
Home » Posts tagged 'GLAXO'
Tag Archives: GLAXO
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Elacridar
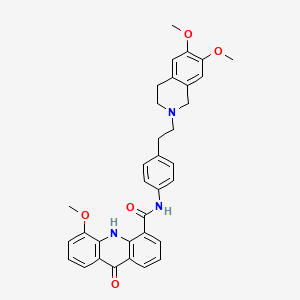
Elacridar
C34H33N3O5, 563.6 g/mol
依克立达;gw0918
UNII-N488540F94
143851-98-3 (monoHCl)
N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide
GF120918
Elacridar (GF120918)
GF-120918
GG-918
GW-120918
GW-918
GF-120918A (HCl)
GlaxoSmithKline (previously Glaxo Wellcome ) was developing elacridar, an inhibitor of the multidrug resistance transporter BCRP (breast cancer resistant protein), as an oral bioenhancer for the treatment of solid tumors.
Elacridar is an oral bioenhancer which had been in early clinical trials at GlaxoSmithKline for the treatment of cancer, however, no recent development has been reported. It is a very potent inhibitor of P-glycoprotein, an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells.
SYN

The condensation of 2-(4-nitrophenyl)ethyl bromide with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline by means of K2CO3 and KI in DMF at 100 C gives 6,7-dimethoxy-2-[2-(4-nitrophenyl)ethyl]-1,2,3,4-tetrahydroisoquinoline,
Which is reduced with H2 over Pd/C in ethanol to yield the corresponding amine . Finally, this compound is condensed with 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acid by means of DCC and HOBt in DMF to afford the target carboxamide.
The intermediate 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acidhas been obtained as follows: The condensation of 2-amino-3-methoxybenzoic acid with 2-bromobenzoic acid by means of K2CO3 and copper dust give the diphenylamine , which is cyclized to the target acridine Elacridar by means of POCl3 in refluxing acetonitrile.
PATENT
WO-2019183403
Deuterated analogs of elacridar as P-gp/BCRP inhibitor by preventing efflux useful for treating cancer.
Elacridar, previously referred to as GF120918, is a compound with the structure of 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1 ,2,3,4-tetrahydro- 6,7-dimethoxy-2-isoquinolinyl)ethyl] phenyl]-4-acridine-carboxamide or, as sometimes written, N-4-[2-(1 ,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy- 9-oxo-4-acridine carboxamide. Elacridar was originally described as a P-gp selective inhibitor but is now recognized as a dual P-gp/BCRP inhibitor. (Matsson P, Pedersen JM, Norinder U, Bergstrom CA, and Artursson P 2009 Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res 26:1816-1831 ).
003 Elacridar has been examined with some success both in vitro and in vivo as a P-gp and BCRP inhibitor. By way of example, in cancer patients, coadministration of elacridar with therapeutic agents such as paclitaxel (P-gp substrate) and topotecan (BCRP substrate) improved their oral absorption – presumably by preventing efflux into the intestinal lumen by P-gp/BCRP pumps located in the Gl tract. Similarly, in rodents, elacridar has been coadministered with some success with pump substrates such as morphine, amprenavir, imatinib, dasatinib, gefitinib, sorafenib, and sunitinib to increase drug levels in the brain (by blocking efflux mediated by P-gp and BCRP at the blood brain barrier). A summary of some of these studies can be found in a study report by Sane et al. (Drug Metabolism And Disposition 40:1612-1619, 2012).
004 Administration of elacridar has several limitations. By way of example, elacridar has unfavorable physicochemical properties; it is practically insoluble in water, making it difficult to formulate as, for example, either an injectable or oral dosage form. Elacridar’s poor solubility and high lipophilicity result in dissolution rate-limited absorption from the gut lumen.
005 A variety of approaches have been pursued in order to increase efficacy of elacridar. For example, United States Patent Application Publication 20140235631 discloses a nanoparticle formulation in order to increase oral bioavailability.
006 Sane et al. (Journal of Pharmaceutical Sciences, Vol. 102, 1343-1354 (2013)) report a micro-emulsion formulation of elacridar to try and overcome its dissolution-rate-limited bioavailability.
007 Sawicki et al. (Drug Development and Industrial Pharmacy, 2017 VOL. 43, NO. 4, 584-594) described an amorphous solid dispersion formulation of freeze dried elacridar hydrochloride-povidone K30-sodium dodecyl sulfate. However, when tested in healthy human volunteers, extremely high doses (e.g. 1000 mg) were required to achieve a Cmax of 326 ng/ml. (Sawicki et al. Drug Deliv. and Transl.
Res. Published online 18 Nov 2016).
008 Montesinos et al. (Mol Pharm. 2015 Nov 2; 12(11 ):3829-38) attempted several PEGylated liposome formulations of elacridar which resulted in a partial increase in half life, but without an increase in efficacy when co-administered with a therapeutic agent.
009 Because of the great unpredictability in the art and poor correlations in many cases between animal and human data, the value of such formulation attempts await clinical trial.
0010 Studies of the whole body distribution of a microdose of 11C elacridar after intravenous injection showed high level accumulation in the liver (Bauer et al. J Nucl Med. 2016;57:1265-1268). This has led some to suggest that systemic levels of elacridar are also substantially limited by clearance in the liver.
0011 A potentially attractive strategy for improving metabolic stability of some drugs is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the rate of formation of inactive metabolites by replacing one or more hydrogen atoms with deuterium atoms.
Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the absorption, distribution, metabolism, excretion and/or toxicity (‘ADMET’) properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
0012 Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, M I et al, J Pharm Sci, 1975, 64:367-91 ; Foster, A B, Adv Drug Res 1985, 14:1 -40 (“Foster”); Kushner, D J et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, M B et al, Curr Opin Drug Discov Devel, 2006, 9:101 -09 (“Fisher”)). The results have been variable and unpredictable. For some compounds, deuteration indeed caused decreased metabolic clearance in vivo. For others, no change in metabolism was observed. Still others demonstrated increased metabolic clearance. The great unpredictability and variability in deuterium effects has led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting metabolism (see Foster at p. 35 and Fisher at p. 101 ).
0013 The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem. 1991 , 34, 2871 -76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
