
Home » Posts tagged 'FDA 2015' (Page 4)
Tag Archives: FDA 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Isavuconazole – Basilea reports positive results from study

This post is updated in sept 2015……..
Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
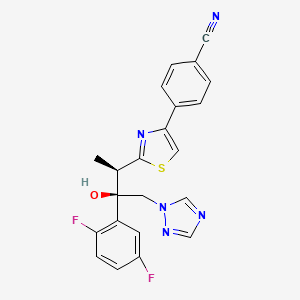
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.

-

-
Scheme 1.
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

-
Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
-
Figure options

-
Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).

read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
Clinical trials
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
References
- [1]
- Saboo, Alok. “Basilea Announces Global Partnership With Astellas for Its Antifungal Isavuconazole.” FierceBiotech. N.p., 24 Feb. 2010. Web.
- “Basilea reports isavuconazole orphan drug designation by U.S. FDA.” Market Wired. 28 May 2013.
- “FDA Grants Orphan Drug Designation to Astellas for Isavuconazole for the Treatment of Invasive Candidiasis.” News Releases. Astellas. 3 Nov 2014.
- Cresemba (isovuconazole sulfate) [prescribing information]. Astella Pharma US, Inc. Revised March 2015.
- Jump up^ “Aspergillosis.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 08 Sept. 2014.
- Jump up^ “Astellas Receives FDA Approval for CRESEMBA® (isavuconazonium Sulfate) for the Treatment of Invasive Aspergillosis and Invasive Mucormycosis.” PR Newswire. N.p., 6 Mar. 2015.
- Jump up^ “Isavuconazonium.” Micromedex Solutions. Truven Health Analytics, n.d. Web. <www.micromedexsolutions.com>.
- Jump up^ Pettit, Natasha N.; Carver, Peggy L. (2015-07-01). “Isavuconazole A New Option for the Management of Invasive Fungal Infections”. Annals of Pharmacotherapy 49 (7): 825–842.doi:10.1177/1060028015581679. ISSN 1060-0280. PMID 25940222.
- Mujais, A. “2014: M-1756. A Phase 3 Randomized, Double-Blind, Non-Inferiority Trial Evaluating Isavuconazole (ISA) vs. Voriconazole (VRC) for the Primary Treatment of Invasive Fungal Disease (IFD) Caused by Aspergillus spp. or other Filamentous Fungi (SECURE): Outcomes by Malignancy Status”. http://www.icaaconline.com. Retrieved 2015-06-19.
- “Abstract: An Open-Label Phase 3 Study of Isavuconazole (VITAL): Focus on Mucormycosis (IDWeek 2014)”. idsa.confex.com. Retrieved 2015-06-19.
- Ltd., Basilea. “Basilea Pharmaceutica – Portfolio – Isavuconazole”. http://www.basilea.com. Retrieved 2015-06-19.
- “Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2015-06-19.
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
Takeda’s Ixazomib, Multiple Myeloma Drug

CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
UPDATES AT THE BOTTOM OF PAGE
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
http://www.pharmalive.com/takeda-begins-phase-iii-trial-of-multiple-myeloma-drug
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.

http://www.cancernetwork.com/conference-reports/ash2012/content/article/10165/2119611
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitor MLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive to MLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
PATENT

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom or heteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
PATENT
http://www.google.com/patents/WO2009154737A1?cl=en
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French] [ACD/IUPAC Name]
MLN9708
UPDATES………..
Ixazomib (trade name Ninlaro) is a drug for the treatment of multiple myeloma, developed by Takeda Pharma. It acts as aproteasome inhibitor and has orphan drug status in the US. In November 2015, the U.S. Food and Drug Administration approved ixazomib for use in combination with lenalidomide and dexamethasone for the treatment of multiple myeloma after at least one prior therapy.[2]
Mechanism
Ixazomib is a peptide analogue that reversibly inhibits the protein proteasome subunit beta type-5 (PSMB5), which is part of the 20Sproteasome complex.[3]
Chemistry

