
Home » Posts tagged 'GlaxoSmithKline' (Page 2)
Tag Archives: GlaxoSmithKline
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
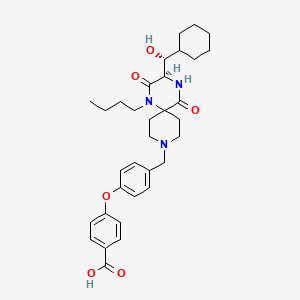
Aplaviroc, AK602, GSK-873140

Aplaviroc
4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo- 1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid
for the treatment of HIV infection
461023-63-2 of hydrochloride
461443-59-4 (free base)
873140
AK-602
GW-873140
ONO-4128
ono…….innovator
| Ono Pharmaceutical Co., Ltd. |
| Identifiers | |
|---|---|
| CAS number | 461023-63-2 |
| ATC code | None |
| PubChem | CID 3001322 |
| ChemSpider | 2272720 |
| UNII | 98B425P30V |
| KEGG | D06557 |
| ChEMBL | CHEMBL1255794 |
| Chemical data | |
| Formula | C33H43N3O6 |
| Mol. mass | 577.711 g/mol |
Aplaviroc (INN, codenamed AK602 and GSK-873140) is a CCR5 entry inhibitor developed for the treatment of HIV infection.[1][2] It is developed by GlaxoSmithKline.
In October 2005, all studies of aplaviroc were discontinued due to liver toxicity concerns.[3][4] Some authors have claimed that evidence of poor efficacy may have contributed to termination of the drug’s development;[5] the ASCENT study, one of the discontinued trials, showed aplaviroc to be under-effective in many patients even at high concentrations.[6]
Aplaviroc hydrochloride, an orally-effective, long-acting chemokine CCR5 receptor antagonist, had been under development by Ono and GlaxoSmithKline for the treatment of HIV infection. In early 2006, the companies discontinued development of the antagonist based on reports of elevated liver function test values from clinical studies.
Originally developed at Ono, aplaviroc was licensed to GlaxoSmithKline in 2003 for development, manufacturing and marketing. GlaxoSmithKline also obtained rights to evaluate the agent in non-HIV conditions worldwide with the exception of Japan, South Korea and Taiwan.
A low-molecular-weight compound, aplaviroc prevents HIV viral infection by blocking the binding of the virus to the CCR5 receptor
……………….
WO 2002074770
0r
http://www.google.com/patents/EP1378510A1?cl=en
Reference example 3(3)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0136]

TLC:Rf 0.32 (butanol:acetic acid:water = 4:2:1);
NMR (CD3OD): δ 4.16 (d, J = 2.0 Hz, 1H), 3.95 (m, 1H), 3.70 (m, 1H), 3.52 (m, 1H), 3.37 (m, 1H), 3.28 (m, 1H), 3.22-3.13 (m, 2H), 2.46-1.93 (m, 6H), 1.80-1.64 (m, 5H), 1.48-1.15 (m, 6H), 1.02-0.87 (m, 5H);
Optical rotation:[α]D +1.22 (c 1.04, methanol, 26°C).

Example 9(54)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexylmethyl)-9-(4-(4-carboxyphenyloxy)phenylmethyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0359]

TLC:Rf 0.43(chloroform:methanol = 5:1);
NMR (CD3OD):δ 8.05 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 4.38 (s, 2H), 4.17 (d, J = 2.1 Hz, 1H), 4.02 (m, 1H), 3.78 (m, 1H), 3.60-3.40 (m, 3H), 3.30-3.10 (m, 2H), 2.56-1.86 (m, 6H), 1.82-1.60 (m, 5H), 1.52-1.16 (m, 6H), 1.06-0.82 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H).

