
SR48692 (Meclinertant)
Reminertant; SR 48692
CAS [146362-70-1]
- Molecular FormulaC32H31ClN4O5
- Average mass587.065
SEE…...https://newdrugapprovals.org/2014/12/31/meclinertant-sr48692/
2-[[1-(7-chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)pyrazole-3-carbonyl]amino]adamantane-2-carboxylic acid
- Originatorsanofi-aventis
- ClassAnalgesics; Antineoplastics; Antipsychotics
- Mechanism of ActionNeurotensin antagonists
Meclinertant (SR-48692) is a drug which acts as a selective, non-peptide antagonist at the neurotensin receptor NTS1, and was the first non-peptide antagonist developed for this receptor.[1][2] It is used in scientific research to explore the interaction between neurotensin and other neurotransmitters in the brain,[3][4][5][6][7][8] and produces anxiolytic, anti-addictive and memory-impairing effects in animal studies.[9][10][11][12]
CLIP
Methods for the synthesis of pharmaceuticals have improved over the years, however, the technology and tools used to perform synthetic operations have remained the same. Batch-mode processes are still common but many improvements can be made by using modern technologies. Recently, the use of machine-assisted protocols has increased, with flow-based chemical synthesis being extensively investigated. Under dynamic flow regimes, mixing and heat transfer can be more accurately controlled, the use of solid-phase reagents and catalysts can facilitate purification, and tedious downstream processes (workup, extraction, and purification) are reduced.
Steven V. Ley and co-workers, University of Cambridge, UK, have been evaluating the utility of flow-based syntheses to accelerate multistep routes to highly complex, medically relevant compounds, in this case Meclinertant (SR48692, pictured). They show that new technologies can help to overcome many synthetic issues of the existing batch process. In this case, flow chemistry has allowed control of exothermic events, controlled the superheating of solvents, and streamlined the synthesis by allowing reaction telescoping. It has also helped to prevent back mixing and the accumulation of byproducts. The use of polymer-supported reagents has simplified downstream processing and enhanced the safety of reactions, and in-line monitoring can track hazardous intermediates.
These new technologies have been shown to be powerful synthetic tools, although care must be taken not to convert them to expensive solutions to nonexistent problems.
http://community.dur.ac.uk/i.r.baxendale/papers/ChemEurJ2013.19.7917.pdf
A Machine-Assisted Flow Synthesis of SR48692: A Probe for the Investigation of Neurotensin Receptor-1,
Claudio Battilocchio, Benjamin J. Deadman, Nikzad Nikbin, Matthew O. Kitching, Ian R. Baxendale, Steven V. Ley,
Chem. Eur. J. 2013.
DOI: 10.1002/chem.201300696


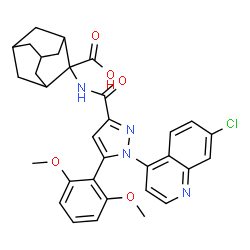
2-[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido]adamantane-2-carboxylic acid (1):
Polymer-supported sulfonic acid (QP-SA; 0.6 g, 2.4 mmol) was added to a solution of tert-butyl 2-[1- (7-chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxamido]adamantane-2-carboxylate (13; 30 mg, 0.05 mmol) in dichloromethane and the reaction was stirred at RT for 18 h. The QP-SA was filtered off and the filtrate concentrated in vacuo to provide the title compound as white crystals (yield 25 mg, 0.04 mmol, 86%).
