
Home » Posts tagged 'clinical trials' (Page 4)
Tag Archives: clinical trials
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Orexigen files obesity drug Contrave for approval in Europe

Orexigen files obesity drug Contrave for approval in Europe
Orexigen Therapeutics has submitted the Marketing Authorization Application (MAA) for Contrave, an investigational weight-loss drug to the European Medicines Agency (EMA).
 The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
read all at
http://www.pharmatopics.com/2013/10/orexigen-files-obesity-drug-contrave-approval-europe/
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Isavuconazole – Basilea reports positive results from study

This post is updated in sept 2015……..
Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
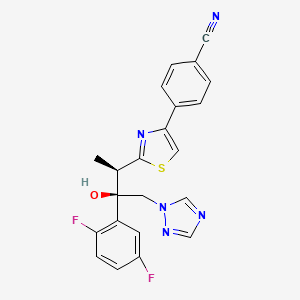
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.

-

-
Scheme 1.
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

-
Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
-
Figure options

-
Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).

read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
Clinical trials
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
References
- [1]
- Saboo, Alok. “Basilea Announces Global Partnership With Astellas for Its Antifungal Isavuconazole.” FierceBiotech. N.p., 24 Feb. 2010. Web.
- “Basilea reports isavuconazole orphan drug designation by U.S. FDA.” Market Wired. 28 May 2013.
- “FDA Grants Orphan Drug Designation to Astellas for Isavuconazole for the Treatment of Invasive Candidiasis.” News Releases. Astellas. 3 Nov 2014.
- Cresemba (isovuconazole sulfate) [prescribing information]. Astella Pharma US, Inc. Revised March 2015.
- Jump up^ “Aspergillosis.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 08 Sept. 2014.
- Jump up^ “Astellas Receives FDA Approval for CRESEMBA® (isavuconazonium Sulfate) for the Treatment of Invasive Aspergillosis and Invasive Mucormycosis.” PR Newswire. N.p., 6 Mar. 2015.
- Jump up^ “Isavuconazonium.” Micromedex Solutions. Truven Health Analytics, n.d. Web. <www.micromedexsolutions.com>.
- Jump up^ Pettit, Natasha N.; Carver, Peggy L. (2015-07-01). “Isavuconazole A New Option for the Management of Invasive Fungal Infections”. Annals of Pharmacotherapy 49 (7): 825–842.doi:10.1177/1060028015581679. ISSN 1060-0280. PMID 25940222.
- Mujais, A. “2014: M-1756. A Phase 3 Randomized, Double-Blind, Non-Inferiority Trial Evaluating Isavuconazole (ISA) vs. Voriconazole (VRC) for the Primary Treatment of Invasive Fungal Disease (IFD) Caused by Aspergillus spp. or other Filamentous Fungi (SECURE): Outcomes by Malignancy Status”. http://www.icaaconline.com. Retrieved 2015-06-19.
- “Abstract: An Open-Label Phase 3 Study of Isavuconazole (VITAL): Focus on Mucormycosis (IDWeek 2014)”. idsa.confex.com. Retrieved 2015-06-19.
- Ltd., Basilea. “Basilea Pharmaceutica – Portfolio – Isavuconazole”. http://www.basilea.com. Retrieved 2015-06-19.
- “Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2015-06-19.
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
US FDA grants breakthrough therapy designation to Boehringer Ingelheim’s volasertib to treat patients with AML

Volasertib
755038-65-4
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
CODE DESIGNATION BI 6727
| Ingelheim, Germany Thursday, September 19, 2013, 16:00 Hrs [IST] |
|
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to Boehringer Ingelheim’s volasertib, a selective and potent polo-like kinase (Plk) inhibitor, for the treatment of patients with acute myeloid leukaemia (AML), a type of blood cancer. |
http://www.pharmabiz.com/NewsDetails.aspx?aid=77733&sid=2

