



The latest set of opinions from advisors to the European Medicines Agency include recommendations to approve six new medicines, including Bristol-Myers Squibb’s new hepatitis C drug and Eli Lilly’s biosimilar of the Sanofi diabetes blockbuster Lantus.
Home » Posts tagged 'eli lilly'
Tag Archives: eli lilly
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
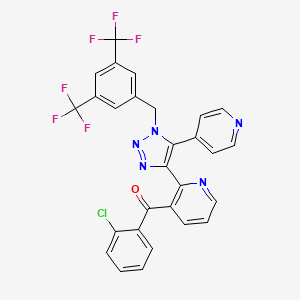
ATICAPRANT


ATICAPRANT
1174130-61-0
BENZAMIDE, 4-(4-(((2S)-2-(3,5-DIMETHYLPHENYL)-1-PYRROLIDINYL)METHYL)PHENOXY)-3-FLUORO-
C26H27FN2O2, 418.512
- 4-[4-[[(2S)-2-(3,5-Dimethylphenyl)-1-pyrrolidinyl]methyl]phenoxy]-3-fluorobenzamide (ACI)
- (S)-4-(4-((2-(3,5-Dimethylphenyl)pyrrolidin-1-yl)methyl)phenoxy)-3-fluorobenzamide
- 4-(4-{[(2S)-2-(3,5-dimethylphenyl)pyrrolidin-1-yl]methyl}phenoxy)-3-fluorobenzamide
- Aticaprant
- CERC 501
- JNJ 67953964
- JNJ 67953964AAA
- LY 2456302
- S-Aticaprant
- CERC-501
- JSPA 0658 JSPA-0658 JSPA0658
- LY 2456302 LY-2456302 , LY2456302
- OriginatorEli Lilly and Company
- DeveloperAvalo Therapeutics; Eli Lilly and Company; Johnson & Johnson Innovative Medicine
- ClassAntidepressants; Benzamides; Benzene derivatives; Drug withdrawal therapies; Fluorinated hydrocarbons; Pyrrolidines; Smoking cessation therapies
- Mechanism of ActionOpioid kappa receptor antagonists
- Phase III Major depressive disorder
- DiscontinuedAlcoholism; Cocaine-related disorders; Smoking withdrawal
- 26 Jun 2024Janssen Research & Development initiates a phase III VENTURA-7 trial for Major depressive disorder (Adjunctive treatment) in USA (PO, Tablet) (NCT06514742) (EudraCT2024-511557-21-00)
- 01 Oct 2023Janssen Pharmaceuticals is now called Johnson & Johnson Innovative Medicine (Janssen Pharmaceuticals website, October 2023)
- 19 May 2023Chemical structure information added
Aticaprant, also known by its developmental codes JNJ-67953964, CERC-501, and LY-2456302, is a κ-opioid receptor (KOR) antagonist which is under development for the treatment of major depressive disorder.[2][3][4] A regulatory application for approval of the medication is expected to be submitted by 2025.[2] Aticaprant is taken by mouth.[1]
Side effects of aticaprant include itching, among others.[4][5] Aticaprant acts as a selective antagonist of the KOR, the biological target of the endogenous opioid peptide dynorphin.[3] The medication has decent selectivity for the KOR over the μ-opioid receptor (MOR) and other targets, a relatively long half-life of 30 to 40 hours, and readily crosses the blood–brain barrier to produce central effects.[4][6]
Aticaprant was originally developed by Eli Lilly, was under development by Cerecor for a time, and is now under development by Janssen Pharmaceuticals.[2] As of July 2022, it is in phase 3 clinical trials for major depressive disorder.[2] Like other kappa opioid antagonists currently under clinical investigation for the treatment of major depression, its efficacy may be compromised by the countervailing activation of pro-inflammatory cytokines in microglia within the CNS.[7]
Aticaprant was also under development for the treatment of alcoholism, cocaine use disorder, and smoking withdrawal, but development for these indications was discontinued.[2]
Pharmacology
Pharmacodynamics
Aticaprant is a potent, selective, short-acting (i.e., non-“inactivating”) antagonist of the KOR (Ki = 0.81 nM vs. 24.0 nM and 155 nM for the μ-opioid receptor (MOR) and δ-opioid receptor (DOR), respectively; approximately 30-fold selectivity for the KOR).[8][9][10] The drug has been found to dose-dependently block fentanyl-induced miosis at 25 mg and 60 mg in humans (with minimal to no blockade at doses of 4 to 10 mg), suggesting that the drug significantly occupies and antagonizes the MOR at a dose of at least 25 mg but not of 10 mg or less.[10] However, a more recent study assessing neuroendocrine effects of the drug in normal volunteers and subjects with a history of cocaine dependence reported observations consistent with modest MOR antagonism at the 10 mg dose.[11] In animal models of depression, aticaprant has been found to have potent synergistic efficacy in combination with other antidepressants such as citalopram and imipramine.[12]
Positron emission tomography imaging revealed that brain KORs were almost completely saturated by the drug 2.5 hours following a single dose of 10 mg, which supported the 4 mg to 25 mg dosages that aticaprant is being explored at in clinical trials.[13][14] Occupancy was 35% for a 0.5 mg dose and 94% for a 10 mg dose.[15][14] At 24 hours post-dose, receptor occupancy was 19% for 0.5 mg and 82% for 25 mg.[15][14] No serious side effects were observed, and all side effects seen were mild to moderate and were not thought to be due to aticaprant.[14]
Pharmacokinetics
The oral bioavailability of aticaprant is 25%.[1] The drug is rapidly absorbed, with maximal concentrations occurring 1 to 2 hours after administration.[1] It has an elimination half-life of 30 to 40 hours in healthy subjects.[1] The circulating levels of aticaprant increase proportionally with increasing doses.[1] Steady-state concentrations are reached after 6 to 8 days of once-daily dosing.[1] Aticaprant has been shown to reproducibly penetrate the blood–brain barrier.[13][14]
History
Aticaprant was originally developed by Eli Lilly under the code name LY-2456302.[2] It first appeared in the scientific literature in 2010 or 2011.[16][17] The compound was first patented in 2009.[18]
In February 2015, Cerecor Inc. announced that they had acquired the rights from Eli Lilly to develop and commercialize LY-2456302 (under the new developmental code CERC-501).[19]
As of 2016, aticaprant has reached phase II clinical trials as an augmentation to antidepressant therapy for treatment-resistant depression.[20][12] A phase II study of aticaprant in heavy smokers was commenced in early 2016 and results of the study were expected before the end of 2016.[14] Aticaprant failed to meet its main endpoint for nicotine withdrawal in the study.[21]
In August 2017, it was announced that Cerecor had sold its rights to aticaprant to Janssen Pharmaceuticals.[22][21] Janssen was also experimenting with esketamine for the treatment of depression as of 2017.[21]
Research
In addition to major depressive disorder, aticaprant was under development for the treatment of alcoholism, cocaine use disorder, and smoking withdrawal.[2] However, development for these indications was discontinued.[2]
See also
κ-Opioid receptor § Antagonists
SCHEME

SYNTHESIS
WO/2024/178082COMPOSITION OF OPIOID RECEPTOR MODULATOR AND MDMA FOR USE THEREOF
WO/2024/173843QUINOLINE DERIVATIVES WHICH ACT AS KAPPA-OPIOID RECEPTOR ANTAGONISTS
20240238245COMPOSITIONS AND METHODS FOR THE TREATMENT OF DEPRESSION
20240189274Compositions And Methods For The Treatment Of Depression
WO/2024/102802ZELATRIAZIN FOR THE TREATMENT OF DEPRESSION
WO/2024/100285TREATMENT OF A COGNITIVE DISORDER WITH AN AGENT THAT INCREASES THE..
117615757Compositions and methods for treating depression
117142999Racemization method of drug intermediate
20230348377PURE FORMS OF CRYSTALLINE ATICAPRANT
WO/2023/170550POLYMORPH FORMS OF ATICAPRANT FOR USE IN TREATING MAJOR DEPRESSIVE DISORDER
WO/2023/170547PURE FORMS OF CRYSTALLINE ATICAPRANT
20230277499Forms of aticaprant
20230277500COMPOSITIONS COMPRISING ATICAPRANT
WO/2023/164385NEUROACTIVE STEROIDS FOR TREATMENT OF GASTROINTESTINAL DISEASES OR CONDITIONS
20090186873Kappa selective opioid receptor antagonist
WO/2009/094260KAPPA SELECTIVE OPIOID RECEPTOR ANTAGONIST
20100197669Kappa selective opioid receptor antagonist
2252581KAPPA SELECTIVE OPIOID RECEPTOR ANTAGONIST
201500053151-substituted 4-arylpiperazine as kappa opioid receptor antagonists
WO/2013/0864961-SUBSTITUTED 4-ARYLPIPERAZINE AS KAPPA OPIOID RECEPTOR ANTAGONISTS
101925576Kappa selective opioid receptor antagonist
PAPERS
ACS Omega (2020), 5(41), 26938-26945 https://pubs.acs.org/doi/full/10.1021/acsomega.0c04329


REF https://pubs.acs.org/doi/suppl/10.1021/acsomega.0c04329/suppl_file/ao0c04329_si_001.pdf
N-Methoxy-N-methyl-4-chlorobutyramide (S1). To a mixture of N,O-dimethylhydroxylamine hydrochloride (95.0 mmol, 9.27 g) in CH2Cl2 (150 mL) was
added 2 M NaOH (300 mmol, 150 mL) and 4-chlorobutyryl chloride (100 mmol,
11.2 mL) at 0 ˚C. The mixture was stirred for 42 h at room temperature. The
organic phase was separated, and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was washed with 2 M NaOH (100 mL), dried over Na2SO4, filtered, and concentrated
to afford the title comlund in 75% yield as a colorless liquid.
1H NMR (400 MHz, CDCl3) : 2.08-2.15
(m, 2H), 2.63 (t, J = 7.0 Hz, 2H), 3.19 (s, 3H), 3.64 (t, J = 6.3 Hz, 2H), 3.71 (s, 3H).
13C{
1H} NMR (100
MHz, CDCl3) : 27.1, 28.6, 32.1, 44.6, 61.1. IR (max/cm-1
): 2965, 2940, 2821, 1656, 14421, 1417, 1387,
1178, 1107, 997. HRMS (ESI+): calculated for [M+Na]+
: 188.0449, found: 188.0450.
4-Chloro-1-(3,5-dimethylphenyl)butan-1-one (S2). To a mixture of N-methoxy-N-methyl-4-chlorobutyramide (S1, 65.0 mmol, 10.8 g) in anhydrous Et2O
(100 mL) was added dropwise 3,5-dimethylphenylmagnesium bromide (ca. 1 M
in Et2O, ca. 130 mmol, prepared from 1-bromo-3,5-dimethylbenzene (130 mmol,
17.7 mL) and Mg turnings (169 mmol, 4.11 g) in anhydrous Et2O (130 mL)) over 1 h at -40 ˚C under Ar.
The reaction mixture was stirred at room temperature for 20 h. After cooling to 0 ˚C, saturated NH4Cl
solution (200 mL) was added. The organic phase was separated, washed with water (100 mL) and brine
(100 mL), dried over Na2SO4, and filtered. After concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to afford the title compound in 91% yield as a greenish
yellow liquid.
1H NMR (400 MHz, CDCl3) : 2.18-2.25 (m, 2H), 2.38 (s, 6H), 3.15 (t, J = 7.0 Hz, 2H),
3.67 (t, J = 6.3 Hz, 2H), 7.21 (s, 1H), 7.58 (s, 2H). 13C{
1H} NMR (100 MHz, CDCl3) : 21.2, 26.8, 35.4,
44.7, 125.8, 134.8, 136.8, 138.3, 199.4. IR (max/cm-1
): 3047, 3006, 2961, 2920, 2868, 1443, 1411, 1322,
1303, 1181, 1159, 844, 785, 687. HRMS (APCI+): calculated for [M+H]+
: 211.0884, found: 211.0884.
(RS)-N-(4-Chloro-1-(3,5-dimethylphenyl)butylidene)-tertbutanesulfinamide (S3). Ti(OEt)4 (100 mol, 21.0 mL) was added to a mixture
of (RS)-tert-butanesulfinamide (1.0 M in THF, 50 mmol, 50 mL) and 4-chloro1-(3,5-dimethylphenyl)butan-1-one (S2, 50.0 mmol, 10.5 g) under N2. The mixture was refluxed for 48 h. After cooling to room temperature, brine (100 mL)
was added, and the resulting mixture was filtered over Celite using EtOAc (ca.
300 mL). The organic was separated, dried over Na2SO4, and filtered. After concentration under reduced
pressure, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent) to
afford the title compound in 57% yield as a brown viscous liquid.
1H NMR (400 MHz, CDCl3) : 1.33
(s, 9H), 2.10-2.22 (m, 2H), 2.36 (s, 6H), 3.27 (s, 1H), 3.43 (s, 1H), 3.64 (t, J = 6.5 Hz, 2H), 7.13 (s, 1H),
7.47 (s, 2H).
13C{
1H} NMR (100 MHz, CDCl3) : 21.3, 22.7, 30.2, 31.6, 44.7, 57.7, 125.2, 133.4, 137.6,
138.2, 178.6. IR (max/cm-1
): 3046, 2958, 2922, 2866, 1599, 1577, 1455, 1361, 1320, 1308, 1069, 856.
HRMS (ESI+): calculated for [M+H]
+
: 314.1340, found: 314.1344. []D
20 +11.0 (c = 1.01, CH2Cl2).
(RS,S)-1-tert-Butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4). To a solution of (RS)-N-(4-chloro-1-(3,5-dimethylphenyl)butylidene)-tert-butanesulfinamide
(S3, 25.6 mmol, 8.06 g) in anhydrous THF (100 mL) at -78 °C was added LiBEt3H
(28 mmol, 0.5 M in THF, 28.2 mL) under Ar. The reaction was stirred at -78 °C for
1 h, subsequently allowed to warm up to room temperature and stirred for additional
20 h. Saturated NaHCO3 solution (80 mL) was slowly added. The mixture was filtered and extracted
with EtOAc (3 × 100 mL). The combined organic phase was dried over Na2SO4 and filtered. After
concentration, the residue was purified by column chromatography (silica gel, hexane/EtOAc as eluent)
to afford the title compound in 72% yield as pale yellow solid. mp.: 56 ˚C. 1H NMR (400 MHz, CDCl3)
: 1.12 (s, 9H), 1.74-1.90 (m, 3H), 1.93-2.02 (m, 1H), 2.18-2.27 (m, 1H), 2.30 (s, 6H), 2.94-3.02 (m, 1H),
3.85-3.91 (m, 1H), 4.55-4.59 (m, 1H), 6.88 (s, 1H), 6.90 (s, 2H).
13C{
1H} NMR (100 MHz, CDCl3) :
21.3, 23.8, 26.3, 36.0, 42.1, 57.2, 69.2, 125.0, 128.7, 137.7, 143.2. IR (max/cm-1
): 3023, 2957, 2920,
2866, 1607, 1471, 1360, 1061, 957, 847. HRMS (ESI+): calculated for [M+Na]+
: 302.1549, found:
302.1548. []D
20
-137 (c = 0.49, CH2Cl2)
(S)-2-(3,5-Dimethylphenyl)pyrrolidine hydrochloride (1j•HCl). To a solution
of (RS,S)-1-tert-butylsulfinyl-2-(3,5-dimethylphenyl)pyrrolidine (S4, 14.7 mmol,
4.12 g) in dioxane (250 mL) was added dropwise HCl (ca. 150 mmol, 4 M in dioxane, 38 mL). The mixture was stirred for 1 h at room temperature under N2, and
then the mixture was concentrated under reduced pressure. Then, Et2O (200 mL) was added to the residue
and the mixture was cooled to 0 ˚C. The precipitate was collected by filtration, washed with Et2O (40
mL), and dried under reduced pressure to afford the title compound in 94% yield as white solid. mp.: 198
˚C. 1H NMR (400 MHz, D2O) : 2.00-2.15 (m, 3H), 2.18 (s, 6H), 2.27-2.35 (m, 1H), 3.27-3.36 (m, 2H),
4.45 (t, J = 8.0 Hz, 1H), 6.97 (s, 2H), 7.01 (s, 1H). 13C{
1H} NMR (100 MHz, D2O) : 20.9, 24.19, 30.9,
46.0, 63.8, 119.79, 125.6, 131.4, 135.3, 140.1. IR (max/cm-1
): 3033, 3012, 2970, 2855, 2743, 2571, 2480,
1608, 1590, 1414, 850. HRMS (ESI+): calculated for [M-Cl]
+
: 176.1434, found: 176.1435. []D
20 +7.1
(c = 1.01, MeOH).
(S)-2-(3,5-Dimethylphenyl)pyrrolidine (1j). To a suspension of (S)-2-(3,5-dimethylphenyl)pyrrolidine hydrochloride (1j•HCl, 13.5 mmol, 2.86 g) in anhydrous Et2O
(200 mL) was added a saturated solution of NaHCO3 (200 mL). The resulting mixture
was stirred for 20 min at room temperature. The organic was separated and the aqueous
phase was extracted with Et2O (2 × 100 mL). The combined organic phase was dried over MgSO4 and
filtered. The solvent was removed under reduced pressure to afford the title compound as a pale yellow
liquid in 99% yield.
1H NMR (400 MHz, CDCl3) : 1.60-1.71 (m, 1H), 1.78-1.96 (m, 2H), 1.98 (s, 1H),
2.11-2.19 (m, 1H), 2.30 (s, 6H), 2.95-3.02 (m, 1H), 3.17-3.23 (m, 1H), 4.03 (t, J = 7.7 Hz, 1H), 6.87 (s,
1H), 6.97 (s, 2H). 13C{
1H} NMR (100 MHz, CDCl3) : 21.3, 25.5, 34.2, 46.9, 62.6, 124.2, 128.4, 137.8,
144.7. IR (max/cm-1
): 3332, 3010, 2960, 2915, 2869, 1605, 1458, 1101, 845. HRMS (ESI+): calculated
for [M+H]+
: 176.1434, found: 176.1436. []D
20
-30.5 (c = 1.01, MeOH). Chiral HPLC (ChiralPak ODH, 4.6 mm × L 250 mm, hexane:2-propanol = 90:10, 0.5 mL/min, = 254 nm): tR/min = 18.7 (1%),
19.8 (99%).

