

![(7R)-7-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carbonitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=134405105&t=l)
| Patent ID | Date | Patent Title |
|---|---|---|
| US7378424 | 2008-05-27 | Derivatives of pyrimido[6, 1-A]isoquinolin-4-one |
| US7105663 | 2006-09-12 | Derivatives of pyrimido[6, 1-a]isoquinolin-4-one |
| US6794391 | 2004-09-21 | Derivatives of pyrimido[6.1-a]isoquinolin-4-one |
| US2004001895 | 2004-01-01 | Combination treatment for depression and anxiety |
| US2003235631 | 2003-12-25 | Combination treatment for depression and anxiety |
Home » Posts tagged 'inflammation'
Tag Archives: inflammation
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WXFL-10203614
WXFL-10203614
CAS 2054932-34-0 R isomer, (S isomer 2054932-33-9 )
C15 H15 N7, 293.33
(7R)-7-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carbonitrile
- (7R)-5,6,7,8-Tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)imidazo[1,2-a]pyridine-2-carbonitrile
-
Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-
Wuxi Fortune Pharmaceutical Co Ltd
Jak1 tyrosine kinase inhibitor
Wuxi Fuxin Pharmaceutical Research and Development , in collaboration with Wuxi Apptec , is investigating a tablet formulation of WXFL-10203614 , a JAK1 tyrosine kinase inhibitor, for the oral treatment of rheumatoid arthritis. In January 2019, a phase I trial was planned.
- Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-, 4-methylbenzenesulfonate, hydrate (1:1:1)
- cas 2226936-85-0
- Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-, 2,2,2-trifluoroacetate (1:1)
- cas 2226936-87-2
syn

PATENT
WO2018095345 claiming novel crystalline salt forms of similar compound
PATENT
WO 2016192563
PATENT
US-20190218231
Novel crystalline forms of 7h-pyrrolo[2,3-D]pyrimidine compounds (designated as forms A to E) useful as JAK1 and JAK2 inhibitors for treating arthritis, inflammation and autoimmune diseases.
JAK belongs to the family of tyrosine kinases involved in inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6. The present invention also provides a method for preparing the compound or a pharmaceutical composition comprising the compound, and a method for preventing and/or treating inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6 by administrating the compound of the present invention.
Janus kinase (JAK) is a cytoplasmic tyrosine kinase that transduces a cytokine signal from a membrane receptor to an STAT transcription factor. The prior art has described four members of the JAK family: JAK1, JAK2, JAK3 and TYK2. When cytokines bind to their receptors, JAK family members are auto-phosphorylated and/or trans-phosphorylated from each other, followed by STATs phosphorylation, and then are migrated into the cell nucleus to regulate the transcription. JAK-STAT intracellular signal transduction is suitable for interferons, most interleukins, as well as various cytokines and endocrine factors, such as EPO, TPO, GH, OSM, LIF, CNTF, GM-CSF and PRL (Vainchenker W. et al. (2008)).
A combinatorial study of a genetic model and a small molecule JAK inhibitor has revealed the therapeutic potential of several JAKs. It has been confirmed by mouse and human genetics that JAK3 is an immunosuppressive target (O’Shea J. et al. (2004)). A JAK3 inhibitor has been successfully used in clinical development. At first, it was used in organ transplant rejection, and later also used in other immunoinflammatory indications such as rheumatoid arthritis (RA), psoriasis and Crohn’s disease (http://clinicaltrials.gov/). It has been confirmed by human genetics and mouse knockout studies that TYK2 is a potential target for immunoinflammatory diseases (Levy D. and Loomis C. (2007)). JAK1 is a new target in the field of immunoinflammatory diseases. The heterodimerization of JAK1 and other JAKs arouses a transduction of cytokine-driven pro-inflammatory signaling. Thus, it is expected that inhibition of JAK1 and/or other JAKs has a therapeutic benefit for a series of inflammatory diseases and other diseases driven by JAK-mediated signal transduction.
transduction.
Example 1: Preparation of Compound 1
Step 1: 2-chloro-4-nitro-1-oxo-pyridin-1-ium (40.0 g, 229.2 mmol) and (4-methoxyphenyl)methylamine (63 g, 458.4 mmol) were dissolved in EtOH (400 mL), and the resulting solution was stirred at reflux for 5 hours. TLC (PE:EA=2:1) showed that the reaction was complete. The EtOH was concentrated to half of its volume and was cooled in an ice bath for 2-3 hours. The resulting cold mixture was filtered, and the isolated solid was washed with PE (60 mL*3) and ice water (60 mL*3), respectively. Drying in vacuum given an orange solid, N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (2) (38.6 g, 140.2 mmol, with a yield of 61.2%). MS (ESI) calcd. For r C 13H 13N 3O 4 [M+H] + 275, found 276.
Step 2: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (5.0 g, 18.16 mmol) in CHCI 3 (50 mL) was dropwise added PCI 3 (8.4 g, 60.8 mmol) at 0° C. After the addition, the reaction mixture was heated to 25° C. and stirred vigorously for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was filtered, and the resulting solid was washed with PE (30 mL*3) to give a yellow solid compound, N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (3) (4.2 g, a crude product) which was directly used in the next step without further purification. MS (ESI) calcd. For C 15H 18N 6 [M+H] +259, found 260.
Step 3: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (4.2 g, 16.2 mmol) in toluene (10 mL) was dropwise added TFA (5.0 mL) at atmospheric temperature. Then, the mixture was stirred at 80° C. for 2 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The mixture was concentrated under reduced pressure to remove the solvent. The residue was diluted with H 2O (50 mL), and its pH was adjusted to be neutral with solid NaHCO 3. The aqueous phase was extracted with EA (50 mLE*3). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica, petroleum ether/ethyl acetate=1/0-1:1) to obtain an orange solid compound, 4-nitropyridine-2-amine (4) (700 mg, 5.0 mmol, with a yield of 31.1%). MS (ESI) calcd. For C 5H 5N 3O 2 [M+H] + 139, found 140.
Step 4: to a solution of 4-nitropyridine-2-amine (200 mg, 1.4 mmol) in DME (5 mL) was added 3-bromo-2-oxo-propanoate (280 mg, 1.4 mmol) at atmospheric temperature. The resulting mixture was stirred at 25° C. for 1 hour, and then was concentrated under reduced pressure to remove the solvent. The residue was dissolved with EtOH (10 mL); and then was refluxed for 3 hours. TLC showed that the reaction was complete. The reaction solution was cooled to room temperature, and the solvent was concentrated under reduced pressure. The residue was basified with saturated NaHCO 3 aqueous solution (25 mL). The aqueous phase was extracted with DCM (15 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (EA:PE=10-60%) to obtain a light yellow solid compound, ethyl 7-nitroimidazo[1,2-]pyridin-2-carboxylate (5) (302 mg, with a yield of 88.9%). MS (ESI) calcd. For C 10H 9N 3O 4 [M+H] + 235, found 236.