Volasertib (also known as BI 6727) is a small molecule inhibitor of the PLK1 (polo-like kinase 1) protein being developed byBoehringer Ingelheim for use as an anti-cancer agent. Volasertib is the second in a novel class of drugs called dihydropteridinone derivatives.[1]
Mechanism of action
Volasertib is a novel small-molecule targeted therapy that blocks cell division by competitively binding to the ATP-binding pocket of the PLK1 protein. PLK1 proteins are found in the nuclei of all dividing cells and control multiple stages of the cell cycle and cell division.[2] [3] [4] The levels of the PLK1 protein are tightly controlled and are raised in normal cells that are dividing. Raised levels of the PLK1 protein are also found in many cancers including; breast, non-small cell lung, colorectal, prostate, pancreatic, papillary thyroid, ovarian, head and neck and Non-Hodgkin’s Lymphoma.[5] [3] [6] [4] [7] [8] Raised levels of PLK1 increase the probability of improper segregation of chromosomes which is a critical stage in the development of many cancers. Raised levels of PLK1 have been associated with a poorer prognosis and overall survival in some cancers[4][9] [10] In addition to its role in cell division, there is evidence that PLK1 also interacts with components of other pathways involved in cancer development including the K-Ras oncogene and the retinoblastoma and p53 tumour suppressors[11] These observations have led to PLK1 being recognised as an important target in the treatment of cancer.
Volasertib can be taken either orally or via intravenous infusion, once circulating in the blood stream it is distributed throughout the body, crosses the cell membrane and enters the nucleus of cells where it binds to its target; PLK1. Volasertib inhibits PLK1 preventing its roles in the cell-cycle and cell division which leads to cell arrest and programmed cell death.[2] Volasertib binds to and inhibits PLK1 at nanomolar doses however, it has also been shown to inhibit other PLK family members; PLK2 and PLK3 at higher; micromolar doses. The roles of PLK2 and PLK3 are less well understood; however they are known to be active during the cell cycle and cell division.[12]
Volasertib inhibits PLK1 in both cancer and normal cells; however it only causes irreversible inhibition and cell death in cancer cells, because inhibition of PLK1 in cancer cells arrests the cell cycle at a different point to normal, non-cancer cells. In cancer cells PLK1 inhibition results in G2/M cell cycle arrest followed by programmed cell death, however, in normal cells inhibition of PLK1 only causes temporary, reversible G1 and G2 arrest without programmed cell death.[13] This specificity for cancer cells improves the efficacy of the drug and minimizes the drug related toxicity.
Clinical uses
Volasertib is currently undergoing investigation in phase 1 and 2 trials and has yet to be licensed by the FDA. Volasertib may be effective in several malignancies evidenced by the fact that its target PLK1 is overexpressed in up to 80% of malignancies, where it has been associated with a poorer treatment outcome and reduced overall survival.[1][4][9]Further phase 1 and 2 trials are active, investigating the effects of Volasertib both as a single agent and in combination with other agents in solid tumours and haematological malignancies including; ovarian cancer, urothelial cancer and acute myeloid leukaemia.[14]
Studies
Preclinical studies on volasertib have demonstrated that it is highly effective at binding to and blocking PLK1 function and causing programmed cell death in colon and non-small cell lung cancer cells both in vitro and in vivo. Volasertib can also cause cell death in cancer cells that have are no longer sensitive to existing anti-mitotic drugs such as vinca alkaloids and taxanes.[13] This suggests that volasertib may be effective when used as a second line treatment in patients who have developed resistance to vinca alkaloid and taxane chemotherapeutics.
A first in man trial of volasertib in 65 patients with solid cancers reported that the drug is safe to administer to patients and is stable in the bloodstream. This study also reported favourable anti-cancer activity of the drug; three patients achieved a partial response, 48% of patients achieved stable disease and 6 patients achieved progression free survival of greater than 6 months.[15] A further phase 1 trial of volasertib in combination with cytarabine in patients with relapsed / refractory acute myeloid leukaemiareported that 5 of 28 patients underwent a complete response, 2 achieved a partial response and a further 6 patients no worsening of their disease.[16]
- Schoffski, P. (2009). “Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology”. Oncologist 14 (6): 559–70. ISSN (Electronic) 1083-7159 (Linking) 1549-490X (Electronic) 1083-7159 (Linking).
