

| Patent ID
|
Title
|
Submitted Date
|
Granted Date
|
|---|---|---|---|
| US9790208 | CRYSTALLINE SALT FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1-YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AS OREXIN RECEPTOR ANTAGONIST |
2014-12-02
|
|
| US2016368901 | CRYSTALLINE FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1 -YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AND ITS USE AS OREXIN RECEPTOR ANTAGONISTS |
2014-12-02
|
Home » Posts tagged 'Insomnia'
Tag Archives: Insomnia
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DIMDAZENIL


DIMDAZENIL
CAS 308239-86-3
WeightAverage: 372.81
Monoisotopic: 372.1101515
Chemical FormulaC17H17ClN6O2
EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201
7-Chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-4,5-dihydro-5-methyl-6H-imidazo[1,5-a]
[1,4]benzodiazepin-6-one
7-chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-5-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-6-one
EVT 201 is a novel partial positive allosteric modulator of the GABAA receptor complex which is being developed as a treatment for insomnia. It is being developed by Evotec Inc.
- OriginatorRoche
- DeveloperEvotec SE; Zhejiang Jingxin Pharmaceutical
- ClassBenzodiazepines; Chlorobenzenes; Dimethylamines; Imidazoles; Ketones; Oxadiazoles; Sleep disorder therapies; Small molecules
- Mechanism of ActionGABA A receptor modulators
- RegisteredInsomnia
- 29 Nov 2023Registered for Insomnia in China (PO) – First global approval
- 24 Oct 2023Efficacy and adverse events data from a phase III trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
- 21 Oct 2023Efficacy and adverse events data from a phase II trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
Dimdazenil, sold under the brand name Junoenil, is a medication used in the treatment of insomnia in China.[1] It is a benzodiazepine derivative and a partial positive allosteric modulator of the GABAA receptor[2] with two- to four-fold higher functional affinity for the α1 subunit relative to the α2, α3, and α5 subunits.
Medical use
Dimdazenil shows effectiveness in the treatment of insomnia, but has less intrinsic activity in comparison to currently-marketed benzodiazepines and the Z-drugs;[3] however, it is thought that the lower efficacy may result in fewer side effects, such as motor incoordination.[3] In China, dimdazenil is approved for short-term treatment of insomnia.[4]
History
Dimdazenil was originally developed by Roche, based on preclinical data, as a non-sedating anxiolytic, but was found to produce sedation in humans in phase I clinical trials. For this reason, it was subsequently licensed to Evotec, which is now developing it for the treatment of insomnia.[3] By 2007, dimdazenil completed phase II clinical trials for this indication, with positive findings reported.[5] In China, the drug was developed by Zhejiang Jingxin Pharmaceutical.
SCHEME

PATENT
CN111620834
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN306317338&_cid=P10-MAWAJX-84923-1
| Example 16 |
| |
| 1M lithium bis(trimethylsilyl)amide (320 mL, 0.32 mol, 3 eq, 1 Mol/liter) was added to the flask, nitrogen was passed through, the temperature was lowered to -15°C, and the compound K1 (22.6 g, 0.11 mol, 1 eq) obtained in Example 11 was added dropwise. After the addition, the mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the compound b (26 g, 0.11 mol, 1 eq) obtained by the method of Example 15 was added dropwise. The mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the mixture was naturally heated to room temperature, and glacial acetic acid was slowly added dropwise. The temperature was controlled to be below 35°C. After completion, the temperature was raised to 55-60°C, and the reaction was kept warm for 2 hours. Then, the mixture was transferred to a rotary evaporator, and the mixture was concentrated under reduced pressure at 45-50°C in batches. The temperature was lowered to 25-30°C, and water and dichloromethane were added in batches. The layers were stirred and separated, and the organic layer was collected. The aqueous layer was extracted once more with dichloromethane, and the organic layers were combined. The layers were washed with a saturated aqueous solution of sodium bicarbonate and water. After washing, the organic layers were collected and transferred to a rotary evaporator for concentration to obtain a solid. The solid was slurried with ethanol at -15°C to -5°C for 15 minutes, filtered, rinsed with cold ethanol, and dried under reduced pressure at 55-60°C to obtain a compound of formula I (36 g, 96.6%), MS: M ++ 1=373.1, HPLC purity 99.85%. |
| 1 H-NMR data: 1 H NMR (400 MHz, DMSO-d 6 δ8.57(s,1H),7.69(d,J=1.9Hz,3H),4.60(d,J=3.7Hz,2H),3.61(s,2H),3.05(s,3H),2.16(s,6H). |
| 13 C-NMR data: 13 C NMR (101 MHz, DMSO) δ 163.35, 163.25, 161.50, 138.88, 134.17, 133.15, 132.81, 130.95, 128.29, 122.67, 114.56, 110.52, 61.10, 46.6 (2), 41.77, 34.48. |
PATENT
WO2000069858
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2000069858&_cid=P10-MAWAOS-90001-1


EXAMPLE
a) 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5(lH)-dione (III).
25.0 g 6-chloro-isatoic anhydride (II) and 12.4 g sarcosine were suspended under stirring and argon atmosphere in 100 ml p-xylene and heated at reflux for two hours. The suspension was cooled to room temperature and further stirred 1 hour, then filtered off. The precipitate was washed with 25 ml p-xylene twice and dried at 50°C under vacuum. The solid so obtained (6-chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (II)) was digested in 75 ml deionized water at 0°C for 1 hour, filtered off, washed with 25 ml deionized water and dried under vacuum 18 hours at 80°C. Crude product: 25.2 g as a beige powder, m.p. 230-232°C
b) Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ 1,4] benzodiazepine- 3-carboxylate (V).
25.0 g 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (III) were suspended under stirring and argon atmosphere in 200 ml toluene and 32.1 ml N,N-dimethyl-p-toluidine. The suspension was heated to 100°C and 11.2 ml phosphorus oxychloride were added over 30 minutes and stirring was pursued two and an half hours at 100°C. The dark-orange solution was cooled to 40°C and toluene was removed under reduced pressure to give 82 g of a dark-orange oil.
