
Home » Posts tagged 'flozin'
Tag Archives: flozin
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LIK 066, Licogliflozin diprolinate
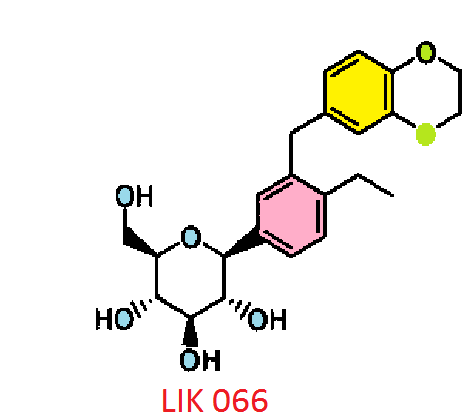
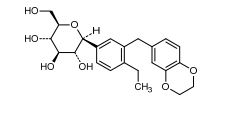
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
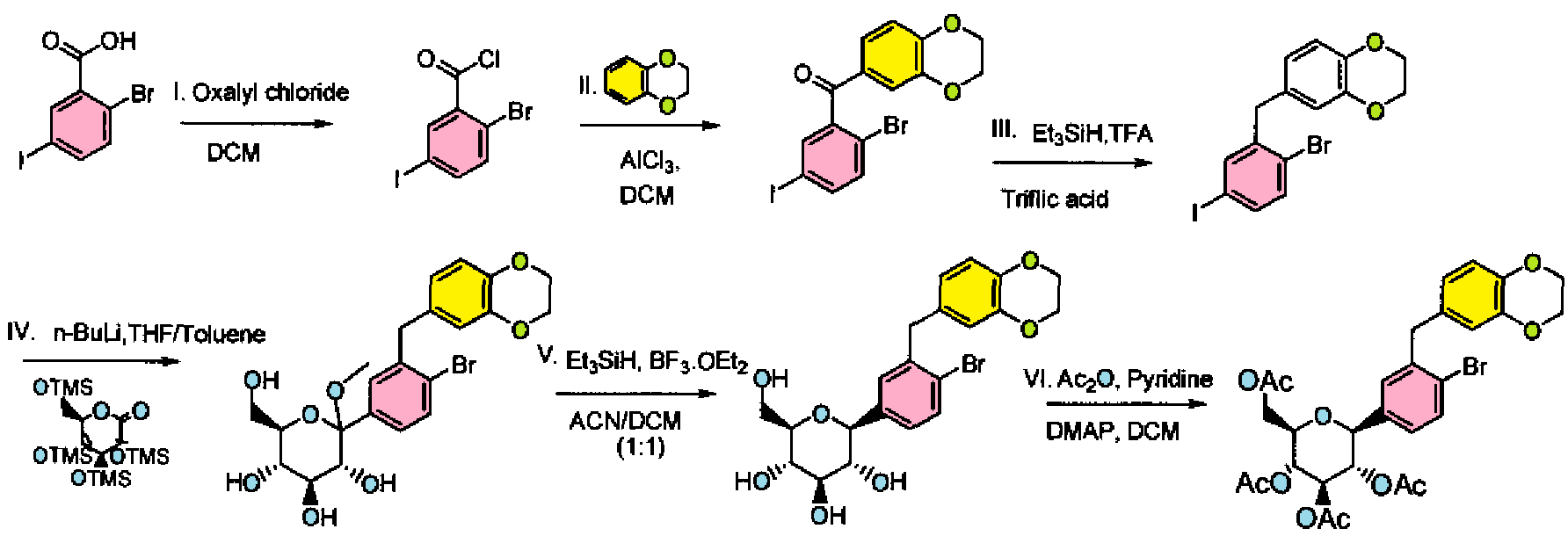
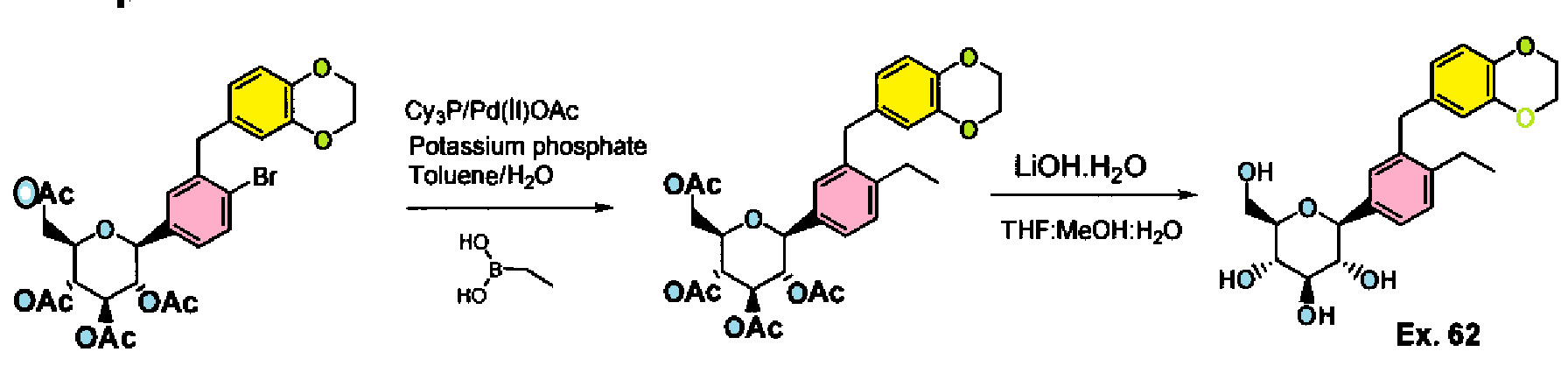
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
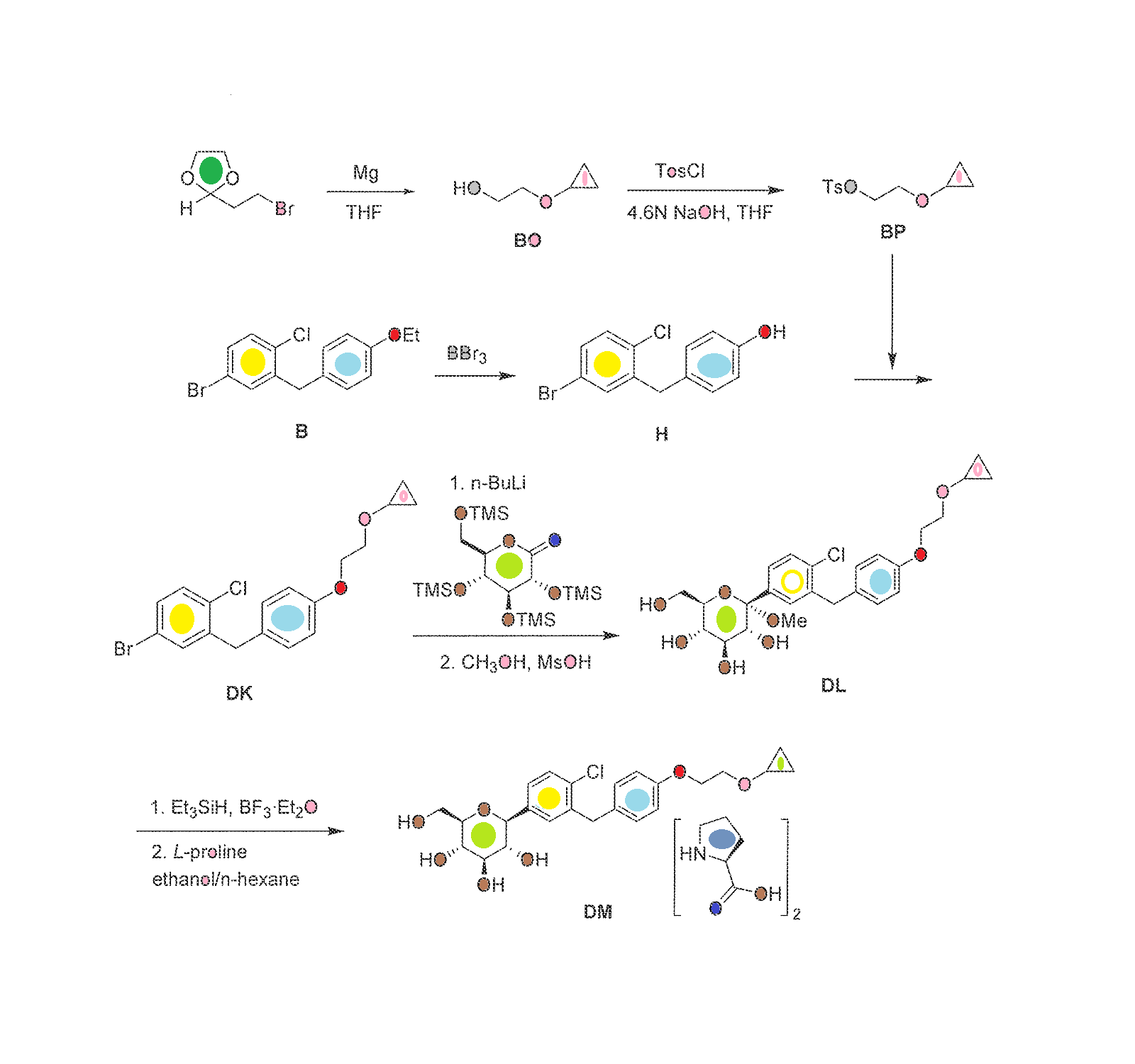
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
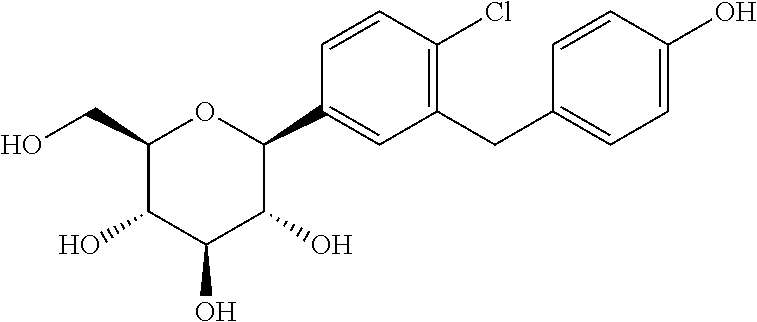
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
Bexagliflozin (Brenzavvy)
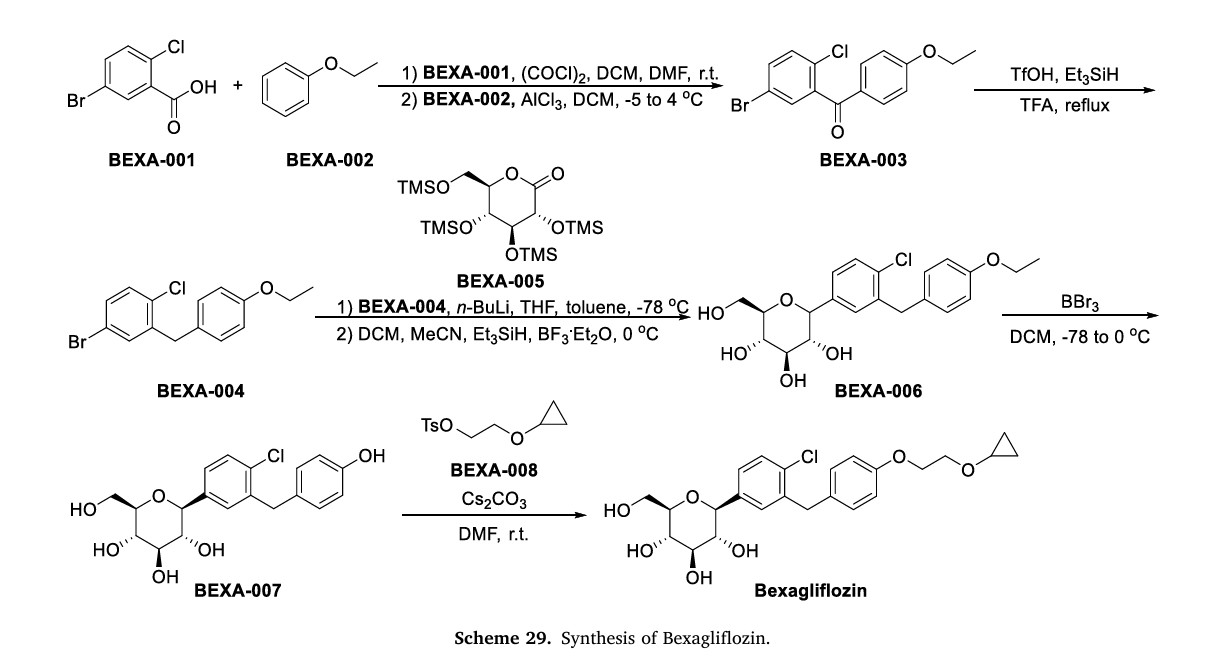
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
What is SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem !!
A new “flozin” seems to me appearing on the horizon in form of SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem, picked up a list from WO 2012160218, from TFChem…….see link , Sirona Biochem Announces SGLT2 Inhibitor and Skin Lightening Patent Granted, 29 Jun 2015, Patent entitled “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
This led me to search, “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
WO 2012160218 A1, IN 2013-DN10635, CN 103649033Tf化学公司
| Applicant | Tfchem |

