
Home » Posts tagged 'EMA 2021'
Tag Archives: EMA 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
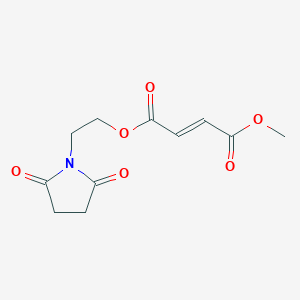
Diroximel fumarate

Diroximel fumarate
ジロキシメルフマル酸エステル;
| Formula |
C11H13NO6
|
|---|---|
| CAS | 1577222-14-0 |
| Mol weight |
255.224
|
2021/11/15 EMA APPROVED, VUMERITY
Treatment of multiple sclerosis
Diroximel fumarate, sold under the brand name Vumerity, is a medication used for the treatment of relapsing forms of multiple sclerosis (MS).[1][3][4]
Diroximel fumarate was approved for medical use in the United States in October 2019,[5] and in the European Union in November 2021.[2]
History
This drug was formulated by Alkermes in collaboration with Biogen.[6]
Society and culture
Legal status
Diroximel fumarate was approved for medical use in the United States in October 2019.[5]
On 16 September 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vumerity, intended for the treatment of adults with relapsing remitting multiple sclerosis.[7] The applicant for this medicinal product is Biogen Netherlands B.V.[7] It was approved for medical use in the European Union in November 2021.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
US 8669281
https://patents.google.com/patent/US8669281B1/en
PATENT
WO 2014152494
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152494
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (14)
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate 14 was synthesized following general procedure 1 (1.03 g, 35 %).
1H NMR (400 MHz, DMSO): δ 6.81 (2H, dd, J = 15.8 Hz); 4.36 (2H, t, J = 5.3 Hz); 3.84 (2H, t, J = 5.1 Hz); 3.80 (3H, s); 2.73 (4H, s). [M+H]+ = 256.07.
General Procedure 1
To a mixture of monomethyl fumarate (MMF) (1.0 equivalent) and HBTU (1.5 equivalents) in DMF (25 ml per g of MMF) was added Hünigs base (2.0 equivalents). The dark brown solution was stirred for 10 minutes, where turned into a brown suspension, before addition of the alcohol (1.0 – 1.5 equivalents). The reaction was stirred for 18 hours at room temperature. Water was added and the product extracted into ethyl acetate three times. The combined organic layers were washed with water three times, dried with magnesium sulphate, filtered and concentrated in vacuo at 45 ºC to give the crude product. The crude product was purified by silica chromatography and in some cases further purified by trituration with diethyl ether to give the clean desired ester product. All alcohols were either commercially available or made following known literature procedures.
As an alternative to HBTU (N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1 -yl)uronium hexafluorophosphate), any one of the following coupling reagents can be used: EDCI/HOBt (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride/hydroxybenzotriazole hydrate); COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate); TBTU (O-(benzotriazol-1 -yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TATU (O-(7-azabenzotriazole-1-yl)-1,1 ,3,3-tetramethyluronium tetrafluoroborate); Oxyma (ethyl (hydroxyimino)cyanoacetate); PyBOP ((benzotriazol-1 -yloxy)tripyrrolidinophosphonium hexafluorophosphate); HOTT (5-(1-oxido-2-pyridyl)-N,N,N’,N’-tetramethylthiuronium hexafluorophosphate); FDPP (pentafluorophenyl diphenylphosphinate); T3P (propylphosphonic anhydride); DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-y1)-4-methylmorpholinium tetrafluoroborate); PyOxim ([ethyl
cyano(hydroxyimino)acetato-O2]tri-1-pyrrolidinylphosphonium hexafluorophosphate); TSTU (N,N,N’,N’-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate); TDBTU (O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TPTU (O-(2-oxo-1(2H)pyridyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TOTU (O-[(ethoxycarbonyl)cyanomethylenamino]-N,N,N’,N’-tetramethyluronium tetrafluoroborate); IIDQ (isobutyl 1,2-dihydro-2-isobutoxy- 1-quinolinecarboxylate); or PyCIU
(chlorodipyrrolidinocarbenium hexafluorophosphate),
As an alternative to Hünig’s base (diisopropylethylamine), any one of the following amine bases can be used: triethylamine; tributylamine; triphenylamine; pyridine; lutidine (2,6-dimethylpyridine); collidine (2,4,6-trimethylpyridine); imidazole; DMAP (4-(dimethylamino)pyridine); DABCO (1 ,4-diazabicyclo[2.2.2]octane); DBU (1 ,8-
diazabicyclo[5.4.0]undec-7-ene); DBN (1,5-diazabicyclo[4.3.0]non-5-ene); or proton sponge® (N,N,N’,N’-tetramethyl-1 ,8-naphthalenediamine).

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
PATENT
WO 2016124960
PATENT
WO 2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Example 3b: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester
Procedure A:
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (3 g; 20.96 mmol) and maleic acid anhydride (2.26 g; 23.1 mmol) in toluene (10 mL) were heated to 60°C under stirring for 29 hours. The temperature was raised to 80°C and heated for another 19 hours. Acetyl chloride (0.3 mL; 4.2 mmol) was added and heating (80°C) was continued for 24 hours. The reaction mixture was cooled to RT. The biphasic system was separated, the upper layer was discarded. The lower layer (viscous oil) crystallized. The crystallized compound was suspended in acetone (50 mL) and stirred for 15 minutes before being filtrated off. The product was dried at 50°C for 5 hours and 8 mbar to yield the 1st crop (1.65 g). The mother liquor was evaporated and the obtained oil/solid was suspended in acetone (5 mL) and stirred overnight at RT. The product was filtrated off and dried at 50°C for 5 hours and 8 mbar to yield the 2nd crop (1.41 g). The mother liquor was evaporated and the obtained oil/solid was suspended in a mixture of diethylether/acetone (5 mL/1 mL) and stirred overnight at RT. The product was filtrated off and dried at 8mbar/50°C for 3 hours (3rd crop, 0.37 g).<a name=”
Yield: 3.43 g (68% of theory)
Purity: 1st crop 96.8 area%; 2nd crop 96.0 area-%; 3rd crop 85.4 area-% (HPLC/UV, method A, λ=200nm; tr: 3.8 min.)