Meanwhile, 81.2 ml hexamethyldisilazane and 265 ml tetrahydrofuran were mixed and cooled to -35°C. 229.5 ml Butyllithium were added over 45 minutes and, after stirring 30 minutes at -35°C, a solution of 35.2 g ethyl(dimethylamino-methylenamino)acetate in 70.4 ml tetrahydrofuran was added over 30 minutes. The orange solution obtained was stirred one more hour at -35°C and a solution of the crude iminochloride in 100 ml
tetrahydrofuran was added over 1 hour at -15°C. The dark red solution was stirred one hour at -15°C, then 18 hours at room temperature (r.t.). 75 ml Acetic acid were added in 10 minutes, then 75 ml deionized water were added in one portion and the orange suspension was heated at reflux for two hours. Tetrahydrofuran was removed under reduced pressure and the residue was partitioned between 200 ml dichloromethane and 100 ml deionized water. The phases were separated and the organic phase was washed with 100 ml aqueous HC1 IN twice and with 100 ml deionized water. The aqueous phases were extracted twice with 100 ml dichloromethane. The combined organic extracts were dried (Na2S04) and evaporated. The residue was digested in 200 ml n-heptane 30 minutes at r.t. and filtered off. The sticky crystals obtained were digested at reflux for 30 minutes in 213.5 ml ethanol, then stirred 3 hours to r.t. and 2 hours at -20°C. The precipitate (ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxylate (V)) was filtered off, washed three times with 20 ml ethanol and dried under reduced pressure 16 hours at 60°C. Crude product: 23.4 g as a beige powder, m.p. 225.5-226.5 °C c) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo [ 1 ,5-a] [ 1 ,4]benzodiazepine-3- carboxamide (VI).
22.8 g Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [l,4]- benzodiazepine-3-carboxylate (V)were suspended under stirring and argon atmosphere in 91.2 ml 1 ,4-dioxane. 14.1 ml Formamide and 13.9 ml sodium methanolate were successively added to yield a clear light-orange solution, which turned to a white suspension after 10 minutes. This suspension was stirred two hours at 30°C. 200 ml deionized water were added in one portion and 1,4-dioxane was distilled off at 40°C under reduced pressure. The remaining white suspension was stirred two hours at 0°C and filtered. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxamide (VI)) was washed with 50 ml deionized water three times and dried under reduced pressure for 18 hours at 80°C. Crude product: 19.43 g as a white powder. m.p.>250°C
d) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carbonitrile (VII).
19.0 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carboxamide (VI) were suspended under stirring and argon atmosphere in 95 ml 1,4- dioxane and 6.58 phosphorous oxychloride were added in one portion. The reaction mixture was heated to reflux for one hour giving a yellow solution, which was concentrated at 50°C under reduced pressure. The residue was digested in 100 ml deionized water for two hours at r.t.. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5- a] [ l ,4]benzodiazepine-3-carbonitrile (VII)) was filtered off, washed three times with 30 ml deionized water and dried under vacuum at 80°C for 18 hours. Crude product: 17.3 g as a light yellow powder, m.p. 238.5-239.5°C
_ e) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carboxamidoxime (VIII).
16.8 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carbonitrile (VIII) were suspended under stirring and argon atmosphere in 101 ml N,N- dimethylformamide and 13.48 g hydroxylamine hydrochloride was added in one portion. 34.2 ml Sodium methanolate were then added over 60 minutes to the yellow suspension, which turned to a colorless suspension. It was stirred one more hour at r.t., then cooled to 0-2°C and 202 ml deionized water were added over 30 minutes. After stirring one more hour at 0°C, the precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[l,5-a] [l,4]benzodiazepine-3-carboxamidoxime (VIII) was filtered off, washed twice with 40 ml deionized water and dried under vacuum at 70°C for 18 hours Crude product 17.84 g as a white powder m.p.>250°C
f) 7-Chloro-3- (5-chloromethyl- [ 1 ,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro- imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one (IX).
8.0 g 7-chloro-5,6-dιhydro-5-methyl-6-oxo-4H-ιmιdazo[ 1,5-a] [ l,4]benzodιazepιne-3-carboxamidoxime (VIII) and 1.0 g magnesium oxide were suspended under stirring and argon atmosphere in 160 ml 1,4-dioxane. 2 7 ml Chloracetyl chloride were added in one portion and the white thick gel obtained was stirred 4 hours at r.t. and then 17 hours at reflux to give a lightly orange fluid suspension 100 ml Dioxane were distilled off and the reaction mixture was cooled to room temperature. 180 ml Deionized water were added within 15 minutes and the suspension was stirred 1 hour at r.t . The precipitate was filtered off, washed with 50 ml deionized water twice and dried under vacuum at 80°C for 18 hours Crude product: 8.3 g as a light pink powder. This crude product was dissolved in 120 ml tetrahydrofuran at reflux and 0.83 g active charcoal Darco G 60 were added. The system was refluxed 1 hour, then filtered on 25 g Dicaht-Speedex and the filter cake was washed with three portions of 50 ml warm tetrahydrofuran. The filtrate was concentrated at 40°C under reduced pressure The residue was digested in 80 ml ethanol 1 hour at reflux, then stirred 16 hours at r.t. and finally 2 hours at 2°C. The precipitate (7-chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo [ 1,5-a] [ l,4]benzo-dιazepιn-6-one (IX)) was filtered off, washed with 2 portions of 25 ml cold tert-butyl ethvl- ether and dried under vacuum 5 hours at 80°C Crude product: 7.6 g as a light beige powder, m p. 234-238°C
g) 7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5- dιhydro-imidazo[l,5-a] [l,4]benzodιazepin-6-one (I).