………………….
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
Owing to the special properties of piperazines (increased solubility and H-bond acceptor capability etc.) it is often considered to be a privileged structure and therefore occurs widely, for instance in GlaxoSmithKlines investigational anti-HIV drug aplaviroc (4.37) which, despite being a promising CCR5 receptor antagonist, was discontinued due to hepatotoxicity concerns. In this compound the spirodiketopiperazine unit (4.35) was designed to mimic a type-1 β-turn (4.36) as present in G-protein coupled receptors (Figure 14) [117].
![[1860-5397-9-265-14]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-14.png?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of aplaviroc and its analogues can be accomplished via the use of an Ugi multicomponent reaction (Ugi-MCR) [118]. The procedure involved the condensation of piperidone 4.38 and butylamine (4.39) followed by reaction of the resulting imine with isocyanide 4.41 and interception of the nitrilium intermediate with the amino acid4.40 (Scheme 47) [119]. This sequence was completed by structural rearrangement and acid-mediated ring closure to produce the spirocyclic diketopiperazine 4.43. Following debenzylation this material was subjected to a reductive amination finally affording aplaviroc analogues (Scheme 47).
![[1860-5397-9-265-i47]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i47.png?scale=2.0&max-width=1024&background=FFFFFF)
- 117 Habashita, H.; Kokubo, M.; Hamano, S.; Hamanaka, N.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H. J. Med. Chem. 2006, 49, 4140–4152. doi:10.1021/jm060051s
- Dömling, A.; Huang, Y. Synthesis 2008, 2859–2883. doi:10.1055/s-0030-1257906
ref 118 - Nishizawa, R.; Nishiyama, T.; Hisaichi, K.; Matsunaga, N.; Minamoto, C.; Habashita, H.; Takaoka, Y.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H.Bioorg. Med. Chem. Lett. 2007, 17, 727–731. doi:10.1016/j.bmcl.2006.10.084
ref 119
| Patent | Submitted | Granted |
|---|---|---|
| Triazaspiro[5.5]undecane derivative and pharmaceutical composition comprising the same as active ingredient [US7262193] | 2005-09-29 | 2007-08-28 |
| Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient [US7285552] | 2004-06-03 | 2007-10-23 |
| Triazaspiro[5.5]undecane derivatives and drugs containing the same as the active ingredient [US7053090] | 2004-04-29 | 2006-05-30 |
| WO1998031364A1 * | Jan 20, 1998 | Jul 23, 1998 | Timothy Harrison | 3,3-disubstituted piperidines as modulators of chemokine receptor activity |
| WO2000014086A1 * | Jan 21, 1999 | Mar 16, 2000 | Kyowa Hakko Kogyo Kk | Chemokine receptor antagonists and methods of use therefor |
| WO2002074769A1 * | Mar 18, 2002 | Sep 26, 2002 | Kenji Maeda | Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient |
References
- Maeda, Kenji; Ogata, Hiromi; Harada, Shigeyoshi et al. (2004). “Determination of binding sites of a unique CCR5 inhibitor AK602 / ONO-4128/ GW873140 on human CCR5” (PDF). Conference on Retroviruses and Opportunistic Infections. Archived from the original on November 3, 2005.
- Nakata, Hirotomo; Maeda, Kenji; Miyakawa, Toshikazu et al. (February 2005). “Potent Anti-R5 Human Immunodeficiency Virus Type 1 Effects of a CCR5 Antagonist, AK602/ONO4128/GW873140, in a Novel Human Peripheral Blood Mononuclear Cell Nonobese Diabetic-SCID, Interleukin-2 Receptor γ-Chain-Knocked-Out AIDS Mouse Model”. Journal of Virology 79 (4): 2087–96.doi:10.1128/jvi.79.4.2087-2096.2005.
- “Aplaviroc (GSK-873,140)”. AIDSmeds.com. October 25, 2005. Retrieved September 5, 2008.[dead link]
- Nichols WG, Steel HM, Bonny T et al. (March 2008). “Hepatotoxicity Observed in Clinical Trials of Aplaviroc (GW873140)”.Antimicrobial Agents and Chemotherapy 52 (3): 858–65. doi:10.1128/aac.00821-07. PMC 2258506. PMID 18070967.
- Moyle, Graeme (December 19, 2006). “The Last Word on Aplaviroc: A CCR5 Antagonist With Poor Efficacy”. The Body.Archived from the original on 6 October 2008. Retrieved September 5, 2008.
- Currier, Judith; Lazzarin, Adriano; Sloan, Louis et al. (2008). “Antiviral activity and safety of aplaviroc with lamivudine/zidovudine in HIV-infected, therapy-naive patients: the ASCENT (CCR102881) study”. Antiviral Therapy (Lond.) 13 (2): 297–306.PMID 18505181.
Further reading
- Horster, S; Goebel, FD (April 2006). “Serious doubts on safety and efficacy of CCR5 antagonists: CCR5 antagonists teeter on a knife-edge”. Infection 34 (2): 110–13. doi:10.1007/s15010-006-6206-1. PMID 16703305.
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor…….

AMPRENAVIR
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.

| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | C (US) |
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Mol. mass | 505.628 g/mol |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available
………………….
New approaches to the industrial synthesis of HIV protease inhibitors
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f/unauth#!divAbstract
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are
available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB
The US Food and Drug Administration (FDA) has approved the GlaxoSmithKline vaccine Flulaval Quadrivalent, used to treat seasonal influenza.

FDA backs second GSK flu vaccine
The US Food and Drug Administration (FDA) has approved the GlaxoSmithKline vaccine Flulaval Quadrivalent, used to treat seasonal influenza.
READ ALL AT
http://www.pharmaceutical-technology.com/news/newsfda-backs-second-gsk-flu-vaccine?WT.mc_id=DN_News


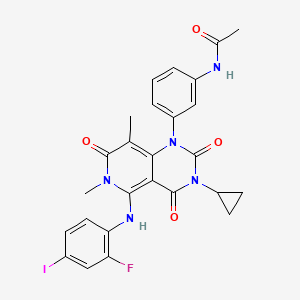
FDA Approves Mekinist (trametinib) for Advanced Melanoma

Mekinist (trametinib)
JTP-74057, GSK212, GSK1120212
N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4, 3-d] pyrimidin-1-yl] phenyl}acetamide
| Molecular Weight | 615.39 |
| Formula | C26H23FIN5O4 |
| CAS Number | 871700-17-3 |

Trametinib (GSK1120212) is experimental cancer drug. It is a MEK inhibitor drug with anti-cancer activity.[1]
It inhibits MEK1 and MEK2.[1]
Trametinib had good results for V600E mutated metastatic melanoma in a phase III clinical trial.[2]
- Trametinib, NCI Drug Dictionary
- METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM).
GSK1120212 (JTP-74057) is a potent and selective allosteric inhibitor of the MEK1 and MEK2 (MEK1/2) enzymes with promising antitumor activity in a phase I clinical trial (ASCO 2010). GSK1120212 (JTP-74057) inhibits MEK1/2 kinase activity and prevents Raf-dependent MEK phosphorylation (S217 for MEK1), producing prolonged p-ERK1/2 inhibition. Potent cell growth inhibition was evident in most tumor lines with mutant BRAF or Ras. In xenografted tumor models, GSK1120212 orally dosed once daily had a long circulating half-life and sustained suppression of p-ERK1/2 for more than 24 hours; GSK1120212 also reduced tumor Ki67, increased p27 (Kip1/CDKN1B), and caused tumor growth inhibition in multiple tumor models.
May 29, 2013 —
GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Mekinist (trametinib) as a single-agent oral treatment for unresectable or metastatic melanoma in adult patients with BRAF V600E or V600K mutations. Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Mekinist is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma. It is also approved for patients with the V600K mutation, which makes up approximately 10 percent of all BRAF V600 mutations in metastatic melanoma.
Melanoma is the most serious and deadly form of skin cancer.[iii] According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States.[iv] When melanoma spreads in the body, the disease is called metastatic melanoma.[v] Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.2 One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis,while in the U.S., the five-year survival rate was 16 percent (2003-2009). The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Mekinist (trametinib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E and V600K mutations as detected by an FDA-approved test. Limitation of use: Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy.
(WO2005121142A1). Aniline a reaction with CDI was added cyclopropylamine get two , two and malonic acid cyclization get 3 . 3 chlorination with phosphorus oxychloride reaction with methylamine 4 , as well as byproducts 5 (ratio of 2:1). Mixture 4 + 5 and acid 6 crystals obtained after cyclization compound 7 (pure substance). 7 with activated trifluoromethanesulfonyl chloride to the amide 8 SNAr reaction occurs 9 , 9 in alkaline conditions rearrangement trimetazidine imatinib.