Step 5: a solution of ethyl 7-nitroimidazo[1,2-a]pyridin-2-carboxylate (150 mg, 637.8 mmol) in ethanol (20 mL) was added HCl (7 mg, 0.2 mmol) and PtO 2 (15 mg, 0.6 mmol) at atmospheric temperature. The reaction system was repeatedly vacuumed and filled with N 2 for three times, then filled with H 2(50 psi), and was stirred at 50° C. for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was concentrated to half of its volume, and filtered to obtain a white solid compound, ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (6) (120 mg, a crude product). MS (ESI) calcd. For C 10H 15N 3O 2 [M+H] + 209, found 210.
Step 6: ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (100 mg, 0.4 mmol) and 4-chloro-7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidine (137 mg, 0.4 mmol) were dissolved in n-BuOH (5 mL), and DIEA (158 mg, 1.2 mmol) were added to the above solution. The resulting mixture was stirred under reflux for 16 hours. LC-MS showed that the reaction was complete. The reaction mixture was concentrated under reduced pressure, and the resulting residue was diluted with H 2O (10 mL). The aqueous phase was extracted with EA (20 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (PE:EA=0:1) to obtain a light yellow solid compound, ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl] amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (7) (55 mg, 0.11 mmol, with a yield of 28.1%). MS (ESI) calcd. For C 23H 24N 6O 4S [M+H] + 480, found 481.
Step 7: to a solution of ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (3.0 g, 6.2 mmol) in THF (150 mL) was added NaH (499 mg, 12.5 mmol) in portions under N 2 atmosphere at 0° C. The mixture was stirred at that temperature for 1 hour, and then was dropwise added MeI (7.1 g, 50.2 mmol). After the addition, the mixture was stirred at atmospheric temperature for 1 hour. TLC showed that the reaction was complete. The reaction was quenched by the addition of saturated NH 4Cl (10 mL), and then was diluted by the addition of ice water (50 mL). The aqueous phase was extracted with a mixed solvent of DCM/MeOH (3:1, 50 mL*3). The combined organic phase was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude product was purified by flash column chromatography (DCM:MeOH=10:1) to obtain a light yellow solid, ethyl 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (8) (1.5 g, with a yield of 45%). MS (ESI) calcd. For C 24H 26N 6O 4S [M+H] + 494, found 495.
Step 8: to a solution of 7-[methyl-[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (4.0 g, 8.1 mmol) in THF (40 mL) and H 2O (8 mL) was added LiOH.H 2O (509 mg, 12.1 mmol), and the mixture was stirred at 20° C. for 10 hours. TLC showed that the reactants were completely consumed. THF in the reaction mixture was removed under reduced pressure; and the pH of the residue was adjusted to 2-3 with 2M HCl (4 mL) to form a white solid. The solid was filtered out, and was concentrated under reduced pressure to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (9) as a white solid (3.6 g, with a yield of 95.4%). MS (ESI) calcd. For C 22H 22N 6O 4S [M+H] + 466, found 467.
Step 9: to a solution of 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (1.8 g, 3.9 mmol) in DMF (20 mL) was added CDI (751 mg, 4.6 mmol) at 0° C. The reaction solution was heated to 25° C. and stirred for 2 hours, and after that, solid ammonium chloride (2.1 g, 38.6 mmol) was added, and then the reaction was kept overnight at atmospheric temperature. LC-MS showed that the reactants were completely consumed. The reaction mixture was poured into ice water (50 mL), and a white solid was precipitated. The solid was filtered out, washed with water (20 mL), and was dried under reduced pressure in a rotating manner to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (10) as a white solid (2.5 g, a crude product) which product was directly used in the next step. MS (ESI) calcd. For C 22H 23N 7O 3S [M+H] + 465, found 466.
Step 10: 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxamide (2.5 g, 5.4 mmol) was dissolved in a mixture of THF (20 mL), MeOH (10 mL) and H 2O (6 mL), and NaOH (429.6 mg, 10.7 mmol) was added. The mixture was heated to 60° C. and stirred for 30 minutes. LC-MS showed that the reactants were completely consumed. The reaction mixture was concentrated under reduced pressure to obtain 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxamide (11) as a white solid (2.5 g, a crude product) which was directly used in the next step. MS (ESI) calcd. For C 15H 17N 7O [M+H] + 311, found 312.
Step 11: to a solution of 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (2.0 g, 6.4 mmol) and triethylamine (3.9 g, 38.5 mmol) in THF (20 mL) was dropwise added TFAA (4.1 g, 19.3 mmol) at 0° C. After the addition, the reaction solution was stirred at atmospheric temperature for 30 minutes. LC-MS showed the starting materials were completely consumed. The reaction mixture was poured into ice water (20 mL), and extracted with DCM/MeOH (5:1, 100 mL*2). The combined organic layer was washed with saturated saline (20 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a residue. The residue was purified by column chromatography (DCM/MeOH=40/1 to 20:1) to obtain 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (12,378 mg, with a yield of 19.8%). MS (ESI) calcd. For C 15H 15N 7 [M+H] + 293, found 294. 1H NMR (400 MHz, DMSO-d6) 11.44-11.71 (m, 1H), 7.99-8.17 (m, 2H), 7.11-7.20 (m, 1H), 6.63 (dd, J=1.76, 3.26 Hz, 1H), 5.33 (br. s., 1H), 4.21-4.21-4.31 (m, 1H), 4.13 (dt, J=4.14, 12.49 Hz, 1H), 3.27 (s, 3H), 2.91-3.11 (m, 2H), 2.31-2.44 (m, 1H), 2.07 (d, J=11.54 Hz, 1H).
Step 12: racemic 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (30 mg, 102.3 umol) was separated by a chiral column to obtain the compound 1 (10 mg, with a yield of 32.8%).