7.0 g 7-Chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo-[ 1,5-a] [ l,4]benzodιazepιn-6-one (IX) were suspended under stirring and argon
atmosphere in 70 ml 1,4-dioxane and 25.7 ml dimethylamine (33% in ethanol) were added over 60 minutes The reaction mixture was stirred one more hour at r.t. and then the solvents were removed under reduced pressure at 35°C. The residue was partitioned between 50 ml dichloromethane and 20 ml deionized water. The phases were separated and the organic phase was washed twice with 20 ml deionized water. The aqueous phases were extracted separately with the same portion of 25 ml dichloromethane, twice. The combined organic extracts were dried (Na2SO4) and the solvent was removed under reduced pressure Crude product: 8.0 g as a light yellow foam Purification
The crude product was dissolved in 40 ml ethanol at reflux and 400 mg active charcoal Darco G 60 were added. The system was stirred 1 hour at reflux, then filtered on a hot pad of Dicalit Speedex, which was washed with two portions of 40 ml hot ethanol. The filtrate was concentrated to 14 g under reduced pressure, heated to reflux and at this temperature and 40 ml terf-butyl-methylether were added over 5 minutes. The suspension was cooled slowly to r.t., stirred 16 hours, further cooled to 2°C. After stirring 1 hour at 2°C, the precipitate was filtered off, washed with 20 ml tert-butyl-methylether and dried 1 hour at 60°C under vacuum. The so obtained powder was dissolved at reflux in 26 ml ethyl acetate. 6.5 ml Ethyl acetate were then distilled off and the turbid solution obtained was slowly cooled to r.t., then to 0°C. After 1 hour stirring at 0°C, the precipitate was filtered off, washed with 10 ml cold tert-butyl-methylether and dried under vacuum at 60°C for 16 hours. The so obtained powder (7-chloro-3-(5-dimethylaminomethyl-[ 1,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [l,4]benzodiazepin-6-one (I)) was crystallized a second time in 24.3 ml ethyl acetate according to the procedure described above. Product: 5.5 g as a white powder, m.p. 151.5-153°C
7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one maleate (1:1)
373 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [ l,4]benzodiazepin-6-one (I) and 116 mg maleic acid were dissloved in 3 ml hot ethanol. The salt crystalized on cooling. The suspension was stirred for 10 min at 0°C. Filtration and drying afforded 460 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[l,5-a] [ l,4]benzodiazepin-6-one maleate (1:1) as a white solid, m.p. 182-184°C
References
- ^ Huang Z, Zhan S, Chen C, Zhang R, Zhou Y, He J, et al. (February 2024). “Efficacy and safety of Dimdazenil in adults with insomnia disorder: results from a multicenter, randomized, double-blind, placebo-controlled phase III trials”. Sleep. 47 (2). doi:10.1093/sleep/zsad272. PMC 10851846. PMID 37875349.
- ^ Guilleminault C (2010). Sleep Medicine. Elsevier Health Sciences. pp. 574–. ISBN 978-1-4377-1836-2.
- ^ Jump up to:a b c Monti JM, Pandi-Perumal SR, Möhler H (28 September 2010). GABA and Sleep: Molecular, Functional and Clinical Aspects. Springer Science & Business Media. pp. 50–51. ISBN 978-3-0346-0226-6.
- ^ Syed YY (March 2024). “Dimdazenil: First Approval”. Drugs. doi:10.1007/s40265-024-02020-9. PMID 38546956.
- ^ Plunkett JW (September 2007). Plunkett’s Biotech & Genetics Industry Almanac 2008: Biotech & Genetics Industry Market Research, Statistics, Trends & Leading Companies. Plunkett Research, Ltd. pp. 311–. ISBN 978-1-59392-087-6.
External links
| Clinical data | |
|---|---|
| Trade names | Junoenil |
| Other names | EVT-201; EVT201 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 308239-86-3 |
| PubChem CID | 9885841 |
| DrugBank | DB05721 |
| ChemSpider | 8061514 |
| UNII | 6J8AF7CLE4 |
| ChEMBL | ChEMBL5095096 |
| CompTox Dashboard (EPA) | DTXSID301032055 |
| Chemical and physical data | |
| Formula | C17H17ClN6O2 |
| Molar mass | 372.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////DIMDAZENIL, EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201, CHINA 2023, INSOMNIA
Daridorexant

Daridorexant
- Molecular FormulaC23H23ClN6O2
- Average mass450.921 Da
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1[RN]
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
ACT-541468, , Nemorexant
FDA APPROVED 2022, 1/7/2022, To treat insomnia,

Daridorexant HCl
CAS#: 1792993-84-0 (HCl)
Chemical Formula: C23H24Cl2N6O2
Molecular Weight: 487.39
Elemental Analysis: C, 56.68; H, 4.96; Cl, 14.55; N, 17.24; O, 6.57
Methanone, ((2S)-2-(6-chloro-7-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)-, hydrochloride (1:1)
Daridorexant HCl; Daridorexant hydrochloride; ACT541468A; ACT 541468A; ACT-541468A; ACT541468 hydrochloride; ACT 541468 hydrochloride; ACT-541468 hydrochloride
Daridorexant HCl is used in the treat of Insomnia Disorder in Adult Patients
Daridorexant, sold under the brand name Quviviq, is a medication used for the treatment of insomnia.[1] Daridorexant is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals.[3][4] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[3][4] The medication has a relatively short elimination half-life of 6 to 10 hours.[2] As of April 2020, daridorexant has passed its first phase III clinical trial for the treatment of insomnia.[3]Daridorexant was approved for medical use in the United States in January 2022.[1][5][6]
Daridorexant, formerly known as nemorexant, is a selective dual orexin receptor antagonist used to treat insomnia. Insomnia is characterized by difficulties with sleep onset and/or sleep maintenance and impairment of daytime functioning. It chronically affects the person’s daily functioning and long-term health effects, as insomnia is often associated with comorbidities such as hypertension, diabetes, and depression. Conventional treatments for insomnia include drugs targeting gamma-aminobutyric acid type-A (GABA-A), serotonin, histamine, or melatonin receptors; however, undesirable side effects are frequently reported, such as next-morning residual sleepiness, motor incoordination, falls, memory and cognitive impairment. Novel drugs that target orexin receptors gained increasing attention after discovering the role of orexin signalling pathway in wakefulness and almorexant, an orexin receptor antagonist that improved sleep. Daridorexant was designed via an intensive drug discovery program to improve the potency and maximize the duration of action while minimizing next-morning residual activity.1
Daridorexant works on orexin receptors OX1R and OX2R to block the binding of orexins, which are wake-promoting neuropeptides and endogenous ligands to these receptors. Daridorexant reduces overactive wakefulness: in the investigational trials, daridorexant reportedly improved sleep and daytime functioning in patients with insomnia.1 It was approved by the FDA on January 10, 2022, under the name QUVIVIQ.6 as the second orexin receptor antagonist approved to treat insomnia following suvorexant.2
QUVIVIQ
- Generic Name: daridorexant tablets
- Brand Name: Quviviq
QUVIVIQ contains daridorexant, an orexin receptor antagonist. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5- methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
 |
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains 27 mg or 54 mg of daridorexant hydrochloride equivalent to 25 mg or 50 mg of daridorexant, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
Dosage Forms And Strengths
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with “25” on one side and “i” (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with “50” on one side and “i” (Idorsia logo) on the other side, containing 50 mg daridorexant.