- Barr, F. A.; H. H. Sillje, E. A. Nigg (2004). “Polo-like kinases and the orchestration of cell division”. Nat Rev Mol Cell Biol 5 (6): 429–40. ISSN (Print) 1471-0072 (Linking) 1471-0072 (Print) 1471-0072 (Linking).
- Garland, L. L.; C. Taylor, D. L. Pilkington, J. L. Cohen, D. D. Von Hoff (2006). “A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors”. Clin Cancer Res 12 (17): 5182–9. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- Santamaria, A.; R. Neef, U. Eberspacher, K. Eis, M. Husemann, D. Mumberg, S. Prechtl, V. Schulze, G. Siemeister, L. Wortmann, F. A. Barr, E. A. Nigg (2007). “Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis”. Mol Biol Cell 18 (10): 4024–36. ISSN (Print) 1059-1524 (Linking) 1059-1524 (Print) 1059-1524 (Linking).
- Fisher, R.A.H.; D.K. Ferris (2002). “The functions of Polo-like kinases and their relevance to human disease.”. Curr Med Chem 2: 125–134.
- Holtrich, U.; G. Wolf, A. Brauninger, T. Karn, B. Bohme, H. Rubsamen-Waigmann, K. Strebhardt (1994). “Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors”. Proc Natl Acad Sci U S A 91 (5): 1736–40. doi:10.1073/pnas.91.5.1736. ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 43238. PMID 8127874.
- Steegmaier, M.; M. Hoffmann, A. Baum, P. Lenart, M. Petronczki, M. Krssak, U. Gurtler, P. Garin-Chesa, S. Lieb, J. Quant, M. Grauert, G. R. Adolf, N. Kraut, J. M. Peters, W. J. Rettig (2007). “BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo”. Curr Biol 17 (4): 316–22. doi:10.1016/j.cub.2006.12.037. ISSN (Print) 0960-9822 (Linking) 0960-9822 (Print) 0960-9822 (Linking). PMID 17291758.
- Winkles, J. A.; G. F. Alberts (2005). “Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues”. Oncogene 24 (2): 260–6.doi:10.1038/sj.onc.1208219. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640841.
- Eckerdt, F.; J. Yuan, K. Strebhardt (2005). “Polo-like kinases and oncogenesis”. Oncogene 24 (2): 267–76. doi:10.1038/sj.onc.1208273. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640842.
- Weichert, W.; A. Ullrich, M. Schmidt, V. Gekeler, A. Noske, S. Niesporek, A. C. Buckendahl, M. Dietel, C. Denkert (2006). “Expression patterns of polo-like kinase 1 in human gastric cancer”. Cancer Sci 97 (4): 271–6. ISSN (Print) 1347-9032 (Linking) 1347-9032 (Print) 1347-9032 (Linking).
- Liu, X.; R. L. Erikson (2003). “Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells”. Proc Natl Acad Sci U S A 100 (10): 5789–94. doi:10.1073/pnas.1031523100.ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 156279. PMID 12732729.
- Schmit, T. L.; N. Ahmad (2007). “Regulation of mitosis via mitotic kinases: new opportunities for cancer management”. Mol Cancer Ther 6 (7): 1920–31. ISSN (Print) 1535-7163 (Linking) 1535-7163 (Print) 1535-7163 (Linking).
- Rudolph, D.; M. Steegmaier, M. Hoffmann, M. Grauert, A. Baum, J. Quant, C. Haslinger, P. Garin-Chesa, G. R. Adolf (2009). “BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity”. Clin Cancer Res 15 (9): 3094–102. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- ClinicalTrials.gov (2011). “Clinical Trials.gov Search of: Volasertib”. Missing or empty
|url=(help) - Gil, T.; P. Schöffski, A. Awada, H. Dumez, S. Bartholomeus, J. Selleslach, M. Taton, H. Fritsch, P. Glomb, Munzert G.M. (2010). “Final analysis of a phase I single dose-escalation study of the novel polo-like kinase 1 inhibitor BI 6727 in patients with advanced solid tumors”. J Clin Oncol 28.
- Bug, G.; R. F. Schlenk, C. Müller-Tidow, M. Lübbert, A. Krämer, F. Fleischer, T. Taube, O. G. Ottmann, H. Doehner (2010). “Phase I/II Study of BI 6727 (volasertib), An Intravenous Polo-Like Kinase-1 (Plk1) Inhibitor, In Patients with Acute Myeloid Leukemia (AML): Results of the Dose Finding for BI 6727 In Combination with Low-Dose Cytarabine”. 52nd ASH Annual Meeting and Exposition. Orange County Convention Centre, Florida: American Society of Haematology.
VOLASERTIB TRIHYDROCHLORIDE
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-, hydrochloride (1:3)
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
trihydrochloride
MOLECULAR FORMULA C34H50N8O3 . 3 HCl
MOLECULAR WEIGHT 728.2
SPONSOR Boehringer Ingelheim Pharmaceuticals, Inc.
CODE DESIGNATION BI 6727 CL3
CAS REGISTRY NUMBER 946161-17-7
Volasertib is a highly potent and selective inhibitor of the serine-threonine Polo like kinase 1 (Plk1), a key regulator of cell-cycle progression. Volasertib is a dihydropteridinone derivative with distinct pharmacokinetic (PK) properties. The problem underlying this invention was to develop improved dosage schedules for combination therapy of advanced and/or metastatic solid tumours.
Volasertib (I) is known as the compound N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-methoxy-benzamide,

