
Home » Posts tagged 'European Union'
Tag Archives: European Union
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Regulation of Herbal (Traditional) Medicinal Products in the European Union

Regulation of Herbal (Traditional) Medicinal Products in the European Union
| Introduction | |
| The European Union (EU) regulatory framework for medicinal products is complex and is based on the need of a marketing authorization before placing medicines in the market. The main objective is to protect public health by assuring quality, efficacy and safety. The requirements and procedures to obtain a marketing authorization are laid down in regulations, directives and scientific guidelines which are contained in the “Rules Governing Medicinal Products in the European Union”. Several volumes are included which are supported by other publications with complementary information such as scientific or Good Manufacturing Practice (GMP) guidelines, between others [1]. | |
 |
|
| Medicinal plants have been used since Ancient times in all parts of the world. Nonetheless, regulation of herbal medicines in a legal environment was introduced in the 20th century. The EU regulatory framework includes specific requirements for herbal medicinal products (HMP) which are independent from their legal status: traditional herbal medicinal product (THMP) or products based on clinical evidence – well established use (WEU). | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full marketing authorization application, well-established use marketing authorization application or Traditional use marketing registration (Table 1).
|
|
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| EU Pharmaceutical Legislation for Herbal Medicinal Products for Human Use | |
 |
|
| Quality requirements | |
| The principles to assure quality of medicinal products are defined mainly in two Directives of volume 1: Directive 2001/83/EC (which was emended by Directive 2004/24/EC) and Directive 2003/63/EC. | |
| The basic legislation lay down in Directive 2001/83/EC describes the general requirements and provides legal definitions of herbal substances, herbal preparations and herbal medicinal products (Table 2). These concepts are essential for setting quality standards for HMP, as they are by definition complex in nature and so quality requirements set for purified compounds are not suitable for herbal products. | |
| According to the Directive 2001/83/EC, monographs in the European Pharmacopoeia (Eur. Ph.) are legally binding and applicable to all substances which are included in it. For substances which do not have a Eur. Ph. monograph, each Member State (MS) may apply its own national pharmacopoeia. Constituents which are not given in any pharmacopoeia shall be described in the form of a monograph under the same headings included in any monograph in the Eur. Ph., i.e., the name of the substance supplemented by any trade or scientific synonyms; the definition of the substance, set down in a form similar to that used in the European Pharmacopoeia; methods of identification and purity tests. | |
| Moreover, all medicinal products have to be manufactured according to the principles and guidelines of GMP for medicinal products. GMP are applicable to both finished HMP and active substances and, according to Article 46 (f) of Directive 2001/83/EC as amended, marketing authorization holders are required to use as starting materials only active substances which have been manufactured in accordance with the guidelines on the GMP for starting materials as adopted by the Community and distributed in accordance with good distribution practices for active substances. | |
| |
|
| Additional requirements are found in the Directive 2003/63/EC, as Herbal medicinal products differ substantially from conventional medicinal products in so far as they are intrinsically associated with the very particular notion of herbal substances and herbal preparations. It is therefore appropriate to determine specific requirements in respect of these products with regards to the standardized marketing authorization requirements. Then, detailed information on the herbal medicinal product, herbal substances and herbal preparations has to be included, such as the name, address and responsibility of each herbal substance supplier or description of the plant production process, geographical source or drying and storage conditions. The application dossier of a HMP should include specifications and details of all the analytical methods used for testing herbal substances and herbal preparations, results of batch analyses and analytical validation, together with the justification for the specifications. | |
| Most of the quality requirements for HMP are laid down in soft laws (considered as EU measures such as scientific guidelines) which do not have legal force but provide practical harmonization between the MS and the European Medicines Agency (EMA). | |
| The guideline on quality of HMP/THMP covers the general quality aspects of HMP for human and veterinary use, including THMP for human use (EMA, 2014) and indicates which information has to be included in the application dossier. It provides definitions to be taken in account such as genuine (native) herbal preparations, markers, drug to extract ratio (DER) and specifications. Which is more important, it states that the herbal substance or herbal preparation is considered as the whole active substance. In consequence, the quality control of these products has to include appropriate fingerprint analysis to cover not only the content of markers or constituents with known therapeutic activity but also a wider range of chemical constituents. | |