M.p. 219–222 deg C;
1 H NMR (400 MHz, CDCl3, 25 deg C): d=8.91 (d, 1H, J=4.6 Hz), 8.15 (d, 1H, J=2.1 Hz), 7.78 (d, 1H, J=9.1 Hz), 7.68 (dd, 1H, J=2.1, 9.1 Hz), 7.28 (d, 1H, J=4.7 Hz), 7.24 (t, 1H, J=8.5 Hz), 7.91 (s, 1H), 6.52 (d, 2H, J=8.5 Hz), 3.42 (s, 6H), 2.64–2.56 (m, 2H), 2.17–2.05 (m, 2H), 2.04–1.92 (m, 2H), 1.82–1.71 (m, 2H), 1.71–1.61 (m, 4H), 1.61–1.50 ppm (m, 2H);
13C NMR (100 MHz, CDCl3, 25 deg C): d=173.3(C), 159.9 (C), 157.5 (C), 157.5 (C), 151.8 (CH), 149.1 (C), 143.4 (C), 139.2 (C), 134.8 (C), 131.9 (CH), 128.0 (CH), 127.7 (CH), 125.9 (CH), 122.2 (C), 118.6 (CH), 109.6 (CH), 105.8 (C), 104.0 (CH), 55.4 (CH3), 55.3 (C), 37.4 (CH2), 33.6 (CH2), 32.8 (CH2), 31.9 (CH), 26.5 (CH), 26.2 ppm (CH);
FT-IR (neat): 3405, 2922, 1728, 1674, 1591, 1527, 1474, 1433, 1379, 1357, 1288, 1251, 1206, 1101, 1077, 1031, 1006, 957, 882, 865, 823, 779, 725, 682 cm1 ;
LCMS: tR =5.29 min, m/z [M+H]+: 587.46;
HRMS (ESI): m/z calcd for C32H32N4O5Cl+: 587.2061, found 587.2053; the structure was unambiguously confirmed by single X-ray crystallography; space group P1¯: a= 10.249, b=11.718, c=12.634 ; a=76.6, b=72.9, g=76.4o
CLIP AND ITS OWN REFERENCES
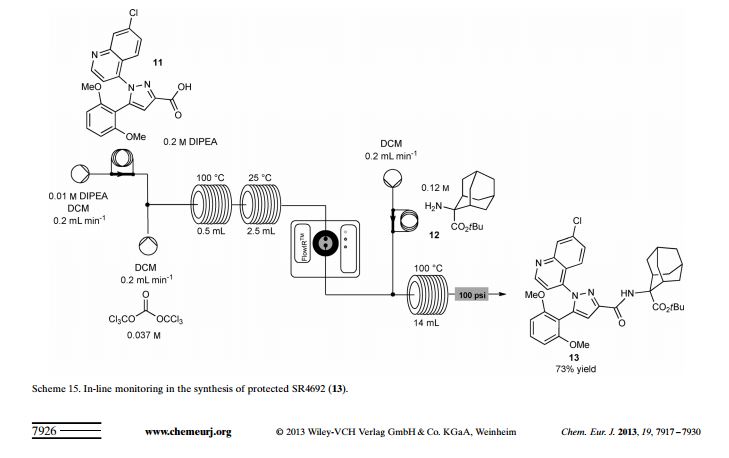
Although batch processes remain the most used procedure for running chemical reactions, the use of machine-assisted flow methodologies(24) enables an improved efficiency and high throughput. A direct comparison between conventional batch preparation and flow multistep synthesis of selective neurotensine probe SR48692 (Meclinertant) was reported by Ley and co-workers in 2013 (Scheme 6).(25)
In this case study, the authors investigated whether flow technology could accelerate a multistep synthesis (i.e., higher yields or lower reaction times) and overcome many synthetic issues (i.e., solid precipitation or accumulation of byproducts). The initial Claisen condensation between ketone 31 and ethyl glyoxalate in the presence of NaOEt as base and EtOH as solvent in batch is run at room temperature and product 32 is obtained in 60% yield after 3 h stirring.
Superheating (heat above solvent boiling point) the reaction in flow provided a faster alternative: using a 52 mL PFA reactor coil at 115 °C with a residence time of 22 min gave the corresponding product 32 in 74% yield. In order to solve some problems of solid accumulation an ad-hoc pressurized stainless-steel tank (5 bar, nitrogen) was designed; it allowed to run the reaction continuously without any precipitation or blockage.