…………………
http://www.google.com/patents/WO2014039375A1?cl=en
The term “trametinib” as used herein means the MEK inhibitor represented by the structure of formula (I):

or a pharmaceutically acceptable salt or solvate thereof. Trametinib is preferably administered as a solvate in the form of N-{3-[3- cyclopropyl-5-(2-fluoro-4-iodo-phenylamino)6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydro- 2H-pyrido[4,3-d]pyrimidin-1 -yl]phenyl}acetamide dimethyl sulfoxide (solvate).
Depending on naming convention, the compound of formula (I) may also properly be referred to as N-{3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7- trioxo-3,4,6,7-tetrahydropyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl}acetamide.
Trametinib is disclosed and claimed, along with pharmaceutically acceptable salts thereof, and also as solvates thereof, as being useful as an inhibitor of MEK activity, particularly in treatment of cancer, in WO 2005/121 142. Trametinib can be prepared as described in WO 2005/121 142.
Suitably, trametinib is in the form of a dimethyl sulfoxide solvate. Suitably, trametinib is in the form of a sodium salt. Suitably, trametinib is in the form of a solvate selected from: hydrate, acetic acid, ethanol, nitromethane, chlorobenzene, 1 -pentancol, isopropyl alcohol, ethylene glycol and 3-methyl-1 -butanol. These solvates and salt forms can be prepared by one of skill in the art from the description in WO2005/121 142.
…………………….
http://www.google.com/patents/WO2005121142A1?cl=en
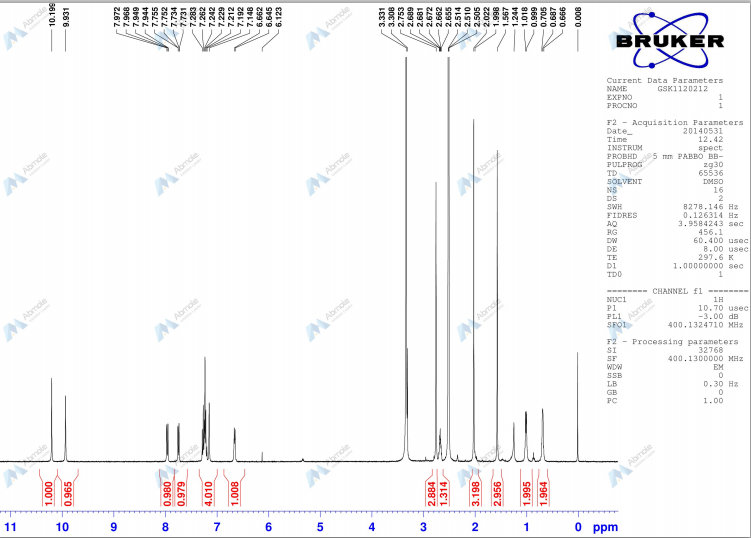
Example 3-10 By treating N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4 , 3-d]pyrimidin-1-yl] phenyl Jmethanesulfonamide 46 according to conventional methods, sodium salt and potassium salt thereof were obtained.
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide sodium salt:
^-NMR (DMSO-de, 300 MHz) δ 0.47 (brs, 2H) , 0.70-0.90 (m, 2H) , 1.23(s, 3H) , 2.35(brs, IH) , 2.82(s, 3H) , 3.22(s, 3H) , 6.69(t, J=8.8Hz, IH) , 6.81 (d, J=8.1Hz , IH) , 6.98 (s, IH) , 7.02 (d, J=8.8Hz , IH) , 7.10-7.30 (m, 2H) , 7.38(d, J=9.2Hz , IH) , 10.22(brs, IH) . MS (ESI) m/z 652 [MH]+.
N-{3-[3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide potassium salt: Example 4-1
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 ,8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1-yl] – phenyl}-acetamide Step 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) rea

47 48 Under a nitrogen atmosphere, to N,N-carbonyldiimidazole (39.9 g) were added N,N-dimethylformamide (200 ml) and triethylamine (34.3 ml) and a solution of 2-fluoro-4-iodoaniline 47 (48.5 g) in N,N-dimethylformamide (50 ml) was added dropwise with stirring under ice-cooling. After the completion of the dropwise addition, the mixture was stirred at room temperature for 18 hrs. The reaction mixture was ice-cooled, and cyclopropylamine (21.3 ml) was added dropwise. The reaction mixture was stirred at room temperature for 1 hr and added dropwise to water-toluene [2:1 (volume ratio), 750 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4-iodophenyl) urea 48 (61.3 g, yield 93.4%) as colorless crystals. Step 2 Synthesis of l-cyclopropyl-3- (2-fluoro-4- iodophenyl) pyrimidine-2 , 4 , 6-trione