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.61 (s, 4 H) 3.66 (t, J=5.47 Hz, 2 H) 4.23 (t, J=5.47 Hz, 2 H) 6.51 – 6.72 (m, 2 H) 6.60 (s, 1 H) 6.63 (s, 1 H) 13.21 (br s, 1 H)
Procedure B:
Reaction performed in a reactor (Mettler Toledo, Optimax):
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (20 g; 0.14 mol) and maleic acid anhydride (15 g; 0.15 mol) in toluene (70 mL) were heated to 80°C under stirring (150 rpm) for 29 hours. Acetyl chloride (2 mL; 0.03 mol) was added and heating (80°C) was continued overnight. Stirring speed was raised to 200 rpm) after 15.5 hours (at 80°C) (product precipitated upon raising stirring speed. The reaction mixture was cooled to 20°C within 1 hour, directly after highering stirring speed. The reaction mixture was stirred for 4 hours, before being filtrated off. The filtrated precipitate was washed with toluene (30 mL) and then with heptane (70 mL), the product was dried at 60°C and 18 mbar. The crude product (26.26 g) with -90% purity was suspended in a mixture of acetone (30 mL)/heptane (30 mL) and stirred at RT for 2 days. The product was filtrated off, washed with heptane (30 mL) and dried at 50°C and 7 mbar.
Yield: 24.12 g (72% of theory)
Purity: 97.4 area-% at 200 nm
Procedure C:
a) Ethylene carbonate (8.89 g; 0.1 mol), succinimide (10 g; 0.1 mol) and sodium carbonate (0.53 g, 5 mmol) were heated to 100°C, the temperature was hold overnight. The product was cooled down yielding a brownish solid (13.73 g) which was grinded in a mortar.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g, 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (33 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (0.5 mL; 7 mmol) was added and heating<a name=”
(80°C) was continued overnight. Heating was stopped and after stirring for another 2 hours the product was filtered off. The product was dried for 2 hours at 60°C and 8 mbar, yielding 15.82 g of crude product.
purity: 63 area-% at 200nm; 80 area-% at 220 nm
Procedure D
a) Ethylene carbonate (44.43 g; 0.5 mol), succinimide (50 g; 0.5 mol) and sodium carbonate (2.67 g; 25 mmol) were heated to 100°C. The reaction mixture was stirred at 100°C for overnight. The mixture was cooled to RT, yielding 72.4 g of the raw product.
40 g of the raw product were suspended in ethylacetate (40 mL) and heated to reflux for 30 minutes. The turbid mixture was cooled to RT and left stirring O/N. The product was filtrated off and dried under vacuum at RT to yield 29.19 g.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g; 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (30 mL) were heated to 80°C under stirring. Acetyl chloride (0.5 mL; 7 mmol) was added after 19 hours and heating (80°C) was continued overnight. Heating was stopped and stirring was continued for 2 days. The product was filtrated off and dried at 23 mbar and 60°C.
purity: 82 area% at 200 nm; 91 area-% at 220 nm
Procedure E:
a) Succinimide (500 g; 5.0 mol), ethylene carbonate (444.34 g; 5.0 mol) and sodium carbonate (26.74 g; 0.25 mol) were mixed and slowly heated to 130°C under stirring for 7 hours. The product was distilled via vacuum distillation to yield the product as colourless substance (628.14 g; 87% of theory)
b) The distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (150 g; 1.05 mol) from sequence a) and maleic acid anhydride (102.76 g; 1.05 mol) in toluene (350 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (7 mL; 0.01 mol) was added and heating (80°C) was continued. After 6 hours, the reaction mixture was cooled to 20°C within 30 minutes. The product was filtrated off and washed with toluene (200 mL), yielding 221.8 g of a white crystalline product (crude product).
purity: 91 area% at 200 nm; 92 area-% at 220 nm<a name=”
Procedure F:
a) Ethylene carbonate (9.78 g; 0.11 mol), succinimide (10 g; 0.10 mol) and triethylamine (0.7 mL; 5mmol) were heated to 98°C. The reaction mixture was stirred at this temperature overnight. The mixture was cooled to RT, yielding a colourless liquid, which crystallizes upon standing at RT to a colorless solid (14.89 g).
b) The crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione from sequence a) (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (25 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene and dried at 50°C and 8 mbar for 3 hours. Yield: 6.52 g (77%)purity: 93 area% at 200 nm; 94 area-% at 220 nm
Procedure F’
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in a reactor, succinimide (173.07 g, 1.747 mol) and Et3N (12.2 mL, 87.350 mmol) were added and the reaction mixture was warmed up to 90°C and stirred for 24h. Reaction mixture was cooled to 50°C, 500 mL of acetone was added, followed by addition of maleic anhydride (164.19 g, 1.674 mol) and Et3N (10.15 mL, 72.772 mmol). Reaction mixture was stirred at 50-55°C for 4h, cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid.
Yield: 274 g (65%)
Purity: 97.23 area % at 200 nm
Procedure F”
(Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (250 g, 1.036 mol) was suspended in acetone (500 mL) in 1-L reactor, acetyl chloride (5.53 mL, 77.736 mmol) was added drop wise at 20-25°C and reaction mixture was warmed up to 50-55°C and stirred for 20h. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone <a name=”(2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II).
Yield: 231.3 g (92.5%)
Purity: 99.47 area % at 200 nm
Summary:
Procedure B and E, using distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, showed purities of -90-91 area-% of the crude product, ongoing crystallization of the target compound could improve the purity to -97% also shown in procedure A. Distillation of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione needs harsh conditions (Ex. 3a; procedure A). Using the crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, produced with Na2CO3 lead to low product purities of 63 area-% (procedure C).
Crystallization of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (procedure D) lead to product purities comparable to procedure A, B and E with distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, but crystallization is compounded by a significant product loss of – 25%.
