
Home » Posts tagged 'immunosuppressant'
Tag Archives: immunosuppressant
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Laporolimus
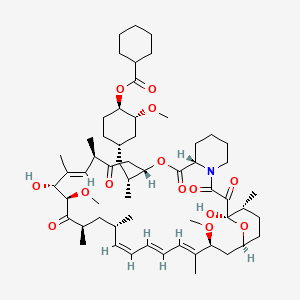
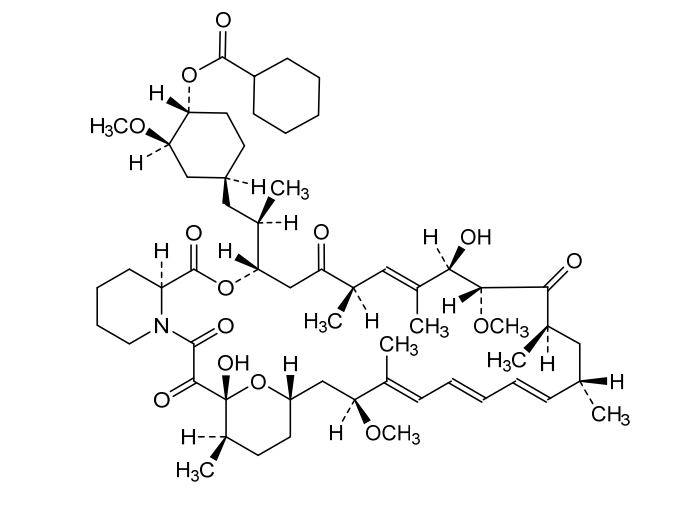
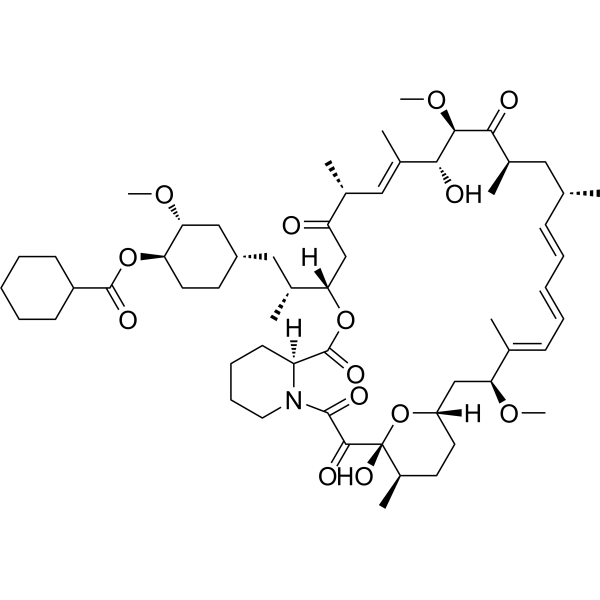
Laporolimus
Rapamycin, 42-cyclohexanecarboxylate
CAS 1504576-27-5
MF C58H89NO14 MW 1024.3 g/mol
[(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl] cyclohexanecarboxylate
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,3
1,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl
cyclohexanecarboxylate
immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
Laporolimus is an experimental immunosuppressant compound that acts as an mTOR (mechanistic target of rapamycin) pathway inhibitor. It is chemically classified as a macrolide derivative and is also known by its chemical synonym, rapamycin 42-cyclohexanecarboxylate.
Currently, Laporolimus is designated for research use only and has not been approved for clinical medical applications in humans or animals.
Key Technical Details
- Mechanism of Action: It blocks the mTOR signaling pathway, which is responsible for regulating cell growth, proliferation, and immune cell activation.
Distinguishing Laporolimus from Clinical Alternatives
Because it ends with the suffix -limus, it shares structural and nomenclature similarities with widely used clinical immunosuppressants. However, its legal status and development stage differ significantly:
| Drug Name | Clinical Availability | Primary Mechanism | Primary Uses |
|---|---|---|---|
| Laporolimus | None (Research Only) | mTOR Inhibitor | Laboratory research |
| Sirolimus (Rapamycin) | Approved | mTOR Inhibitor | Transplant rejection, coating coronary stents |
| Tacrolimus | Approved | Calcineurin Inhibitor | Organ transplant prophylaxis, severe eczema |
If you are researching this compound for a laboratory study, you can review its structural data and biochemical properties via the PubChem Laporolimus Compound Page
Laporolimus (CAS 1504576-27-5) is an immunosuppressive agent and mTOR inhibitor structurally derived from rapamycin as a cyclohexanecarboxylate derivative. Its total chemical synthesis is highly complex, typically achieved via semisynthesis starting from natural macrolides produced by Streptomyces fermentation.
Semisynthetic Pathway
Because the core macrocyclic lactone (a 36-membered polyketide ring) is incredibly challenging to build from scratch, researchers and pharmaceutical manufacturers rely on a derivatization approach:
- Fermentation: The baseline macrolide is produced via large-scale fermentation of Streptomyces hygroscopicus (similar to the base rapamycin process).
- Purification: The naturally produced macrocyclic core is isolated and purified from the fermentation broth using column chromatography.
- Esterification: The C-42 hydroxyl group of the macrolide core is selectively protected and subjected to acylation with a cyclohexanecarboxylic acid derivative (or reactive cyclohexanecarbonyl chloride).
- Deprotection & Purification: The C-42 cyclohexanecarboxylate is then deprotected and purified via preparative chromatography to yield pure laporolimus.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

PAT
/////////laporolimus, immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
EVEROLIMUS
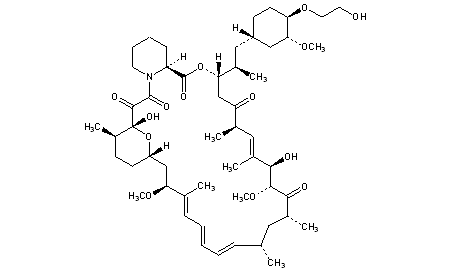
Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.

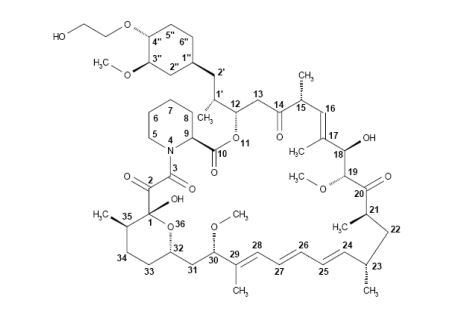
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2

Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4

Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5

Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6

Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
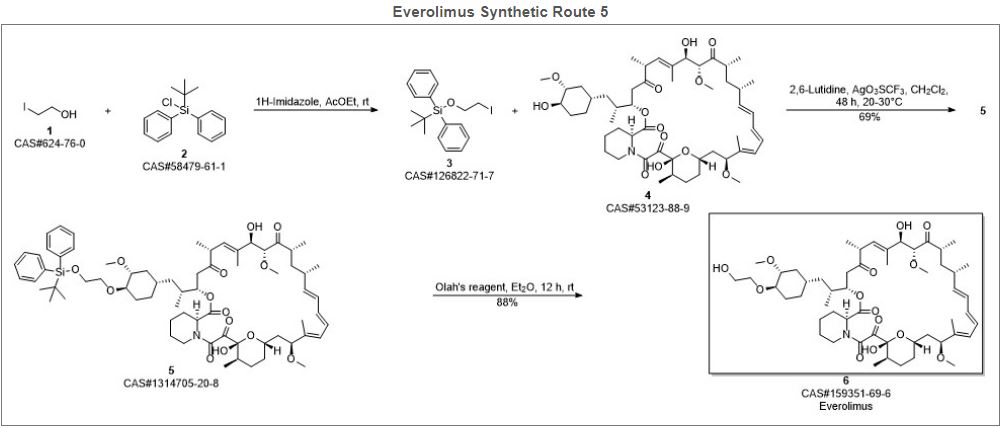
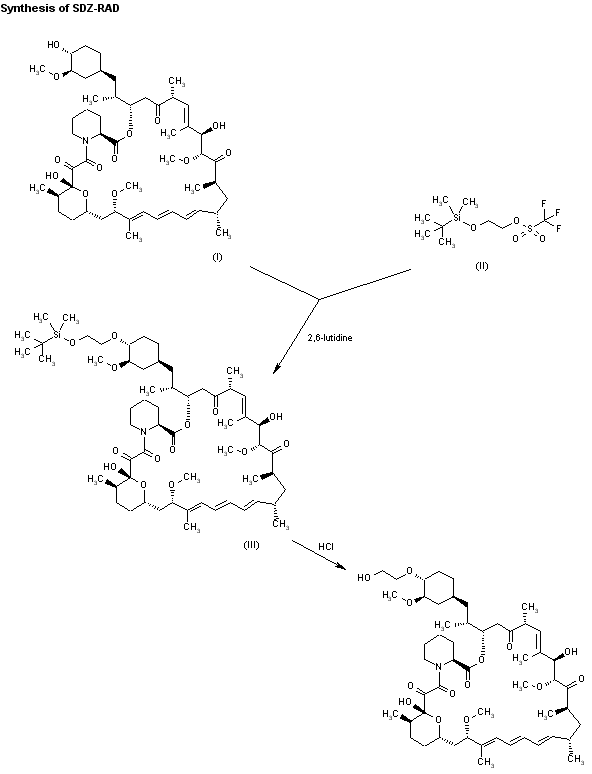
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
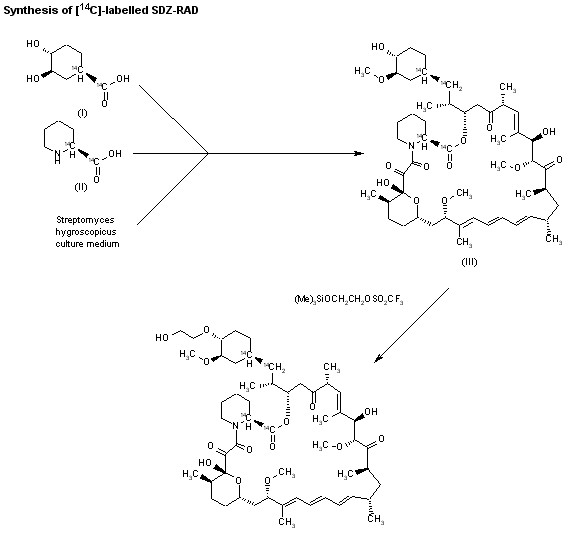
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
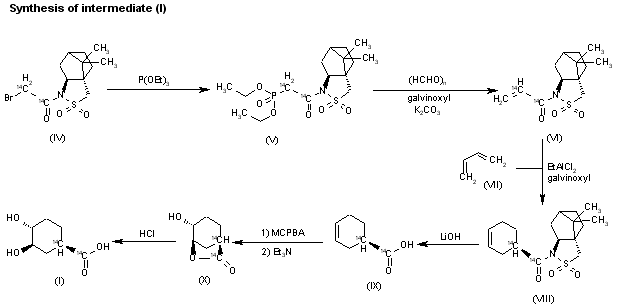
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
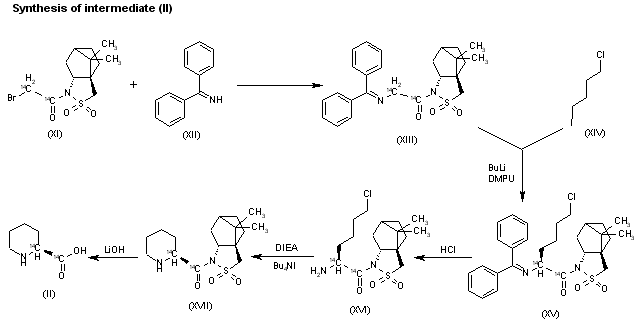
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),

It is a derivative of sirolimus of formula III),

and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),

wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),

was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche

Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),

as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),

wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Clip


References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
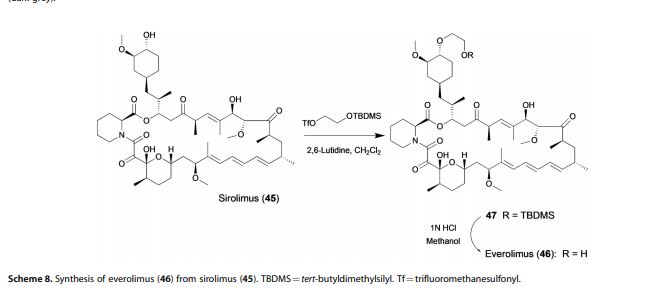
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]

Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.

“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.

When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References

- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,

Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
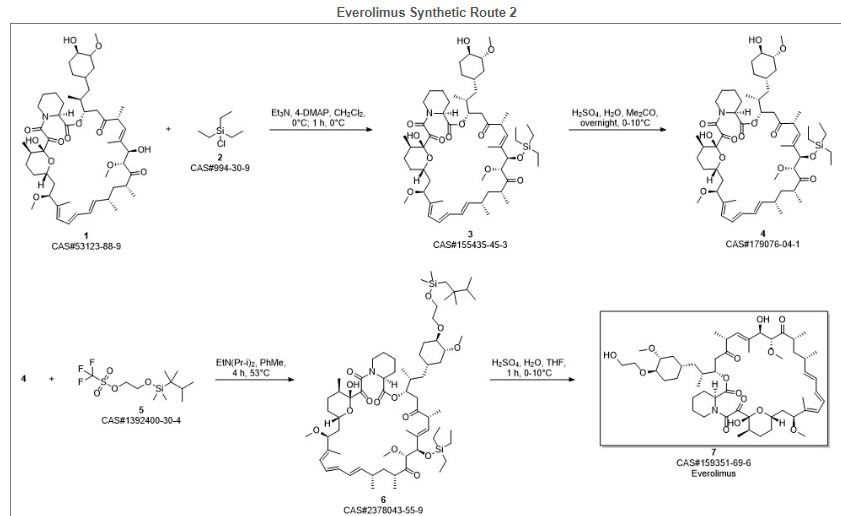
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
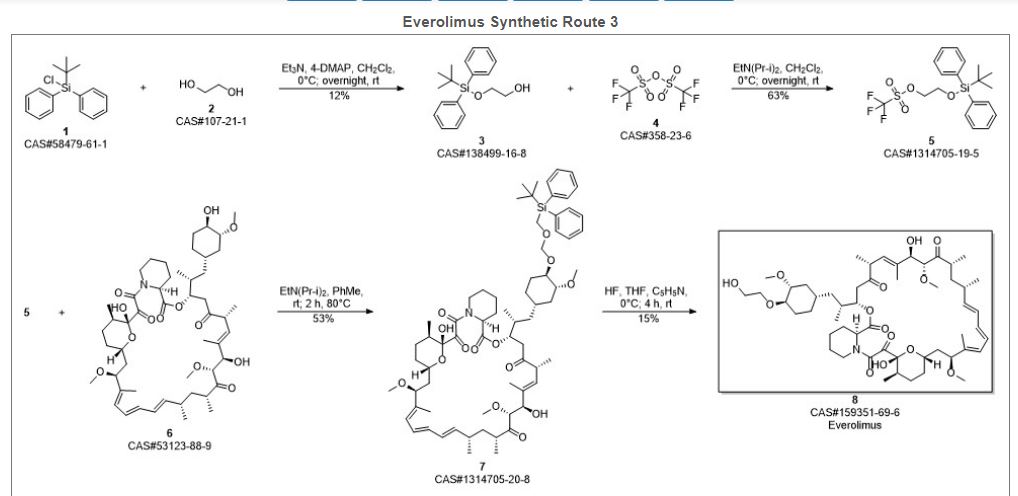
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
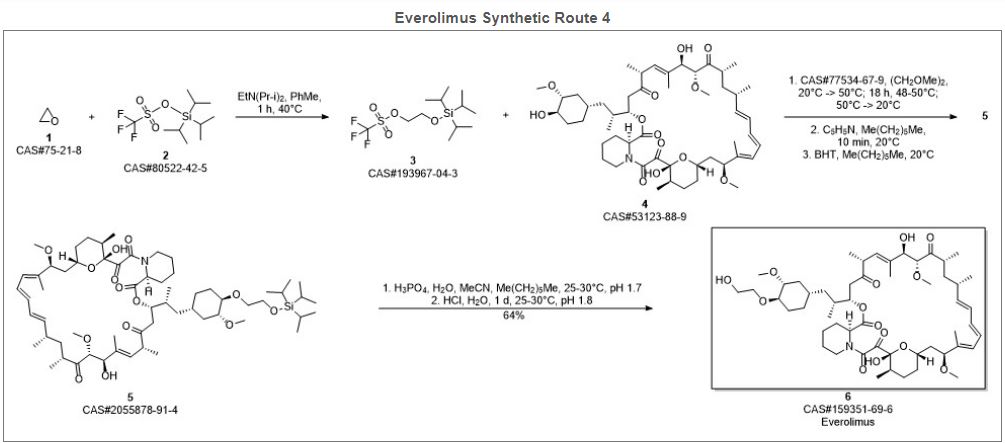
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
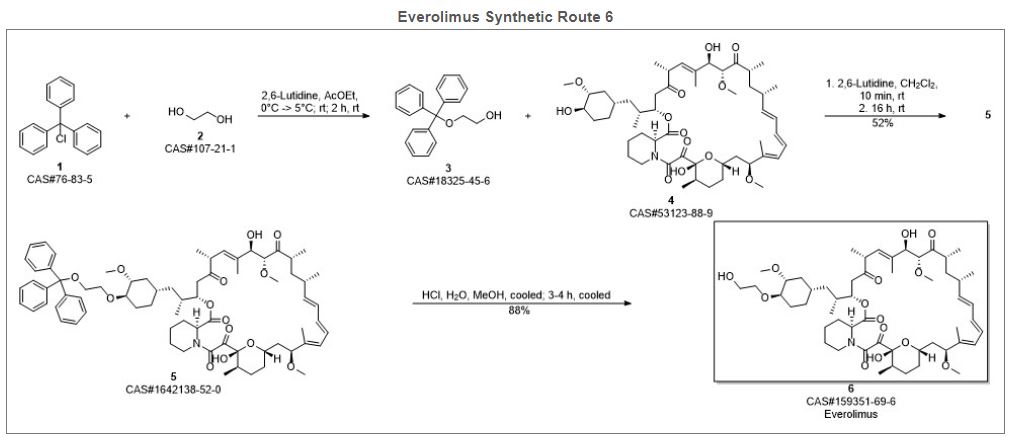
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Enzymes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Certican | Novartis ,2004 | |
| F | Certican | Novartis | |
| I | Certican | Novartis | |
| J | Certican | Novartis |
Formulations
- tabl. 0.25 mg, 0.5 mg, 0.75 mg
References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.