This compound is disclosed in WO 04/076454. Furthermore, trihydrochloride salt forms and hydrates thereof are known from WO 07/090844. They possess properties which make those forms especially suitable for pharmaceutical use. The above mentioned patent applications further disclose the use of this compound or its monoethanesulfonate salt for the preparation of pharmaceutical compositions intended especially for the treatment of diseases characterized by excessive or abnormal cell proliferation.
U.S. 8,188,086
Several dihydropteridione derivatives effectively prevent cell proliferation. G. Linz and co-inventors report a comprehensive method for preparing pharmacologically active crystalline and anhydrous forms of compound 1 (Figure 1) that are suitable for drug formulations.

The inventors list several criteria for the properties of 1 and its manufacturing procedure:
- favorable bulk characteristics such as drying times, filterability, solubility in biologically acceptable solvents, and thermal stability;
- purity of the pharmaceutical composition;
- low hygroscopicity;
- no or low tendency toward polymorphism; and
- scalability to a convenient commercial process.
They describe their finding that the tri-HCl salt of 1 satisfies these criteria as “surprising”.
Free base 1 is prepared by condensing cyclopropylmethylpiperazine derivative 2 with pteridinone 3 in the presence of p-toluenesulfonic acid (TsOH), as shown in Figure 1. After the reaction is complete, the crude free base 1 is recovered as a viscous oil. It is then treated with HCl in an organic solvent to form 1·3HCl, isolated in 91% yield. Alternatively, the free base is not isolated; instead, concd HCl is added to the reaction mixture, followed by acetone. The crude salt is recovered in 92% yield.
The salt is purified by crystallization from refluxing EtOH, adding water, and cooling to precipitate the crystals. The inventors do not report the purity of this or any other reaction product.
The inventors obtained a hydrated form of the tri-HCl salt by dissolving the free base in EtOH at room temperature, followed by adding concd HCl and cooling to 2 °C. An anhydrous form can be recovered by drying the hydrate at 130 °C. The solubility of the hydrated salt in aqueous and organic media is reported, as are X-ray diffraction data for the hydrated form. The hydrated salt has good solid-state stability.
The patent also contains the syntheses of reactants 2 and 3 (Figures 2 and 3). The preparation of 2 begins with the formation of amide 7. Acid 4 is treated with SOCl2–DMF to form acid chloride 5; the crude product is added to a suspension of chiral difunctionalized cyclohexane 6 in THF and aq K2CO3 to produce 7. The crude product is recovered in 98% yield and oxidized to 8 with RuCl3 and N-methylmorpholine N-oxide (NMMO) in 91% yield.