Compound 1: retention time 6.407 min; MS (ESI) calcd. For C 15H 15N 7 [293, found 294 M+H]+. Purity 98.8%, e.e. was 98.9%; [α] D 20=+78.4° (c=0.6, DMSO). MS ESI calcd. For C 15H 15N 7 [M+H] + 294, found 294. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.02-2.15 (m, 1H) 2.39 (qd, J=12.42, 5.90 Hz, 1H) 2.92-3.12 (m, 2H) 3.28 (s, 3H) 4.05-4.36 (m, 2H) 5.20-5.45 (m, 1H) 6.64 (dd, J=3.39, 1.88 Hz, 1H) 7.17 (dd, J=3.26, 2.51 Hz, 1H) 8.02-8.17 (m, 2H) 11.69 (br s, 1H).
//////////WXFL-10203614, WXFL 10203614 , WXFL10203614, Wuxi Fuxin, arthritis, inflammation, autoimmune diseases, Wuxi Apptec, JAK1, JAK2 inhibitors
N#Cc1cn2CC[C@H](Cc2n1)N(C)c4ncnc3nccc34
RPL 554, Ensifentrine


RPL-554, Ensifentrine
- Molecular FormulaC26H31N5O4
- Average mass477.555
FDA 6/26/2024, Ohtuvayre, To treat chronic obstructive pulmonary disease
Drug Trials Snapshot
RPL 554
Urea, N-[2-[(2E)-6,7-dihydro-9,10-dimethoxy-4-oxo-2-[(2,4,6-trimethylphenyl)imino]-2H-pyrimido[6,1-a]isoquinolin-3(4H)-yl]ethyl]-
(2-[(2E)-9,10-DIMETHOXY-4-OXO-2-[(2,4,6-TRIMETHYLPHENYL)IMINO]-2H,3H,4H,6H,7H-PYRIMIDO[4,3-A]ISOQUINOLIN-3-YL]ETHYL)UREA
2-[9,10-dimethoxy-4-oxo-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[6,1-a]isoquinolin-3-yl]ethylurea
{2-[(2E)-9,10-dimethoxy-4-oxo-2-[(2,4,6-trimethylphenyl)imino]-2H,3H,4H,6H,7H-pyrimido[4,3-a]isoquinolin-3-yl]ethyl}urea
2-[4-keto-9,10-dimethoxy-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[4,3-a]isoquinolin-3-yl]ethylurea
2-[9,10-dimethoxy-4-oxo-2-(2,4,6-trimethylphenyl)imino-6,7-dihydropyrimido[4,3-a]isoquinolin-3-yl]ethylurea
298680-25-8 CAS
UNII:3E3D8T1GIX
CFTR stimulator; PDE 3 inhibitor; PDE 4 inhibitor
RPL-554 is a mixed phosphodiesterase (PDE) III/IV inhibitor in phase II clinical development at Verona Pharma for the treatment of asthma, allergic rhinitis, chronic obstructive pulmonary disease (COPD) and inflammation.
RPL-554 is expected to have long duration of action and will be administered nasally thereby preventing gastrointestinal problems often resulting from orally administered PDE4 antiinflammatory drugs.
The company is now seeking licensing agreements or partnerships for the further development and commercialization of the drug.
RPL-554 (LS-193,855) is a drug candidate for respiratory diseases. It is an analog of trequinsin, and like trequinsin, is a dual inhibitor of the phosphodiesterase enzymes PDE-3 and PDE-4.[1] As of October 2015, inhaled RPL-554 delivered via a nebulizer was in development for COPD and had been studied in asthma.[2]
PDE3 inhibitors act as bronchodilators, while PDE4 inhibitors have an anti-inflammatory effect.[1][3]
RPL554 was part of a family of compounds invented by Sir David Jack, former head of R&D for GlaxoSmithKline, and Alexander Oxford, a medicinal chemist; the patents on their work were assigned to Vernalis plc.[4][5]:19-20
In 2005, Rhinopharma Ltd, acquired the rights to the intellectual property from Vernalis.[5]:19-20 Rhinopharma was a startup founded in Vancouver, Canada in 2004 by Michael Walker, Clive Page, and David Saint, to discover and develop drugs for chronic respiratory diseases,[5]:16 and intended to develop RPL-554, delivered with an inhaler, first for allergic rhinitis, then asthma, then forCOPD.[5]:16-17 RPL554 was synthesized at Tocris, a contract research organization, under the supervision of Oxford, and was studied in collaboration with Page’s lab at King’s College, London.[1] In 2006 Rhinopharma recapitalized and was renamed Verona Pharma plc.[5]
This was first seen in April 2015 when it was published as a France national. Verona Pharma (formerly Rhinopharma), under license from Kings College via Vernalis, is developing the long-acting bronchodilator, RPL-554 the lead in a series dual inhibitor of multidrug resistant protein-4 and PDE 3 and 4 inhibiting trequinsin analogs which included RPL-565, for treating inflammatory respiratory diseases, such as allergic rhinitis, asthma, and COPD.
RPL554
Verona Pharma’s lead drug, RPL554, is a “first-in-class” inhaled drug under development for chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis. The drug is an inhibitor of the phosphodiesterase 3 (PDE3) and phosphodiesterase 4 (PDE4) enzymes, two enzymes known to be of importance in the development and progression of immunological respiratory diseases. The drug has the potential to act as both a bronchodilator and an anti-inflammatory which would significantly differentiate it from existing drugs.
RPL554 was selected from a class of compounds co-invented by Sir David Jack, the former Director of Research at Glaxo who led the team that discovered many of the commercially successful drugs in the respiratory market.
Verona Pharma has successfully completed two double-blind placebo controlled randomised Phase 2b studies of RPL554: one in mild to moderate asthma and another in mild to moderate COPD. The drug was found to be well tolerated, free from drug-related adverse effects (especially cardiovascular and gastro-intestinal effects) and generated significant bronchodilation. Additionally, double-blind placebo controlled exploratory studies in healthy volunteers challenged with an inhaled irritant also generated consistent, clinically meaningful anti-inflammatory effects.
Verona Pharma is also carrying out exploratory studies to investigate the potential of RPL554 as a novel treatement for cystic fibrosis. In November 2014, the Company received a Venture and Innovation Award from the UK Cystic Fibrosis Trust to further such studies.
For further information on the potential of RPL554 for the treatment of respiratory diseases, refer to the peer-reviewed paper available on-line in the highly-respected medication journal, The Lancet Respiratory Medicine, entitled “Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials”.
The competitive advantages of RPL554 include the following:
- combining bronchodilator (PDE 3) and anti-inflammatory actions (PDE 4) in a single drug, something that is currently only achieved with a combination LABA and glucocorticosteroid inhaler,
- unique in not using steroids or beta agonists, which have known side effects,
- planned to be administered by nasal inhalation, thereby reducing the unwanted gastrointestinal side effects of many orally administered drugs.