[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,
#RAD-001, #SDZ RAD, #Certican, #Novartis, #Immunosuppressant, #Everolimus, #Afinitor, #эверолимус , #إيفيروليموس , #依维莫司 ,
TACROLIMUS

Tacrolimus, Fujimycin
104987-11-3 CAS, 804.0182, C44H69NO12
- Astagraf XL
- FK 506
- FR 900506
- FR900506
- LCP-Tacro
- Prograf
- Protopic
- Tacrolimus
- Tacrolimus hydrate
- Tsukubaenolide hydrate
- UNII-WM0HAQ4WNM
3S-[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5, 19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c] [1,4] oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone, monohydrate
17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetraone
Astellas Pharma (Originator), LAUNCHED 1993
CTK8E6891, 109581-93-3 MONOHYDRATE TACROLIMUS
Tacrolimus (also FK-506 or Fujimycin) is an immunosuppressive drug whose main use is after organ transplant to reduce the activity of the patient’s immune system and so the risk of organ rejection. It is also used in a topical preparation in the treatment of severe atopic dermatitis, severe refractory uveitis after bone marrow transplants, and the skin condition vitiligo. It was discovered in 1984 from the fermentation broth of a Japanese soil sample that contained the bacteria Streptomyces tsukubaensis. Tacrolimus is chemically known as a macrolide. It reduces peptidyl-prolyl isomerase activity by binding to the immunophilin FKBP-12 (FK506 binding protein) creating a new complex. This FKBP12-FK506 complex interacts with and inhibits calcineurin thus inhibiting both T-lymphocyte signal transduction and IL-2 transcription.

PATENT
| Canada | 2037408 | 2002-12-17 | EXPIRY 2011-03-01 |
| Canada | 1338491 | 1996-07-30 | 2013-07-30 |
| United States | 5665727 | 1994-09-09 | 2014-09-09 |
| United States | 5260301 | 1994-02-28 | 2011-02-28 |
Pan Sup Chang, Hoon Cho, “Water soluble polymer-tacrolimus conjugated compounds and process for preparing the same.” U.S. Patent US5922729, issued April, 1997.