Amide 8 reacts with cyclopropylmethylpiperazine 9 in the presence of methanesulfonic acid (MsOH). The solvent is evaporated, and the reaction mixture is treated with NaBH4. After further workup, product 10 is isolated in 46% yield. The nitro group is then hydrogenated over Raney Ni to give 2 in 90% yield. An alternative method for preparing10 is also described.
To prepare 3, readily available amino acid 11 is esterified and alkylated to form 12. In a multistep, one-pot procedure, 11 is first treated with HC(OMe)3 and SOCl2. Further reaction with NaBH(OAc)3, acetone, and NH4OH produces 12 as its HCl salt in 90% yield. The salt is treated with aq NaOH to form the free base, which reacts with pyrimidine 13 in the presence of NaHCO3 to form 14 in 79% isolated yield.

The pteridinone system is formed by hydrogenating 14 over a Pt/C catalyst in the presence of V(acac)3. Precursor 15 is recovered in 90% yield and methylated with (MeO)2CO and K2CO3 to give 3 in 82% isolated yield.
The inventors succeeded in developing a route for making a crystalline salt that is suitable for preparing pharmaceutical formulations. The many synthetic steps, however, use a large number of solvents that are frequently evaporated to dryness. [This observation implies that the processes have a significant environmental burden. —Ed.] (Boehringer Ingelheim International [Ingelheim am Rhein, Germany]. US Patent U.S. 8,188,086,
Novel Drug Shows Promise for Early Stage Breast Cancer

pertuzumab
TUESDAY Sept. 10, 2013 — A drug already used to treat advanced breast cancer also appears to shrink early stage breast tumors, potentially offering women a first-of-its-kind treatment option, U.S. health regulators say.
read all at
http://www.drugs.com/news/novel-shows-promise-early-stage-breast-cancer-47311.html
FDA Advisory Committee Recommends Approval in U.S. of Umeclidinium/Vilanterol for the Treatment of COPD
umeclidinium

vilanterol
09/10/13 — GlaxoSmithKline plc (LSE: GSK) and Theravance, Inc. (NASDAQ: THRX) today announced that the Pulmonary-Allergy Drugs Advisory Committee (PADAC) to the US Food and Drug Administration (FDA) voted 11 yes to 2 no that the efficacy and safety data provide substantial evidence to support approval of umeclidinium/vilanterolumeclidinium (UMEC/VI, 62.5/25mcg dose) for the long-term, once-daily, maintenance bronchodilator treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema.
Anoro Ellipta is the proposed proprietary name for UMEC/VI, a combination of two investigational bronchodilator molecules — GSK573719 or umeclidinium bromide (UMEC), a long-acting muscarinic antagonist (LAMA) and vilanterol (VI), a long-acting beta2 agonist (LABA), administered using the Ellipta inhaler.
The FDA Advisory Committee also voted that the safety of the investigational medicine has been adequately demonstrated at the 62.5/25mcg dose for the proposed indication (10 yes, 3 no), and the efficacy data provided substantial evidence of a clinically meaningful benefit for UMEC/VI 62.5/25mcg once daily for the long-term, maintenance treatment of airflow obstruction in COPD (13 yes, 0 no).
Patrick Vallance, GSK’s President of Pharmaceuticals R&D, said: “Today’s recommendation is good news and a reflection of our commitment to giving an alternative treatment option for patients living with COPD — a disease that affects millions of Americans. If approved, Anoro Ellipta will be the first, once-daily dual bronchodilator available in the US, marking another significant milestone for GSK’s portfolio of medicines to treat respiratory disease. We will continue to work with the FDA as they complete their review.”
“We are pleased with the Advisory Committee’s support of UMEC/VI,” said Rick E Winningham, Chief Executive Officer of Theravance. “This is a transformative year for Theravance and today’s positive recommendation brings the second major respiratory medicine in our GSK collaboration closer to approval and becoming an important therapeutic option for COPD patients.”
In December 2012, a New Drug Application (NDA) was submitted to the FDA for the use of UMEC/VI administered by the Ellipta™ inhaler for the long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. UMEC/VI is not proposed for the relief of acute bronchospasm or for the treatment of asthma in any of the regulatory applications.
The FDA Advisory Committee provides non-binding recommendations for consideration by the FDA, with the final decision on approval made by the FDA. The Prescription Drug User Fee Act (PDUFA) goal date for UMEC/VI is 18 December 2013.
UMEC/VI is an investigational medicine and is not currently approved anywhere in the world.
Safety Information
Across the four pivotal COPD studies for UMEC/VI, the most frequently reported adverse events across all treatment arms, including placebo, were headache, nasopharyngitis, cough, upper respiratory tract infection, and back pain. COPD exacerbation was the most common serious adverse event reported. In addition, in the four pivotal COPD studies, a small imbalance was observed in cardiac ischemia which was not observed in the long term safety study.
The UMEC/VI clinical development programme involved over 6,000 COPD patients.
About COPD
Chronic obstructive pulmonary disease (COPD) is a term referring to two lung diseases, chronic bronchitis and emphysema, that are characterized by obstruction to airflow that interferes with normal breathing. COPD is the third most common cause of death in the US and The National Heart, Lung and Blood Institute (NHLBI) estimates that nearly 15 million US adults have COPD and another 12 million are undiagnosed or developing COPD(1).
According to the NHLI, long-term exposure to lung irritants that damage the lungs and the airways are usually the cause of COPD and in the United States, the most common irritant that causes COPD is cigarette smoke. Breathing in second hand smoke, air pollution, or chemical fumes or dust from the environment or workplace also can contribute to COPD. Most people who have COPD are at least 40 years old when symptoms begin.
Bayer seeks EMA approval for marketing of regorafenib to treat GIST