The following reaction between 32 and commercially available hydrazine 33 was performed in DMF in the presence of concentrated H2SO4. After 52 min of residence time at 140 °C into a 52 mL PFA reactor coil the crude mixture was treated with an Na2CO3 aq. and then inline extracted through a semipermeable membrane with CH2Cl2. After crystallization, pyrazole ester 34 was isolated in 89% yield.
The corresponding reaction in batch was conducted in DMF under microwaves irradiation at 140 °C for 2 h. Running the reaction in batch on the same scale as in flow (3.58 mmol) gave product 34 in a lower yield (70%). The subsequent hydrolysis was performed combining a THF solution of ester 34 and 3 M aqueous KOH. The reaction was performed inside a 14 mL PFA reactor coil heated at 140 °C with a residence time of 14 min.
Upon treatment with 3 M HCl aq., acid 35 precipitated, and it was isolated by filtration in 90% yield. In this case, the corresponding batch hydrolysis afforded product 35 with the same yield (90%); however, a longer reaction time (1.5 h) was required. The final amide formation was performed by reacting acid 35 (activated as acyl chloride) and protected amino alcohol 37through a telescoped synthesis. Triphosgene 36 (a safer substitute for phosgene) was found to be the best acid activator.
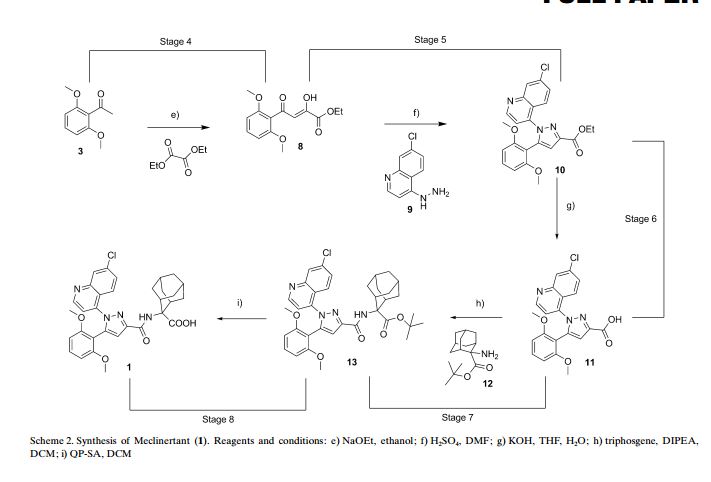
Triphosgene decomposition occurred in the presence of DIPEA at 100 °C into a stainless steel heat exchanger, where phosgene was generated. The crude mixture, containing also acid 35, then passed into a 2.5 mL stainless steel reactor coil at 25 °C, to complete the formation of the corresponding acyl chloride. An inline Flow-IR spectrometer(26)was used to monitor the formation of phosgene without exposing the operator to the toxic gas during analysis. As soon as acyl chloride was formed it was reacted with protected amino alcohol 37.
The amide formation took place into a 14 mL stainless steel reactor coil at 100 °C with a residence time of 75 s. Amide 38 was isolated in 85% yield after quenching with NH4Cl and extraction with AcOEt. For obvious safety concerns, avoiding the handling of phosgene and the isolation of highly reactive acyl chloride intermediate represent a remarkable improvement with respect to batch procedure.
Finally, meclinertant 39 was obtained after deprotection of ester38 by using a polymer-supported sulfonic acid. The last synthetic step was conducted in batch on a small scale; however, it could be easily transferred to flow mode by using a column packed with commercially available polymer-supported sulfonic acid.