To l-cyclopropyl-3- (2-fluoro-4-iodophenyl) urea 48 (61.0 g) obtained in Step 1 and malonic acid 4 (19.9 g) were added acetic anhydride (300 ml) and acetyl chloride (27.2 ml), and the mixture was stirred under a nitrogen atmosphere at 60°C for 3 hrs. After allowing to cool to room temperature, the reaction mixture was added dropwise to water-toluene [2:1 (volume ratio), 900 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4- iodophenyl)pyrimidine-2,4, 6-trione 49 (60.9 g, yield 82%) as pale-yellow crystals.
Step 3 Synthesis of 6-chloro-3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -lH-pyrimidine-2 , -dione

49 50 51 To l-cyclopropyl-3- (2-fluoro-4-iodophenyl) -pyrimidine-
2, 4, 6-trione 49 (59.0 g) obtained in Step 2 were added phosphorus oxychloride (85.0 ml) and dimethylaniline (29.0 ml), and water (8.3 ml) was added dropwise to the mixture at room temperature with stirring. After the completion of the dropwise addition, the mixture was stirred with heating at 110°C for 1 hr. After allowing to cool to room temperature, the reaction mixture was added dropwise to ice water-toluene [2:1 (volume ratio), 900 ml] with stirring. The mixture was stirred at room temperature for 1 hr. The organic layer was separated, and washed successively with water (300 ml) and brine (300 ml) . Anhydrous magnesium sulfate and activated carbon were added and the mixture was stirred. Anhydrous magnesium sulfate and activated carbon were filtered off, and the filtrate was concentrated under reduced pressure to give a 1:2 mixture (62.9 g) of 6-chloro-3-cyclopropyl-l- (2- fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 50 and 6-chloro-l- cyclopropyl-3- (2-fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 51 as a yellow foamy oil, which was used for the next step without purification.
Step 4 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6- methylamino-lH-pyrimidine-2 , 4-dione

To a 1:2 mixture (62.9 g) of 6-chloro-3-cyclopropyl-l- (2- fluoro-4-iodophenyl) -lH-pyrimidine-2 ,4-dione 50 and 6-chloro-l- cyclopropyl-3- (2-fluoro-4-iodophenyl) -lH-pyrimidine-2 , -dione 51 obtained in Step 3 were added methanol (189 ml) and a solution (126 ml) of 40% methylamine in methanol, and the mixture was stirred at room temperature for 2 hrs . The precipitated crystals were filtered off and the filtrate was concentrated under reduced pressure. The residue was extracted with chloroform (200 ml) and water (200 ml) , and the organic layer was washed with brine (200 ml) and dried over anhydrous magnesium sulfate. Anhydrous magnesium sulfate was filtered off and the filtrate was concentrated under reduced pressure to give a 2:1 mixture (34.55 g) of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6-methylamino-lH- pyrimidine-2 ,4-dione 52 and l-cyclopropyl-3- (2-fluoro-4- iodophenyl) -6-methylamino-lH-pyrimidine-2,4 ,-dione 53 as yellow crystals, which were used for the next step without purification. Step 5 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -5- hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 , 3-d] pyrimidine-2 , 4 , 7-trione

To a 2:1 mixture (34.6 g) of 3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6-methylamino-lH-pyrimidine-2, 4-dione 52 and 1- cyclopropyl-3- (2-fluoro-4-iodo-phenyl) 6-methylamino-lH- pyrimidine-2,4,-dione 53 obtained in Step 4, and 2-methylmalonic acid 54 (10.2 g) was added acetic anhydride (173 ml) , and the mixture was stirred at 100°C for 2 hrs. After allowing to cool to room temperature, the reaction mixture was concentrated under reduced pressure. Acetone (104 ml) was added to the residue, and the mixture was stirred with heating under reflux for 30 min. After allowing to cool to room temperature, the precipitated crystals were collected by filtration and dried to give 3- cyclopropyl-1- (2-fluoro-4-iodophenyl) -5-hydroxy-6 , 8-dimethyl- lH,8H-pyrido [2, 3-d] pyrimidine-2, 4, 7-trione 55 (15.1 g, yield from 48, 21%) as colorless crystals.
Step 6 Synthesis of trifluoromethanesulfonic acid 3-cyclopropyl- 1- (2-fluoro-4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

55 56 43 Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro-
4-iodophenyl) -5-hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2,3- d] pyrimidine-2 ,4 ,7-trione 55 (33.0 g) obtained in Step 5 were added chloroform (165 ml) and 2 , 6-lutidine (10.4 ml), and trifluoromethanesulfonic anhydride 56 (14.4 ml) was added dropwise under ice-cooling with stirring. After the completion of the dropwise addition, the mixture was stirred at same temperature for 30 min and at room temperature for 2 hrs. The reaction mixture was washed successively with aqueous sodium hydrogen carbonate (165 ml) , IN hydrochloric acid (165 ml) and brine (165 ml) and dried over anhydrous magnesium sulfate. Anhydrous magnesium sulfate was filtered off and the filtrate was concentrated under reduced pressure. 2-Propanol (198 ml) was added to the residue, and the mixture was stirred with heating under reflux, and allowed to return to room temperature. The crystals were collected by filtration and dried to give trifluoromethanesulfonic acid 3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (31.9 g, yield 93%) as colorless crystals.
Step 7 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 ,4 , 7-trioxo-l ,2,3,4,7 , 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-ylamino] phenyl } acetamide

To trifluoromethanesulfonic acid 3-cyclopropyl-l- (2-fluoro-
4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (25.0 g) obtained in Step 6 and 3 ‘-aminoacetanilide 57 (7.33 g) were added N,N- dimethylacetamide (50.0 ml) and 2,6-lutidine (5.68 ml), and the mixture was stirred at 130°C for 5 hrs. After allowing to cool to room temperature, methanol-water [1:2 (volume ratio), 150 ml] was added with stirring. The crystals were collected by filtration and dried to give N- {3- [3-cyclopropyl-l- (2-fluoro-4-iodophenyl) – 6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro-pyrido [2,3- d]pyrimidin-5-ylamino] phenyl}acetamide 58 (24.8 g, yield 99%) as colorless crystals. Step 8 Synthesis of N- { 3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4″, 7-trioxo-3 , 4 , 6 , 7-tetrahydro-2H- pyrido [4 , 3 -d]pyrimidin-l-yl] phenyl} acetamide