Tacrolimus is a naturally-occurring macrolide isolated from the fermentation broth of Streptomyces tsukubaensis that was originally discovered by Fujisawa (now Astellas Pharma) in 1984. Tacrolimus possesses immunosuppressive properties and suppresses IL-2 production from helper T-cells, resulting in inhibition of the activation and proliferation of cytotoxic T-cells. In the cell, tacrolimus binds to an immunophilin called FKBP-12 and forms a tacro-immunophilin complex that, in turn, binds to calcineurin and prevents the dephosphorylation of cytoplasmic NF-AT thus disallowing it from reaching the nucleus, thereby strongly inhibiting IL-2 gene transcription. As a result, T-cell activation and proliferation is inhibited.
In 1993, Prograf(R) (tacrolimus capsules and injection) received clearance from the Japanese Ministry of Health and Welfare and was introduced in Japan the same year for the treatment of kidney and liver transplant rejection. Based on two large phase III comparative clinical trials, the product received clearance from the FDA in April 1994, and was made available two months later for commercial use in the U.S. The product is available extensively for transplant rejection. Prograf(R) was also launched in Japan for the treatment of myasthenia gravis and for the treatment of heart transplant rejection; the latter indication was approved in the U.S. in 2006 and launched in 2007. In 2008, Astellas Pharma preregistered the compound in Japan for the oral treatment of all cases of myasthenia gravis. The same year, Senju launched the product in Japan for the treatment of vernal and perennial allergic conjunctivitis in patients unresponsive to anti-allergic drugs. In 2009, the product was approved and commercialized in Japan for the treatment of ulcerative colitis. In 1999, Astellas Pharma launched Protopic(R) (tacrolimus ointment) in Japan for the treatment of atopic dermatitis and in 2001, Protopic(R) was commercialized in the U.S. and Europe. In April 2005, tacrolimus (capsules) was commercialized again by Astellas Pharma in Japan for the treatment of rheumatoid arthritis (RA) in patients who respond insufficiently to current therapies. The following year, Senju received approval in Japan for the use of tacrolimus for the treatment of vernal conjunctivitis and perennial allergic conjunctivitis. A once-daily capsule was approved in the E.U. in 2006. The compound was launched in 2007 in Japan for lupus nephritis. In 2009, the product was approved in US for the prophylaxis of organ rejection in allogeneic kidney transplantation in combination with mycophenolate mofetil and, in the E.U., for the prophylaxis of transplant rejection in adult and pediatric, kidney, liver or heart allograft recipients. In 2011, the compound was launched in Japan for the prophylaxis of organ rejection in patients receiving allogeneic small bowel transplants. In 2013, the indication for interstitial pneumonia associated with polymyositis/dermatomyositis was approved in Japan and an extended release formulation was approved in the U.S. for the prophylaxis of organ rejection in adult patients receiving kidney transplants. This extended release formulation was launched in the U.S. in August 2013. Veloxis Pharmaceuticals (formerly LifeCycle Pharma) is developing a once-daily tablet formulation of tacrolimus (Envarsus®) with improved bioavailability and reduced variability compared with the modified-release version of the compound. Envarsus® has been pre-registered in E.U. and the U.S. for the prevention of transplant rejection in kidney transplant patients. The company is also evaluating the compound in phase II trials for the treatment of autoimmune hepatitis.
In terms of clinical development, the National Cancer Institute (NCI) is developing tacrolimus in phase III for the treatment of graft-versus-host disease (GVHD). Phase III trials are also underway at Astellas Pharma for the treatment of psoriasis, ulcerative colitis and chronic focal encephalitis (Rasmussen’s encephalitis), while early clinical trials are ongoing for asthma. In 2009, Astellas Pharma withdrew an NDA seeking approval in the U.S. based on potential clinical challenges that would result from FDA requirements to conduct additional clinical studies. Kyoto University had been conducting phase II clinical studies for the treatment of Crohn’s disease; however, no recent development has been reported for this research.
In 2003, Sucampo Pharmaceuticals obtained a license from Astellas Pharma to develop and market tacrolimus for ophthalmic indications in the U.S. and Europe, however, in June 2005, the company voluntarily discontinued its tacrolimus eye drops development program due to FDA safety concerns. In 2005, Senju and Astellas Pharma established an agreement to codevelop an eye drop formulation of tacrolimus in Japan. Also, Astellas Pharma granted Senju exclusive manufacturing and marketing rights of the compound. In 2003, Astellas Pharma and GlaxoSmithKline signed an agreement for the copromotion of Protopic(R) in the U.S for atopic dermatitis. An additional agreement for the copromotion of Protopic(R) in South America for the same indication was signed in 2004 between Astellas Pharma and Roche. Tacrolimus was designated orphan drug status in Japan in 1993 and in 2005 for the suppression of organ rejection in allogenic kidney transplantation and for the treatment of vernal conjunctivitis, respectively, in patients unresponsive to anti-allergic drugs. In the E.U., the latter indication was assigned orphan drug designation in 2004. The product was withdrawn from the community register of designated orphan medicinal products in the E.U. in April 2010 on request of the sponsor. In 1998 and 2005, the FDA assigned orphan drug designation for the prophylaxis of GVHD and for the prophylaxis of organ rejection in patients receiving heart transplants. Finally, in 2008, orphan designation was received in Japan for the treatment of myasthenia gravis. In 2012, an additional orphan drug designation was assigned in the U.S. for the treatment of hemorrhagic cystitis. This designation was granted in Japan in 2012 for the treatment of interstitial pneumonia accompanied with polymyositis/dermatomyositis complex. In 2012, orphan drug designation was assigned in Japan for the treatment of interstitial pneumonia accompanied with polymyositis/dermatomyositis complex. In 2012, the product was licensed by Veloxis Pharmaceuticals to Chiesi on an exclusive basis for the commercialization and distribution in Europe, Turkey and CIS countries for the prevention of rejection in kidney transplant recipients. In 2013, an additional orphan drug designation was assigned in the U.S. for the prophylaxis of organ rejection in patients receiving allogeneic kidney transplant.
Tacrolimus, also known as FK-506 or FR-900506, has the chemical tricyclic structure shown below:

corresponding to C44H69NO-|2- Tacrolimus appears in the form of white crystals or crystalline powder. It is practically insoluble in water, freely soluble in ethanol and very soluble in methanol and chloroform. The preparation of tacrolimus is described in EP-A-0 184 162 and analogues of tacrolimus are disclosed e.g. in EP-A-0444659 and US 6,387,918
Tacrolimus is an immunosuppressive agent produced by Streptomyces tsukubaensis No. 9993 and is the compound of formula (I) wherein R.sub.1 and R.sub.2 are both hydrogen. Tacrolimus, which is also called FK-506, has first discovered by Tanaka, Kuroda and their colleague in Japan see, J. Am. Chem. Soc., 1987, 109, 5031 and U.S. Pat. No. 4,894,366 issued on Jan. 16, 1990!.
July 19, 2013 /PRNewswire/ — Astellas Pharma US, Inc. (“A.stellas”), a U.S. subsidiary of Tokyo-based Astellas Pharma Inc., announced today that the U.S. Food and Drug Administration (FDA) has approved Astagraf XL (tacrolimus extended-release capsules) for the prophylaxis of organ rejection in patients receiving a kidney transplant with mycophenolate mofetil (MMF) and corticosteroids, with or without basiliximab induction.
“Each transplant recipient is different and requires a personalized treatment approach. The approval of Astagraf XL marks an important milestone in post-transplant care as it provides physicians with a new treatment option for kidney t recipients,” said Sef Kurstjens, M.D., PhD., chief medical officer, Astellas Pharma, Inc. “Astellas is pleased to continue our more than 20-year commitment to the field of transplant immunology.”


PROTOPIC (tacrolimus) Ointment contains tacrolimus, a macrolide immunosuppressant produced by Streptomyces tsukubaensis. It is for topical dermatologic use only. Chemically, tacrolimus is designated as [3S[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*]]5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10, 12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c][1,4] oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone,monohydrate. It has the following structural formula:
 |
Tacrolimus has an empirical formula of C44H69NO12•H2O and a formula weight of 822.03. Each gram of PROTOPIC Ointment contains (w/w) either 0.03% or 0.1% of tacrolimus in a base of mineral oil, paraffin, propylene carbonate, white petrolatum and white wax.
FK-506 (also Tacrolimus or fujimycin) is a potent calcineurin (protein phosphatase 2B) inhibitor that requires FK 506-binding protein 12 (FKBP12) for activity (IC50 = 3 nM). FK-506 inhibits secretion of IL-1, IL-2 (IC50 = 1 nM), IL-3, IL-4, IL-6 (IC50 = 35 nM), GM-CSF, TNFα (IC50 = 10 nM), IFNγ and Myc from activated T-cells in vitro. FK-506 exhibits potent immunosuppressive, neuroprotective and anticonvulsant activity in vivo. The physiological effects of FK-506 also include regulation of nitric oxide neurotoxicity, neurotransmitter release, and regulation of Ca2+ release via the ryanodine and inositol-(1,4,5)-trisphosphate (IP3) receptors. Furthermore, it has become clear that, predominantly as a result of CaN inhibition, FK506 alters multiple biochemical processes in a variety of cells besides lymphocytes. FK506 and ascomycin inhibit signaling pathways in astrocytes and change the pattern of cytokine and neurotrophin gene expression.
Tacrolimus (also FK-506 or fujimycin, trade names Prograf, Advagraf, Protopic) is an immunosuppressive drug that is mainly used after allogeneic organ transplant to reduce the activity of the patient’s immune system and so lower the risk of organ rejection. It is also used in a topical preparation in the treatment of atopic dermatitis (eczema), severe refractory uveitis after bone marrow transplants, exacerbations of minimal change disease, and the skin condition vitiligo.
It is a 23-membered macrolide lactone discovered in 1984 from the fermentation broth of a Japanese soil sample that contained the bacteria Streptomyces tsukubaensis. It reduces interleukin-2 (IL-2) production by T-cells.
 Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from ‘Tsukuba macrolide immunosuppressant’.
Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from ‘Tsukuba macrolide immunosuppressant’.