| |
|
| Efficacy requirements | |
| Before a HMP is placed in the market, it must be approved by a MS or by the European Commission by one of the existing types of application: full authorization application, well-established use authorization application or Traditional use registration (Table 3). | |
| The applicant has to submit adequate quality, non-clinical and clinical documentation of the product, irrespectively of the procedure used. Quality requirements of the pharmaceutical product are the same, regardless of the type of application, while efficacy documentation differs between them. The full marketing application is chosen for new medicinal products (new chemical entity) and it has to be completed with the results of pharmaceutical tests (quality documentation), nonclinical (toxicological and pharmacological) studies and clinical trials. Safety data have to be of sufficient size according the existing guidelines; efficacy is demonstrated by results from the clinical trials which have to be in conformity with the guidelines of the corresponding therapeutic area. This type of application is open for HMP, but only a few examples of herbal products have obtained a marketing authorization in the EU in this way. | |
| The well-established medicinal use (WEU) in the EU can be applied to medicinal products for which there exists a wide clinical experience within the EU (not only HMP). The assessment may be based in published controlled clinical trials, non-clinical studies and epidemiological studies. In this type of application, there are no limitations to the therapeutic indication, as this will be derived from the available documentation. | |
| |
|
| (Traditional) Herbal Medicinal Products | |
| Under the Traditional use registration for herbal medicinal product (article 16e), there exist some herbal products that not fulfill the efficacy requirements for a marketing authorization but are endorsed with a long tradition of use. In this case, no clinical trials on these products have been conducted and the efficacy is based on the long-standing use and experience. This simplified registration procedure is limited to products which are intended for use without medical supervision, with a specified strength and posology, to be used by oral, external or inhalation ways, and which can demonstrate a period of use equal or superior to 30 years, including at least 15 years within the EU. In this case, therapeutic indications are limited to those which can be considered safe for use without the supervision of a physician such as minor disorders or symptoms that are benign or self- limiting. In case the applicant should consider another kind of indication, the product must be documented with results of clinical and non-clinical studies, so a full application would be necessary. | |
| Simplified registration of THMP is described in Chapter 2a of Directive 2004/24/EC with three main objectives: a) to protect public health by allowing access to safe and high-quality HMP; b) to allow European citizens the access to medicines of their choice, even those HMP with a long tradition of use and which efficacy hasn’t been proved by clinical trials performed according the modern standards; c) to facilitate movement of medicinal products on the European market. | |
| Directive 2004/24/EC has two different dimensions: the evaluation by National Competent Authorities (NCA) of applications submitted by companies at any MS in the EU and at the EMA, and the establishment of advisory scientific opinions on the medicinal use of herbal substances or preparations. The directive on THMP also established a new scientific committee, the Herbal Medicinal Products Committee (HMPC) at the EMA in London, in 2004, to replace the previous Working Party on Herbal Medicinal Products (CPMP) with the following aims: to elaborate Community monographs and List entries for herbal substances/preparations; to publish scientific guidelines useful for the application of European legal framework; to publish its scientific opinion on questions related to herbal medicinal products and coordinate its work with the European Quality group. The HMPC is made up by 33 members, one member (and one alternate) nominated by each MS of the EU and by Iceland and Norway (the EFAEFTA states). Among them, also five experts are included, representing specific fields of expertise as clinical and non-clinical pharmacology, toxicology or pediatrician medicine. | |
| The guidelines and the monographs developed and approved by the HMPC are accepted by both companies and NCAs and are used for TUR and WEU marketing authorizations. This committee plays a key role in the harmonization of the regulation of HMP whereby Community herbal monographs have a fundamental role. | |
| Usage of Community herbal monographs in the EU regulation of traditional HMP | |
