
Home » Posts tagged 'EMA'
Tag Archives: EMA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sodium zirconium cyclosilicate, ナトリウムジルコニウムシクロケイ酸塩

Sodium zirconium cyclosilicate
ZS-9, ZS 9, UZSi-9
CAS 242800-27-7, H2 O3 Si . x H2 O . 2/3 Na . 1/3 Zr, Sodium zirconium cyclosilicate; Silicic acid (H2SiO3), Sodium zirconium(4+) salt (3:2:1), hydrate
USAN CAS 17141-74-1, H6 O9 Si3 . 2 Na . Zr, Silicic acid (H2SiO3), sodium zirconium(4+) salt (3:2:1), hydrate, Sodium zirconium silicate (Na2ZrSi3O9) hydrate
ナトリウムジルコニウムシクロケイ酸塩
ZrH4O6. 3H4SiO4. 2H2O. 2Na, 561.6068, AS IN kegg
Molecular Formula, H6-O9-Si3.2Na.Z, Molecular Weight, 371.5004 as in chemid plus
APPROVED FDA 2018/5/18, LOKELMA, NDA 207078
APPROVED EMA 2018/3/22, LOKELMA
ATC code: V03AE10
UNII-D652ZWF066
| TREATMENT |
selective cation exchanger
Treatment of hyperkalemia |
|---|
Sodium zirconium cyclosilicate (ZS-9) is a selective oral sorbent that traps potassium ions throughout the gastrointestinal tract. It is being developed by ZS Pharma and AstraZeneca for the treatment of hyperkalemia (elevated serum potassium levels).[1]
The product was originated at ZS Pharma, a wholly owned subsidiary of AstraZeneca. In 2015, ZS Pharma was acquired by AstraZeneca.
Hyperkalaemia is the presence of an abnormally high concentration of potassium in the blood. Most data on the occurrence of hyperkalaemia have been obtained from studies of hospitalised patients, and the incidence ranges from 1 to 10%. There is no agreed definition of hyperkalaemia, since the raised level of potassium at which a treatment should be initiated has not been established. The European Resuscitation Council guidelines consider hyperkalaemia to be a serum potassium (S-K) level > 5.5 mmol/L, with mild elevations defined as 5.5 to 5.9 mmol/L, moderate as 6.0-6.4 mmol/L, and severe as ≥ 6.5 mmol/L. The guidelines also note that extracellular potassium levels are usually between 3.5 and 5.0 mmol/L, which is considered the normal range for adults. However, a number of recent retrospective studies have shown the risk of mortality is increased even with only modest elevations of S-K. Mortality risk has been shown to be significantly higher in chronic kidney disease (CKD) patients with S-K levels > 5.0 mmol/L. In acute myocardial infarction patients, a mean postadmission S-K ≥ 5.5 mmol/L during hospitalisation corresponded to a 12-fold increase in death compared with S-K levels between 3.5 and 4.5 mmol/L but, more importantly, S-K levels between 4.5 and 5.0 mmol/L, which is within the normal range, were associated with a 2-fold increased risk of mortality compared with S-K between 3.5 and 4.5 mmol/L.
Sodium zirconium cyclosilicate (ZS) has been developed as treatment for hyperkalaemia. The indication applied for is: Treatment of hyperkalaemia in adult patients, acute and extended use. ZS is an inorganic cation exchange crystalline compound. ZS has a high capacity to selectively entrap monovalent cations, specifically excess potassium and ammonium ions, over divalent cations such as calcium and magnesium, in the gastrointestinal tract. The high specificity of ZS for potassium is attributable to the chemical composition and diameter of the micro pores, which act in an analogous manner to the selectivity filter utilized by physiologic potassium channels. The exchange with potassium ions occurs throughout the gastrointestinal tract with onset in the upper part of the gastrointestinal tract. The trapped potassium ions are excreted from the body via the faeces, thereby reducing any excess and resolving hyperkalaemia. As claimed by the applicant, ZS demonstrates improved capacity, selectivity, and speed for entrapping excess potassium over currently available options for the treatment of hyperkalaemia. The proposed commercial formulation of ZS is a non-absorbed, insoluble, white crystalline powder for suspension with a specific particle size distribution profile. The proposed starting dose of ZS for reversal of hyperkalaemia (when serum potassium is > 5.0 mmol/l) is up to 10 g/day, divided in 3 doses (TID) to achieve normokalaemia.
EMA
The chemical name of the active substance is hydrogen sodium zirconium (IV) silicate hydrate. Due to the natural variability in the manufacturing process of the active substance, it is expected to have the formula Na~1.5H~0.5ZrSi3O9 • 2–3 H2O and relative molecular mass in the range of 390.5 – 408.5. The WHO chose not to designate an INN for the active substance, and a USAN sodium zirconium cyclosilicate is used throughout the dossier and this CHMP AR. The active substance has the following structure:
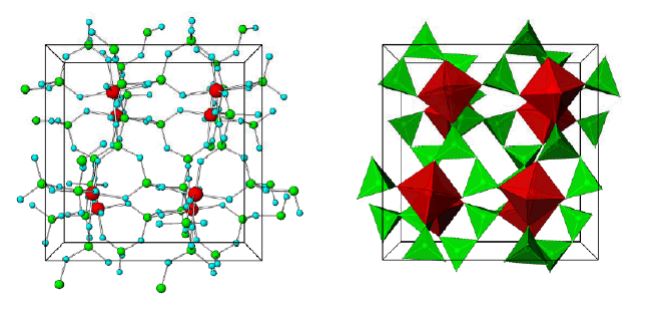
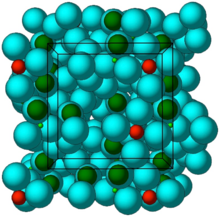
Figure 1. Stick-and-ball (left) and polyhedral (right) unit cell structural representation of the main framework of the microporous sodium zirconium cyclosilicate active substance. Red = zirconium, green = silicon, blue = oxygen atoms. Cations are not pictured.
The structure of sodium zirconium cyclosilicate is a cubic cell arrangement of octahedrally coordinated Zr and tetrahedrally coordinated Si units that interconnect through oxygen bridges as Zr–O–Si and Si–O–Si. The two types of units are observed in a ratio 1:3, respectively, and repeat orderly to form a three-dimensional framework characteristic of the compound. The framework acquires its negative charge from the octahedral fractions, [ZrO6]2– , and features channels and cavities that interconnect and locate the positive ions that counter-balance the negative charge of the framework. Electrostatic interactions between the framework and the cations allow for mobility and possibility of exchange with other cations that would fit and pass the free pore openings of ~ 3.0 Å. The uniform micropore structure allows a high exchange capacity and selectivity for potassium (K+) and ammonium (NH4 +) cations, providing the compound with its distinctive ion-exchange selectivity features responsible for its mode of action. In vitro characterisation of ion selectivity of sodium zirconium cyclosilicate was provided by the applicant and considered satisfactory
The structure of sodium zirconium cyclosilicate was confirmed using synchrotron powder diffraction, standard X-ray powder diffraction, 29Si magic angle spinning solid nuclear magnetic resonance studies (29Si-MASNMR), Fourier transform infrared spectroscopy, inductive coupled plasma-optical emission spectrometry, wave dispersive X-ray microprobe analysis and thermo-gravimetric analysis. Calculations using proprietary software were also used for structure elucidation. The active substance is a white crystalline powder. Bonding interactions in the main framework are considered primarily of covalent nature, with some ionic contribution due to the difference in electronegativity between Si–O and Zr–O. The covalent bonding interactions in all directions within the crystals make sodium zirconium cyclosilicate a compound insoluble in water or in organic solvents. It is neither hygroscopic nor sensitive to light and it is resistant to heat. During the hydrothermal synthesis, the possibility that other crystalline phases are formed exists. The observed crystalline forms are controlled by the manufacturing process parameters and release specifications. Sodium zirconium cyclosilicate is considered to be a new active substance. The applicant demonstrated that neither it, nor its derivatives have ever been active substances in medicinal products authorised in the EU………http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004029/WC500246776.pdf
TGA
DOC]Australian Public Assessment Report for Sodium zirconium … – TGA
Jan 29, 2018 – The sponsor has submitted an application to register a new chemical entity Lokelma,sodium zirconium cyclosilicate hydrate powder for …
The chemical formula of sodium zirconium cyclosilicate hydrate is Na~1.5H~0.5ZrSi3O9.2-3H2O.
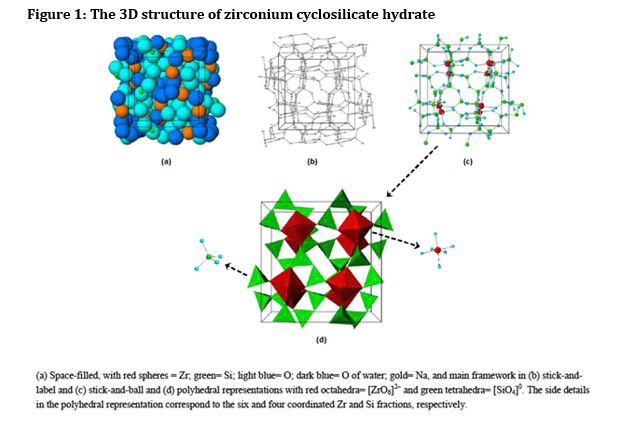

