
Home » Posts tagged 'CEP-37440'
Tag Archives: CEP-37440
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TEV-37440, CEP-37440
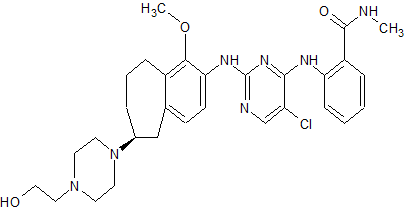
- Molecular Weight, 580.12
2-[[5-chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin- 1 -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methyl-benzamide, and is also known as CEP-37440.
![]()
| Applicants: | CEPHALON, INC. [US/US]; 41 Moores Road P.O. Box 4011 Frazer, Pennsylvania 19355 (US) |
| Inventors: | COURVOISIER, Laurent; (US). JACOBS, Martin J.; (US). OTT, Gregory R.; (US). ALLWEIN, Shawn P.; (US) |
Anaplastic Lymphoma Kinase (ALK) is a cell membrane-spanning receptor tyrosine kinase, which belongs to the insulin receptor subfamily. The most abundant expression of ALK occurs in the neonatal brain, suggesting a possible role for ALK in brain development (Duyster, J. et al, Oncogene, 2001, 20, 5623-5637).
ALK is also implicated in the progression of certain tumors. For example, approximately sixty percent of anaplastic large cell lymphomas (ALCL) are associated with a chromosome mutation that generates a fusion protein consisting of nucleophosmin (NPM) and the intracellular domain of ALK. (Armitage, J.O. et al., Cancer: Principle and Practice of Oncology, 6th edition, 2001, 2256-2316; Kutok J.L. & Aster J.C., J. Clin. Oncol, 2002, 20, 3691-3702). This mutant protein, NPM- ALK, possesses a constitutively active tyrosine kinase domain that is responsible for its oncogenic property through activation of downstream effectors. (Falini, B. et al, Blood, 1999, 94, 3509-3515; Morris, S.W. et al, Brit. J. Haematol, 2001, 113, 275-295; Duyster et al; Kutok & Aster). In addition, the transforming EML4-ALK fusion gene has been identified in non-small-cell lung cancer (NSCLC) patients (Soda, M., et al, Nature, 2007, 448, 561 – 566) and represents another in a list of ALK fusion proteins that are promising targets for ALK inhibitor therapy. Experimental data have demonstrated that the aberrant expression of constitutively active ALK is directly implicated in the pathogenesis of ALCL and that inhibition of ALK can markedly impair the growth of ALK+ lymphoma cells (Kuefer, Mu et al. Blood, 1997, 90, 2901-2910; Bai, R.Y. et al, Mol. Cell Biol, 1998, 18, 6951-6961; Bai, R.Y. et al, Blood, 2000, 96, 4319-4327; Ergin, M. et al, Exp. Hematol, 2001, 29, 1082-1090; Slupianek, A. et al, Cancer Res., 2001, 61, 2194-2199; Turturro, F. et al, Clin. Cancer Res., 2002, 8, 240-245). The constitutively activated chimeric ALK has also been demonstrated in about 60% of inflammatory myofibroblastic tumors (IMTs), a slow-growing sarcoma that mainly affects children and young adults. (Lawrence, B. et al., Am. J. Pathol, 2000, 157, 377-384; Duyster et al).
In addition, ALK and its putative ligand, pleiotrophin, are overexpressed in human glioblastomas (Stoica, G. et al, J. Biol. Chem., 2001, 276, 16772-16779). In mouse studies, depletion of ALK reduced glioblastoma tumor growth and prolonged animal
survival (Powers, C. et al, J. Biol. Chem., 2002, 277, 14153-14158; Mentlein, R. et al, J. Neurochem., 2002, 83, 747-753).
An ALK inhibitor would be expected to either permit durable cures when combined with current chemotherapy for ALCL, IMT, proliferative disorders,
glioblastoma and possible other solid tumors, or, as a single therapeutic agent, could be used in a maintenance role to prevent cancer recurrence in those patients. Various ALK inhibitors have been reported, such as indazoloisoquinolines (WO 2005/009389), thiazole amides and oxazole amides (WO 2005/097765), pyrrolopyrimidines (WO 2005080393), and pyrimidinediamines (WO 2005/016894).