Ixazomib citrate—a prodrug for ixazomib
U.S. FDA Approves Takeda’s NINLARO® (ixazomib), the First and Only Oral Proteasome Inhibitor to Treat Multiple Myeloma
NINLARO Provides a New Option for Patients Living with Multiple Myeloma Who Have Received at Least One Prior Therapy
Cambridge, Mass. and Osaka, Japan, November 20, 2015 – Takeda Pharmaceutical Company Limited (TSE: 4502) today announced that the U.S. Food and Drug Administration (FDA) has approved NINLARO®(ixazomib) capsules, the first and only oral proteasome inhibitor, indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is a once-weekly pill. More information is available at www.NINLARO.com.
Takeda submitted a New Drug Application for NINLARO to the FDA in July 2015, and in September NINLARO was granted Priority Review status with a PDUFA date of March 10, 2016, reflecting the profound and continuing unmet need for new treatments for multiple myeloma, a devastating, relapsing and incurable rare cancer.
“With the approval of NINLARO, we can now offer patients a once-weekly oral proteasome inhibitor as part of a highly active triplet therapy,” said Paul Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center Institute Physician at Dana-Farber Cancer Institute, and investigator for TOURMALINE-MM1, the pivotal Phase 3 trial on which today’s approval is based. “We, as investigators of the TOURMALINE-MM1 trial, felt it was vital to conduct a comprehensive ‘real world’ evaluation of this combination that included some of the most common patient types in the relapsed/refractory multiple myeloma setting, such as older patients, patients with moderate renal impairment, light chain disease, and high risk cytogenetics. Further, we treated patients until disease progression to determine the sustainability of NINLARO in treating their relapsed/refractory disease. The TOURMALINE-MM1 data demonstrate convincingly that oral NINLARO-based triplet treatment is effective at extending progression-free survival, over and above the clinical benefit seen with lenalidomide and dexamethasone, with a tolerable safety profile.”
“We introduced the first proteasome inhibitor for multiple myeloma, VELCADE, into clinical research approximately 20 years ago. Since that time, we’ve significantly advanced scientific understanding of this rare cancer, culminating in the introduction of NINLARO,” said Andy Plump, M.D., Ph.D, Takeda Chief Medical and Scientific Officer. “NINLARO is an entirely new molecule that offers the efficacy of this proteasome inhibitor in a convenient once-weekly pill with a tolerable safety profile. Takeda is delighted to bring this significant innovation to multiple myeloma patients today, and we continue to examine the potential of NINLARO through a robust clinical development program.”
Dr. Brian Durie, Chairman of the International Myeloma Foundation, said, “The IMF is pleased by the approval of ixazomib. This opens the door for a fully oral proteasome inhibitor-based triplet combination therapy. Having worked in multiple myeloma for decades, I’ve seen notable progress, yet significant unmet needs remain. With today’s approval, we now have another attractive option for many patients living with multiple myeloma.”
The FDA approval of NINLARO is based on results from the TOURMALINE-MM1 Phase 3 clinical trial, the first double-blind, placebo-controlled trial with a proteasome inhibitor. TOURMALINE-MM1 is the first of five ongoing Phase 3 clinical trials with study results available. The TOURMALINE program has enrolled approximately 3,000 patients to date in 40 countries. Data from the NINLARO Phase 3 TOURMALINE-MM1 pivotal trial will be presented at the upcoming 57th Annual Meeting of the American Society of Hematology on December 7, 2015.
“The approval of ixazomib offers a much-needed additional option in the multiple myeloma treatment landscape. It is developments such as these that help us to better understand the disease and provide continued hope for patients,” said Kathy Giusti, Founder and Executive Chairman of the Multiple Myeloma Research Foundation (MMRF). “A cancer diagnosis today is different from what it was just a few years ago and it’s exciting to see continued progress. As a patient, I understand the urgent need for advancing research through partnerships that bring new treatment options, as we’ve done with Takeda.”
