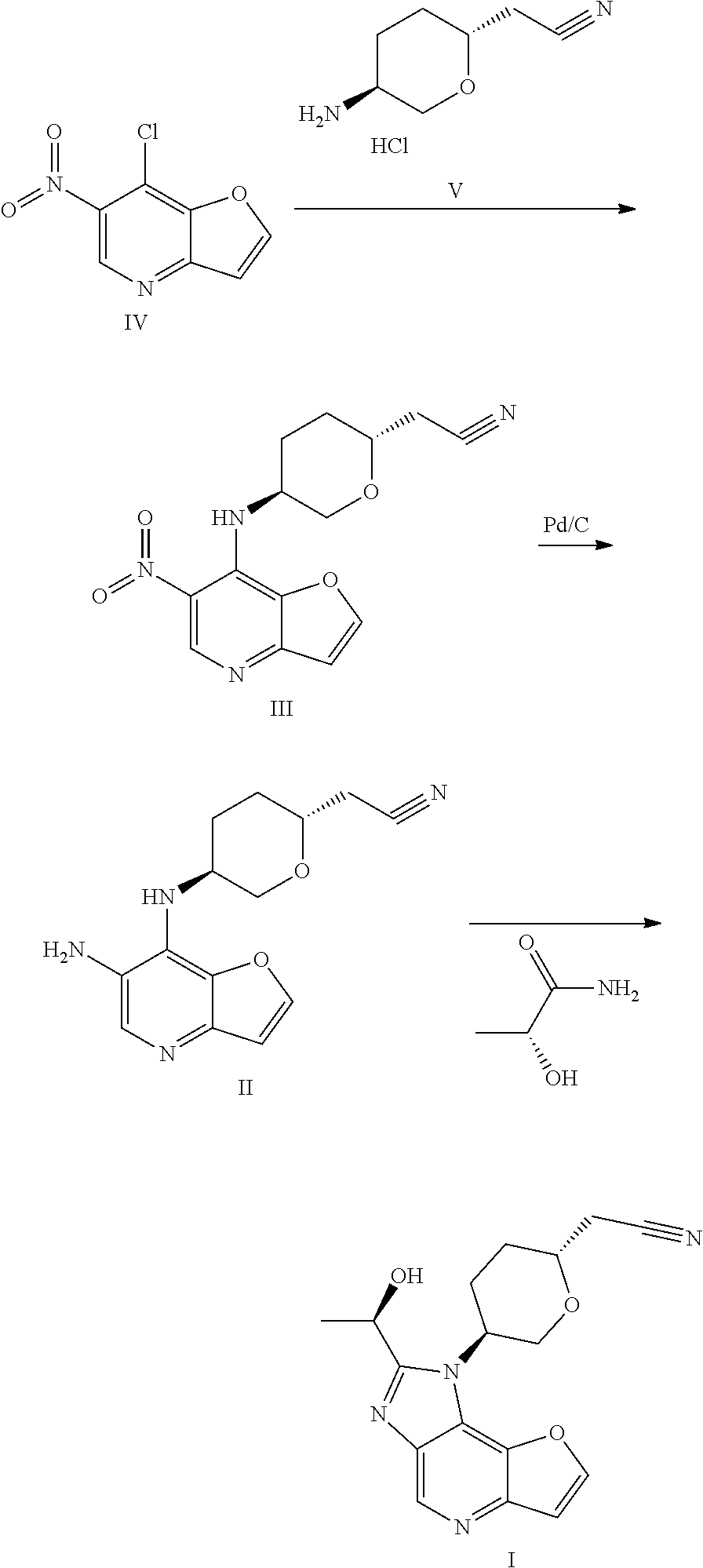
International patent application WO2018067422A1 discloses 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives as selective JAK1 kinase inhibitors and preparation methods thereof, wherein compound I and its preparation method is disclosed.