WO 2008/051547 discloses fused bicyclic derivatives of 2,4-diaminopyrimidine as ALK and c-Met inhibitors. The lead drug candidate disclosed in the ‘547 application is CEP-28122, a potent ALK inhibitor with oral efficacy against SUP-M2 and Karpas-299 ALK-dependent tumors in mouse xenograft models. CEP-28122 progressed to IND-enabling studies until its development was terminated due to the unexpected occurrence of severe lung toxicity in CEP-28122-treated monke s.

CEP-28122
Focal adhesion kinase (FAK) is an evolutionarily conserved non-receptor tyrosine kinase localized at focal adhesions, sites of cellular contact with the ECM (extra-cellular matrix) that functions as a critical transducer of signaling from integrin receptors and multiple receptor tyrosine kinases, including EGF-R, HER2, IGF-R1, PDGF-R and VEGF-R2 and TIE-2 (Parsons, JT; Slack-Davis, J; Tilghman, R; Roberts, WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin. Cancer Res., 2008, 14, 627-632; Kyu-Ho Han, E; McGonigal, T. Role of focal adhesion kinase in human cancer – a potential target for drug discovery. Anti-cancer Agents Med. Chem., 2007, 7, 681-684). The integrin-activated FAK forms a binary complex with Src which can phosphorylate other substrates and trigger multiple signaling pathways. Given the central role of FAK binding and phosphorylation in mediating signal transduction with multiple SH2- and SH3- domain effector proteins (Mitra, SK; Hanson, DA; Schlaeper, DD. Focal adhesion kinase: in command and control of cell motility. Nature Rev. Mol.
Cell Biol, 2005, 6, 56-68), activated FAK plays a central role in mediating cell adhesion, migration, morphogenesis, proliferation and survival in normal and malignant cells (Mitra et al. 2005; McLean, GW; Carragher, NO; Avizzienyte, E; et al. The role of focal adhesion kinase in cancer – a new therapeutic opportunity. Nature Reviews Cancer, 2005, 5, 505-515; and Kyu-Ho Han and McGonigal, 2007). In tumors, FAK activation mediates anchorage-independent cell survival, one of the hallmarks of cancer cells. Moreover, FAK over expression and activation appear to be associated with an enhanced invasive and metastatic phenotype and tumor angiogenesis in these malignancies (Owens, LV; Xu, L; Craven, RJ; et al. Over expression of the focal adhesion kinase (pi 25 FAK) in invasive human tumors. Cancer Res., 1995, 55, 2752-2755; Tremblay, L; Hauck, W. Focal adhesion kinase (ppl25FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer, 1996, 68, 164-171; Kornberg, IJ. Focal adhesion kinase in oral cancers. Head and Neck, 1998, 20: 634-639; Mc Clean et al 2005; Kyu-Ho Han and McGonigal, 2007) and correlated with poor prognosis and shorter metastasis-free survival.
Multiple proof-of-concept studies conducted in various solid tumors using siRNA
(Haider, J; Kamat ,AA; Landen, CN; et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin. Cancer Res., 2006, 12, 4916-4924), dominant-negative FAK, and small molecule FAK inhibitors (Haider, J; Lin, YG; Merritt, WM; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor, TAE226 in ovarian carcinoma. Cancer Res., 2007, 67, 10976-10983; Roberts, WG; Ung, E; Whalen, P; et al. Anti-tumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res., 2008, 68, 1935-1944; Bagi CM; Roberts GW; and Andersen CJ. Dual focal adhesion kinse/Pyk2 inhibitor has positive effects on bone tumors – implications for bone metastases. Cancer, 2008, 112, 2313-2321) have provided pre-clinical support for the therapeutic utility of FAK inhibition as an anti-tumor/anti-angiogenic strategy, particularly for androgen-independent prostate cancers, breast cancers, and HNSCCs. In preclinical models of human breast cancer (MDA-MB-231) in nude rats, administration of a small molecule FAK inhibitor (PF-562,271) inhibited primary tumor growth and intra-tibial tumor spread, and restored tumor-induced bone loss (Bagi et al, 2008). Roberts et al, (2008) showed that PF-562,271 inhibited bone metastases, prevented bone resorption, and increased osteogenesis in breast and androgen-independent prostate cancer patients with and without bone metastases, supporting an additional benefit of FAK inhibition in these specific malignancies.