The drug substance ‘sodium zirconium cyclosilicate hydrate’ (abbreviated to ZS) is a white crystalline powder. The structure of ZS is summarised as a cubic cell arrangement of octahedrally coordinated zirconium Zr ([ZrO6]2-) and tetrahedrally coordinated silicon Si ([SiO4]0) units that interconnect through oxygen bridges as Zr-O-Si and Si-O-Si. The two types of units are observed in a ratio of 1:3, respectively, and repeat orderly to form a three dimensional framework characteristic of the compound. The framework acquires its negative charge from the octahedral fractions, [ZrO6]2- and features channels and cavities that interconnect and locate the positive ions (sodium, Na+, and hydrogen, H+) that counter balance the negative charge of the framework.
The manufacturing process is tightly controlled in terms of order of addition of starting material, reaction and crystallisation temperatures, mixing speeds and times, and minimum number of rinses, in order to meet expected yields of the drug substance of an expected quality. In process quality control tests [information redacted] are applied during the manufacturing process to ensure the formation of the correct crystalline structure and batch to batch consistency.
Sodium zirconium cyclosilicate hydrate is completely insoluble.
The drug substance forms part of a family of zirconium silicates that have specific ion exchange properties. Its mechanism of action is based on the cations within its porous crystalline structure, and their ability to freely exchange with a select group of monovalent cations, most specifically the potassium (K+) and ammonium (NH4+) cations. The pore size within the three dimensional crystalline structure has been measured at ~3Å (2.4 x 3.5 Å[1]), which is sufficiently wide enough to trap the potassium monovalent cations which have an approximate ionic diameter of 2.98Å.
The particle size of the drug substance is controlled to maintain a non-systemic mode of action. The sponsor adequately justified not routinely controlling the size of larger particles in the drug substance as differences in particle size were shown to not affect performance as measured by potassium ion exchange capacity (KEC), and there was no correlation between KEC and D90 for clinical lots manufactured.
There are two alternate zirconium silicate crystalline phases which may be formed in the reaction process; Crystalline Phase A (CPA) and Crystalline Phase B (CPB). These layered, two-dimensional structures also exhibit ion exchange properties, although their ion selectivity is less specific for the potassium K+ cations compared to the desired drug substance. PXRD techniques are used to differentiate between the desired drug substance and levels of CPA and CPB. Appropriate limits are applied in the drug substance specification to limit the content of these crystalline phases in the drug substance/drug product.
The quality of the drug substance is controlled by an acceptable specification that includes test and limits for Appearance, Identification (by FTIR and PXRD), KEC , Crystalline Phase A , Crystalline Phase B , Zirconium content , Silicon content , Hafnium content , Moisture content , Particle Size , and Elemental Impurities.
[1] 1 Å = 0.1 nm.