History of Clinical Trials
- Following completion in May 2008 of toxicological studies of RPL554, the Company commenced in February 2009 a Phase I/IIa clinical trial of the drug at the Centre for Human Drug Research (CHDR) at Leiden in the Netherlands. In September 2009, the Company announced that it had successfully completed the trial, demonstrating that RPL554 has a good safety profile and has beneficial effects in terms of bronchodilation and bronchoprotection in asthmatics and a reduction in the numbers of inflammatory cells in the nasal passages of allergic rhinitis patients.
- In November 2010, the Company successfully completed a further trial that examined the safety and bronchodilator effectiveness of the drug administered at higher doses.
- In August 2011, the Company demonstrated that bronchodilation is maintained over a period of 6 days with daily dosing of RPL554 in asthmatics.
- In November 2011, the Company successfully demonstrated safety and bronchodilation of RPL554 in patients with mild to moderate forms of COPD.
- In March 2013, the Company demonstrated positive airway anti-inflammatory activity with respect to COPD at a clinical trial carried out at the Medicines Evaluation Unit (MEU) in Manchester, UK.
![]()
Synthesis
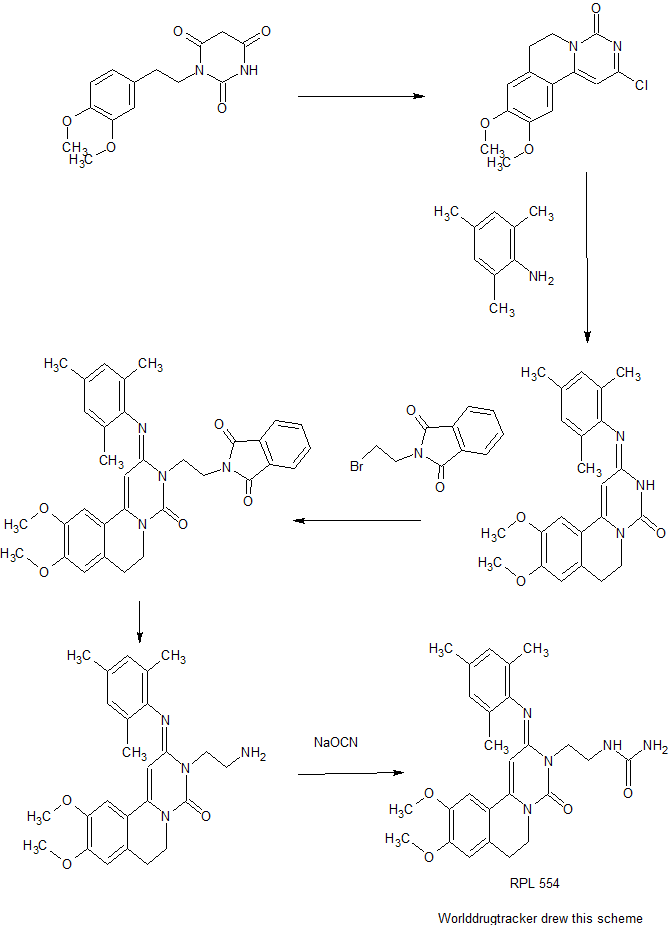
Cyclization of 1-(3,4-dimethoxyphenethyl)barbituric acid in refluxing POCl3 produces the pyrimidoisoquinolinone , which is further condensed with 2,4,6-trimethylaniline in boiling isopropanol to afford the trimethylphenylimino derivative . Subsequent alkylation of with N-(2-bromoethyl)phthalimide in the presence of K2CO3 and KI, followed by hydrazinolysis of the resulting phthalimidoethyl compound yields the primary amine . This is finally converted into the title urea RPL 554 by reaction with sodium cyanate in aqueous HCl.
Example 1 : 9 Λ 0-Dimethoxy-2-(2.4-6-trimethy-phen yliminoY-3-(N-carbamoyl-2- aminoethylV3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one

Sodium cyanate (6.0g, 0.092 mol) in water (100 ml) was added dropwise to a stirred solution of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-aminoethyl)-3,4,6,7- tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 4 above (20.0g, 0.046 mol) in water (600 ml) and IN ΗC1 (92 ml) at 80°C. After stirring for 2h at 80°C the mixture was cooled in an ice-bath and basified with 2N NaOH. The mixture was extracted with dichloromethane (3 x 200 ml) and the combined extract was dried (MgSO- ) and evaporated in vacuo. The resulting yellow foam was purified by column chromatography on silica gel eluting with CH2CI2 / MeOH (97:3) and triturated with ether to obtain the title compound as a yellow solid, 11.9g, 54%.
M.p.: 234-236°C m/z: C26H31N5O4 requires M=477 found (M+l) = 478
HPLC: Area (%) 99.50 Column ODS (150 x 4.6 mm)
MP pH3 KH2PO4 / CH3CN (60/40)
FR (ml/min) 1.0 RT (min) 9.25 Detection 250 nm
lK NMR (300 MHz, CDCI3): δ 1.92 (1H, br s, NH), 2.06 (6H, s, 2xCH3), 2.29 (3H, s, CH3), 2.92 (2H, t, CH2), 3.53 (2H, m, CH2), 3.77 (3H, s, OCH3), 3.91 (3H, s, OCH3), 4.05 (2H, t, CH2), 4.40 (2H, t, CH2), 5.35 (2H, br s, NH2), 5.45 (1H, s, C=CH), 6.68 (1H, s, ArH), 6.70 (1H, s, ArH), 6.89 (2H, s, 2xArH).