Preparation of a Compound of Formula I
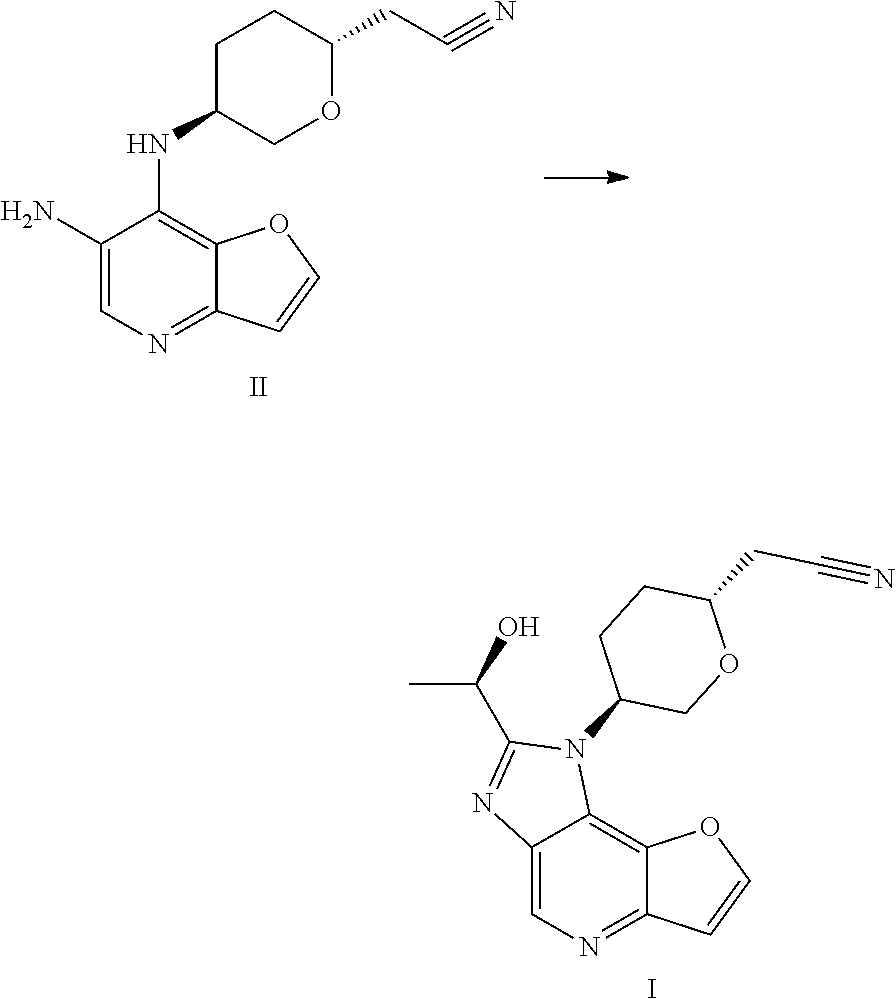
- [0204]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5.0 g, 1.0 eq) and ethanol (80 mL, 16 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 with a syringe dropwise within 10-20 minutes; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (80 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (50 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (60 mL) and EA (10 mL), respectively; the filter cake was dried under vacuum at 50° C. for 16 hours; 4.3 g of faint yellow solid was obtained, with a purity of 95.0%; the solid was dissolved with methanol (30 mL); 4.1 g of silicon based metal eliminator and 1.0 g of activated carbon were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (30 mL); the filtrate was concentrated with rotary evaporator until there was basically no fraction flowing out; methanol (10 mL) and MTBE (25 mL) were added to the residue, the system was heated to 50° C., and was stirred for 0.5 hour, then was cooled, the system was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (25 mL); the filter cake was dried under vacuum at 50° C. for 16 hours, 3.2 g of faint yellow solid was obtained, with a purity of 97.9%.
- [0205]MS-ESI: [M+1]+: 327.6
- [0206]1H NMR (400 MHz, CDCl3): 8.988 (s, 1H), 7.922 (d, 1H), 7.175 (d, 1H), 5.200-5.265 (m, 1H), 4.859-4.942 (m, 1H), 4.350-4.406 (t, 1H), 4.020-4.108 (m, 2H), 3.067 (d, 1H), 2.619-2.779 (m, 3H), 2.108-2.269 (m, 2H), 1.790-1.895 (m, 3H).