Background
Hyperkalemia occurs in 3 to 10% of hospitalized patients[2] but is often mild. Hyperkalemia can arise from impaired renal function, potassium-sparing diuretics and renin–angiotensin system blockers (e.g., ACE inhibitors, angiotensin receptor blockers, spironolactone) and diabetes mellitus.[2][3][4][5]
There is no universally accepted definition of what level of hyperkalemia is mild, moderate, or severe.[6] However, if hyperkalemia causes any ECG change it is considered a medical emergency[6] due to a risk of potentially fatal abnormal heart rhythms (arrhythmia) and is treated urgently.[6] serum potassium concentrations greater than 6.5 to 7.0 mmol/L in the absence of ECG changes are managed aggressively.[6]
Hyperkalemia, particularly if severe, is a marker for an increased risk of death.[2] However, there is disagreement regarding whether a modestly elevated serum potassium level directly causes significant problems. One viewpoint is that mild to moderate hyperkalemia is a secondary effect that denotes significant underlying medical problems.[2] Accordingly, these problems are both proximate and ultimate causes of death,[2] and adjustment of potassium may not be helpful. Alternatively, hyperkalemia may itself be an independent risk factorfor cardiovascular mortality.[7]
Several approaches are used in the treatment of hyperkalemia.[6] In October 2015, the U.S. Food and Drug Administration (FDA) approved patiromer which works by binding free potassium ions in the gastrointestinal tract and releasing calcium ions for exchange. Previously, the only approved product was sodium polystyrene sulfonate (Kayexalate),[8] an organic ion-exchange resin that nonspecifically binds cations (e.g., calcium, potassium, magnesium) in the gastrointestinal tract. The effectiveness of sodium polystyrene sulfonate has been questioned: a study in healthy subjects showed that laxatives alone were almost as effective in increasing potassium secretion as laxatives plus Kayexalate.[9] In addition, use of sodium polystyrene sulfonate, particularly if formulated with high sorbitol content, is uncommonly but convincingly associated with colonic necrosis.[6][8][10][11]
Mechanism of action