List above as in http://www.google.com/patents/WO2012160218A1?cl=en
FROM THE ABOVE LIST, SBM-TFC-039 MAY BE PREDICTED/OR AS SHOWN BELOW
COMPD 16 as in/WO2012160218
COMPD 16, PREDICTED/LIKELY SBM-TFC-039 has CAS 1413373-30-4, name D-myo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,2,3-trideoxy-2,2-difluoro-3-(hydroxymethyl)-
Just scrolling through the patent gave me more insight
MORE EVIDENCE….http://www.google.com/patents/WO2012160218A1?cl=en, this patent descibes compd 16 as follows
Compound 16 according to the invention has been compared to Dapaglifozin to underline the improvement of the duration of action, i.e. the longer duration of glucosuria, of the compound when the intracyclic oxygen atom of the glucose moiety is replaced by a CF2 moiety.

This assay has been carried out at a dose of 3 mg/ kg.
The results obtained are presented on Figure 5. It appears thus that 16 (3 mg/kg) triggered glucosuria that lasted beyond 24 hours compared to Dapagliflozin.
• Compound 16 according to the invention has been compared to the compound 9 of WO 2009/1076550 to underline the improvement of the duration of action of the compound when a mimic of glucose bearing a CH-OH moiety instead of the intracyclic oxygen atom is replaced by a mimic of glucose bearing a CF2 in place of the CH-OH moiet .

%7D)
| Company | Sirona Biochem Corp. |
| Description | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Molecular Target | Sodium-glucose cotransporter 2 (SGLT2) |
| Mechanism of Action | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
| Regulatory Designation | |
| Partner | Shanghai Fosun Pharmaceutical Group Co. Ltd. |
SBM-TFC-039
PATENT
WO 2012160218
http://www.google.com/patents/WO2012160218A1?cl=en
Examples within this first subclass include but are not limited to:

Synthesis of compound 8
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)

A.

Procedure A:
To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340

The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
C15H14B1CIO M = 325.63 g.mof1

10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)

Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was charged into a three necked flask, followed by addition of a portion of 1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene (2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol % of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the reaction was initiated (exothermic and consuming of Mg), the remaining solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was added dropwise. The mixture was then allowed to react for another one hour under gentle reflux until most of the Mg was consumed.
The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)

Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3, O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous solution of sodium chloride was added to quench the reaction. The mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04, filtrated and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)

Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF, 16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol, leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then refluxed for lh,cooled to 0°C and treated carefully with sodium hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20, 3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture was allowed to react overnight at room temperature (~25°C) before a saturated aqueous solution of ammonium chloride was added to quench the reaction. The mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried over Na2S04, filtered, and concentrated. The residue was purified by silica gel chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the desired compound 13 (1.05g; 43%) as a yellowish oil.
Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)

13 14
Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)

14 15
A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)

o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10% (0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg, 0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The reaction was placed under hydrogen atmosphere and stirred at room temperature for 2h. The reaction mixture was filtered and concentrated before being purified on silica gel chromatography (dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16 (105mg, 83% yield).
Sirona Biochem’s SGLT Inhibitor Performs Better Than Johnson and Johnson’s SGLT Inhibitor, According to Study
Vancouver, British Columbia – December 7, 2012 – Sirona Biochem Corp. (TSX-V: SBM), announced its sodium glucose transporter (SGLT) inhibitor for Type 2 diabetes reduced blood glucose more effectively than Johnson and Johnson’s canagliflozin, an advanced SGLT inhibitor being considered for market approval in Europe and the U.S. Studies compared Sirona Biochem’s SGLT Inhibitor, SBM-TFC-039, with canagliflozin and were conducted on Zucker Diabetic Fatty (ZDF) rats.
In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.
![]()
Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64



……………………………………………………………………………….
Shanghai Fosun Pharmaceutical Group Co. Ltd.

![]()
//////
Luseogliflozin, TS 071…………. strongly inhibited SGLT2 activity,

LUSEOGLIFLOZIN, CAS 898537-18-3
An antidiabetic agent that inhibits sodium-dependent glucose cotransporter 2 (SGLT2).
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol
(1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
Taisho Pharmaceutical Co., Ltd
Taisho (Originator), PHASE 3
Click to access 2013041801-e.pdf
TS-071
| Taisho Pharmaceutical Holdings Co. Ltd. | |
| Description | Oral sodium-glucose cotransporter 2 (SGLT2) inhibitor |

TS-071, an SGLT-2 inhibitor, is in phase III clinical development at Taisho for the oral treatment of type 1 and type 2 diabetes
In 2012, the product was licensed to Novartis and Taisho Toyama Pharmaceutical by Taisho in Japan for comarketing for the treatment of type 2 diabetes.
Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels. The number of people with diabetes worldwide is expected to rise from 285 million in 2010 to 438 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic β-cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
Diabetes which results from complete deficiency in insulin secretion is Type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is Type 2 diabetes. Type 2 diabetes (Non insulin dependent diabetes) accounts for 90-95 % of all diabetes. An early defect in Type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes. A key component of the pathophysiology of Type 2 diabetes mellitus involves an impaired pancreatic β-cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The β-cell compensates for insulin resistance by increasing the insulin secretion, eventually resulting in reduced β-cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in Type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith. Various antidiabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity). These therapies have various side effects: biguanides cause lactic acidosis, sulfonylurea compounds cause significant hypoglycemia, a-glucosidase inhibitors cause abdominal bloating and diarrhea, and thiazolidinediones cause edema and weight gain. Recently introduced line of therapy includes inhibitors of dipeptidyl peptidase-IV (DPP-IV) enzyme, which may be useful in the treatment of diabetes, particularly in Type 2 diabetes. DPP-IV inhibitors lead to decrease in inactivation of incretins glucagon like peptide- 1 (GLP-1) and gastric inhibitory peptide (GIP), thus leading to increased production of insulin by the pancreas in a glucose dependent manner. All of these therapies discussed, have an insulin dependent mechanism.
Another mechanism which offers insulin independent means of reducing glycemic levels, is the inhibition of sodium glucose co-transporters (SGLTs). In healthy individuals, almost 99% of the plasma glucose filtered in the kidneys is reabsorbed, thus leading to only less than 1% of the total filtered glucose being excreted in urine. Two types of SGLTs, SGLT-1 and SGLT-2, enable the kidneys to recover filtered glucose. SGLT-1 is a low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose ” transporter, located mainly in the S 1 segment of the proximal tubule of the kidney. SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases. The inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
Humans lacking a functional SGLT-2 gene appear to live normal lives, other than exhibiting copious glucose excretion with no adverse effects on carbohydrate metabolism. However, humans with SGLT-1 gene mutations are unable to transport glucose or galactose normally across the intestinal wall, resulting in condition known as glucose-galactose malabsorption syndrome.
Hence, competitive inhibition of SGLT-2, leading to renal excretion of glucose represents an attractive approach to normalize the high blood glucose associated with diabetes. Lower blood glucose levels would, in turn, lead to reduced rates of protein glycation, improved insulin sensitivity in liver and peripheral tissues, and improved cell function. As a consequence of progressive reduction in hepatic insulin resistance, the elevated hepatic glucose output which is characteristic of Type 2 diabetes would be expected to gradually diminish to normal values. In addition, excretion of glucose may reduce overall caloric load and lead to weight loss. Risk of hypoglycemia associated with SGLT-2 inhibition mechanism is low, because there is no interference with the normal counter regulatory mechanisms for glucose.
The first known non-selective SGLT-2 inhibitor was the natural product phlorizin
(glucose, 1 -[2-P-D-glucopyranosyloxy)-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)- 1 – propanone). Subsequently, several other synthetic analogues were derived based on the structure of phlorizin. Optimisation of the scaffolds to achieve selective SGLT-2 inhibitors led to the discovery of several considerably different scaffolds.
C-glycoside derivatives have been disclosed, for example, in PCT publications
W.O20040131 18, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033, WO2007136116, WO2007000445, WO2007093610, WO2008069327, WO2008020011, WO2008013321, WO2008013277, WO2008042688, WO2008122014, WO2008116195, WO2008042688, WO2009026537, WO2010147430, WO2010095768, WO2010023594, WO2010022313, WO2011051864, WO201 1048148 and WO2012019496 US patents US65151 17B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630. The compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb’s “Dapagliflozin” with Formula A, Mitsubishi Tanabe and Johnson & Johnson’s “Canagliflozin” with Formula B, Lexicon’s “Lx-421 1″ with Formula C, Boehringer Ingelheim and Eli Lilly’s “Empagliflozin” with Formula D, Roche and Chugai’s “Tofogliflozin” with Formula E, Taisho’s “Luseogliflozin” with Formula F, Pfizer’ s “Ertugliflozin” with Formula G and Astellas and Kotobuki’s “Ipragliflozin” with Formula H.