24 Ley, S. V.; Fitzpatrick, D. E.; Myers, R. M.; Battilocchio, C.; Ingham, R. J. Angew. Chem., Int. Ed. 2015, 54, 2, DOI: 10.1002/anie.201501618
25.Battilocchio, C.; Deadman, B. J.; Nikbin, N.; Kitching, M. O.; Baxendale, I. C.; Ley, S. V. Chem. – Eur. J. 2013, 19, 7917, DOI: 10.1002/chem.201300696
Org. Process Res. Dev., 2016, 20 (1), pp 2–25
DOI: 10.1021/acs.oprd.5b00325
CLIP AND ITS OWN REFERENCES
The choice of the flow reactor also plays a key role in the synthesis of meclinertant (SR48692, 103), which is a potent probe for investigating neurotensin receptor-1 [92]. The flow synthesis of this challenging compound was reported in 2013 and aims to evaluate the benefits of flow chemistry in order to avoid shortcomings of previous batch synthesis efforts particularly in regard to scale up [93].
The investigation first involved the preparation of the key acetophenone starting material 112 which although commercially available was expensive and could be generated from 1,3-cyclohexadione (104). The sequence consisted of O-acetylation, a Steglich rearrangement, oxidation and a final methylation reaction.
As the use of flow chemistry had already improved the O-acetylation during scale-up tests (130 mmol) by avoiding exotherms, it was anticipated that the subsequent Steglich rearrangement could be accomplished in flow using catalytic DMAP instead of stoichiometric AlCl3 as precedented (Scheme 19).
This was eventually realised by preparing a monolithic flow reactor functionalised with DMAP that proved far superior to commercially available DMAP on resin. Employing the monolithic reactor cleanly catalysed the rearrangement step when a solution of 106 was passed through the reactor at elevated temperature (100 °C, 20 min residence time).
The resulting triketone 107 was telescoped into an iodine mediated aromatisation, followed by high temperature mono-methylation using dimethyl carbonate/dimethylimidazole as a more benign alternative to methyl iodide at scale.
Scheme 19: First stage in the flow synthesis of meclinertant (103).
The subsequent Claisen condensation step between ketone 112 and diethyl oxalate (113) was reportedly hampered by product precipitation and clogging problems, thus a pressure chamber was developed [94] that would act as a pressure regulator allowing this step to be scaled up in flow in order to provide 114 on multigram scale (134 g/h).
A Knorr pyrazole formation between 114 and commercially available hydrazine 115 had previously been found difficult to scale up in batch (the yield dropped from 87% to 70%) and was thus translated into a high temperature flow protocol (140 °C) delivering the desired product 116 in 89% yield (Scheme 20).
Ester hydrolysis and a triphosgene (118) mediated amide bond formation between acid 117 and adamantane-derived aminoester119 [95] completed this flow synthesis. Meclinertant (103) was subsequently obtained after batch deprotection using polymer supported sulfonic acid.
Overall, this study showcases how flow chemistry can be applied to gain benefits when faced with problems during mesoscale synthesis of a complex molecule. However, despite the successful completion of this campaign, it could be argued that the development time required for such a complex molecule in flow can be protracted; therefore both synthetic route and available enabling technologies should be carefully examined before embarking upon such an endeavour.
Scheme 20: Completion of the flow synthesis of meclinertant (103).
| 92 Myers, R. M.; Shearman, J. W.; Kitching, M. O.; Ramos-Montoya, A.; Neal, D. E.; Ley, S. V. ACS Chem. Biol. 2009, 4, 503–525. doi:10.1021/cb900038e |
| 93. |
Battilocchio, C.; Deadman, B. J.; Nikbin, N.; Kitching, M. O.; Baxendale, I. R.; Ley, S. V.Chem. – Eur. J. 2013, 19, 7917–7930. doi:10.1002/chem.201300696 |
| 94. |
Deadman, B. J.; Ley, S. V.; Browne, D. L.; Baxendale, I. R.; Ley, S. V.Chem. Eng. Technol. 2015, 38, 259–264. doi:10.1002/ceat.201400445 |
| 95. |
Battilocchio, C.; Baxendale, I. R.; Biava, M.; Kitching, M. O.; Ley, S. V.Org. Process Res. Dev. 2012, 16, 798–810. doi:10.1021/op300084z |
The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry
Marcus Baumann
and
Ian R. Baxendale
Department of Chemistry, Durham University, South Road, DH1 3LE Durham, United Kingdom
Corresponding author email
Associate Editor: J. A. Murphy
Beilstein J. Org. Chem.2015,11, 1194–1219.