Bayer seeks EMA approval for marketing of regorafenib to treat GIST
Bayer HealthCare has submitted an application to the European Medicines Agency (EMA) for marketing authorisation regarding the oral multi-kinase inhibitor, regorafenib.
read all at
Novartis Ilaris Approved for SJIA in Europe

Novartis Ilaris Approved for SJIA in Europe
Zacks.com
The EC cleared Ilaris for the treatment of active systemic juvenile idiopathic arthritis (SJIA) in patients aged 2 years and above in the EU, who did not respond adequately to previous therapy with non-steroidal anti-inflammatory drugs (NSAIDs) and …http://www.zacks.com/stock/news/108513/novartis-ilaris-approved-for-sjia-in-europe
Canakinumab (INN, trade name Ilaris, previously ACZ885)[1] is a human monoclonal antibody targeted at interleukin-1 beta. It has no cross-reactivity with other members of the interleukin-1 family, including interleukin-1 alpha.[2]
Canakinumab was approved for the treatment of cryopyrin-associated periodic syndromes (CAPS) by the U.S. Food and Drug Administration (FDA) on June 2009[3] and by the European Medicines Agency in October 2009.[4] CAPS is a spectrum of autoinflammatory syndromes including familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Canakinumab was being developed by Novartis for the treatment of rheumatoid arthritis but this trial has been discontinued.[5] Canakinumab is also in phase I clinical trials as a possible treatment for chronic obstructive pulmonary disease,[6] gout and coronary artery disease.