| These documents are established for HMP with regards to bibliographic applications (art. 10 a Directive 2001/83/EC) as well as THPMs. Community monographs reflect the scientific opinion of the HMPC on safety and efficacy data concerning a herbal substance. Any single plant or herbal preparation is assessed individually, according to the available information and includes qualitative and quantitative composition, pharmaceutical form(s), therapeutic indication(s), posology and method of administration, contraindications, special warnings and precautions of use, interactions, use in special population (pregnancy, lactation), effects on ability to drive and use machines, undesirable effects, overdose, pharmacological,pharmacodynamics, pharmacokinetic properties and preclinical safety data. | |
| Community list entry | |
| In the EU, a community list of herbal substances, preparations and combinations thereof for use in THMPs has been established. This list is based in the proposals form HMPC and is gradually developed. Substances or preparations which are included in the list have the main advantage that applicants do not need to provide evidence on the safe or traditional use for its registration at the NCA in the intended use and indication. | |
| A Public statement for one herbal substance/preparation is published because of safety reasons or lack of data to comply with the conditions in the Directive 2004/24/EC (the assessment work didn’t allow a monograph to be published) [2]. | |
| Community monographs are published by the EMA while list entries are approved and published by the European Commission because they are endorsed with a wider legal status: list entries are legally binding and NCAs should not request additional data on safety and traditional use. | |
| The establishment of monographs and list entries is based on the assessment of the published scientific data, together with the existing products in the market. Most of the assessment work is developed by the Monograph and List Working Party (MLWP) at the HMPC, which was established in 2006. In this working group, a member is designed as rapporteur and is responsible of drafting a monograph and/or list entry which will be later on considered and approved by the HPC and then, by the EMA. The documents are published on the EMA website: Community monographs have to be taken in account by the MS when assessing the application of any company. Monographs are note legally binding and MS are not obliged to follow the monographs. | |
| More than 100 species are included in the priority list with the following data: a) scientific data being assessed (R- Rapporteur assigned); b) evaluation report in progress and discussion in the MLWP (D- Draft under discussion); c) scientific opinion under public consultation (PDraft Published); d) comments after public consultation period being evaluated (PF- Assessment close to finalization – pre-final); e) final opinion adopted (F- Final opinion adopted). | |
| MLWP is also responsible of developing guidelines related to legal requirements for TU and WEU, as well as evaluating hazards and problems related to HMP. For the latter, coordination is established with the Safety Working Party (SWP) from the Committee for Medicinal Products for Human Use (CHMP). | |
| Community herbal monographs to support HMPC authorization | |
| A community monograph reflects the scientific opinion from the HMPC in relation to safety and efficacy of one herbal substance/ preparation for medicinal use. AS stated before, a community monograph may be used by a company for a TU or WEU application. That’s the reason why monographs are divided in to two columns: Well Established Use and Traditional Use (simplified application) (Figure 1). WEU is based in the existence of safety data of sufficient size and efficacy data derived from good-quality clinical trials. Traditional use is accepted for those applications which fulfill the criteria shown in the Directive 2004/24/EC. | |
| Each herbal substance/preparation is assessed individually, as the available information may be different for each one. As a result, some substances/preparations may be included in the WEU side, while others will be included in the TU side. If no enough data are available for the substance/preparation, it won’t be included in the monograph. | |
| The approved draft art he HMPC is published for public consultation for 3 month at the EMA website. Comments received are discussed and taken in account when necessary to achieve the final version of the monograph which will be finally published at the MA website. | |
| By the end of 2014, 126 monographs have been adopted and published by the EMA: 104 of them for TU only; 9 of the monographs refer only to WEU (Aloe vera, Cimicifuga racemosa, Rhamnus frangula, Plantago ovata – seed and tegumentum-, Plantago afra, Rheum palmatum, Cassia senna – leaves and fruits-.among them 13 monograph include both TU and WEU. | |
| The main application of a community monograph is to serve as a reference material for the marketing application, both for TU or WEU. Simplified registration is carried on at a national level, so the company gives the dossier to the NCA. With the aim of improving harmonization, the other MSs should recognize the first authorization granted in the first MS, considering that this is based in the European list. | |
| Directive 2004/24/CE established an adaptation period for those herbal products which were on the European market at the moment the Directive was approved. This seven-year period finalized last April 30th, 2011 and implies that nowadays those herbal preparations that not fulfill the actual legislation will not be marketed any more. | |