- [0207]THF (650 mL, 12 V), (R)-lactamide (70.6 g, 4.0 eq) and Et3O—BF4 (150.6 g, 4.0 eq) were added to a 1000 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (54 g, 1.0 eq) and ethanol (860 mL, 16 V) were added to another 2000 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 1 hour; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (450 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (270 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (540 mL); MTBE (270 mL) was added to the filter cake, the system was stirred at room temperature for 0.5 hour, filtered, the filter cake was washed with MTBE (108 mL); the filter cake was dried under vacuum at 50° C. for 16 hours; 49.2 g of light yellow solid was obtained, with an HPLC purity of 94.2%; the solid was dissolved with methanol (380 mL); silicon based metal eliminator (44 g) and activated carbon (5.4 g) were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (430 mL); the filtrate was concentrated with a rotary evaporator to (80-110 mL, 1.5 V-2 V); MTBE (540 mL) was added to the residue, the system was heated to 50° C., and was stirred for 1 hour, then was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (270 mL); 42.4 g of filter cake was obtained, with an HPLC purity of 96.9%; the filter cake was dried under vacuum at 50° C. for 16 hours, 41.0 g of light yellow solid was obtained, with an HPLC purity of 96.7%, a yield of 63.3%.
- [0208]Purification of a Compound of Formula I:
- [0209]A compound of formula I (41 g) was dissolved with methanol; silica gel (50 g) was added to the solution, the system was concentrated to dryness for later use; silica gel (200 g) was added to the chromatographic column, the column was compacted with an air pump; a compound of formula I mixed with silica gel was added to the chromatographic column, the column was compacted with an air pump; the chromatographic column was eluted with an eluent (VMeOH:VDCM=1:100-1:30); qualified components were collected, concentrated to dryness; the product was dried under vacuum at 50° C. for 16 hours; 36 g of off-white solid was obtained, with an HPLC purity of 98.5%.
- [0210]The MS-ESI and 1H NMR data are consistent with example 21.
- [0211]THF (60 mL, 6 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.9 g, 4.0 eq) were added to a 100 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (100 mL, 10 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 20 minutes; the system was heated to 80±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 0.5 hour; the system was cooled to room temperature 20-30° C.; the reaction liquid was concentrated to about 50-80 mL with a rotary evaporator between 30-40° C.; water (100 mL, 10 V) was added to the system, then the system was concentrated with a rotary evaporator between 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (5.5 g) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with saturated potassium carbonate solution (23 g); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and MTBE (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 18 g of earth yellow solid was obtained, with an HPLC purity of 93.5%.
- [0212]The MS-ESI and 1H NMR data are consistent with example 21.
- [0213]THF (120 mL, 12 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.8 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (140 mL, 14 V) were added to another 500 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 1 hour; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 4.5 hours; the system was cooled to room temperature, and water (20 mL, 2V) was added; the system was concentrated with a rotary evaporator at 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (3 mL) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with 50% potassium carbonate solution (15 mL); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the crude product was triturated and stirred with water (50 mL) at 20-25° C. for 1 hour; the system was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 17.8 g of khaki solid was obtained, with an HPLC purity of 95.3%.
- [0214]The MS-ESI and 1H NMR data are consistent with example 21.