3-Fluoro-4-(4-formylphenoxy)benzonitrile2
(S5). A mixture of 3,4-
difluorobenzonitrile (35.0 mmol, 4.87 g), 4-hydroxybenzaldehyde (35.0
mmol, 4.27 g), and K2CO3 (70.0 mmol, 9.67 g) in N,N-dimethylacetamide
(90 mL) was stirred at 100 ˚C for 2 h under N2. After cooling, the reaction
mixture was poured into ice water. White precipitate was collected by filtration, washed with water, and dried under reduced pressure to afford the title compound as pale yellow
solid in 82% yield. mp.: 101 ˚C. 1H NMR (400 MHz, CDCl3) : 7.11-7.15 (m, 2H), 7.20 (t, J = 8.2 Hz,
1H), 7.49-7.51 (m, 1H), 7.54 (dd, J = 9.7, 1.9 Hz, 1H), 7.91-7.94 (m, 2H), 9.98 (s, 1H).
13C{
1H} NMR
(100 MHz, CDCl3) : 109.1 (d, 3
JC-F = 8.2 Hz), 117.1 (d, 4
JC-F = 2.5 Hz), 117.9, 121.3 (d, 2
JC-F = 21.3 Hz),
122.5 (d, 4
JC-F = 1.6 Hz), 129.6 (d, 3
JC-F = 4.1 Hz), 132.1, 132.7, 147.0 (d, 2
JC-F = 11.5 Hz), 153.6 (d, 1
JCF = 254.8 Hz), 160.7, 190.4. IR (max/cm-1
): 3100, 3060, 2846, 2812, 2761, 2232, 1697, 1687, 1585, 1497,
1277, 1216, 1166, 1114, 836. HRMS (APCI+): calculated for [M+H]+
: 242.0612, found: 242.0616.
3-Fluoro-4-(4-formylphenoxy)benzamide2
(2f). To a mixture of 3-
fluoro-4-(4-formylphenoxy)benzonitrile (S5, 26.0 mmol, 6.27 g) and
K2CO3 (13.0 mmol, 1.80 g) in DMSO (24 mL) was added dropwise 35%
H2O2 (ca. 29 mmol, 3.1 mL) at 10 ˚C over 5 min. The reaction mixture
was stirred at room temperature for 2 h. The reaction mixture was
poured into ice water. White precipitate was collected by filtration, washed with water, and dried under
reduced pressure to afford the title compound as white solid in 92% yield. mp. 129 ˚C. 1H NMR (400
MHz, (D3C)2SO) : 9.96 (s, 1H), 8.12 (s, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.93 (dd, J = 1.9, 10.0 Hz, 1H),
7.85-7.82 (m, 1H), 7.58 (s, 1H), 7.42 (t, J = 8.2 Hz, 1 H), 7.20 (d, J = 8.2 Hz, 2H).
13C{
1H} NMR (100
MHz, (D3C)2SO) : 116.6 (d, 2
JC-F = 19.7 Hz), 116.9, 122.6, 125.1 (d, 4
JC-F = 3.3 Hz), 131.9 (d, 2
JC-F =
21.3 Hz), 132.1, 132.7 (d, 3
JC-F = 5.7 Hz), 143.7 (d, 3
JC-F = 12.3 Hz), 153.1 (d, 1
JC-F = 248.2 Hz), 161.3,
165.8, 191.5. IR (max/cm-1
): 3356, 3185, 2844, 1668, 1598, 1504, 1433, 1382, 1269, 1218, 1156, 1128,
- HRMS (ESI+): calculated for [M+Na]
+
: 282.0537, found: 282.0541. HRMS (APCI+): calculated
for [M+H]+
: 260.0717, found: 260.0716.
NEXT
Reaction Chemistry & Engineering (2022), 7(8), 1779-1785
Journal of Medicinal Chemistry (2011), 54(23), 8000-8012
| Clinical data | |
|---|---|
| Other names | JNJ-67953964; CERC-501; LY-2456302 |
| Routes of administration | By mouth[1] |
| Pharmacokinetic data | |
| Bioavailability | 25%[1] |
| Elimination half-life | 30–40 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1174130-61-0 |
| PubChem CID | 44129648 |
| IUPHAR/BPS | 9194 |
| DrugBank | DB12341 |
| ChemSpider | 28424203 |
| UNII | DE4G8X55F5 |
| KEGG | D11831 |
| ChEMBL | ChEMBL1921847 |
| CompTox Dashboard (EPA) | DTXSID90151777 |
| Chemical and physical data | |
| Formula | C26H27FN2O2 |
| Molar mass | 418.512 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
^ Jump up to:a b c d e f g h i Li W, Sun H, Chen H, Yang X, Xiao L, Liu R, et al. (2016). “Major Depressive Disorder and Kappa Opioid Receptor Antagonists”. Translational Perioperative and Pain Medicine. 1 (2): 4–16. PMC 4871611. PMID 27213169.
- ^ Jump up to:a b c d e f g h “CERC 501”. Adis Insight. 30 January 2018.
- ^ Jump up to:a b Browne CA, Wulf H, Lucki I (2022). “Kappa Opioid Receptors in the Pathology and Treatment of Major Depressive Disorder”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 493–524. doi:10.1007/164_2020_432. ISBN 978-3-030-89073-5. PMID 33580854. S2CID 231908782.
- ^ Jump up to:a b c Reed B, Butelman ER, Kreek MJ (2022). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Mood and Substance Use Disorders”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 473–491. doi:10.1007/164_2020_401. ISBN 978-3-030-89073-5. PMID 33174064. S2CID 226305229.
- ^ Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J, Lisanby SH, et al. (May 2020). “A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia”. Nature Medicine. 26 (5): 760–768. doi:10.1038/s41591-020-0806-7. PMC 9949770. PMID 32231295. S2CID 256839849.
- ^ Dhir A (January 2017). “Investigational drugs for treating major depressive disorder”. Expert Opinion on Investigational Drugs. 26 (1): 9–24. doi:10.1080/13543784.2017.1267727. PMID 27960559. S2CID 45232796.
- ^ Missig G, Fritsch EL, Mehta N, Damon ME, Jarrell EM, Bartlett AA, et al. (January 2022). “Blockade of kappa-opioid receptors amplifies microglia-mediated inflammatory responses”. Pharmacology, Biochemistry, and Behavior. 212: 173301. doi:10.1016/j.pbb.2021.173301. PMC 8748402. PMID 34826432.
- ^ Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. (February 2014). “LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders”. Neuropharmacology. 77: 131–144. doi:10.1016/j.neuropharm.2013.09.021. PMID 24071566. S2CID 3230414.
- ^ Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. (September 2014). “Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects”. Journal of Clinical Pharmacology. 54 (9): 968–978. doi:10.1002/jcph.286. PMID 24619932. S2CID 14814449.
- ^ Jump up to:a b Rorick-Kehn LM, Witcher JW, Lowe SL, Gonzales CR, Weller MA, Bell RL, et al. (October 2014). “Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human”. The International Journal of Neuropsychopharmacology. 18 (2): pyu036. doi:10.1093/ijnp/pyu036. PMC 4368892. PMID 25637376.
- ^ Reed B, Butelman ER, Fry RS, Kimani R, Kreek MJ (March 2018). “Repeated Administration of Opra Kappa (LY2456302), a Novel, Short-Acting, Selective KOP-r Antagonist, in Persons with and without Cocaine Dependence”. Neuropsychopharmacology. 43 (4): 928. doi:10.1038/npp.2017.245. PMC 5809790. PMID 29422497.
- ^ Jump up to:a b Urbano M, Guerrero M, Rosen H, Roberts E (May 2014). “Antagonists of the kappa opioid receptor”. Bioorganic & Medicinal Chemistry Letters. 24 (9): 2021–2032. doi:10.1016/j.bmcl.2014.03.040. PMID 24690494.
- ^ Jump up to:a b “Publication Reports Human Brain Penetration and Target Engagement of Cerecor’s Oral Kappa Opioid Receptor Antagonist, CERC-501”. BusinessWire. 11 December 2015.
- ^ Jump up to:a b c d e f Naganawa M, Dickinson GL, Zheng MQ, Henry S, Vandenhende F, Witcher J, et al. (February 2016). “Receptor Occupancy of the κ-Opioid Antagonist LY2456302 Measured with Positron Emission Tomography and the Novel Radiotracer 11C-LY2795050”. The Journal of Pharmacology and Experimental Therapeutics. 356 (2): 260–266. doi:10.1124/jpet.115.229278. PMC 4727157. PMID 26628406.
- ^ Jump up to:a b Placzek MS (August 2021). “Imaging Kappa Opioid Receptors in the Living Brain with Positron Emission Tomography”. In Liu-Chen LY, Inan S (eds.). The Kappa Opioid Receptor. Handbook of Experimental Pharmacology. Vol. 271. pp. 547–577. doi:10.1007/164_2021_498. ISBN 978-3-030-89073-5. PMID 34363128. S2CID 236947969.
- ^ Zheng MQ, Nabulsi N, Kim SJ, Tomasi G, Lin SF, Mitch C, et al. (March 2013). “Synthesis and evaluation of 11C-LY2795050 as a κ-opioid receptor antagonist radiotracer for PET imaging”. Journal of Nuclear Medicine. 54 (3): 455–463. doi:10.2967/jnumed.112.109512. PMC 3775344. PMID 23353688.
- ^ Mitch CH, Quimby SJ, Diaz N, Pedregal C, de la Torre MG, Jimenez A, et al. (December 2011). “Discovery of aminobenzyloxyarylamides as κ opioid receptor selective antagonists: application to preclinical development of a κ opioid receptor antagonist receptor occupancy tracer”. Journal of Medicinal Chemistry. 54 (23): 8000–8012. doi:10.1021/jm200789r. PMID 21958337.
- ^ “WO2009094260A1 – Kappa selective opioid receptor antagonist”. Google Patents. 13 January 2009. Retrieved 29 August 2022.
- ^ “Cerecor Bolsters Clinical Pipeline with Acquisition of Phase 2-ready Kappa Opioid Receptor Antagonist from Eli Lilly and Company”. cerecor.com. February 20, 2015. Archived from the original on 2015-02-23. Retrieved March 18, 2015.
- ^ Rankovic Z, Hargreaves R, Bingham M (2012). Drug Discovery for Psychiatric Disorders. Royal Society of Chemistry. pp. 314–317. ISBN 978-1-84973-365-6.
- ^ Jump up to:a b c Bushey R (August 2017). “J&J Adds New Depression Drug to Portfolio”. Drug Discovery and Development Magazine.
- ^ “Cerecor Announces Divestiture of CERC-501 to Janssen Pharmaceuticals, Inc”. Marketwired. August 2017. Archived from the original on 2017-09-01. Retrieved 2017-09-01.
Further reading
- Carlezon WA, Krystal AD (October 2016). “Kappa-Opioid Antagonists for Psychiatric Disorders: From Bench to Clinical Trials”. Depression and Anxiety. 33 (10): 895–906. doi:10.1002/da.22500. PMC 5288841. PMID 27699938.
- Li W, Sun H, Chen H, Yang X, Xiao L, Liu R, et al. (2016). “Major Depressive Disorder and Kappa Opioid Receptor Antagonists”. Translational Perioperative and Pain Medicine. 1 (2): 4–16. PMC 4871611. PMID 27213169.
- Dhir A (January 2017). “Investigational drugs for treating major depressive disorder”. Expert Opinion on Investigational Drugs. 26 (1): 9–24. doi:10.1080/13543784.2017.1267727. PMID 27960559. S2CID 45232796.
- Reed B, Butelman ER, Kreek MJ (2017). “Endogenous opioid system in addiction and addiction-related behaviors”. Current Opinion in Behavioral Sciences. 13: 196–202. doi:10.1016/j.cobeha.2016.12.002. ISSN 2352-1546. S2CID 53149180.
- Rakesh G, Pae CU, Masand PS (August 2017). “Beyond serotonin: newer antidepressants in the future”. Expert Review of Neurotherapeutics. 17 (8): 777–790. doi:10.1080/14737175.2017.1341310. PMID 28598698. S2CID 205823807.
- Helal MA, Habib ES, Chittiboyina AG (December 2017). “Selective kappa opioid antagonists for treatment of addiction, are we there yet?”. European Journal of Medicinal Chemistry. 141: 632–647. doi:10.1016/j.ejmech.2017.10.012. PMID 29107424.
- McHugh KL, Kelly JP (2018). “Modulation of the central opioid system as an antidepressant target in rodent models”. The Opioid System as the Interface between the Brain’s Cognitive and Motivational Systems. Progress in Brain Research. Vol. 239. pp. 49–87. doi:10.1016/bs.pbr.2018.07.003. ISBN 9780444641670. PMID 30314569.
- Bailey SJ, Husbands SM (June 2018). “Targeting opioid receptor signaling in depression: do we need selective κ opioid receptor antagonists?”. Neuronal Signaling. 2 (2): NS20170145. doi:10.1042/NS20170145. PMC 7373229. PMID 32714584.
- Chavkin C (August 2018). “Kappa-opioid antagonists as stress resilience medications for the treatment of alcohol use disorders”. Neuropsychopharmacology. 43 (9): 1803–1804. doi:10.1038/s41386-018-0046-4. PMC 6046055. PMID 29752444.
- Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. (December 2018). “The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development”. Nature Reviews. Drug Discovery. 18 (1): 82–84. doi:10.1038/nrd.2018.222. PMC 6816017. PMID 30591715.
- Lazar MA, McIntyre RS (2019). “Novel Therapeutic Targets for Major Depressive Disorder”. Neurobiology of Depression. pp. 383–400. doi:10.1016/B978-0-12-813333-0.00034-2. ISBN 9780128133330. S2CID 86782597.
- Browne CA, Lucki I (September 2019). “Targeting opioid dysregulation in depression for the development of novel therapeutics”. Pharmacology & Therapeutics. 201: 51–76. doi:10.1016/j.pharmthera.2019.04.009. PMC 6859062. PMID 31051197.
- Banks ML (2020). “The Rise and Fall of Kappa-Opioid Receptors in Drug Abuse Research”. In Nader MA, Hurd YL (eds.). Substance Use Disorders. Handbook of Experimental Pharmacology. Vol. 258. pp. 147–165. doi:10.1007/164_2019_268. ISBN 978-3-030-33678-3. PMC 7756963. PMID 31463605.
- Browne CA, Jacobson ML, Lucki I (2020). “Novel Targets to Treat Depression: Opioid-Based Therapeutics”. Harvard Review of Psychiatry. 28 (1): 40–59. doi:10.1097/HRP.0000000000000242. PMID 31913981. S2CID 210120636.
- Jacobson ML, Browne CA, Lucki I (January 2020). “Kappa Opioid Receptor Antagonists as Potential Therapeutics for Stress-Related Disorders”. Annual Review of Pharmacology and Toxicology. 60: 615–636. doi:10.1146/annurev-pharmtox-010919-023317. PMID 31914893. S2CID 210121357.
- Mercadante S, Romualdi P (2020). “The Therapeutic Potential of Novel Kappa Opioid Receptor-based Treatments”. Current Medicinal Chemistry. 27 (12): 2012–2020. doi:10.2174/0929867326666190121142459. PMID 30666905. S2CID 58558833.
External links
Aticaprant – Eli Lilly and Company/Janssen Pharmaceuticals – AdisInsight
//////ATICAPRANT, CERC-501, JSPA 0658, JSPA-0658, JSPA0658, LY 2456302, LY-2456302, LY2456302, Phase 3, ELI LILLY, Major depressive disorder, JNJ-67953964, WHO 10582
FDA approves new treatment for certain advanced or metastatic breast cancers