Formula G Formula H
In spite of all these molecules in advanced stages of human clinical trials, there is still no drug available in the market as SGLT-2 inhibitor. Out of the potential candidates entering the clinical stages, many have been discontinued, emphasizing the unmet need. Thus there is an ongoing requirement to screen more scaffolds useful as SGLT-2 inhibitors that can have advantageous potency, stability, selectivity, better half-life, and/ or better pharmacodynamic properties. In this regard, a novel class of SGLT-2 inhibitors is provided herein
………………………
SYNTHESIS
- Example 5

Synthesis of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose
-
Five drops of 1,2-dibromoethane were added to a mixture of magnesium (41 mg, 1.67 mmol), 1-bromo-3-(4-ethoxybenzyl)-6-methoxy-4-methylbenzene (0.51 g, 1.51 mmol) and tetrahydrofuran (2 mL). After heated to reflux for one hour, this mixture was allowed to stand still to room temperature to prepare a Grignard reagent. A tetrahydrofuran solution (1.40 mL) of 1.0 M i-propyl magnesium chloride and the prepared Grignard reagent were added dropwise sequentially to a tetrahydrofuran (5 mL) solution of 2,3,4,6-tetra-O-benzyl-5-thio-D-glucono-1,5-lactone (0.76 g, 1.38 mmol) while cooled on ice and the mixture was stirred for 30 minutes. After the reaction mixture was added with a saturated ammonium chloride aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate =4:1) to obtain (0.76 g, 68%) a yellow oily title compound.
1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.37 (t, J=6.92 Hz, 3 H) 2.21 (s, 3 H) 3.51 – 4.20 (m, 12 H) 3.85 – 3.89 (m, 3 H) 4.51 (s, 2 H) 4.65 (d, J=10.72 Hz, 1 H) 4.71 (d, J=5.75 Hz, 1 H) 4.78 – 4.99 (m, 3 H) 6.59 – 7.43 (m, 26 H)
Example 6
-
[0315]


Synthesis of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol
-
An acetonitrile (18 mL) solution of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose (840 mg, 1.04 mmol) was added sequentially with Et3SiH (0.415 mL, 2.60 mmol) and BF3·Et2O (0.198 mL, 1.56 mmol) at -18°C and stirred for an hour. After the reaction mixture was added with a saturated sodium bicarbonate aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and then dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=4:1) to obtain the title compound (640 mg, 77%).
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.35 (t, J=6.88 Hz, 3 H) 2.21 (s, 3 H) 3.02 – 3.21 (m, 1 H) 3.55 (t,J=9.40 Hz, 1 H) 3.71 (s, 1 H) 3.74 – 3.97 (m, 10 H) 4.01 (s, 1 H) 4.45 – 4.56 (m, 3 H) 4.60 (d, J=10.55 Hz, 2 H) 4.86 (s, 2 H) 4.90 (d, J=10.55 Hz, 1H) 6.58 – 6.76 (m, 5 H) 6.90 (d, J=7.34 Hz, 1 H) 7.09 – 7.19 (m, 5 H) 7.23 – 7.35 (m, 15 H).
ESI m/z = 812 (M+NH4).
Example 7

Synthesis of (1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
-
A mixture of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol (630 mg, 0.792 mmol), 20% palladium hydroxide on activated carbon (650 mg) and ethyl acetate (10 mL) – ethanol (10 mL) was stirred under hydrogen atmosphere at room temperature for 66 hours. The insolubles in the reaction mixture were filtered off with celite and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography (chloroform:methanol =10:1) to obtain a colorless powdery title compound (280 mg, 81%) as 0.5 hydrate. 1H NMR (600 MHz, METHANOL- d4) δ ppm 1.35 (t, J=6.9 Hz, 3 H) 2.17 (s, 3 H) 2.92 – 3.01 (m, 1 H) 3.24 (t, J=8.71 Hz, 1 H) 3.54 – 3.60 (m, 1 H) 3.72 (dd, J=11.5, 6.4 Hz, 1 H) 3.81 (s, 3 H) 3.83 (s, 2 H) 3.94 (dd, J=11.5, 3.7 Hz, 1 H) 3.97 (q, J=6.9 Hz, 2 H) 4.33 (s, 1 H) 6.77 (d, J=8.3 Hz, 2 H) 6.76 (s, 1 H) 6.99 (d, J=8.3 Hz, 2 H) 7.10 (s, 1 H). ESI m/z = 452 (M+NH4+), 493 (M+CH3CO2-). mp 155.0-157.0°C. Anal. Calcd for C23H30O6S·0.5H2O: C, 62.28; H, 7.06. Found: C, 62.39; H, 7.10.
………………………………..
PAPER
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (TS-071) is a Potent, Selective Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for Type 2 Diabetes Treatment
(Journal of Medicinal Chemistry) Saturday March 20th 2010
Author(s): ,
DOI:10.1021/jm901893x
GO TO: [Article]
http://pubs.acs.org/doi/abs/10.1021/jm901893x

(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (3p)
 3p is compd
3p is compd
| cmpds | R1 | R2 | R3 | SGLT2 (nM) mean (95% CI) | SGLT1 (nM) mean (95% CI) | T1/T2 selectivity |
|---|---|---|---|---|---|---|
| 1 | 27.8 (21.8−35.3) | 246 (162−374) | 8.8 | |||
| 3a | H | H | OEt | 73.6 (51.4−105) | 26100 (20300−33700) | 355 |
| 3b | H | OH | OEt | 283 (268−298) | 14600 (11500−18500) | 51.6 |
| 3c | H | OMe | OEt | 13.4 (11.3−15.8) | 565 (510−627) | 42.2 |
| 3d | H | F | OEt | 9.40 (5.87−15.0) | 7960 (7180−8820) | 847 |
| 3e | H | Me | OEt | 2.29 (1.76−2.99) | 671 (230−1960) | 293 |
| 3f | H | Cl | OEt | 1.77 (0.95−3.30) | 1210 (798−1840) | 684 |
| 3g | OH | H | OEt | 17.4 (15.9−19.0) | 4040 (1200−13600) | 232 |
| 3h | OMe | H | OEt | 37.9 (26.4−54.4) | 100000 (66500−151000) | 2640 |
| 3i | OMe | OMe | OEt | 10.8 (6.84−17.1) | 4270 (1560−11600) | 395 |
| 3j | H | Cl | OMe | 1.68 (1.08−2.60) | 260 (72.5−931) | 155 |
| 3k | H | Cl | Me | 1.37 (0.97−1.95) | 209 (80.2−545) | 153 |
| 3l | H | Cl | Et | 1.78 (0.88−3.63) | 602 (473−767) | 338 |
| 3m | H | Cl | iPr | 4.01 (1.75−9.17) | 8160 (4860−13700) | 2040 |
| 3n | H | Cl | tBu | 18.8 (11.0−32.1) | 35600 (31900−39800) | 1890 |
| 3o | H | Cl | SMe | 1.16 (0.73−1.85) | 391 (239−641) | 337 |
| 3p | OMe | Me | OEt | 2.26 (1.48−3.43) | 3990 (2690−5920) | 1770 |
| 3q | OMe | Me | Et | 1.71 (1.19−2.46) | 2830 (1540−5200) | 1650 |
| 3r | OMe | Me | iPr | 2.68 (2.15−3.34) | 17300 (14100−21100) | 6400 |
| 3s | OMe | Cl | Et | 1.51 (0.75−3.04) | 3340 (2710−4110) | 2210 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2004014930A1 * | Aug 8, 2003 | Feb 19, 2004 | Asanuma Hajime | PROCESS FOR SELECTIVE PRODUCTION OF ARYL 5-THIO-β-D- ALDOHEXOPYRANOSIDES |
| Reference | ||
|---|---|---|
| 1 | * | AL-MASOUDI, NAJIM A. ET AL: “Synthesis of some novel 1-(5-thio-.beta.-D-glucopyranosyl)-6-azaur acil derivatives. Thio sugar nucleosides” NUCLEOSIDES & NUCLEOTIDES , 12(7), 687-99 CODEN: NUNUD5; ISSN: 0732-8311, 1993, XP008091463 |
| 2 | * | See also references of WO2006073197A1 |
| EP2419097A1 * | Apr 16, 2010 | Feb 22, 2012 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2455374A1 * | Oct 15, 2009 | May 23, 2012 | Janssen Pharmaceutica N.V. | Process for the Preparation of Compounds useful as inhibitors of SGLT |
| EP2601949A2 * | Apr 16, 2010 | Jun 12, 2013 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2668953A1 * | May 15, 2009 | Dec 4, 2013 | Bristol-Myers Squibb Company | Pharmaceutical compositions comprising an SGLT2 inhibitor with a supply of carbohydrate and/or an inhibitor of uric acid synthesis |
| WO2009143020A1 | May 15, 2009 | Nov 26, 2009 | Bristol-Myers Squibb Company | Method for treating hyperuricemia employing an sglt2 inhibitor and composition containing same |
| WO2010043682A2 * | Oct 15, 2009 | Apr 22, 2010 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of sglt |
| WO2010119990A1 | Apr 16, 2010 | Oct 21, 2010 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| WO2013152654A1 * | Mar 14, 2013 | Oct 17, 2013 | Theracos, Inc. | Process for preparation of benzylbenzene sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
-
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin regulatory update 05/13/2013
Week in Review, RegulatoryTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Last month, Taisho’s Taisho Pharmaceutical Co. Ltd. subsidiary submitted a regulatory … -
Strategy: Doubling down in diabetes 05/06/2013
Merck shoring up slowing diabetes franchise with Pfizer, Abide dealsBioCentury on BioBusiness, StrategyAs sales flatten for Merck’s sitagliptin franchise and a new class of oral diabetes drugs comes to market, the pharma has tapped Pfizer and Abide to shore up its position.
see
SEE
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-132352


ANTHONYFLOZIN………Find one if you can in this review

find here
http://medcheminternational.blogspot.in/p/flozin-series.html
1 TOFOGLIFLOZIN
2 SERGLIFLOZIN
3 DAPAGLIFLOZIN
4 IPRAGLIFLOZIN
5 EMPAGLIFLOZIN
6 LUSEOGLIFLOZIN
7 REMOGLIFLOZIN
8 ERTUGLIFLOZIN
9 SOTAGLIFLOZON

 DR ANTHONY
DR ANTHONY
BLOGS………





Sotagliflozin, LX 4211 in phase 2 For type 1, 2 diabetes
LX 4211, Sotagliflozin, LP-802034 , lex 1287
UNII-6B4ZBS263Y
Methyl (5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-thio-beta-L-xylopyranoside
β-L-Xylopyranoside, methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-, (5S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol,
(5S)-Methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-beta-L-xylopyranoside
1018899-04-1
C21H25ClO5S, 424.94, LP-802034
LX-4211 is a dual SGLT2/1 inhibitor; Antidiabetic agents.
LX-4211 is a SGLT-2 inhibitor being evaluated in phase II clinical studies at Lexicon Pharmaceuticals for the oral treatment of type 2 diabetes.