0014 Considering elacridar’s challenging physicochemical and ADMET properties in humans, in spite of recent formulation advancements, there remains a need in the art for elacridar analogs that can achieve higher, less variable levels in the systemic circulation, at the blood-brain barrier, and elsewhere to optimize efflux inhibition.
Example 1 : Synthesis of Instant Analogs and Compositions
00179 This example demonstrates a synthetic method for making elacridar analogs, deuterium substitutions based upon the deuteration of the starting compounds. The synthesis and the analog numbers refer to Figure 4.
00180 Step 1
00181 A 12L three-neck flask was charged with compound 1 (270.5 g, 1.618 mol), compound 2 (357.8 g, 1.78 mol, 1.1 eq.), K2C03 (447 g, 3.236 mol, 2.0 eq), Cu (20.6 g, 0.324 mol, 0.2 eq.) and ethanol (2.7 L) and the resulting mixture was heated to reflux under nitrogen for 1 hour. The reaction mixture was cooled to room
temperature after the reaction progress was checked with LC-MS. Water (2.7 L) was added and the mixture was filtered through a pad of Celite. The Celite was washed with water (1.35L) and the combined filtrate was adjusted to pH~2 by addition of concentrated HCI (~410 mL) over 15 min. The resulting suspension was stirred at 10°C for 1.5 hours and the solid was filtered, washed with water (2.7 L) and dried at 45°C using a vacuum oven for 2 days to give compound 3 (465 g, ~100%) as a yellow solid.
00182 Step 2
00183 A suspension of compound 3 (498 g, 1.734 mol) in acetonitrile (4.0 L) was heated to reflux under stirring. To the suspension was added POCb (355.5 mL,
3.814 mol, 2.2 eq.) drop-wise over 2h. The mixture was heated at reflux for 2.5h and then cooled to 30 °C. To the mixture was slowly added water (3.0 L) and the resultant thick slurry was heated to reflux for 1 5h. The slurry was cooled to 10 °C and filtered. The solid was washed with water (2 X 1.0 L), acetonitrile (2 X 1.0 L) and dried using a vacuum oven overnight at 45 °C to afford compound 4 (426 g, 91.3%) as a yellow solid.
00184 Step 3:
00185 A 12L three-neck flask was charged with compound 5 (475g, 2.065 mol), compound 6 (474.8g, 2.065 mol), K2C03 (314g, 2.273 mol), Kl (68.6g, 0.413 moL) and DMF (2.5L) and the resulting mixture was heated to 70 °C and stirred for 2.5 hours. After LC-MS showed that the reaction was complete, the mixture was cooled to 50 °C and methanol (620 ml_) was added. Then the mixture was cooled to 30 °C and water (4.75 L) was added. The resulting suspension was cooled to 10 °C and for 1 hour. The solid was filtered, washed with water (2 X 2.5 L) and air dried for 2 days to afford the compound 7 (630 g, 89.1 %) as a yellow solid.
00186 Step 4
00187 To a solution of compound 7 (630 g, 1.84 mol) in THF/ethanol (8 L at 1 :1 ) was added Pd/C (10%, 50% wet, 30 g). The mixture was stirred under an
atmosphere of hydrogen (1 atm, balloon) at 15-20 °C for 4h. The reaction mixture was filtered through a pad of Celite and the pad was washed with TFIF (1.0 L). The filtrate was concentrated to 3 volumes under vacuum and hexanes (4.0 L) was added. The resulting slurry was cooled to 0 °C and stirred for 1 h. The solid was filtered and washed with hexanes (2 X 500 ml_) and air dried overnight to afford the compound 8 (522 g, 90.8%) as an off -white solid.
00188 Step 5
00189 A 5L three-neck flask was charged with compound 4 (250 g, 0.929 mol, 1 eq.), compound 8 (290 g, 0.929mol, 1 eq.) and DMF (2.5 L) and the resulting mixture was stirred at room temperature until it became a clear solution. To the solution was added TBTU (328 g, 1.021 mol, 1.1 eq.), followed by triethylamine (272 ml_, 1.95 mol, 2.1 eq.) and the resulting mixture was stirred at room temperature under nitrogen overnight. The mixture was poured slowly into water (7.5 L) with stirring and the resulting suspension was stirred for 1 hour at room temperature. The solid was filtered and washed with water (2 X 7 L). The solid thus obtained was dried using a vacuum oven at 50 °C for two days and 509.0 g (97.3%) of compound 9 was obtained as yellow solid.
00190 Step 6
00191 300.0 g (0.532 mol) of compound 9 was suspended in acetic acid (1.2 L) and heated to 70 °C. The resultant solution was hot filtered and heated to 70°C again. Preheated ethanol (70 °C, 3.6 L) was then added. To this solution was added concentrated HCI (66.0 ml_, 0.792 mol, 1.5 eq.) dropwise over 30 min. The resulting solution was stirred at 70°C until crystallization commenced (~about 20 min). The suspension was cooled to room temperature over 3h, filtered, washed with ethanol (2 X 1.8 L) and dried using a vacuum oven at 60°C over the weekend to afford compound 10 (253.0 g, 79.2%) as a brown solid.
Example 2 Manufacture of a Deuterated Elacridar analog EE60.
00192 EE60 is synthesized by the procedure shown in Figure 4 and as continued in Figure 5.
00193 The structure of EE60 is confirmed as follows: Samples of 5 pi are measured using an LC system comprising an UltiMate 3000 LC Systems (Dionex, Sunnyvale, CA) and an 2996 UV diode array detector (Waters). Samples are injected on to a 100 x 2mm (ID) 3.5 pm ZORBAX Extend-C18 column (Agilent, Santa Clara, CA). Elution is done at a flow rate of 0.4 mL/min using a 5 minute gradient from 20% to 95% B (mobile phase A was 0.1 % FICOOFI in water (v/v) and mobile phase B was methanol). 95% B is maintained for 1 min followed by re-equilibration at 20% B. Chromeleon (v6.8) is used for data acquisition and peak processing.
Example 3: Manufacture of a Deuterated Elacridar analog EE59
00194 EE59 was synthesized by the procedure shown in Figure 6.
00195 The resulting yellowish brown precipitate was removed by filtration and the filter cake was dried overnight (72 mg). Analysis of the filter cake by LCMS indicated the presence of a single peak at multiple wavelengths (215 nm, 220 nm, 254 nm,
280 nm); each peak confirmed the presence of the desired product (LC retention time, 5.3 min; m/z = 575 [(M+FI)+]).
00196 1H NMR of EE598 revealed 1H NMR (400 MHz, DMSO-d6) d 12.3 ( s , 1H), 10.6 (s, 1H), 8.51-8.46 (m, 2H), 7.80 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 7.6 Hz, 2H), 7.45-7.38 (m, 2H), 7.32-7.25 (m, 3H), 6.66 (d, J = 6.8 Hz, 2H), 3.62 (s, 2H), 2.86 (t, J = 6.8 Hz, 2H), 2.66 (m, 4H).
Example 4: Demonstration of superior properties of instant analogs and compositions: in vivo ADMET.
00197 Pharmacologic studies are performed according to Ward KW et al (2001 Xenobiotica 317783-797) and Ward and Azzarano (JPET 310:703-709, 2004).