FDA approves new treatment for certain advanced or metastatic breast cancers
|
|||
https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm578071.htm
(abemaciclib)
September 28, 2017
Release
The U.S. Food and Drug Administration today approved Verzenio (abemaciclib) to treat adult patients who have hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer that has progressed after taking therapy that alters a patient’s hormones (endocrine therapy). Verzenio is approved to be given in combination with an endocrine therapy, called fulvestrant, after the cancer had grown on endocrine therapy. It is also approved to be given on its own, if patients were previously treated with endocrine therapy and chemotherapy after the cancer had spread (metastasized).
“Verzenio provides a new targeted treatment option for certain patients with breast cancer who are not responding to treatment, and unlike other drugs in the class, it can be given as a stand-alone treatment to patients who were previously treated with endocrine therapy and chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
Verzenio works by blocking certain molecules (known as cyclin-dependent kinases 4 and 6), involved in promoting the growth of cancer cells. There are two other drugs in this class that are approved for certain patients with breast cancer, palbociclib approved in February 2015 and ribociclib approved in March 2017.
Breast cancer is the most common form of cancer in the United States. The National Cancer Institute at the National Institutes of Health estimates approximately 252,710 women will be diagnosed with breast cancer this year, and 40,610 will die of the disease. Approximately 72 percent of patients with breast cancer have tumors that are HR-positive and HER2-negative.
The safety and efficacy of Verzenio in combination with fulvestrant were studied in a randomized trial of 669 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and who had not received chemotherapy once the cancer had metastasized. The study measured the length of time tumors did not grow after treatment (progression-free survival). The median progression-free survival for patients taking Verzenio with fulvestrant was 16.4 months compared to 9.3 months for patients taking a placebo with fulvestrant.
The safety and efficacy of Verzenio as a stand-alone treatment were studied in a single-arm trial of 132 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and chemotherapy after the cancer metastasized. The study measured the percent of patients whose tumors completely or partially shrank after treatment (objective response rate). In the study, 19.7 percent of patients taking Verzenio experienced complete or partial shrinkage of their tumors for a median 8.6 months.
Common side effects of Verzenio include diarrhea, low levels of certain white blood cells (neutropenia and leukopenia), nausea, abdominal pain, infections, fatigue, low levels of red blood cells (anemia), decreased appetite, vomiting and headache.
Serious side effects of Verzenio include diarrhea, neutropenia, elevated liver blood tests and blood clots (deep venous thrombosis/pulmonary embolism). Women who are pregnant should not take Verzenio because it may cause harm to a developing fetus.
The FDA granted this application Priority Review and Breakthrough Therapydesignations.
The FDA granted the approval of Verzenio to Eli Lilly and Company.
//////////Verzenio, abemaciclib, fda 2017, metastatic breast cancers, Eli Lilly , Priority Review, Breakthrough Therapy designations, antibodies
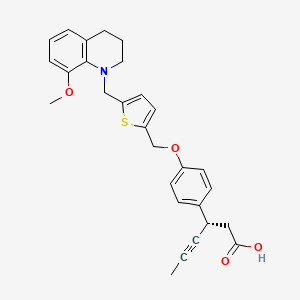
Tradipitant, традипитант , تراديبيتانت , 曲地匹坦 ,

Tradipitant
VLY-686, LY686017
традипитант
تراديبيتانت [Arabic]
曲地匹坦 [Chinese]
- Molecular Formula C28H16ClF6N5O
- Average mass 587.903 Da
622370-35-8 CAS
Methanone, [2-[1-[[3,5-bis(trifluoromethyl)phenyl]methyl]-5-(4-pyridinyl)-1H-1,2,3-triazol-4-yl]-3-pyridinyl](2-chlorophenyl)-
(2-(1-(3,5-bis(trifluoromethyl)benzyl)-5-(pyridin-4-yl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)(2-chlorophenyl)methanone
[2-[1-[[3,5-bis(trifluoromethyl)phenyl]methyl]-5-(4-pyridinyl)-1H-1,2,3-triazol-4-yl]-3-pyridinyl](2-chlorophenyl)methanone
PHASE 2, Gastroparesis; Pruritus
FDA 2025, APPROVALS 2025, 12/30/2025, To treat vomiting associated with motion
pyridine-containing NK-1 receptor antagonist ie tradipitant, useful for treating anxiety, pruritus and alcoholism.
Vanda Pharmaceuticals, under license from Eli Lilly, was developing tradipitant, a NK1 antagonist, for treating anxiety disorder, pruritus and alcohol dependence. The company was also investigating the drug for treating gastroparesis. In February 2017, tradipitant was reported to be in phase 2 clinical development for treating anxiety and pruritus.
- Originator Eli Lilly
- Developer Eli Lilly; National Institute on Alcohol Abuse and Alcoholism; Vanda Pharmaceuticals
- Class Antipruritics; Anxiolytics; Chlorobenzenes; Pyridines; Small molecules; Triazoles
- Mechanism of Action Neurokinin 1 receptor antagonists; Substance P inhibitors
Highest Development Phases
- Phase II Gastroparesis; Pruritus
- Discontinued Alcoholism; Social phobia
- The drug had been in phase II clinical trials at Lilly and the National Institute on Alcohol Abuse and Alcoholism for the treatment of alcoholism; however, no recent development has been reported for this research.
- A phase II clinical trial for the treatment of social phobia has been completed by Lilly.
PATENT WO 2003091226
SYNTHESIS

Condensation of 2-chloropyridine with thiophenol in the presence of K2CO3 in DMF at 110ºC yields sulfide intermediate,
which is then oxidized by means of NaOCl in AcOH to give 2-(benzenesulfonyl)pyridine.
This is treated with (iPr)2NH and n-BuLi in THF at -60 to -70°C and subsequently couples with 2-chlorobenzaldehyde in THF at -60 to -70°C to furnish (2-(phenylsulfonyl)pyridin-3-yl)-(2-chlorophenyl)methanone.
Ketone couples with the enolate of 4-acetylpyridine (formed by treating 4-acetylpyridine (VII) with t-BuOK in DMSO) in the presence of LiOH in DMSO and subsequently is treated with PhCOOH in iPrOAc to give rise to pyridine benzoate derivative.
This finally couples with 1-azidomethyl-3,5-bistrifluoromethylbenzene (obtained by treating 3,5-bis(trifluoromethyl)benzylchloride with NaN3 ini DMSO) in the presence of K2CO3 in t-BuOH to afford the title compound Tradipitant.
Tradipitant (VLY-686 or LY686017) is an experimental drug that is a neurokinin 1 antagonist. It works by blocking substance P, a small signaling molecule. Originally, this compound was owned by Eli Lilly and named LY686017. VLY-686 was purchased by Vanda Pharmaceuticals from Eli Lilly and Company in 2012.[1] Vanda Pharmaceuticals is a U.S. pharmaceutical company that as of November 2015 only has 3 drugs in their product pipeline: tasimelteon, VLY-686, and iloperidone.[2]
Tachykinins are a family of peptides that are widely distributed in both the central and peripheral nervous systems. These peptides exert a number of biological effects through actions at tachykinin receptors. To date, three such receptors have been characterized, including the NK-1 , NK-2, and NK-3 subtypes of tachykinin receptor.
The role of the NK-1 receptor subtype in numerous disorders of the central nervous system and the periphery has been thoroughly demonstrated in the art. For instance, NK-1 receptors are believed to play a role in depression, anxiety, and central regulation of various autonomic, as well as cardiovascular and respiratory functions. NK- 1 receptors in the spinal cord are believed to play a role in pain transmission, especially the pain associated with migraine and arthritis. In the periphery, NK-1 receptor activation has been implicated in numerous disorders, including various inflammatory disorders, asthma, and disorders of the gastrointestinal and genitourinary tract.
There is an increasingly wide recognition that selective NK-1 receptor antagonists would prove useful in the treatment of many diseases of the central nervous system and the periphery. While many of these disorders are being treated by new medicines, there are still many shortcomings associated with existing treatments. For example, the newest class of anti-depressants, selective serotonin reuptake inhibitors (SSRIs), are increasingly prescribed for the treatment of depression; however, SSRIs have numerous side effects, including nausea, insomnia, anxiety, and sexual dysfunction. This could significantly affect patient compliance rate. As another example, current treatments for chemotherapy- induced nausea and emesis, such as the 5-HT3receptor antagonists, are ineffective in managing delayed emesis. The development of NK-1 receptor antagonists will therefore greatly enhance the ability to treat such disorders more effectively. Thus, the present invention provides a class of potent, non-peptide NK-1 receptor antagonists, compositions comprising these compounds, and methods of using the compounds.
Indications
Pruritus
It is being investigated by Vanda Pharmaceuticals for chronic pruritus (itchiness) in atopic dermatitis. In March 2015, Vanda announced positive results from a Phase II proof of concept study.[3] A proof of concept study is done in early stage clinical trials after there have been promising preclinical results. It provides preliminary evidence that the drug is active in humans and has some efficacy.[4]
Alcoholism
VLY-686 reduced alcohol craving in recently detoxified alcoholic patients as measured by the Alcohol Urge Questionnaire.[5] In a placebo controlled clinical trial of recently detoxified alcoholic patients, VLY-686 significantly reduced alcohol craving as measured by the Alcohol Urge Questionnaire. It also reduced the cortisol increase seen after a stress test compared to placebo. The dose given was 50 mg per day.
Social anxiety disorder
In a 12-week randomized trial of LY68017 in 189 patients with social anxiety disorder, 50 mg of LY68017 did not provide any statistically significant improvement over placebo.[6]
PATENT
WO03091226,
https://www.google.com/patents/WO2003091226A1?cl=en
PATENT
The compound {2-[l-(3,5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]- pyridin-3-yl}-(2-chlorophenyl)-methanone, depicted below as the compound of Formula I, was first described in PCT published application WO2003/091226.

(I)
Because the compound of Formula I is an antagonist of the NK-I subtype of tachykinin receptor, it is useful for the treatment of disorders associated with an excess of tachykinins. Such disorders include depression, including major depressive disorder; anxiety, including generalized anxiety disorder, panic disorder, obsessive compulsive disorder, and social phobia or social anxiety disorder; schizophrenia and other psychotic disorders, including bipolar disorder; neurodegenerative disorders such as dementia, including senile dementia of the Alzheimer’s type or Alzheimer’s disease; disorders of bladder function such as bladder detrusor hyper-reflexia and incontinence, including urge incontinence; emesis, including chemotherapy-induced nausea and acute or delayed emesis; pain or nociception; disorders associated with blood pressure, such as hypertension; disorders of blood flow caused by vasodilation and vasospastic diseases, such as angina, migraine, and Reynaud’s disease; hot flushes; acute and chronic obstructive airway diseases such as adult respiratory distress syndrome, bronchopneumonia, bronchospasm, chronic bronchitis, drivercough, and asthma; inflammatory diseases such as inflammatory bowel disease; gastrointestinal disorders or diseases associated with the neuronal control of viscera such as ulcerative colitis, Crohn’s disease, functional dyspepsia, and irritable bowel syndrome (including constipation-predominant, diarrhea- -?-
predominant, and mixed irritable bowel syndrome); and cutaneous diseases such as contact dermatitis, atopic dermatitis, urticaria, and other eczematoid dermatitis.
In PCT published application, WO2005/042515, novel crystalline forms of the compound of Formula I, identified as Form IV and Form V, are identified. Also described in WO2005/042515 is a process for preparation of the compound of Formula I, comprising reacting (2-chlorophenyl)-[2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone or a phosphate salt thereof with l-azidomethyl-3,5- bistrifluoromethylbenzene in the presence of a suitable base and a solvent. Use of this procedure results in several shortcomings for synthesis on a commercial scale. For example, use of the solvent DMSO, with (2- chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone phosphate, requires a complex work-up that has a propensity to emulsify. This process also requires extraction with CH2CI2, the use of which is discouraged due to its potential as an occupational carcinogen, as well as the use of MgSC>4 and acid-washed carbon, which can generate large volumes of waste on a commercial scale. Conducting the reaction with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone in isopropyl alcohol, as also described in WO2005/042515, is also undesirable due to the need to incorporate a free base step. Furthermore, variable levels of residual l-azidomethyl-3,5-bistrifluoromethylbenzene, a known mutagen, are obtained from use of the procedures described in WO2005/042515.
An improved process for preparing the compound of Formula I would control the level of 1- azidomethyl-3,5-bistrifluoromethylbenzene impurity, and improve the yield. We have discovered that use of the novel salt, (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, as well as use of tert-butanol as the reaction solvent, improves reaction times and final yield, and decreases impurities in the final product. In addition, a novel process for the preparation of (2-chlorophenyl)- [2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, in which a pre-formed enolate of 4-acetyl pyridine is added to (2-phenylsulfonyl-pyridine-3-yl)-(2-chlorophenyl)methanone, results in an overall improved yield and improved purity, and is useful on a commercial scale.
EXAMPLES
Example 1 {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone (Form IV)