Tacrolimus 0.1%
| Indication | For use after allogenic organ transplant to reduce the activity of the patient’s immune system and so the risk of organ rejection. It was first approved by the FDA in 1994 for use in liver transplantation, this has been extended to include kidney, heart, small bowel, pancreas, lung, trachea, skin, cornea, and limb transplants. It has also been used in a topical preparation in the treatment of severe atopic dermatitis. |
|---|---|
| Pharmacodynamics | Tacrolimus is a macrolide antibiotic. It acts by reducing peptidyl-prolyl isomerase activity by binding to the immunophilin FKBP-12 (FK506 binding protein) creating a new complex. This inhibits both T-lymphocyte signal transduction and IL-2 transcription. Although this activity is similar to cyclosporine studies have shown that the incidence of acute rejection is reduced by tacrolimus use over cyclosporine. Tacrolimus has also been shown to be effective in the topical treatment of eczema, particularly atopic eczema. It suppresses inflammation in a similar way to steroids, but is not as powerful. An important dermatological advantage of tacrolimus is that it can be used directly on the face; topical steroids cannot be used on the face, as they thin the skin dramatically there. On other parts of the body, topical steroid are generally a better treatment. |
| Mechanism of action | The mechanism of action of tacrolimus in atopic dermatitis is not known. While the following have been observed, the clinical significance of these observations in atopic dermatitis is not known. It has been demonstrated that tacrolimus inhibits T-lymphocyte activation by first binding to an intracellular protein, FKBP-12. A complex of tacrolimus-FKBP-12, calcium, calmodulin, and calcineurin is then formed and the phosphatase activity of calcineurin is inhibited. This prevents the dephosphorylation and translocation of nuclear factor of activated T-cells (NF-AT), a nuclear component thought to initiate gene transcription for the formation of lymphokines. Tacrolimus also inhibits the transcription for genes which encode IL-3, IL-4, IL-5, GM-CSF, and TNF-, all of which are involved in the early stages of T-cell activation. Additionally, tacrolimus has been shown to inhibit the release of pre-formed mediators from skin mast cells and basophils, and to downregulate the expression of FceRI on Langerhans cells. |
Tacrolimus was first approved by the Food and Drug Administration (FDA) in 1994 for use in liver transplantation; this has been extended to include kidney, heart, small bowel, pancreas, lung, trachea, skin, cornea, bone marrow, and limb transplants.
The branded version of the drug is owned by Astellas Pharma, and is sold under the trade names Prograf given twice daily, Advagraf, a sustained release formulation allowing once daily dosing, and Protopic (Eczemus in Pakistan by Brookes Pharma), the topical formulation. Advagraf is available in 0.5, 1, 3 and 5 mg capsules, the ointment is concentrations of 0.1% and 0.03%.
A second once-daily formulation of tacrolimus is in Phase 3 clinical trials in the U.S. and Europe. This formulation also has a smoother pharmacokinetic profile that reduces the peak-to-trough range in blood levels compared to twice-daily tacrolimus.Data from the first Phase 3 trial in stable kidney transplant patients showed that this once-daily formulation was non-inferior in efficacy and safety compared to twice-daily tacrolimus. A second Phase 3 trial in de novo patients is ongoing.

Tacrolimus, which is also referred to as FK-506 (Fermentek catalogue number 506), is a 23-membered macrolide lactone and belongs to the group of polyketides. Tacrolimus was first isolated in the 1980’s from the fermentation broth of the soil bacteria Streptomyces tsukubaensis. The antibiotic macrolide compound tacrolimus was e.g. reported in 1984 by Kino et al. (J. Antibiotics 40, 1249-1255, 1984). Later on tacrolimus was prepared as a microbial natural product by using different microorganisms, i.e. soil bacteria such as Streptomyces sp. MA6858 (US 5,116,756) ATCC 55098, Streptomyces tsukubaensis NRRL 18488 (EP-B 0 356 399 and US 5,200,41 1 ), Streptomyces clavuligerus CKD 1119 (KR-B 100485877) or Streptomyces glaucescens MTCC 5115 (US 2007191415).
The product tacrolimus exhibits immunosuppressive activities which are due to its effect to reduce the activity of the enzyme peptidyl-propyl isomerase and to the binding to the protein immunophilin FKBP12 (FK506 binding protein). Tacrolimus and the structurally similar polyketides ascomycin and rapamycin require initial binding to the highly conserved protein cyclophilin FKBP12 in order to be physiologically active. The rapamycin/FKBP12 complex binds to mTOR (mammalian target of rapamycin), a serine- threonine kinase that appears to act as a central controller for sensing the cellular environment and regulating translation initiation (see e.g. Easton J. B. and Houghton P.J., 2004, Expert Opin Ther Targets; 8(6):551-64). However, the tacrolimus/FKBP12 complex was found to bind to a different cellular target and inhibits the phosphatase activity of calcineurin, in analogy to cyclosporine (see Allison A.C., 2000, Immunopharmacology; 47(2-3):63-83).
Tacrolimus is often used for immunosuppression following e.g. organ transplantation. Furthermore, tacrolimus and its derivatives have been shown to be effective in treating a number of diseases such as asthma, inflammatory diseases and hyperproliferative skin disease. Tacrolimus and other immunosuppressant such as rapamycin, cyclosporine, or a combination thereof are also useful in the treatment of various auto-immmune diseases. For many years calcineurin inhibitors (e.g. cyclosporine and tacrolimus) have been the mainstay of immunosuppressive therapy. These two compounds are potent suppressors of cellular immune response and have significantly improved the outcome of organ transplants during the past two decades (see Allison A.C., 2000, Immunopharmacology; 47(2-3):63-83). Gene clusters encoding the biosynthetic pathways of a great number of medically important drugs of microbial origin have already been cloned and sequenced, including the gene cluster of macrolides rapamycin, ascomycin and tacrolimus. With respect to cloning of the tacrolimus gene cluster, a partial sequence, mostly encompassing genes encoding polyketide synthase (PKS), was reported in the literature (see Motamedi H. and Shafiee A. 1998, Eur J Biochem; 256(3):528-34). On the other hand, scientists reported cloning of the ascomycin gene cluster in 2000 (see Wu K et al. 2000, Gene; 251(1 ):81- 90, US 6,503,737). Tacrolimus structurally and by the biosynthetic origin resembles ascomycin (FK520) and rapamycin (see Reynolds et al.; Drugs and the Pharmaceutical Sciences, 1997, 82, 497-520. They all can be synthesised by combined polyketide (PKS) and non-ribosomal peptide biosynthetic pathways (NRPS) (see McDaniel R et al. 2005, Chem Rev; 105(2):543-58).
Tacrolimus and ascomycin are structurally similar. As only structural difference, the allyl side chain at carbon 21 of tacrolimus is replaced by an ethyl side chain in ascomycin. The structures of tacrolimus (FK506) and ascomycin (FK520) compounds are shown as formulae (Ia) and (Ib). The structures of ascomycin and tacrolimus already suggest complex biosynthetic pathways which can be divided into four steps considering the biosynthetic mechanism:
1. chain initiation using the unusual shikimate derived starter,
2. chain elongation common to most PKS derived compounds,
3. chain termination and cyclization by incorporation of L-pipecolic acid and
4. post-PKS processing.
During the tacrolimus fermentation process, undesired ascomycin (FK520) product is also produced as an impurity, thus lowering the final yield of tacrolimus and causing significant additional costs to the downstream isolation processes of tacrolimus.

(Ia) FK506, R = -CH2-CH = CH2
(Ib) FK520, R = CH2-CH3
For oral administration, tacrolimus is currently formulated and marketed as soft gelatine capsules comprising the equivalent of 0.5, 1 or 5 mg anhydrous tacrolimus and marketed under the trade name Prograf® and Protropic®. The recommended initial oral dose is from about 0.1 to 0.2 mg/kg/day in patients. The dose aims at a certain trough plasma level from about 5 to about 20 ng/ml. Prograf® is indicated for the prophylaxis of organ rejection in patients receiving allogeneic liver or kidney transplants. There remains a need for novel pharmaceutical compositions and/or dosage forms comprising tacrolimus exhibiting enhanced bioavailability. An increased bioavailability may allow a reduction in the dosage units taken by a patient, e.g. down to a single dose daily, and may also reduce or negate the need for food to be takes simultaneously with the dosage form thereby allowing patients more freedom on when the drug is taken. Furthermore, it is contemplated that fluctuations in the plasma concentration versus time profile may be significantly reduced. Further, enhanced bioavailability may also result in a more reproducible (i.e. less variable compared to that of Prograf®) release profile….
………………….
h) Determination of tacrolimus and ascomycin production with HPLC of thiostrepton resistant ccr disrupted mutants derived by secondary homologous recombination using pKC1 139-ccrTs.:
Method for tacrolimus and ascomycin determination: The analysis for determination of tacrolimus or ascomycin production thereof was carried out by isocratic reversed phase HPLC using an appropriate column and running conditions: column Nucleosil-100 C18 (150×4.0 mm, particle size 3 μm), flow 1.5 ml/min, T°C=60°C, mobile phase: 560 ml water, 335 ml acetonitrile, 70 ml MTBE and 0.2 ml 85% H3PO4, detection 210 nm, sample injection 20 μl.
The tacrolimus and ascomycin content in samples quantification was performed by using external standards of tacrolimus and ascomycin, where tacrolimus was eluted at 12.5 min and ascomycin at 11.5 min. Results are expressed as % of ascomycin production compared to tacrolimus production in samples.
……………………….
http://www.drugfuture.com/synth/syndata.aspx?ID=124071