QUVIVIQ tablets are available as:
25 mg, light purple, arc-triangle shaped film-coated tablets debossed with “25” on one side, and “i” on the other side. NDC 80491-7825-3, bottle of 30 with child-resistant closure
50 mg: light orange, arc-triangle shaped film-coated tablets debossed with “50” on one side, and “i” on the other side. NDC 80491-7850-3, bottle of 30 with child-resistant closure
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202000453
Since its discovery in 1998, the orexin system has been of interest to the research community as a potential therapeutic target for the treatment of sleep/wake disorders. Herein we describe our efforts leading to the identification of daridorexant, which successfully finished two pivotal phase 3 clinical trials for the treatment of insomnia disorders.

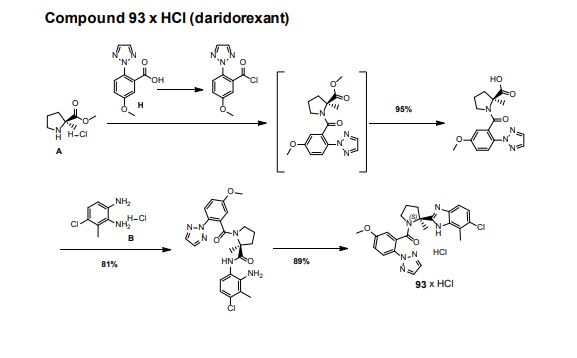
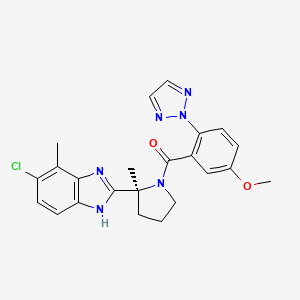
Step 3. Amide (S7) (1000 g, 2.13 mmol) was dissolved in EtOH (5 L) and 32% aqueous HCl (500 mL) was added at 23 °C. The solution was filtered through a Whatman filter (5 µm). The filtrate was heated to 75 °C for 4h. The resulting suspension was cooled to 0 °C and filtered. The product was dried under reduced pressure to yield 93 x HCl (922 g, 89%) as a white solid.
LC-MS B: tR = 0.78 min; [M+H]+ = 451.19, mp 280 °C.
1H NMR (500 MHz, D6-DMSO) δ: 15.05- 15.65 (m, 1 H), 8.06 (s, 2 H), 7.79 (s, 1 H), 7.75 (d, J = 8.9 Hz, 2 H), 7.66 (m, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.15 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 4.06-4.10 (m, 1 H), 3.92 (s, 3 H), 3.35 (s, 1 H), 2.78 (s, 3 H), 2.54-2.67 (m, 1 H), 2.23-2.31 (m, 1 H), 2.06-2.20 (m, 2 H), 1.97 (s, 3 H),
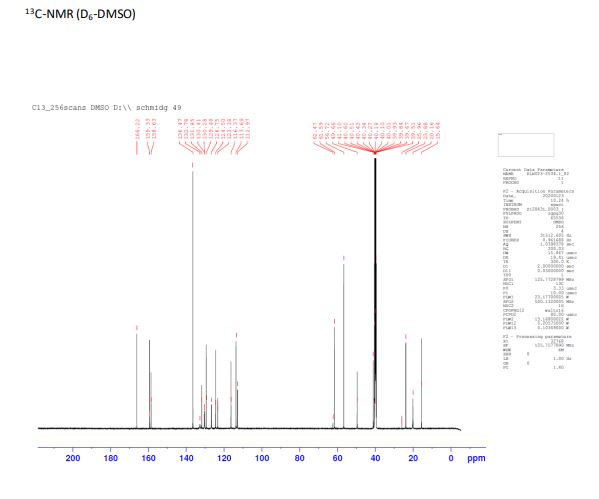
13C NMR (125 MHz, D6-DMSO) δ: 166.2, 159.3, 158.6, 136.5, 132.7, 131.9, 130.4, 130.3, 129.4, 126.8, 124.5, 123.4, 116.4, 113.7, 113.0, 61.6, 56.8, 49.7, 41.1, 23.9, 20.2, 15.7.

SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.201900618
Abstract
DORA explorers: The orexin system plays an important role in regulating the sleep-wake cycle. Herein we report our optimization efforts toward a novel dual orexin receptor antagonist (DORA) with improved properties over compound 6. Replacing the oxadiazole by a triazole resulted in compounds (e. g. compound 33) with improved properties, such as higher intrinsic metabolic stability, lower plasma protein binding, higher brain free fraction, and increased solubility. Further optimization was needed to decrease the compounds P-glycoprotein susceptibility. Our work led to the identification of compound 42, a potent, brain-penetrating DORA with improved in vivo efficacy in dogs compared with compound 6.

Abstract
The orexin system is responsible for regulating the sleep-wake cycle. Suvorexant, a dual orexin receptor antagonist (DORA) is approved by the FDA for the treatment of insomnia disorders. Herein, we report the optimization efforts toward a DORA, where our starting point was (5-methoxy-4-methyl-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-[5-(2-trifluoromethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-pyrrolidin-1-yl}methanone (6), a compound which emerged from our in-house research program. Compound 6 was shown to be a potent, brain-penetrating DORA with in vivo efficacy similar to suvorexant in rats. However, shortcomings from low metabolic stability, high plasma protein binding (PPB), low brain free fraction (fu brain), and low aqueous solubility, were identified and hence, compound 6 was not an ideal candidate for further development. Our optimization efforts addressing the above-mentioned shortcomings resulted in the identification of (4-chloro-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-methyl-2-[5-(2-trifluoromethoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-pyrrolidin-1-yl}l-methanone (42), a DORA with improved in vivo efficacy compared to 6.
PAT
WO 2015083071
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PAT
WO 2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689
Examples
Reference Example 1
Synthesis of 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
4,5-dibromo-2-(4-methoxy-2-nitrophenyl)-2H-1,2,3-triazole
4- Fluoro-3-nitroanisole (3.44 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (4.56 g, 1 eq.)1, K2C03 (2.78 g, 1 eq.) and DMF (30 mL) are heated to 1 10 °C for 32 h. The reaction mixture is cooled to 22 °C and treated with water (70 mL). The resulting suspension is filtered, washed with water (15 mL). The product is slurried in isopropanol (40 mL), filtered and dried under reduced pressure to yield a white solid. Yield: 6.42 g, 84%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, CDCI3) δ: 7.71 (d, J = 8.9 Hz, 1 H), 7.47 (d, J = 2.8 Hz, 1 H), 7.25 (dd, Ji = 2.8 Hz, J2 = 8.9 Hz, 1 H), 3.97 (s, 3 H).