Suspend (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl] methanone benzoate (204.7 g; 1.04 equiv; 445 mmoles) in t-butanol (614 mL) and treat the slurry with potassium carbonate (124.2 g; 898.6 mmoles). Heat to 7O0C with mechanical stirring for 1 hour. Add l-azidomethyl-3,5- bistrifluoromethylbenzene (115.6 g; 1.00 equiv; 429.4 mmoles) in a single portion, then heat the mixture to reflux. A circulating bath is used to maintain a condenser temperature of 3O0C. After 18 hours at reflux, HPLC reveals that the reaction is complete (<2% l-azidomethyl-3,5-bistrifluoromethylbenzene remaining). The mixture is cooled to 7O0C, isopropanol (818 mL) is added, then the mixture is stirred at 7O0C for 1 hour. The mixture is filtered, and the waste filter cake is rinsed with isopropanol (409 mL). The combined filtrate and washes are transferred to a reactor, and the mechanically stirred contents are heated to 7O0C. To the dark purple solution, water (1.84 L) is added slowly over 35 minutes. The solution is cooled to 6O0C, then stirred for 1 hour, during which time a thin precipitate forms. The mixture is slowly cooled to RT, then the solid is filtered, washed with 1 : 1 isopropanol/water (614 mL), subsequently washed with isopropanol (410 mL), then dried in vacuo at 450C to produce 200.3 g of crude {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone as a white solid. Crude {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin- 3-yl}-(2-chlorophenyl)-methanone (200.3 g) and isopropyl acetate (600 mL) are charged to a 5L 3-neck jacketed flask, then the contents heated to 750C. After dissolution is achieved, the vessel contents are cooled to 550C, then the solution polish filtered through a 5 micron filter, and the filter rinsed with a volume of isopropyl acetate (200 mL). After the polish filtration operation is complete, the filtrates are combined, and the vessel contents are adjusted to 5O0C. After stirring for at least 15 minutes at 5O0C, 0.21 grams of {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2- chlorophenyl)-methanone Form IV seed (d90 = 40 microns) is added, and the mixture stirred at 5O0C for at least 2 h. Heptanes (1.90 L) are then added over at least 2 h. After the heptanes addition is completed, the slurry is stirred for an hour at 5O0C, cooled to 230C at a rate less then 2O0C per hour, then aged at 230C for an hour prior to isolation. The mixture is then filtered in portions through the bottom outlet valve in the reactor into a 600 mL filter. The resulting wetcake is washed portionwise with a solution containing heptanes (420 mL) and isopropyl acetate (180 mL), which is passed directly through the 5L crystallization vessel. The wetcake is blown dry for 5 minutes with nitrogen, then transferred to a 500 mL plastic bottle. The product is dried at 5O0C for 4 h. to produce 190.3g of pure {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone, Form IV in 75.0% yield with 100% purity, as determined by HPLC analysis. Particle size is reduced via pin or jet mill. 1H NMR (400 MHz, CDCl3): 5.46 (s, 2H); 7.19 (m, 5H); 7.36 (dd, IH, J = 4.9, 7.8); 7.45 (s, 2H); 7.59 (m, IH); 7.83 (s, IH); 7.93 (dd, IH, J = 1.5, 7.8); 8.56 (dd, IH, J= 1.5, 4.9); 8.70 (d, 2H, J= 5.9).
Preparation 1-A (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate Charge powdered KOfBu (221.1 g, 1.93 moles, 1.40 eq.) to Reactor A, then charge DMSO (2 L) at
250C over 10 min. The KOfBu/DMSO solution is stirred for 30 min at 230C, then a solution of 4-acetyl pyridine (92 mL, 2.07 moles, 1.50 eq) in DMSO (250 mL) is prepared in reactor B. The contents of reactor B are added to Reactor A over 10 minutes, then the Reactor A enolate solution is stirred at 230C for Ih. In a separate 12-L flask (Reactor C), solid LiOH (84.26 g, 3.45 moles, 2.0 eq) is poured into a mixture of (2- phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone (500.0 g, 1.34 moles, 1.0 eq) and DMSO (2L), with stirring, at 230C. The enolate solution in reactor A is then added to Reactor C over a period of at least 15 minutes, and the red suspension warmed to 4O0C. The reaction is stirred for 3h, after which time HPLC analysis reveals less than 2% (2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone. Toluene (2.5 L) is charged, and the reactor temperature cooled to 3O0C. The mixture is quenched by addition of glacial acetic acid (316 mL, 5.52 moles, 4.0 eq), followed by 10 % NaCl (2.5 L). The biphasic mixture is transferred to a 22-L bottom-outlet Morton flask, and the aqueous layer is removed. The aqueous layer is then extracted with toluene (750 mL). The combined organic layers are washed with 10 % NaCl (750 mL), then concentrated to 4 volumes and transferred to a 12-L Morton flask and rinsed with isopropyl acetate (4 vol, 2 L). The opaque amber solution is warmed to 75 degrees to 750C over 40 min. Benzoic acid (171. Ig, 1.34 moles, 1.0 eq) is dissolved in hot isopropyl acetate (1.5 L), and charged to the crude free base solution over at least 30 min. The crude solution containing benzoate salt is stirred for 0.5 h at 750C then cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate (2.25g) at 750C, followed by stirring for 0.5 h at 750C, then cooling to 230C over at least 1.5 h. The mixture is then cooled to <5 0C, then filtered through paper on a 24cm single-plate filter. The filtercake is then rinsed with cold z‘-PrOAc (750 mL) to produce granular crystals of bright orange-red color. The wet solid is dried at 550C to produce 527.3 g (83% yield) with 99.9% purity. (2-chlorophenyl)-[2-(2-hydroxy-2- pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate. Anal. Calcd. for C26Hi9N2ClO4: C, 68.05; H, 4.17; N, 7.13. Found: C, 67.89; H, 4.15; N 6.05. HRMS: calcd for C19H13ClN2O2, 336.0666; found 336.0673.
The synthesis of(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate proceeds optimally when the potassium enolate of 4-acetyl pyridine is pre-formed using KOfBu in DMSO. Pre-formation of the enolate allows the SNAR (nucleophilic aromatic substitution) reaction to be performed between room temperature and 4O0C, which minimizes the amount of degradation. Under these conditions, the SNAR is highly regioselective, resulting in a ratio of approximately 95:5 preferential C – acylation. In all cases, less polar solvents such as THF or toluene, or co-solvents of these solvents mixed with DMSO, results in a substantial increase of acylation at the oxygen in the SNAR, and leads to a lower yield of product. This is a substantial improvement over the procedures described in WO2005/042515 for synthesis of the free base or the phosphate salt, in which the SNAR is performed at 60-700C, resulting in a substantial increase in chemical impurity. Using the conditions described in WO2005/042515, when scaled to 2kg, results in maximum yields of 55%, with sub-optimal potency. In comparison, the improved conditions described herein can be run reproducibly from 0.4 to 2kg scale to give yields of 77-83%, with >99% purity. In addition, the reaction can be held overnight at 4O0C with minimal degradation, whereas holding the reaction for 1 h past completion at 60-70°C results in substantial aromatized impurity. The reaction may also be performed using sodium tert-amylate as the base, in combination with an aprotic solvent, such as DMSO or DMF.
The title compound exists as a mixture of tautomers and geometric isomers. It is understood that each of these forms is encompassed within the scope of the invention.

Preparation 1-B
(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate The procedure described in Preparation 1-A is followed, with the following exception. Solid toluic acid (1.0 eq) is added to the crude free base solution at 550C, then the solution cooled to 45 0C. The solution is stirred for one hour at 45 0C, then slowly cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded, aged for 3 h at 450C , then cooled to O0C over 4 h. The isolation slurry is filtered, and the wetcake washed with MeOH (3 volumes). The wetcake is dried at 5O0C to provide 14.0 g (76.4%) of (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl- vinyl)pyridin-3-yl]methanone toluate as a light red powder.
As with the benzoate salt, the toluate salt can also exist as a mixture of tautomers and geometric isomers, each of which is encompassed within the scope of the invention. (2-chlorophenyl)-[2-(2-hydroxy- 2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate . 13C NMR (125 MHz,DMS0-d6) δ 194.5, 167.8, 167.4, 155.5, 150.7 (2C), 147.4, 144.0, 143.4, 142.7, 138.6, 133.0, 130.8, 130.7, 130.5, 129.8(2C), 129.5(2C), 128.5, 128.0, 127.9, 119.9 (2C), 118.6, 92.6, 21.5.
Preparation 1-C
(2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone
A solution of 1.3 eq of diisopropylamine (based on 2-benzenesulfonyl pyridine) in 5 volumes of THF in a mechanically stirred 3 -necked flask is cooled to -70 to -75 0C. To this solution is added 1.05 eq of w-butyllithium (1.6M in hexanes) at such a rate as to maintain the temperature below -6O0C. The light yellow solution is stirred at -60 to -70 0C for 30 minutes. Once the temperature has cooled back down to – 60 to -650C, 1.0 eq of 2-benzene-sulfonyl pyridine, as a solution in 3 volumes of THF, is added at the fastest rate that will maintain the reaction temperature under -6O0C. A yellow suspension forms during the addition that becomes yellow-orange upon longer stirring. This mixture is stirred for 3 hours at -60 to – 750C, and then 1.06 eq of 2-chlorobenzaldehyde, as a solution in 1 volume of THF, is added dropwise at a sufficient rate to keep the temperature under -55 0C. The suspension gradually turns orange-red, thins out, and then becomes a clear red solution. The reaction mixture is allowed to stir at -60 to -7O0C for 1 hour, 3N aqueous HCl (7 volumes) is added over 20-30 minutes, and the temperature is allowed to exotherm to 0-100C. The color largely disappears, leaving a biphasic yellow solution. The solution is warmed to at least 1O0C, the layers are separated, and the aqueous layer is back-extracted with 10 volumes of ethyl acetate. The combined organic layers are washed with 10 volumes of saturated sodium bicarbonate solution and concentrated to about 2 volumes. Ethyl acetate (10 volumes) is added, and the solution is once again concentrated to 2 volumes. The thick solution is allowed to stand overnight and is taken to the next step with no purification of the crude alcohol intermediate. The crude alcohol intermediate is transferred to a 3 -necked flask with enough ethyl acetate to make the total solution about 10 volumes. The yellow solution is treated with 3.2 volumes of 10% aqueous (w/w) potassium bromide, followed by 0.07 eq of 2,2,6,6-Tetramethylpiperidine-N-oxide (TEMPO). The orange mixture is cooled to 0-50C and treated with a solution of 1.25 eq of sodium bicarbonate in 12% w/w sodium hypochlorite (9 volumes) and 5 volumes of water over 30-60 minutes while allowing the temperature to exotherm to a maximum of 2O0C. The mixture turns dark brown during the addition, but becomes yellow, and a thick precipitate forms. The biphasic light yellow mixture is allowed to stir at ambient temperature for 1-3 hours, at which time the reaction is generally completed. The biphasic mixture is cooled to 0-50C and stirred for 3 hours at that temperature. The solid is filtered off, washed with 4 volumes of cold ethyl acetate, followed by 4 volumes of water, and dried in vacuo at 450C to constant weight. Typical yield is 80-83% with a purity of greater than 98%. 1H NMR (600 MHz, CDCl3-^) δ ppm 7.38 (td, ./=7.52, 1.28 Hz, 1 H) 7.47 (dd, ./=7.80, 1.30 Hz, 1 H) 7.51 (td, ./=7.79, 1.60 Hz, 1 H) 7.51 (t, ./=7.89 Hz, 2 H) 7.50 – 7.54 (m, J=7.75, 4.63 Hz, 1 H) 7.60 (t, J=7.43 Hz, 1 H) 7.73 (dd, J=7.75, 1.60 Hz, 1 H) 7.81 (dd, J=7.79, 1.56 Hz, 1 H) 8.00 (dd, ./=8.44, 1.10 Hz, 2 H) 8.76 (dd, ./=4.63, 1.61 Hz, 1 H).
Preparation 1-D 1 -azidomethyl-3,5-bistrifluoromethyl-benzene
Sodium azide (74.3 g, 1.14 mol) is suspended in water (125 mL), then DMSO (625 mL) is added. After stirring for 30 minutes, a solution consisting of 3,5-Bis(trifluoromethyl)benzyl chloride (255.3 g, 0.97 moles) and DMSO (500 mL) is added over 30 minutes. (The 3,5-Bis(trifluoromethyl)benzyl chloride is heated to 350C to liquefy prior to dispensing (MP = 30-320C)). The benzyl chloride feed vessel is rinsed with DMSO (50 mL) into the sodium azide solution, the mixture is heated to 4O0C, and then maintained for an hour at 4O0C, then cooled to 230C.
In Process Analysis: A drop of the reaction mixture is dissolved in d6-DMSO and the relative intensities of the methylene signals are integrated (NMR verified as a 0.35% limit test for 3,5- Bis(trifluoromethyl)benzyl Chloride). Work-up: After the mixture reaches 230C , it is diluted with heptanes (1500 mL), then water (1000 mL) is added, and the mixture exotherms to 350C against a jacket setpoint of 230C. The aqueous layer is removed (-2200 mL), then the organic layer (approximately 1700 mL) is washed with water (2 X 750 mL). The combined aqueous layers (-3700 mL) are analyzed and discarded.
The solvent is then partially removed via vacuum distillation with a jacket set point of 850C, pot temperature of 60-650C and distillate head temperature of 50-550C to produce 485g (94.5% yield) of 51 Wt% solution title compound as a clear liquid. Heptanes can be either further removed by vacuum distillation or wiped film evaporation technology. 1H NMR (400 MHz, CDCl3): 4.58 (s, 2H); 7.81 (s, 2H); 7.90 (s, IH).
Preparation 1-E 2-benzene-sulfonyl pyridine Charge 2-chloropyridine (75 mL, 790 mmol), thiophenol (90 mL, 852 mmol), and DMF (450 mL) to a 2L flask. Add K2CO3 (134.6 g, 962 mmol), then heat to HO0C and stir for 18 hours. Filter the mixture, then rinse the waste cake with DMF (195 mL). The combined crude sulfide solution and rinses are transferred to a 5-L flask, and the waste filtercake is discarded. Glacial acetic acid (57 mL, 995 mmol) is added to the filtrate, then the solution is heated to 4O0C, and 13 wt % NaOCl solution (850 mL, 1.7 mol) is added over 2 hours. After the reaction is complete, water (150 mL) is added, then the pH of the mixture adjusted to 9 with 20 % (w/v) NaOH solution (250 mL). The resulting slurry is cooled to <5 0C, stirred for 1.5 h, then filtered, and the cake washed with water (3 x 200 mL). The product wetcake is dried in a 550C vacuum oven to provide 2-benzene-sulfonyl pyridine (149 g, 676 mmol) in 86 % yield: 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 5.5 Hz, IH), 8.19 (d, J = 1.1 Hz, IH), 8.05 (m, 2H), 7.92 (ddd, J= 9.3, 7.7, 1.6 Hz, IH), 7.60 (m, IH), 7.54 (m, 2H), 7.44 (m, IH); IR (KBr) 788, 984, 1124, 1166, 1306, 1424, 1446, 1575, 3085 cm“1; MS (TOF) mlz 220.0439 (220.0427 calcd for C11H10NO2S, MH); Anal, calcd for C11H9NO2S: C, 60.26; H, 4.14; N, 6.39; S, 14.62. Found: C, 60.40; H, 4.02; N, 6.40; S, 14.76.
As noted above, use of the improved process of the present invention results in an improved habit of the crystalline Form IV compound of Formula I. The improved habit reduces surface area of the crystal, improves the filtration, and washing, and improves the efficiency of azide mutagen rejection. These improvements are described in greater detail below.
In patent application WO2005/042515, the polish filtration is carried out in 7 volumes (L/kg) of isopropanol near its boiling point (65-83 0C), a process that is difficult and hazardous to execute in commercial manufacturing because of the high risk of crystallization on the filter and/or vessel transfer lines due to supersaturation. In the preferred crystallization solvent, isopropyl acetate, the polish filtration is conducted in four volumes of isopropyl acetate at temperatures from 45 to 55 0C. This temperature range is 35 to 45 0C lower than the boiling point of isopropyl acetate, which provides a key safety advantage.
PATENT
PATENT
WO 2017031215
EXAMPLES
Example 1: Preparation of Compound (I) via Negishi Coupling Route
Example 1 provides a scheme including preparations 1A-1D, described below, for the synthesis of the compound of Formula (I) and intermediates used in the route. An overview of the scheme is as follows:

80 on ma s ale
Example 1A: Preparation of Compound (I)

Zinc dust (200 mg, 3.06 mmol) combined with 2.0 mL of dimethylformamide was treated with 0.010 mL of 1,2-dibromoethane and heated to 65°C for 3 minutes. The mixture was cooled to ambient temperature and treated with 0.010 mL of trimethylsilyl chloride. After 5 minutes, 1.26 mL of 1M zinc chloride in diethyl ether was added to the mixture followed by Compound (Ila) (600 mg, 1.20 mmol). The mixture was heated to 65°C and further treated with 0.020 mL each of 1,2-dibromoethane and trimethylsilyl chloride. After 2.5 hours, via HPLC chromatogram, the reaction showed some formation of the zincate and was allowed to stir at ambient temperature for 16 hours. At this time
tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.06 mmol), Compound (Ilia) (357 mg, 1.20 mmol) were added to the reaction and the mixture heated to 65°C. HPLC analysis showed the formation of Compound (I) in the reaction.
IB: Preparation of Comp

To a solution of Compound (IV) (8.00 g, 18 mmol) in 40 mL of 1,2-dichloroethane was added a solution of iodine monochloride (10.7 g, 65.9 mmol) in 40 mL of 1,2-dichloroethane resulting in a slurry. The slurry was heated to 75°C for 4 hours then cooled to ambient temperature. The solids were collected by filtration, washed with heptane, then combined with 90 mL of ethyl acetate and 80 mL of saturated sodium thiosulfate solution. The organic phase was washed with saturated sodium chloride solution and dried with sodium sulfate. The mixture was concentrated to yield 7.80 g (87%) of Compound (Ila) as a yellow solid. The product could be further purified by silica gel chromatography. Thus 2.0 g of yellow solid was dissolved in dichloromethane and charged onto a silica gel column. The product was eluted using tert-butyl methyl ether to provide 1.87 g (93% recovery) of Compound (Ila) as a white powder. Analytical data: Iodine monochloride complex: ¾ NMR (500 MHz, DMSO-de) δ 8.80 (2 H), 8.05 (1 H), 7.77 (2 H), 7.59 (2 H), 5.86 (2 H).
Uncomplexed: ¾ NMR (500 MHz, DMSO-de) δ 8.71 (2 H), 8.03 (1 H), 7.74 (2 H), 7.44 (2 H), 5.86 (2 H).
It was observed that the iodination proceeded smoothly as a suspension in 1,2-dichloroethane with IC1 (4.0 equiv) at 75°C. An ICl-Compound (Ila) complex was initially isolated by filtration. Compound (Ila) was then obtained in approximately 85% yield by treatment of the ICl-Compound (Ila) complex with sodium thiosulfate. This protocol provided a viable means of isolation of Compound (Ila) without the use of DMF.
Example 1C: Preparation of silyl substituted triazole (Compound IV)

A mixture of Compound (V) (8.07 g, 30.0 mmol) and Compound (VI) (5.12 g, 29.2 mmol) was heated to 100°C for 18 hours. To the mixture was added 40 mL of heptane and the reaction was allowed to cool with rapid stirring. After 1 hour the solids were collected by filtration and washed with heptane then dried to 9.30 g (72%) of Compound (IV) as a tan solid. Analytical data: ¾ NMR (500 MHz, DMSO-de) δ 8.66 (2 H), 8.04 (1 H), 7.67 (2 H), 7.32 (2 H), 5.72 (2 H), 0.08 (9 H).
It was further found that combining Compound (V) and Compound (VI) (neat) and heating at 95 – 105°C afforded a 92: 8 mixture of regioisomers as shown below:

Crystallization of the mixture from heptane afforded Compound (IV) in 62-72% yield, thus obviating the need for chromatography to isolate Compound (IV).
Example ID: Preparation of starting material Compound (VI)

Zinc bromide (502 g, 2.23 mole) was added in approximately 100 g portions to 2.0 L of tetrahydrofuran cooled to between 0 and 10°C. To this cooled solution was added 4-bromopyridine hydrochloride (200 g, 1.02 mol), triphenylphosphine (54 g, 0.206 mol), and palladium (II) chloride (9.00 g, 0.0508 mol). Triethylamine (813 g, 8.03 mol) was then added at a rate to maintain the reaction temperature at less than 10°C, and finally
trimethylsilylacetylene (202 g, 2.05 mol) was added. The mixture was heated to 60°C for 4.5 hours. The reaction was cooled to -5°C and combined with 2.0 L of hexanes and treated with 2 L of 7.4 M NH4OH. Some solids were formed and were removed as much as possible with the aqueous phase. The organic phase was again washed with 2.0 L of 7.4 M NH4OH, followed by 2 washes with 500 mL of water, neutralized with 1.7 L of 3 M hydrochloric acid, dried with sodium sulfate, and concentrate to a thick slurry. The slurry was combined with 1.0 L of hexanes to give a precipitate. The precipitate was removed by filtration and the filtrate was concentrated to 209 g of dark oil. The product was purified by distillation (0.2 torr, 68°C) to give 172 g (96%) of Compound (VI) as colorless oil. Analytical data: ¾ NMR (500 MHz, DMDO-de) δ 8.57 (2 H), 7.40 (2 H), 0.23 (9 H).
EXAMPLE 2 – Preparation of Compound (Ilia)
Example 2 provides a morpholine amide route for the synthesis of Compound (Ilia). In this approach, morpholine amide (Compound VII) was prepared from 2-chlorobenzoyl chloride (Preparation 2A). Metallation of 2-bromopyridine with LDA (1.09 equiv.) in THF at -70°C followed by addition of (Compound VII) afforded Compound (Ilia) in 37% yield after crystallization from IP A/heptane (Preparation 2B). This sequence provides a direct route to Compound (Ilia), and a means to isolate Compound (Ilia) without the use of
chromatography. Compound (Ilia) may then be used to form Compound (I) as shown in Example 1A above (Preparation 2C).
Preparation 2A: Preparation of Compound (VII)

Toluene (1.5 L) was added to Compound (IX) (150 g, 0.86 mol) and cooled to 10°C. Morpholine (82 mL, 0.94 mol) was added to the clear solution over 10 minutes. The resulting white slurry was stirred for 20 minutes then pyridine (92 mL, 1.2 mol) was added dropwise over 20 minutes. The cloudy white mixture was stirred in a cold bath for 1 hour. Water (600 mL) was added in a single portion and the cold bath removed. The mixture was stirred for 20 minutes and the layers are separated. The organic layer was washed with a mixture of 1 N HC1 and water (2: 1, 500 mL:250 mL). The pH of the aqueous layer was ~ 2. The organic layer was washed with a mixture of saturated NaHCCb and water (1 : 1, 100 mL: 100 mL). The pH of the aqueous layer was ~ 9. The layers were separated. The organic layer was concentrated in vacuo to an oil. The oil was dissolved in IPA (70 mL) and heated at 60°C for 30 min. The clear solution was allowed to cool to 30°C, then heptane (700 mL, 4.7 v) was added dropwise. The resulting slurry was stirred at RT for 2 hours then cooled to 0°C for 1 hour. The slurry was filtered at RT, washed with heptane then dried under vacuum at 30°C overnight. Compound (VII) (156.2 g, 81%) was obtained as a white solid. Analytical data: ¾ NMR (500 MHz, CDCh) δ 7.42-7.40 (m, 1 H), 7.35-7.29 (m, 3 H), 3.91-3.87 (m, 1 H), 3.80-3.76 (m, 3 H), 3.71 (ddd, J= 11.5, 6.8, 3.3 Hz, 1 H), 3.60 (ddd, J = 11.2, 6.4, 3.4 Hz, 1 H), 3.28 (ddd, J= 13.4, 6.3, 3.2 Hz, 1 H), 3.22 (ddd, J= 13.7, 6.8, 3.3 Hz, 1 H); LRMS (ES+) calcd for CnHi3F6ClN02 (M+H)+ 226.1, found 225.9 m/z.
Preparation 2B: Preparation of Compound (Ilia)

THF (75 mL) was added to diisopropyl amine (4.9 mL, 34.8 mmol) and cooled to a
temperature of -70°C under N2 atmosphere. 2.5 M w-BuLi in hexanes (13.9 mL, 34.8 mmol) was added in a single portion (a 30-40°C exotherm) to the clear solution and cooled back to -70°C. Compound (VIII) (5.0 g, 31.6 mmol) was added neat to the LDA solution (a 2 to 5°C exotherm) followed by a THF (10 mL) rinse, keeping T< -65°C. This clear yellow solution was stirred at -70°C for 15 min. Compound (VII) (7.1 g, 31.6 mmol) in THF (30 mL) was added keeping T< -65°C. The resulting clear orange solution was stirred at -70°C for 3 hours. MeOH (3 mL) was added to quench reaction mixture and the cold bath was removed. 5 N HC1 (25 mL) was added to the reaction solution. MTBE (25 mL) was added, and the layers were separated. The organic layer was washed with water (25 mL X 2). The organic layer was dried over MgS04 and filtered. The organic layer was concentrated in vacuo to an orange oil. The oil was dissolved in IPA (15 mL, 3 vol) at ambient temperature. Heptane (25 mL) was added dropwise and the resulting slurry was stirred at RT for 1 hour. The slurry was cooled to 0°C for 1 hour and filtered. The filter cake was rinsed with chilled heptane (20 mL) and dried under vacuum at 30°C overnight. Compound (Ilia) (4.25 g, 45%) was obtained as a yellow solid.
Several reactions were run at different temperatures and with different addition rates of Compound (VII). If the reaction temperature was maintained below -65°C and Compound (VII) was added in <5 min, it was found that the reaction worked well. If the temperature was increased and/or the addition time of Compound (VII) was increased, then yields suffered, and the work-up was complicated by emulsions.
Preparation 2C: Preparation of Compound (I)
Compound (Ilia) may then reacted with Compound (Ila) to produce Compound (I) as shown in Preparation 1A.
EXAMPLE 3
Example 3 describes a new route for the synthesis of an intermediate free base, which may be used to form Compound (I) as described further below.
Example 3A: Preparation of starting material (Compound X) from 2-Chloronicotinonitrile

A mixture of NaH (40.0 g, 1 mol, 60% dispersion in mineral oil) and 2-chloronicotinonitrile (69.3 g, 500 mmol) in THF (1 L) was heated to reflux. A solution of 4-acetylpyridine (60.6 g, 500 mmol) in THF (400 mL) was added over a period of 40 min. The resulting dark brown mixture was stirred at reflux for ~ 2 h. The heating mantle was then removed, and AcOH (58 mL, 1 mol) was added. EtOAc (1 L) and H2O (1 L) were then added, and the layers were separated. The organic layer was concentrated to afford an oily solid. CH3CN (500 mL) was added, and the mixture was stirred for 30 min. H2O (1 L) was then added. The mixture was stirred for 1 h then filtered. The solid was rinsed with 2: 1
CH3CN-H2O (900 mL) and hexanes (400 mL) then dried under vacuum at 45°C overnight to afford 61.4 g (55% yield) of Compound (X) as yellow solid. Compound (X) exists as an approximate 95:5 enol-ketone mixture in CDCI3. Analytical data for enol: IR (CHCI3): 3024, 2973, 2229, 1631, 1597, 1579, 1550, 1497; ¾ NMR (500 MHz, CDCI3) δ 8.69 (dd, J= 4.4,
1.7 Hz, 2H), 8.55 (dd, J = 5.2, 1.8 Hz, 1H), 7.97 (dd, J= 7.9, 1.8 Hz, 1H), 7.70 (dd, J= 4.6, 1.5 Hz, 2H, 7.17 (dd, J = 7.8, 5.0 Hz, 1H), 6.59 (s, 1H); LRMS (ES+) calcd for C13H10N3O (M+H)+ 224.1, found 224.0 m/z.
Preparation 3B: Preparation of Compound (XI)
Preparation 3B(1):

(X) (XI)
Compound (XI) may be prepared using Compound (X).
Preparation 3B(2):
Alternatively, the following procedure for the conversion of nitrile into an acid which may also yield compound (XI). A mixture of Compound (X) (1 eq) and NaOH (1.5 eq) in 1 : 1 fhO-EtOH (3.5 mL/g of Compound (X)) was heated at 65°C overnight. The reaction mixture was cooled to RT then added to CH2C12 (12.5 mL/g of Compound (X)) and H20 (12.5 mL/g of Compound (X)). Cone. HC1 (2.5 mL/g of Compound (X)) was then added, and the layers were separated. The aqueous layer was extracted with CH2CI2 (10 mL/g of Compound (X)). The combined organic extracts were washed with H2O (12.5 ml/g of Compound (X)), dried (MgS04), filtered and concentrated to afford Compound (XI).
Preparation 3C
Compound Compound (XI) may then be converted into a Stage C intermediate free base, with observed 87% conversion in Grignard reaction as shown above. A complete synthesis route for Com ound (I) starting from compound Compound (XI) is depicted below.