| In the public report form the EMA last June 2014, the status of updating the medicines registration in the EU was shown. The number Traditional use registrations (TUR) and Well-established use marketing authorizations (WEU) grouped for mono component and combination products has increased in the last years (Figure 2). | |
| The European market for HMP is increasing during the last years and even exceeds prescription medicines. The indications approved cover a wide range of therapeutic areas, most of them characteristic of self-medication diseases: the main therapeutic areas are respiratory tract disorders (cough and cold), mental stress and mood disorders, urinary tract and gynecology disorders, sleep disorders and temporary insomnia (Figure 3). Most of the approved THMP until now were updates of existing authorizations and were based on Community monographs. The Summary of Product Characteristics (SoPC) reflects the items in the corresponding monograph [3]. | |
| A good correlation between the HMPC work and the evaluation of the dossiers from the companies was detected. The relevance of these documents (as shown by the accepted dossiers) is reflected in the HMPC working plan; as an example, last December 2012, 54 among the 56 species with more than 3 marketing authorizations were listed in the priority list. | |
| Conclusión | |
| European legal framework for medicinal products does also include herbal medicinal products to assure their quality, efficacy and safety. The specific characteristics of these products led to the development of a simplified procedure to assure pharmaceutical quality, while keeping safety and efficacy criteria according to marketing authorization granted. | |
| Although the starting point was quite different for the MS, nowadays there exist Community monographs for most of the herbal substances/ preparations that are used in the European market and which form the basis for a harmonization scenario. Moreover, HMPC acts as an International Regulatory Body for herbal medicinal products in order to achieve global standards for this type of medicines, according to other International organization such as the International Conference on Harmonization (ICH). The main tasks the HMPC has to face are those related to herbal medicinal products which have been previously marketed abroad the EU and the increasing existence of combination products within the MS. | |
| References | |
|
|
|
|
||||||||||||
| Table 1: Types of applications for marketing authorization for a HMP in the EU according the Directive 2001/83/EC. |
|
||||||
| Table 2: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
|
||||||
| Table 3: Definitions applicable to herbal medicinal products (Directive 2001/83/EC). |
 |
| Figure 1: European Community Monograph for Valeriana officinalis L., radix, for WEU and TU |
Ruiz-Poveda OMP*
Department of Pharmacology, Faculty of Pharmacy, Universidad Complutense de Madrid, 28040 Madrid, Spain
Ruiz-Poveda OMP
Department of Pharmacology, Faculty of Pharmacy
Universidad Complutense de Madrid, Madrid
Ciudad Universitaria s/n. 28040 Madrid, Spain
Tel: 913 941 767
Fax: 913 941 726
E-mail: olgapalomino@farm.ucm.es
Citation: Ruiz-Poveda OMP (2015) Regulation of Herbal (Traditional) Medicinal Products in the European Union. Pharmaceut Reg Affairs 4:142. doi: 10.4172/2167-7689.1000142
/////////Herbal medicines, Good manufacturing practices, Traditional uses
EMA: European Medicines Agency; DER: Drug to Extract Ratio; HMP: Herbal Medicinal Products; THMP: Traditional Herbal Medicinal Products
The pharmacovigilance system in the European Union (EU)
DRUG REGULATORY AFFAIRS INTERNATIONAL

The pharmacovigilance system in the European Union (EU) operates with the management and involvement of regulatory authorities in Member States, the European Commission and the European Medicines Agency. In some Member States, regional centres are in place under the coordination of the national competent authority.
Within this system, the Agency’s role is to coordinate the EU pharmacovigilance system and to ensure the provision of advice for the safe and effective use of medicines.
More information
- Pharmacovigilance legislation
- Good pharmacovigilance practices
- Risk-management plans
- Medication errors
- Signal management
- Post-authorisation safety studies
- Medicines under additional monitoring
- Periodic safety update reports
- European Risk Management Strategy
- EudraVigilance
- Incident management plan
- Regulatory and procedural guidance

Pharmacovigilance (PV or PhV), also known as Drug Safety, is the pharmacologicalscience relating to the collection, detection, assessment, monitoring, and prevention ofadverse effects with pharmaceutical products.[1] The etymological roots for the word “pharmacovigilance” are:
View original post 7,655 more words
Pixantrone
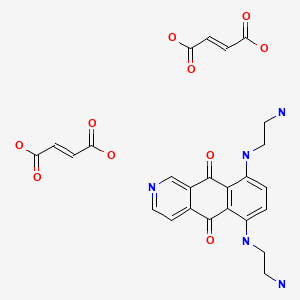

Pixantrone
BBR-2778 , CTK0H5262
- Pixolti
- Pixuvri
- UNII-P0R64C4CR9
An immunosuppressant.
144510-96-3 [RN]
5,8-Bis((2-aminoethyl)amino)-2-aza-anthracene-9,10-dione
6,9-Bis((2-aminoethyl)amino)benz(g)isoquinoline-5,10-dione
5,8-Bis((2-aminoethyl)amino)-2-aza-anthracene-9,10-dione
6,9-Bis((2-aminoethyl)amino)benz(g)isoquinoline-5,10-dione
CTI BioPharma receives Israeli approval for aggressive B-cell non-Hodgkin’s lymphoma therapy
CTI BioPharma has obtained Israeli Ministry of Health’s approval for Pixuvri (pixantrone), as a monotherapy to treat adult patients with multiply relapsed or refractory aggressive B-cell non-Hodgkin’s lymphoma who have received up to three previous courses of treatment.