Preparation 1 : Synthesis of 2-Chloro-6.7-d-hydro-9.10-Dimethoxy-4H-pyrimido- [6,l-a]isoquinoHn-4-one (shown as (1) in Figure 1

A mixture of l-(3,4-dimethoxyphenyl) barbituric acid (70g, 0.24mol), prepared according to the method described in B. Lai et al. J.Med.Chem. 27 1470-1480 (1984), and phosphorus oxychloride (300ml, 3.22mol) was refluxed for 2.5h. The excess phosphorous oxychloride was removed by distillation (20mmHg) on wa ming. After cooling the residue was slurried in dioxan (100ml) and cautiously added to a vigorously stirred ice/water solution (11). Chloroform (11) was added and the resulting mixture was basified with 30% sodium hydroxide solution. The organic layer was separated and the aqueous phase further extracted with chloroform (2x750ml). The combined organic extracts were washed with water (1.51), dried over magnesium sulphate and concentrated in vacuo to leave a gummy material (90g). This was stirred in methanol for a few minutes, filtered and washed with methanol (200ml), diethyl ether (2x200ml) and dried in vacuo at 40°C to yield the title compound as a yellow/orange solid. 47g, 62%
(300MHz, CDCI3) 2.96(2H, t, C(7) H2); 3.96(6H, s, 2xOCH3; 4.20(2H, t, C(6) H2); 6.61(1H, s, C(1) H); 6.76(1H, s, Ar-H); 7.10(1H, s, Ar-H). Preparation 2: 9.10-Dimethoxy-2-(2.4.6-trimethylphenyliminoV3.4.6.7- tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one (shown as (2) in Figure 1
2-Chloro-9,10-dimethoxy-6,7-dihydro-4H-pyrimido[6,l-a]isoquinolin-4-one, prepared according to Preparation 1, (38.5g, 0.13 mol) and 2,4,6-trimethylaniline (52.7g, 0.39 mol) in propan-2-ol (3 1) was stirred and heated at reflux, under nitrogen, for 24h. After cooling to room temperature, the solution was evaporated in vacuo and the residue was purified by column chromatography on silica gel, eluting with CΗ2CI2 /
MeOH, initially 98:2, changing to 96:4 once the product began to elute from the column. The title compound was obtained with a slight impurity, (just above the product on tic). Yield 34.6g, 67%.
Preparation 3: 9.10-Dimethoxy-2-(2.4.6-trimethylphenyliminoV3-(2-N- phthalimidoethyπ-3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquinolin-4-one
(shown as (3 in Figure 1)
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3,4,6,7-tetrahydro-2H- pyrimido[6,l-a]isoquinolin-4-one (which was prepared according to Preparation 2) (60.0g, 0.153 mol), potassium carbonate (191g, 1.38 mol), sodium iodide (137g, 0.92 mol) and N-(2-bromoethyl)phthalimide (234g, 0.92 mol) in 2-butanone (1500 ml) was stirred and heated at reflux, under nitrogen, for 4 days. After cooling to room temperature the mixture was filtered and the filtrate was evaporated in vacuo. The residue was treated with methanol (1000 ml) and the solid filtered off, washed with methanol and recrystallised from ethyl acetate to obtain the title compound as a pale yellow solid in yield 40. Og, 46%. Evaporation of the mother liquor and column chromatography of the residue on silica gel (CΗ2C-2 / MeOH 95:5) provided further product 11.7g, 13.5%. Preparation 4: 9.10-Dimethoxy-2-(2A6-trimethylphenylimino)-3-(2-arninoethyO- 3.4.6.7-tetrahydro-2H-pyrimido[6.1-a]isoquino-in-4-one (shown as (4) in Figure 1)
A mixture of 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(2-N- phthalimidoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,l-a]isoquinolin-4-one (22. Og, 0.039 mol), prepared according to Preparation 3, and hydrazine hydrate (11.3g, 0.195 mol) in chloroform (300 ml) and ethanol (460 ml) was stined at room temperature, under nitrogen, for 18h. Further hydrazine hydrate (2.9g, 0.05 mol) was added and the mixture was stirred a further 4h. After cooling in ice / water, the solid was removed by filtration and the filtrate evaporated in vacuo. The residue was dissolved in dichloromethane and the insoluble material was removed by filtration. The fitrate was dried (MgSO-i) and evaporated in vacuo to afford the title compound as a yellow foam in yield 16.2g, 96%.
PATENT
Novel crystalline acid addition salts forms of RPL-554 are claimed, wherein the salts, such as ethane- 1,2-disulfonic acid, ethanesulfonic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid. .
RPL554 (9, 10-dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(/V-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6, l-a]isoquinolin-4-one) is a dual PDE3/PDE4 inhibitor and is described in WO 00/58308. As a combined PDE3/PDE4 inhibitor, RPL554 has both antiinflammatory and bronchodilatory activity and is useful in the treatment of respiratory disorders such as asthma and chronic obstructive pulmonary disease (COPD). The structure of RPL554 is shown below.

Owing to its applicability in the treatment of respiratory disorders, it is often preferable to administer RPL554 by inhalation. Franciosi et al. disclose a solution of RPL554 in a citrate-phosphate buffer at pH 3.2 (The Lancet: Respiratory Medicine 11/2013; l(9):714-27. DOI: 10.1016/S2213-2600(13)70187-5). The preparation of salts of RPL554 has not been described.
PATENT
http://www.google.ch/patents/WO2000058308A1?cl=en&hl=de
PATENT
http://www.google.ch/patents/WO2012020016A1?cl=en
U.S. Pat. No. 6,794,391, 7,378,424, and 7,105,663, which are each incorporated herein by reference, discloses compound RPL-554 (N-{2-[(2iT)-2-(mesityiimino)-9,10- dimethoxy-4-oxo-6,7-dihydro-2H-pyrimido[6,l-a]-isoquinolin-3 4H)-yl]ethyl}urea).

It would be beneficial to provide a composition of a stable polymorph of RPL-554, that has advanrtages over less stable polymorphs or amorphous forms, including
stability, compressibility, density, dissolution rates, increased potency or. lack toxicity.