Detailed experimental procedures for the synthesis of benzoate salt and final step are given in
International Patent Application Publication WO 2008/079600 Al .
References
- “Company Overview of Eli Lilly & Co., Worldwide License to Develop and Commercialize VLY-686”. Bloomberg Business. Retrieved 16 November 2015.
- [1]
- “Vanda Pharmaceuticals Announces Tradipitant Phase II Proof of Concept Study Results for Chronic Pruritus in Atopic Dermatitis”. PR Newswire. Retrieved 16 November 2015.
- Schmidt, B (2006). “Proof of principle studies”. Epilepsy Res. 68 (1): 48–52. doi:10.1016/j.eplepsyres.2005.09.019. PMID 16377153.
- George, DT; Gilman, J; Hersh, J; et al. (2008). “Neurokinin 1 receptor antagonism as a possible therapy for alcoholism.”. Science. 6: 1536–1539. doi:10.2147/SAR.S70350. PMC 4567173
 . PMID 26379454.
. PMID 26379454. - Tauscher, J; Kielbasa, W; Iyengar, S; et al. (2010). “Development of the 2nd generation neurokinin-1 receptor antagonist LY686017 for social anxiety disorder”. European Neuropsychopharmacology. 20 (2): 80–87. doi:10.1016/j.euroneuro.2009.10.005. PMID 20018493.
George, D.T.; Gilman, J.; Hersh, J.; Thorsell, A.; Herion, D.; Geyer, C.; Peng, X.; Kielbasa, W.; Rawlings, R.; Brandt, J.E.; Gehlert, D.R.; Tauscher, J.T.; Hunt, S.P.; Hommer, D.; Heilig, M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism, Science 2008, 319(5869): 1536
Gackenheimer, S.L.; Gehlert, D.R.In vitro and in vivo autoradiography of the NK-1 antagonist (3H)-LY686017 in guinea pig brain39th Annu Meet Soc Neurosci (October 17-21, Chicago) 2009, Abst 418.16
Tonnoscj, K.; Zopey, R.; Labus, J.S.; Naliboff, B.D.; Mayer, E.A.
The effect of chronic neurokinin-1 receptor antagonism on sympathetic nervous system activity in irritable bowel syndrome (IBS) Dig Dis Week (DDW) (May 30-June 4, Chicago) 2009, Abst T1261
Kopach, M.E.; Kobierski, M.E.; Coffey, D.S.; et al.
Process development and pilot-plant synthesis of (2-chlorophenyl)[2-(phenylsulfonyl)pyridin-3-yl]methanone
Org Process Res Dev 2010, 14(5): 1229
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016060250 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2015-11-10 | 2016-03-03 |
| US2015320866 | PHARMACEUTICAL COMPOSITION COMPRISING ANTIEMETIC COMPOUNDS AND POLYORTHOESTER | 2013-12-13 | 2015-11-12 |
| US2014206877 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2014-03-27 | 2014-07-24 |
| US2012225904 | New 7-Phenyl-[1, 2, 4]triazolo[4, 3-a]Pyridin-3(2H)-One Derivatives | 2010-11-09 | 2012-09-06 |
| US2010056795 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2010-03-04 | |
| US7381826 | Crystalline forms of {2-[1-(3, 5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-1H-[1, 2, 3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone | 2007-04-05 | 2008-06-03 |
| US7320994 | Triazole derivatives as tachykinin receptor antagonists | 2005-10-27 | 2008-01-22 |
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C28H16ClF6N5O |
| Molar mass | 587.90 g/mol |
| 3D model (Jmol) | |
Tradipitant
Tradipitant is being evaluated in a Phase II study in treatment resistant pruritus in atopic dermatitis.
Tradipitant is an NK-1 receptor antagonist licensed from Eli Lilly in 2012. Tradipitant has demonstrated proof-of-concept in alcohol dependence in a study published by the NIH1. In that study tradipitant was shown to reduce alcohol cravings and voluntary alcohol consumption among patients with alcohol dependence. NK-1R antagonists have been evaluated in a number of indications including chemotherapy-induced nausea and vomiting (CINV), post-operative nausea and vomiting (PONV), alcohol dependence, anxiety, depression, and pruritus.
The NK-1R is expressed throughout different tissues of the body, with major activity found in neuronal tissue. Substance P (SP) and NK-1R interactions in neuronal tissue regulate neurogenic inflammation locally and the pain perception pathway through the central nervous system. Other tissues, including endothelial cells and immune cells, have also exhibited SP and NK-1R activity2. The activation of NK-1R by the natural ligand SP is involved in numerous physiological processes, including the perception of pain, behavioral stressors, cravings, and the processes of nausea and vomiting1,2,3. An inappropriate over-expression of SP either in nervous tissue or peripherally could result in pathological conditions such as substance dependence, anxiety, nausea/vomiting, and pruritus1,2,3,4. An NK-1R antagonist may possess the ability to reduce this over-stimulation of the NK-1R, and as a result address the underlying pathophysiology of the symptoms in these conditions.
References
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, Peng X, Keilbasa W, Rawlings R, Brandt JE, Gehlert DR, Tauscher JT, Hunt SP, Hommer D, Heilig M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008; 319(5869):1536-9
- Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Current Medicinal Chemistry. 2004;11:2045-2081.
- Hargreaves R, Ferreira JC, Hughes D, Brands J, Hale J, Mattson B, Mill S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Annals of the New York Academy of Sciences. 2011; 1222:40-48.
- Stander S, Weisshaar E, Luger A. Neurophysiological and neurochemical basis of modern pruritus treatment. Experimental Dermatology. 2007;17:161-69.
///////////////////tradipitant, PHASE 2, VLY-686, LY686017, традипитант , تراديبيتانت , 曲地匹坦 , VANDA, ELI LILLY, Gastroparesis Pruritus, FDA 2025, APPROVALS 2025, vomiting associated with motion
LY 2922470

LY 2922470

LY 2922470
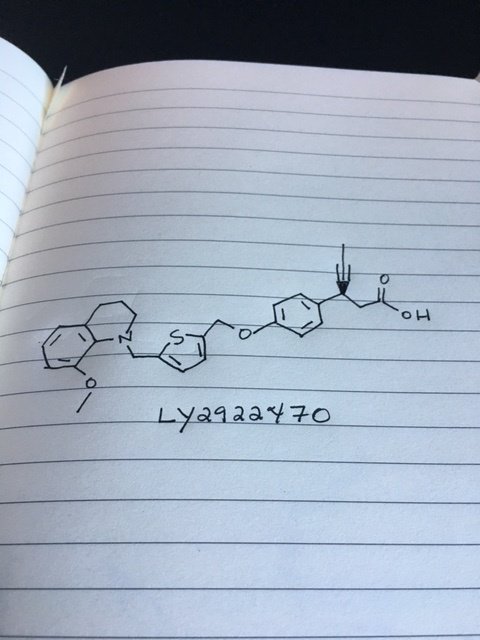
Picture credit….Bethany Halford
(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophen-2-yl]methoxy]phenyl]hex-4-ynoic acid
Benzenepropanoic acid, 4-[[5-[(3,4-dihydro-8-methoxy-1(2H)-quinolinyl)methyl]-2-thienyl]methoxy]-β-1-propyn-1-yl-, (βS)-
Glucose Lowering Agents, Signal Transduction Modulators
| CAS | 1423018-12-5 |
|---|---|
| Molecular Formula: | C28H29NO4S |
| Molecular Weight: | 475.59916 g/mol |
|---|
https://clinicaltrials.gov/ct2/show/NCT01867216
- Phase I Type 2 diabetes mellitus
Eli Lilly
Antihyperglycaemics
- 28 Jan 2014 Eli Lilly completes a phase I trial in Type-2 diabetes mellitus in USA (NCT01867216)
- 30 Jun 2013 Phase-I clinical trials in Type-2 diabetes mellitus in USA (PO)
- 14 Jun 2013 Eli Lilly plans a phase I trial for Type-2 diabetes mellitus in USA (NCT01867216)
PATENT
WO 2013025424
https://www.google.com/patents/US20130045990?cl=de
| Also published as | CA2843474A1, CA2843474C, CN103687856A, CN103687856B, EP2744806A1, US8431706, WO2013025424A1, Less « |
| Inventors | Chafiq Hamdouchi |
| Original Assignee | Eli Lilly And Company |



Preparation 18-Methoxyquinoline
Add potassium hydroxide (435 g, 7.76 mol) to a solution of 8-hydroxy quinoline (250 g, 1.724 mol) in THF (10 L) at ambient temperature and stir. Add methyl iodide (435 g, 2.58 mol) dropwise and stir overnight. Filter the reaction mixture and wash the solid with THF (2 L). Concentrate the solution to dryness; add water; extract with dichloromethane (2×3 L); combine the organic layers; and wash with brine. Collect the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a red oil, which solidifies on standing, to give the title compound (281 g, 102%), which can be used without further purification. ESI (m/z) 160(M+H).
Preparation 2
8-Methoxy-1,2,3,4-tetrahydroquinoline
Add sodium cyanoborohydride (505 g, 8.11 mol) in EtOH (1 L) to a solution of 8-methoxy quinoline (425 g, 2.673 mol) in EtOH (9 L), and stir. Cool the reaction mixture to an internal temperature of 0° C. and add HCl (35%, 1.12 L, 10.962 mol) dropwise over 60 min so that the internal temperature did not rise above 20° C. Allow the reaction mixture to warm to ambient temperature and then heat to reflux for 2.5 hours. Cool to ambient temperature and stir overnight. Add ammonium hydroxide (25%, 1 L); dilute with water (15 L); and extract the mixture with dichloromethane (3×10 L). Combine the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography, eluting with ethyl acetate: hexane (1:10) to give the title compound (357 g, 82%). ESI (m/z) 164(M+H).
Preparation 3
Methyl-5-methylthiophene-2-carboxylate
Add thionyl chloride (153 ml, 2.1 mol) dropwise over 20 min to a solution of 5-methylthiophene-2-carboxylic acid (100 g, 0.703 mol) in MeOH (1 L) at 0° C. and stir. After the addition is complete, heat the reaction mixture to reflux for 3.5 hours. Cool and concentrate in vacuo to give a thick oil. Dilute the oil with EtOAc (500 ml) and sequentially wash with water (300 ml) then brine (300 ml). Dry the organic layer over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give the title compound (106 g, 97%), which is used without further purification. ESI (m/z) 156(M+H).
Preparation 4
Methyl 5-(bromomethyl)thiophene-2-carboxylate
Add freshly recrystallised NBS (323.8 g, 1.81 mol) to a solution of methyl-5-methylthiophene-2-carboxylate (258 g, 1.65 mol) in chloroform (2.6 L) at room temperature, and stir. Add benzoyl peroxide (3.99 g, 0.016 mol) and heat the reaction mixture to reflux for 7 hours. Cool the reaction mixture to ambient temperature and filter through diatomaceous earth. Wash the filter cake with chloroform (250 ml). Collect the organic layers and remove the solvent to give the title compound (388 g, 100%), which is used without further purification. ESI (m/z) 236(M+H).
Preparation 5
Methyl-5-[8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophene-2-carboxylate
Add methyl-5-(bromoethyl)thiophene-2-carboxylate (432.5 g, 1.84 mol) in EtOH (500 ml) to a solution of 8-methoxy-1,2,3,4-tetrahydroquinoline (300 g 1.84 mol) in EtOH (1 L) and stir. Add DIPEA (641 ml, 3.67 mol) dropwise and stir at room temperature overnight. After completion of the reaction, remove the EtOH in vacuo, and add water (5 L). Extract the aqueous with EtOAc (3×3 L); combine the organic layers; and dry over sodium sulfate. Filter the solution and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography eluting with ethyl acetate: hexane (6:94) to give the title compound (325 g, 56%). ESI (m/z) 318(M+H).
Preparation 6
[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methanol
Add DIBAL-H (1 M in toluene 2.7 L, 2.66 mol) slowly via a cannula over a period of 1.5 h to a stirred solution of methyl-5-(8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-carboxylate (281 g, 0.886 mol) in THF (4 L) at −70° C. Monitor the reaction via thin layer chromatography (TLC) for completion. After completion of the reaction, allow the reaction mixture to warm to 20° C. and add a saturated solution of ammonium chloride. Add a solution of sodium potassium tartrate (1.3 Kg in 5 L of water), and stir overnight. Separate the organic layer; extract the aqueous phase with EtOAc (2×5 L); then combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration. Remove the solvent from the filtrate under reduced pressure to give the title compound as a white solid (252 g, 98%). ESI (m/z) 290(M+H).
Preparation 7
Ethyl(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoate
Add tributylphosphine (50% solution in EtOAc, 543 ml, 1.34 mol) to a solution of ADDP (282.5 g, 1.5 eq) in THF (3 L) and cool the mixture to an internal temperature of 0° C., then stir for 15 minutes. Add (S)-ethyl 3-(4-hydroxyphenyl)hex-4-ynoate (173.5 g, 0.747 mol) in THF (3 L) dropwise over 15 min; then add 5-((8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-yl)methanol (216 g, 0747 mol) in THF (5 L) dropwise. Allow the reaction mixture to warm to ambient temperature and stir overnight. Filter the reaction mixture through diatomaceous earth and wash the filter cake with ethyl acetate (2 L). Concentrate the organic filtrate to dryness. Add water (4 L); extract with ethyl acetate (3×5 L); combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration and concentrate under reduced pressure to give an oil. Purify the residue by silica gel flash chromatography by eluting with ethyl acetate: hexane (6:94) to give the title compound (167 g, 44%). ESI (m/z) 504(M+H).
Example 1
(3S)-3-[4-[[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoic acid

Add a solution of potassium hydroxide (49.76 g, 0.88 mol) in water (372 ml) to a solution of (S)-ethyl-3-(4-((5-8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophen-2-yl)methoxy) phenyl)hex-4-ynoate (149 g, 0.296 mol) in EtOH (1.49 L) at room temperature and stir overnight. Concentrate the reaction mixture to dryness and add water (1.3 L). Extract the resulting solution with EtOAc (2×300 ml) and separate. Adjust the pH of the aqueous layer to pH=6 with 2 N HCl. Collect the resulting solids. Recrystallise the solids from hot MeOH (298 ml, 2 vol) to give the title compound (91 g, 65%). ESI (m/z) 476(M+H).
Abstract
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260


Presenter

Chafiq Hamdouchi
Senior Research Advisor at Eli Lilly and Company
https://www.linkedin.com/in/chafiq-hamdouchi-4988126
Summary
Dr. Hamdouchi earned his bachelor’s degree and doctorate in organic chemistry from Louis Pasteur University, Strasbourg-France.
Following two postdoctoral fellowships, sponsored by the National Science Foundation-USA and Ministerio de Educación y Ciencia-Spain, he joined Eli Lilly and Company in 1995.
Throughout his 20 years of career at Lilly, he has contributed to a sustainable drug discovery portfolio from preclinical hypothesis to clinical proof-of-concept that spans the oncology, neuroscience and endocrinology therapeutic areas. He has led multidisciplinary (chemistry, pharmacology, ADMET, PK, medical) scientific teams in USA, Europe and Asia to deliver a number of compounds that achieved first human dose.
He is a co-inventor of six innovative molecules being pursued in clinical development for the treatment of Diabetes, Cancer and Neurodegenerative Diseases.
He has an extensive patent and publication record and deep experience in conducting drug discovery and development in Asia through effective partnership and mentorship.
SEE AT…………ONE ORGANIC CHEMIST ONE DAY BLOG
LINK……http://oneorganichemistoneday.blogspot.in/2016/03/chafiq-hamdouchi-senior-research.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US8431706 | 2013-04-30 | 1,2,3,4-tetrahydroqinoline derivative useful for the treatment of diabetes |
References
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260
//////Phase 1, LY2922470, LY 2922470, Eli Lilly, Type 2 diabetes mellitus, 1423018-12-5, Chafiq Hamdouchi
CC#CC(CC(=O)O)C1=CC=C(C=C1)OCC2=CC=C(S2)CN3CCCC4=C3C(=CC=C4)OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
OLANZEPINE VISITED PART 1/3



Olanzapine, LY170053
CAS : 132539-06-1
2-Methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine
2-Methyl-4-(4-methyl-1-piperazinyl)-10Hthieno[2,3-b][1,5]benzodiazepine
Manufacturers’ Codes: LY-170053
Trademarks: Zyprexa (Lilly)
Molecular Formula: C17H20N4S
Molecular Weight: 312.43
Percent Composition: C 65.35%, H 6.45%, N 17.93%, S 10.26%
PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/
Properties: Crystals from acetonitrile, mp 195°. Practically insol in water.
Melting point: mp 195°
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Other Tricyclics; Serotonin-Dopamine Antagonist.