In summary, there is clear genetic and biological evidence that links aberrant ALK activation and constitutive activation of FAK with the onset and progression of certain types of cancer in humans. Considerable evidence indicates that ALK- and FAK-positive tumor cells require these oncogenes to proliferate and survive, and in the case of FAK, to invade and metastasize to distant sites, while inhibition of both ALK and FAK signaling leads to tumor cell growth arrest or apoptosis, resulting in objective cytoreductive effects. Inhibition of FAK also results in attenuation of tumor motility, invasiveness, and metastatic spread, particularly in specific cancers characterized by bone metastatic dissemination and osteolytic disease. FAK activation protects tumor cells from
chemotherapy-induced apoptosis, contributing to tumor resistance; modulation of FAK activity (by siRNA or pharmacologically) potentiates efficacy of chemotherapeutic agents in vivo (e.g., doxorubicin, docetaxel and gemcitabine), suggesting the utility for rational combination therapies in specific cancers. ALK and FAK are minimally expressed in most normal tissues in the healthy adult and are activated and/or dysregulated in specific cancers during oncogenesis and/or during early stages of malignant progression.
Consequently, the on-target effects of treatment with a dual ALK and FAK inhibitor against normal cells should be minimal, creating a favorable therapeutic index.
A need exists for additional safe and effective ALK and/or FAK inhibitors for the treatment of cancer.




The present invention rovides a compound of formula (I)

or a salt form thereof.
The compound of formula (I) has ALK and FAK inhibitory activity, and may be used to treat ALK- or FAK-mediated disorders or conditions.
The present invention further provides a pharmaceutical composition comprising at least one compound of the present invention together with at least one pharmaceutically acceptable excipient.
CEP-37440
The synthesis of 2-(5-chloro-2-{(S)-6-[4-(2-hydroxy-ethyl)-piperazin-l-yl]-l-methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide can be carried out according Fig. 1, following the procedures outlined in Steps 1-8.
Stepl : 5-Methoxy-l-methylene-l,2,3,4-tetrahydro-naphthalene: To a slurry of 5- Methoxy-3,4-dihydro-2H-naphthalen-l-one (25 g, 0.14 mol) and methyltriphenylphos-phonium iodide (1.13 eq) in THF (250 mL) at RT was added potassium t-butoxide (1.6 eq) at such a rate as to maintain a temperature no higher than warm to the touch. The reaction was stirred for one hour and concentrated. The reaction was then azeotroped with three volumes of hexane to remove excess t-butanol. Fresh hexane was added the solution was let to stand overnight to effect trituration. The red-brown solid was removed by filtration and the filtratewas washed twice with water and was concentrated. Purification by
chromatography on ISCO (330g Si02 cartridge: stepwise hexane and then DCM) affords the title compound as a pale yellow oil (24 g, 99%). 1H-NMR (400 MHz, CDC13) 7.29 (d, J = 8.0 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 5.49 (s, 1H), 4.98 (s, 1H), 3.85 (s, 3H), 2.77 (t, J = 6.4 Hz, 2H), 2.53-2.50 (m, 2H), 1.93-1.87 (m, 2H).