The raw 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione could be used without any disadvantageous impact on product quality by substituting Na2CO3 with triethylamine as shown in procedure F with a purity of 93 area-%.
Procedure G
Two experiments were performed in parallel:
Each with 1 g (7 mmol) 1-(2-hydroxy-ethyl)-pyrrolidine-2,5-dione and 0.75 g (7.7 mmol) maleic acid anhydride in 6 mL acetonitrile in screw capped vials. To one of the reaction mixtures was given 0.1 mL triethylamine. Both mixtures were stirred at RT. Samples were taken and investigated by NMR (in DMSO).
product formation after 1 hour (quantified by NMR):
mixture without triethylamine: 0%
mixture with triethylamine: 55%<a name=”
product formation after 2 hours:
mixture without triethylamine: 0%
mixture with triethylamine: 71 %
Procedure H (isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for -24 hours. The reaction was cooled to RT, first a biphasic layer was observed, then the product solidified (sticking to glass wall and stirrer). The product was filtrated off after 2.5 hours of stirring, washed with toluene (50 mL) and dried under vacuum. The dried product was milled and suspended again in toluene (60 mL) at RT, after 30 minutes the product was filtrated off and dried under atmospheric conditions to yield 7.24 g of the cis intermediate (86% of theory). The intermediate product was suspended in toluene (30 mL) and heated to 80°C, acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for 5 hours. The reaction mixture was cooled to RT and stirred for 2 hours. The product was filtrated off, washed with toluene (30 mL) and dried at 50°C and 8 mbar O/N.
purity: 95.6 area-% at 200nm; (0.2% of Impurity I)
Procedure H (without isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene (30 mL) and dried at 50°C and 8 mbar for 3 hours.
purity: 93.2 area-% at 200nm; (1.3% of Impurity I)
Procedure I (scale-up without cis isolation)
Maleic acid (959.09 g; 9.8 mol) was added to a reactor under stirring, which was already loaded with toluene (7 L), then 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione<a name=”
(1400 g; 9.8 mol) was added. Then the mixture was heated to 76°C within ~1 h (up to ~50°C the mixture is a suspension with the tendency of conglomeration of solids, very difficult consistency) at 50°C a turbid solution resulted. Stirring was continued at 80°C for 2 days. Acetyl chloride (138 mL; 1.96 mol) was added under enhanced stirring at 80°C. After -5-10 minutes a crystalline precipitate was formed, which transformed into a pasty/syrupy solid, sticking to reactor walls (difficult handling). Heating was continued overnight (reaction completed after 5 hours as IPC showed). Mixture is still an emulsion, seeding was added and the product precipitated. Stirring at 80°C was continued for ~2 hours then the mixture was cooled to RT. The solid was filtrated off and dried at 50°C and 12 mbar overnight to yield 1818.74 g of the product.
purity: 96.34 area-% at 218 nm; (1.5% of Impurity I)
Procedure J:
2L flask (reaction volume ~1 L): Succinimide (460 g; 4.6 mol), ethylene carbonate (450 g; 5.1 mol) and triethylamine (32 mL; 0.23 mol) were heated to 85°C under stirring overnight. Temperature was raised to 95°-97C and heating was continued O/N. The mixture was cooled to 50°C. Acetonitrile (1600 mL) was charged into a 10 L reactor. To the reaction mixture was added acetonitrile (1000 mL) at 50°C and the solution was transferred to the reactor (reactor T ~22°C), triethylamine (35 mL) was added, then maleic acid anhydride (500.81 g; 5.1 mol). The mixture was heated to 55°C for 5.5 hours. A part of the solvent was distilled off (~1200 mL). Then toluene (1200 mL) was added. The mixture was heated to 90°C. The mixture was cooled to 50°C. At 60°C (clear solution), seeding was added ~300 mg, after -3 minutes a suspension resulted. The mixture was further cooled down to 20°C within 10 hours and kept on stirring O/N. The white crystalline product was filtrated off, washed with toluene (1000 mL) and dried at 55°C and 9 mbar for 2 h to yield 908.99 g (81% yield).
905 g of the isolated, crystallized product was suspended in acetonitrile (2.9 L). Acetyl chloride (23 mL) was added and the mixture was heated to 80°C (clear, colorless solution) for 4 hours. Toluene (1000 mL) was added and the mixture was cooled to RT within 2 hours (linear). The mixture was further cooled to 0°C within 60 minutes. The <a name=”product was filtrated off and washed with toluene (1000 mL). The product was dried overnight at 9 mbar and 50°C.
Yield: 742.06 g (66%)
purity: 99.9 area% at 200 nm
Summary:
Isolation of the cis intermediate leads to a significantly lower content of impurities, in particular of Impurity I. Toluene as solvent leads to disadvantageous conditions regarding consistency of the reaction mixture (procedure H). The use of acetonitrile or acetone (procedure I/F) leads to improved reaction conditions and product quality.
Example 3c
Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) from ethylene carbonate and succinimide (without isolation of
intermediates)
Procedure
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in an 1-L reactor, succinimide (173.07 g, 1.747 mol) and Et3N (24.4 mL, 0.175 mol) were added and the reaction mixture was warmed up to 90-92°C and stirred for 24h. Distillation column<a name=”
was set up on the reactor and the remaining Et3N was distilled off. Reaction mixture was cooled to 40-45°C, 500 mL of acetone was added, followed by addition of maleic anhydride (184 g, 1.878 mol) and Et3N (10.96 mL, 78.615 mmol). Reaction was stirred at 40°C for 6h (precipitation occurred after 3h), cooled to 20-25°C and acetyl chloride (20.86 mL, 0.293 mol) was added drop wise. Reaction mixture was then warmed up to 50-55°C and stirred for 20h. Orange solution crystallized upon seeding. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid. Yield: 352.8 g (83.7%)
Purity: 99.69 area % at 200 nm
Example 4: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester
Procedure A:
The starting material (E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (5 g; 20 mmol) was suspended in dichloromethane (60 mL) and cooled to 0°C, triethylamine (3.16 mL; 22.8 mmol) was added, resulting a clear solution. To this solution methylchloroformate (3.3 mL; 20.7 mmol) was carefully added within 30 minutes via syringe (reaction very exothermic). After 15 min of stirring at 0°C, DMAP (0.25 g; 2.1 mmol) was added into the reaction mixture at 0°C, stirring was continued for 3 hours at 0°C. The reaction mixture was poured into water (200 mL) and additional dichloromethane (100 mL) was added. The organic layer was separated and the aqueous layer was extracted once again with dichloromethane (50 mL). The combined organic layers were washed with brine (50 mL). The solvent was evaporated at 52°C. To the brown oil, which solidified, was added acetone (20 mL) and the mixture was stirred overnight. The product was filtrated off (white solid, part I) (2.73 g) and to the mother <a name=”liquor silica was added, the mixture was evaporated. Acetone (50 mL) was added and silica was filtrated off. The solvent was evaporated and diethylether (30 mL) was added to the solid, the mixture was stirred for ~1 hour. The product was filtrated off (part II) (1.6 g).