Olanzapine (sold under the brand names Zyprexa, Zypadhera and Lanzek or in combination with fluoxetine, Symbyax) is anatypical antipsychotic. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia and bipolar disorder.[4]
Olanzapine is structurally similar to clozapine and quetiapine, but is classified as a thienobenzodiazepine. The olanzapine formulations are manufactured and marketed by the pharmaceutical company Eli Lilly and Company; the drug went generic in 2011. Sales of Zyprexa in 2008 were $2.2B in the US, and $4.7B worldwide.[5]

Zyprexa (olanzapine) 10 mg tablets (AU)
Olanzapine has a higher affinity for 5-HT2A serotonin receptors than D2 dopamine receptors, which is a common property of all atypical antipsychotics, aside from the benzamide antipsychotics such as amisulpride. Olanzapine also had the highest affinity of any second-generation antipsychotic towards the P-glycoprotein in one in vitro study.[60] P-glycoprotein transports a number of drugs across a number of different biological membranes including the blood-brain barrier, which could mean that less brain exposure to olanzapine results from this interaction with the P-glycoprotein.[61]
Olanzapine is a potent antagonist of the muscarinic M3 receptor,[65] which may underlie its diabetogenic side effects.[64][66] Additionally, olanzapine also exhibits a relatively low affinity for serotonin 5-HT1, GABAA, beta-adrenergic receptors, and benzodiazepine binding sites.[67] [27]
Dosage forms
Olanzapine is marketed in a number of countries, with tablets ranging from 2.5 to 20 milligrams. Zyprexa (and generic olanzapine) is available as an orally-disintegrating “wafer” which rapidly dissolves in saliva. It is also available in 10 milligram vials for intramuscular injection.[4]
Research
Olanzapine has been investigated for use as an antiemetic, particularly for the control of chemotherapy-induced nausea and vomiting (CINV). A 2007 study demonstrated its successful potential for this use, achieving a complete response in the acute prevention of nausea and vomiting in 100% of patients treated with moderately and highly-emetogenic chemotherapy, when used in combination with palonosetron and dexamethasone.[85]
Olanzapine has been considered as part of an early psychosis approach for schizophrenia. The Prevention through Risk Identification, Management, and Education (PRIME) study, funded by the National Institute of Mental Health and Eli Lilly, tested the hypothesis that olanzapine might prevent the onset of psychosis in people at very high risk forschizophrenia. The study examined 60 patients with prodromalschizophrenia, who were at an estimated risk of 36–54% of developing schizophrenia within a year, and treated half with olanzapine and half with placebo.[86] In this study, patients receiving olanzapine did not have a significantly lower risk of progressing to psychosis. Olanzapine was effective for treating the prodromal symptoms, but was associated with significant weight gain.[87]
1H NMR PREDICT

13C NMR PREDICT


COSY

HMBC
…………………………………..WILL BE UPDATED
UV
IR
1H NMR
13C NMR
MASS
…………………………………….
INTERMEDIATE/S USED IN SYNTHESIS AND REFERENCE


LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;


Apotex Pharmachem Inc. Patent: US2008/319189 A1, 2008 ; Location in patent: Page/Page column 2 ;

LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;

Leyva-Perez, Antonio; Cabrero-Antonino, Jose R.; Corma, Avelino Tetrahedron, 2010 , vol. 66, # 41 p. 8203 – 8209
SEE
WATSON PHARMACEUTICALS, INC. Patent: WO2004/94390 A1, 2004 ; Location in patent: Page 15 ;

WO2006/6180 A1, ; Page/Page column 11 ;
 Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
WO2006/6180 A1, ; Page/Page column 11 ;
 US5605897 A1, ;
US5605897 A1, ;
………………………………………
PATENT
http://www.google.com.na/patents/EP1730153B1?cl=en


Figure 2 shows the NMR spectrum of the solvate according to the invention. The peaks were assigned as follows (1H NMR; CDCl3, 300 MHz) :

| Chemical shift δ | Assignement |
| 1.20 (3H, d) | CH3 – isopropanol |
| 2.30 (3H, s) | 4′-CH3 |
| 2.34 (3H, s) | 2- CH3 |
| 2.20-2.40 (2H, br s) | H – water |
| 2.49 (4H, m) | 3′-CH2 |
| 3.52 (4H, m) | 2′-CH2 |
| 4.03 (0.5H, dq) | CH – isopropanol |
| 5.02 (H, broad s) | 10-NH |
| 6.29 (H, broad s) | 3-CH |
| 6.29-7.05 (4H, m) | 6, 7, 8, 9-H |
-
Olanzapine has shown to have high activity with regard to the central nervous system and is also useful for the treatment of schizophrenia, schizophreniform disorders, acute mania, mild anxiety states and psychosis.
-
Various polymorphic and pseudopolymorphic forms, such as solvates, of olanzapine have become known. Some of them are useful for conversion to other desirable forms.
-
The British patent GB 1 533 235 discloses antipsychotically effective thienobenzodiazepines by a generic formula which also covers olanzapine.
-
US patent 5,229,382 discloses olanzapine explicitly. The described process for its synthesis involves a crystallization from acetonitrile.
-
EP-B-733 635 claims crystalline form II olanzapine, and this polymorphic form is said to be more stable than the material obtained according to US 5,229,382 which is designated “form I olanzapine”. Both the form I and the form II of olanzapine are characterized by e. g. X-ray data. The preparation of the more stable form II of olanzapine is effected by dissolving technical grade olanzapine in ethyl acetate and crystallization from the resulting solution by any conventional process such as seeding, cooling, scratching the glass of the reaction vessel or other common techniques.
-
WO 02/18390 discloses the monohydrate form I and the dihydrate form I of olanzapine, a process for production thereof and a process for production of form I of olanzapine which comprises the steps of stirring olanzapine monohydrate form I or crude olanzapine or form II of olanzapine in methylene chloride at reflux, cooling, filtering and drying. It is also described that a repeating of the process described in US 5,229,382 Example 1, subexample 4 did not lead to formation of form I of olanzapine.
-
WO 03/101997 relates to processes for preparation of form I of olanzapine by regulation of the pH-value of the solution.
-
WO 03/055438 discloses the preparation of form I olanzapine by crystallization from ethanol and subsequent conversion of the obtained ethanol solvate.
-
US patent 5,637,584 discloses the (mono)methylene chloride solvate form of olanzapine and a method for its conversion to the polymorphic form I of olanzapine.
-
EP-B-733 634 discloses three specific solvates of olanzapine, namely the methanol, ethanol and 1-propanol solvates and a process for production of form II olanzapine by drying such a solvate.
-
WO 03/097650 describes two new, mixed solvate forms, the water/methylene chloride solvate and the water/DMSO solvate, methods for preparing them, and their transformation to polymorphic form I.
-
WO 2004/006933 discloses a process for the preparation of form I olanzapine, as well as various pseudopolymorphic forms, namely the isopropanol solvate, and the acetonitrile/methylene chloride/water and acetonitrile/water mixed solvates of olanzapine, and the polymorphic form A.

Preparation of the water-isopropanol mixed solvate of olanzapine Example 1
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (120 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 200 ml were distilled off under vacuum. The residue was cooled to room temperature, isopropanol (180 ml) was added, and the solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C and after completion of the crystallization the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 22.84 g. Loss on drying (140°C): 13.6%. Water content (Karl Fischer): 5.12%.
Example 2
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 80 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35 °C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 21.98 g. Loss on drying (140°C): 13.2 %. Water content (Karl Fischer): 5.09%. Assay of isopropanol (GC): 8.55 %.
Example 3
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 120 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After completion of the crystallization, the precipitate.was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon completion of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 24.35 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.05%.
Example 4
-
Anhydrous olanzapine (10 g) was suspended in isopropanol (108 ml) and heated to reflux to obtain a clear solution. The solution was slowly cooled. Water (6 ml) was added at 57°C to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (5 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 10.97 g. Loss on drying (140°C): 13.3%. Water content (Karl Fischer): 5.13%.
Example 5
-
60 g of olanzapine obtained from mother liquors was suspended in isopropanol (650 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (7.9 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (16 ml) was added to start crystallization. The suspension was cooled to 0°C and, upon completion of the crystallization, the product was filtered off and washed with isopropanol (50 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 57.64 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.26%.
Example 6
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (41.86 g, 0.11 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (55.29 g, 0.22 mmol) and sulfur (11.99 g, 0.374 mmol) in benzonitrile (1100 mL) was stirred at 140°C for 11 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with dichloromethane and isopropanol (250 mL, 1 : 1). The precipitate was filtered off and washed with dichloromethane and isopropanol (20 ml, 1 : 1). The filtrate was extracted with HCl (250 ml, 2 M). The organic phase was further extracted with HCl (2 X 100 ml, 1 M). The combined aqueous phases were cooled in an ice bath and made alkaline by using 5 M NaOH. The obtained turbid solution was left in a refrigerator over night resulting in a suspension. This was separated by filtration and washed with isopropanol (2 X 25 ml). The wet material was suspended in isopropanol (215 ml) and heated to reflux to obtain a clear solution. The solution was hot filtered. Water (6.5 ml) was added to induce crystallization. The obtained’suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 18.61 g. Loss on drying (140°C): 12.8 %. Water content (Karl Fischer): 5.29 %.
Example 7
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (3.805 g, 10 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (5.026 g, 20 mmol) and sulfur (1.122 g, 35 mmol) in benzonitrile (100 ml.) was stirred at 140°C for 8.5 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with isopropanol (50 ml) and dimethyl sulfoxide (5 ml). The precipitate was filtered off and washed with isopropanol (5 ml). Water (10 ml) and sodium hydroxide (1.00 g, 25 mmol) were added to the filtrate. The mixture was stirred at room temperature until the sodium hydroxide had dissolved. The turbid solution was left in a refrigerator over night resulting in a suspension. This was filtered off and washed with isopropanol (5 mL). The wet material was suspended in isopropanol (25 mL) and the suspension was heated to reflux. Then solids were hot filtered. Water (0.75 mL) was added to the filtrate to induce crystallization. The resulting suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (1 mL). The product was then dried at room temperature under vacuum to a constant weight. Yield: 0.738 g.
………………………………..
PAPER
http://www.biomedcentral.com/1471-2210/12/8

Scheme 1
Synthesis of compounds 8a, 8b, and 8c. Reagents and conditions: (i) 1-fluoro-2-nitrobenzene, NaH, THF, rt, 20 h, 60%; (ii) SnCl2, EtOH, 80°C, 1 h, 80%; (iiia) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 80°C, 4 h,65%; (iiib) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 120°C, 3 h, 55%; (iiic) N-methylpiperazine (10 equiv), N-methylpiperazine hydrochloride (10 equiv), DMSO, 110–120°C, 20 h, 56%.
References
- Burton, Michael E.; Shaw, Leslie M.; Schentag, Jerome J.; Evans, William E. (May 1, 2005). Applied Pharmacokinetics & Pharmacodynamics: Principles of Therapeutic Drug Monitoring (4th ed.). Lippincott Williams & Wilkins. p. 815. ISBN 978-0-7817-4431-7.
- “PRODUCT INFORMATION OLANZAPINE SANDOZ® 2.5mg/5mg/7.5mg/10mg/15mg/20mg FILM-COATED TABLETS” (PDF). TGA eBusiness Services. Sandoz Pty Ltd. 8 June 2012. Retrieved 26 November 2013.
- “Zyprexa, Zyprexa Relprevv (olanzapine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 November 2013.
- ^ Jump up to:a b c “Olanzapine Prescribing Information” (PDF). Eli Lilly and Company. 2009-03-19. Retrieved 2009-09-06.
- “Lilly 2008 Annual Report” (PDF). Lilly. 2009. Retrieved 2009-08-06.
- National Collaborating Centre for Mental Health (25 March 2009). “Schizophrenia: Full national clinical guideline on core interventions in primary and secondary care”. Retrieved 25 November 2009.
- Duggan L, Fenton M, Dardennes RM, El-Dosoky A, Indran S (2005). Duggan, Lorna, ed. “Olanzapine for schizophrenia”. Cochrane Database of Systematic Reviews (2): CD001359.doi:10.1002/14651858.CD001359.pub2. PMID 10796640.
- “Psychosis and schizophrenia in adults: treatment and management | Guidance and guidelines | NICE”. National Institute for Health and Care Excellence.
- Barnes TR (2011). “Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology”. J. Psychopharmacol. (Oxford) 25 (5): 567–620. doi:10.1177/0269881110391123.PMID 21292923.
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ (2013). “World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects”. World J. Biol. Psychiatry 14 (1): 2–44. doi:10.3109/15622975.2012.739708. PMID 23216388.
- Abou-Setta, AM; Mousavi, SS; Spooner, C; Schouten, JR; Pasichnyk, D; Armijo-Olivo, S; Beaith, A; Seida, JC; Dursun, S; Newton, AS; Hartling, L (August 2012).PMID 23035275. Missing or empty
|title=(help) - Zhang JP, Gallego JA, Robinson DG, Malhotra AK, Kane JM, Correll CU (July 2013).“Efficacy and safety of individual second-generation vs. first-generation antipsychotics in first-episode psychosis: a systematic review and meta-analysis”. Int. J. Neuropsychopharmacol. 16 (6): 1205–18. doi:10.1017/S1461145712001277.PMC 3594563. PMID 23199972.
- Citrome L (August 2012). “A systematic review of meta-analyses of the efficacy of oral atypical antipsychotics for the treatment of adult patients with schizophrenia”. Expert Opin Pharmacother 13 (11): 1545–73. doi:10.1517/14656566.2011.626769.PMID 21999805.
- Lepping P, Sambhi RS, Whittington R, Lane S, Poole R (May 2011). “Clinical relevance of findings in trials of antipsychotics: systematic review”. Br J Psychiatry 198 (5): 341–5.doi:10.1192/bjp.bp.109.075366. PMID 21525517.
- Désaméricq G, Schurhoff F, Meary A et al. (February 2014). “Long-term neurocognitive effects of antipsychotics in schizophrenia: a network meta-analysis”. Eur. J. Clin. Pharmacol. 70 (2): 127–34. doi:10.1007/s00228-013-1600-y. PMID 24145817.
- Leucht S, Cipriani A, Spineli L et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”.Lancet 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harv Rev Psychiatry 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760.
- [+http://www.nice.org.uk/guidance/cg185/chapter/1-recommendations “Bipolar disorder: the assessment and management of bipolar disorder in adults, children and young people in primary and secondary care | 1-recommendations | Guidance and guidelines | NICE”].
- Yatham LN, Kennedy SH, O’Donovan C et al. (December 2006). “Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007”. Bipolar Disord 8 (6): 721–39. doi:10.1111/j.1399-5618.2006.00432.x. PMID 17156158.
- Selle V, Schalkwijk S, Vázquez GH, Baldessarini RJ (March 2014). “Treatments for acute bipolar depression: meta-analyses of placebo-controlled, monotherapy trials of anticonvulsants, lithium and antipsychotics”. Pharmacopsychiatry 47 (2): 43–52.doi:10.1055/s-0033-1363258. PMID 24549862.
- Maglione M, Maher AR, Hu J et al. (2011). “Off-Label Use of Atypical Antipsychotics: An Update”. PMID 22132426.
- Review of olanzapine in the management of bipolar disorders Neuropsychiatr Dis Treat. 2007 October; 3(5): 579–587.
- Scott, Lisa (Winter 2006). “Genetic and Neurological Factors in Stuttering”. Stuttering Foundation of America.
- “Important Safety Information for Olanzapine”. Zyprexa package insert. Eli Lilly & Company. 2007. Archived from the original on 2007-11-23. Retrieved 2007-12-03.
Elderly patients with dementia-related psychosis treated with atypical antipsychotic drugs are at an increased risk of death compared to placebo. […] ZYPREXA (olanzapine) is not approved for the treatment of elderly patients with dementia-related psychosis.
- “Doctors ‘ignoring drugs warning'”. BBC News. 17 June 2008. Retrieved 2008-06-22.
- L-Z\In re Zyprexa\Documents Leak\Injunction Memo & Order\FINAL INJUNCTION MEMO 2.13.07.wpd
- EFF.org
- Press Releases: January, 2007 | Electronic Frontier Foundation
- Eli Lilly was Concerned by Zyprexa Side-Effects from 1998,[dead link] The Times (London), January 23, 2007
- Navari RM, Einhorn LH, Loehrer PJ, Passik SD, Vinson J, McClean J, Chowhan N, Hanna NH, Johnson CS (2007). “A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: A Hoosier oncology group study”. Supportive Care in Cancer 15 (11): 1285–91. doi:10.1007/s00520-007-0248-5. PMID 17375339.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller TJ, Woods SW, Hawkins KA, Hoffman R, Lindborg S, Tohen M, Breier A (2003). “The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis”. Schizophrenia Research 61 (1): 7–18.doi:10.1016/S0920-9964(02)00439-5. PMID 12648731.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller T, Woods SW, Hawkins KA, Hoffman RE, Preda A, Epstein I, Addington D, Lindborg S, Trzaskoma Q, Tohen M, Breier A (2006). “Randomized, Double-Blind Trial of Olanzapine Versus Placebo in Patients Prodromally Symptomatic for Psychosis”. American Journal of Psychiatry 163 (5): 790–9.
Literature References:
Serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. Prepn: J. K. Chakrabarti et al., EP 454436; eidem, US 5229382 (1991, 1993 both to Lilly). PRODUCT PATENT
Comparative pharmacology: N. A. Moore et al., Curr. Opin. Invest. Drugs 2, 281 (1993). HPLC determn in human plasma: J. T. Catlow et al., J. Chromatogr. B 668, 85 (1995).
Clinical evaluation in schizophrenia: D. S. Baldwin, S. A. Montgomery, Int. Clin. Psychopharmacol. 10, 239 (1995); in mania of bipolar disorder: M. Tohen et al., Am. J. Psychiatry 156, 702 (1999).
Review of pharmacology and clinical experience: B. C. Lund, P. J. Perry, Expert Opin. Pharmacother. 1, 305-323 (2000).
External links
- Zyprexa.com – official Zyprexa brand website from Eli Lilly and Company
- Zyprexa package insert
- Berenson, Alex (December 17, 2006). “Lilly Settles With 18,000 Over Zyprexa”. New York Times.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
EDIVOXETINE REVISITED