Under a nitrogen atmosphere, to a solution (1.57 g) of 28% sodium methoxide in methanol was added tetrahydrofuran (40 ml) , N- {3- [3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro-pyrido [2 , 3-d] pyrimidin-5- ylamino]phenyl}acetamide 58 (5.00 g) obtained in Step 7 was added, and the mixture was stirred at room temperature for 4 hrs. Acetic acid (0.56 ml) was added, and the mixture was stirred at room temperature for 30 min. Water (40 ml) was added and the mixture was further stirred for 1 hr. The crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4, 3-d] pyrimidin-1-yl] phenyl}acetamide 59 (4.75 g, yield 95%) as colorless crystals. MS ESI m/e: 616 (M+H) , 614 (M-H) .
1H-NMR(DMSO-d6, 400MHz) δ 0.63-0.70 (m, 2H) , 0.91-1.00 (m, 2H) , 1.25(s, 3H) , 2.04(s, 3H) , 2.58-2.66(m, IH) , 3.07(s, 3H) , 6.92(t,
J=8.8Hz, IH) , 7.00-7.05 (m, IH) , 7.36 (t, J=8.2Hz , IH) , 7.52-7.63 (m,
3H) , 7.79(dd, J=2.0 , 10.4Hz, IH) , 10.10(s, IH) , 11.08(s, IH) .
Example 4-1 (alternative method)
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d] pyrimidin-1-yl] – phenyl } -acetamide
Step 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -urea

48 47 Under a nitrogen atmosphere, to N,N-carbonyldiimidazole (82.1 g) were added N, N-dimethylformamide (400 ml) and triethylamine (70.5 ml) , and a solution of 2-fluoro-4-iodoaniline 47 (100 g) in N, N-dimethylformamide (100 ml) was added dropwise under ice-cooling. After the completion of the dropwise addition, the mixture was stirred at room temperature for 5 hrs . The reaction mixture was ice-cooled, and cyclopropylamine (44.0 ml) was added dropwise. The mixture was stirred at room temperature for 1 hr, and the reaction mixture was added dropwise to water- toluene [2:1 (volume ratio) , 1500 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -urea 48 (129 g, yield 95.5%) as colorless crystals. Step 2 Synthesis of 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro- 4-iodo-phenyl) -urea

48 73 74 Under a nitrogen atmosphere, to l-cyclopropyl-3- (2-fluoro- 4-iodo-phenyl) -urea 48 (167 g) and cyanoacetic acid 73 (80.0 g) , was added N, N-dimethylformamide (836 ml) , and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at room temperature. The mixture was stirred at room temperature for 4 hrs. The reaction mixture was cooled with water, and water- isopropanol [2:1 (volume ratio) , 1670 ml] was added dropwise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) – urea 74 (192 g) .
Step 3 Synthesis of 6-amino-3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -lH-pyrimidine-2 , 4-dione

74 75 To 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro-4-iodo- phenyl)-urea 74 (192 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml) , and the mixture was stirred with heating at 80°C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -1H- pyrimidine-2, 4-dione 75 (178g, yield from 48, 88%) as pale-yellow crystals . Step 4 Synthesis of N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) – 2 , 6-dioxo-l ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N ,N-dimethyl- formamidine

75 76 Under a nitrogen atmosphere, to 6-amino-3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -lH-pyrimidine-2 , 4-dione 75 (178 g) were added N,N-dimethylformamide (356 ml) and N,N-dimethylformamide dimethylacetal (178 ml) , and the mixture was stirred at room temperature for 2 hrs. Isopropanol (178 ml) was added with stirring at room temperature, and water (1068 ml) was added dropwise. The mixture was stirred at room temperature for 2 hrs, and the precipitated crystals were collected by filtration and dried to give N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -2,6- dioxo-1 ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N,N-dimethyl-formamidine 76 (188 g, yield 92%) as yellow crystals.
Step 5 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6- methylamino- lH-pyrimidine-2 ,4-dione

76 52 Under a nitrogen atmosphere, to t-butanol-ethanol [2:1 (volume ratio) , 250 ml] was added sodium borohydride (6.41 g) , and the mixture was stirred at room temperature for 1 hr. Under water-cooling, N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -2 ,6- dioxo-1 ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N,N-dimethy1-formamidine 76 (50.0 g) was added, and the mixture was stirred for 2.5 hrs. Under water-cooling, water (225 ml) and 10% aqueous citric acid solution (175 ml) were successively added dropwise, and the mixture was stirred for 3 hrs . The precipitated crystals were collected by filtration and dried to give crude crystals (34.5 g, LC purity 91%) of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6- methylamino- lH-pyrimidine-2 ,4-dione 52, which were used for the next reaction without purification. Step 6 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -5- hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 , 3-d] pyrimidine-2 ,4 ,7-trione

Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro- 4-iodo-phenyl) -6-methylamino-lH-pyrimidine-2, -dione 52 (34.4 g) and 2-methyl-malonic acid 54 (15.2 g) was added acetic anhydride (34.4 ml) , and the mixture was stirred with heating at 100°C for 3 hrs. After allowing to cool to 50°C, acetone (68.8 ml) was added dropwise, and the mixture was stirred as it was for 30 min. Water (172 ml) was further added dropwise, and the mixture was stirred for 1 hr. After allowing to cool to room temperature with stirring, the precipitated crystals were collected by filtration and dried to give crude crystals (37.7 g, LC purity 91%) of 3- cyclopropyl-1- (2-fluoro-4-iodo-phenyl) -5-hydroxy-6 , 8-dimethyl- lH,8H-pyrido [2 ,3-d] pyrimidine-2 ,4 ,7-trione 55. Isopropanol (92.0 ml) was added to the obtained crude crystals (30.7 g) , and the mixture was stirred at room temperature for 4 hrs . The crystals were collected by filtration and dried to give 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -5-hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 ,3- d] pyrimidine-2 ,4 ,7-trione 55 (25.9 g, yield from 76, 58%) as pale-yellow crystals.
Step 7 Synthesis of p-toluenesulfonic acid 3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 , , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

55 11 77 Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro- 4-iodo-phenyl) -5-hydroxy-6 ,8-dimethyl-lH,8H-pyrido [2,3- d] pyrimidine-2 ,4,7-trione 55 (23.9 g) was added acetonitrile (167 ml) , and the mixture was stirred under ice-cooling. Triethylamine (11.0 ml) and trimethylamine hydrochloride (2.37 g) were added, and a solution of p-toluenesulfonyl chloride 11 (12.3 g) in acetonitrile (72.0 ml) was added dropwise. The mixture was stirred under ice-cooling for 1 hr, and stirred at room temperature for 3 hrs. Methanol (239 ml) was added, and the mixture was stirred at room temperature for 1 hr. The crystals were collected by filtration and dried to give p-toluenesulfonic acid 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro-pyrido [2 ,3-d]pyrimidin-5-yl ester 77 (28.7 g, yield 91%) as colorless crystals.
Step 8 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 ,3-d]pyrimidin-5-ylamino] -phenyl}-acetamide

To p-toluenesulfonic acid 3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro- pyrido [2,3-d]pyrimidin-5-yl ester 77 (28.0 g) and 3′- aminoacetanilide 57 (13.2 g) were added N,N-dimethylacetamide (84.0 ml) and 2,6-lutidine (15.3 ml), and the mixture was stirred at 130°C for 4 hrs. After allowing to cool with stirring, methanol (196 ml) was added dropwise, and the mixture was stirred at room temperature. The crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) – 6 , 8-dimethyl-2 , , 7-trioxo-l ,2,3,4,7, 8-hexahydro-pyrido [2,3- d]pyrimidin-5-ylamino] -phenyl}-acetamide 58 (25.2 g, yield 93%) as colorless crystals. Step 9 Synthesis of N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodo- phenylamino) -6 , 8-dimethyl-2 ,4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4 ,3-d] pyrimidin-1-yl] -phenyl}-acetamide

58 59 Under a nitrogen atmosphere, to N-{3- [3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 ,4 , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 ,3-d]pyrimidin-5-ylamino] -phenyl }-acetamide 58 (45.7 g) was added tetrahydrofuran (366 ml), and a solution (15.7 g) of 28% sodium methoxide in methanol was added dropwise with stirring at room temperature and the mixture was stirred at room temperature for 4 hrs. Acetic acid (5.61 ml) was added, and the mixture was stirred at room temperature for 30 min. With stirring at 70°C in an oil bath, water (366 ml) was added dropwise, and the mixture was stirred for 1 hr. After allowing to cool with stirring, the crystals were collected by filtration and dried to give crystal 1 (46.0 g) of N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodo-phenylamino) -6 , 8-dimethyl-2 ,4 , 7-trioxo-3 ,4,6, 7-tetrahydro- 2H-pyrido [4 , 3-d]pyrimidin-1-yl] -phenyl}-acetamide 59. N,N-Dimethylacetamide (184 ml) was added to crystal 1 (46.0 g) , and the mixture was stirred with heating at 130°C. After complete dissolution, the solution was filtered by suction using with paper (5B) , and washed with N,N-dimethylacetamide (92.0 ml).
The filtrate was stirred under heating at 130°C, 1-butanol (138 ml) and water (96.0 ml) were successively added dropwise, and the mixture was stirred for 30 min. Water (46.0 ml) was further added dropwise, and the mixture was stirred for 30 min allowed to cool with stirring. The crystals were collected by filtration and dried to give crystal 2 (41.7 g) of N-{3- [3-cyclopropyl-5- (2- fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [ ,3-d]pyrimidin-l-yl] -phenyl}-acetamide 59 as colorless crystals. To crystal 2 (41.5 g) was added 1-butanol-water [19:1 (volume ratio) , 415 ml] , and the mixture was stirred at 130°C for 18 hrs. After allowing to cool with stirring, the crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [4 ,3-d]pyrimidin-1-yl] -phenyl}-acetamide 59 (40.7 g, yield 89%) as colorless crystals
Combination Small Molecule MEK and PI3K Inhibition Enhances Uveal Melanoma Cell Death in a Mutant GNAQ- and GNA11-Dependent Manner.
Khalili JS et al. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. PMID: 22733540.
Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212.
Jing J et al. Mol Cancer Ther. 2012 Mar;11(3):720-9. PMID: 22169769.
Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo.
Yamaguchi T et al. Int J Oncol. 2011 Jul;39(1):23-31. PMID: 21523318.
GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition.
Gilmartin AG et al. Clin Cancer Res. 2011 Mar 1;17(5):989-1000. PMID: 21245089.
//////

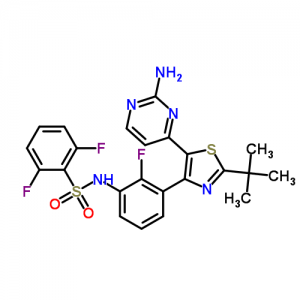
FDA Approves Tafinlar (dabrafenib mesylate, GSK 2118436) for Advanced Melanoma