1 X. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake, P. Wipf Organic Letters 2010 12 (20), 4632-4635.
5- methoxy-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(4-methoxy-2-nitrophenyl)-2/-/-1 ,2,3-triazole (2 g, 1 eq.), sodium acetate (1.3 g, 3 eq.), and 10% Pd/C 50% water wet (0.3 g) is suspended in EtOAc (10 mL). The mixture is heated to 50 °C and set under hydrogen until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with 1 N NaOH (10 mL) and water (15 mL). The organic layer is concentrated under reduced pressure to yield an oil. Yield: 0.95 g, 94%. Purity: 96% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.05 (s, 2 H), 7.53 (d, J = 8.9 Hz, 1 H), 6.49 (d, J = 2.7 Hz, 1 H), 6.30 (dd, Ji = 2.7 Hz, J2 = 8.9 Hz, 1 H), 5.94 (s, 2 H), 3.74 (s, 3 H).
5-methoxy-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline (455 g, 1 eq ) is dissolved in isopropanol (3 L). To the solution is added cone. H2SO4 (235 g, 1 eq.) below 40 °C. The suspension is cooled to
20 °C and filtered. The cake is washed with isopropanol (700 mL) and TBME (1.5 L). The product is dried to obtain a white solid. Yield: 627 g, 91 %. Purity: 100% a/a (LC-MS method 2).
2-(2-iodo-4-methoxyphenyl)-2H-1,2,3-triazole
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (200 g, 1 eq.) is dissolved in 2 M aq. H2SO4 soln. (1.4 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (62 g, 1.3 eq.) in water (600 mL) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of Kl (161 g, 1.4 eq.) in water (700 mL) at 65 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (27 g, 0.4 eq.) in water (120 mL). The mixture is extracted with isopropyl acetate (2 L). The organic layer is washed with a mixture of 2 N NaOH (500 mL) and 40% NaHS03 soln. (100 mL), and a mixture of 1 N HCI (50 mL) and water (500 mL). The organic layer is concentrated to dryness. The residue is dissolved in isopropanol (700 mL) and cooled to 0 °C. The resulting suspension is filtered. The solid is dried under reduced pressure. Yield: 164 g, 79%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.08 (s, 2 H), 7.57 (d, J = 2.8 Hz, 1 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.13 (dd, Ji = 2.8 Hz, J2 = 8.8 Hz, 1 H), 3.85 (s, 3 H).
5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-4-methoxyphenyl)-2/-/-1 ,2,3-triazole (200 g, 1 eq.) is dissolved in THF (2 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (350 mL, 1.05 eq.) is added at 0 °C. The mixture is cooled to -20 °C and C02 (gas) is bubbled into the solution over 30 min until the exothermicity is ceased. To the mixture is added 2 N HCI (600 mL) at 8 °C and concentrated under reduced pressure to remove 2.4 L solvent. The residue is extracted with TBME (1.6 L). The organic layer is washed with 1 N HCI (200 mL) and extracted with 1 N NaOH (600 mL and 200 mL). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (160 mL). The resulting suspension is filtered and washed with water (200 mL). Yield: 127 g, 87%. Purity: 100% a/a (LC-MS method 2); MP: 130 °C (DSC goldpan). The obtained product may be re-crystallized from toluene (MP: 130.9 °C) or water (MP: 130 °C).
Table Ref 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 2 (recrystallization from toluene)
Technique Data Summary Remarks
XRPD Crystalline see Fig. 8
Reference Example 2
Synthesis of 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
4,5-Dibromo-2-(5-methyl-2-nitrophenyl)-2H-1 ,2,3-triazole
3- Fluoro-4-nitrotoluene (1367 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (1999 g, 1 eq.), K2C03 (1340 g, 1.1 eq.) and DMF (1 1 L) is heated to 75 °C for 15 h. The reaction mixture is cooled to 22 °C and treated with water (18 L). The resulting suspension is filtered, washed with water (4 L). The product is washed with isopropanol (5 L), and dried under reduced pressure to yield a white solid. Yield: 281 1 g, 88%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.10 (d, J = 8.3 Hz, 1 H), 7.86 (d, J = 1.0 Hz, 1 H), 7.66 (dd, J1 = 0.9 Hz, J2 = 8.3 Hz, 1 H), 2.51 (s, 3 H).
4- Methyl-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(5-methyl-2-nitrophenyl)-2/-/-1 ,2,3-triazole (205 g, 1 eq.), sodium acetate (149 g, 3.2 eq.), and 5% Pd/C 50% water wet (37.8 g) is suspended in EtOAc (0.8 L). The mixture is heated to 40-50 °C and set under hydrogen (2 bar) until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with water (300 mL), 2N NaOH (300 ml_+250 mL) and water (300 mL). The organic layer is concentrated under reduced pressure to yield a yellow oil. Yield: 132 g, 90%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.09 (s, 2 H), 7.48 (d, J = 1.3 Hz, 1 H), 6.98 (dd, J1 = 1.8 Hz, J2 = 8.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 5.79 (s, 2 H), 2.23 (s, 3 H).
4-Methyl-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl) aniline (199 g, 1 eq ) is dissolved in isopropanol (1.7 L). To the solution is added cone. H2SO4 (118 g, 1.05 eq.) below 40 °C. The suspension is cooled to 20 °C and filtered. The cake is washed with isopropanol (500 mL). The product is dried to obtain a white solid. Yield: 278 g, 89%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.21 (s, 2 H), 7.70 (s, 1 H), 7.23 (s, 2 H), 2.35 (s, 3 H).
2-(2-iodo-5-methylphenyl)-2H-1 ,2,3-triazole
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (1553 g, 1 eq.) is dissolved in 1 M aq. H2S04 Soln. (1 1 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (433 g, 1.1 eq.) in water (4 L) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of potassium iodide (1325 g, 1.4 eq.) in water (4 L) at 55-70 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (220 g, 0.4 eq.) in water (900 mL). The mixture is extracted with isopropyl acetate (13 L). The organic layer is washed with a mixture of 2 N NaOH (3.5 L) and 40% NaHSOs soln. (330 g), and a mixture of 1 N HCI (280 mL) and water (3.5 L). The
organic layer is concentrated to dryness. Yield: 1580 g, 97%. Purity: 91 % a/a (LC-MS method 2). 1 H NMR (400 MHz, CDCI3) <5: 7.90 (s, 2 H), 7.87 (d, J = 8.1 Hz, 1 H), 7.34 (d, J = 1 .6 Hz, 1 H), 7.03-7.06 (m, 1 H), 2.40 (s, 3 H).