| EP 0477049; FR 2665898; JP 1992244065; US 5420141; US 5607958; US 5616592; US 5635526; US 5744491; US 5744493 |
 |
| The condensation of 2′,6′-dimethoxyacetophenone (I) with diethyl oxalate (II) by means of sodium methoxide in refluxing methanol gives the dioxobutyrate (III), which is cyclized with 7-chloroquinoline-4-hydrazine (IV) in refluxing acetic acid yielding the pyrazole derivative (V). The hydrolysis of the ester group of (V) with KOH in refluxing methanol/water affords the corresponding carboxylic acid (VI), which is finally treated with SOCl2 in refluxing toluene and condensed with 2-aminoadamantane-2-carboxylic acid. |
| Patent ID |
Date |
Patent Title |
| US8642566 |
2014-02-04 |
Therapeutic approaches for treating neuroinflammatory conditions |
| US7927613 |
2011-04-19 |
Pharmaceutical co-crystal compositions |
| US7790905 |
2010-09-07 |
Pharmaceutical propylene glycol solvate compositions |
| US2007243257 |
2007-10-18 |
PHARMACEUTICAL COMPOSITION COMPRISING A SOLID DISPERSION WITH A POLYMER MATRIX CONTAINING A CONTINUOUS POLYDEXTROSE PHASE AND A CONTINUOUS PHASE OF A POLYMER OTHER THAN POLYDEXTROSE |
| US6284277 |
2001-09-04 |
Stable freeze-dried pharmaceutical formulation |
| US6172239 |
2001-01-09 |
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharamaceutical compositions containing them |
| US5965579 |
1999-10-12 |
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharmaceutical compositions containing them |
| US5955474 |
1999-09-21 |
Use of neurotensin antagonists for the treatment of edematous conditions |
| US5939449 |
1999-08-17 |
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharmaceutical compositions containing them |
| US5936123 |
1999-08-10 |
Hydrazine derivative compounds as intermediates for preparing substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors |
| Patent ID |
Date |
Patent Title |
| US5925661 |
1999-07-20 |
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharmaceutical compositions containing them |
| US5744491 |
1998-04-28 |
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them |
| US5744493 |
1998-04-28 |
3-amidopyrazole derivatives and pharmaceutical compositions containing them |
| US5723483 |
1998-03-03 |
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharmaceutical compositions containing them |
| US5635526 |
1997-06-03 |
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them |
| US5616592 |
1997-04-01 |
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them |
| US5607958 |
1997-03-04 |
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them |
| US5585497 |
1996-12-17 |
Substituted 1-naphthyl-3-pyrazolecarboxamides which are active on neurotensin |
| US5561234 |
1996-10-01 |
1-(7-chloroquinolin-4-yl)pyrazole-3-carboxamide N-oxide derivatives, method of preparing them, and their pharmaceutical compositions |
| US5523455 |
1996-06-04 |
Substituted 1-naphthyl-3-pyrazolecarboxamides which are active on neurotensin, their preparation and pharmaceutical compositions containing them |
| Patent ID |
Date |
Patent Title |
| EP0699438 |
1996-03-06 |
Use of neurotensin antagonists for the preparation of diuretic drugs Use of neurotensin antagonists for the preparation of diuretic drugs |
| US5420141 |
1995-05-30 |
3-amidopyrazole derivatives, process for preparing these and pharmaceutical composites containing them |
References
- Gully D, Canton M, Boigegrain R, Jeanjean F, Molimard JC, Poncelet M, Gueudet C, Heaulme M, Leyris R, Brouard A (January 1993).“Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor”. Proceedings of the National Academy of Sciences of the United States of America. 90 (1): 65–9. doi:10.1073/pnas.90.1.65. PMC 45600
 . PMID 8380498.
. PMID 8380498.