Cross-sections of ZS-9 pores with three different ions (K⁺ = potassium, Na⁺ = sodium, Ca²⁺ = calcium). The specificity for potassium is thought to be caused by the diameter and composition of the pores, which resembles potassium channels.
ZS-9 is a zirconium silicate. Zirconium silicates have been extensively used in medical and dental applications because of their proven safety.[12] 11 zirconium silicates were screened by an iterative optimization process. ZS-9 selectively captures potassium ions, presumably by mimicking the actions of physiologic potassium channels.[13] ZS-9 is an inorganic cation exchanger crystalline with a high capacity to entrap monovalent cations, specifically potassium and ammonium ions, in the GI tract. ZS-9 is not systemically absorbed; accordingly, the risk of systemic toxicity may be minimized.
Clinical studies
A phase 2 clinical trial in 90 patients with chronic kidney disease and mild-to-moderate hyperkalemia found a significantly greater reduction in serum potassium with ZS-9 than placebo. ZS-9 was well tolerated, with a single adverse event (mild constipation).[14]
A double-blind, phase 3 clinical trial in 753 patients with hyperkalemia and underlying chronic kidney disease, diabetes, congestive heart failure, and in patients on renin–angiotensin system blockers compared ZS-9 with placebo.[15] Patients were randomly assigned to receive either ZS-9 (1.25 g, 2.5 g, 5 g, or 10 g) or placebo 3 times daily for 48 hours (acute phase). Patients who achieved normokalemia (serum potassium of 3.5-4.9 mmol/L) were randomly assigned to receive ZS-9 or placebo once daily for 12 additional days (maintenance phase). At the end of the acute phase, serum potassium significantly decreased in the 2.5 g, 5 g, and 10 g ZS-9 groups. During the maintenance phase, once daily 5 g or 10 g ZS-9 maintained serum potassium at normal levels. Adverse events, including specifically gastrointestinal effects, were similar with either ZS-9 or placebo.[15]
A double-blind, phase 3 clinical trial in 258 patients with hyperkalemia and underlying chronic kidney disease, diabetes, congestive heart failure, and in patients on renin–angiotensin system blockers compared ZS-9 with placebo.[16] All patients received 10 g ZS-9 three times daily for 48 hours in the initial open-label phase. Patients who achieved normokalemia (serum potassium 3.5-5.0 mEq/L) were randomly assigned to receive either ZS-9 (5 g, 10 g, or 15 g) or placebo once daily for 28 days (double-blind phase). 98% of patients (n=237) achieved normokalemia during the open-label phase. During the double-blind phase, once daily 5 g, 10 g, and 15 g ZS-9 maintained serum potassium at normal levels in a significantly higher proportion of patients (80%, 90%, and 94%, respectively) than placebo (46%). Adverse events were generally similar with either ZS-9 or placebo. Hypokalemiaoccurred in more patients in the 10 g and 15 g ZS-9 groups (10% and 11%, respectively), versus none in the 5 g ZS-9 or placebo groups.[16]
Regulatory
In the United States, regulatory approval of ZS-9 was rejected by the Food and Drug Administration in May 2016 due to issues associated with manufacturing.[17] On May 18th, 2018, the FDA approved ZS-9 (now known as Lokelma®) for treatment of adults with hyperkalemia.[18]
PATENT
WO 2012109590
PATENT
WO 2015070019
https://patents.google.com/patent/WO2015070019A1/en
The present invention relates to novel zirconium silicate (“ZS”) compositions which are preferably sodium zirconium cyclosilicates having an elevated level of ZS-9 crystalline form relative to other forms of zirconium cyclosilicates (i.e., ZS-7) and zirconium silicates (i.e., ZS-8, ZS-11). The ZS compositions are preferably sodium zirconium cyclosilicate compositions where the crystalline form has at least 95% ZS-9 relative to other crystalline forms of zirconium silicate. The ZS compositions of the present invention unexpectedly exhibit a markedly improved in vivo potassium ion absorption profile and rapid reduction in elevate levels of serum potassium.