The crude product, together with a second batch (141 1 g) is purified by distillation on a short path distillation equipment at 120 °C jacket temperature, feeding tank (70 °C), cooling finger (20 °C) and at a pressure of 0.004 mbar. Yield: 2544 g (78%), Purity: 100 % a/a ()LC-MS method 2).
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-5-methylphenyl)-2/-/-1 ,2,3-triazole (1250 g, 1 eq.) is dissolved in THF (13 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (2.2 L, 1 eq.) is added at 0 °C. The mixture is cooled to -25 °C and CO2 (gas) is bubbled into the solution over 60 min until the exothermicity is ceased. To the mixture is added 2 N HCI (5 L) at 4 °C and concentrated under reduced pressure to remove 14.5 L solvent. The residue is extracted with TBME (10 L). The organic layer is extracted with 1 N NaOH (6 L and 3 L). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (1 .23 L). The resulting suspension is filtered and washed with water (5 L). Yield: 796 g, 89%. Purity: 100% a/a (LC-MS method 2); MP: 125 °C (DSC goldpan).
The following examples illustrate the invention.
Example 1 :
Example 1.1: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methoxybenzoic acid (21 .5 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (600 mL) and water (8.4 mL). To the mixture were added 1 H-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-/V,/V-dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 3.5 h. IPC showed full conversion. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 84: 16. The mixture was cooled to 40 °C and filtered. The cake was washed with dioxane (100 mL). The solid was dried to obtain 50.6 g of a blue solid. The ratio of N{2) to Λ/(1 ) isomer of was 98.6: 1 .4.
Table 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 1.2: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.1 was dissolved in water (300 mL). TBME (200 mL) and 32% aq. HCI (35 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (20 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 optionally seed crystals were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.4 g, 61 %. Purity: 100% a/a, tR 0.63 min. Seed crystals may be obtained by careful crystallization according to the above procedure.
MP: 80 °C (DSC).
1H NMR (400 MHz, DMSO) & 3.87 (s, 3 H), 7.26 (m, 2 H), 7.64 (d, J = 8.7 Hz, 1 H), 8.02 (s, 2 H), 13.01-13.22 (br, 1 H).
Table 2: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Example 1.3: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid, e.g. obtained according to the procedure of Reference Example 1 (5 g, 0.0228 mol) and KHCO3 (1.61 g, 0.7 eq) were suspended in dioxane (100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.56 g, 44%. 1H NMR (400 MHz, D20) & 3.80 (s, 3 H), 7.04 (m, 2 H), 7.46 (d, J = 8.7 Hz, 1 H), 7.82 (s, 2 H). MP: 279.5°C (DSC shows additionally a broad endothermic event at about 153 °C to 203 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
Table 3: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 2
Example 1.4: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
In an alternative procedure, 2-Bromo-5-methoxybenzoic acid (20 g, 0.086 mol, 1 eq.) copper (I) iodide (0.824 g, 0.05 eq.), and K2C03 powder (26.9 g, 2.25 eq.) were suspended in dioxane (494 mL). To the mixture was added 1 H-1 ,2,3-triazole (12 g, 2 eq.). The mixture was heated at reflux for 1 h. To the mixture was added water (12.5 g, 8 eq.). The mixture was heated at reflux for 2 h. Solvent (100 mL) was removed by distillation. The residue was cooled to 45 °C in 8 min, filtered and washed with dioxane (50 mL).
XRPD corresponds to crystalline form 1 (see Fig. 1 , Example 1.1 ).
Example 1.5: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.4 was dissolved in water (200 mL). The mixture was heated to 50 °C and 20% aq. H2SO4 (40 mL) was added to adjust the pH to 5. The mixture was filtered over Celite. The filtrate was treated at 45 °C with 20% aq. H2S04 (40 mL). At pH 3 seeds (obtained for example using the procedure of reference example 1 ) were added. The suspension was stirred at 45 °C and filtered. The product was washed with water (20 mL) and dried at 60 °C and 10 mbar to yield a white solid. Yield: 10.8 g, 57%. Purity: 100% a/a, tR 0.63 min.
Characterisation of 5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid obtained according to Example 1.5:
XRPD corresponds to crystalline form 1 (see Fig. 2, Example 1.2).
Example 2:
Example 2.1: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-4-methylbenzoic acid (20 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 A7-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-Λ/,ΛΑ-
dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of 98.5%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 75:25. The mixture was concentrated at normal pressure and external temperature of 130 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 48.8 g of a blue solid. The ratio of N(2) to Λ/(1 ) isomer was 98.7: 1 .3.
Table 4: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 2.2: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid
The solid of Example 2.1 was dissolved in water (300 mL) and filtered. To the filtrate were added TBME (200 mL) and 32% aq. HCI (30 mL). The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (10 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 1 1 .7 g, 62%. Purity: 100% a/a. tR 0.66 min.
MP: 125 °C (DSC).
1H NMR (400 MHz, DMSO) & 2.44 (s, 3 H), 7.41 (d, J = 7.9 Hz, 1 H), 7.56 (s, 1 H), 7.68 (d, J = 7.9 Hz, 1 H), 8.06 (s, 2 H), 12.53-13.26 (br, 1 H)
Table 5: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Technique Data Summary Remarks
XRPD Crystalline see Fig. 5
Example 2.3: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and KHC03 (1 .74 g , 0.7 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.47 g, 42% . MP: 277 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.28 (d, J = 7.9 Hz, 1 H), 7.39 (m, 2 H), 7.84 (s, 2 H).
MP: 276.8 °C (DSC shows additionally a broad endothermic event at about 140 °C to 208 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
XRPD corresponds to crystalline form 1 (see Fig. 4, Example 2.1 ).
Reference Example 3:
Reference Example 3.1: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid sodium salt (sodium 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), Na2CC>3 powder (24.6 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 5 h. IPC showed a conversion of >99%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 78:22. The mixture was concentrated at normal pressure and external temperature of 135 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 36.2 g of a yellow solid. The ratio of N(2) to Λ/( 1 ) isomer of was 99: 1 .
Table 6: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid sodium salt in crystalline form 1
Reference Example 3.2: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid obtaind in Reference Example 3.1 was dissolved in water (300 mL) and filtered. To the filtrate was added TBME (200 mL) and 32% aq. HCI (30 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with 1 N aq. HCI ( 100 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed
under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of Reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.1 g, 64%. Purity: 100% a/a. tR 0.67 min.