Summary
- Co-administration of LX4211 led to a nearly one-third reduction in mealtime insulin for Type 1 diabetics.
- Although there was no reduction in basal insulin use, the LX4211 group saw better glucose control, lower HbA1c, and weight loss.
- Partnering LX4211 is still management’s top priority but independent development in Type 1 diabetes is at least an option.
Lexicon Pharmaceuticals (LXRX) continues to generate data on its SGLT-1/2 inhibitor LX4211 that suggest this is an effective and promising medication for treating not only Type 2 diabetes (the common target for non-insulin medications for diabetes), but also Type 1 as well. Lexicon’s most recent update, a small short-term Phase II study in Type 1 diabetics is certainly a positive update, but it’s not what investors really want to see. Lexicon still needs to find a development partner for LX4211 and the ongoing delays don’t help sentiment or the long-term prospects for the drug.
A Potentially Meaningful Addition To Type 1 Care
On Monday morning, Lexicon released top-line data from a small (33-patient) Phase II study of LX4211 in Type 1 diabetics on insulin. The results support the notion that SGLT inhibition can play a valuable role in improving glucose control for Type 1 diabetics.
This small study enrolled generally well-controlled patients (HbA1c levels ranging from 7 to 9, with an average of 7.9) and the addition of LX4211 led to 32% reduction in bolus (mealtime) insulin versus a 6% reduction in the placebo group. Even with the lower bolus insulin, patients in the LX4211 group showed a 0.55% reduction in HbA1c versus a 0.06% reduction in the placebo group. Patients taking LX4211 demonstrated better glucose control (more time spent in the target range of 70-180 mg/dL) and saw a 1.7kg weight loss versus a 0.5kg weight gain in the placebo group
……………………..
Scheme 1 :




3(a) 3(b)


4(a) 4(b)

Scheme 3:

…………………
http://www.google.com/patents/EP2332947A1?cl=en
EXAMPLES
-
Aspects of this invention can be understood from the following examples.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone
-
To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt% water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86% yield, 96.7 area% pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 – 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73.
-
To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H).
-
To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahvdrofuro[2.3-d][1,3]dioxol-5-yl)(morpholino)methanone
-
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL),∼1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25°C. Additional charges of TEMPO (14.3g, 0.5 mol%) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to < 10°C and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15°C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L x 3). The combined organic layer was concentrated under vacuo (∼100-120 torr) at < 35°C (28-32°C vapor, 45-50°C bath) to low volume (- 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached < 1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5°C and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ5.96 (d, J = 3.6 Hz, 1H), 4.5 8 (d, J = 3.6 Hz, 1H), 4.53 (d, J =3.2Hz,1H), 4.30 (d, J= 3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) 8 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25. 1.
-
The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (-80 L) to remove excess morpholine. When final volume reached -12 L, heptane (14 L) was added slowly at 60-70°C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136°C (DSC). 1H NMR (CDCl3), δ 6.02 (d, J = 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:

6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
-
A 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF = 0.003 wt% water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step.
-
A jacketed 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF = 0.003 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 6°C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9°C. The resulting orange solution was diluted with dichloromethane (75mL) and was cooled to -7°C. Then a solution of 2-chloro-5-iodobenzoyl chloride (≤ 0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3°C. The reaction mixture was warmed slightly and held at +5°C for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450mL pre-cooled (∼5°C) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10min with internal temperature remaining below 28°C. The quenched biphasic mixture was stirred at 20°C for 45min and the lower organic phase was washed with 1N aq. HCl (200mL), twice with saturated aq sodium bicarbonate (200mL per wash), and with saturated aq sodium chloride (200mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93g, 99.0 area% by HPLC at 220nm, 1.0 area% regioisomer at 200nm, 98.5 % “as-is” yield).
-
A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300mL, KF = 0.004 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 5°C.Then triethylsilane (28mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24mL, 194.46mmo1,2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150mL) followed by saturated aq sodium bicarbonate (150mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq sodium bicarbonate (100 mL), and with saturated aq sodium chloride (50mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45°C was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45°C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
-
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500mag, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5°C, and the mixture was stirred for 1.5 h at 0-5°C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5°C and the mixture was kept stirring for 1h, warmed to 20°C and stirred at 20°C for 2 hours. The reaction was quenched with saturated aq NH4CI, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7. 88 (dd, J= 8.4, 2.0 Hz, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.12 (d, J= 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.07 (d, J = 3.2 Hz, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.16 (d, J = 7.2 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone
-
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4X to the morpholinoamide) at room temperature and cooled to -5°C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at -5°C over 3 hours. This Grignard solution was used in the ketone formation below.
-
[0055]To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity = 97 wt%, 2.01 kg, 7.34 mol) and THF (11 L, 5.5X) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30°C. The homogeneous solution was then cooled to – 25°C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at -25°C over 3 hours. Then the above Grignard solution was added to this solution at -20 over 41 minutes. The resulting solution was further stirred at -20°C before quench. The reaction mixture was added to 10 wt% aqueous NH4Cl (10 L, 5X) at 0°C with vigorous stirring, and stirred for 30 minutes at 0°C. To this mixture was added slowly 6 N HCl (4 L, 2X) at 0°C to obtain a clear solution and stirred for 30 minutes at 10°C. After phase split, the organic layer was washed with 25 wt% aq NaCl (5 L, 2.5X). Then the organic layer was concentrated to a 3X solution under the conditions (200 mbar, bath temp 50°C). EtOAc (24 L, 12X) was added, and evaporated to a 3X solution under the conditions (150 mbar, bath temp 50°C). After removed solids by a polish filtration, EtOAc (4 L, 2X) was added and concentrated to dryness (150 mbar, bath temp 50°C). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70°C to obtain a 2.5X homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5X) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5X) was added slowly to a little cloudy solution at 70°C. After stirred for 0.5 h at 70°C, the suspension was slowly cooled to 60°C and stirred for 1 h at 60°C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4X), dried under vacuum at 45°C to give the desired ketone as fluffy solids (2.57 kg, 100 wt% by HPLC, purity-adjusted yield: 81%).
(2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)-((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17X) was added CeCl3.7H2O (118.5g, 1.2 equiv) and the mixture was stirred at 20°C until all solids were dissolved. The mixture was then cooled to -78°C and NaBH4 (12.03g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed -70°C. The mixture was stirred at – 78°C for 1 hour, slowly warmed to 0°C and quenched with saturated aq NH4Cl (550 mL, 5X). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1L, 10X x2) and washed with brine (550 mL, 5X). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100°C and stirred for 15 hours. The mixture was then cooled to room temperature (20°C) and concentrated under vacuum to give a yellow oil (crude, ∼118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0°C. Then, Ac2O (195 mL, -8.0 equiv) was added and the mixture was warmed to 20°C and stirred at 20°C for 2h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, x2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (-133g).
-
To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4X) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80°C for 3.5 hours. The mixture was cooled to 20°C and Mel (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20°C for 3h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1.3 L, 10X) and washed with H2O (650 mL, 5X x2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5X) and the mixture was reslurried at 60°C for 2h and then cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 70 mL, x3). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) δ 7.37 (d, J= 8.0 Hz, 1H), 7.20 (dd, J= 8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J = 9.6 Hz, 1H), 5.20 (t, J = 9.6 Hz, 1H), 5.05 (t, J= 9.6 Hz, 1H), 4.51 (d, J=9.6Hz, 1H), 4.38 (d, J= 9.6Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2.11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19X). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25°C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt% aq NH4Cl (2.5 L, 1X) and the mixture was concentrated under vacuum to 5X, diluted with water (10 L, 4X) and MTBE (12.5L, 5X). The mixture was cooled to 10°C and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2X). The combined aqueous layer was extracted with MTBE (12.5 L, 5X). The combined organic layers were washed with brine (2.5 L, 1X) and concentrated under vacuum to 3X. MeCN (15 L, 6X) was added. The mixture was concentrated again to 10 L (4X) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
-
The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol% aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80°C for 2 hours and then cooled to 20°C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2X) and diluted with MTBE (15 L, 6X). The organic layer was separated, washed with brine (5 L, 2X) and concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the mixture was concentrated to 7.5 L (3X).
-
The above MeCN solution of (3S,4R,SR,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10°C, added with dimethylaminopyridine (17.53 g, 2.5 mol%), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2X, 6.0 equiv) so that the temperature of the mixture was kept below 20°C. The reaction was then warmed to 20°C and stirred for 1 hour and diluted with MTBE (15 L, 6X). The mixture was slowly quenched with water (7.5 L, 3X). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2X), 1 N NaHSO4 (5 L, 2X), and brine (5 L, 2X) in sequence.
-
The organic layer was then concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the solution was concentrated to 7.5 L (3X) (KF = 0.08%). Dioxane (12.5 L, 5X) was added and the solution was concentrated to 7.50 L (3X) (KF = 0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL).
-
To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80°C for 3 hours (>97% conversion). The mixture was cooled to 20°C and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20°C for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20°C for 1 hours. The mixture was then diluted with MTBE (25 L, 10X) and washed with water (12.5 L, 5X x2). The organic layer was separated and concentrated under vacuum to -5 L (2X). MeOH (12.5 L, 5X) was added and the mixture was concentrated to 5X to afford a slurry. The mixture was then heated at 60°C for 1 hour and cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 2.5 L, 1X x2, 1.0 L, 0.4X). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
-
To a slurry of (2S,3S,4R,SS,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mo1) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 20°C and the mixture was stirred at 20°C for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.0g, 95%). 1H NMR (CDCl3) δ 7.38 (d, J = 8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, 1H), 3.50 (m, 2H), 3.42 (br s, 1H), 2.95 (br s, 1H), 2.57 (br s, 1H), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
…………
http://www.google.com/patents/WO2010009197A1?cl=en
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H- pyran-3,4,5-triol:

LEX-1287 The compound is an inhibitor of the sodium glucose co-transporter 2, and may be useful in the treatment of diabetes and a variety of other diseases and conditions. See U.S. patent application no. 11/862,690, filed September 28, 2007.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran-3,4,5-triol To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-
(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mol) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. The mixture was then
18
LEX-1287 concentrated to 300 mL, added to H2O (IL) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.Og, 95%). IH NMR (CDC13) δ 7.38 (d, J = 8.4 Hz, IH), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, IH), 4.15 (d, J = 9.6 Hz, IH), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, IH), 3.50 (m, 2H), 3.42 (br s, IH), 2.95 (br s, IH), 2.57 (br s, IH), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
6.9. Preparation of Crystalline Anhydrous (2S,3R,4R,5S,6R)-2-(4-chloro-
3-f4-ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran- 3,4,5-triol Form 1
Under slightly positive nitrogen pressure, to a 50 L reactor was charged MeOH (12 L) and the triacetate (1.70 Kg, 3.09 mol). Methanol (5L) was added as a rinse. The slurry was then added NaOMe in MeOH (25 wt%, 340 mL, 0.2X) in 15 minutes at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. To the mixture was added slowly water (25.5 L, 15X) in 45 minutes with 5 g seeding (DSC123°C). Solids crashed out and the mixture was stirred at 200C for 1 hour, cooled to 00C and stirred for 30 minutes. The solid was filtered and washed with water (1.7 L, IX, x2) and the cake was dried under vacuum at 45°C overnight to afford the title compound (m.p. ~ 123 0C by DSC peak; 1.28 Kg, 97.7% yield).
…………..
http://www.google.com/patents/US20090030198

EXAMPLES
Aspects of this invention can be understood from the following examples, which do not limit its scope.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone

To a 12 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(−)-xylose (504.40 g, 3.360 mol), acetone (5 L, reagent grade) and anhydrous MgSO4 powder (811.23 g, 6.740 mol/2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol/0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24° C. over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH =8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25 L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt % water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23 g, 86% yield, 96.7 area % pure by GC). 1H NMR (400 MHz, DMSO-d6) δ 1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93-4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 3C NMR (101 MHz, DMSO-d6) δ 26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0 g, 131 mmol) in acetone (375 mL, 15×) and H2O (125 mL, 5×) was added NaHCO3 (33.0 g, 3.0 equiv), NaBr (2.8 g, 20 mol %) and TEMPO (0.40 g, 2 mol %) at 20° C. The mixture was cooled to 0-5° C. and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20° C. for 24h. Methanol (20 mL) was added and the mixture was stirred at 20° C. for 1 h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2×). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12× ×3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5×) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0 g, 79%).1H NMR (methanol-d4), δ 6.00 (d, J=3.2 Hz, 1H), 4.72 d, J=3.2 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.38 (d, J=3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0 g, 24.5 mmol) in THF (100 ML, 20×) was added TBTU (11.8 g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20° C. for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20° C. for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2× ×2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4: 1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10° C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (˜100-120 torr) at <35° C. (28-32° C. vapor, 45-50° C. bath) to low volume (˜6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5° C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) δ 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (˜80 L) to remove excess morpholine. When final volume reached 12 L, heptane (14 L) was added slowly at 60-70° C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136° C. (DSC). 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene

A 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF=0.003 wt % water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step. A jacketed 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF=0.003 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 6° C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9° C. The resulting orange solution was diluted with dichloromethane (75 mL) and was cooled to −7° C. Then a solution of 2-chloro-5-iodobenzoyl chloride (<0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3° C. The reaction mixture was warmed slightly and held at +5° C. for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450 mL pre-cooled (˜5° C.) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10 min with internal temperature remaining below 28° C. The quenched biphasic mixture was stirred at 20° C. for 45 min and the lower organic phase was washed with 1N aq. HCl (200 mL), twice with saturated aq. sodium bicarbonate (200 mL per wash), and with saturated aq. sodium chloride (200 mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93 g, 99.0 area % by HPLC at 220 nm, 1.0 area % regioisomer at 200 nm, 98.5 % “as-is” yield). A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300 mL, KF=0.004 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 5° C. Then triethylsilane (28 mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24 mL, 194.46 mmol, 2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30 min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150 mL) followed by saturated aq sodium bicarbonate (150 mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq. sodium bicarbonate (100 mL), and with saturated aq. sodium chloride (50 mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45° C. was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45° C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220 nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone

To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500 mg, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5° C., and the mixture was stirred for 1.5 h at 0-5° C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5° C. and the mixture was kept stirring for 1 h, warmed to 20° C. and stirred at 20° C. for 2 hours. The reaction was quenched with saturated aq NH4Cl, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7.88 (dd, J=8.4, 2.0 Hz, 1H), 7.82 (d, J=2.0 Hz, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.12 (d, J=8.4 Hz, 2H), 6.86 (d, J=8.4 Hz, 2H), 6.07 (d, J=3.2 Hz, 1H), 5.21 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 4.56 (d, J=3.2 Hz, 1H), 4.16 (d, J=7.2 Hz, 2H), 4.03 (q, J=7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J=7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d]1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4× to the morpholinoamide) at room temperature and cooled to −5° C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at −5° C. over 3 hours. This Grignard solution was used in the ketone formation below. To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity=97 wt %, 2.01 kg, 7.34 mol) and THF (11 L, 5.5×) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30° C. The homogeneous solution was then cooled to −25° C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at −25° C. over 3 hours. Then the above Grignard solution was added to this solution at −20 over 41 minutes. The resulting solution was further stirred at −20° C. before quench. The reaction mixture was added to 10 wt % aqueous NH4Cl (10 L, 5×) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C. To this mixture was added slowly 6 N HCl (4 L, 2×) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C. After phase split, the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5×). Then the organic layer was concentrated to a 3× solution under the conditions (200 mbar, bath temp 50° C.). EtOAc (24 L, 12×) was added, and evaporated to a 3× solution under the conditions (150 mbar, bath temp 50° C.). After removed solids by a polish filtration, EtOAc (4 L, 2×) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5× homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5×) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5×) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C. and stirred for 1 h at 60° C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4×), dried under vacuum at 45° C. to give the desired ketone as fluffy solids (2.57 kg, 100 wt % by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate

To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17×) was added CeCl3.7H2O (118.5 g, 1.2 equiv) and the mixture was stirred at 20° C. until all solids were dissolved. The mixture was then cooled to −78° C. and NaBH4 (12.03 g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed −70° C. The mixture was stirred at −78° C. for 1 hour, slowly warmed to 0° C. and quenched with saturated aq NH4Cl (550 mL, 5×). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1 L, 10× ×2) and washed with brine (550 mL, 5×). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115 g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100° C. and stirred for 15 hours. The mixture was then cooled to room temperature (20° C.) and concentrated under vacuum to give a yellow oil (crude, 118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0° C. Then, Ac2O (195 mL, ˜8.0 equiv) was added and the mixture was warmed to 20° C. and stirred at 20° C. for 2 h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, ×2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (˜133 g). To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4×) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80° C. for 3.5 hours. The mixture was cooled to 20° C. and MeI (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20° C. for 3 h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1. 3 L, 10×) and washed with H2O (650 mL, 5× ×2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5×) and the mixture was reslurried at 60° C. for 2 h and then cooled to 0C and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 70 mL, ×3). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) 6 7.37 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J=9.6 Hz, 1H), 5.20 (t, J=9.6 Hz, 1H), 5.05 (t, J =9.6 Hz, 1H), 4.51 (d, J =9.6 Hz, 1H), 4.38 (d, J=9.6 Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2. 11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J=7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19×). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25° C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt % aq NH4Cl (2.5 L, 1×) and the mixture was concentrated under vacuum to 5×, diluted with water (10 L, 4×) and MTBE (12.5 L, 5×). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2×). The combined aqueous layer was extracted with MTBE (12.5 L, 5×). The combined organic layers were washed with brine (2.5 L, 1×) and concentrated under vacuum to 3×. MeCN (15 L, 6×) was added. The mixture was concentrated again to 10 L (4×) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN. The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80° C. for 2 hours and then cooled to 20° C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2×) and diluted with MTBE (15 L, 6×). The organic layer was separated, washed with brine (5 L, 2×) and concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the mixture was concentrated to 7.5 L (3×). The above MeCN solution of (3S,4R,5R,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10° C., added with dimethylaminopyridine (17.53 g, 2.5 mol %), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2×, 6.0 equiv) so that the temperature of the mixture was kept below 20° C. The reaction was then warmed to 20° C. and stirred for 1 hour and diluted with MTBE (15 L, 6×). The mixture was slowly quenched with water (7.5 L, 3×). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2×), 1 N NaHSO4 (5 L, 2×), and brine (5 L, 2×) in sequence. The organic layer was then concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the solution was concentrated to 7.5 L (3×) (KF=0.08%). Dioxane (12.5 L, 5×) was added and the solution was concentrated to 7.50 L (3×) (KF=0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL). To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80° C. for 3 hours (>97% conversion). The mixture was cooled to 20° C. and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20° C. for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20° C. for 1 hours. The mixture was then diluted with MTBE (25 L, 10×) and washed with water (12.5 L, 5× ×2). The organic layer was separated and concentrated under vacuum to ˜5 L (2×). MeOH (12.5 L, 5×) was added and the mixture was concentrated to 5× to afford a slurry. The mixture was then heated at 60° C. for 1 hour and cooled to 0° C. and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 2.5 L, 1× ×2, 1.0 L, 0.4×). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol

To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0. 164 mol) in MeOH (900 mL, 10×) was added NaOMe in MeOH (25 wt %, 18 mL, 0.2×) at 20° C. and the mixture was stirred at 20° C. for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1 L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, ×3) and the cake was dried under vacuum at 45° C. overnight to afford the desired methyl thiolate (67.0 g, 95%). 1H NMR (CDCl3) 6 7.38 (d, J=8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J=8.8 Hz, 2H), 6.83 (d, J=8.8 Hz, 2H), 4.35 (d, J=9.6 Hz, 1H), 4.15 (d, J=9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J=8.8 Hz, 1H), 3.50 (m, 2H), 2.73 (br s, 3H), 2.17 (s, 3H), 1.40 (t, J=7.2 Hz, 3H).
…………..
SGLT inhibitors: a novel target for diabetes.
Kanwal A, Banerjee SK.
Pharm Pat Anal. 2013 Jan;2(1):77-91. doi: 10.4155/ppa.12.78.
clinical trials………..http://clinicaltrials.gov/search/intervention=LX-4211+OR+LX4211
On the importance of synthetic organic chemistry in drug discovery: reflections on the discovery of antidiabetic agent ertugliflozin. , Med. Chem. Commun., 2013, 4, 101
ERTUGLIFLOZIN

ERTUGLIFLOZIN, PFIZER
THERAPEUTIC CLAIM Treatment of type 2 diabetes
CHEMICAL NAMES
1. β-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-(hydroxymethyl)-
2. (1S,2S,3S,4R,5S)-5-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
PF-04971729, MK 8835
M. Wt: 436.88
Formula: C22H25ClO7
CAS No:. 1210344-57-2
Diabetes looms as a threat to human health worldwide. As a result, considerable research efforts are devoted to identify new and efficacious anti-diabetic agents lacking the side effects associated with some of the current drugs (hypoglycemia, weight gain).Inhibition of sodium-dependent glucose cotransporter 2 (SGLT2), a transporter located in the kidney, is a mechanism that promotes glucosuria and therefore, reduction of plasma glucose concentration. Since the mechanism operates in a glucose-dependent and insulin-independent manner, and is associated with weight loss, it has emerged as a very promising approach to the pathophysiologic treatment of type 2 diabetes. Ertugliflozin (PF-04971729), an anti-diabetic agent currently in development (Phase 3 clinical trials) and belonging to a new class of SGLT2 inhibitors bearing a dioxa-bicyclo[3.2.1]octane bridged ketal motif.