A new total synthesis of FK-506 is described: This synthesis has been performed by previous construction of two building fragments (XXIV) and (LI), which later were coupled and cyclized. (Schemes 1-3): 1) (1R*S*,3R,5S,6R,7S,9R)-6-(tert-butyldimethylsilyloxy)-9-(1,3-dithian-2-yl)-5,7-dimethoxy-1-methyldecyl diphenyl phosphine oxide (XXIV). The Sharpless asymetric epoxidation of 1,4-pentadien-3-ol (I) with (-)-diisopropyltartrate and tert-butylhydroperoxide gives the epoxy alcohol (II) with high optical purity, which is benzylated in the usual way to (III). The reaction of (III) with lithioacetonitrile and then HCl yields lactone (IV), which is methylated with lithium diisopropylamide and methyl iodide to lactone (V) as major isomer (separated by chromatography on SiO2). The reduction of (V) with LiAlH4 affords the diol (VI), which is converted into the bis(tert-butyl carbonate) (VII) with 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile (BOC-N). The reaction of (VII) with Br2 and K2CO3 in dichloromethane gives the bromocarbonate (VIII), which by selective saponification of the cyclic carbonate with NaOCH3 in methanol yields the epoxy alcohol (IX). Methylation of (IX) with NaH and methyl iodide affords the methyl ether (X), which is converted into the butyrolactone (XI) with lithioacetonitrile as before. The protection of the OH group of (XI) with TBS-Cl gives the silyl ether (XII), which by trans-selective methylation with lithium diisopropylamide and methyl iodide yields lactone (XIII). The reduction of (XIII) with LiAlH4 affords diol (XIV) as major isomer (separated by column chromatography). The selective esterification of the primary OH group of (XIV) with pivaloyl chloride gives the hydroxy ester (XV), which is methylated with NaH and methyl iodide as usual to the methoxy derivative (XVI). Debenzylation of (XVI) by hydrogenolysis with H2 over Pd/C yields the hydroxy ester (XVII), which is silylated with TBS-SO3CF3 to the fully protected compound (XVIII).

Selective deprotection of (XVIII) with trifluoroacetic acid in THF – water affords the primary alcohol (XIX), which is oxidized with oxalyl chloride and DMSO in dichloromethane to the aldehyde (XX). The protection of the aldehyde group of (XX) with propane-1,3-dithiol and BF3 gives the dithiane derivative (XXI), which is resilylated with TBS-SO3CF3 as before to the dithiane (XXII). The pivaloyl group of (XXII) is eliminated with LiAlH4 in THF yielding the alcohol (XXIII), which is finally treated with benzenesulfonyl chloride and then with ethyl diphenylphosphine oxide and butyllithium in THF to obtain the first building group, the phosphine derivative (XXIV).

2) [2S,3S,5S,6R,7S,8E,9(1’R,3’R,4’R)]-2-Allyl-3-(tert-butyldimethylsilylox y)-6,8-dimethyl-7-(triethylsilyloxy)-5-(triisopropylsilyloxy)-9-[3-meth oxy-4-(triisopropylsilyloxy)cyclohexyl]-8-nonenal (LI). Quinic acid (XXV) is converted into the lactone (XXVI) by known methods. Then this lactone is treated with thiocarbonyldiimidazole in refluxing dichloroethane yielding the bis(thiocarbonyl)lactone (XXVII), which by reaction with tributyltin hydride and AIBN in refluxing xylene is converted into the lactone (XXIX), either directly or through the intermediate thiocarbonyl-lactone (XXVIII). The silylation of (XXIX) with TIPS-SO3CF3 as usual affords the protected lactone (XXX). Opening of the lactone ring with methylchloroaluminum N-methoxy-N-methylamide gives the methoxyamide (XXXI), which is methylated with methyl trifluoromethylsulfonate to the methoxy-N-methoxyamide (XXXII). The reduction of (XXXII) with diisobutylaluminum hydride gives the aldehyde (XXXIII), which is condensed with 2-lithio-2-(triethylsilyl)propanal (XXXIV), yielding unsaturated aldehyde (XXXV). The condensation of (XXXV) with the boron enolate of oxazolidone (XXVI) affords the oxazolidone derivative (XXXVII), which is treated with methylchloroaluminum N-methoxy-N-methylamide to give the methoxyamide (XXXVIII). The silylation of (XXXVIII) with TES-SO3CF3 as usual yields the silylated amide (XXXIX), which is reduced with diisobutylaluminum hydride to the aldehyde (XL). The condensation of (XL) with chiral acetate (XLI) by means of lithium diisopropylamide in THF affords the hydroxy ester (XLII). Transesterification of (XLII) with NaOCH3 and methanol gives methyl ester (XLIII).
http://www.drugfuture.com/synth/syndata.aspx?ID=124071
…………..
Use of the microorganism streptomyces tsukubaensis No. 9993 for the production of the FR-900506 substance of the formula:

- [II]
Synthetic Processes
- :(1)
Process 1
- (Introduction of common Hydroxy-Protective Group)

(2)
Process 2
- (Introduction of common Hydroxy-Protective Group)

(3)
Process 3
- (Formation of Double Bond)

(4)
Process 4
- (Oxidation of Hydroxyethylene Group)

(5)
Process 5
- (Reduction of Allyl Group)
-

in which
R¹, R², R³, n and the symbol of a line and dotted line are each as defined above,
R 1 a
and R 2 a

are each commonly protected hydroxy, and
R 2 b
is a common leaving group.
- THE MICROORGANISM
-
The microorganism which can be used for the production of the FR-900506, FR-900520 and/or FR-900525 substances is FR-900506 FR-900520 and/or FR-900525 substance(s)-producing strain belonging to the genusStreptomyces, among which Streptomyces tsukubaensis No. 9993 has been newly isolated from a soil sample collected at Toyosato-cho, Tsukuba-gun, Ibaraki Prefecture, Japan.
-
A lyophilized sample of the newly isolated Streptomyces tsukubaensis No. 9993 has been deposited with the Fermentation Research Institute, Agency of Industrial Science and Technology (No. 1-3, Higashi 1-chome, Yatabemachi Tsukuba-gun, Ibaraki Prefecture, Japan) under the deposit number of FERM P-7886 (deposited date: October 5th, 1984), and then converted to Budapest Treaty route of the same depository on October 19, 1985 under the new deposit number of FERM BP-927.
-
The Streptomyces tsukubaensis No. 9993 has the following morphological, cultural, biological and physiological characteristics.
-

-
This white powder of the FR-900506 substance could be transformed into a form of crystals by recrystallization thereof from acetonitrile, which possess the following physical and chemical properties.
(1) Form and Color:
colorless prisms
(2)Elemental Analysis: C: 64.30 %, H: 8.92 %, N: 1.77 % 64.20 %, 8.86 %, 1.72 %, (3) Melting Point:
127 – 129 °C
(4) Specific Rotation:
[α] 23 D
: -84.4° (c = 1.02, CHCl₃)
(5) ¹³C Nuclear Magnetic Resonance Spectrum:
the chart of which being shown in Figure 3,
(6) ¹H Nuclear Magnetic Resonance Spectrum:
the chart of which being shown in Figure 4. -
Other physical and chemical properties, that is, the color reaction, solubility, ultraviolet absorption spectrum, infrared absorption spectrum, thin layer chromatography and property of the substance of the colorless prisms of the FR-900506 substance were the same as those for the white powder of the same under the identical conditions.
-
From the above physical and chemical properties and the analysis of the X ray diffraction, the FR-900506 substance could be determined to have the following chemical structure.