- [0215]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5 g, 1.0 eq) and ethanol (70 mL, 14 V) were added to another 250 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 20 minutes; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 3 hours; the system was cooled to room temperature and was filtered, the filter cake was washed with THF (10 mL); water (10 mL, 2V) was added to the filtrate; the filtrate was concentrated with a rotary evaporator to 10-20 mL (2V-4V), the concentrated residue was exchanged with ethyl acetate (25 mL×2) and concentrated to 10-20 mL (2V-4V); water (50 mL, 10V) was added to the concentrated residue; the internal temperature was controlled at 20-25° C., 12M HCl (4.1 g) was used to adjust the pH of the system to 1-2; activated carbon (0.5 g) was added to the system, and the system was stirred at room temperature for 2 hours, and was filtered, the filter cake was washed with water (10 mL) and 1M HCl (10 mL); the combined filtrate was extracted with ethyl acetate (25 mL×2), the organic phase was discarded; the internal temperature was controlled at 20-25° C., the pH of the system was adjusted to 9-10 with saturated potassium carbonate solution (15 g); the internal temperature was controlled at 15-20° C., the system was stirred for 1 hour, and was filtered, the filter cake was washed with water (10 mL); the filter cake was triturated with acetone aqueous solution (50 mL, V/V=1:1) for 1 hour; the system was filtered, the filter cake was washed with acetone aqueous solution (10 mL, V/V=1:1); the filter cake was dried with an air blower at 50° C. for 24 hours; 5.0 g of pale gray solid was obtained, with an HPLC purity of 95.6%, and a yield of 83.5%;
- [0216]Purification of a Compound of Formula I:
- [0217]5.0 g of the obtained solid and methanol (40 mL) were added to a flask, and were stirred for 10 minutes at room temperature, the materials were basically dissolved and the solution was clear; activated carbon (0.5 g) and silica gel (4.0 g) were added to the system; the system was heated to 50-55° C., the temperature was maintained and the system was stirred for 2 hours, then was filtered with silica gel (5 g), the filter cake was washed with methanol (50 mL); the filtrate was concentrated with a rotary evaporator to 5-10 mL; MTBE (50 mL) was added to the concentrated residue; the system was heated to reflux, and was allowed for reflux for 1 hour; the system was cooled to 5-10° C., the temperature was maintained and the system was stirred for 1 hour and was filtered, the filter cake was washed with MTBE; the filter cake was dried with a drying oven under vacuum at 50° C. for 16 hours; 3.0 g of off-white solid was obtained, with a yield of 60% and a purity of 97.9%; the filtrate was concentrated to dryness to obtain 1.4 g of yellow solid.