SYNTHESIS
Scheme 1 outlines the general procedures one could use to provide compounds of the present invention.

Scheme 1 AIIyI 2,3,4-tιϊ-O-benzyl-D-glucopyranoside (La, where Pg1 is a benzyl group) can be prepared by procedures described by Shinya Hanashima, et al., in Bioorganic & Medicinal Chemistry, 9, 367 (2001 ); Patricia A. Gent et al. in Journal of the Chemical Society, Perkin 1, 1835 (1974); Hans Peter Wessel in the Journal of Carbohydrate Chemistry, 7, 263, (1988); or Yoko Yuasa, et al., in Organic Process Research & Development, 8, 405-407
(2004). In step 1 of Scheme 1 , the hydroxymethylene group can be introduced onto the glycoside by means of a Swern oxidation followed by treatment with formaldehyde in the presence of an alkali metal hydroxide (e.g., sodium hydroxide). This is referred to as an aldol-Cannizzaro reaction. The Swern oxidation is described by Kanji Omura and Daniel Swern in Tetrahedron, 34, 1651 (1978). Modifications of this process known to those of skill in the art may also be used. For example, other oxidants, like stabilized 2- iodoxybenzoic acid described by Ozanne, A. et al. in Organic Letters, 5, 2903 (2003), as well as other oxidants known by those skilled in the art can also be used. The aldol Cannizzaro sequence has been described by Robert Schaffer in the Journal of The American Chemical Society, 81 , 5452 (1959) and Amigues, E.J., et al., in Tetrahedron, 63,10042 (2007).
In step 2 of Scheme 1 , protecting groups (Pg2) can be added by treating intermediate (MD) with the appropriate reagents and procedures for the particular protecting group desired. For example, p-methoxybenzyl (PMB) groups may be introduced by treatment of intermediate (MD) with p-methoxybenzyl bromide or p-methoxybenzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide (DMF). Conditions involving para-methoxybenzyltrichloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. Benzyl (Bn) groups may be introduced by treatment of intermediate (MD) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide. Conditions involving benzylthchloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. In step 3 of Scheme 1 , the allyl protection group is removed (e.g., by treatment with palladium chloride in methanol; cosolvent like dichloromethane may also be used; other conditions known by those skilled in the art could also be used, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991 ) to form the lactol (Ld).
In step 4 of Scheme 1 , oxidation of the unprotected hydroxyl group to an oxo group (e.g., Swern oxidation) then forms the lactone (l-e).
In step 5 of Scheme 1 , the lactone (Le) is reacted with Λ/,O-dimethyl hydroxylamine hydrochloride to form the corresponding Weinreb amide which may exist in equilibrium in a closed/opened form, (l-f/l-g). The “Weinreb amide” (LgJ can be made using procedures well known to those of skill in the art. See, Nahm, S., and S. M. Weinreb, Tetrahedron Letters. 22 (39), 3815-1818 (1981 ). For example, intermediate (l-f/l-α) can be prepared from the commercially available Λ/,O-dimethylhydroxylamine hydrochloride and an activating agent (e.g., trimethylaluminum). In step 6 of Scheme 1 , the arylbenzyl group (Ar) is introduced using the desired organometallic reagent (e.g., organo lithium compound (ArLi) or organomagnesium compound (ArMgX)) in tetrahydrofuran (THF) at a temperature ranging from about -780C to about 2O0C followed by hydrolysis (upon standing in protic conditions) to the corresponding lactol (N) which may be in equilibrium with the corresponding ketone (Ni). The bridged ketal motif found in (A) and (B) can be prepared by removing the protecting groups (Pg2) using the appropriate reagents for the protecting groups employed. For example, the PMB protecting groups may be removed by treatment with trifluoroacetic acid in the presence of anisole and dichloromethane (DCM) at about O0C to about 230C (room temperature). The remaining protecting groups (Pg1) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final products (A) and (B). When R1 is CN, the use of a Lewis acid like boron trichloride at a temperature ranging from about -780C to about room temperature in a solvent like dichloromethane or 1 ,2-dichloroethane may also be used to remove benzyl protective and/or para- methoxybenzyl protective groups. When R1 is CN and R2 is (Ci-C4)alkoxy in intermdediate (l-i) or in products (A) or (B), upon treatment with a Lewis acid such as boron trichloride or boron tribomide, partial to complete de-alkylation to the corresponding phenol may occur to lead to the corresponding compound (A) or (B) where R1 is CN and R2 is OH. If this occurs, the (d- C4)alkoxy group may be re-introduced via selective alkylation using a (CrC4) alkyl iodide under mildly basic conditions, for example, potassium carbonate in acetone at a temperature ranging from about room temperature to about 56 degrees Celsius.
When R1 and/or R2 is (CrC4)alkyl-SO2- it is understood by one skilled in the art that the organometallic addition step 6 (Scheme 1 ) will be carried out on the corresponding (d- C4)alkyl-S- containing organometallic reagent. The thio-alkyl is then oxidized at a later stage to the corresponding sulfone using conventional methods known by those skilled in the art.
The compounds of the present invention may be prepared as co-crystals using any suitable method. A representative scheme for preparing such co-crystals is described in Scheme 2.

Scheme 2
In Scheme 2, wherein Me is methyl and Et is ethyl, in step 1 , 1-(5-bromo-2- chlorobenzyl)-4-ethoxybenzene is dissolved in 3:1 , toluene: tetrahydrofuran followed by cooling the resulting solution to <-70°C. To this solution is added hexyllithium while maintaining the reaction at <-65°C followed by stirring for 1 hour. (3R,4S,5R,6R)-3,4,5- ths(thmethylsilyloxy)-6-((trimethylsilyloxy)methyl)-tetrahydropyran-2-one (ll-a) is dissolved in toluene and the resulting solution is cooled to -150C. This solution is then added to the – 7O0C aryllithium solution followed by stirring for 1 hour. A solution of methanesulfonic acid in methanol is then added followed by warming to room temperature and stirring for 16 to 24 hours. The reaction is deemed complete when the α-anomer level is < 3%. The reaction is then basified by the addition of 5 M aqueous sodium hydroxide solution. The resulting salts are filtered off followed by concentration of the crude product solution. 2- methyltetrahydrofuran is added as a co-solvent and the organic phase is extracted twice with water. The organic phase is then concentrated to 4 volumes in toluene. This concentrate is then added to a 5:1 , heptane: toluene solution causing precipitate to form. The solids are collected and dried under vacuum to afford a solid.
In step 2 of Scheme 2, to (ll-b) in methylene chloride is added imidazole followed by cooling to O0C and then addition of trimethylsilylchlohde to give the persilylated product.
The reaction is warmed to room temperature and quenched by the addition of water, and the organic phase is washed with water. This crude methylene chloride solution of (ll-c) is dried over sodium sulfate and then taken on crude into the next step.
In step 3 of Scheme 2, the crude solution of (ll-c) in methylene chloride is concentrated to low volume and then the solvent is exchanged to methanol. The methanol solution of (ll-c) is cooled to O0C, then 1 mol% of potassium carbonate is added as a solution in methanol followed by stirring for 5 hours. The reaction is then quenched by addition of 1 mol% acetic acid in methanol, followed by warming to room temperature, solvent exchange to ethyl acetate, and then filtration of the minor amount of inorganic solids. The crude ethyl acetate solution of (ll-d) is taken directly into the next step.
In step 4 of Scheme 2, the crude solution of (ll-d) is concentrated to low volume, then diluted with methylene chloride and dimethylsulfoxide. Triethylamine is added followed by cooling to 1O0C and then sulfur trioxide pyridine complex is added in 3 portions as a solid at 10 minute intervals. The reaction is stirred an additional 3 hours at 1O0C before quenching with water and warming to room temperature. The phases are separated followed by washing the methylene chloride layer with aqueous ammonium chloride. The crude methylene chloride solution of (ll-e) is taken directly into the next step.
In step 5 of Scheme 2, the crude solution of (ll-e) is concentrated to low volume and then the solvent is exchanged to ethanol. Thirty equivalents of aqueous formaldehyde is added followed by warming to 550C. An aqueous solution of 2 equivalents of potassium phosphate, tribasic is added followed by stirring for 24 hours at 550C. The reaction temperature is then raised to 7O0C for an additional 12 hours. The reaction is cooled to room temperature, diluted with te/t-butyl methyl ether and brine. The phases are separated followed by solvent exchange of the organic phase to ethyl acetate. The ethyl acetate phase is washed with brine and concentrated to low volume. The crude concentrate is then purified by silica gel flash chromatography eluting with 5% methanol, 95% toluene. Product containing fractions are combined and concentrated to low volume.
Methanol is added followed by stirring until precipitation occurs. The suspension is cooled and the solids are collected and rinsed with heptane followed by drying. Product (ll-f) is isolated as a solid.
In step 6 of Scheme 2, compound (ll-f) is dissolved in 5 volumes of methylene chloride followed by the addition of 1 mol% SiliaBonc/® tosic acid and stirring for 18 hours at room temperature. The acid catalyst is filtered off and the methylene chloride solution of (ll-g) is taken directly into the next step co-crystallization procedure.
In step 7 of Scheme 2, the methylene chloride solution of (ll-g) is concentrated and then the solvent is exchanged to 2-propanol. Water is added followed by warming to 550C. An aqueous solution of L-pyroglutamic acid is added followed by cooling the resulting solution to room temperature. The solution is then seeded and granulated for 18 hours. After cooling, the solids are collected and rinsed with heptane followed by drying. Product (ll-h) is isolated as a solid.
An alternative synthesis route for compounds (A) of the present invention is depicted in Scheme 3 and described below.