“NINLARO is a first-of-its-kind innovation that is supported by a global development program, unprecedented for us at Takeda Oncology, and we would like to express our immense appreciation for all patients involved for their incredible strength and invaluable participation. The introduction of NINLARO marks an important step forward, as its efficacy and safety profile – coupled with its completely oral administration – potentially can reduce some logistical burdens, and help enable patients to reap the full benefits of this sustainable therapy,” explained Christophe Bianchi, M.D., President, Takeda Oncology. “As part of our unwavering 20-year commitment, Takeda will continue to pursue advances for these patients, and we look forward to introducing and expanding access to NINLARO in other markets around the world.”
About the TOURMALINE-MM1 Trial
TOURMALINE-MM1 is an international, randomized, double-blind, placebo-controlled clinical trial of 722 patients, designed to evaluate NINLARO plus lenalidomide and dexamethasone compared to placebo plus lenalidomide and dexamethasone in adult patients with relapsed and/or refractory multiple myeloma. Results showed NINLARO is effective in extending Progression Free Survival (PFS) and has a manageable safety profile. The trial achieved its primary endpoint and demonstrated a clinically meaningful and statistically significant prolongation in PFS at this analysis, which showed that patients treated in the NINLARO arm lived without their disease worsening for a significantly longer time compared to patients in the control arm. Patients continue to be treated to progression in this trial and will be evaluated for long term outcomes.
In the TOURMALINE-MM1 trial, the most common adverse reactions (≥20%) in patients receiving NINLARO included diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting and back pain. Serious adverse reactions reported in ≥2% patients included thrombocytopenia (2%) and diarrhea (2%).
Efficacy and safety data were reviewed by an Independent Data Monitoring Committee (IDMC), who recommended the study be continued in blinded fashion to allow further maturation of long term outcomes, including overall survival (OS) and long-term safety.
About NINLARO (ixazomib) capsules
NINLARO (ixazomib) is the first and only oral proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is administered orally, once-weekly on days 1, 8, and 15 of a 28-day treatment cycle. NINLARO is currently under review by the European Medicines Agency (EMA) and was granted an accelerated assessment by the Committee for Medicinal Products for Human Use (CHMP). NINLARO also received Breakthrough Therapy status by the U.S. FDA for relapsed or refractory systemic light-chain (AL) amyloidosis, a related ultra orphan disease, in 2014.
The TOURMALINE clinical development program further reinforces Takeda’s ongoing commitment to developing innovative therapies for people living with multiple myeloma worldwide and the healthcare professionals who treat them. Five global Phase 3 trials are ongoing:
- TOURMALINE-MM1, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in relapsed and/or refractory multiple myeloma
- TOURMALINE-MM2, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma
- TOURMALINE-MM3, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma following induction therapy and autologous stem cell transplant (ASCT)
- TOURMALINE-MM4, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma who have not undergone ASCT
- TOURMALINE-AL1, investigating ixazomib plus dexamethasone vs. physician choice of selected regimens in patients with relapsed or refractory AL amyloidosis
In addition to the TOURMALINE program, a large number of investigator initiated studies are evaluating ixazomib for patients globally.
For additional information on the ongoing Phase 3 studies please visit www.clinicaltrials.gov. To learn more about NINLARO, please visit www.NINLARO.com or call 1-844-N1POINT (1-844-617-6468).
![]()
References
- “Ninlaro (ixazomib) Capsules, for Oral Use. Full Prescribing Information” (PDF). NINLARO (ixazomib) For Healthcare Professionals. Takeda Pharmaceutical Company Limited Cambridge, MA 02139. Retrieved 21 November 2015.
- “FDA Okays Ixazomib, Another Multiple Myeloma Drug”. November 20, 2015.
- KEGG: Ixazomib
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N2-(2,5-Dichlorobenzoyl)-N-[(1R)-1-(dihydroxyboryl)-3-methylbutyl]glycinamide
|
|
| Clinical data | |
| Trade names | Ninlaro |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 58%[1] |
| Protein binding | 99% |
| Metabolism | hepatic, CYP3A4 (42%),CYP1A2 (26%) and others |
| Biological half-life | 9.5 days |
| Excretion | urine (62%), feces (22%) |
| Identifiers | |
| CAS Number | 1072833-77-2 |
| ATC code | L01XX50 |
| PubChem | CID 25183872 |
| ChemSpider | 25027391 |
| UNII | 71050168A2 |
| KEGG | D10130 |
| ChEBI | CHEBI:90942 |
| Synonyms | MLN2238 |
| Chemical data | |
| Formula | C14H19BCl2N2O4 |
| Molar mass | 361.03 g·mol−1 |