The company also announced that the Dutch Healthcare Authority and the College voor zorgverzekeringen of the Netherlands have approved funding for Pixuvri as an add-on drug for patients who need a third or fourth-line treatment option for aggressive B-cell lymphoma.
Tel Aviv University faculty of medicine Dr Abraham Avigdor said: “The approval of PIXUVRI in Israel provides patients with aggressive B-cell NHL who have failed second or third-line therapy a new approved option, where none existed before, that can effectively treat their disease with manageable side-effects.
read at
| Pixantrone | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 144510-96-3 |
| PubChem | 134019 |
| ChemSpider | 118174 |
| KEGG | D05522 |
| ChEMBL | CHEMBL167731 |
| ATC code | L01 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C17H19N5O2 |
| Molar mass | 325.365 g/mol |
| Appearance | Blue solid |
| Pharmacology | |
| Routes of administration |
Intravenous |
| Elimination half-life |
9.5–17.5 hours |
| Excretion | Fecal (main route of excretion) and renal (4–9%) |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |

Pixantrone dimaleate [USAN]
CAS 144675-97-8
Molecular Formula
-
C17-H19-N5-O2.2C4-H4-O4
Molecular Weight
- 441.4417
-
Benz(g)isoquinoline-5,10-dione, 6,9-bis((2-aminoethyl)amino)-, (2Z)-2-butenedioate (1:2)
On May 10, 2012, the European Commission issued a conditional marketing authorization valid throughout the European Union for pixantrone for the treatment of adult patients with multiply relapsed or refractory aggressive non-Hodgkin’s B-cell lymphoma (NHL). Pixantrone is a cytotoxic aza-anthracenedione that directly alkylates DNA-forming stable DNA adducts and cross-strand breaks. The recommended dose of pixantrone is 50 mg/m2 administered on days 1, 8, and 15 of each 28-day cycle for up to 6 cycles. In the main study submitted for this application, a significant difference in response rate (proportion of complete responses and unconfirmed complete responses) was observed in favor of pixantrone (20.0% vs. 5.7% for pixantrone and physician’s best choice, respectively), supported by the results of secondary endpoints of median progression-free and overall survival times (increase of 2.7 and 2.6 months, respectively). The most common side effects with pixantrone were bone marrow suppression (particularly of the neutrophil lineage) nausea, vomiting, and asthenia. This article summarizes the scientific review of the application leading to approval in the European Union. The detailed scientific assessment report and product information, including the summary of product characteristics, are available on the European Medicines Agency website (http://www.ema.europa.eu).
Pixantrone (rINN; trade name Pixuvri) is an experimental antineoplastic (anti-cancer) drug, an analogue of mitoxantrone with fewertoxic effects on cardiac tissue.[1] It acts as a topoisomerase II poison and intercalating agent.[2][3] The code name BBR 2778 refers topixantrone dimaleate, the actual substance commonly used in clinical trials.[4]
History
Anthracyclines are important chemotherapy agents. However, their use is associated with irreversible and cumulative heart damage. Investigators have attempted to design related drugs that maintain the biological activity, but do not possess the cardiotoxicity of the anthracyclines.[5] Pixantrone was developed to reduce heart damage related to treatment while retaining efficacy.[1]
Random screening at the US National Cancer Institute of a vast number of compounds provided by the Allied Chemical Company led to the discovery of ametantrone as having significant anti-tumor activity. Further investigation regarding the rational development of analogs of ametantrone led to the synthesis of mitoxantrone, which also exhibited marked anti-tumor activity[5] Mitoxantrone was considered as an analog of doxorubicin with less structural complexity but with a similar mode of action. In clinical studies, mitoxantrone was shown to be effective against numerous types of tumors with less toxic side effects than those resulting from doxorubicin therapy. However, mitoxantrone was not totally free of cardiotoxicity. A number of structurally modified analogs of mitoxantrone were synthesized and structure-activity relationship studies made.[5] BBR 2778 was originally synthesized by University of Vermont researchers Miles P. Hacker and Paul A. Krapcho[5] and initially characterized in vitro for tumor cell cytotoxicity and mechanism of action by studies at the Boehringer Mannheim Italia Research Center, Monza, and University of Vermont, Burlington.[4]Other studies have been completed at the University of Texas M. D. Anderson Cancer Center, Houston, the Istituto Nazionale Tumori,Milan, and the University of Padua.[2][6][4] In the search for novel heteroanalogs of anthracenediones, it was selected as the most promising compound. Toxicological studies indicated that BBR 2778 was not cardiotoxic, and US patents are held by the University of Vermont. An additional US patent application was completed in June 1995 by Boehringer Mannheim, Italy.[5]
Novuspharma, an Italian company, was established in 1998 following the merger of Boehringer Mannheim and Hoffmann-La Roche, and BBR 2778 was developed as Novuspharma’s leading anti-cancer drug, pixantrone.[7] A patent application for the injectable preparation was filed in May 2003.[8]
In 2003, Cell Therapeutics, a Seattle biotechnology company, acquired pixantrone through a merger with Novuspharma.[9]
Clinical trials
Pixantrone is a substance that is being studied in the treatment of cancer. It belongs to the family of drugs called antitumor antibiotics.[10] phase III clinical trials of pixantrone have been completed.[11][12] Pixantrone is being studied as an antineoplastic for different kinds of cancer, including solid tumors and hematological malignancies such as non-Hodgkin lymphomas.