[004] Preferably ZS compositions of the present invention are specifically formulated at particular dosages to remove select toxins, e.g., potassium ions or ammonium ions, from the gastrointestinal tract at an elevated rate without causing undesirable side effects. The preferred formulations are designed to remove and avoid potential entry of particles into the bloodstream and potential increase in pH of urine in patients. The formulation is also designed to release less sodium into the blood. These compositions are particularly useful in the therapeutic treatment of hyperkalemia and kidney disease. The present invention also relates to pharmaceutical granules, tablets, pill, and dosage forms comprising the microporous ZS as an active ingredient. In particular, the granules, tablets, pills or dosage forms are compressed to provide immediate release, delayed release, or specific release within the subject. Also disclosed are microporous ZS compositions having enhanced purity and potassium exchange capacity (“KEC”). Methods of treating acute, sub-acute, and chronic hyperkalemia have also been investigated. Disclosed herein are particularly advantageous dosing regimens for treating different forms of hyperkalemia using the microporous ZS compositions noted above. In addition, the present invention relates to methods of co-administering microporous ZS compositions in combination with other pharmacologic drugs that are known to induce, cause, or exacerbate the hyperkalemic condition.
Patent
References
- Jump up^ “ZS-9. A selective potassium binder”. ZS-Pharma.
- ^ Jump up to:a b c d e Elliott, M. J.; Ronksley, P. E.; Clase, C. M.; Ahmed, S. B.; Hemmelgarn, B. R. (2010). “Management of patients with acute hyperkalemia”. Canadian Medical Association Journal. 182 (15): 1631–5. doi:10.1503/cmaj.100461. PMC 2952010
 . PMID 20855477.
. PMID 20855477. - Jump up^ Stevens, M. S.; Dunlay, R. W. (2000). “Hyperkalemia in hospitalized patients”. International Urology and Nephrology. 32 (2): 177–80. doi:10.1023/A:1007135517950. PMID 11229629.
- Jump up^ Navaneethan, S. D.; Yehnert, H.; Moustarah, F.; Schreiber, M. J.; Schauer, P. R.; Beddhu, S. (2009). “Weight Loss Interventions in Chronic Kidney Disease: A Systematic Review and Meta-analysis”. Clinical Journal of the American Society of Nephrology. 4 (10): 1565–74. doi:10.2215/CJN.02250409. PMC 2758256 . PMID 19808241.
- Jump up^ Tamirisa, K. P.; Aaronson, K. D.; Koelling, T. M. (2004). “Spironolactone-induced renal insufficiency and hyperkalemia in patients with heart failure”. American Heart Journal. 148(6): 971–8. doi:10.1016/j.ahj.2004.10.005. PMID 15632880.
- ^ Jump up to:a b c d e f Taal, M.W.; Chertow, G.M.; Marsden, P.A.; Skorecki, K.; Yu, A.S.L.; Brenner, B.M. (2012). Brenner and Rector’s The Kidney (Chapter 17, page 672, 9th ed.). Elsevier. ISBN 978-1-4160-6193-9.
- Jump up^ Fang, J.; Madhavan, S.; Cohen, H.; Alderman, M. H. (2000). “Serum potassium and cardiovascular mortality”. Journal of General Internal Medicine. 15 (12): 885–90. doi:10.1046/j.1525-1497.2000.91021.x. PMC 1495719 . PMID 11119186.
- ^ Jump up to:a b Watson, M.; Abbott, K. C.; Yuan, C. M. (2010). “Damned if You Do, Damned if You Don’t: Potassium Binding Resins in Hyperkalemia”. Clinical Journal of the American Society of Nephrology. 5 (10): 1723–6. doi:10.2215/CJN.03700410. PMID 20798253.
- Jump up^ Emmett, M.; Hootkins, R. E.; Fine, K. D.; Santa Ana, C. A.; Porter, J. L.; Fordtran, J. S. (1995). “Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion”. Gastroenterology. 108 (3): 752–60. doi:10.1016/0016-5085(95)90448-4. PMID 7875477.
- Jump up^ Sterns, R. H.; Rojas, M.; Bernstein, P.; Chennupati, S. (2010). “Ion-Exchange Resins for the Treatment of Hyperkalemia: Are They Safe and Effective?”. Journal of the American Society of Nephrology. 21 (5): 733–5. doi:10.1681/ASN.2010010079. PMID 20167700.
- Jump up^ Kamel, K. S.; Schreiber, M. (2012). “Asking the question again: Are cation exchange resins effective for the treatment of hyperkalemia?”. Nephrology Dialysis Transplantation. 27(12): 4294–7. doi:10.1093/ndt/gfs293. PMID 22989741.
- Jump up^ Denry I, Kelly JR. State of the art of zirconia for dental applications. Dental Materials. Volume 24, Issue 3, March 2008, Pages 299–307
- Jump up^ =Stavros, F (2014). “Characterization of Structure and Function of ZS-9, a K⁺ Selective Ion Trap”. PLOS ONE. 9 (12): e114686. doi:10.1371/journal.pone.0114686. PMC 4273971 . PMID 25531770.
- Jump up^ Ash SR, et al. “Safety and efficacy of ZS-9, a novel selective cation trap, for treatment of hyperkalemia in CKD patients.” American Society of Nephrology 2013 conference, Late-Breaking Abstract.
- ^ Jump up to:a b Packham DK, et al. (2014). “Sodium zirconium cyclosilicate in hyperkalemia”. New England Journal of Medicine. 372 (3): 222–31. doi:10.1056/NEJMoa1411487. PMID 25415807.
- ^ Jump up to:a b Kosiborod M, et al. (2014). “Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia”. Journal of the American Medical Association. 312 (21): 2223–33. doi:10.1001/jama.2014.15688. PMID 25402495.
- Jump up^ Ben Adams (May 27, 2016). “AstraZeneca’s $2.7B hyperkalemia drug ZS-9 rejected by FDA”. FierceBiotech.
- Jump up^ https://www.drugs.com/history/lokelma.html