| WO2000058308A1 * | Mar 29, 2000 | Oct 5, 2000 | Vernalis Limited | DERIVATIVES OF PYRIMIDO[6,1-a]ISOQUINOLIN-4-ONE |
| US6794391 | Sep 26, 2001 | Sep 21, 2004 | Vernalis Limited | Derivatives of pyrimido[6.1-a]isoquinolin-4-one |
| US7105663 | Feb 24, 2004 | Sep 12, 2006 | Rhinopharma Limited | Derivatives of pyrimido[6,1-a]isoquinolin-4-one |
| US7378424 | Feb 24, 2004 | May 27, 2008 | Verona Pharma Plc | Derivatives of pyrimido[6, 1-A]isoquinolin-4-one |
| WO2012020016A1 * | 9. Aug. 2011 | 16. Febr. 2012 | Verona Pharma Plc | Crystalline form of pyrimidio[6,1-a]isoquinolin-4-one compound |
| WO2014140647A1 | 17. März 2014 | 18. Sept. 2014 | Verona Pharma Plc | Drug combination |
| WO2014140648A1 | 17. März 2014 | 18. Sept. 2014 | Verona Pharma Plc | Drug combination |
| WO2015173551A1 * | 11. Mai 2015 | 19. Nov. 2015 | Verona Pharma Plc | New treatment |
| US8883857 | 8. März 2013 | 11. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8883858 | 23. Juli 2014 | 11. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8895626 | 23. Juli 2014 | 25. Nov. 2014 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US8987337 | 23. Juli 2014 | 24. März 2015 | Baylor College Of Medicine | Small molecule xanthine oxidase inhibitors and methods of use |
| US9061983 | 23. Juli 2014 | 23. Juni 2015 | Baylor College Of Medicine | Methods of inhibiting xanthine oxidase activity in a cell |
| US9062047 | 9. Aug. 2011 | 23. Juni 2015 | Verona Pharma Plc | Crystalline form of pyrimido[6,1-A] isoquinolin-4-one compound |
References
- Boswell-Smith V et al. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquinolin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. PMID 16682455
- Nick Paul Taylor for FierceBiotech. October 1, 2015 Verona sets sights on PhIIb after COPD drug comes through early trial
- Turner MJ et al. The dual phosphodiesterase 3 and 4 inhibitor RPL554 stimulates CFTR and ciliary beating in primary cultures of bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2016 Jan 1;310(1):L59-70. PMID 26545902
- Jump up^ see US20040171828, identified in the citations of PMID 16682455
- ISIS Resources, PLC. August 23, 2006 Proposed Acquisition of Rhinopharma
REFERENCES
1: Calzetta L, Cazzola M, Page CP, Rogliani P, Facciolo F, Matera MG. Pharmacological characterization of the interaction between the dual phosphodiesterase (PDE) 3/4 inhibitor RPL554 and glycopyrronium on human isolated bronchi and small airways. Pulm Pharmacol Ther. 2015 Jun;32:15-23. doi: 10.1016/j.pupt.2015.03.007. Epub 2015 Apr 18. PubMed PMID: 25899618.
2: Franciosi LG, Diamant Z, Banner KH, Zuiker R, Morelli N, Kamerling IM, de Kam ML, Burggraaf J, Cohen AF, Cazzola M, Calzetta L, Singh D, Spina D, Walker MJ, Page CP. Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med. 2013 Nov;1(9):714-27. doi: 10.1016/S2213-2600(13)70187-5. Epub 2013 Oct 25. PubMed PMID: 24429275.
3: Wedzicha JA. Dual PDE 3/4 inhibition: a novel approach to airway disease? Lancet Respir Med. 2013 Nov;1(9):669-70. doi: 10.1016/S2213-2600(13)70211-X. Epub 2013 Oct 25. PubMed PMID: 24429260.
4: Calzetta L, Page CP, Spina D, Cazzola M, Rogliani P, Facciolo F, Matera MG. Effect of the mixed phosphodiesterase 3/4 inhibitor RPL554 on human isolated bronchial smooth muscle tone. J Pharmacol Exp Ther. 2013 Sep;346(3):414-23. doi: 10.1124/jpet.113.204644. Epub 2013 Jun 13. PubMed PMID: 23766543.
5: Gross N. The COPD pipeline XX. COPD. 2013 Feb;10(1):104-6. doi: 10.3109/15412555.2013.766103. PubMed PMID: 23413896.
6: Gross NJ. The COPD Pipeline XIV. COPD. 2012 Feb;9(1):81-3. doi: 10.3109/15412555.2012.646587. PubMed PMID: 22292600.
7: Boswell-Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP. The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6, 7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquino lin-4-one]. J Pharmacol Exp Ther. 2006 Aug;318(2):840-8. Epub 2006 May 8. PubMed PMID: 16682455.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-{2-[(2E)-2-(mesitylimino)-9,10-dimethoxy-4-oxo-6,7-dihydro-2H-pyrimido[6,1-a]-isoquinolin-3(4H)-yl]ethyl}urea