17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetraone
……………………

The total synthesis of FK-506 is described: This synthesis was performed by previously constructing three building fragments (XX), (XXXII) and (XLVI), which later were coupled sequentially. First the synthesis of these fragments will be presented, and afterwards their sequential coupling will be described. 1) (2RS,4R,6S,7R,8S,10R)-2-(Bis(dimethylamino)phosphono)-7-(tert-butyldimethylsilyloxy)-6,8-dimethoxy-10-(1,3-dithian-2-yl)-4-methylundecane (XX). The reaction of L-arabitol (I) with 2-acetoxyisobutyryl chloride in acetonitrile gives the diacetoxycompound (II), which by treatment with sodium methoxide in THF yields (2S,4S)-1,2:4,5-diepoxy-3-pentanol (III). The protection of (III) with TBS-Cl in THF affords the protected compound (IV), which is condensed with ethoxyacetylene (V) by means of butyllithium and boron trifluoride ethearate in THF giving the diacetylenic alcohol (VI). Cyclization of (VI) by means of HgCl2 and p-toluenesulfonic acid in refluxing ethanol yields the dilactone (VII), which is methylated by means of methyl iodide and lithium diisopropylamide in THF affording the methylated dilactone (VIII). The deprotection of (VIII) with HF in acetonitrile gives the hydroxydilactone (IX), which is benzylated with benzyl trichloroacetimidate and trifluoromethanesulfonic acid in dichloromethane-cyclohexane yielding the benzyl protected dilactone (X). The methanolysis of (X), followed by methylation with NaH and methyl iodide in DMF affords the nonanedioic ester (XI), which is debenzylated by hydrogenolysis with H2 over Pd/C in ethyl acetate giving the hydroxy diester (XII). The lactonization of (XII) with pyridinium p-toluenesulfonate in dichloromethane yields the lactone-methyl ester (XIII), which is selectively reduced with L-Selectride in THF affording the lactol-methyl ester (XIV). The reaction of (XIV) with propane-1,3-dithiol and boron trifluoride ethearate in dichloromethane gives the 1,3-dithiane derivative (XV), which by reduction of its lactone group with LiAlH4 in THF yields (2R,4S,5R,6S,8R)-8-(1,3-dithian-2-yl)-4,6-dimethoxy-2-methylnonane-1,5-diol (XVI). The reaction of (XVI) with I2, pyridine and triphenylphosphine in benzene affords the 1-iodo derivative (XVII), which is protected with TBS trifluoromethanesulfonate and triethylamine in dichloromethane giving the protected iodide (XVIII). Finally, this compound is condensed with ethylphosphonic acid bis(dimethylamide) (XIX) by means of butyllithium in THF to afford the first building fragment (XX).
SEE
http://www.drugfuture.com/synth/syndata.aspx?ID=124071
………………
Isolation and Purification
The cultured broth thus obtained was filtered with an aid of diatomaseous earth (5 kg). The mycelial cake was extracted with acetone (50 liters), yielding 50 liters of the extract. The acetone extract from mycelium and the filtrate (135 liters) were combined and passed through a column of a non-ionic adsorption resin “Diaion HP-20” (Trade Mark, maker Mitsubishi Chemical Industries Ltd.) (10 liters). After washing with water (30 liters) and 50% aqueous acetone (30 liters), elution was carried out with 75% aqueous acetone. The eluate (30 liters) was evaporated under reduced pressure to give residual water (2 liters). This residue was extracted with ethyl acetate (2 liters) three times. The ethyl acetate extract was concentrated under reduced pressure to give an oily residue. The oily residue was mixed with twice weight of acidic silica gel (special silica gel grade 12, maker Fuji Devision Co.), and this mixture was slurried in ethyl acetate. After evaporating the solvent, the resultant dry powder was subjected to column chromatography of the same acidic silica gel (800 ml) which was packed with n-hexane. The column was developed with n-hexane (3 liters), a mixture of n-hexane and ethyl acetate (4:1 v/v, 3 liters) and ethyl acetate (3 liters). The fractions containing the object compound were collected and concentrated under reduced pressure to give an oily residue. The oily residue was dissolved in a mixture of n-hexane and ethyl acetate (1:1 v/v, 30 ml) and subjected to column chromatography of silica gel (maker Merck Co., Ltd. 230-400 mesh) (500 ml) packed with the same solvents system. Elution was carried out with a mixture of n-hexane and ethyl acetate (1:1 v/v, 2 liters and 1:2 v/v, 1.5 liters) and ethyl acetate (1.5 liters).
Fractions containing the first object compound were collected and concentrated under reduced pressure to give crude FR-900506 substance (3 g) in the form of yellowish powder.
…………