Briefly, instant analogs are administered solutions in 10% aqueous polyethylene glycol-300 (PEG-300) or 6% Cavitron with 1 % dimethyl sulfoxide, or as well triturated suspensions in 0.5% aqueous HPMC containing 1 % Tween 80. Blood samples are collected at various times up to 48 h after drug administration; plasma samples are prepared and at “70°C until analysis.
00198 Mice. Instant analogs are administered to four groups of animals by oral gavage (10 ml/kg dose volume). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and the fourth group receive instant analogs as a solution in Cavitron at 3 mg/kg. Blood sampling in mice is performed via a tail vein at 0.5, 1 , 2, 4, 8, 24, and 32 h postdose.
00199 Rats. A total of seven groups of animals receive instant analogs by oral gavage (10 ml/kg). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and a fourth and fifth group each receive instant analogs as a solution in Cavitron or PEG-300, respectively, at 3 mg/kg. A sixth and seventh group of rats with indwelling hepatic portal vein catheters receive instant analogs by oral gavage (10 ml/kg) as a suspension at 3 or 30 mg/kg, respectively. Blood sampling in rats are performed via a lateral tail vein; samples are also obtained from the hepatic portal vein catheter. Blood samples are obtained before dosing and at 5, 15, 30, and 45 min, and 1 ,1.5, 2, 3, 4, 6, 8, 10, 24, and 32 h postdose.
00200 Dogs. Dogs receive instant analogs by lavage (4 ml/kg) on three separate occasions with dosages at 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a cephalic vein and from the hepatic portal vein catheter before dosing and at 5, 15, 30, and 45 min and 1 , 1.5, 2, 3, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00201 Monkeys. Monkeys receive instant analogs by oral gavage (8 ml/kg dose volume) on three separate occasions at dosages of 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a femoral vein via an indwelling catheter and from the hepatic portal vascular access port
before dosing and at 5, 15, and 30 min and 1 , 1.5, 2, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00202 Humans. Healthy volunteers receive instant analogs orally at doses ranging from 25 mg to 1000 mg. Blood samples are obtained and analyzed for analog concentrations at 0, 15 min, 30 min, 45 min, 60 min, 90 min, 120 min, 180 min, 2 hr, 4 hr, 6hr, 8 hr, 12 hr, 24 hr, and 48 h after administration .
Analytical Methods
00203 Instant analogs are isolated from samples by precipitation with acetonitrile and quantified by LC/MS/MS coupled with an atmospheric pressure chemical ionization interface (475°C). Internal standards [in acetonitrile/10 mM ammonium formate, pH 3.0; 95:5 (v/v)] are added to 50 pi samples and vortexed and centrifuged for 30 min at 4000 rpm. The supernatants are injected onto the LC/MS/MS system using an HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland) coupled to an Aria TX2 high-throughput liquid chromatographic system using turbulent flow technology (Cohesive Technologies, Franklin, MA) in focus mode. The mobile phase consists of a mixture of 0.1 % formic acid in water and 0.1 % formic acid in
acetonitrile. The turbulent flow column is a 0.5 X 50-mm Cyclone P column
(Cohesive Technologies) in series to a 2 X 20 mm, 4 pm Polar RP (Phenomenex, Torrance, CA) analytical column. Positive-ion multiple reaction monitoring is used for the detection of instant analogs and internal standard and the selected precursor and product ions are mlz 564 and 252, respectively. Using a (1/x) weighted linear regression analysis of the calibration curve, linear responses in analyte/internal standard peak area ratios are observed for instant analog concentrations ranging from 2 to 10,000 ng/ml.
00204 Alternatively, useful analytical methods to demonstrate the surprising and superior properties of the instant elacridar analogs are the methods as described by Stokvis et al, J Mass Spectr 2004: 39: 1122-1130.
PATENT
WO2014018932
claiming nano-particle composition comprising breast cancer resistance protein inhibitor (eg elacridar). Family member of the elacridar
PAPER
J Med Chem 1995, 38(13): 2418
PATENT
Product PATENT WO9212132
PATENT
US5604237
NMR includes d 2.60-2.95 (m,8H,CH2); 3.58 (s,2H,N–CH2 –Ph); 3.72 (s,6H,OMe); 4.05 (s,3H,OMe acridone); 6.78 (2s,2H,Ar.isoquinoline), 7.20-7.88 (m,8H,Ar.), 8.48 (t,2H,H1 and H8 acridone), 10.60 (s, 1H,CONH), 12.32 (s, 1H,NH acridone)
///////////Elacridar, GF-120918, GG-918 , GW-120918, GW-918, GF-120918A (HCl), solid tumors, GSK, GLAXO
[11C]-elacridar
GSK 2793660, Trying to crack the structure
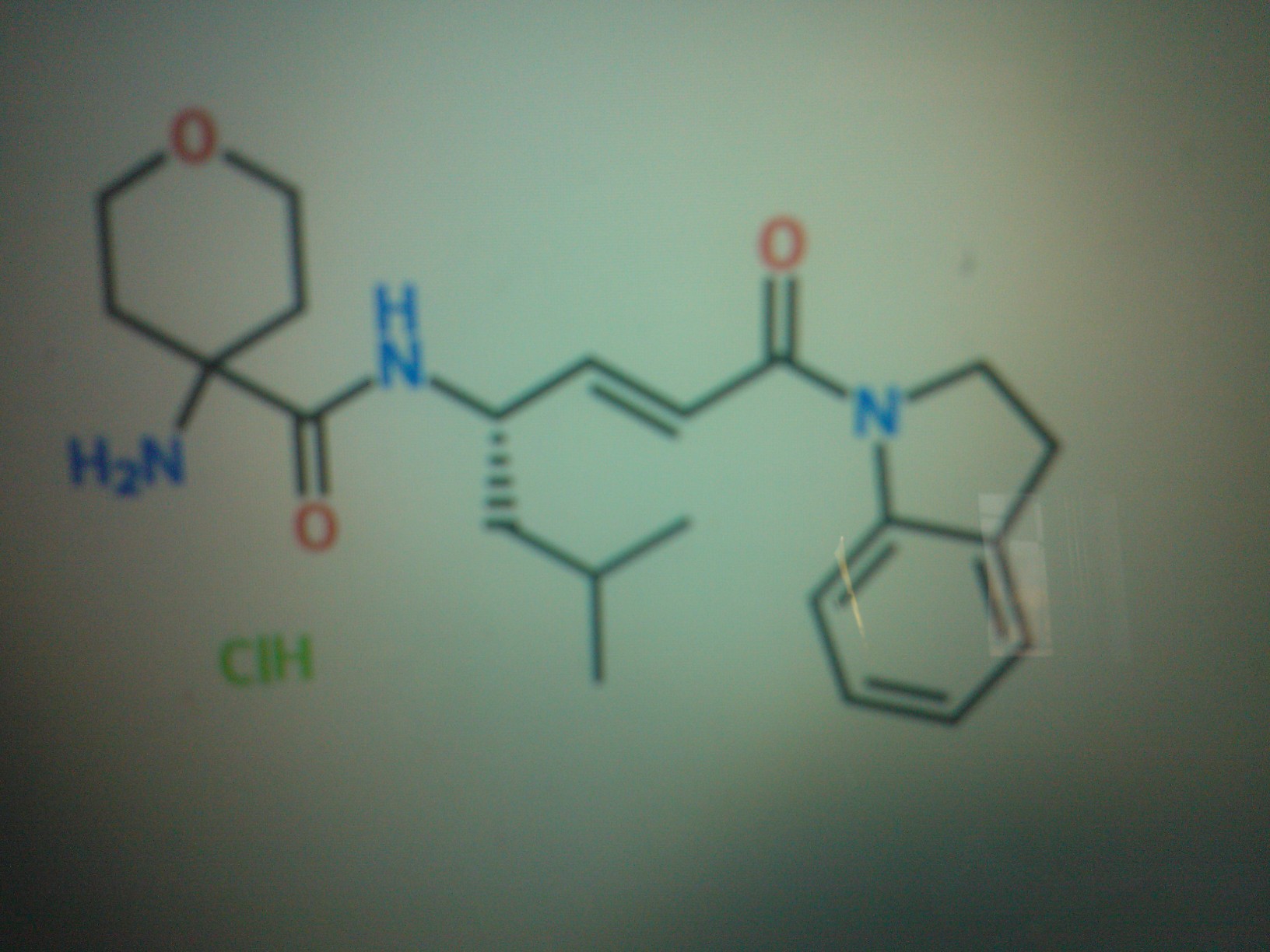
 COMPD A
COMPD A
 COMPD B
COMPD B
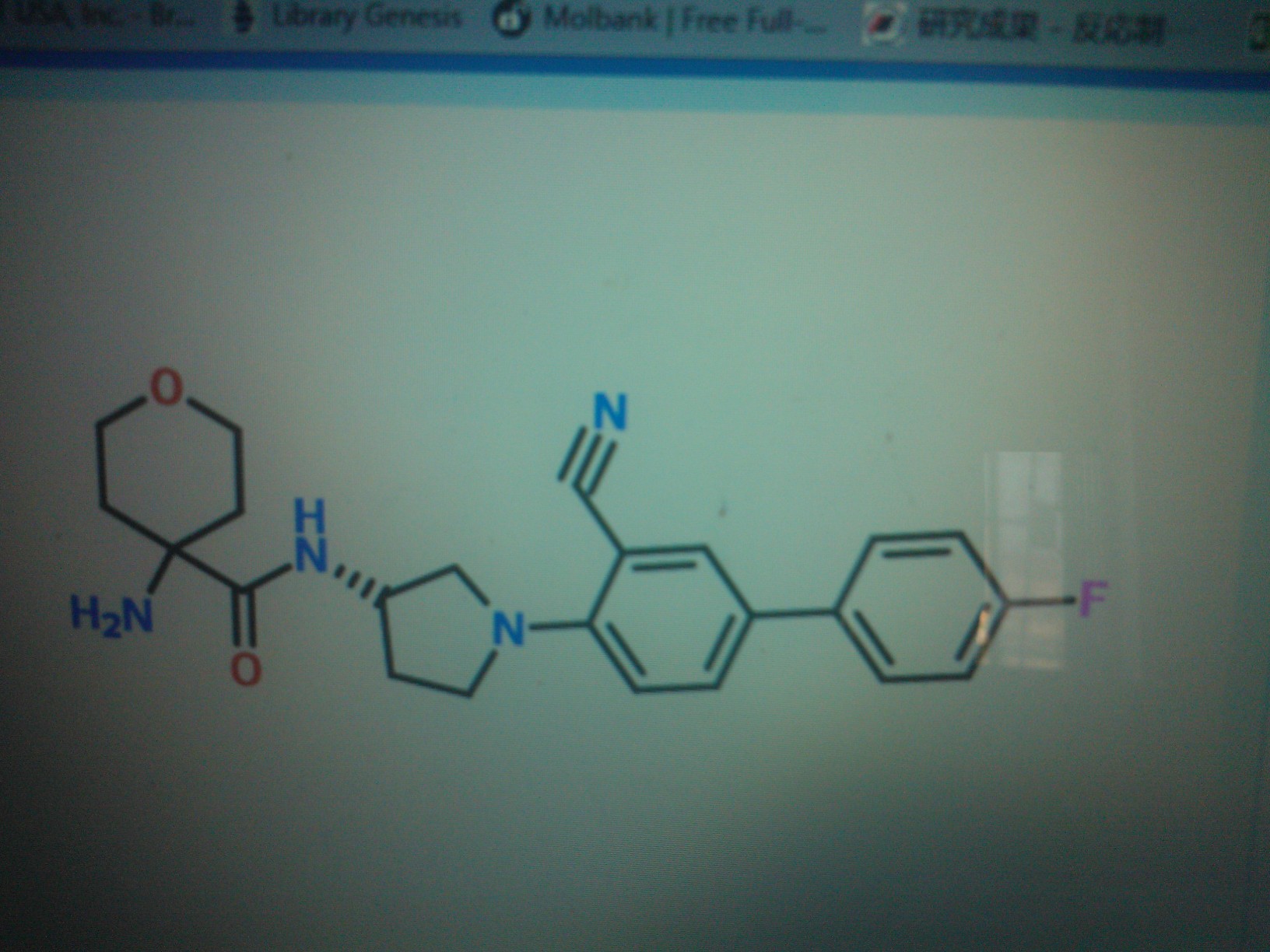
 COMPD C
COMPD C
 COMPD D
COMPD D
 A
A B
B C
C D
DGSK 2793660
DATA FOR A
HCL SALT CAS 1613458-78-8
BASE CAS 1613458-70-0
C20 H27 N3 O3 . Cl H
MW OF BASE…..357.45
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
2H-Pyran-4-carboxamide, 4-amino-N-[(1S,2E)-4-(2,3-dihydro-1H-indol-1-yl)-1-ethyl-4-oxo-2-buten-1-yl]tetrahydro-, hydrochloride (1:1)
DATA FOR B
1613458-79-9 HCL SALT
1613458-71-1 BASE
C22 H31 N3 O3 . Cl H
MW 385.50 OF BASE
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
C24 H27 F N4 O . Cl H, MW 442.957
CAS OF BASE 1394001-73-0
CAS OF HCL 1394001-71-8
DATA FOR D
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
CAS OF BASE 1394001-74-1
CAS OF HCL 1394001-72-9
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Bronchiectasis
Dipeptidyl peptidase I inhibitor
http://www.gsk.com/media/280387/product-pipeline-2014.pdf
This study is the first administration of GSK2793660 to humans and will evaluate the safety, tolerability, PK and PD of single oral ascending doses of GSK2793660, and of repeat oral doses of GSK2793660 in healthy subjects. The study will comprise two parts (Part A and Part B). Part A will consist of two cohorts of subjects, each taking part in a three-way cross over study, with ascending doses of GSK2793660 and placebo. Available safety, PK and PD data will be reviewed before each dose escalation. This will be followed by a food-effect arm in the cohort that received what is deemed to be the target clinical dose. Part B is planned to consist of up to two cohorts of subjects, each taking part in one 14 day repeat dose study period. Subjects will be dosed on Day 1 and then on Days 3-15. It is planned that two doses will be evaluated. The dose(s) to be tested will be selected based on safety, PK, and PD from Part A. The study is intended to provide sufficient confidence in the safety profile of the molecule and information on target engagement to allow progression to further studies………..https://clinicaltrials.gov/ct2/show/NCT02058407
