
Home » Posts tagged 'CTP-656'
Tag Archives: CTP-656
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Deutivacaftor
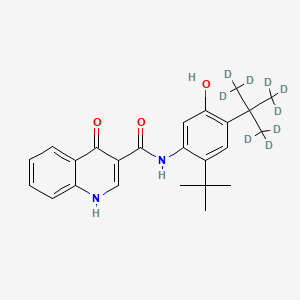

Deutivacaftor
RN: 1413431-07-8
UNII: SHA6U5FJZL,
N-[2-tert-butyl-4-[1,1,1,3,3,3-hexadeuterio-2-(trideuteriomethyl)propan-2-yl]-5-hydroxyphenyl]-4-oxo-1H-quinoline-3-carboxamide
Molecular Formula, C24-H28-N2-O3, Molecular Weight, 401.552
Synonyms
-
CTP-656
-
D9-ivacaftor
-
Deutivacaftor
-
Ivacaftor D9
-
UNII-SHA6U5FJZL
-
VX-561
-
WHO 10704
Treatment of Cystic Fibrosis
- Originator Concert Pharmaceuticals
- Class Amides; Aminophenols; Antifibrotics; Organic deuterium compounds; Quinolones; Small molecules
- Mechanism of Action Cystic fibrosis transmembrane conductance regulator stimulants
- Orphan Drug Status Yes – Cystic fibrosis
- Phase II Cystic fibrosis
- 15 Apr 2019 Vertex Pharmaceuticals plans a phase II trial for Cystic fibrosis in April 2019 , (EudraCT2018-003970-28), (NCT03911713)
- 11 Apr 2019 Vertex Pharmaceuticals plans a phase II trial for Cystic Fibrosis (Combination therapy) in May 2019 (NCT03912233)
- 24 Oct 2018 Vertex Pharmaceuticals plans a phase II trial for Cystic fibrosis (with gating mutation) in the US in the first half of 2019
Patent
WO 2012158885
Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, nonradioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res, 1985, 14: 1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol, 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9: 101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p. 35 and Fisher at p. 101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem., 1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
[9] This invention relates to novel derivatives of ivacaftor, and pharmaceutically acceptable salts thereof. This invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of treating diseases and conditions that are beneficially treated by administering a CFTR (cystic fibrosis transmembrane conductance regulator) potentiator.
[10] Ivacaftor, also known as VX-770 and by the chemical name, N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide, acts as a CFTR potentiator. Results from phase III trials of VX-770 in patients with cystic fibrosis carrying at least one copy of the G551D-CFTR mutation demonstrated marked levels of improvement in lung function and other key indicators of the disease including sweat chloride levels, likelihood of pulmonary exacerbations and body weight. VX-770 is also currently in phase II clinical trials in combination with VX-809 (a CFTR corrector) for the oral treatment of cystic fibrosis patients who carry the more common AF508-CFTR mutation. VX-770 was granted fast track designation and orphan drug designation by the FDA in 2006 and 2007, respectively.
[11] Despite the beneficial activities of VX-770, there is a continuing need for new compounds to treat the aforementioned diseases and conditions.
Patent
US 20140073667
Patent
JP 2014097964
PATENT
WO 2018183367
The use according to embodiment 1, comprising administering to the patient an effect amount of (N-(2-(tert-butyl)-5-hydroxy-4-(2-(methyl-d3)propan-2-yl-l, 1, 1,3, 3,3-d6)phenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (Compound Il-d):
Il-d
PATENT
WO 2019018395,

CONTD…………………………..

//////////////////deutivacaftor, Orphan Drug Status, Cystic fibrosis, CTP-656, D9-ivacaftor, Deutivacaftor, Ivacaftor D9, UNII-SHA6U5FJZL, VX-561, WHO 10704, PHASE 2
[2H]C([2H])([2H])C(c1cc(c(NC(=O)C2=CNc3ccccc3C2=O)cc1O)C(C)(C)C)(C([2H])([2H])[2H])C([2H])([2H])[2H]