DABRAFENIB
1195765-45-7
Benzenesulfonamide, N-[3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluoro-
MW 519.56
MF C23 H20 F3 N5 O2 S2
- Dabarefenib
- Dabrafenib
- GSK 2118436
- Tafinlar
Dabrafenib (trade name Tafinlar) is a drug for the treatment of cancers associated with a mutated version of the gene BRAF. Dabrafenib acts as an inhibitor of the associated enzyme B-Raf, which plays a role in the regulation of cell growth. Dabrafenib has clinical activity with a manageable safety profile in clinical trials of phase 1 and 2 in patients with BRAF(V600)-mutated metastatic melanoma.[1][2]
The Food and Drug Administration approved dabrafenib as a single agent treatment for patients with BRAF V600E mutation-positive advanced melanoma on May 30, 2013.[3] Clinical trial data demonstrated that resistance to dabrafinib and other BRAF inhibitors occurs within 6 to 7 months.[4] To overcome this resistance, the BRAF inhibitor dabrafenib was combined with the MEK inhibitor trametinib.[4] As a result of this research, on January 8, 2014, the FDA approved the combination of dabrafenib and trametinib for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma.[5]

Inhibitor of BRAF(V600) mutants
May 29, 2013 — GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Tafinlar (dabrafenib). Tafinlar is indicated as a single-agent oral treatment for unresectable melanoma (melanoma that cannot be removed by surgery) or metastatic melanoma (melanoma which has spread to other parts of the body) in adult patients with BRAF V600E mutation. Tafinlar is not indicated for the treatment of patients with wild-type BRAF melanoma. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread
Tafinlar is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma.
GSK will be making Tafinlar available for prescription no later than in the early third quarter of 2013.
In 2010, GSK entered a collaboration with bioMérieux to develop a companion diagnostic test to detect BRAF V600 (V600E and V600K) gene mutations found in several cancers, including melanoma. bioMérieux has received FDA pre-market approval of THxID™-BRAF. Currently, it is the only FDA-approved test that detects the V600K mutation.
The primary outcome measure was the estimation of the overall intracranial response rate (OIRR) in each cohort. The OIRR for Cohort A was 18 percent (95% CI: 9.7, 28.2). For Cohort B, the OIRR was also 18 percent (95% CI: 9.9, 30.0). The median duration of response was 4.6 months (95% CI: 2.8, Not Reached) and 4.6 months (95% CI: 1.9, 4.6) in Cohort A and Cohort B, respectively.
Melanoma is the most serious and deadly form of skin cancer. According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States. When melanoma spreads in the body, the disease is called metastatic melanoma.Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread. One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis, while in the U.S., the five-year survival rate was 16 percent (2003-2009).The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Tafinlar (dabrafenib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. Limitation of use: Tafinlar is not recommended for use in patients with wild-type BRAF melanoma.
Tafinlar is not approved or licensed in Europe and may not be approved in other parts of the world for the treatment of patients with BRAF V600 mutation-positive unresectable melanoma or metastatic melanoma.
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure:
 |
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50-mg and 75-mg capsules for oral administration. Each 50-mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75-mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
Dabrafenib mesylate
1195768-06-9 cas of mesylate
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide;methanesulfonic acid
Chemical structure

………………….
PATENT
http://www.google.com/patents/WO2009137391A2?cl=en
WO 2009137391
Example 58a: Λ/-{3-r5-(2-Amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Following a procedure analogous to the procedure described in Example 51, Step B using Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) and heating to 90 0C for 24 h, the title compound, Λ/-{3- [5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 –
7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Example 58b: Λ/-{3-r5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
19.6 mg of Λ/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 μl_ of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-400C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Example 58c: Λ/-{3-r5-(2-amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Step A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 ml_, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 ml_, 1.1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 ml_, 1.0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-250C and held overnight. Ethyl acetate (150 ml.) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50- 55°C with stirring until all solids dissolve. The layers were settled and separated. The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes. Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 0C. The solids were filtered and rinsed with cyclohexane (2 x 100 ml_). The solids were air dried overnight to obtain methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2- fluorobenzoate (94.1 g, 91 %).
Step B: Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 0C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2-chloro-4- methylpyrimidine (238 g, 1.3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded. The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 0C overnight. The solids were dried in vacuum oven at 30 0C over weekend to obtain Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 ml_). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 ml_. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 ml_. Dimethylacetamide (270 ml.) was then added to the reaction mixture and cooled to -1O0C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for -5 minutes between additions. The reaction was warmed to 20-25 0C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 300C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-250C. The organic layers were then concentrated under heat and vacuum to 120 ml_. The vessel contents were then heated to 500C and heptanes (120 ml.) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for -2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 300C to obtain Λ/-{3-[5-(2-chloro-4-pyrimidinyl)- 2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Step D: Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1- dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 0C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 0C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 0C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1vol). The aqueous layer was removed and discarded at 60-70 0C. The organic layer was washed with water (1 vol) at 60-70 0C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 0C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 0C for 10 min , then cooled to 2O0C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at -65-7O0C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 0C. The slurry was stirred at 65-70 0C for 2-3 hours and then cooled slowly to 0-50C. The product was filtered, washed with EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain Λ/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Example 58d: Λ/-{3-r5-(2-amino-4-pyrimidinvn-2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate
 MESYLATE
MESYLATETo a solution of Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield).
1H NMR (400 MHz, DMSO-d6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
…………………………………………………………………
PAPER
ACS Medicinal Chemistry Letters (2013), 4(3), 358-362.
http://pubs.acs.org/doi/abs/10.1021/ml4000063
http://pubs.acs.org/doi/suppl/10.1021/ml4000063/suppl_file/ml4000063_si_001.pdf

…………………………………………………………….
Patent
http://www.google.com/patents/WO2014158467A1?cl=en