- [0218]The MS-ESI and 1H NMR data are consistent with example 21.
PAT
- NEW SELECTIVE JAK1 INHIBITORS AND THEIR USEPublication Number: HR-P20211965-T1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: KR-102399848-B1Priority Date: 2016-10-03Grant Date: 2022-05-19
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-BPriority Date: 2016-10-03Grant Date: 2022-10-28
- Jak1 selective inhibitors and uses thereofPublication Number: US-RE49834-EPriority Date: 2016-10-03Grant Date: 2024-02-13
- Novel jak1 selective inhibitors and uses thereofPublication Number: US-2019256523-A1Priority Date: 2016-10-03
- JAK1 selective inhibitors and uses thereofPublication Number: US-10738060-B2Priority Date: 2016-10-03Grant Date: 2020-08-11
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-108366994-BPriority Date: 2016-10-03Grant Date: 2021-10-01
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-APriority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-B1Priority Date: 2016-10-03Grant Date: 2021-11-17
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof.Publication Number: MX-2024006688-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-12195476-B2Priority Date: 2019-06-06Grant Date: 2025-01-14
- Novel jak1 selective inhibitors and uses thereofPublication Number: CA-3039178-A1Priority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-A1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: JP-2019537559-APriority Date: 2016-10-03
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: JP-2023089169-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-113906035-BPriority Date: 2019-06-06Grant Date: 2023-11-10
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-117327083-APriority Date: 2019-06-06
- METHOD OF SYNTHESIS OF FUROIMIDAZOPYRIDINE COMPOUND, CRYSTAL FORM OF FUROIMIDAZOPYRIDINE COMPOUND, AND CRYSTAL FORM OF ITS SALT.Publication Number: MX-2024004146-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-2022227777-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-B2Priority Date: 2019-06-06Grant Date: 2023-05-11
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: WO-2020244348-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound and crystal form of salt thereofPublication Number: CN-113906035-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-3981771-A1Priority Date: 2019-06-06