- Gully D, Jeanjean F, Poncelet M, Steinberg R, Soubrié P, Le Fur G, Maffrand JP (1995). “Neuropharmacological profile of non-peptide neurotensin antagonists”. Fundamental & Clinical Pharmacology. 9 (6): 513–21. doi:10.1111/j.1472-8206.1995.tb00528.x.PMID 8808171.
- Rostene W, Azzi M, Boudin H, Lepee I, Souaze F, Mendez-Ubach M, Betancur C, Gully D (April 1997). “Use of nonpeptide antagonists to explore the physiological roles of neurotensin. Focus on brain neurotensin/dopamine interactions”. Annals of the New York Academy of Sciences. 814: 125–41. doi:10.1111/j.1749-6632.1997.tb46151.x. PMID 9160965.
- Jump up^ Jolas T, Aghajanian GK (August 1997). “Neurotensin and the serotonergic system”. Progress in Neurobiology. 52 (6): 455–68.doi:10.1016/S0301-0082(97)00025-7. PMID 9316156.
- Jump up^ Dobner PR, Deutch AY, Fadel J (June 2003). “Neurotensin: dual roles in psychostimulant and antipsychotic drug responses”. Life Sciences.73 (6): 801–11. doi:10.1016/S0024-3205(03)00411-9. PMID 12801600.
- Jump up^ Chen L, Yung KK, Yung WH (September 2006). “Neurotensin selectively facilitates glutamatergic transmission in globus pallidus”.Neuroscience. 141 (4): 1871–8. doi:10.1016/j.neuroscience.2006.05.049. PMID 16814931.
- Jump up^ Petkova-Kirova P, Rakovska A, Della Corte L, Zaekova G, Radomirov R, Mayer A (September 2008). “Neurotensin modulation of acetylcholine, GABA, and aspartate release from rat prefrontal cortex studied in vivo with microdialysis”. Brain Research Bulletin. 77 (2–3): 129–35. doi:10.1016/j.brainresbull.2008.04.003. PMID 18721670.
- Jump up^ Petkova-Kirova P, Rakovska A, Zaekova G, Ballini C, Corte LD, Radomirov R, Vágvölgyi A (December 2008). “Stimulation by neurotensin of dopamine and 5-hydroxytryptamine (5-HT) release from rat prefrontal cortex: possible role of NTR1 receptors in neuropsychiatric disorders”.Neurochemistry International. 53 (6–8): 355–61. doi:10.1016/j.neuint.2008.08.010. PMID 18835308.
- Jump up^ Griebel G, Moindrot N, Aliaga C, Simiand J, Soubrié P (December 2001). “Characterization of the profile of neurokinin-2 and neurotensin receptor antagonists in the mouse defense test battery”. Neuroscience and Biobehavioral Reviews. 25 (7–8): 619–26. doi:10.1016/S0149-7634(01)00045-8. PMID 11801287.
- Jump up^ Tirado-Santiago G, Lázaro-Muñoz G, Rodríguez-González V, Maldonado-Vlaar CS (October 2006). “Microinfusions of neurotensin antagonist SR 48692 within the nucleus accumbens core impair spatial learning in rats”. Behavioral Neuroscience. 120 (5): 1093–102. doi:10.1037/0735-7044.120.5.1093. PMID 17014260.
- Felszeghy K, Espinosa JM, Scarna H, Bérod A, Rostène W, Pélaprat D (December 2007). “Neurotensin receptor antagonist administered during cocaine withdrawal decreases locomotor sensitization and conditioned place preference”. Neuropsychopharmacology. 32 (12): 2601–10. doi:10.1038/sj.npp.1301382. PMC 2992550. PMID 17356568.
- Lévesque K, Lamarche C, Rompré PP (October 2008). “Evidence for a role of endogenous neurotensin in the development of sensitization to the locomotor stimulant effect of morphine”.European Journal of Pharmacology. 594 (1–3): 132–8. doi:10.1016/j.ejphar.2008.07.048. PMID 18706409.