//////////
see….http://apisynthesisint.blogspot.in/2016/02/takedas-ixazomib-multiple-myeloma-drug.html
Cangrelor, AR-C69931MX Shows Improvement Over Plavix in Phase III Trial

Cangrelor, AR-C69931MX
[dichloro-[[[(2R,3S,4R,5R)-3,4-dihydroxy-5-[6-(2-methylsulfanylethylamino)-2-(3,3,3-trifluoropropylsulfanyl)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]methyl]phosphonic acid
N-[2-(Methylthio)ethyl]-2-[(3,3,3-trifluoropropyl)thio]-5¢-adenylic acid monoanhydride with (dichloromethylene)bis[phosphonic acid]
163706-06-7 cas no
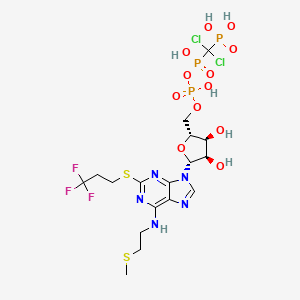 Cangrelor
CangrelorUPDATE
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Kengreal (cangrelor)
MAR 09, 2013
The Medicines Company said yesterday it will pursue marketing approvals for its anti-clotting drug candidate Cangrelor after it met its primary efficacy endpoint in a Phase III clinical trial of improvement compared with Plavix (clopidogrel).
The intravenous small molecule antiplatelet agent reduced by 22% the likelihood of patients experiencing death, myocardial infarction, ischemia-driven revascularization, or stent thrombosis within 48 hours of taking it—to 4.7% from 5.9% of subjects randomized during CHAMPION PHOENIX. The Phase III trial compared Cangrelor to oral Plavix in 11,145 patients undergoing percutaneous coronary intervention.
Cangrelor also showed a 38% reduction (0.8% compared with 1.4%) over Plavix in the likelihood of patients experiencing the key secondary endpoint, incidence of stent thrombosis at 48 hours.
Cangrelor is designed to prevent platelet activation and aggregation that leads to thrombosis in acute care settings, including in patients undergoing percutaneous coronary intervention. During CHAMPION PHOENIX, Cangrelor made its best showing in patients with Q-wave myocardial infarction (QMI), lowering by 39% (to 0.2% compared with 0.3%) the incidence of QMI. Cangelor’s most disappoint showing was its inability to lower the odds of death compared with Clopidogrel; both drugs showed a likelihood of 0.3%.
“Our next step is to submit for market approvals in the U.S. and Europe. We anticipate submitting these data for a new drug application to the U.S. Food and Drug Administration in the second quarter with findings of prior trials, including the BRIDGE trial in patients awaiting open heart surgery,” Simona Skerjanec, PharmD, senior vp and innovation leader for antiplatelet therapies at The Medicines Company, said in a statement.
Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug[1] for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation.[1] Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion.
Poor interim results led to the abandonment of the two CHAMPION clinical trials in mid 2009.[2] The BRIDGE study, for short term use prior to surgery, continues.[3] The CHAMPION PHOENIX trial was a randomized study of over 11,000 patients published in 2013. It found usefulness of cangrelor in patients getting cardiac stents. Compared with clopidogrel given around the time of stenting, intravenous ADP-receptor blockade with cangrelor significantly reduced the rate of stent thrombosis and myocardial infarction.[4] Reviewers have questioned the methodology of the trial.[5]

One particularly preferred example of a reversible, short-acting P2Y12 inhibitor is cangrelor. Cangrelor is a potent, direct, and reversible antagonist of the platelet P2Y12 receptor. Cangrelor has a half-life of approximately less than 10 minutes, allowing for a return to normal platelet function in a very short period of time upon discontinuation of the drug. By reducing the need for a compound to be metabolized for activity, and by having a relatively short half-life, reversible, short-acting P2Y12 inhibitors are considered “reversible,” meaning that full platelet functionality may return rather quickly as compared to thienopyridines.
The binding of cangrelor to the P2Y12 receptor inhibits platelet activation as well as aggregation when mediated in whole or in part via this receptor. Cangrelor can be derived completely from synthetic materials, and is an analogue of adenosine triphosphate (ATP). ATP is a natural antagonist of the P2Y12 receptor sites and is found in humans.
The chemical structure for cangrelor is depicted below as Formula I.