MP: 173 °C (DSC)
1 H NMR (400 MHz, DMSO) & 2.42 (s, 3 H), 7.50-7.52 (m, 1 H), 7.58 (s, 1 H), 7.63 (m, 1 H), 8.05 (s, 2 H), 13.01 (s, 1 H).
Table 7: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Reference Example 3.3: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid sodium salt
5-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and Na2C03 (1 .05 g, 0.4 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.79 g, 50%. MP: 341 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.30 (m, 2 H), 7.43 (m, 1 H), 7.83 (s, 2 H).
XRPD corresponds to crystalline form 1 (see Fig. 6, Reference Example 3.1 ).
Reference Example 3.4: 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.1 g, 2.5 eq.) were suspended in dioxane (600 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of >94%. The ratio of the desired N(2) to the regioisomeric Λ/( 1 ) isomer was 78:22. The mixture was cooled to 35 °C and filtered. The cake was washed with dioxane (100 mL). The products were dissolved in water and a LC-MS was recorded. The ratio of N(2) to Λ/(1 ) isomer of was 83: 17.
Reference Example 4.1: Methyl (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoic acid (100 g, 0.46 mol) was suspended in DCM (650 mL) and DMF (10 mL) at 20 °C. To this suspension was added oxalyl chloride (51 mL, 0.59 mol) over a period of 30 min. LC-MS showed 60% conversion to acid chloride intermediate. Oxalyl chloride (17.6 mL, 0.45 eq.) was added dropwise. LC-MS showed full conversion to acid chloride intermediate.
Methyl (S)-2-methylpyrrolidine-2-carboxylate hydrochloride (84 g, 0.47 mol) was suspended in DCM (800 mL) in a second flask. The suspension was cooled to 10 °C. Triethylamine (200 mL, 1.41 mol) was added over 15 min. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. The reaction mixture was washed with 1 M HCI (500 mL), 1 N NaOH (500 mL) and water (500 mL). The organic layer was concentrated to dryness to give a light-yellow solid as product. Yield: 157 g, 100%, 99% a/a (LC-MS), M+1 =345. 1H NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.79 (d, J = 8.9 Hz, 1 H), 7.21 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 6.85 (d, J = 1.9 Hz, 1 H), 3.89 (s, 3 H), 3.66 (s, 3 H), 3.29 (m, 1 H), 3.03 (m, 1 H), 2.08 (m, 1 H), 1.82 (m, 3 H), 1.50 (s, 3 H).
Reference Example 4.2: (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid
Methyl (S)-1-(5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate (157 g, 0.46 mol) was dissolved in MeOH (750 mL) at 20 °C. To this solution was added 16% NaOH (300 mL). The resulting solution was heated up to 80 °C and stirred for 60 min. Solvent was distilled off under reduced pressure (850 mL). The residue was taken up in DCM (1500 mL) and water (450 ml) at 20 °C. 32% HCI (200 mL) was added. Layers were separated and the organic layer was washed with water (450 mL). The organic layer was concentrated to the minimum stirring volume under reduced pressure. Toluene (750 mL) was added and solvent was further distilled under vacuum (150 mL distilled). The mixture was cooled to 20 °C and stirred for 15 min. The suspension was filtered at 20 °C. The cake was rinsed with toluene (150 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 128 g, 85%, 94% a/a (LC-MS), M+1 =331. Melting point: 178 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 12.3 (s, 1 H), 8.04 (s, 2 H), 7.79 (d, 1 H), 7.20 (dd, J1 = 2.8 Hz, J2 = 8.9 Hz, 1 H), 6.84 (m, 1 H), 3.88 (s, 3 H), 3.29 (m, 1 H), 2.99 (m, 1 H), 2.1 1 (m, 1 H), 1.81 (m, 3 H), 1.47 (s, 3 H).
Reference Example 4.3: (S)-N-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide
(S)-1-(5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid (128 g, 0.39 mol) was suspended in DCM (850 mL) and DMF (6 mL) at 20 °C. To this suspension was added oxalyl chloride (39 mL, 0.45 mol) over a period of 30 min. 4-Chloro-3-methylbenzene-1 ,2-diamine hydrochloride (75 g, 0.39 mol) was suspended in DCM (1300 mL) in a second flask. The suspension was cooled down to 10 °C. Triethylamine (180 mL, 1.27 mol) was added. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. Water (650 mL) was added to the reaction mixture. Layers were separated and the organic phase was concentrated under reduced pressure (1900 mL distilled out). TBME (1000 mL) was added and solvent was further distilled under vacuum (400 mL distilled). The mixture was finally cooled down to 20 °C and stirred for 15 min. The resulting suspension was filtered off at 20 °C. The cake was rinsed with TBME (250 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 145 g, 80%, 97% a/a (LC-MS), M+1=469. Melting point: 185 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 9.10-9.14 (m, 1 H), 7.88-8.12 (m, 2 H), 7.81-7.82 (m, 1 H), 7.38-7.44 (m, 1 H), 7.21 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 6.84 (d, J = 7.8 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 1 H), 5.01 (brs, 2 H), 3.88 (s, 3 H), 3.61-3.73 (m, 1 H), 3.14-3.26 (m, 1 H), 2.25-2.30 (m, 1 H), 2.13 (s, 3 H), 1.97 (m, 3 H), 1.47-1.61 (m, 3 H).
Reference Example 4.4: (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl) (5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride
(S)-/V-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1 ,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide (145 g, 0.31 mol) was dissolved in isopropanol (870 mL) at 20 °C. To this solution was added carefully 5-6 N HCI in isopropanol (260 mL) over 10 min. the reaction mixture was then heated up to 90 °C and stirred for 4 hours. Water (28 mL) was added and the reaction mixture was stirred for an additional one hour. The reaction mixture was cooled to 20 °C. A light brown suspension was obtained which was filtered. The cake was rinsed with isopropanol (220 mL). The solid was finally dried under reduced pressure at 60 °C to give a beige solid. Yield: 133 g, 88%, 100% a/a (LC-MS), M+1 =451. Melting point: 277 °C (DSC). Ή NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.76 (d, J = 8.9 Hz, 1 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.55 (m, 1 H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 3.98 (m, 1 H), 3.90 (s, 3 H), 3.33 (m, 2H), 3.32 (m, 1 H), 2.74 (s, 3 H), 2.55 (m, 1 H), 2.23 (m, 1 H), 2.10 (m, 2 H), 1.95 (s, 3 H).