|
|
| Identifiers | |
| PubChem | CID 9934746 |
| ChemSpider | 8110374 |
| Synonyms | 9,10-Dimethoxy-2-(2,4,6-trimethylphenylimino)-3-(N-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one |
| Chemical data | |
| Formula | C26H31N5O4 |
| Molar mass | 477.554 g/mol |
///////////RPL-554, LS-193,855, 298680-25-8, UNII:3E3D8T1GIX, RPL554, RPL 554, phase 2, Chronic Obstructive Pulmonary Diseases , COPD, Allergic Rhinitis, Asthma Therapy, Cystic Fibrosis, Inflammation, Bronchodilators
Cc3cc(C)cc(C)c3N=c2cc1-c(cc4OC)c(cc4OC)CCn1c(=O)n2CCNC(N)=O
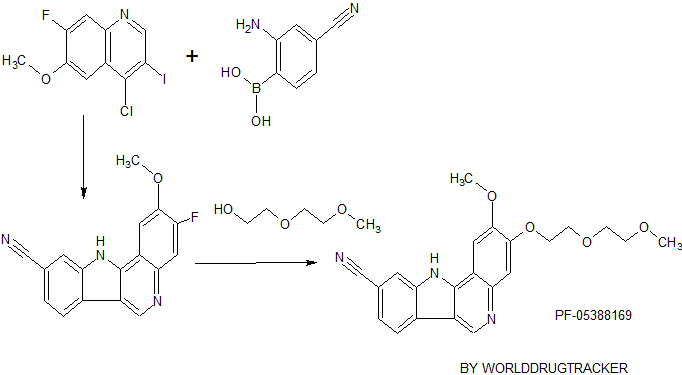
PF-05387252
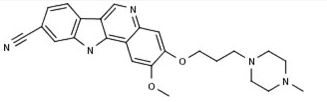
PF-05387252
CAS 1604034-71-0
| C25H27N5O2 | |
| MW | 429.51418 g/mol |
|---|
2-methoxy-3-[3-(4-methylpiperazin-1-yl)propoxy]-11H-indolo[3,2-c]quinoline-9-carbonitrile
IRAK4 inhibitor
Rheumatoid arthritis;
SLE
Preclinical
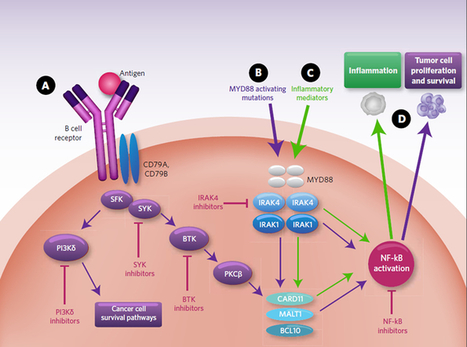
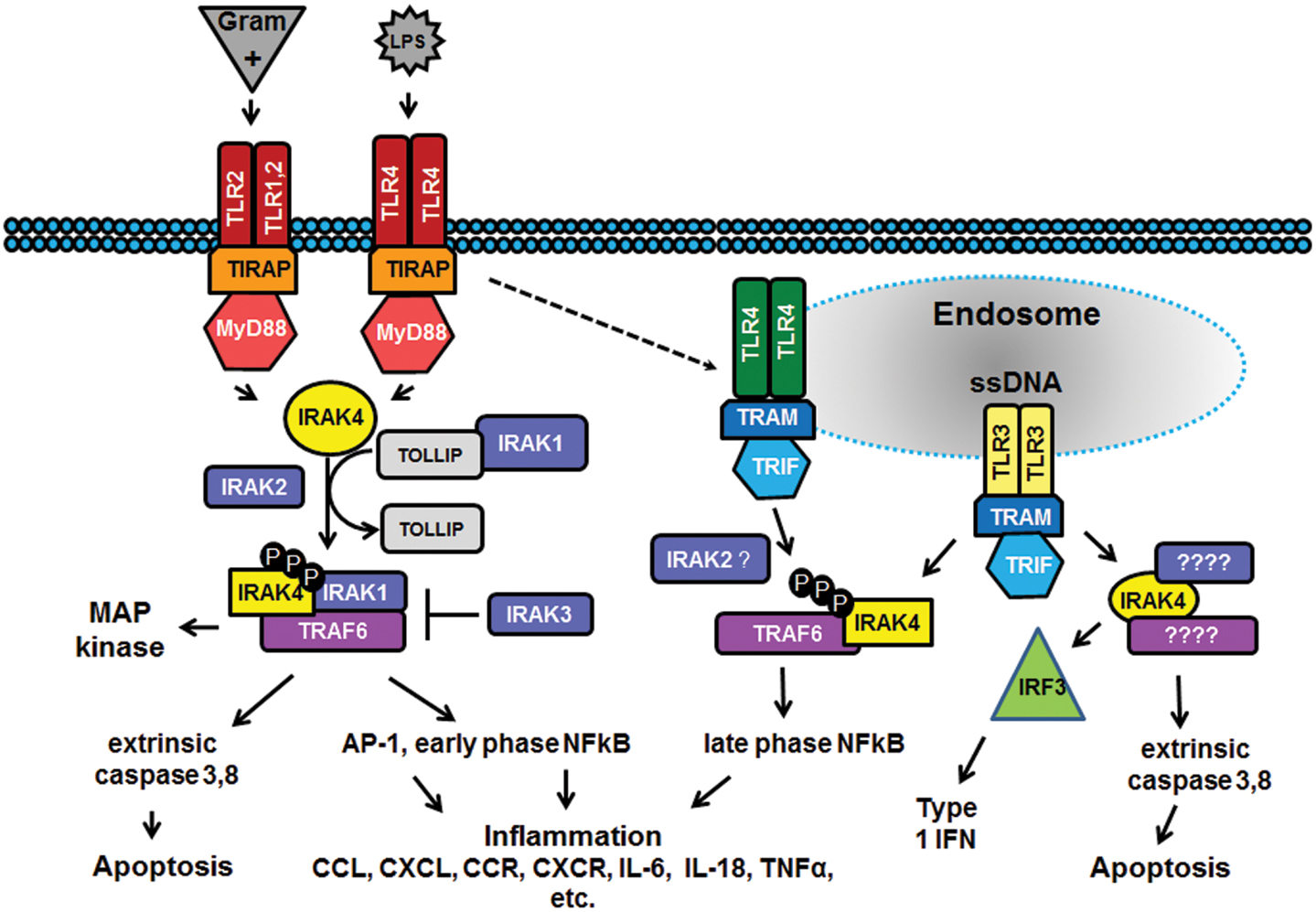
In the past decade there has been considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFα, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as HMGB1 and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system.
This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
Interleukin-1 receptor associated kinase (IRAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity. IRAK4 is responsible for initiating signaling from TLRs and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice lead to reductions in TLR and IL-1 induced pro-inflammatory cytokines. and 7 IRAK-4 kinase-dead knock-in mice have been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models. Likewise, humans deficient in IRAK4 also display the inability to respond to challenge by TLR ligands and IL-1
However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age implying redundant or compensatory mechanisms for innate immunity in the absence of IRAK4.These data suggest that inhibitors of IRAK4 kinase activity will have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be a viable strategy for the treatment of other inflammatory pathologies such as atherosclerosis.
Indeed, the therapeutic potential of IRAK4 inhibitors has been recognized by others within the drug-discovery community as evidenced by the variety of IRAK4 inhibitors have been reported to-date.12, 13, 14, 15 and 16 However, limited data has been published about these compounds and they appear to suffer from a variety of issues such as poor kinase selectivity and poor whole-blood potency that preclude their advancement into the pre-clinical models. To the best of our knowledge, no in vivo studies of IRAK4 inhibitors have been reported to-date in the literature. Herein we report a new class of IRAK4 inhibitors that are shown to recapitulate the phenotype observed in IRAK4 knockout and kinase-dead mice.
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
doi:10.1016/j.bmcl.2014.03.056
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- a Pfizer Global R&D, 445 Eastern Point Rd., Groton, CT 06340, USA
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
![]()

Abstract
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.



SYNTHESIS

////////PF-05387252, 1604034-71-0, PF 05387252, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL
N1(CCN(CC1)CCCOc3c(cc2c4nc5cc(ccc5c4cnc2c3)C#N)OC)C
OR
CN1CCN(CC1)CCCOC2=C(C=C3C(=C2)N=CC4=C3NC5=C4C=CC(=C5)C#N)OC
PF-05388169
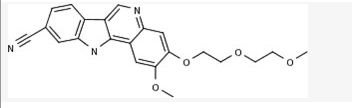
PF-05388169
CAS 1604034-78-7, MF C22 H21 N3 O4
MW 391.42
11H-Indolo[3,2-c]quinoline-9-carbonitrile, 2-methoxy-3-[2-(2-methoxyethoxy)ethoxy]-
- IRAK4 inhibitor
Rheumatoid arthritis;
SLE
Preclinical
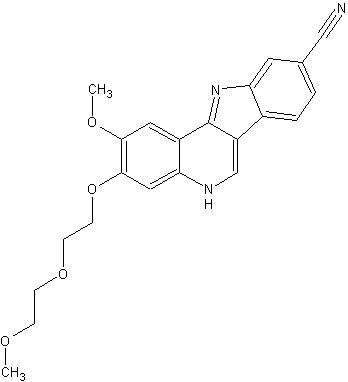
![]()
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- a Pfizer Global R&D, 445 Eastern Point Rd., Groton, CT 06340, USA
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.