Cangrelor is clinically well tolerated and safe and has no drug-drug interaction with aspirin, heparin or nitroglycerin. Unlike orally dosed thienopyridines, cangrelor can be administered intravenously and binds directly to P2Y12 receptor sites of platelets. In each of the embodiments of the present invention, the term “cangrelor” encompasses the compound of Formula I as well as tautomeric, enantiomeric and diastereomeric forms thereof, and racemic mixtures thereof, other chemically active forms thereof, and pharmaceutically acceptable salts of these compounds, including a tetrasodium salt. These alternative forms and salts, processes for their production, and pharmaceutical compositions comprising them, are well known in the art and set forth, for example, in U.S. Pat. No. 5,721,219. Additional disclosure relevant to the production and use of cangrelor may be found in U.S. Pat. Nos. 5,955,447, 6,130,208 and 6,114,313, as well as in U.S. Appln. Publication Nos. 2006/0270607 and 2011/0112030.
Invasive procedures means any technique where entry to a body cavity is required or where the normal function of the body is in some way interrupted by a medical procedure and/or treatment that invades (enters) the body, usually by cutting or puncturing the skin and/or by inserting instruments into the body. Invasive procedures can include coronary artery bypass grafting (CABG), orthopedic surgeries, urological surgeries, percutaneous coronary intervention (PCI), other general invasive procedures, such as endarterectomy, renal dialysis, cardio-pulmonary bypass, endoscopic procedures or any medical, surgical, or dental procedure that could result in excessive bleeding or hemorrhage to the patient.
Perioperative means the period of a patient’s invasive procedure which can occur in hospitals, surgical centers or health care providers’ offices. Perioperative includes admission, anesthesia, surgery, to recovery.
Thrombosis is the formation of a blood clot (thrombus) inside a blood vessel obstructing the flow of blood through the circulatory system. When a blood vessel is injured, the body uses platelets and fibrin to form a blood clot to prevent blood loss. Some examples of the types of thrombosis include venous thrombosis which includes deep vein thrombosis, portal vein thrombosis, renal vein thrombosis, jugular vein thrombosis, Budd-Chiari syndrome, Paget-Schroetter disease, cerebral venous sinus thrombosis, cerebral venous sinus thrombosis and arterial thrombosis which includes stroke and myocardial infarction.
The compound cangrelor from the Medicines Company is represented by the structure

 OR
ORRN: 163706-36-3



…………
J. Med. Chem., 1999, 42 (2), pp 213–220
http://pubs.acs.org/doi/full/10.1021/jm981072s
10l (AR-C69931MX)
N6–(2-Methylthioethyl)-2-(3,3,3-trifluoropropylthio)-5‘-adenylic Acid, Monoanhydride withDichloromethylenebis(phosphonic acid) (10l). Prepared as the triammonium salt in 4% yield from 3l: 1H NMR δ(D2O) 8.30 (1H, s, H8), 5.97 (1H, d, J = 5.5 Hz, H1‘), 4.65 (1H, m, H2‘), 4.47 (1H, m, H3‘), 4.28 (1H, m, H4‘), 4.17 (2H, m, H5‘a and H5‘b), 3.67 (br s, NHCH2), 3.21 (2H, t, J = 7.6 Hz, SCH2), 2.72 (2H, t, J = 6.6 Hz, SCH2CH2CF3), 2.58 (2H, m, NCH2CH2), 2.04 (3H, s, SCH3);31P NMR δ(D2O) 8.80 (d, 1P, J = 18.6 Hz, Pγ), 0.42 (dd, 1P, J1 = 18.9 Hz, J2 = 28.9 Hz, Pβ), −9.41 (d, 1P, J = 29.0 Hz, Pα). Anal. (C17H34Cl2F3N8O12P3S2·3H2O) H, N, S; C: calcd, 23.16; found, 23.66.
References
- Cangrelor Attenuates Coated-Platelet Formation
- CHAMPION Trials With Cangrelor Stopped for Lack of Efficacy
- What Cangrelor Failure Means to Medicines
- Effect of Platelet Inhibition with Cangrelor during PCI on Ischemic Events (2013) Bhatt, DL etal. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMoa1300815 (published initially online).
- The Duel between Dual Antiplatelet Therapies (2013) Lange, RA and Hillis, LD. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMe1302504
- 15th European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (Sept 6 1998, Edinburgh)1998,:Abst P.281
- Specific P2Y12 purinoceptor antagonist; inhibits ADP-induced platelet aggregation. Prepn: A. H. Ingall et al., WO 9418216 (1994 to Fisons); eidem, US 5721219 (1998 to Astra); and in vivo antithrombotic activity: idem et al., J. Med. Chem. 42, 213 (1999).
- In vivo antithrombotic effects in canine arterial thrombosis: J. Huang et al., J. Pharmacol. Exp. Ther. 295, 492 (2000).
- Mechanism of action study: A. Ishii-Watabe et al., Biochem. Pharmacol. 59, 1345 (2000).
- Clinical safety assessment and evaluation in acute coronary syndromes: R. F. Storey et al., Thromb. Haemostasis 85, 401 (2001); in angina pectoris and non-Q-wave myocardial infarction: F. Jacobsson et al., Clin. Ther. 24, 752 (2002).
- Clinical pharmacodynamics compared with clopidogrel: R. F. Storey et al., Platelets 13, 407 (2002).
- Review of clinical development: S. C. Chattaraj, Curr. Opin. Invest. Drugs2, 250-255 (2001).
- WO2013/103567 A2,
- Journal of Medicinal Chemistry, 1999 , vol. 42, 2 p. 213 – 220