//////////////////////Flow synthesis, Meclinertant, SR48692, Reminertant, SR 48692, 146362-70-1
COC1=C(C(=CC=C1)OC)C2=CC(=NN2C3=C4C=CC(=CC4=NC=C3)Cl)C(=O)NC5(C6CC7CC(C6)CC5C7)C(=O)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











![[1860-5397-11-134-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i19.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-134-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i20.png?scale=2.0&max-width=1024&background=FFFFFF)
















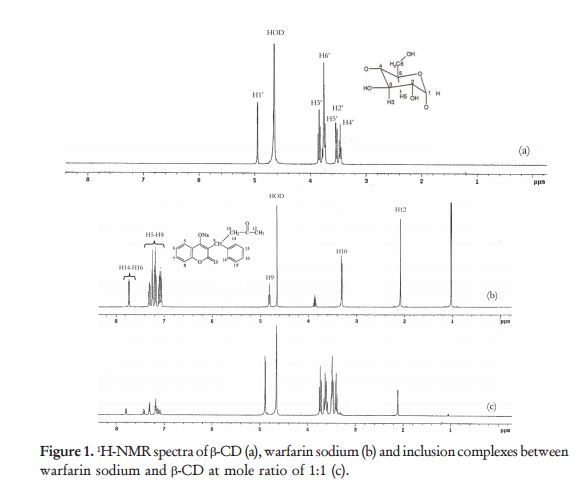











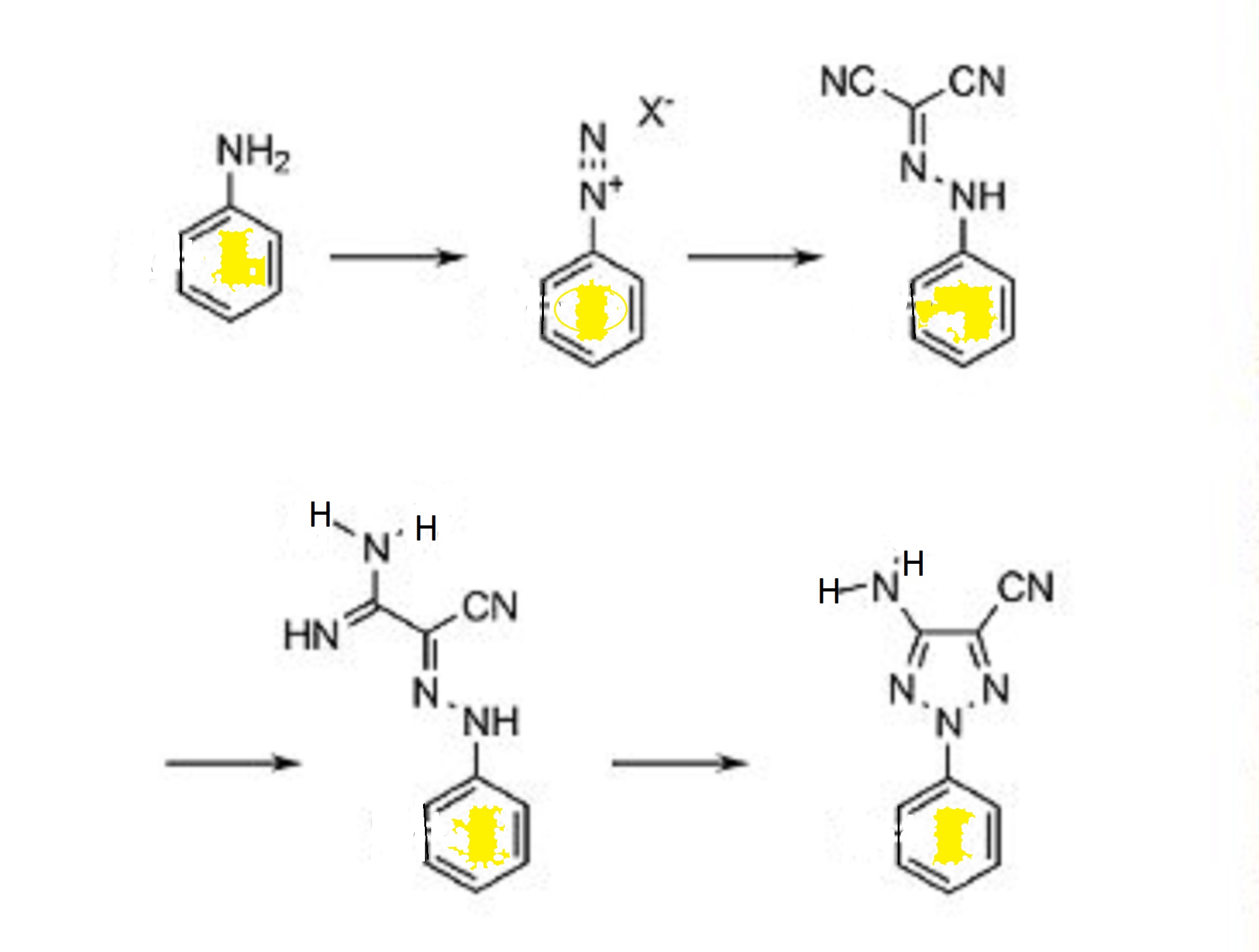
 Reaction of 4-hydroxycoumarin with benzylacetone under
Reaction of 4-hydroxycoumarin with benzylacetone under


 Vitamin K is a cofactor in the synthesis of blood clotting factors II, VII, IX and X*, this step occurs in the liver and involves the gammacarboxylation of the first 10 glutamic acid residues in the amino-terminal region of the prothrombin clotting factor to generategamma-carboxyglutamate. The gamma-carboxyglutamatee amino acid groups can chelate Ca2+ better than ten replaced glutamate residues, thus providing binding sites for four Vitamin Ks onto the phospholipid membrane during coagulation. The clotting occurs via a cascade*, a kind of biochemical chain reaction. {See
Vitamin K is a cofactor in the synthesis of blood clotting factors II, VII, IX and X*, this step occurs in the liver and involves the gammacarboxylation of the first 10 glutamic acid residues in the amino-terminal region of the prothrombin clotting factor to generategamma-carboxyglutamate. The gamma-carboxyglutamatee