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Cathepsins are a family of enzymes included in the papain superfamily of cysteine proteases. Cathepsins B, C, F, H, K, L, S, V, and X have been described in the scientific literature. Cathepsin C is also known in the literature as Dipeptidyl Peptidase I or “DPPI.”
A number of recently published studies have begun to describe the role cathepsin C plays in certain inflammatory processes. See e.g. Adkison et al., The Journal of Clinical Investigation 109:363-371 (2002); Tran et al., Archives of Biochemistry and Biophysics 403 : 160-170 (2002); Thiele et al., The Journal of Immunology 158: 5200-5210 (1997);
Bidere et al., The Journal of Biological Chemistry 277: 32339-32347 (2002); Mabee et al., The Journal of Immunology 160: 5880-5885 (1998); McGuire et al., The Journal of
Biological Chemistry, 268: 2458-2467 (1993); and Paris et al., FEBS Letters 369: 326-330 (1995). From these studies, it appears that cathepsin C is co-expressed in granules of neutrophils and other leukocytes with certain serine proteases and cathepsin C functions to process the pro-forms of the serine proteases to active forms. Serine proteases are released from the granules of leukocytes recruited to sites of inflammation. Once activated, these proteases have a number of functions including degradation of various extracellular matrix components, which together can propagate tissue damage and chronic inflammation.
Studies in both cathepsin C deficient mice, and the human cathepsin C deficiency
Papillon-Lefevre syndrome clearly demonstrate that cathepsin C is required for the
activation of the neutrophil serine proteases in azurophilic granules such as neutrophil elastase (NE), cathepsin G, and proteinase 3. See Pham, C. T. et al., J. Immunol. 173 :
7277-7281 (2004).
A number of respiratory diseases are associated with an overabundant
acculumation of neutrophils and the presence of increased levels of at least some
neutrophil serine proteases. These enzymes are believed to play a role in the pathology of several respiratory diseases, such as Chronic Obstructive Pulmonary Disease (“COPD”), cystic fibrosis (CF), and non-cystic fibrosis (non-CF) bronchiectasis. Each of these diseases is associated with increased levels of E in particular, and E at least is considered to play a role in the progression of disease. See Ranes, J. and Stoller, J. K., Semin. Respir. Crit. Care Med 26: 154-166 (2005); Saget, S. D. et al., Am. J. Resp. Crit. Care Med. 186: 857-865 (2012); Tsang, K. W. et al., Chest 117: 420-426 (2000).
Additional roles of the other proteases is emerging. See Hartl, D. et al., Nature Med. 13 : 1423-1430 (2007); Korkmaz, B. et al., Pharm. Rev. 62: 726-759 (2010).
Cigarette smoking is a significant risk factor for developing COPD. Exposure to cigarette smoke and other noxious particles and gases may result in chronic inflammation of the lung. In response to such exposure, inflammatory cells such as CD8+ T cells, macrophages, and neutrophils are recruited to the area. These recruited inflammatory cells release proteases, which are believed to play a major role in the disease etiology by a number of mechanisms. Proteases released from recruited cells include the serine proteases NE as above; granzymes A and B, released from cytotoxic T cells or natural killer cells; and chymases, released from mast cells. Cathepsin C appears to be involved in activating all of these enzymes to some extent.
A number of studies with cathepsin C deficient mice have suggested roles for cathepsin C in disease models. Cathepsin C knockout mice are resistant to lung airspace enlargement and inflammatory cell infiltration in both cigarette smoke and ozone exposure models of COPD. See Guay et al., Current Topics in Medicinal Chemistry, 2010, 10, 708- 716; See also Podolin et al. (2008), Inflammation Research, 57(Suppl 2) S104.
In a model of rheumatoid arthritis (“RA”), another chronic inflammatory disease where cathepsin C may play a role, neutrophils are recruited to the site of joint
inflammation and release cathepsin G, NE, and proteinase 3, which are believed to be responsible in part for cartilage destruction associated with RA (Hu, Y. and Pham, C. T. Arthritis Rheum. 52: 2553-2558 (2005); Zen, K. et al, Blood 117:4885-4894 (2011)). Other models where cathepsin C may play a role include osteoarthritis, asthma, Multiple Sclerosis, and Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-related diseases (e.g. ANCA-associated vasculitis). See e.g. Matsui, K., Yuyama, N., Akaiwa, M., Yoshida, N. L., Maeda, M., Sugita, Y., Izuhara, K., Gene 293(1-2): 1-7 (2002); Wolters, P. J., Laig- Webster, M., Caughey, G. H., American Journal of Respiratory Cell & Molecular Biology 22(2): 183-90 (2000); Schreiber et al., J. Am. Soc. Nephrol. 23 :470-482 (2012). Cathepsin C has been demonstrated to have a role in neutrophil migration in the development of aortic aneurysms by a mechanism which has not been clearly elucidated (Pagano, M. B. et al., PNAS 104: 2855-2860 (2007)).
One approach to treating these conditions is to inhibit the activity of the serine proteases involved in the inflammatory process, especially NE activity. See e.g.,
Ohbayashi, Expert Opin. Investig. Drugs 11(7): 965-980 (2002); Shapiro, Am. J. Respir. Cell Mol. Biol. 26: 266-268 (2002). Indeed, a potent and selective inhibitor of NE was found to improve lung function in patients with bronchiectasis (Stockley, R. et al. Respir. Med. 107, 524-533 (2013)). In light of the role cathepsin C plays in activating certain serine proteases, especially NE, it is desirable to prepare compounds that inhibit its activity, which thereby inhibit serine protease activity. Thus, there is a need to identify compounds that inhibit cathepsin C, which can be used in the treatment of a variety of conditions mediated by cathepsin C.
There are additional activities of cathepsin C that may also be related to disease etiology. Cathepsin C is highly expressed in the lung epithelium where it may play a role in the processing of other enzymes not yet identified. Cathepsin C has also been reported to cleave kallikrein-4, which is believed to play a role in dental enamel maturation (Tye, C. E. et al. J. Dental Res. 88: 323-327 (2009)). Finally, cathepsin C is itself released from cells and may play a direct role in the degradation of matrix proteins.
DATA FOR A
WO 2014091443
http://www.google.com/patents/WO2014091443A1?cl=en