Overall yield: 4.33 g (82%)
Purity: part I 100 area-% at 200 nm; part II 97.96 area-% at 200 nm
Procedure A’
(E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) (200 g, 0.829 mol) was suspended in acetone (2000 mL) in 3-L reactor at 20-25°C and cooled to 0°C. Et3N (150.31 mL, 1.078 mol) was added drop wise at 0-5°C. Into resulting solution, methyl chloroformate (83.27 mL, 1.072 mol) was added drop wise at 0-5°C. Reaction mixture was warmed up to 45°C and stirred for 2h. Upon completion, reaction mixture was cooled to 20-25°C and water (600 mL) was added drop wise with maintaining the temperature at 20-25°C resulting with off white to yellowish solution. pH was adjusted to 7 with 1M HCl. One more volume of water was added and pH corrected if needed. Part of acetone from the reaction mixture (5 volumes or 1000 mL) was distilled off under diminished pressure and reactor walls were washed with 1 more volume of water (200 mL), thus resulting in a solution of acetone/water mixture 1:1 (total 10 volumes). Reaction mixture was gradually cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold water (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (Formula I).
Yield: 183.7 g (86.8%)
Purity: 100.00 area % at 200 nm
Crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (170 g) was suspended in acetone (850 mL) at 20-25°C and warmed up to 50°C resulting with colorless solution. Water (850 mL) was added in portions at 50°C and solution was cooled gradually. Crystallization started at 32°C. Reaction mixture was stirred at crystallization temperature for 30 minutes and cooled further to 0°C, stirred at 0°C for 2h and resulting <a name=”white suspension was filtered off and solid was washed with cold water (2×170 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate.
Yield: 152.5 g (89.7%)
Purity: 100.00 area % at 200 nm
Procedure B:
The starting material (5 g, 20 mmol) was suspended in toluene (25 mL). Acetyl chloride (0.29 mL) and methanol (2.5 mL) were added, the reaction mixture was heated to 55°C and stirred for 3 hours. The reaction mixture was poured into water (100 mL) and extracted with ethylacetate (100 mL). The organic layer was separated and dried over sodium sulfate. The solvent was evaporated (crude product 4.7 g, main impurities dimetylfumarate (13%) and fumaric acid (1%) (HPLC at 200 nm)).
Yield: 4.7 g (88%)
Purity: 82.1 area-% at 200 nm
Procedure C:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form A; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (without isolation of (Z)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (30 g; 0.12 mol) was suspended in dichloromethane (DCM, 160 mL) and cooled to 0°C, triethylamine (TEA, 19 mL; 0.14 mol) was added, resulting a clear solution. To this solution methyl chloroformate (19.74 mL; 0.12 mol) was added carefully within 30 minutes via syringe. Stirring was continued for ~2 hours. Water (200 mL) was added to the reaction mixture and stirring was continued for 5-10 minutes. The organic layer was separated and the aqueous layer was washed with another portion of DCM (100 mL). The combined organic layers were dried over sodium sulfate, before being evaporated. To the crude product was added acetone (50 mL) and the mixture was stirred for 3 hours before being filtered off. The product was washed with heptane (50 mL) and dried at 50°C and 21 mbar for 1 h.<a name=”
Yield: 20.52 g (65%)
Purity: 98.7 area-% at 220 nm; (0.3% of Impurity I)
XRPD diffraction peaks: 7.1, 11.6, 13.5, 13.7, 16.3, 16.7, 18.0, 18.4, 21.1, 22.1, 23.1, 23.9, 24.4, 25.5, 27.0, 27.5, 28.0, 28.6, 30.8, 31.2, 31.9, 32.3, 33.7, 34.2, 34.4, 34.9, 35.1, 35.7, 36.0, 36.8, 38.3, 40.1, 40.5, 41.7, 42.4, 43.0, 43.4, 45.0, 45.3, 46.2, 46.4, 47.0, 48.6, 49.4, 49.9, 52.0 + 0.2 degrees two theta.
The Form A according to Procedure C showed a habitus as depicted in Figure 7a
Procedure D:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A The starting material (10 g; 41.5 mmol) was suspended in toluene (70 mL) at 23°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added. Methyl chloroformate (6.58 mL; 41.5 mmol) was slowly added within -30 minutes. After stirring for 2 hours water (40 mL) was added and shortly after acetone (110 mL), stirring was continued for ~2 minutes. The organic layer was separated and washed with brine (15 mL). After drying over sodium sulfate, the solvent was evaporated, yielding a slightly grey solid as crude product (9.42 g). The raw product was suspended in acetone (20 mL) and heptane (20 mL). The mixture was heated to reflux for 15 minutes resulting in a clear solution with just a small amount of solid. The mixture was cooled to RT and stirred overnight (precipitation started at 45°C, cooling: flask left in cooling oil bath ~lh to RT). The resulting product showed polymorphic form A.
Yield: 7.83 g (74%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure D showed a habitus as depicted in Figure 7b<a name=”
Procedure E:
The starting material (1 g; 4.15 mmol) was suspended in dichloromethane (50 mL) at RT. Methyl chloroformate (0.64 mL; 8.3 mmol) was added and stirring was continued overnight, in process control by HPLC showed no conversion.