Crystal structure of ZS-9. Blue spheres = oxygen atoms, red spheres = zirconium atoms, green spheres = silicon atoms.
|
|
| Clinical data | |
|---|---|
| Trade names | Lokelma |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | Not absorbed |
| Excretion | Stool |
| Identifiers | |
| CAS Number | |
| UNII | |
| KEGG | |
//////////////Sodium zirconium cyclosilicate, ナトリウムジルコニウムシクロケイ酸塩 , FDA 2018, EMA, 2018, EU 2018, ZS 9, UZSi-9
O[Si]1(O[Si](O[Si](O1)(O)O)(O)O)O.[Na+].[Na+].[Zr
When can a Chemical Substance be qualified as a “New Active Substance”? The New Reflection Paper of the EMA gives Information

When can a Chemical Substance be qualified as a “New Active Substance”? The New Reflection Paper of the EMA gives Information
A chemical structure with a therapeutic moiety for which no authorisation dossier has been submitted so far and which is – from a chemical structure point of view – not related to any other authorised substances is per se a “NAS” (New Active Substance). But what about a physiologically active molecule present for example in different salts or esters? In which cases do the different derivatives of an effective substance have the NAS status?
The EMA provides clarification to these questions in a new Reflection Paper which was published on 19 January this year. The document entitled “Reflection paper on the chemical structure and properties criteria to be considered for the evaluation of new active substance (NAS) status of chemical substances” describes the criteria according to which isomers, mixtures of isomers, complexes, derivatives, esters, ethers, salts and other solid forms of physiologically active molecules can be classified as “NAS “. If an applicant claims the NAS status of a substance to the regulatory authority in the centralised (CP) or decentralised procedure (MRP/DCP), the authority will first check whether the claim is justified. Afterwards – in case of a positive decision – the usual review of the application dossier will be performed.
The applicant can refer to the criteria described in this Reflection Paper to substantiate his/ her claim of a NAS status. In general, the evidence has to be brought for the derivative in question that it differs significantly in properties with regard to efficacy and /or safety from the already approved active substance.
The scope of this Reflection Papers covers neither biological and biotechnological active substances nor active substances to be included in radiopharmaceuticals.
//////
EMA approves AstraZeneca’s lesinurad to treat gout patients

EMA approves AstraZeneca’s lesinurad to treat gout patients
British-Swedish drugmaker AstraZeneca has received approval from European Medicines Agency (EMA) for its lesinurad 200mg tablets to treat gout patients. READ AT…..[LINK]

“The company submitted a MAA based on data from the Clear1, Clear2 and Crystal pivotal Phase III combination therapy studies.”
AstraZeneca’s subsidiary Ardea Biosciences carried out Clear1, Clear2 and Crystal trials.


EMA Guideline on similar Biological Medicinal Products adopted
![]()

EMA Guideline on similar Biological Medicinal Products adopted
On 23 October, the CHMP adopted the revised Guideline on similar biological medicinal products. Get more details here.

Last year the “Draft Guideline on Similar Biological Medicinal Products” was published by EMA.
After agreement of the revised draft by the Biosimilar Medicinal Products Working Party and Biologics Working Party in July, the CHMP adopted and published the final Guideline on 23 October 2014. They summarized the outline of the document as follows:
“This Guideline outlines the general principles to be applied for similar biological medicinal products (also known as biosimilars) as referred to in Directive 2001/83/EC, as amended, where it is stated that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.
This Guideline describes and addresses the application of the biosimilar approach, the choice of the reference product and the principles for establishing biosimilarity.