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Girocitinib, Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
Frevecitinib
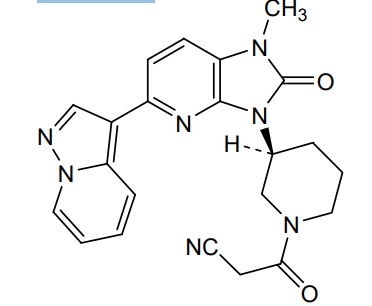
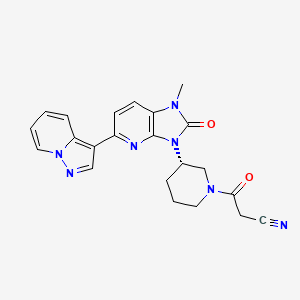
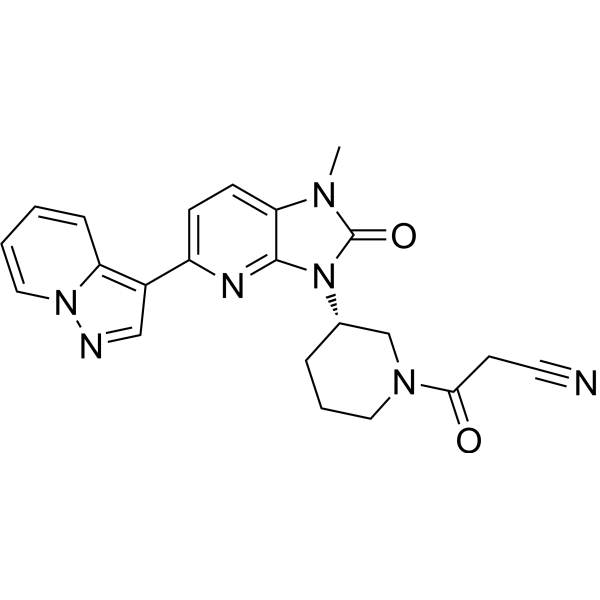
Frevecitinib
CAS 1299417-07-4
MF C22H21N7O2 MW 415.4 g/mol
3-[(3S)-3-(1-methyl-2-oxo-5-pyrazolo[1,5-a]pyridin-3-ylimidazo[4,5-b]pyridin-3-yl)piperidin-1-yl]-3-oxopropanenitrile
3-{(3S)-3-[1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridin-3-
yl)-1,2-dihydro-3H-imidazo[4,5-b]pyridin-3-yl]piperidin1-yl}-3-oxopropanenitrile
Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002
Single and Multiple Ascending Dose Study of KN-002
CTID: NCT05006521
Phase: Phase 1
Status: Completed
Date: 2024-08-07
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011157397&_cid=P11-MH2TVG-48083-1
SYN
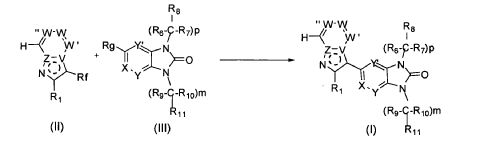
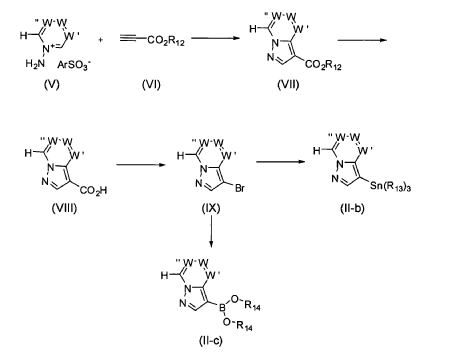
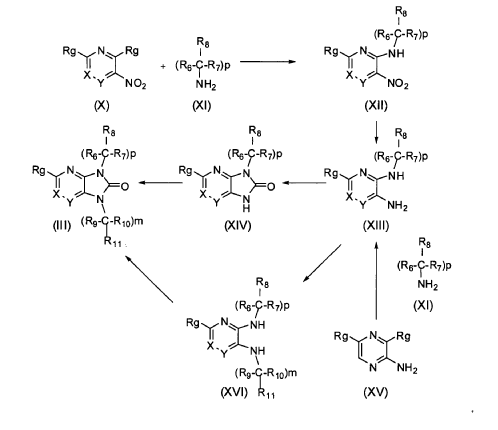
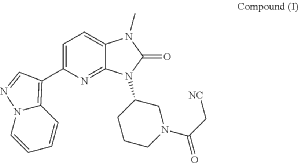
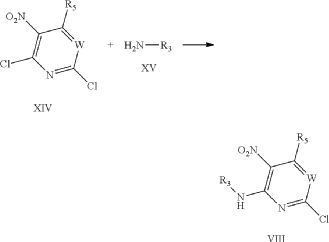
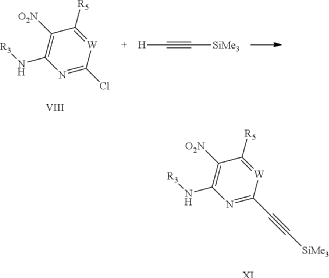
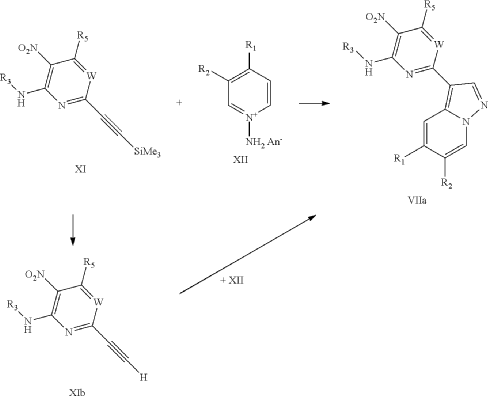
It has now been found that a drug substance disclosed in WO2011/051452, namely the compound (S)-3-(3-(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3-yl)-1H-imidazo[4,5-b]pyridine-3(2H)-yl)piperidin-1-yl)-3-oxopropanenitrile having the structure shown below and known herein as compound (I) can be prepared in different polymorphic forms. Surprisingly one form exists as a polymorph with particularly advantageous stability properties. Compound (I) as prepared following the process in WO2011/051452 is known as Form I herein.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76222175&_cid=P11-MH2U0A-51623-1
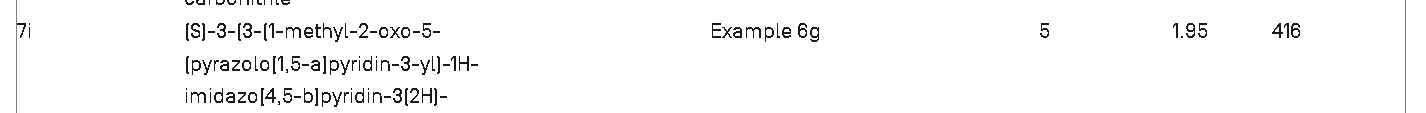
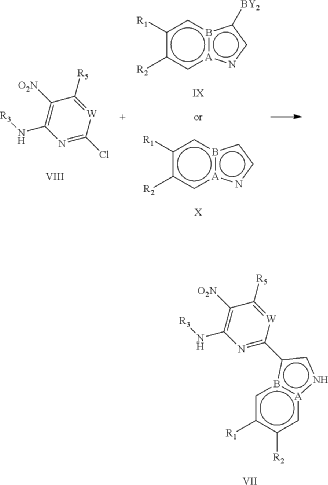
PAT
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: KR-101675614-B1Priority Date: 2009-10-29Grant Date: 2016-11-11
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2012245140-A1Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2013131038-A9Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8501735-B2Priority Date: 2009-10-29Grant Date: 2013-08-06
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8946257-B2Priority Date: 2009-10-29Grant Date: 2015-02-03
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: EP-2582703-A1Priority Date: 2010-06-15