Animal studies demonstrated that pixantrone does not worsen pre-existing heart muscle damage, suggesting that pixantrone may be useful in patients pretreated with anthracyclines. While only minimal cardiac changes are observed in mice given repeated cycles of pixantrone, 2 cycles of traditional anthracyclines doxorubicin or mitoxantrone result in marked or severe heart muscle degeneragion.[1]
Clinical trials substituting pixantrone for doxorubicin in standard first-line treatment of patients with aggressive non-Hodgkin’s lymphoma, had a reduction in severe side effects when compared to patients treated with standard doxorubicin-based therapy. Despite pixantrone patients receiving more treatment cycles, a three-fold reduction in the incidence of severe heart damage was seen as well as clinically significant reductions in infections and thrombocytopenia, and a significant reduction in febrile neutropenia. These findings could have major implications for treating patients with breast cancer, lymphoma, and leukemia, where debilitating cardiac damage from doxorubicin might be prevented.[13]Previous treatment options for multiply relapsed aggressive non-Hodgkin lymphoma had disappointing response rates.[14]
The completed phase II RAPID trial compared the CHOP-R regimen of Cyclophosphamide, Doxorubicin, Vincristine, Prednisone, and Rituximab to the same regimen, but substituting Doxorubicin with Pixantrone. The objective was to show that Pixantrone was not inferior to Doxorubicin and less toxic to the heart.[15]
Pixantrone was shown to have potentially reduced cardiotoxicity and demonstrated promising clinical activity in these phase II studies in heavily pretreated non-Hodgkin lymphomapatients.[14]
The pivotal phase III EXTEND (PIX301) randomized clinical trial studied pixantrone to see how well it works compared to other chemotherapy drugs in treating patients with relapsed non-Hodgkin’s lymphoma.[16] The complete response rate in patients treated with pixantrone has been significantly higher than in those receiving other chemotherapeutic agents for treatment of relapsed/refractory aggressive non-Hodgkin lymphoma.[14]
Administration
It can be administered through a peripheral vein rather than a central implanted catheter as required for other similar drugs.[8][14]
Regulatory approval
U.S. Food and Drug Administration
The FDA granted fast track designation for pixantrone in patients who had previously been treated two or more times for relapsed or refractory aggressive NHL. Study sponsor Cell Therapeutics announced that Pixantrone achieved the primary efficacy endpoint. The minutes of the Oncologic Drugs Advisory Committee meeting of March 22, 2010[17]show that this had not in fact been achieved with statistical significance and this combined with major safety concerns lead to the conclusion that the trial was not sufficient to support approval. In April 2010 the FDA asked for an additional trial.[18]
European Medicines Agency
On May 5, 2009, Pixantrone became available in Europe on a Named-Patient Basis. A named-patient program is a compassionate use drug supply program under which physicians can legally supply investigational drugs to qualifying patients. Under a named-patient program, investigational drugs can be administered to patients who are suffering from serious illnesses prior to the drug being approved by the European Medicines Evaluation Agency. “Named-patient” distribution refers to the distribution or sale of a product to a specific healthcare professional for the treatment of an individual patient. In Europe, under the named-patient program the drug is most often purchased through the national health system.[19] In 2012 pixantrone received conditional marketing authorization in the European Union as Monotherapy to Treat Adult Patients with Multiply Relapsed or Refractory Aggressive Non-Hodgkin B-Cell Lymphomas.