The scope of the guideline is to fulfil the requirement of section 4, Part II, Annex I to Directive 2001/83/EC, as amended, which states that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.”
The date for coming into effect is 30 April 2015 (with the advice: After adoption by CHMP applicants may apply some or all provisions of this guideline in advance of this date.). The document replaces the Guideline on similar biological medicinal products (CHMP/437/04).
For further infromation please see the complete “Guideline on similar biological medicinal products“.
European Medicines Agency …Clinical trials in human medicines
![]()
The European Medicines Agency relies on the results of clinical trials carried out by pharmaceutical companies to reach its opinions on the authorisation of medicines. Although the authorisation of clinical trials occurs at Member State level, the Agency plays a key role in ensuring that the standards of good clinical practice (GCP) are applied across the European Economic Area in cooperation with the Member States. It also manages a database of clinical trials carried out in the European Union.
Clinical trials are studies that are intended to discover or verify the effects of one or more investigational medicines. The regulation of clinical trials aims to ensure that the rights, safety and well-being of trial subjects are protected and the results of clinical trials are credible.
Regardless of where they are conducted, all clinical trials included in applications for marketing authorisation for human medicines in the European Economic Area (EEA) must have been carried out in accordance with the requirements set out in Annex 1 ofDirective 2001/83/EC![]() . This means that:
. This means that:
- clinical trials conducted in the EEA have to comply with European Union (EU) clinical-trial legislation (Directive 2001/20/EC
 );
); - clinical trials conducted outside the EEA have to comply with ethical principles equivalent to those set out in the EEA, including adhering to international good clinical practice and the Declaration of Helsinki.
In the EEA, approximately 4,000 clinical trials are authorised each year. This equals approximately 8,000 clinical-trial applications, with each trial involving two Member States on average. Approximately 61% of clinical trials are sponsored by the pharmaceutical industry and 39% by non-commercial sponsors, mainly academia.
Role of the Agency
Clinical-trial data is included in clinical-study reports that form a large part of the application dossiers submitted by pharmaceutical companies applying for a marketing authorisation via the Agency.
The Agency’s Committee for Medicinal Products for Human Use (CHMP) is responsible for conducting the assessment of a human medicine for which an EU-wide marketing authorisation is sought. As part of its scientific evaluation work, the CHMP reviews the clinical-trial data included in the application.
Assessments are based on purely scientific criteria and determine whether or not the medicines concerned meet the necessary quality, safety and efficacy requirements in accordance with EU legislation, particularly Directive 2001/83/EC![]() .
.
Good clinical practice
The Agency plays a central role in ensuring application of good clinical practice (GCP). GCP is the international ethical and scientific quality standard for designing, recording and reporting clinical trials that involve the participation of human subjects.
The Agency works in cooperation with GCP inspectors from medicines regulatory authorities (‘national competent authorities’) in EEA Member States on the harmonisation and coordination of GCP-related activity at an EEA level.
The Agency does not have a role in the approval of clinical-trial applications in the EEA. The approval of clinical-trial applications is the responsibility of the national competent authorities.
EudraCT database and the EU Clinical Trials Register
The Agency is responsible for the development, maintenance and coordination of the EudraCT database. This is a database used by national competent authorities to enter clinical-trial data from clinical trial sponsors and paediatric-investigation-plan (PIP) addressees.
A subset of this data is made available through the European Union Clinical Trials Register, which the Agency manages on behalf of EU Member States and forms part ofEudraPharm![]() , the EU database of medicines.
, the EU database of medicines.
Users are able to view:
- the description of phase-II to phase-IV adult clinical trials where the investigator sites are in the EEA;
- the description of any clinical trials in children with investigator sites in the EU and any trials that form part of a PIP including those where the investigator sites are outside the EU.
As of 21 July 2014, it will be mandatory for sponsors to post clinical trial results in the EudraCT database. A subset of the data included in EudraCT is made available to the public in the European Union Clinical Trials Register. The content and level of detail of these summary results is set out in a European Commission guideline and in its technical guidance. A typical set of summary results provides information on the objectives of a given study, explains how it was designed and gives its main results and conclusions.
The Agency is also working towards the proactive publication of data from clinical trials carried out on the medicines that it authorises. For more information, see release of data from clinical trials.
Clinical trials conducted in countries outside the EU
Clinical trials conducted outside the EU but submitted in an application for marketing authorisation in the EU have to follow the principles which are equivalent to the provisions of the Directive 2001/20/EC![]() .
.
In April 2012, the Agency published the final version of this paper:
This paper aims to strengthen existing processes to provide assurance that clinical trials meet the required ethical and GCP standards, no matter where in the world they have been conducted.
The number of clinical trials and clinical-trial subjects outside Western Europe and North America has been increasing for a number of years. More information is available in this document:
Revision of EU clinical trial legislation
In July 2012, the European Commission published a proposal on a regulation to revise the EU clinical trial legislation.
More information is available at: Revision of the clinical trials directive![]() .
.
Clinical Trials Facilitation Group
The Clinical Trials Facilitation Group![]() (CTFG) is a working group of the Heads of Medicines Agencies that:
(CTFG) is a working group of the Heads of Medicines Agencies that:
- acts as forum for discussion to agree on common principles and processes to be applied throughout the European medicines regulatory network;
- promotes harmonisation of clinical-trial-assessment decisions and administrative processes by national competent authorities;
- operates the voluntary harmonisation procedure for assessment of clinical-trial applications involving several Member States.
The Group is composed of representatives from the clinical-trial departments of the national competent authorities.

World Hepatitis Day – European Medicines Agency uses regulatory tools to facilitate patient access to innovative medicines
DRUG REGULATORY AFFAIRS INTERNATIONAL

25/07/2014
World Hepatitis Day – European Medicines Agency uses regulatory tools to facilitate patient access to innovative medicines
According to the World Health Organization![]() (WHO), viral hepatitis kills 1.4 million people worldwide every year. That is as many as are killed by AIDS/HIV infections.
(WHO), viral hepatitis kills 1.4 million people worldwide every year. That is as many as are killed by AIDS/HIV infections.
Viral hepatitis is caused by five different types of hepatitis viruses, hepatitis A, B, C, D and E, which can lead to the development of acute or chronic inflammation of the liver.
In Europe, hepatitis C virus (HCV) infection is a major public-health challenge. It occurs in between 0.4% and 3.5% of the population in different European Union (EU) Member States and is the most common reason for liver transplantation in the EU.
The treatment paradigm for chronic hepatitis C is currently shifting rapidly with the development of several new classes of direct-acting antivirals. These new medicines display high efficacy rates allowing patients with chronic HCV…
View original post 462 more words
EMA publishes Document on the Validation of analytical Methods

Is it possible to use the results of collaborative trials for analytical methods to prove the laboratory- and product-specific validation of a method? From the perspective of this EMA reflection paper the concrete specifications are missing. These will be developed in the future. Find out more in this news.