- Heteroaryl Imidazolone Derivatives as Jap InhibitorsPublication Number: KR-20130113331-APriority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: US-2013089512-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: WO-2011157397-A1Priority Date: 2010-06-15
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: EP-2493895-B1Priority Date: 2009-10-29Grant Date: 2017-04-26
- Novel polymorphsPublication Number: US-2018016284-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: US-2019031687-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: WO-2016124464-A1Priority Date: 2015-02-05
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: CA-2802588-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as JAK inhibitorsPublication Number: CN-102933583-APriority Date: 2010-06-15
- Novel polymorphsPublication Number: EP-3053927-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: EP-3253769-B1Priority Date: 2015-02-05Grant Date: 2019-03-13
- New polymorphPublication Number: JP-2018502929-APriority Date: 2015-02-05
- New polymorphPublication Number: JP-6685326-B2Priority Date: 2015-02-05Grant Date: 2020-04-22
- PolymorphsPublication Number: US-10087196-B2Priority Date: 2015-02-05Grant Date: 2018-10-02
- Crystalline form of a JAK3 kinase inhibitorPublication Number: US-10155757-B2Priority Date: 2015-03-10Grant Date: 2018-12-18
- Crystalline form of a jak3 kinase inhibitorPublication Number: US-2018044336-A1Priority Date: 2015-03-10
- Crystalline form of a jak3 kinase inhibitorPublication Number: WO-2016142201-A1Priority Date: 2015-03-10
- Polymorphic forms of (s)-3-(3(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3(2h)-yl)piperidin-1-yl)-3-oxopropanenitrilePublication Number: CA-2972977-CPriority Date: 2015-02-05Grant Date: 2019-04-09
- polymorphPublication Number: CN-107207533-BPriority Date: 2015-02-05Grant Date: 2019-04-16
- Formulation of a pan-jak inhibitorPublication Number: TW-202440105-APriority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: US-2024261224-A1Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A2Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A3Priority Date: 2022-12-02
- Crystalline form of a jak3 kinase inhibitorPublication Number: EP-3268364-A1Priority Date: 2015-03-10
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Frevecitinib, Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002
Envudeucitinib
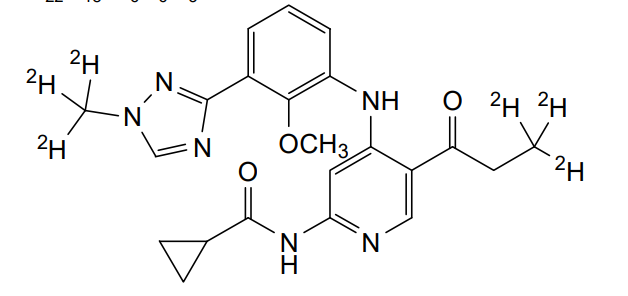
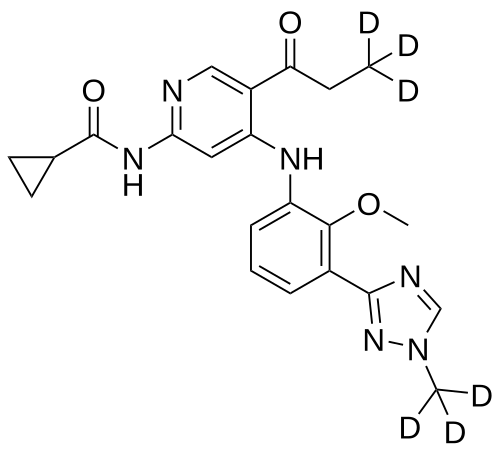
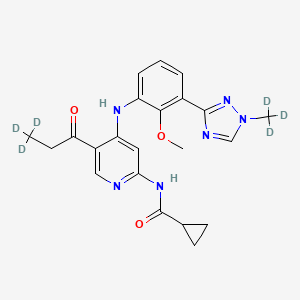
Envudeucitinib
CAS 2417135-66-9