amino acid groups can chelate Ca2+ better than ten replaced glutamate residues, thus providing binding sites for four Vitamin Ks onto the phospholipid membrane during coagulation. The clotting occurs via a cascade*, a kind of biochemical chain reaction. {See  To work, the Vitamin K must be reduced to its quinol or hydroquinone form. This is achieved with Vitamin K Oxide reductase, which is the step inhibited by S-warfarin, being some three times more potent than R-warfarin. S-warfarin is metabolized primarily by the CYP2C9 enzyme of the cytochrome P450 system. The R-warfarin is metabolized by the two cytochrome P450 enzymes, CP1A4Y and CYP3A4. Warfarin is very soluble in water, and is absorbed into the blood stream within 90 minutes of taking the pills.
To work, the Vitamin K must be reduced to its quinol or hydroquinone form. This is achieved with Vitamin K Oxide reductase, which is the step inhibited by S-warfarin, being some three times more potent than R-warfarin. S-warfarin is metabolized primarily by the CYP2C9 enzyme of the cytochrome P450 system. The R-warfarin is metabolized by the two cytochrome P450 enzymes, CP1A4Y and CYP3A4. Warfarin is very soluble in water, and is absorbed into the blood stream within 90 minutes of taking the pills.
![[1860-5397-11-134-i8]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-134-i8.png?scale=2.0&max-width=1024&background=FFFFFF)















 Halifax survey names South Cambridgeshire as best place to live in rural Britain
Halifax survey names South Cambridgeshire as best place to live in rural Britain






