Scheme 3
The synthesis of (lll-a), where R3 is an alkyl or fluoro substituted alkyl (except for the carbon adjacent to the oxygen atom) can be prepared in a similar way as described in step 1 of Scheme 2. In step 1 of Scheme 3, the primary hydroxyl group is selectively protected by an appropriate protective group. For example, a trityl group (Pg3 = Tr) can be introduced by treatment of intermediate (lll-a) with chlorotriphenylmethane in presence of a base like pyridine in a solvent like toluene, tetrahydrofuran or dichloromethane at a temperature ranging from about 0 degrees Celsius to about room temperature. Additional examples of such protective groups and experimental conditions are known by those skilled in the art and can be found in T. W. Greene, Protective Groups in Organic Synthesis. John Wiley & Sons, New York, 1991.
In step 2 of Scheme 3, the secondary hydroxyl groups can be protected by the appropriate protecting groups. For example, benzyl groups (Pg4 is Bn) can be introduced by treatment of intermediate (lll-b) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius. Acetyl or benzoyl groups (Pg4 = Ac or Bz) may be introduced by treatment of intermediate (lll-b) with acetyl chloride, acetyl bromide or acetic anhydride or benzoyl chloride or benzoic anhydride in the presence of a base like triethylamine, Λ/,Λ/-diisopropylethylamine or 4-
(dimethylamino)pyridine in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or dichloromethane at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius.
In step 3 of Scheme 3, the primary hydroxyl group is deprotected to lead to intermediate (lll-d). When Pg3 is Tr, intermediate (lll-c) is treated in the presence of an acid like para-toluenesulfonic acid in a alcoholic solvent like methanol at a temperature ranging from about -20 degrees Celsius to about room temperature to provide intermediate (lll-d). Cosolvents like chloroform may be used.
In step 4 of Scheme 3, a hydroxymethylene group is introduced through a process similar to the one already described in Scheme 1 (step 1 ) and Scheme 2 (steps 4 and 5).
Other sources of formaldehyde, like paraformaldehyde in a solvent like ethanol at a temperature ranging from about room temperature to about 70 degrees Celsius in the presence of an alkali metal alkoxide can also be used in this step. When Pg4is Bn, this step provides intermediate (lll-e) and when Pg4 is Ac or Bz, this step provides intermediate (lll-f).
In step 5 of Scheme 3, intermediate (lll-e) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (lll-g).
In step 6 of Scheme 3, the remaining protecting groups (Pg4) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final product (A).
In step 7 of Scheme 3, intermediate (lll-f) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A). Another alternative scheme for synthesizing product (A) is depicted in Scheme 4 and described below.

Scheme 4 In step 1 of Scheme 4, intermediate (lll-a) is treated with the appropriate arylsulfonyl chloride R4SO2CI or arylsulfonic anhydride R4S(O)2OS(O)2R4 (wherein R4 is an optionally substituted aryl group, such as found in the arylsulfonyl chlorides 4-methyl-benzenesulfonyl chloride, 4-nitro-benzenesulfonyl chloride, 4-fluoro-benzenesulfonyl chloride, 2,6-dichloro- benzenesulfonyl chloride, 4-fluoro-2-methyl-benzenesulfonyl chloride, and 2,4,6-trichloro- benzenesulfonyl chloride, and in the arylsulfonic anhydride, p-toluenesulfonic anhydride) in presence of a base like pyridine, triethylamine, Λ/,Λ/-diisopropylethylamine in a solvent like tetrahydrofuran, 2-methyltetrahydrofuran at a temperature ranging from about -20 degrees Celsius to about room temperature. Some Lewis acids like zinc(ll) bromide may be used as additives. In step 2 of Scheme 4, intermediate (IV-a) is submitted to a Kornblum-type oxidation
(see, Kornblum, N., et al., Journal of The American Chemical Society, 81 , 4113 (1959)) to produce the corresponding aldehyde which may exist in equilibrium with the corresponding hydrate and/or hemiacetal form. For example intermediate (IV-a) is treated in the presence of a base like pyridine, 2,6-lutidine, 2,4,6-collidine, Λ/,Λ/-diisopropylethylamine, A- (dimethylamino)pyridine in a solvent like dimethyl sulfoxide at a temperature ranging from about room temperature to about 150 degrees Celsius. The aldehyde intermediate produced is then submitted to the aldol/Cannizzaro conditions described for step 1 (Scheme 1 ) and step 5 (Scheme 2) to produce intermediate (IV-b). In step 3 of Scheme 4, intermediate (IV-b) is treated with an acid like thfluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A).
When R2 is (C2-C4)alkynyl the process may be performed using Scheme 5, wherein R6 is H or (CrC2)alkyl.

Scheme 5
In step 1 of Scheme 5, which provides intermediate (V-i), the organometallic addition step is carried out in a similar way to the one described in Schemel , step 6, using the organometallic reagent derived from (V-a), where Pg5 is a suitable protective group for the hydroxyl group. For instance Pgs can be a te/t-butyldimethylsilyl group (TBS) (see
US2007/0054867 for preparation of for instance {4-[(5-bromo-2-chloro-phenyl)-methyl]- phenoxy}-te/t-butyl-dimethyl-silane).
In step 2 of Scheme 5, when Pg2 = PMB, intermediate (V-i) is treated with an acid like trifluoroacetic acid, methanesulfonic acid or an acidic resin in presence of anisole in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (V-j).
In step 3 of Scheme 5, protecting groups (Pg5) and (Pg1) can be removed to provide (V-k). Typically (Pg5) is TBS and Pg1 is Bn. In this circumstance, the protecting groups are removed by sequential treatment of (V-j) with 1 ) tetrabutylammonium fluoride in a solvent like tetrahydrofuran or 2-methyltetrahydrofuran at a temperature ranging from 0 degrees
Celsius to about 40 degrees Celsius and 2) treatment with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature. In this sequence, the order of the 2 reactions is interchangeable.
In step 4 of Scheme 5, intermediate (V-k) is treated with N,N-bis- (trifluoromethanesulfonyl)-aniline in presence of a base like triethylamine or 4- dimethyaminopyridine in a solvent like dichloromethane or 1 ,2-dichloroethane at a temperature ranging from 0 degrees Celsius to about 40 degrees Celsius to produce intermediate (V-I).
In step 5 of Scheme 5, intermediate (V-I) is subjected to a Sonogashira-type reaction (see, Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In
Comprehensive Organic Synthesis (eds. Trost, B. M., Fleming, I.), 3, 521-549, (Pergamon, Oxford, 1991 )).

IS ERTUGLIFLOZIN
Example 4
(1 S.2S.3S.4R.5S)-5-[4-chloro-3-(4-ethoxy-benzyl)-Dhen yll- 1 -h vdroxymeth yl-6.8-dioxa- bicvclo[3.2.1loctane-2,3Λ-triol (4A) and (1S,2S,3SΛS,5S)-5-[4-chloro-3-(4-ethoxy- benzvD-phen yll- 1 -h vdroxymeth yl-6, 8-dioxa-bicvclo[3.2.1 loctane-2, 3, 4-triol (4B):

To a solution of {(2S,3S)-2,3,4-tris-benzyloxy-5-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6,8- dioxa-bicyclo[3.2.1]oct-1-yl}-methanol (l-4k: 335 mg) in ethanol/tetrahydrofuran (10 ml_, 4/1 volume) was added successively formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) and the resulting mixture was stirred at room temperature. After 1 hour, additional formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) were added and the mixture was allowed to stir for an additional hour at room temperature. The palladium was filtered and the crude mixture obtained after evaporation of solvent was purified by HPLC preparative.
HPLC preparative: reverse phase C18 Gemini column 5 micrometer 30 x 100 mm, 40 mL/minute, gradient of acetonitrile/0.1 % formic acid : water/0.1 % formic acid; 25 to 50% of acetonitrile/0.1 % formic acid over 18 minutes; UV detection: 220 nm. The HPLC indicated a ratio of diastereomers of 1.1 :1 (4A:4B).
4A: (60 mg, 29% yield); Rt = 12.4 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode); 481.3 (M+HCO2 ~; negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.43 (d, 1 H, J = 1.9 Hz), 7.36 (dd, 1 H, J = 8.3 and 2Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m, 2H), 4.12 (d, 1 H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d, 1 H, J = 12.5 Hz), 3.75 (dd, 1 H, J = 8.3 and 1.3 Hz), 3.65 (d, 1 H, J = 12.5 Hz), 3.63 (t, 1 H, J = 8.2 Hz), 3.57 (dd, 1 H, J = 7.5 and 1.3 Hz), 3.52 (d, 1 H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1360.
4B: (30 mg, 15% yield); Rt = 13.2 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode) 481.3 (M+HCO2 “, negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.48 (d, 1 H, J = 1.9 Hz) 7.40 (dd, 1 H, J = 8.1 and 1.9 Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m, 2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1 H, J = 12.5 Hz), 3.49 (d, 1 H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1358.
Merck & Co., Inc. and Pfizer Enter Worldwide Collaboration Agreement to Develop and Commercialize Ertugliflozin, an Investigational Medicine for Type 2 Diabetes
 ERTUGLIFLOZIN
ERTUGLIFLOZIN
Merck & Co., Inc. (NYSE: MRK), known as MSD outside the United States and Canada (“Merck”), and Pfizer Inc. (NYSE:PFE) today announced that they have entered into a worldwide (except Japan) collaboration agreement for the development and commercialization of Pfizer’s ertugliflozin (PF-04971729), an investigational oral sodium glucose cotransporter (SGLT2) inhibitor being evaluated for the treatment of type 2 diabetes. Ertugliflozin is Phase III ready, with trials expected to begin later in 2013.
“We are pleased to join forces with Merck in the battle against type 2 diabetes and the burden that it poses on global health,” said John Young, president and general manager, Pfizer Primary Care. “Through this collaboration, we believe we can build on Merck’s leadership position in diabetes care with the introduction of ertugliflozin, an innovative SGLT2 inhibitor discovered by Pfizer scientists.”
Under the terms of the agreement, Merck, through a subsidiary, and Pfizer will collaborate on the clinical development and commercialization of ertugliflozin and ertugliflozin-containing fixed-dose combinations with metformin and JANUVIA® (sitagliptin) tablets. Merck will continue to retain the rights to its existing portfolio of sitagliptin-containing products. Pfizer has received an upfront payment and milestones of $60 million and will be eligible for additional payments associated with the achievement of pre-specified future clinical, regulatory and commercial milestones. Merck and Pfizer will share potential revenues and certain costs on a 60/40 percent basis.
“Merck continues to build upon our leadership position in the oral treatment of type 2 diabetes through our own research and business development,” said Nancy Thornberry, senior vice president and Diabetes and Endocrinology franchise head, Merck Research Laboratories. “We believe ertugliflozin has the potential to complement our strong portfolio of investigational and marketed products, and we look forward to collaborating with Pfizer on its development.”
……………….
Development of an Early-Phase Bulk Enabling Route to Sodium-Dependent Glucose Cotransporter 2 Inhibitor Ertugliflozin