GMP News: EMA publishes Document on the Validation of analytical Methods
On 26 June 2014, the European Medicines Agency (EMA) published the concept paper “Transferring quality control methods validated in collaborative trials to a product/laboratory specific context”.
To accept a method an authority always requires a scientific validation. The same applies when existing methods are to be replaced, reduced or to be optimized (3R = replacement, reduction, refinement). Many of these new methods principally represent an improvement compared to the old “standard” methods and therefore are acceptable from a regulatory perspective.
The scientific proof of validation also includes the evidence of the concept and the possibility to transfer a method between different laboratories as well as large scale collaborative studies indicating that a method is suitable for the intended purpose. After completing these steps successfully, it can ultimately result in a monograph of the European Pharmacopoeia (Ph. Eur.) or also in a guidance document for the WHO or the EMA.
This method’s validity has to be proven by the laboratory that proposes the new method. Moreover, this validation also needs to be proven specifically for the medicinal products it is supposed to be used for. Laboratories that participated in large scale collaborative studies before, usually already created plenty of data telling something about the function of this method.
This EMA concept paper now suggests that more guidance documents should be developed on this subject: how can these data from large scale collaborative studies be used to easier implement the laboratory- and product-specific validation of 3R methods (3R – see above)? The concrete specifications for this are currently still missing.
The issue is also to introduce an alternative method without necessarily having to show that the new method correlates with the existing Pharmacopoeia method.
To get additional details please see the complete Reflection Paper “Transferring quality control methods validated in collaborative trials to a product/laboratory specific context“.
The deadline for submission of comments is on 31 October 2014.
EMA publishes final QP Declaration Template

EMA publishes final QP Declaration Template
The European Medicines Agency (EMA) has published the Template for the Qualified Person’s declaration concerning GMP compliance of the active substance used as starting material and verification of its supply chain – “The QP declaration template”.
Read more.
Newly approved drugs: EMA presents figures
In the EU, the number of approved drugs is rising with a new active ingredient; however, stagnated, the number of newly approved generics.
read at
EMA publishes New Process Validation Guideline
![]()
EMA publishes New Process Validation Guideline
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. What’s new? click here
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. The objective of the revision was to integrate modern GMP aspects:
- Integration of the ICH Q8, Q9 and Q10 Guidelines
- Incorporation of Process Analytical Technology (PAT), Quality by Design (QbD) and Real-Time Release Testing (RTRT).
- Extension with regard to an “enhanced approach” and integration of “continuous process verification”
- Integration of the Annexes to the current Note for Guidance
- Harmonisation with the current FDA Guidance on Process Validation
The deadline for comments on the draft for the revision of the process validation guideline ended in October 2012 already. Now, elements in accordance with the Annex 15 have also flowed into the final document. In the following, you will read a short evaluation of the document with regard to the original draft from March 2012, the (still) applicable Note for Guidance on Process Validation and FDA’s Guidance on Process Validation. The GMP relevant aspects of the documents will also be addressed.
The original 7-page long Note for Guidance on Process Validation has more than doubled and now contains 15 pages. Even the original revision draft had only 11 pages. The change in the title to “Guideline on process validation for finished products- information and data to be provided in regulatory submissions” is noticeable. The title itself gives indication about the content of the document, namely marketing authorisation matters.
Like in the draft, the document is composed of 8 numerated chapters, a summary, definitions, references, an Annex I (Process validation scheme) and an Annex II (Standard/non-standard processes) which is a new part compared to the draft. A sub section on “Design space verification” has been newly added to the chapter on process validation.
There haven’t been big changes to the draft document released in 2012. Only the chapter “Design space verification” is brand new, all other parts have been mostly updated. The chapter on ongoing process validation has been removed. Compared to the draft, indications about standard/ non-standard processes are now available in the Annex II – like in the currently applicable Note for Guidance.
What are the changes to the currently applicable Note for Guidance on Process Validation?
Compared to the current Note for Guidance, the revision remains in its final version pretty difficult to read and rather general. This is a marketing authorisation document, which is clearly addressed in the title and only applies to finished dosage forms of chemical medicinal products for human and veterinary use but not for old ones, which are already authorised and on the market. The introduction of a validation life cycle and the integration of continued process verification (CPV) are completely new although this approach is already acquainted from ICH Q8. The “traditional approach” remains accepted. Like in the Annex 15 draft the hybrid approach remains here in the final document “nebulous”. The idea to integrate modern elements from ICH Q8, Q10 (and Q11) into the document is clearly noticeable. Yet, far less concrete references are made to ICH Q9.
A stronger overlap of the FDA Guidance would have been desirable. FDA’s Guidance also deals with APIs and biologicals, and the process validation life cycle runs like a thread through the whole FDA document. FDA’s Guidance also contains GMP aspects. The FDA Guidance explicitly addresses old products which should be integrated to stage 3 of the life cycle. Yet, there is another big difference. The revised document doesn’t highlight statistical methods like the FDA Guidance.
Before the finalisation, a comparison with the Annex 15 has been made which is a nice thing. This explains the long period between the publication of the draft (March 2012) and that of the finalisation (February 2014).
What is significant for the GMP world? On the one hand almost nothing, on the other hand quite a lot: one may wonder why? Direct references to the Annex 15 can be found with regard to the “ongoing process verification” and “concurrent validation”, which is almost nothing looking at the whole document. Moreover, validation in general is required to be executed according to the GMP regarding “continuous process verification” and “change control”; these are the essential parts of the document, and (almost) the complete document should therefore be seen from a GMP perspective.
The new EMA guideline on process validation will apply by the end of August 2014.