Procedure F:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (7 g; 0.03 mol) and Na2CO3 were suspended in ethylacetate (50 mL). To the suspension was added methyl chloroformate (3.37 mL; 0.04 mol) in one portion. The reaction mixture was heated to 70°C. The temperature was kept for 15.5 h. The reaction mixture was cooled to 20°C and ethyl acetate (70 mL) was added to the white suspension. The solids were filtrated off and the ethyl acetate layer was washed with water (40 mL), dried over Na2S04 and evaporated to yield 6.4 g of the white crystalline crude product.
The crude product was suspended in a mixture of ethylacetate (10 mL) and heptane (10 mL). The suspension was heated to reflux for 30 minutes, then cooled to 23°C and stirred overnight. The product was filtrated off and dried at 8 mbar and 50°C overnight.
Yield: 5.62 g (75%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure E showed a habitus as depicted in Figure 7c
Procedure G:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form B; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B
(A) 9 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methylester was heated to 115°C. The melted compound was stirred for -20 minutes and then dropped into a precooled mortar (0°C).<a name=”
Purity of form B: 98.8 area-% at 200nm
XRPD-pattern: (Figure 4)
(B) 3.00 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester (form A) was suspended in 150 mL of dibutyl ether. Suspension was heated to 120° C while mixing. Solution was left at 25°C for 2 days. Crystallized material was filtered and dried at 23 °C at 12 mbar.
XRPD-pattern: (Figure 4′)
A measure of the relative volume change of a solid as a response to pressure change is called compressibility. An API should exhibit good compressibility which is dependent on the polymorphic state.
Experimental data:
The compressibility of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester form B and form A was assessed using a die and a flat-faced punch fitted on a TA-XT2 Texture analyser (Stable Micro Systems Ltd., Godalming, UK). 200 mg of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester sample is compressed in a steel mould (with the rate of displacement 0.03 mm/s). Cyclic procedure (similar to tapping) was performed: compressing, then retracting, relaxation for 15 s and then repeated compressive steps (altogether 10 steps). Each step exerts 0.2 MPa pressure on to the sample. Sample density is calculated by dividing the weight by the sample volume for each cycle. Maximum density is reached within 10 steps. Measurements were performed in duplicates for each sample, results are expressed as an average of duplicate measurements.
Results:
<a name=”
Form B of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester exhibits a higher density at compression, indicating superior compressibility compared to form A.
Procedure H:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form C; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form C
(E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (10 g; 41.5 mmol) was suspended in dichloromethane (DCM; 100 mL) and cooled to 0°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added, resulting a clear solution. To the reaction mixture was added methyl chloroformate (6.58 mL; 41.5 mmol) within 30 minutes via a syringe pump. After 15 min of stirring at 0°C, DMAP (0.51 g; 4 mmol) was added into the reaction mixture at 0°C. The resulting solution was stirred at 0°C for 2.5 hours, then the cold suspension was poured into water (70 mL), the reactor was washed with further DCM (20 mL), which was added also to the DCM/water mixture. The organic layer was separated and washed with HCl (32% aq) (5 mL) in water (60 mL), then with water (50 mL) and finally with brine (50 mL). To the obtained deep red to brown solution was added silica (40-63 um) and the mixture was stirred for 5 minutes, before being filtered off to yield a colorless solution, which was evaporated to yield a colorless oil (crude product). The obtained oil was dissolved in a mixture of ethyl acetate/heptane (1/4) (20 mL). The mixture was stirred for 2 days before being filtered off. The product was dried under vacuum.
Yield: 2.87 g (26%)
Purity: 90.9 area-% at 200 nm
XRPD diffraction peaks: 11.2, 11.8, 13.0, 13.6, 13.6, 16.8, 18.1, 19.6, 20.6, 21.2, 21.5, 22.3, 23.2, 23.7, 24.3, 24.4, 25.2, 25.6, 26.5, 27.6, 28.4, 29.1, 30.3, 31.1, 32.0, 33.1, 33.8, 36.1, 36.7, 37.5, 38.4, 38.9, 41.6, 42.5, 43.2, 44.8, 46.5, 48.7, 49.6, 49.9 + 0.2 degrees two theta.<a name=”
Procedure I:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form D; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form D
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B (1 g) was suspended in acetonitrile (3 mL). The suspension was stirred for 7 days in a closed screw cap vials followed by slow evaporation of the solvent under ambient conditions within 3 days.
Purity of form D: 96.3 area-% at 200 nm
XRPD diffraction peaks: 6.9, 11.7, 13.6, 13.9, 16.4, 16.9, 18.2, 20.9, 21.3, 22.3, 23.3, 24.0, 24.6, 25.7, 27.5, 27.7, 31.0, 31.3, 32.1, 32.4, 33.9, 35.3, 35.7, 38.4, 41.9, 42.7, 43.1, 43.6, 44.4, 46.5, 48.9 + 0.2 degrees two theta.
Procedure J:
The starting material (obtained via isolation of (Z)-But-2-enedioic acid mono-[2-(2, 5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (400 g; 1.7 mol) and Na2CO3 (264 g; 2.5 mol) were suspended in ethylacetate (2.7 L). To the suspension was added methyl chloroformate (193 mL; 2.5 mol) at 20°C. The reaction mixture was heated to 45°C within 90 minutes (linear heated). The mixture was kept on stirring for 5.5 hours. Ethylacetate (4 L) was added to the white suspension (at 45 °C). The suspension was stirred for 15 minutes before being filtrated off (45 °C suspension). The reactor was rinsed with another portion of ethylacetate (1 L). The filtrated solids were discarded. To the ethylacetate solution was added a mixture of HClaq (32%) (50 mL) and water (1 L) and the mixture was vigorously stirred for 10 minutes (at 35°C). Then the ethylacetate layer was separated (at ~35°C). The ethylacetate layer was transferred back to the reactor and stirred over sodium sulfate for 30 minutes, sodium sulfate was filtrated off and the ethylacetate layer was reduced to 900 mL. The suspension was transferred into a 3 L flask, equipped with a KPG stirrer and reflux condenser. The mixture was heated to reflux (stirring speed 160 rpm), the suspension was stirred until a clear solution was obtained (-30 minutes). Then heptane (550 mL) was added dropwise within 30 minutes<a name=”
under reflux conditions. Then the mixture (still solution) was slowly cooled to RT. The mixture was stirred O/N. The product was filtrated off and the filter cake was rinsed with heptane (500 mL) to yield the crystalline product (362.56 g; 86%).
purity: 99.8 area% at 218 nm (no Impurity I).