EDIVOXETINE, LY 2216684
(1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol
UNII-3W9N3F4JOO, 1194508-25-2, Edivoxetine [USAN], Edivoxetine (USAN/INN), Edivoxetine [USAN:INN], 3W9N3F4JOO
Molecular Formula:C18H26FNO4
Molecular Weight:339.401743 g/mol
Edivoxetine (INN; LY-2216684) is a drug which acts as a selective norepinephrine reuptake inhibitor and is currently under development by Eli Lilly for attention-deficit hyperactivity disorder (ADHD) and as an antidepressant treatment.[1][2] It was in phase IIIclinical trials, in 2012, for major depressive disorder, but failed to get approval.[1][3]
Effectiveness
In a study published in 2010, edivoxetine failed to prove superiority over placebo, as measured by Hamilton Depression Rating Scale. However, effectiveness could be observed using the Self-Rated Quick Inventory of Depressive Symptomatology.[4]
In a study published in 2011, using the Montgomery-Åsberg Depression Rating Scale and the Sheehan Disability Scale, edivoxetine showed superiority over placebo, with higher response and remission rates.[5]
In December 2013, Eli Lilly announced that the clinical development of edivoxetine will be stopped due to lack of efficacy compared to SSRI alone in three separate clinical trials.[6]
Side effects
Side effects significantly associated with edivoxetine are headache, nausea, constipation, dry mouth and insomnia.[4]
The above mention studies report increases of the cardiac rhythm, and one also increases of diastolic and systolic blood pressures.[4][5]


Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op5003825
There is a growing trend in Ireland toward greater collaboration between academia and the pharmaceutical industry. This is an activity encouraged at a national policy level as a means of providing researchers from academic institutions the opportunity to gain important first-hand experience in a commercial research environment, while also providing industry access to expertise and resources to develop new and improved processes for timely medicines. The participating company benefits in terms of its growth, the evolution of its strategic research and development, and the creation of new knowledge that it can use to generate commercial advantage. The research institute benefits in terms of developing skill sets, intellectual property, and publications, in addition to access to identified current industry challenges. A case study is provided describing the collaborative partnership between a synthetic chemistry research team at University College Cork (UCC) and Eli Lilly and Company.
Department of Chemistry and School of Pharmacy, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre,University College Cork, Cork, Ireland
E-mail: a.maguire@ucc.ie.
![]()
University College Cork

| Systematic (IUPAC) name | |
|---|---|
| (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(tetrahydro-2H-pyran-4-yl)ethanol | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 1194508-25-2 1194374-05-4 (hydrochloride) |
| ATC code | None |
| PubChem | CID 11186829 |
| ChemSpider | 9361913 |
| Chemical data | |
| Formula | C18H26FNO4 |
| Molecular mass | 339.402 g/mol |
References
- Jun Yan (March 2012). “Pipeline for new antidepressants flowing slowly”. Psychiatric News (American Psychiatric Association) 47 (5): 1b-29. Retrieved 2012-04-27.
- “Statement on a nonproprietary name adopted by the USAN council – Edivoxetine” (Press release). American Medical Association. 2012. Retrieved 2012-04-12.
- Chancellor D (November 2011). “The depression market”. Nature Reviews. Drug Discovery 10 (11): 809–10. doi:10.1038/nrd3585. PMID 22037032.
- Dubé S, Dellva MA, Jones M, Kielbasa W, Padich R, Saha A, Rao P (April 2010). “A study of the effects of LY2216684, a selective norepinephrine reuptake inhibitor, in the treatment of major depression”. Journal of Psychiatric Research 44 (6): 356–363. doi:10.1016/j.jpsychires.2009.09.013. PMID 19909980.
- Pangallo P, Dellva MA, D’Souza DN, Essink B, Russell J, Goldberger C (June 2011). “A randomized, double-blind study comparing LY2216684 and placebo in the treatment of major depressive disorder”. Journal of Psychiatric Research 45 (6): 748–755. doi:10.1016/j.jpsychires.2011.03.014. PMID 21511276.
- https://investor.lilly.com/releasedetail.cfm?ReleaseID=811751
H-NMR spectral analysis
![(1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol NMR spectra analysis, Chemical CAS NO. 1194508-25-2 NMR spectral analysis, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/570/465/1194508-25-2-1h.png) CAS NO. 1194508-25-2, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol H-NMR spectral analysis |
C-NMR spectral analysis
![(1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol NMR spectra analysis, Chemical CAS NO. 1194508-25-2 NMR spectral analysis, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/570/465/1194508-25-2-13c.png) CAS NO. 1194508-25-2, (1R)-2-(5-fluoro-2-methoxyphenyl)-1-[(2S)-morpholin-2-yl]-1-(oxan-4-yl)ethanol C-NMR spectral analysis |
/////////////
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Mahabalipuram
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.



Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …




http://www.weather-forecast.com/locations/Mamallapuram
 Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
 Moonraikers Restaurant, Mamallapuram
Moonraikers Restaurant, Mamallapuram


 Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
 .
.
A carving at the Varaha Temple, Mahabalipuram

/////////////
EU approves Lilly diabetes drug Trulicity, dulaglutide
Regulators in Europe have given the green light to Eli Lilly’s Trulicity, its once-weekly glucagon-like peptide-1 receptor agonist for type 2 diabetes.
Read more at: http://www.pharmatimes.com/Article/14-11-25/EU_approves_Lilly_diabetes_drug_Trulicity.aspx
Dulaglutide is a glucagon-like peptide 1 receptor agonist (GLP-1 agonist) for the treatment of type 2 diabetes that can be used once weekly.[1][2]GLP-1 is a hormone that is involved in the normalization of level of glucose in blood (glycemia). The FDA approved dulaglutide for use in the United States in September 2014.[3] The drug is manufactured by Eli Lilly under the brand name Trulicity.[3]

Mechanism of action
Dulaglutide binding to glucagon-like peptide 1 receptor, slows gastric emptying and increases insulin secretion by beta cells in the pancreas. Simultaneously the compound reduces the elevated glucagon secretion by alpha cells of the pancreas, which is known to be inappropriate in the diabetic patient. GLP-1 is normally secreted by L cells of the gastrointestinal mucosa in response to a meal.[4]
Medical uses[
The compound is indicated for adults with type 2 diabetes mellitus as an adjunct to diet and exercise to improve glycemic control. Dulaglutide is not indicated in the treatment of subjects with type 1 diabetes mellitus or patients with diabetic ketoacidosis. Dulaglutide can be used either stand-alone or in combination with other medicines for type 2 diabetes, in particular metformin, sulfonylureas, thiazolidinediones, and insulin taken concomitantly with meals.[5]
Side effects
The most common side effects include gastrointestinal disorders, such as dyspepsia, decreased appetite, nausea, vomiting, abdominal pain, diarrhea.[6] Some patients may experience serious adverse reactions: acute pancreatitis (symptoms include persistent severe abdominal pain, sometimes radiating to the back and accompanied by vomiting),hypoglycemia, renal impairment (which may sometimes require hemodialysis). The risk of hypoglycemia is increased if the drug is used in combination with sulfonylureas orinsulin.[7][8]
Contraindications
The compound is contraindicated in subjects with hypersensitivity to active principle or any of the product’s components. As a precautionary measure patients with a personal or family history of medullary thyroid carcinoma or affected by multiple endocrine neoplasia syndrome type 2 should not take dulaglutide, because for now it is unclear whether the compound can increase the risk of these cancers.[9]
![]()
References
- JCourtney Aavang Tibble, Tricia Santos Cavaiola, Robert R Henry (2013). “Longer Acting GLP-1 Receptor Agonists and the Potential for Improved Cardiovascular Outcomes: A Review of Current Literature”. Expert Rev Endocrinol Metab 8 (3): 247–259.doi:10.1586/eem.13.20.
- “Lilly’s Once-Weekly Dulaglutide Shows Non-Inferiority to Liraglutide in Head-to-Head Phase III Trial for Type 2 Diabetes”. Eli Lilly. Feb 25, 2014.
- “FDA approves Trulicity to treat type 2 diabetes” (Press release). FDA. Sep 18, 2014.
- Nadkarni P, Chepurny OG, Holz GG (2014). “Regulation of glucose homeostasis by GLP-1”. Prog Mol Biol Transl Sci 121: 23–65. doi:10.1016/B978-0-12-800101-1.00002-8.PMC 4159612. PMID 24373234. Retrieved 2014-09-29.
- Terauchi Y, Satoi Y, Takeuchi M, Imaoka T (July 2014). “Monotherapy with the once weekly GLP-1 receptor agonist dulaglutide for 12 weeks in Japanese patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomised, double-blind, placebo-controlled study”. Endocr. J. PMID 25029955. Retrieved 2014-09-29.
- Nauck M, Weinstock RS, Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z (August 2014). “Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5)”. Diabetes Care 37 (8): 2149–58.doi:10.2337/dc13-2761. PMID 24742660.
- Amblee A (April 2014). “Dulaglutide for the treatment of type 2 diabetes”. Drugs Today50 (4): 277–89. doi:10.1358/dot.2014.50.4.2132740. PMID 24918645.
- Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E (February 2014). “Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials”. Diabetes Res. Clin. Pract. 103 (2): 269–75. doi:10.1016/j.diabres.2014.01.010.PMID 24485345.
- Samson SL, Garber A (April 2013). “GLP-1R agonist therapy for diabetes: benefits and potential risks”. Curr Opin Endocrinol Diabetes Obes 20 (2): 87–97.doi:10.1097/MED.0b013e32835edb32. PMID 23403741. Retrieved 2014-09-30.
| Identifiers | |
|---|---|
| CAS number | 923950-08-7 |
| ATC code | None |
| Chemical data | |
| Formula | C2646H4044N704O836S18 |
| Mol. mass | 59669.81 g/mol |
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog as once-weekly treatment for type 2 diabetes.

DULAGLUTIDE
PRONUNCIATION doo” la gloo’ tide
THERAPEUTIC CLAIM Treatment of type II diabetes
CHEMICAL NAMES
1. 7-37-Glucagon-like peptide I [8-glycine,22-glutamic acid,36-glycine] (synthetic
human) fusion protein with peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
2. [Gly8,Glu22,Gly36]human glucagon-like peptide 1-(7-37)-peptidyltetraglycyl-Lseryltetraglycyl-L-seryltetraglycyl-L-seryl-L-alanyldes-Lys229-[Pro10,Ala16,Ala17]human immunoglobulin heavy constant γ4 chain H-CH2-CH3 fragment, (55-55′:58-58′)-bisdisulfide dimer
STRUCTURAL FORMULA
Monomer
HGEGTFTSDV SSYLEEQAAK EFIAWLVKGG GGGGGSGGGG SGGGGSAESK 50
YGPPCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSQEDP 100
EVQFNWYVDG VEVHNAKTKP REEQFNSTYR VVSVLTVLHQ DWLNGKEYKC 150
KVSNKGLPSS IEKTISKAKG QPREPQVYTL PPSQEEMTKN QVSLTCLVKG 200
FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSRLT VDKSRWQEGN 250
VFSCSVMHEA LHNHYTQKSL SLSLG 275
Disulfide bridges location
55-55′ 58-58′ 90-150 90′-150′ 196-254 196′-254′
MOLECULAR FORMULA C2646H4044N704O836S18
MOLECULAR WEIGHT 59.67 kDa
MANUFACTURER Eli Lilly and Company
CODE DESIGNATION LY2189265
CAS REGISTRY NUMBER 923950-08-7
http://www.ama-assn.org/resources/doc/usan/dulaglutide.pdf
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog, is a biologic entity being studied as a once-weekly treatment for type 2 diabetes.
Dulaglatuide works by stimulating cells to release insulin only when blood sugar levels are high.
Gwen Krivi, Ph.D., vice president, product development, Lilly Diabetes, said of the drug, “We believe dulaglutide, if approved, can bring significant benefits to people with type 2 diabetes.”
In fact, it might help to control both diabetics’ blood sugar and their high blood pressure.
Eli Lilly CEO John Lechleiter believes the drug has the potential to be a blockbuster. Lilly could be ready to seek approval by 2013.
For more information on dulaglutide clinical studies, click here.
PRESS RELEASES
Data Preseted at 49th EASD Annual Meeting Show Treatment with Lilly’s Investigational Dulaglutide Resulted in Improved Patient-Reported Health Outcomes – September 26, 2013
Lilly Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – April 16, 2013
Lilly Diabetes Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – October 22, 2012
Ixchelsis, a start-up company that has come out of Pfizer’s former R D site at Sandwich, UK, is progressing a treatment for premature ejaculation boosted by the backing of Eli Lilly.
Lilly, through its venture fund set up with TVM Capital Life Science, has
invested in Ixchelsis, made up of former Pfizer scientists and headed by Gary
Muirhead. The company is based on an oxytocin receptor antagonist called IX-01
originally discovered at Sandwich which the investors say “has the potential to
be the best-in-class pharmacological approach for the treatment of
PE”.
Ixchelsis, which is based at the Sandwich site, now called Discovery
Park, will collaborate with the autonomous early phase virtual drug discovery
arm of Lilly, known as Chorus. Dr Muirhead told PharmaTimes that the
TVM model will fund through to the agreed exit point, which is completion of
proof-of-concept and this requires about $14 million.
read all at
http://www.pharmatimes.com/Article/13-08-08/Lilly_backs_Ixchelsis_a_start-up_born_at_Pfizer.aspx
………..
………….
……………….
…………….

{kind=link}