synthesis







Intermediate 1
1,1-dimethylethyl ((l -l-{[methyl(methyloxy)amino]carbonyl}propyl)carbamate
To a solution of (2,S)-2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)butanoic acid (2.50 g, 12.3 mmol) in THF (15.0 mL) was added Ι,Γ-carbonyldiimidazole (2.39 g, 14.8 mmol) portionwise over about 10 min. After stirring 30 min at RT, a solution of Ν,Ο- dimethylhydroxylamine hydrochloride (1.32 g, 13.5 mmol) and DIPEA (2.36 mL, 13.5 mmol) in DMF (4.0 mL) was added. The reaction mixture was stirred for 2 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HC1 (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.60 g, 88%) as a clear, colorless oil. LC-MS m/z 247 (M+H)+, 0.94 min (ret time).
Intermediate 2
1,1-dimethylethyl [(lS -l-formylpropyl] carbamate
To a solution of L1AIH4 (0.453 g, 11.9 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution of 1, 1-dimethylethyl ((l,S)-l-{[methyl(methyloxy)amino]carbonyl}- propyl)carbamate (2.67 g, 10.8 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6.5 mL) followed by 5% aq. potassium bisulfate (6.5 mL). The reaction mixture was washed with 1 M aq. HC1 (3 x 10 mL), saturated aq. NaHC03 (3 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil.
Intermediate 3
methyl (2E V)-4-({ [(1 , l-dimethylethyl)oxy] car bonyl} amino)-2-hexenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (4.35 g, 13.0 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 2 in Et20 (15 mL). The reaction mixture was stirred at RT overnight. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.44 g, 55% over two steps) as a clear, colorless oil. LC-MS m/z 244 (M+H)+, 0.98 min (ret time). Intermediate 4
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-2-hexenoic acid
Li OH (2.95 g, 123 mmol) was added to a solution of methyl (2£, S 4-({[(1, 1- dimethylethyl)oxy]carbonyl}amino)-2-hexenoate (6 g, 24.66 mmol) in THF (50 mL), MeOH (10.00 mL), and water (50.0 mL). The reaction was stirred overnight at RT. After 18.5 h, the reaction mixture was concentrated under reduced pressure to remove the THF and MeOH. Water (40 mL) was added, and aqueous mixture was adjusted to pH = 3 with 6 M aq. HC1, as measured by pH paper. EtOAc (80 mL) was added, the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried over Na2S04, concentrated under reduced pressure, and dried under high vacuum, giving 6.09 g of the title compound. LC-MS m/z 230 (M+H)+, 0.77 min (ret time).
Intermediate 5
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl] carbamate
A solution of 50 wt% *T3P in EtOAc (22.00 mL, 37.0 mmol) was added dropwise via addition funnel to a solution of (2£,,4,S)-4-({[(l, l-dimethylethyl)oxy]carbonyl}- amino)-2-hexenoic acid (5.65 g, 24.64 mmol), 2,3-dihydro-lH-indole (2.76 mL, 24.64 mmol), and Et3N (11 mL, 79 mmol) in CH2C12 (90 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 30 min, the reaction was quenched by dropwise addition of saturated aq. NaHC03 (50 mL). The layers were separated, and the reaction was washed with 10% citric acid (1 x 50 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 7.21 g (89%) of the title compound. LC-MS m/z 331 (M+H)+, 1.05 (ret time). Intermediate 6
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine
trifluoroacetate
TFA (25 mL, 324 mmol) was added to a solution of 1, 1-dimethylethyl [(1^,2£)-4- (2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]carbamate (7.21 g, 21.82 mmol) in CH2C12 (25 mL). The reaction was stirred at RT. After 3.5 h, CH2C12 (200 mL) was added, and the reaction was concentrated under reduced pressure and dried under high vacuum. LC-MS m/z 231 (M+H)+, 0.69 (ret time).
Intermediate 7
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino carbonyl)tetrahydro-2H-pyran-4-yl]carbamate
A solution of 50 wt% UT3P in EtOAc (1.3 mL, 2.184 mmol) was added dropwise to a solution of [(l,S’,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine trifluoroacetate (500 mg, 1.452 mmol), 4-((tert-butoxycarbonyl)amino)tetrahydro-2H- pyran-4-carboxylic acid (356 mg, 1.452 mmol), and Et3N (1 mL, 7.21 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction mixture was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 251 mg (38%) of the title compound. LC-MS m/z 458 (M+H)+, 0.96 (ret time).
Example 1
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.23 mL, 2.76 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.549 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp) for 1 h 45 min. The solvent was evaporated under reduced pressure. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 193.3 mg (89%) of the title compound. LC-MS m/z 358 (M+H)+, 0.68 (ret time).
1H MR (400 MHz, METHANOL-^) δ ppm 8.14 (br. s., 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.53 Hz, 1 H); 7.02 – 7.09 (m, 1 H); 6.83 (dd, J=15.18, 6.65 Hz, 1 H); 6.49 (d, 7=14.8 Hz, 1 H); 4.56 (d, 7=7.28 Hz, 1 H); 4.22 (br. s., 2 H); 3.95 (d, 7=7.53 Hz, 1 H); 3.88 – 3.94 (m, 1 H); 3.71 – 3.78 (m, 2 H); 3.23 (br. s., 2 H); 2.39 – 2.46 (m, 2 H); 1.79 – 1.86 (m, 2 H); 1.75 (s, 1 H); 1.72 (d, 7=8.28 Hz, 1 H); 1.00 (t, 7=7.40 Hz, 3 H)
DATA FOR B
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride







http://www.google.com/patents/WO2014091443A1?cl=en
Intermediate 8
N -{[(l,l-dimethylet leucinamide
To a solution ofN-(tert-butoxycarbonyl)-L-leucine (3.00 g, 13.0 mmol) in THF (25.0 mL) was added Ι,Γ-carbonyldiimidazole (2.52 g, 15.6 mmol) portionwise over about 10 min. After stirring 1 h at RT, a solution of N,O-dimethylhydroxylamine hydrochloride (1.39 g, 14.3 mmol) and DIPEA (2.49 mL, 14.3 mmol) in DMF (6.0 mL) was added. The reaction mixture was stirred for 2.5 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HCl (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.34 g, 66%) as a clear, colorless oil. LC-MS m/z 275 (M+H)+, 1.17 min (ret time).
Intermediate 9
1,1-dimethylethyl [(lS -l-formyl-3-methylbutyl]carbamate
To a solution of L1AIH4 (0.356 g, 9.38 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution ofN2-{[(l, l-dimethylethyl)oxy]carbonyl}-N1-methyl-N1-(methyloxy)-L- leucinamide (2.34 g, 8.53 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6 mL) followed by 5% aq. potassium bisulfate (6 mL). The reaction mixture was washed with 1 M aq. HCl (2 x 10 mL), saturated aq. NaHC03 (2 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil. Intermediate 10
methyl (2E 4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (3.42 g, 10.2 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 9 in Et20 (15 mL). The reaction mixture was stirred for 15 h at RT. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.74 g, 75% over two steps) as a clear, colorless oil. LC-MS m/z 272 (M+H)+, 1.22 min (ret time).
Intermediate 11
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoic acid
To a solution of methyl (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6- methyl-2-heptenoate (5.00 g, 18.43 mmol) in THF (15 mL), MeOH (15.0 mL), and water (15 mL) was added Li OH (2.206 g, 92.00 mmol). After stirring for 2 h at RT, the reaction mixture was concentrated in vacuo. The reaction mixture was acidified with 6 M aq. HC1 to pH = 5 and then extracted with EtOAc. The organic layer was washed with water, dried over Na2SC”4, filtered, and concentrated in vacuo to afford the title compound (4.7 g, 99%) as a white semi-solid. LC-MS m/z 158 (M+H-Boc)+, 0.94 min (ret time).
Intermediate 12
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]carbamate
To a solution of (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2- heptenoic acid (4.70 g, 18.26 mmol) in DMF (30.0 mL) were added BOP reagent (8.08 g, 18.26 mmol) and DIPEA (6.38 mL, 36.5 mmol). After stirring at RT for 5 min, 2,3-dihydro- lH-indole (2.053 mL, 18.26 mmol) was added and stirring continued overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04, filtered, concentrated in vacuo and purified by flash column chromatography (0-20% EtOAc/hexanes) to afford the title compound (4.83 g, 74%) as a white solid. LC-MS m/z 359 (M+H)+, 1.18 min (ret time).
Intermediate 13
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten-l-yl]amine trifluoroacetate
To a solution of 1, 1-dimethylethyl [(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2- methylpropyl)-4-oxo-2-buten-l-yl]carbamate (3.21 g, 8.95 mmol) in CH2C12 (10.0 mL) was added TFA (10 mL, 130 mmol). The reaction mixture was stirred for 17.5 h at RT and then concentrated under reduced pressure and dried under high vacuum to afford the title compound. LC-MS m/z 259 (M+H)+, 0.76 min (ret time).
Intermediate 14
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l- l]amino}carbonyl)tetrahydro-2H- ran-4-yl]carbamate
A solution of 50 wt% ¾P in EtOAc (1.2 mL, 2.016 mmol) was added dropwise to a solution of [(15′,2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]amine trifluoroacetate (500 mg, 1.343 mmol), 4-((tert- butoxycarbonyl)amino)tetrahydro-2H-pyran-4-carboxylic acid (329 mg, 1.343 mmol), and Et3N (0.93 mL, 6.71 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 204 mg (31%) of the title compound. LC-MS m/z 486 (M+H)+, 1.07 min (ret time).
Example 2
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.22 mL, 2.64 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(1^2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.517 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp). After 1 h 45 min, the solvent was evaporated under reduced pressure at 60 °C. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 130.6 mg (60%) of the title compound. LC-MS m/z 386 (M+H)+, 0.79 (ret time). 1H MR (400 MHz, METHANOL- d4) δ ppm 8.15 (d, J=7.03 Hz, 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.65 Hz, 1 H); 7.06 (t, J=7.91 Hz, 1 H); 6.81 (dd, J=15.18, 6.40 Hz, 1 H); 6.49 (br. s., 1 H); 4.73 – 4.85 (m, 2 H); 4.21 (t, J=8.28 Hz, 2 H); 3.91 – 3.97 (m, 2 H); 3.70 – 3.77 (m, 2 H); 3.25 – 3.21 (m, 2 H); 2.35 – 2.48 (m, 2 H); 1.82 (d, J=14.31 Hz, 2 H); 1.63 – 1.71 (m, 2 H); 1.50 – 1.57 (m, 1 H); 0.98 (dd, J=11.92, 6.40 Hz, 6 H).
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
http://www.google.im/patents/WO2012112733A1?cl=en
Example 1
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride

HCI salt
A solution of 1,1-dimethylethyl [l-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl]amino}carbonyl)cyclohexyl]carbamate (44 mg, 0.087 mmol) in HCI (4 M solution in 1,4-dioxane, 1.0 mL, 4.00 mmol) was stirred at RT for 1 h. The reaction mixture was diluted with Et20 (5 mL), and the mixture was filtered and washed with Et20 (2 x 2 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (33.5 mg, 87%). LC-MS m/z 407 (M+H)+, 0.94 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.65 – 7.72 (m, 2 H), 7.52 – 7.59 (m, 2 H), 7.10 – 7.17 (m, 2 H), 6.89 (d, J=8.53 Hz, 1 H), 4.50 – 4.58 (m, 1 H), 3.94 (dd, J=10.29, 6.53 Hz, 1 H), 3.80 (dt, J=9.41, 7.09 Hz, 1 H), 3.67-3.71 (m, 1 H), 3.64 (dd, J=10.29, 4.52 Hz, 1 H), 2.29 – 2.37 (m, 1 H), 2.04 – 2.16 (m, 3 H), 1.78 – 1.88 (m, 5 H), 1.45 – 1.62 (m, 3 H).
DATA FOR D
http://www.google.im/patents/WO2012112733A1?cl=en
Example 2
4-amino- V-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3-pyrrolidinyl]tetrahydro-2H- pyr -4-carboxamide hydrochloride
HCI salt
A solution of 1,1-dimethylethyl [4-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl] amino }carbonyl)tetrahydro-2H-pyran-4-yl] carbamate (183 mg, 0.360 mmol) in HC1 (4 M solution in 1,4-dioxane, 2.0 mL, 8.00 mmol) was stirred at RT for 0.5 h. The reaction mixture was diluted with Et20 (10 mL), and the mixture was filtered and washed with Et20 (2 x 5 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (122.8 mg, 77%). LC-MS m/z 409 (M+H)+, 0.87 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.66 – 7.72 (m, 2 H), 7.53 – 7.60 (m, 2 H), 7.11 – 7.18 (m, 2 H), 6.89 (d, J=8.78 Hz, 1 H), 4.53 – 4.60 (m, 1 H), 3.87 – 3.97 (m, 3 H), 3.78 – 3.84 (m, 1 H), 3.64 – 3.76 (m, 4 H), 2.30 – 2.44 (m, 3 H), 2.11 – 2.19 (m, 1 H), 1.77 – 1.84 (m, 2 H).
| WO2004002491A1 * | 25 Jun 2003 | 8 Jan 2004 | David J Aldous | Morpholine and tetrahydropyran drivatives and their use as cathepsin inhibitors |
| WO2008121065A1 * | 28 Mar 2008 | 9 Oct 2008 | Astrazeneca Ab | Novel pyrrolidine derivatives as antagonists of the chemokine receptor |
| US20070032484 * | 25 Jul 2006 | 8 Feb 2007 | Roche Palo Alto Llc | Cathepsin K inhibitors |
| US20020107266 * | Dec 11, 2001 | Aug 8, 2002 | Marguerita Lim-Wilby | Amides used particularly in the treatment, prevention or amelioration of one or more symptoms of malaria or Chagas’ disease; inhibiting the activity of falcipain or cruzain |
| US20100286118 * | May 6, 2010 | Nov 11, 2010 | Rhonan Ford | Substituted 1-cyanoethylheterocyclylcarboxamide compounds 750 |
| WO2012109415A1 | Feb 9, 2012 | Aug 16, 2012 | Glaxosmithkline Llc | Cathepsin c inhibitors |
Epelsiban being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.