Research
Pixantrone is as potent as mitoxantrone in animal models of multiple sclerosis.[20] Pixantrone has a similar mechanism of action as mitoxantrone on the effector function of lymphomonocyte B and T cells in experimental allergic encephalomyelitis but with lower cardiotoxicity. Pixantrone inhibits antigen specific and mitogen induced lymphomononuclear cell proliferation, as well as IFN-gamma production.[21] Clinical trials are currently ongoing in Europe.
Pixantrone also reduces the severity of experimental autoimmune myasthenia gravis in Lewis rats,[22] and in vitro cell viability experiments indicated that Pixantrone significantly reduces amyloid beta (A beta(1-42)) neurotoxicity, a mechanism implicated in Alzheimer’s disease.[23]


http://www.chemdrug.com/databases/8_0_mhpyqlxgrykqdwig.html
3,4-Pyridinedicarboxylic acid (I) was converted to the cyclic anhydride (II) upon heating with acetic anhydride. Friedel-Crafts condensation of anhydride (II) with p-difluorobenzene (III) in the presence of AlCl3 gave rise to a mixture of two regioisomeric keto acids, (IV) and (V). Cyclization of this mixture in fuming sulfuric acid at 140 C generated the benzoisoquinoline (VI) (1,2). Subsequent displacement of the fluorine atoms of (VI) with ethylenediamine ( VII) in pyridine provided the target bis (2-aminoethylamino) derivative, which was finally converted to the stable dimaleate salt. Alternatively, ethylenediamine (VII) was protected as the mono-N-Boc derivative (VIII) by treatment with Boc2O. Condensation of the difluoro compound (VI) with the protected ethylenediamine (VIII) furnished (IX). The Boc groups of (IX) were then removed by treatment with trifluoroacetic acid. After adjustment of the pH to 4.2 with KOH, treatment with maleic acid provided BBR-2778.
J Med Chem1994,37, (6): 828
SEE MORE
http://www.chemdrug.com/databases/8_0_mhpyqlxgrykqdwig.html
References
- Cavalletti E, Crippa L, Mainardi P, Oggioni N, Cavagnoli R, Bellini O, Sala F. (2007). “Pixantrone (BBR 2778) has reduced cardiotoxic potential in mice pretreated with doxorubicin: comparative studies against doxorubicin and mitoxantrone”. Invest New Drugs. 25 (3): 187–95. doi:10.1007/s10637-007-9037-8. PMID 17285358.
- De Isabella P, Palumbo M, Sissi C, Capranico G, Carenini N, Menta E, Oliva A, Spinelli S, Krapcho AP, Giuliani FC, Zunino F. (1995). “Topoisomerase II DNA cleavage stimulation, DNA binding activity, cytotoxicity, and physico-chemical properties of 2-aza- and 2-aza-oxide-anthracenedione derivatives”. Mol Pharmacol. 48 (1): 30–8.PMID 7623772.
- Evison BJ, Mansour OC, Menta E, Phillips DR, Cutts SM (2007). “Pixantrone can be activated by formaldehyde to generate a potent DNA adduct forming agent”. Nucleic Acids Res. 35 (11): 3581–9. doi:10.1093/nar/gkm285. PMC 1920253.PMID 17483512.
- Krapcho AP, Petry ME, Getahun Z, Landi JJ Jr, Stallman J, Polsenberg JF, Gallagher CE, Maresch MJ, Hacker MP, Giuliani FC, Beggiolin G, Pezzoni G, Menta E, Manzotti C, Oliva A, Spinelli S, Tognella S (1994). “6,9-Bis[(aminoalkyl)amino]benzo[g]isoquinoline-5,10-diones. A novel class of chromophore-modified antitumor anthracene-9,10-diones: synthesis and antitumor evaluations”. J Med Chem. 37 (6): 828–37. doi:10.1021/jm00032a018. PMID 8145234.
- US patent 5587382, Krapcho AP, Hacker MP, Cavalletti E, Giuliani FC, “6,9-bis[(2-aminoethyl) amino]benzo [g]isoquinoline-5,10- dione dimaleate; an aza-anthracenedione with reduced cardiotoxicity”, issued 1996-12-24, assigned to Boehringer Mannheim Italia, SpA
- Zwelling LA, Mayes J, Altschuler E, Satitpunwaycha P, Tritton TR, Hacker MP. (1993). “Activity of two novel anthracene-9,10-diones against human leukemia cells containing intercalator-sensitive or -resistant forms of topoisomerase II”. Biochem Pharmacol. 46 (2): 265–71. doi:10.1016/0006-2952(93)90413-Q. PMID 8394077.