SYNTHESIS
//////////PF-05388169, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL, 1604034-78-7
C(COC)OCCOc4c(cc3\C2=N\c1cc(ccc1/C2=C/Nc3c4)C#N)OC
PF-05387552
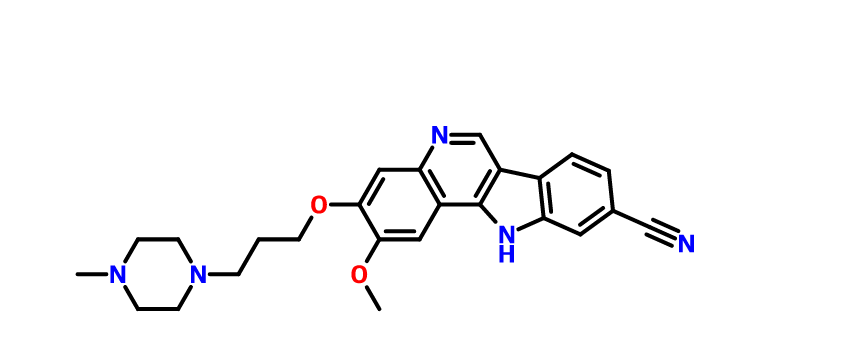
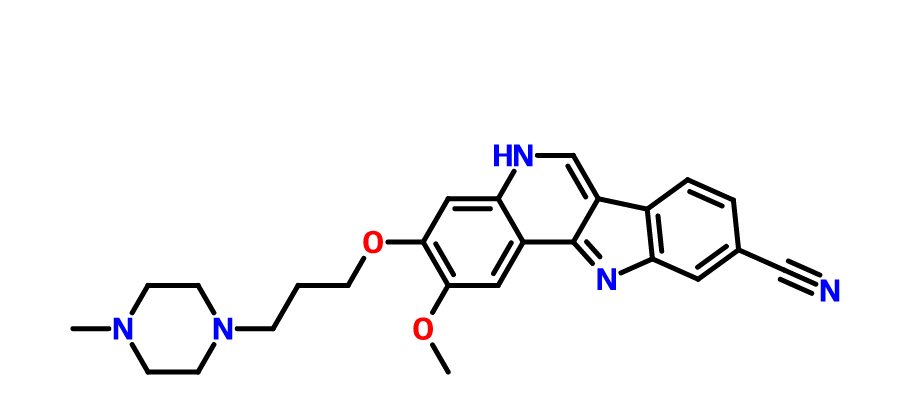
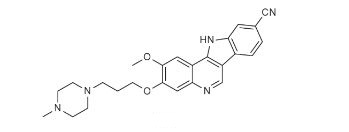
PF-05387552
IRAK4
CAS 1604034-71-0
C25 H27 N5 O2
11H-Indolo[3,2-c]quinoline-9-carbonitrile, 2-methoxy-3-[3-(4-methyl-1-piperazinyl)propoxy]-
2-methoxy-3-[3-(4-methylpiperazin-1-yl)propoxy]-11H-indolo[3,2-c]quinoline-9-carbonitrile
- Molecular Weight429.51
| Molecular Formula: | C25H27N5O2 |
|---|---|
| Molecular Weight: | 429.51418 g/mol |
![]()
Synthesis
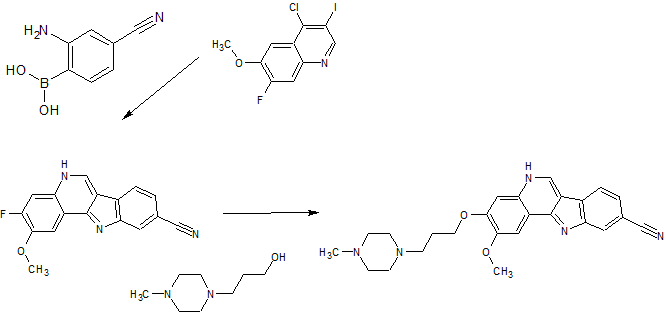
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072
Volume 24, Issue 9, 1 May 2014, Pages 2066–2072

Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- L. Nathan Tumeya, , ,
- Diane H. Boschellia,
- Niala Bhagiratha,
- Jaechul Shima,
- Elizabeth A. Murphyb,
- Deborah Goodwinb,
- Eric M. Bennettc,
- Mengmeng Wangd,
- Lih-Ling Linb,
- Barry Pressa,
- Marina Shenb,
- Richard K. Frisbiea,
- Paul Morganb,
- Shashi Mohanb,
- Julia Shinb,
- Vikram R. Raob
a Pfizer Global R&D, 445 Eastern Point Rd., Groton, CT 06340, USA
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
http://www.sciencedirect.com/science/article/pii/S0960894X14002832?np=y
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis.
Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway.
A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model.
To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.

 L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
REFERENCES

///////////TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PF-05387552, PF 05387552, 1604034-71-0
N#Cc3ccc4c5cnc2cc(OCCCN1CCN(C)CC1)c(OC)cc2c5nc4c3
2,000-year-old herb regulates autoimmunity and inflammation / Chang Shan, from a type of hydrangea that grows in Tibet and Nepal
Researchers discover molecular secrets of ancient Chinese herbal remedy
BOSTON, Mass. (February 12, 2012)—For roughly two thousand years, Chinese herbalists have treated Malaria using a root extract, commonly known as Chang Shan, from a type of hydrangea that grows in Tibet and Nepal. More recent studies suggest that halofuginone, a compound derived from this extract’s bioactive ingredient, could be used to treat many autoimmune disorders as well. Now, researchers from the Harvard School of Dental Medicine have discovered the molecular secrets behind this herbal extract’s power.
It turns out that halofuginone (HF) triggers a stress-response pathway that blocks the development of a harmful class of immune cells, called Th17 cells, which have been implicated in many autoimmune disorders.
“HF prevents the autoimmune response without dampening immunity altogether,” said Malcolm Whitman, a professor of developmental biology at Harvard School of Dental Medicine and senior author on the new study. “This compound…
View original post 635 more words