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Quviviq |
| Other names | Nemorexant; ACT-541468 |
| License data | US DailyMed: Daridorexant |
| Routes of administration | By mouth |
| Drug class | Orexin antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Pharmacokinetic data | |
| Elimination half-life | 6–10 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1505484-82-1 |
| PubChem CID | 91801202 |
| DrugBank | DB15031 |
| ChemSpider | 64854514 |
| UNII | LMQ24G57E9 |
| KEGG | D11886 |
| PDB ligand | NS2 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.93 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
REF
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214985s000lbl.pdf
- ^ Jump up to:a b Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J (November 2020). “Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders”. Expert Opin Drug Metab Toxicol. 16 (11): 1063–1078. doi:10.1080/17425255.2020.1817380. PMID 32901578.
- ^ Jump up to:a b c “Daridorexant – Idorsia Pharmaceuticals – AdisInsight”.
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- ^ “Daridorexant: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 January 2022.
- ^ “Idorsia receives US FDA approval of Quviviq (daridorexant)” (Press release). Idorsia Pharmaceuticals. 10 January 2022. Retrieved 11 January 2022 – via GlobeNewswire.
External links
- “Daridorexant”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03545191 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects With Insomnia Disorder” at ClinicalTrials.gov
- Clinical trial number NCT03575104 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
- Clinical trial number NCT03679884 for “Study to Assess the Long Term Safety and Tolerability of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
///////////////Daridorexant, Quviviq, FDA 2022, APPROVALS 2022, INSOMNIA, ACT541468A, ACT 541468A. ACT-541468A, ACT541468, FDA 2022, APPROVALS 2022
O=C(N1[C@](C)(C2=NC3=CC=C(Cl)C(C)=C3N2)CCC1)C4=CC(OC)=CC=C4N5N=CC=N5.[H]Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
Nemorexant
Nemorexant
ACT-541468, UNII LMQ24G57E9
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1 [RN]
LMQ24G57E9
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
- Originator Actelion Pharmaceuticals
- Developer Idorsia Pharmaceuticals
- Class Sleep disorder therapies
- Mechanism of Action Orexin receptor type 1 antagonists; Orexin receptor type 2 antagonists
- Phase III Insomnia
- 19 Oct 2018 Idorsia Pharmaceuticals plans a phase I trial for Liver disorders (Hepatic impairment) in November 2018 (PO) (NCT03713242)
- 09 Oct 2018 Idorsia Pharmaceuticals completes a phase I trial in Insomnia (In volunteers) in Netherlands (PO) (NCT03609775)
- 27 Sep 2018 Idorsia Pharmaceuticals plans a phase I trial for Hepatic impairment in November 2018 , (NCT03686995)
Nemorexant (developmental code name ACT-541468) is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals for the treatment of insomnia.[1][2] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[1][2] As of June 2018, nemorexant is in phase III clinical trials for the treatment of insomnia.[1]
Idorsia is developing nemorexant, a dual orexin receptor antagonist (DORA), for the oral treatment of insomnia and investigating the program for the treatment of COPD. In May 2018, a phase III study was initiated in subjects with insomnia disorder and in September 2018, a phase I trial was initiated in COPD.
SCHEME
SEE AT END OF PAGE
PATENT
WO2013182972 ,
PATENT
WO2015083094 ,
Patent
WO 2015083070

Synthesis of nemorexant, using 2-methyl-L-proline hydrochloride as the starting material
N-Protection of 2-methyl-L-proline hydrochloride with Boc2O gives N-Boc-2-methyl-L-proline,
Which upon condensation with 4-chloro-3-methylbenzene-1,2-diamine using HATU and DIEA in CH2Cl2 affords the corresponding amide.
Cyclization of diamine in the presence of AcOH at 100 °C provides imidazole derivative,
Whose Boc moiety is removed by means of HCl in dioxane to yield 5-chloro-4-methyl-2-[2(S)-methylpyrrolidin-2-yl]benzimidazole hydrochloride.
N-Acylation of pyrrolidine derivative with 5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid using HATU and DIEA in CH2Cl2 produces Nemorexant
5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid (prepared by the coupling of 2-iodo-5-methoxybenzoic acid with 1,2,3-triazole using CuI and Cs2CO3 in DMF)
PATENT
WO 2016020403
PATENT
WO 2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PATENT
WO-2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689&tab=PCTDESCRIPTION&maxRec=1000
Process for the preparation of a crystalline potassium salt of a 2-(2H-[1,2,3]triazol-2-yl)-benzoic acid derivatives is claimed. Compound is disclosed to be useful for the preparation of pharmaceuticals, especially certain orexin receptor antagonists such as nemorexant .
References
- ^ Jump up to:a b c https://adisinsight.springer.com/drugs/800044843
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- Muehlan, C.; Heuberger, J.; Juif, P.E.; Croft, M.; van Gerven, J.; Dingemanse, J.
Accelerated development of the dual orexin receptor antagonist ACT-541468: Integration of a microtracer in a first-in-human study
Clin Pharmacol Ther 2018, 104(5): 1022 - A Study to Evaluate the Pharmacokinetics of ACT-541468 in Subjects With Mild, Moderate and Severe Hepatic Impairment (NCT03713242)
ClinicalTrials.gov Web Site 2018, October 24 - Boof, M.-.L.; Ufer, M.; Halabi, A.; Dingemanse, J.
Impact of the dual orexin receptor antagonist ACT-541468 on the pharmacokinetics of the CYP3A4 probe drug midazolam and assessment of the effect of food on ACT-541468
119th Annu Meet Am Soc Clin Pharmacol Ther (ASCPT) (March 21-24, Orlando) 2018, Abst PI-043 - Muehlan, C.; Brooks, S.; Zuiker, R.; van Gerven, J.; Dingemanse, J.
Night-time administration of ACT-541468, a novel dual orexin receptor antagonist: Characterization of its pharmacokinetics, next-day residual effects, safety, and tolerability
32nd Annu Meet Assoc Sleep Soc (SLEEP) (June 2-6, Baltimore) 2018, Abst 0008 - Proposed international nonproprietary names (Prop. INN): List 118
WHO Drug Inf 2017, 31(4): 635
External links
from PubChem
 |
|
| Clinical data | |
|---|---|
| Synonyms | ACT-541468 |
| Routes of administration |
By mouth |
| Drug class | Orexin antagonist |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.927 g/mol |
| 3D model (JSmol) | |
///////////////Nemorexant, ACT-541468, Phase III, Insomnia


{kind=link}