Alternative Procedure: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo- pyrrolidin-1-yl) -ethyl ester methyl ester
Procedure A
Monomethylfumarate (20 g, 0.15 mmol) was suspended in dry dichloromethane (400 mL) at RT, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (32.42 g, 0.17 mol), N-(2-hydroxyethyl)succinimide (21.57 g, 0.15 mol) and dimethylaminopyridine (0.94 g, 7.7 mmol) were added. The solution was stirred O/N at RT. The formed yellow solution was diluted with dichloromethane (300 mL) and washed twice with water (2×500 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. To the crude product was added methyl tert. butyl ether (850 mL) and the reaction mixture was refluxed for 2.5 hours, cooled to RT, then filtrated and heated to reflux again for ~2 hours. After cooling to RT, the mixture was stored at ~5°C for 4 days. The white precipitate was filtrated off and washed with isopropylacetate (25 mL). The crystalline product was dried at 50°C and 7 mbar.
Yield: 10.8 g (28%)
Procedure B
Monomethylfumarate (1.5 g; 11.5 mmol) was suspended in dry DCM (30 mL) at 0°C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (2.47 g; 12.8 mmol), N- (2-hydroxyethyl)succinimide (1.62 g; 11.3 mmol) and DMAP (0.07 g; 0.6 mmol) were added. The solution was stirred overnight at RT. The formed yellow solution was diluted with DCM (50 mL) and washed with water twice (2×35 mL). The organic layer<a name=”
was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-heptane:ethyl acetate 1:1->1:2). The final product showed polymorphic form A. The form A according to alternative Procedure B showed a prismatic habitus as depicted in Figure 7d
Yield: 2.3 g (78%)
Purity: 99.5 area-% at 200 nm
Example 5: Kinetic investigations
Monomethyl maleate was prepared in analogy to WO 2014/197860. Samples of 13.2 grams of monomethyl maleate in 50 mL of toluene and 0.1 equivalents of the isomerization catalyst were reacted at 80°C. Samples were taken after the given times and analyzed by HPLC at 200 nm. The absorbance ratio of monomethyl fumarate (3.8 min.) to monomethyl maleate (2.8 min.) was taken as conversion parameter. The results are shown in Figure 1. As it can be seen from Figure 1 the conversion of monomethyl maleate to monomethyl fumarate in the presence of is TMS (trimethylsilylchloride) is advantageously enhanced compared to the one in the presence of AcCl (acetyl chloride).
Example 6: Yield determination
Six samples of 13.2 g (0.1 mol) monomethyl maleate were diluted with toluene (50 mL) and 0.1 eq of the isomerization catalyst (trimethylsilylchloride or acetyl chloride) were added, three samples with trimethylsilylchloride and three samples with acetyl chloride. The resulting reaction mixtures were heated to the temperatures of 45 °C, 51°C and 80°C. After 22 hours the reaction mixtures were cooled to room temperature, the product was filtrated off and dried at 50°C/8-16 mbar overnight. The results are shown in Figure 2 As it can be seen from Figure 2 the isolated yields of the conversion of monomethyl maleate to monomethyl fumarate in the presence of TMS (trimethylsilylchloride) is at any
PATENT
CN 110698442
PATENT
WO 2021053476
PATENT
IN 201921037120
PATENT
WO 2021074842
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021074842
The drug compound having the adopted name “Diroximel fumarate” has chemical name: 2-(2,5-Dioxopyrrolidin-l-yl)ethyl methyl fumarate as below.
Diroximel fumarate is an investigational, novel oral fumarate with a distinct chemical structure and developed by Alkermes pic, for the treatment of relapsing-remitting multiple sclerosis (RRMS) and is currently under review by U.S. Food and Drug Administration. Biogen, under an exclusive license from Alkermes, intends to market Diroximel fumarate under the brand name VUMERITY™.
US 8669281 B1 first disclosed Diroximel fumarate, its preparation, composition and use thereof for treating multiple sclerosis. US 10080733 B2 further discloses the crystalline solid form of Diroximel fumarate having an X-ray powder diffraction pattern comprising 2Q peaks at 11.6, 21.0, 24.3, 27.4, and 27.9 ±0.2 2Q.
WO 2017/108960 A1 also discloses various alternative synthetic approaches to make Diroximel fumarate and crystalline solid forms thereof, designated as Polymorphic forms A to D.
Hence, there remains a need for alternate solid forms of Diroximel fumarate and preparative processes thereof, exhibiting desired bioavailability and stability. Hence, it is desirable to provide a viable solid form of Diroximel fumarate. The known processes for the preparation of Diroximel fumarate are not viable at industrial scale due to the use of expensive reagents and catalyst such as coupling agents disclosed in US 8669281 Bl, with very low yields. Hence, there remains a need for the improved process to make Diroximel fumarate.
In another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of monomethyl fumarate with (2,5-dioxopyrrolidin-l-yl)ethanol in the presence of an acid halide.
n another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid with methylation agent selected from the group consisting of 2,2-dimethoxypropane, trimethyl orthoformate and dimethyl carbonate.
Diroximel Fumarate
Example-7: Preparation of l-(2-hydroxyethyl)pyrrolidine-2,5-dione
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 0 °C; methyl /ert-butyl ether (300 mL) was added and the resulting mixture was stirred for 30 minutes at the same temperature. The solid was filtered and dried under vacuum for 5 minutes. The solid was combined with ethyl acetate (100 mL) at 0 °C and stirred at the same temperature for 30 minutes. The solid was filtered and dried in rotatory vacuum dryer at 40 °C for 30 minutes to obtain 142.5 g of the title compound as off-white solid with HPLC purity of 99.6%.