MF C22H18[2]H6N6O3 MW426.5 g/mol
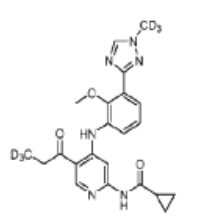
N-[4-{2-methoxy-3-[1-(2H3)methyl-1H-1,2,4-triazol-3-yl]anilino}-5-(3,3,3-2H3)propanoylpyridin-2-yl] cyclopropanecarboxamide
N-(4-(2-methoxy-3-(1-(trideuteriomethyl)-1,2,4-triazol-3-yl)anilino)-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl)cyclopropanecarboxamide
N-[4-[2-methoxy-3-[1-(trideuteriomethyl)-1,2,4-triazol-3-yl]anilino]-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl]cyclopropanecarboxamide
Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637
Envudeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis. It is a selective tyrosine kinase 2 (TYK2) inhibitor developed by Fronthera U.S. Pharmaceuticals LLC and now owned by Alumis, Inc. for the treatment of autoimmune diseases. Envudeucitinib targets the TYK2 signaling pathway, which plays a crucial role in regulating multiple pro-inflammatory cytokines such as IL-12, IL-23, and type I interferons.[1][2]
PAT
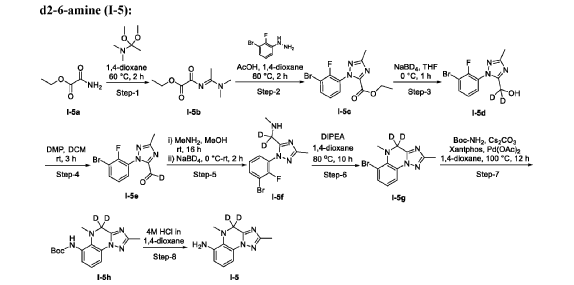
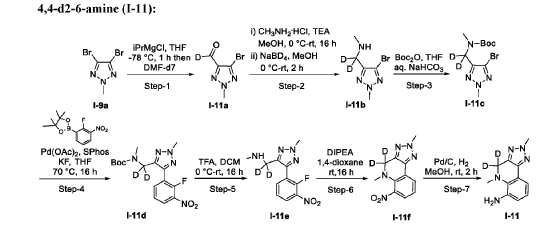
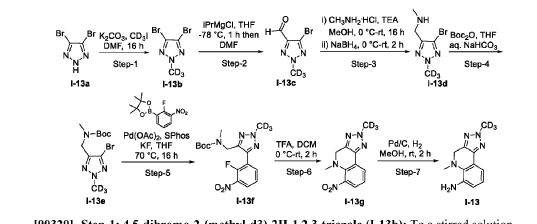
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024081603-A1Priority Date: 2022-10-10
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024059529-A1Priority Date: 2022-09-12
- Tyk2 inhibitors and uses thereofPublication Number: WO-2023227946-A1Priority Date: 2022-05-27
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024081603&_cid=P11-MGGDZU-88200-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023227946&_cid=P11-MGGE36-91523-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FTP-637 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2417135-66-9 |
| PubChem CID | 158715582 |
| IUPHAR/BPS | 13205 |
| UNII | KD2MDJ4GAB |
| KEGG | D13123 |
| Chemical and physical data | |
| Formula | C22H18D6N6O3 |
| Molar mass | 426.506 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Deng L, Wan L, Liao T, Wang L, Wang J, Wu X, et al. (August 2023). “Recent progress on tyrosine kinase 2 JH2 inhibitors”. International Immunopharmacology. 121 110434. doi:10.1016/j.intimp.2023.110434. PMID 37315371.
- Loo WJ, Turchin I, Prajapati VH, Gooderham MJ, Grewal P, Hong CH, et al. (2023). “Clinical Implications of Targeting the JAK-STAT Pathway in Psoriatic Disease: Emphasis on the TYK2 Pathway”. Journal of Cutaneous Medicine and Surgery. 27 (1_suppl): 3S – 24S. doi:10.1177/12034754221141680. PMID 36519621.
////////Envudeucitinib, Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637

{kind=link}
{kind=link}