- Borchmann P, Reiser M (May 2003). “Pixantrone (Novuspharma)”. IDrugs 6 (5): 486–90. PMID 12789604.
- EP patent 1503797, Bernareggi A, Livi V, “Injectable Pharmaceutical Compositions of an Anthracenedione Derivative with Anti-Tumoral Activity”, published 2003-11-27, issued 2008-09-29, assigned to Cell Therapeutics Europe S.R.L.
- Pollack, Andrew (2003-06-17). “Company News; Cell Therapeutics Announces Plan To Buy Novuspharma”. The New York Times. Retrieved 2010-05-22.
- Jump up^ Mosby’s Medical Dictionary, 8th edition. © 2009, Elsevier. “definition of antineoplastic antibiotic”. Free Online Medical Dictionary, Thesaurus and Encyclopedia. Retrieved 2012-01-31.
- Jump up^ “NCT00088530”. BBR 2778 for Relapsed, Aggressive Non-Hodgkin’s Lymphoma (NHL). ClinicalTrials.gov. Retrieved 2012-01-31.
- “NCT00551239”. Fludarabine and Rituximab With or Without Pixantrone in Treating Patients With Relapsed or Refractory Indolent Non-Hodgkin Lymphoma. ClinicalTrials.gov. 2012-01-31. Retrieved 2012-01-31.
- “Pixantrone Combination Therapy for First-line Treatment of Aggressive Non-Hodgkin’s Lymphoma Results in Reduction in Severe Toxicities Including Heart Damage When Compared to Doxorubicin-based Therapy”. Press Release. Retrieved 2012-01-31.
- ^ Jump up to:a b c d Engert A, Herbrecht R, Santoro A, Zinzani PL, Gorbatchevsky I (September 2006). “EXTEND PIX301: a phase III randomized trial of pixantrone versus other chemotherapeutic agents as third-line monotherapy in patients with relapsed, aggressive non-Hodgkin’s lymphoma”. Clin Lymphoma Myeloma 7 (2): 152–4.doi:10.3816/CLM.2006.n.055. PMID 17026830.
- Jump up^ “NCT00268853”. A Trial in Patients With Diffuse Large-B-cell Lymphoma Comparing Pixantrone Against Doxorubicin. ClinicalTrials.gov. Retrieved 2012-01-31.
- Jump up^ “NCT00101049”. BBR 2778 for Relapsed, Aggressive Non-Hodgkin’s Lymphoma (NHL). ClinicalTrials.gov. Retrieved 2012-01-31.
- Jump up^ Vesely N, Eckhardt SG (2010-03-22). “NDA 022-481 PIXUVRI (pixantrone dimaleate) injection” (pdf). Summary Minutes of the Oncologic Drugs Advisory Committee. United States Food and Drug Administration. Retrieved 2012-01-31.
- Jump up^ “Cell Therapeutics Formally Appeals FDA’s Nonapprovable Ruling for Pixantrone”. GEN News. 2010-12-03.
- Jump up^ “Pixantrone Now Available in Europe on a Named-Patient Basis”. Retrieved 2012-01-31.
- Jump up^ Gonsette RE, Dubois B (August 2004). “Pixantrone (BBR2778): a new immunosuppressant in multiple sclerosis with a low cardiotoxicity”. J. Neurol. Sci. 223(1): 81–6. doi:10.1016/j.jns.2004.04.024. PMID 15261566.
- Jump up^ Mazzanti B, Biagioli T, Aldinucci A, Cavaletti G, Cavalletti E, Oggioni N, Frigo M, Rota S, Tagliabue E, Ballerini C, Massacesi L, Riccio P, Lolli F (November 2005). “Effects of pixantrone on immune-cell function in the course of acute rat experimental allergic encephalomyelitis”. J. Neuroimmunol. 168 (1-2): 111–7.doi:10.1016/j.jneuroim.2005.07.010. PMID 16120465.
- Jump up^ Ubiali F, Nava S, Nessi V, Longhi R, Pezzoni G, Capobianco R, Mantegazza R, Antozzi C, Baggi F (February 2008). “Pixantrone (BBR2778) reduces the severity of experimental autoimmune myasthenia gravis in Lewis rats”. J. Immunol. 180 (4): 2696–703. PMID 18250482.
- Jump up^ Colombo R, Carotti A, Catto M, Racchi M, Lanni C, Verga L, Caccialanza G, De Lorenzi E (April 2009). “CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity”. Electrophoresis 30(8): 1418–29. doi:10.1002/elps.200800377. PMID 19306269.