Example-8: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (4.0 g) and dichloromethane (40 mL) at 5 °C, Oxalyl chloride (5.85 g) was added slowly in 10 minutes, then a drop of DMF was added at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (5.06 g) and dichloromethane (35 mL), diisopropylethylamine (DIPEA) (9.93 g) was added and cooled the reaction mixture to 5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane was slowly added to this later mixture at 5 °C for 20 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with saturated ammonium chloride solution and the organic layer was separated. Organic layer was washed with 10% citric acid solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (15 mL) at 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with chilled acetone (3 mL) and then with cyclohexane (4 mL). The wet solid was dried at 40 °C under vacuum to obtain 3.3 g of the title compound with HPLC purity of 99.95 %
Example-9: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (100.0 g) and dichloromethane (1000 mL) at 5 °C, Oxalyl chloride (117 g) was added slowly in 15 minutes, then catalytic DMF (1 mL) was added slowly at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (110 g) and dichloromethane (900 mL), diisopropylethylamine (DIPEA) (139 g) was added and cooled the reaction mixture to -5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane (100 mL) was slowly added to this later mixture at – 5 °C for 60 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with water and the organic layer was separated. Organic layer was washed with 10% citric acid solution, 10% NaHC03 solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (400 mL) at 27 °C. The reaction mixture was heated to 45 °C and stirred at the same temperature for 1 hour. The mixture was cooled to 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with methanol (200 mL). The wet solid was dried at 45 °C under vacuum to obtain 120 g of the title compound with HPLC purity of 99.97 %
Example-10: Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 50 °C and triethylamine was removed by evaporation under vacuum. The reaction mixture was heated to 90 °C to distill out the traces of triethylamine under vacuum. The reaction mixture was cooled to 40 °C. Acetone (300 mL), maleic anhydride (106.2 g) and triethylamine (6.31 mL) were added. The resulting mixture was stirred at 40 °C for 6 hours. The mixture was cooled to 20 °C and acetyl chloride (12 mL) was added slowly over a period of 30 minutes. The mixture was slowly heated to 50 °C and stirred for 20 hours at the same temperature followed 2 hours at 0 °C. The solid was filtered and washed with cold acetone (2 x 120 mL).The wet solid was dried at 40 °C for 2 hours to obtain 194.2 g of the title compound as white solid with HPLC purity of 99.55%
Example-11: Preparation of Diroximel fumarate
Diroximel Fumarate
To a mixture of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid (5 g) and 2,2-dimethoxypropane (50 mL) at 29 °C, concentrated hydrochloric acid (1 mL) and water (5 mL) were added and stirred at the same temperature for 17 hours at the same temperature. The pH of the reaction mixture was adjusted to 7 with a saturated aqueous solution of NaHCCh and the solvent was evaporated completely at 40 °C. To the resultant solid, water (50 mL) was added at 29 °C and stirred for 15 minutes. The solid was filtered and dried under vacuum at 29 °C for 5 hours. The resultant solid was combined with methyl /er/-butyl ether (50 mL) at 29 °C and stirred for 20 hours at the same temperature. The solid obtained was filtered and washed with diethyl ether (20 mL). The wet solid was dried under vacuum for 3 hours at 29 °C to obtain 3.3 g of the title compound as white solid with HPLC purity of 98.11%
Example-12: Preparation of Amorphous solid dispersion of Diroximel fumarate with Copovidone
Diroximel fumarate (100 mg) and Copovidone (500 mg) were dissolved in acetone (30 mL) at 30 °C. The clear solution was filtered to make it particle free and the solvent was evaporated in a rotavapor at 45 °C under reduced pressure to obtain the title amorphous solid dispersion. The solid dispersion (100 mg) obtained was combined with Syloid (500 mg) and ground for 20 minutes to obtain the admixture of title compound. XRPD: Amorphous.
Example-13: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and methyl tert. butyl ether (30 mL) was added to the clear solution. A suspension of crystalline Diroximel fumarate seed (0.25 g) in methyl tert. butyl ether (10 m) was added at 40 °C and stirred the mixture at the same temperature for 30 minutes. Methyl tert. butyl ether (280 mL) was added slowly for 2 hours at 41 °C. The mixture was cooled to 25 °C in 3
hours and then to 0 °C in 1 hour. The mixture was stirred at 0 °C for 1 hour and the solid was filtered. The wet solid was washed with methyl tert. butyl ether (40 mL) and dried at 42 °C for 6 hours to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.776 pm, Dv (50) 31.292 pm & Dv (90) 133.437 pm
Example-14: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and DM water (100 mL) was added to the clear solution. A crystalline Diroximel fumarate seed (0.20 g) was added at 42 °C and stirred the mixture at the same temperature for 10 minutes. DM water (100 mL) was added slowly at 41 °C. The mixture was cooled to 28 °C in 1 hour. The mixture was stirred at 28 °C for 2 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 8.59 pm, Dv (50) 61.08 pm & Dv (90) 187.07 pm
Example-15: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 45 °C and DM water (400 mL) was added to the clear solution. The mixture was cooled to 30 °C in 1 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.22 pm, Dv (50) 45.5 pm &
Dv (90) 136.7 pm
Example-16: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in Isopropyl acetate (360 mL) at 55 °C and cooled to 28 °C. A crystalline Diroximel fumarate seed (0.25 g) was added at 28 °C and cool to 5 °C. The mixture was stirred for 1 hour at the same temperature and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.3 pm, Dv (50) 43.18 pm & Dv (90) 133.56 pm
PATENT
IN 201941042131
References
- ^ Jump up to:a b “Vumerity- diroximel fumarate capsule”. DailyMed. Retrieved 1 February 2021.
- ^ Jump up to:a b c “Vumerity EPAR”. European Medicines Agency. 14 September 2021. Retrieved 24 November 2021.