Epelsiban
557296
GSK-557296
GSK-557296-B
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione
(3R, 6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[(1R)- 1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1-methylpropyl]-2,5- piperazinedione
Glaxo Group Limited INNOVATOR
Epelsiban (GSK-557,296-B)[1][2] is an oral drug which acts as a selective, sub-nanomolar (Ki=0.13 nM) oxytocin receptor antagonist with >31000-fold selectivity over the related vasopressin receptors and is being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.[3][4]


benzenesulfonic acid;(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione,CAS 1159097-48-9
UNII-H629P9T4UN, GSK557296B, Epelsiban besylate (USAN), Epelsiban besylate [USAN], 1159097-48-9, H629P9T4UN
GSK-557296 is being developed in early clinical studies at GlaxoSmithKline for enhancement of embryo and or blastocyst implantation in women undergoing IVF treatment. The product has been in phase II clinical development for the treatment of premature ejaculation.
Preterm labor is a major clinical problem leading to death and disability in newborns and accounts for 10% of all births and causes 70% of all infant mortality and morbidity.
Oxytocin (OT) is a potent stimulant of uterine contractions and is responsible for the initiation of labor via the interaction with the OT receptors in the mammalian uterus. OT antagonists have been shown to inhibit uterine contractions and delay preterm delivery. So there is increasing interest in OT antagonists because of their potential application in the prevention of preterm labor. Although several tocolytics have already been approved in clinical practice, they have harmful maternal or fetal side effects.
The first clinically tested OT antagonist atosiban has a much more tolerable side effect profile and has recently been approved for use in Europe. However, atosiban is a peptide and a mixed OT/vasopressin V1a receptor antagonist that has to be given by iv infusion and is not suitable for long-term maintenance treatment, as it is not orally bioavailable.
Hence there has been considerable interest in overcoming the shortcomings of the peptide OT antagonists by identifying orally active nonpeptide OT antagonists with a higher degree of selectivity toward the vasopressin receptors (V1a, V1b, V2) with good oral bioavailability. Although several templates have been investigated as potential selective OT antagonists, few have achieved the required selectivity for the OT receptor vs the vasopressin receptors combined with the bioavailability and physical chemical properties required for an efficacious oral drug.
Therefore our objective was to design a potent, orally active OT antagonist with high levels of selectivity over the vasopressin receptor with good oral bioavailability in humans that would delay labor safely by greater than seven days and with improved infant outcome, as shown by a reduced combined morbidity score.
| Patent | Submitted | Granted |
|---|---|---|
| Compounds [US7919492] | 2010-12-02 | 2011-04-05 |
| Piperazinediones as Oxytocin Receptor Antagonists [US7550462] | 2007-11-01 | 2009-06-23 |
| Compounds [US8202864] | 2011-06-23 | 2012-06-19 |
| Novel compounds [US2009247541] | 2009-10-01 |
………………………………………
PATENT
https://www.google.com/patents/US7919492
Example 3
Method A

(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
as a white lyophilisate (88 mg, 23%) after freeze-drying from 1,4-dioxane
HPLC Rt=2.70 minutes (gradient 2); m/z [M+H]+=519
1H NMR (CDCl3) δ 7.49 (d, 1H), 7.27-7.15 (m, 4H), 7.10 (d, 1H), 6.68 (s, 1H), 6.40 (d, 1H), 4.10 (dd, 1H), 4.01 (d, 1H), 3.74-3.52 (m, 5H), 3.28-3.07 (m, 5H), 2.97-2.84 (m, 2H), 2.79-2.71 (m, 1H), 2.62 (s, 3H), 2.59 (s, 3H), 1.65-1.53 (m, 1H), 0.98-0.80 (m, 2H), 0.70 (t, 3H), 0.45 (d, 3H).
Example 3
Method B

(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
A suspension of {(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-6-[(1S)-1-methylpropyl]-2,5-dioxo-1-piperazinyl}(2,6-dimethyl-3-pyridinyl)acetic acid hydrochloride (5.0 g, 10.3 mmol) (intermediate 5) in dry dichloromethane (50 ml) was treated with 1,1-carbonyldiimidazole (2.6 g, 16 mmol) and the reaction mixture was stirred under nitrogen for 18 hours. Morpholine (4.8 ml, 55 mmol) was added and the resultant solution was left to stand under nitrogen for 18 hours. The solvent was removed in vacuo and the residue was separated between ethyl acetate and water. The organic phase was washed with brine and dried over anhydrous magnesium sulphate. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This was applied to a basic alumina cartridge (240 g) and eluted using a gradient of 0-7.5% methanol in diethyl ether (9CV), 7.5-10% methanol in diethyl ether (1CV) and 10% methanol in diethyl ether (1CV). The required fractions were combined and evaporated in vacuo to give (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione as a white solid (2.4 g, 45%).
HPLC Rt=2.72 minutes (gradient 2); m/z [M+H]+=519
………………………………………
WO 2011051814
http://www.google.com/patents/WO2011051814A1?cl=en
This invention relates to novel crystalline forms of (3R, 6R)-3-(2,3-dihydro-1 H- inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1 – methylpropyl]-2,5-piperazinedione benzenesulfonate salt, processes for their preparation, pharmaceutical compositions containing them and to their use in medicine. The benzenesulfonate salt of Compound A is represented by the following structure:

In one aspect, the present invention provides a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form provides an X-ray powder diffraction pattern substantially in accordance with Figure 1 .
In another aspect, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

In an additional aspect, the invention includes a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2.
In certain aspects, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2 In one aspect, the invention also provides a crystalline form of {3R, 6R)-3-(2,3- dihydro-1 H-inden-2-yl)-1-[(1 R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]- 6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

Experimental
Process Scheme

Stage 4
Acetone / Water Recrystallisation
Compound A-form I Ste8e 5 Besylate salt
MW 676.83 Acetone / Water
Recrystallisation MW 676.83 Process description for isolation of Compound A-Form 1
Stage 0
methyl d-alloisoleucinate hydrochloride (Compound 2) was charged to ethyl acetate. A solution of potassium carbonate in water was then added. The mixture was then stirred vigorously at room temperature for 1 hour. The two layers were separated and the aqueous layer further extracted with ethyl acetate. The organic layers were combined and washed with brine. The organic layers were then concentrated in vacuo and filtered to yield methyl D-alloisoleucinate (Compound 3) as a pale yellow oil.
Stage 1
2,6-dimethyl-3-pyridinecarbaldehyde (Compound 4) in methanol at ambient temperature was treated with D-alloisoleucinate (Compound 3) in methanol followed by 2,2,2- trifluoroethanol and the reaction mixture was warmed to 40°C. When formation of the intermediate imine (methyl A/-[(2,6-dimethyl-3-pyridinyl)methylidene]-D-alloisoleucine) was complete Compound 5 was added followed by 1-isocyano-2- [(phenylmethyl)oxy]benzene (Compound 6) and the reaction mixture was stirred at 40°C until formation of Compound 7 was deemed complete.
Stage 2
Palladium on carbon catalyst was treated with a solution of Compound 7 in methanol and 2,2,2-trifluoroethanol and diluted with acetic acid. The vessel was purged with nitrogen and the reaction mixture warmed to 50°C and hydrogenated at 4.0-4.5 barg. When the reaction was deemed complete it was cooled to ambient temperature and the catalyst removed by filtration and washed through with methanol. The organic solution of 2- {(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1-piperazinyl}- 2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) was concentrated at reduced pressure and then diluted with /‘so-propyl acetate and concentrated at reduced pressure.
The residue was diluted with /‘so-propyl acetate and washed with aqueous ammonia. The aqueous phase was separated and extracted into another portion of /‘so-propyl acetate. The combined organic phases were washed with water, concentrated by distillation at reduced pressure, diluted with /‘so-propyl acetate and concentrated by distillation at reduced pressure, to leave a concentrated solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8). The product was finally dissolved in 1 ,4-dioxane for the next stage and stored into drums.
Stage 3 Solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) in 1 ,4-dioxane was treated with 1 ,1 ‘-carbonyl diimidazole at ambient temperature to form a solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1 -[1-(2,6-dimethyl-3-pyridinyl)- 2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1 -methylpropyl]-2,5- piperazinedione (Compound 9).
In a separate vessel morpholine in 1 ,4-dioxane was heated to 80-85°C. The solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[1 – (2,6-dimethyl-3-pyridinyl)-2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1- methylpropyl]-2,5-piperazinedione (Compound 9) was slowly added to the morpholine in 1 ,4-dioxane. The reaction mixture was stirred for one hour at 80-85°C and cooled before concentration by distillation at reduced pressure.
The concentrated solution of Compound A was diluted with /‘so-propyl acetate and washed with aqueous sodium hydroxide followed by water. The /so-propyl acetate solution of COMPOUND A was then concentrated by distillation at reduced pressure and cooled to ambient temperature. The concentrated solution of Compound A was then diluted with acetone and treated with benzenesulfonic acid and seed crystals were added and the reaction mixture stirred until crystallisation occurred. The slurry of Compound A besylate was heated to 50°C, a temperature cycle was performed, and finally the slurry was cooled to -10°C and isolated by filtration. The filter cake was washed with cold acetone (-10°C) to give Compound A besylate (intermediate grade) as a wet cake.
Yield: 44% from Compound 5
39% from Compound 5
Stage 4
Compound A besylate (intermediate grade wet cake, Compound A besylate ) was suspended in acetone (17.4 vol including acetone content of wet cake) and heated to 55- 60°C. Water (0.66 vol) was added until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (3.2 vol). The temperature of the reaction mixture was adjusted to 45-50°C before the addition of seed crystals (0.00025wt). When crystallisation was complete the reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins.
The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to -3-2°C over 4.5 h and stirred for at least 1 h before the product was isolated by filtration. The wet cake was washed with acetone at 0°C (3 x 3.1 vol) and blown dry before being unloaded. COMPOUND A besylate was dried at 50°C under vacuum for 3 days. Compound A besylate was then milled. Yield: 66% Stage 5
Compound A besylate (OBU-D-02) was suspended in acetone (8 vol) and water (1 .1 vol) and heated to 48-52°C until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (2 vol). The reaction mixture was cooled to 20-25°C before the addition of Form 1 seed crystals (0.0025wt). When crystallisation was complete the reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins. The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins.
The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to -12— 8°C over 3.5 h and stirred for 15 h before the product was isolated by filtration. The wet cake was washed with acetone at -10°C (2 x 3 vol) and blown dry before being unloaded. Compound A besylate was dried at ambient temperature under vacuum for 6 days with a wet nitrogen bleed to afford Form 1 . Compound A besylate was then milled. Yield: 67%
Recrystallisation of Compound A besylate anhydrate (Form 2)