……………………………
Synthesis pathway
| Synthesis a) |
|---|
                         |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Advagraf | Astellas |
| Prograf | – “- | |
| Protopic | – “- | |
| France | Prograf | – “- |
| Protopic | – “- | |
| United Kingdom | – “- | – “- |
| Italy | Prograf | Fujisawa |
| Japan | – “- | Astellas |
| USA | – “- | – “- |
| Ukraine | Prograf | Astellas Ireland Co.., Ltd., Ireland; Fujisawa Ireland Ltd., Ireland |
| Protopic | Astellas Ireland Co.., Ltd.. (Issue series and packaging), Ireland; Astellas Toyama Co., Ltd.., Plant Toyama, Japan |
|
| Advagraf | Astellas Ireland Co.., Ltd., Ireland |
Formulations
-
ampoules of 5 mg / 1 ml;
-
Capsules 0.5 mg, 1 mg, 5 mg;
-
granules 0.2%;
-
Ointment 0.1%
Links
-
Manufacturing; selection:
-
EP 184 162 (Fujisawa Pharmaceutical; appl. 11/6/1986; GB -prior. 05.02.1985, 1/4/1985).
-
-
synthesis of FK-506:
-
EP 378 318 (Fujisawa Pharmaceutical; appl. 18.7.1990; USA-prior. 11.1.1989, 30.6.1989).
-
Ireland, R. et al .: J. Org. Chem. (JOCEAH) 61, 6856 (1996).
-
-
Synthesis of Intermediates:
-
Danishefsky, SJ et al .: J. Org. Chem. (JOCEAH) 55 (9) 2786 (1990).
-
Schreiber, SL et al .: J. Am. Chem. Soc. (JACSAT) 112 (4), 5583 (1990).
-
US 4,940,797 (Fujisawa Pharmaceutical; 10.7.1990; USA-prior. 23.3.1989).
-
-
Alternative synthesis :
-
Shinkai, I. et al .: J. Am. Chem. Soc. (JACSAT) 111 (3) 1157 (1989).
-
Shinkai, I. et al .: Tetrahedron Lett. (TELEAY) 29 (3), 281 (1988).
-
References
- Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, Kohsaka M, Aoki H, Imanaka H (1987). “FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics.”. J Antibiot (Tokyo) 40 (9): 1249–55. PMID 2445721.
- Pritchard D (2005). “Sourcing a chemical succession for cyclosporin from parasites and human pathogens.”. Drug Discov Today 10 (10): 688–91. doi:10.1016/S1359-6446(05)03395-7.PMID 15896681. Supports source organism, but not team information
- Ponner, B, Cvach, B (Fujisawa Pharmaceutical Co.): Protopic Update 2005
- Healthy Ontario: Tacrolimus topical ointment
- Alloway RR, Germain M, Osama Gaber, A, Bodziak KA, Mulgaonkar SP, Gohh RY, Kaplan B, Katz E, Beckert M, Gordon RD, A Phase II Open-Label, Multi-Center Prospective, Conversion Study in Stable Kidney Transplant Patients to Compare the Pharmacokinetics of LCP-Tacro Tablets Once-A-Day to Prograf Capsules Twice-A-Day. American Transplant Congress, 2008
- http://files.shareholder.com/downloads/ABEA-4J4LWA/1008134289x0x477697/e60eb3d4-849c-41e2-95f3-d8a1eaea3b56/LCP_News_2011_6_21_English_Releases.pdf
- Clinicaltrials.gov identifier: NCT01187953
- William F. Ganong. Review of medical physiology (22nd ed.). Lange medical books. p. 530. ISBN 0-07-144040-2.
- Liu J, Farmer J, Lane W, Friedman J, Weissman I, Schreiber S (1991). “Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes.”. Cell 66 (4): 807–15.doi:10.1016/0092-8674(91)90124-H. PMID 1715244.
- McCauley, Jerry (2004-05-19). “Long-Term Graft Survival In Kidney Transplant Recipients”. Slide Set Series on Analyses of Immunosuppressive Therapies. Medscape. Retrieved 2006-06-06.
- M.M. Abou-Jaoude, R. Naim, J. Shaheen, N. Naufal, S. Abboud, M. AlHabash, M. Darwish, A. Mulhem, A. Ojjeh, and W.Y. Almawi (2005). “Tacrolimus (FK506) versus cyclosporin microemulsion (Neoral) as maintenance immunosuppresion therapy in kidney transplant recipients.”. Transplantation Proceedings 37 (7): 3025–3028. doi:10.1016/j.transproceed.2005.08.040. PMID 16213293.
- Elizabeth Haddad, Vivian McAlister, Elizabeth Renouf, Richard Malthaner, Mette S. Kjaer, and Lise Lotte Gluud (2006). “Cyclosporin versus Tacrolimus for Liver Transplanted Patients”. In McAlister, Vivian. Cochrane Database of Systematic Reviews 4 (CD005161): CD005161. doi:10.1002/14651858.CD005161.pub2. PMID 17054241.
- J.G. O’Grady, A. Burroughs, P. Hardy, D. Elbourne, A. Truesdale, and The UK and Ireland Liver Transplant Study Group (2002). “Tacrolimus versus emulsified cyclosporin in liver transplantation: the TMC randomised controlled trial”. Lancet 360 (9340): 1119–1125. doi:10.1016/S0140-6736(02)11196-2. PMID 12387959.
- Baumgart DC, Pintoffl JP, Sturm A, Wiedenmann B, Dignass AU (2006). “Tacrolimus is safe and effective in patients with severe steroid-refractory or steroid-dependent inflammatory bowel disease–a long-term follow-up”. Am J Gastroenterol 101 (5): 1048–1056. doi:10.1111/j.1572-0241.2006.00524.x. PMID 16573777.
- Baumgart DC, MacDonald JK, Feagan BG (2008). “Tacrolimus (FK506) for induction of remission in refractory ulcerative colitis”. In Baumgart, Daniel C. Cochrane Database Syst Rev 16 (3): CD007216. doi:10.1002/14651858.CD007216. PMID 18646177.
- Silverberg, NB; Lin, P; Travis, L; Farley-Li, J; Mancini, AJ; Wagner, AM; Chamlin, SL; Paller, AS (2004). “Tacrolimus ointment promotes repigmentation of vitiligo in children: a review of 57 cases.”.Journal of the American Academy of Dermatology 51 (5): 760–6. doi:10.1016/j.jaad.2004.05.036. PMID 15523355.
- Naesens M, Kuypers DR, Sarwal M (2009). “Calcineurin inhibitor nephrotoxicity”. Clin. J. Am. Soc. Nephrol. 4 (2): 481–509. doi:10.2215/CJN.04800908. PMID 19218475.
- Miwa Y, Isozaki T, Wakabayashi K, et al. (2008). “Tacrolimus-induced lung injury in a rheumatoid arthritis patient with interstitial pneumonitis”. Mod Rheumatol 18 (2): 208–11. doi:10.1007/s10165-008-0034-3. PMID 18306979.
- O’Donnell MM, Williams JP, Weinrieb R, Denysenko L (2007). “Catatonic mutism after liver transplant rapidly reversed with lorazepam”. Gen Hosp Psychiatry 29 (3): 280–1.doi:10.1016/j.genhosppsych.2007.01.004. PMID 17484951.
- Hanifin JM, Paller AS, Eichenfield L, Clark RA, Korman N, Weinstein G, Caro I, Jaracz E, Rico MJ; US Tacrolimus Ointment Study Group (2005). “Efficacy and safety of tacrolimus ointment treatment for up to 4 years in patients with atopic dermatitis”. J Am Acad Derm 53 (2 suppl 2): S186–94. doi:10.1016/j.jaad.2005.04.062. PMID 16021174.
- N H Cox and Catherine H Smith (December 2002). “Advice to dermatologists re topical tacrolimus” (PDF). Therapy Guidelines Committee. British Association of Dermatologists.
- Fukatsu S, Fukudo M, Masuda S, Yano I, Katsura T, Ogura Y, Oike F, Takada Y, Inui K (2006). “Delayed effect of grapefruit juice on pharmacokinetics and pharmacodynamics of tacrolimus in a living-donor liver transplant recipient”. Drug Metab Pharmacokinet 21 (2): 122–5. doi:10.2133/dmpk.21.122. PMID 16702731.
- Fegan, A; White, B; Carlson, JC; Wagner, CR (Jun 9, 2010). “Chemically controlled protein assembly: techniques and applications.”. Chemical reviews 110 (6): 3315–36. doi:10.1021/cr8002888.PMID 20353181.
- Tacrolimus, which is also called FK-506, has first discovered by Tanaka, Kuroda and their colleague in Japan see, J. Am. Chem. Soc., 1987, 109, 5031
- and U.S. Pat. No. 4,894,366 issued on Jan. 16, 1990!.
- Total synthesis of FK506 and an FKBP probe reagent, (C8,C9-13C2)-FK506
J Am Chem Soc 1990, 112(14): 5583 - A diastereospecific, non-racemic synthesis of the C.10-C.18 segment of FK-506
Tetrahedron Lett 1988, 29(3): 277
- Tacrolimus levels in Liver Transplants-Indian Study by Dr.Pradeep Naik,Dr.Dharmesh Kapoor, Dr.DCS Reddy
- Prograf prescribing information at Fujisawa
- Pimecrolimus (Elidel Cream) FDA adivisory page (for eczema treatment)
- Tacrolimus (FK506) product page from Fermentek
- U.S. National Library of Medicine: Drug Information Portal – Tacrolimus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
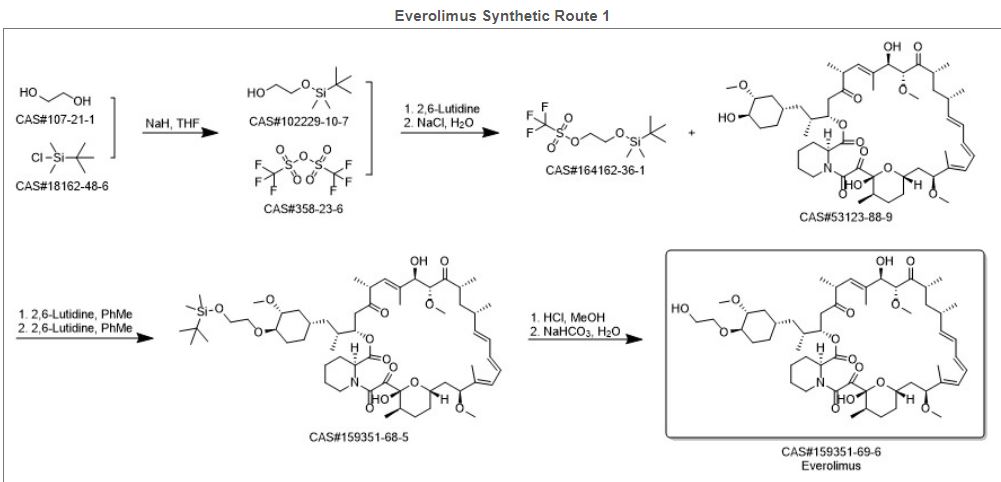{kind=link}