Ilaris neutralises IL-1 beta for a sustained period of time, and reduces inflammation. Image courtesy of Novartis.
- Dhimolea, Eugen (2010). “Canakinumab”. MAbs 2 (1): 3–13. doi:10.4161/mabs.2.1.10328. PMC 2828573. PMID 20065636.
- Lachmann, HJ; Kone-Paut I, Kuemmerle-Deschner JB et al. (4 June 2009). “Use of canakinumab in the cryopyrin-associated periodic syndrome”. New Engl J Med 360 (23): 2416–25. doi:10.1056/NEJMoa0810787. PMID 19494217.
- “New biological therapy Ilaris approved in US to treat children and adults with CAPS, a serious life-long auto-inflammatory disease” (Press release). Novartis. 18 June 2009. Retrieved 28 July 2009.
- Wan, Yuet (29 October 2009). “Canakinumab (Ilaris) and rilonacept (Arcalyst) approved in EU for treatment of cryopyrin-associated periodic syndrome”. National electronic Library for Medicines. Retrieved 14 April 2010.
- “clinicaltrials.gov, Identifier NCT00784628: Safety, Tolerability and Efficacy of ACZ885 (Canakinumab) in Patients With Active Rheumatoid Arthritis”. Retrieved 2010-08-21.
- Yasothan U, Kar S (2008). “Therapies for COPD”. Nat Rev Drug Discov 7 (4): 285. doi:10.1038/nrd2533.
Ilaris Approved by FDA to Treat Active Systemic Juvenile Idiopathic Arthritis

Basel, May 10, 2013 – Novartis announced today that the US Food and Drug Administration (FDA) has approved Ilaris (canakinumab) for the treatment of active systemic juvenile idiopathic arthritis (SJIA) in patients aged 2 years and older. Ilaris is the first interleukin-1 beta (IL-1 beta) inhibitor approved for SJIA and the only treatment approved specifically for SJIA that is given as a once-monthly subcutaneous injection[1]. SJIA is a rare and disabling form of childhood arthritis characterized by spiking fever, rash and arthritis that can affect children as young as 2 years old and can continue into adulthood[2],[3].
This approval was based on two Phase III trials in SJIA patients, aged 2-19, showing significant improvement in the majority of Ilaris-treated patients[1]. Study 1 showed that 84% of patients treated with one subcutaneous dose of Ilaris achieved the primary endpoint of the adapted pediatric American College of Rheumatology 30 (ACR30), compared to 10% achievement of ACR30 for placebo at Day 15[1]. In the open-label part of Study 2, 92 of 128 patients attempted “corticosteroid tapering”. Of those 92 patients, 62% were able to substantially reduce their use of corticosteroids, and 46% completely discontinued corticosteroids[1]. In the controlled portion of Study 2, there was a 64% relative reduction in the risk of flare for patients in the Ilaris group as compared to those in the placebo group (hazard ratio of 0.36; 95% CI: 0.17 to 0.75).
About Ilaris
Ilaris is a selective, fully human, monoclonal antibody that inhibits IL-1 beta, which is an important part of the body’s immune system defenses[1]. Excessive production of IL-1 beta plays a prominent role in certain inflammatory diseases[8]. Ilaris works by neutralizing IL-1 beta for a sustained period of time, therefore inhibiting inflammation[1].
In addition to its approval for SJIA in the US, Ilaris is approved in the EU for the treatment of refractory gouty arthritis, and in more than 60 countries, including in the EU, US, Switzerland and Japan for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), a rare, lifelong, genetic disorder with debilitating symptoms[1]. The approved indication may vary depending upon the individual country
1 Ilaris [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2013.
2. Woo P. Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome. Nat Clin Pract Rheumatol 2006; 2(1):28-34.
3. Ramanan AV, Grom AA. Does systemic-onset juvenile idiopathic arthritis belong under juvenile idiopathic arthritis? Rheumatology (Oxford) 2005; 44(11):1350-3.

Access 4,000+ profiles of new drugs in development!
![]()
Sign up for a 5-day trial and learn why the Drugs in Clinical Trials Database is a cost-effective way to find detailed information on new drug therapies in hundreds of disease conditions worldwide, monitor drug performance, track competitors and find study opportunities.

HIV/AIDS vaccine passes Phase 1 clinical trial in humans

HIV/AIDS vaccine passes Phase 1 clinical trial in humans
DVICE
While other HIV/AIDS vaccines that haven’t used killed whole viruses (relying instead on targeting specific components of HIV) have failed in Phase 3 trials, Sumagen is optimistic about their drug because other successful vaccines (including polio …read all at
http://www.dvice.com/2013-9-4/hivaids-vaccine-passes-phase-1-clinical-trial-humans