Dara Phoenix (Dabrafenib) by the British GlaxoSmithKline (GSK) has developed Sisu threonine protein kinase (BRAF) inhibitor, as monotherapy ro ー kinds of clothes capsules for carrying BRAF V600E mutation surgical unresectable melanoma or metastatic melanoma treatment of adult patients, Dara Phoenix mesylate in May 2013 was approved by the US Food and Drug Administration (FDA), and is listed on the United States, the trade name Tafinlar (Da Feina). Since the European Medicines Agency (EMA) Committee for Medicinal Products for human use (CHMP) positive evaluation of Tafinlar, making the drug is expected to become after Roche’s Weiluofeini (Vemurafinib) to enter the European market, following a second BRAF inhibitors.
The chemical name Phoenix Dallas: N- [3- [5- (2- amino-4-pyrimidinyl) -2_ (tert-butyl) ~ ~ thiazol-4-yl] _2_ fluorophenyl] – 2,6_-difluorobenzenesulfonamide.

World Patent No. W02009137391, No. W02011047238 and W02012148588 number reported Dallas and Phoenix and its medicinal value synthesis method of the composition. According to the structural characteristics of Dara Phoenix and its analogues, the synthesis of such substances currently have A, B and C are three routes.

A more common route is the synthetic route, by reaction of 3-amino-2-fluorobenzoate (IX) first and 2,6_-difluorobenzene sulfonyl chloride (III) to amidation reaction occurs sulfonamide intermediate ( X); intermediate (X) with 2-chloro-4-methyl pyrimidine (XI) The condensation reaction occurs under the action of a strong base to give the intermediate (XII); intermediate (XII) to give the intermediate bromo
(XIII); intermediate (XIII) with 2,2_ dimethyl thiopropionamide (VI) to give the cyclized intermediate (XIV); and finally, the intermediate (XIV) by ammonolysis to afford the title compound Dallas Phoenix (I).

Different [0009] B is the first route by reaction of 3-amino-2-fluorobenzoate (IX) amino group protection, and thus condensation, cyclization, and bromo; then be obtained by deprotection of the amino group and the sulfonamide Intermediate (XIV); similarly, the intermediate
(XIV) obtained by ammonolysis target compound Dara Phoenix (I).

c route design features that first aminolysis reaction, and then give the desired product by deprotection and amino sulfonamide reaction. Clearly, this design is suitable for the route of these substituted amino ー aminolysis reaction, and for compounds such as Dallas Phoenix having pyrimidinylamino structure is not applicable. The reason is that if there are two aromatic amino groups will make the final sulfonamide ー reaction step to lose selectivity.

Example IV: the reaction flask was added N- [3- (5- formyl-2-t-butyl-ko -4_ thiazolyl) -2_ fluorophenyl] -2,6_ difluoro benzenesulfonamide (VIII) (5.4g, 11.5mmol), N, N- dimethylformamide dimethyl acetal (DMF-DMA) (2.74g, 23mmol) and xylene 50mL, heated to 140 ° C. About every four hours methanol was distilled out of the resulting reaction system, the reaction takes about 24 hours in total, the end of the reaction was detected by TLC. Cool, add hexane 40mL, have produced a yellow solid, filtered, and dried solids obtained after January nitrate melon (1.36,11.5mmol), sodium hydroxide (0.46g, 11.5mmol) and n-Ding enjoy 5OmL, warmed to 120 ° C, The reaction for 12 inches, TLC the reaction was complete. Cooling, with a crystal precipitated crystallized slowly for 3 inches, and filtered. The filter cake starched water, filtered and dried to yield an off-white solid Dara Phoenix (I) 3.58g, yield 60%.
References
- Gibney, G. T.; Zager, J. S. (2013). “Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies”. Expert Opinion on Drug Metabolism & Toxicology 9 (7): 1. doi:10.1517/17425255.2013.794220. PMID 23621583.
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. (2013). “B-Raf and the inhibitors: From bench to bedside”. Journal of Hematology & Oncology 6: 30. doi:10.1186/1756-8722-6-30. PMC 3646677. PMID 23617957.
- “GSK melanoma drugs add to tally of U.S. drug approvals”. Reuters. May 30, 2013.
- “Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations” 367 (18). New England Journal of Medicine. November 1, 2012. pp. 1694–703. doi:10.1056/NEJMoa1210093. PMC 3549295. PMID 23020132.
“Dabrafenib/Trametinib Combination Approved for Advanced Melanoma”. OncLive. January 9, 2013.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-{3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide | |
| Clinical data | |
| Trade names | Tafinlar |
|
|
| Legal status | |
| Identifiers | |
| CAS number | 1195765-45-7 |
| ATC code | L01XE23 |
| PubChem | CID 44462760 |
| ChemSpider | 25948204 |
| ChEBI | CHEBI:75045 |
| ChEMBL | CHEMBL2028663 |
| Chemical data | |
| Formula | C23H20F3N5O2S2 |
| Molecular mass | 519.56 g/mol |
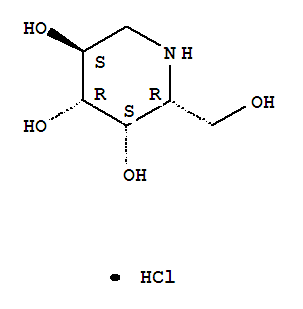
Phase 3 Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease
 CAS Number:75172-81-5
CAS Number:75172-81-5-
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
- Molecular Structure:

- Formula:C6H14ClNO4
- Molecular Weight:199.63
- Synonyms:3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;Migalastat hydrochloride;Galactostatin hydrochloride;(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- Melting Point:260 °C
- Boiling Point:382.7 °C at 760 mmHg
- Flash Point:185.2 °C
end feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2 Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