The development and optimization of a scalable synthesis of sodium-dependent glucose cotransporter 2 inhibitor, ertugliflozin, for the treatment of type-2 diabetes is described. Highlights of the chemistry are a concise, four-step synthesis of a structurally complex API from known intermediate 4 via persilylation–selective monodesilylation, primary alcohol oxidation, aldol-crossed-Cannizzaro reaction, and solid-phase acid-catalyzed bicyclic ketal formation. The final API was isolated as the l-pyroglutamic acid cocrystal.
1= ertugliflozin
| PF-04971729, a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF- 04971729 also were very weak (IC50 900μM). The disposition of PF-04971729, an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. |
| References on PF-04971729: [1]. 1. Amit S. Kalgutkar, Meera Tugnait, Tong Zhu, et al.Preclinical Species and Human Disposition of PF-04971729, a Selective Inhibitor of the Sodium-Dependent Glucose cotransporter 2 and Clinical Candidate for the Treatment of Type 2 . Diabetes Mellitus Drug Metabolism and Diposition, 2011, 39 (9):. 1609-1619 Abstract (1S, 2S, 3S, 4R, 5S) -5 – [4-Chloro-3-(4-ethoxybenzyl) phenyl] -1 -hydroxymethyl-6 ,8-dioxabicyclo [3.2.1] octane-2 ,3,4-triol (PF-04971729), a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. This article describes the preclinical species and in vitro human disposition characteristics of PF-04971729 that were used in experiments performed to support the first-in-human study. Plasma clearance was low in rats (4.04 ml · min? 1 · kg? 1) and dogs (1.64 ml · min? 1 · kg? 1), resulting in half-lives of 4.10 and 7.63 h, respectively. Moderate to good bioavailability in rats (69%) and dogs (94%) was . observed after oral dosing The in vitro biotransformation profile of PF-04971729 in liver microsomes and cryopreserved hepatocytes from rat, dog, and human was qualitatively similar;. prominent metabolic pathways included monohydroxylation, O-deethylation, and glucuronidation No human-specific metabolites of PF-04971729 were detected in in vitro studies. Reaction phenotyping studies using recombinant enzymes indicated a role of CYP3A4/3A5, CYP2D6, and UGT1A9/2B7 in the metabolism of PF-04971729. No competitive or time-dependent inhibition of the major human cytochrome P450 enzymes was discerned with PF-04971729. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF-04971729 also were very weak (IC50 =? 900 μM). Single-species allometric scaling of rat pharmacokinetics of PF-04971729 was used to predict human clearance, distribution volume, and oral bioavailability. Human pharmacokinetic predictions were consistent with the potential for a low daily dose. First-in-human studies after oral administration indicated that the human pharmacokinetics / dose predictions for PF -04971729 were in the range that is likely to yield a favorable pharmacodynamic response.. [2] … Timothy Colin Hardman, Simon William Dubrey Development and potential role of type-2 sodium-glucose transporter Inhibitors for Management of type 2 Diabetes Diabetes Ther 2011 September; 2 (3):. 133-145 Abstract There is a recognized need for new treatment options for type 2 diabetes mellitus (T2DM). Recovery of glucose from the glomerular filtrate represents an important mechanism in maintaining glucose homeostasis and represents a novel target for the management of T2DM. Recovery of glucose from the glomerular filtrate is executed principally by the type 2 sodium-glucose cotransporter (SGLT2). Inhibition of SGLT2 promotes glucose excretion and normalizes glycemia in animal models. First reports of specifically designed SGLT2 inhibitors began to appear in the second half of the 1990s. Several candidate SGLT2 inhibitors are currently under development, with four in the later stages of clinical testing. The safety profile of SGLT2 inhibitors is expected to be good, as their target is a highly specific membrane transporter expressed almost exclusively within the renal tubules. One safety concern is that of glycosuria , which could predispose patients to increased urinary tract infections. So far the reported safety profile of SGLT2 inhibitors in clinical studies appears to confirm that the class is well tolerated. Where SGLT2 inhibitors will fit in the current cascade of treatments for T2DM has yet to be established. The expected favorable safety profile and insulin-independent mechanism of action appear to support their use in combination with other antidiabetic drugs. Promotion of glucose excretion introduces the opportunity to clear calories (80-90 g [300-400 calories] of glucose per day) in patients that are generally overweight, and is expected to work synergistically with weight reduction programs. Experience will most likely lead to better understanding of which patients are likely to respond best to SGLT2 inhibitors, and under what circumstances.[3]. Zhuang Miao, Gianluca Nucci, Neeta Amin. Pharmacokinetics, Metabolism and Excretion of the Anti-Diabetic Agent Ertugliflozin (PF-04971729) in Healthy Male the Subjects. Drug Metabolism and Diposition. Abstract The Disposition of ertugliflozin (PF-04971729) , an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. Mass balance was achieved with approximately 91% of the administered dose recovered in urine and feces. The total administered radioactivity excreted in feces and urine was 40.9% and 50.2%, respectively. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. The principal biotransformation pathway involved glucuronidation of the glycoside hydroxyl groups to yield three regioisomeric metabolites M4a, M4b and M4c (~ 39.3% of the dose in urine) of which M4c was the major regioisomer (~ 31.7% of the dose). The structure of M4a and M4c were confirmed to be PF-04971729-4-O-β-and-3-O-β-glucuronide , respectively, via comparison of the HPLC retention time and mass spectra with authentic standards. A minor metabolic fate involved oxidation by cytochrome P450 to yield monohydroxylated metabolites M1 and M3 and des-ethyl PF-04971729 (M2), which accounted for ~ 5.2% of the dose in excreta. In plasma, unchanged PF-04971729 and the corresponding 4-O-β-(M4a) and 3-O-β-(M4c) glucuronides were the principal components, which accounted for 49.9, 12.2 and 24.1% of the circulating radioactivity. Overall, these data suggest that PF-04971729 is well absorbed in humans, and eliminated largely via glucuronidation.. [4] .. Tristan S. Maurer, Avijit Ghosh, Nahor Haddish-Berhane pharmacodynamic Model of Sodium-Glucose Transporter 2 (SGLT2) Inhibition: Implications for Quantitative Translational Pharmacology AAPS J. 2011; 13 (4): 576-584 Abstract Sodium-glucose co-transporter-2 (SGLT2) inhibitors are an emerging class of agents for use in the treatment of type 2 diabetes mellitus (T2DM). Inhibition of SGLT2 leads to improved glycemic control through increased urinary glucose excretion (UGE). In this study, a biologically based pharmacokinetic / pharmacodynamic (PK / PD) model of SGLT2 inhibitor-mediated UGE was developed. The derived model was used to characterize the acute PK / PD relationship of the SGLT2 inhibitor, dapagliflozin, in rats. The quantitative translational pharmacology of dapagliflozin was examined through both prospective simulation and direct modeling of mean literature data obtained for dapagliflozin in healthy subjects. Prospective simulations provided time courses of UGE that were of consistent shape to clinical observations, but were modestly biased toward under prediction. Direct modeling provided an improved characterization of the data and precise parameter estimates which were reasonably consistent with those predicted from preclinical data. Overall, these results indicate that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be reasonably well predicted from preclinical data through rational accounting of species differences in pharmacokinetics, physiology, and SGLT2 pharmacology. Because these data can be generated at the earliest stages of drug discovery, the proposed model is useful in the design and development of novel SGLT2 inhibitors. In addition, this model is expected to serve as a useful foundation for future efforts to understand and predict the effects of SGLT2 inhibition under chronic administration and in other patient populations.[5]. Yoojin Kim, Ambika R Babu Clinical potential of sodium-glucose cotransporter 2 Inhibitors in the Management of type 2 Diabetes Diabetes Obes Metab Syndr 2012; 5:…. 313-327 Abstract Background The Kidney plays an Important role in glucose metabolism, and has been considered a target for therapeutic intervention. The sodium-glucose cotransporter type 2 (SGLT2) mediates most of the glucose reabsorption from the proximal renal tubule. Inhibition of SGLT2 leads to glucosuria and provides a unique mechanism to lower elevated blood glucose levels in diabetes. The purpose of this review is to explore the physiology of SGLT2 and discuss several SGLT2 inhibitors which have clinical data in patients with type 2 diabetes. Methods We performed a PubMed search using the terms “SGLT2″ and “SGLT2 inhibitor” through April 10, 2012. Published articles, press releases, and abstracts presented at national and international meetings were considered. Results SGLT2 inhibitors correct a novel pathophysiological defect, have an insulin-independent action, are efficacious with glycosylated hemoglobin reduction ranging from 0.5% to 1.5%, promote weight loss, have a low incidence of hypoglycemia, complement the action of other antidiabetic agents, and can be used at any stage of diabetes. They are generally well tolerated. However, due to side effects, such as repeated urinary tract and genital infections, increased hematocrit, and decreased blood pressure, appropriate patient selection for drug initiation and close monitoring after initiation will be important. Results of ongoing clinical studies of the effect of SGLT2 inhibitors on diabetic complications and cardiovascular safety are crucial to determine the risk -benefit ratio. A recent decision by the Committee for Medicinal Products for Human Use of the European Medicines Agency has recommended approval of dapagliflozin for the treatment of type 2 diabetes as an adjunct to diet and exercise, in combination with other glucose-lowering medicinal products , including insulin, and as a monotherapy for metformin-intolerant patients. Clinical research also remains to be carried out on the long-term effects of glucosuria and other potential effects of SGLT2 inhibitors, especially in view of the observed increase in the incidence of bladder and breast cancer SGLT2 inhibitors represent a promising approach for the treatment of diabetes, and could potentially be an addition to existing therapies Keywords:.. sodium-glucose cotransporter type 2, SGLT2, inhibitors, kidney, glucosuria, oral diabetes agent, weight loss.[6]. Clinical Trials with PF-04971729 |
Example 6 Manufacturing Process for Tablets US20130137646
 |
DAPAGLIFLOZIN…FDA approves AZ diabetes drug Farxiga

DAPAGLIFLOZIN, BMS-512148
The US Food and Drug Administration has finally approved AstraZeneca’s diabetes drug Farxiga but is insisting on six post-marketing studies, including a cardiovascular outcomes trial.
The approval was expected given that the agency’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 last month that the benefits of Farxiga (dapagliflozin), already marketed in Europe as Forxiga, outweigh identified risks. The FDA rejected the drug in January 2012 due to concerns about possible liver damage and the potential link with breast and bladder cancer.