- ^ Wang Y, Bhargava P (July 2020). “Diroximel fumarate to treat multiple sclerosis”. Drugs of Today. 56 (7): 431–437. doi:10.1358/dot.2020.56.7.3151521. PMID 32648853. S2CID 220471534.
- ^ Kourakis S, Timpani CA, de Haan JB, Gueven N, Fischer D, Rybalka E (October 2020). “Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility?”. Pharmaceuticals (Basel, Switzerland). 13 (10): 306. doi:10.3390/ph13100306. PMC 7602023. PMID 33066228.
- ^ Jump up to:a b “Drug Approval Package: Vumerity”. U.S. Food and Drug Administration (FDA). 21 April 2020. Retrieved 1 February 2021.
- ^ “Diroximel fumarate”.
- ^ Jump up to:a b “Vumerity: Pending EC decision”. European Medicines Agency. 15 September 2021. Retrieved 17 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Diroximel fumarate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Vumerity |
| Other names | ALKS-8700 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620002 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H13NO6 |
| Molar mass | 255.226 g·mol−1 |
| 3D model (JSmol) | |
/////////Diroximel fumarate, EU 2021, EMA 2021, APPROVALS 2021, VUMERITY, ジロキシメルフマル酸エステル , K0N0Z40J3W, RDC-5108, дироксимела фумарат , ديروكسيميل فومارات , 富马地罗昔美 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab
(Heavy chain)
EVQLLESGGG LVQPGGSLRL SCAASGFTFS SYDMSWVRQA PGKGLEWVST ISGGGSYTYY
QDSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCASPY YAMDYWGQGT TVTVSSASTK
GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS
LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP
PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE QFNSTYRVVS
VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS QEEMTKNQVS
LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS
CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQLTQSPSF LSAYVGDRVT ITCKASQDVG TAVAWYQQKP GKAPKLLIYW ASTLHTGVPS
RFSGSGSGTE FTLTISSLQP EDFATYYCQH YSSYPWTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H130-L214, H143-H199, H222-H’222, H225-H’225, H257-H317, H363-H421, H’22-H’96, H’130-L’214, H’143-H’199, H’257-H’317, H’363-H’421, L23-L88, L134-L194, L’23-L’88, L’194-L’134)
>Heavy Chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYDMSWVRQAPGKGLEWVSTISGGGSYTYY QDSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCASPYYAMDYWGQGTTVTVSSASTK GPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFP PKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVS VLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVS LTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFS CSVMHEALHNHYTQKSLSLSLGK
>Light Chain DIQLTQSPSFLSAYVGDRVTITCKASQDVGTAVAWYQQKPGKAPKLLIYWASTLHTGVPS RFSGSGSGTEFTLTISSLQPEDFATYYCQHYSSYPWTFGQGTKLEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
References:
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
Dostarlimab
Immunoglobulin G4, anti-(programmed cell death protein 1 (PDCD1)) (humanized clone ABT1 γ4-chain), disulfide with humanized clone ABT1 κ-chain, dimer
Protein Sequence
Sequence Length: 1314, 443, 443, 214, 214multichain; modified (modifications unspecified)
- GSK-4057190
- GSK4057190
- TSR 042
- TSR-042
- WBP-285
- ANB 011
| Formula | C6420H9832N1680O2014S44 |
|---|---|
| CAS | 2022215-59-2 |
| Mol weight | 144183.6677 |
Jemperli FDA 2021/4/22 AND EMA 2021/4/21
NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Jemperli | Injection | 50 mg/1mL | Intravenous | GlaxoSmithKline LLC | 2021-04-22 | Not applicable |  |
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
View full prescribing information for Jemperli.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Side effects
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
References[
- ^ Jump up to:a b c d e f g h i j k “FDA grants accelerated approval to dostarlimab-gxly for dMMR endometri”. U.S. Food and Drug Administration(FDA) (Press release). 22 April 2021. Retrieved 22 April 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h i “Jemperli- dostarlimab injection”. DailyMed. Retrieved 28 April 2021.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Dostarlimab, American Medical Association.
- ^ World Health Organization (2018). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 119” (PDF). WHO Drug Information. 32 (2).
- ^ “FDA grants accelerated approval for GSK’s Jemperli (dostarlimab-gxly) for women with recurrent or advanced dMMR endometrial cancer” (Press release). GlaxoSmithKline. 22 April 2021. Retrieved 22 April 2021 – via PR Newswire.
- ^ Jump up to:a b Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
- ^ “Data from GARNET study indicates robust activity of dostarlimab in patients with advanced or recurrent endometrial cancer”. Tesaro (Press release). Retrieved 1 January 2020.
- ^ Scalea B (28 May 2019). “Dostarlimab Effective in Endometrial Cancer Regardless of MSI Status”. Targeted Oncology. Retrieved 1 January 2020.
- ^ “GSK Presents New Data from the GARNET Study Demonstrating Potential of Dostarlimab to Treat a Subset of Women with Recurrent or Advanced Endometrial Cancer – Drugs.com MedNews”. Drugs.com. Retrieved 29 April 2020.
- ^ “FDA Approves New Immunotherapy for Endometrial Cancer”. Medscape. Retrieved 23 April 2021.
- ^ Jump up to:a b “Jemperli: Pending EC decision”. European Medicines Agency (EMA) (Press release). 25 February 2021. Retrieved 22 April 2021.
External links
- “Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
- Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
- Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
- Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
- FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
- FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | PCDP1 |
| Clinical data | |
| Trade names | Jemperli |
| Other names | TSR-042, WBP-285, dostarlimab-gxly |
| License data | US DailyMed: Dostarlimab |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | L01XC40 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2022215-59-2 |
| PubChem SID | 384585344 |
| DrugBank | DB15627 |
| UNII | P0GVQ9A4S5 |
| KEGG | D11366 |
| Chemical and physical data | |
| Formula | C6420H9832N1690O2014S44 |
| Molar mass | 144325.73 g·mol−1 |
/////////Dostarlimab, PEPTIDE, ANTINEOPLASTIC, CANCER, ドスタルリマブ , GSK 4057190, GSK4057190, TSR 042, TSR-042, WBP-285, FDA 2021, EU 2021

{kind=link}