Besylate salt ………………………………………………………………Besylate salt
C30H38 4O4■ C6H603S C30H38 4O4■
MW 676.83 MW 676.83
COMPOUND A besylate is charged to the vessel and treated with methyl ethyl ketone (MEK) (8vol) and water (0.35vol) and the solution heated until dissolution is observed (ca. 55-60°C). The solution is then filtered and recharged to the vessel. Pressure is then reduced to 650mbar and the reaction mixture heated further to distil out solvent. MEK is added at the same rate as solvent is removed by distillation keeping the reaction mixture volume constant. After 4 volumes of MEK have been added the reaction mixture is treated with Form 2 seed crystals (2%wt) and the distillation continued in the same manner until another 7 volumes of MEK has been added. The vacuum is then released to an atmospheric pressure of nitrogen and the temperature of the reaction mixture adjusted to 65°C. The reaction mixture is then filtered and washed with pre heated MEK (2vol at 65°C). The purified COMPOUND A besylate anhydrate is then sucked dry and dried further in a vacuum oven at 65°C at l OOmbar with a nitrogen bleed. Yield 89%
NMR data is the same for Forms 1 and 2.
1 H NMR (500MHz, DMSO-d6) 5ppm 0.71-0.80(m, 6H) 0.87-0.98(m, 1 H) 1 .31 (br. S, 1 H) 1.69(br. S, 1 H) 2.68(s, 3H) 2.69(s, 3H) 2.72-2.79(m, 1 H) 2.80-2.87(m, 1 H) 2.88-3.01 (m, 3H) 3.18-3.25(m, 1 H) 3.27-3.33(m, 1 H) 3.38-3.46(m, 1 H) 3.47-3.52(m, 1 H)3.53-3.57(m, 1 H) 3.60-3.71 (m, 3H) 3.83(dd, J=9.46,3.15 Hz, 1 H) 3.89 (br. S, 1 H)6.10(br. S, 1 H) 7.1 1 – 7.14(m, 2H) 7.19-7.23(m, 2H) 7.30-7.35(m, 3H)7.59-7.63(m, 2H) 7.67(d, J=7.25Hz, 1 H) 8.12(br. S, 1 H) 8.50(d, J=3.78Hz, 1 H)
………………………………………..
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm201287w

A six-stage stereoselective synthesis of indanyl-7-(3′-pyridyl)-(3R,6R,7R)-2,5-diketopiperazines oxytocin antagonists from indene is described. SAR studies involving mono- and disubstitution in the 3′-pyridyl ring and variation of the 3-isobutyl group gave potent compounds (pKi > 9.0) with good aqueous solubility. Evaluation of the pharmacokinetic profile in the rat, dog, and cynomolgus monkey of those derivatives with low cynomolgus monkey and human intrinsic clearance gave 2′,6′-dimethyl-3′-pyridyl R–sec-butyl morpholine amide Epelsiban (69), a highly potent oxytocin antagonist (pKi = 9.9) with >31000-fold selectivity over all three human vasopressin receptors hV1aR, hV2R, and hV1bR, with no significant P450 inhibition. Epelsiban has low levels of intrinsic clearance against the microsomes of four species, good bioavailability (55%) and comparable potency to atosiban in the rat, but is 100-fold more potent than the latter in vitro and was negative in the genotoxicity screens with a satisfactory oral safety profile in female rats.
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione (69 EPELSIBAN)
References
- Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). “Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency”. Journal of Medicinal Chemistry 55 (2): 783–96. doi:10.1021/jm201287w. PMID 205501.
- 2 Borthwick, A. D.; Liddle, J. (January 2013). “Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists”. In Domling, A. Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. ISBN 978-3-527-33107-9.
- 3 World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN): Proposed INN: List 105”. WHO Drug Information 25 (2): 179.
- 4 USAN Council (2011). “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). Retrieved 2011-10-28.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-(morpholin-4-yl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]piperazine-2,5-dione | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 872599-83-2 1159097-48-9 (besylate) |
| ATC code | None |
| PubChem | CID 11634973 |
| ChemSpider | 9809717 |
| KEGG | D10117 |
| Chemical data | |
| Formula | C30H38N4O4 |
| Molecular mass | 518.6 g/mol |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| WO2003053443A1 | Dec 20, 2002 | Jul 3, 2003 | Glaxo Group Ltd | Substituted diketopiperazines as oxytocin antagonists | |
| WO2006000399A1 | Jun 21, 2005 | Jan 5, 2006 | Glaxo Group Ltd | Novel compounds | |
| EP2005006760W | Title not available | ||||
| US6914160 | Jul 31, 2003 | Jul 5, 2005 | Pfizer Inc | Oxytocin inhibitors | |
| US20070254888 | Jun 21, 2005 | Nov 1, 2007 | Glaxo Group Limited | Piperazinediones as Oxytocin Receptor Antagonists |
| US8202864 * | Feb 25, 2011 | Jun 19, 2012 | Glaxo Group Limited | Compounds |
| US8716286 | Oct 28, 2010 | May 6, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US8742099 | May 20, 2013 | Jun 3, 2014 | Glaxo Group Limited | Compounds |
| US8815856 | Mar 18, 2014 | Aug 26, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US20120202811 * | Apr 19, 2012 | Aug 9, 2012 | Glaxo Group Limited | Novel compounds |
Glaxo Plans to File for Malaria Vaccine Approval Next Year

Malaria vaccine candidate reduces disease over 18 months of follow-up in late-stage study of more than 15,000 infants and young children
Malaria is a significant public health burden, claiming 660,000 lives a year – mostly children in sub-Saharan Africa
-Data support plan to submit regulatory application in 2014
Multilateral Initiative on Malaria Pan African Conference, Durban, South Africa — Results from a large-scale Phase III trial, presented today in Durban, show that the most clinically advanced malaria vaccine candidate, RTS,S, continued to protect young children and infants from clinical malaria up to 18 months after vaccination. Based on these data, GSK now intends to submit, in 2014, a regulatory application to the European Medicines Agency (EMA). The World Health Organization (WHO) has indicated that a policy recommendation for the RTS,S malaria vaccine candidate is possible as early as 2015 if it is granted a positive scientific opinion by EMA.
READ ALL AT
http://www.pharmalive.com/glaxo-plans-to-file-for-malaria-vaccine-approval-next-year
![]()