Step 2: l-Methoxy-5,7,8,9-tetrahydro-benzocyclohepten-6-one: 5-Methoxy-l-methylene-l,2,3,4-tetrahydro-naphthalene (23.8 g, 0.137 mol) in 150 mL MeOH added in one portion to freshly prepared solution of thallium(III)nitrate trihydrate (1.0 eq) in 300 mL MeOH. Stirred one minute and 400 mL chloroform added. The solution was filtered and the organics partitioned between dichloromethane and water. The organics were dried (MgS04) and concentrated. Purification by chromatography (ISCO, 330g silica cartridge; stepwise elution hexane (5 min) then 7 minute gradient to 100% dicloromethane (20 min) affords the title compound as the most polar of the products as a pale yellow oil (26g, 97%). 1H-NMR (400 MHz, CDC13) 7.16 (t, J = 7.9 Hz, 1H), 7.84 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 7.5 Hz, 1H), 3.84 (s, 3H), 3.73 (s, 2H), 3.05-3.01 (m, 2H), 2.55 (t, J = 7.0 Hz, 2H), 2.01-1.96 (m, 2H). LC/MS (ESI+) m/z = 191 (M+H)+
Step 3: l-Methoxy-2-nitro-5,7,8,9-tetrahydro-benzocyclohepten-6-one: To potassium nitrate in acetonitrile (50 mL) and trifluoroacetic anhydride (100 mL) at 0°C was added dropwise l-methoxy-5,7,8,9-tetrahydro-benzocyclohepten-6-one (25 g, 0.131 mol) in 50 mL acetonitrile. The reaction was stirred for 2.5 hours while warming to RT. The reaction was concentrated without heat on a rotary evaporator. MeOHwas added and stirred briefly. Reconcentrated and worked-up by partitioning between dichloromethane and sat. sq. sodium bicarbonate solution. The organic layer was separated and dried (Mg2S04), concentrated and purified by chromatography ISCO (330g silica cartridge: gradient elution – 10 to 50% EA:HEX over 60 minutes) affording two isomers. The title compound was the later eluting (10.7 grams, 34.6% yield). 1H-NMR (400 MHz, CDC13) 7.70 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 3.92 (s, 3H), 3.80 (s, 2H), 3.13-3.09 (m, 2H), 2.60 (t, J = 7.0 Hz, 2H), 2.10-2.03 (m, 2H).
Step 4: 2-[4-(l-Methoxy-2-nitro-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol: l-Methoxy-2-nitro-5,7,8,9-tetrahydro-benzocyclohepten-6-one (15.09 g, 64.15 mmol) in methylene chloride (870 ml)treated with 2-Piperazin-l-yl-ethanol (3 eq) followed by acetic acid (10 eq). The mixture was stirred at 50°C for 2 hrs and cooled to 0°C and sodium triacetoxyborohydride (4 eq) was added, then warmed to RT and stirred. After a few hours starting material was still present. Added
0.4 eq further of sodium triacetoxyborohydride, then again after 6 hours. Stirred overnight. Poured into a solution of sat. aq. Sodium bicarbonate and ice and made basic to pH 10 with IN sodium hydroxide, extracted 2X dichloromethane, dried MgS04, filtered and concentrated. This material was taken up into ethanol and HC1/
ethanol was added. The resulting precipitate was triturated for 2 hours then filtered. The solid was free-based using NaOH followed by sodium bicarbonate and extracted into dichloromethane to give the title compound (19g, 85% yield). 1H-NMR (400 MHz, CDCI3) 7.56 (d, J = 8.2 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 3.82 (s, 3H), 3.63-3.06 (m, 2H), 3.29-3.24 (m, 1H), 3.00-2.86 (m, 3H), 2.72-2.67 (m, 2H), 2.60-2.51 (m, 8H), 2.46-2.37 (m, 2H), 2.12-2.07 (m, 2H), 1.87-1.78 (m,lH), 1.37-1.29 (m, 1H). LC/MS (ESI+) m/z = 350 (M+H)+
Step 5 : 2-[4-(2-Amino- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin- 1 -yl]-ethanol. 2-[4-(l -Methoxy-2-nitro-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol (19.0 g, 54.4 mmol) was
split into two batches and dissolved in a total of Ethanol (232 mL). 10 % Pd/C (
1.74 g, 1.64 mmol) was divided in half and the reaction was hydrogenated for 3-4 hours at 50 psi. Each reaction mixture was filtered through celite to remove Pd. The filtrates were combined and then concentrated and the title compound isolated as a foamy solid (17.25g, 99% yield). 1H-NMR (400 MHz, CDC13) 6.76 (d, J = 7.9 Hz, 1H), 6.53 (d, J = 7.9 Hz, 1H), 3.72 (broad s, 3H), 3.71 (s, 3H),3.64 (t, J = 5.4 Hz, 2H), 3.26-3.20 (m, 1H), 2.84- 2.72 (m, 5H), 2.62-2.56 (m, 8H), 2.42-2.35 (m, 2H), 2.40-2.37 (m, 1H), 1.81-1.74 (m, 1H), 1.70 (broad s,lH), 1.41-1.33 (m, 1H). LC/MS (ESI+) m/z = 320 (M+H)+
Step 6 : 2-[4-((S)-2-Amino- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl] -ethanol: 34 grams of racemic 2-[4-(2 -Amino- l-methoxy-6, 7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethanol were separated using SFC (supercritical fluid C02) chromatography using a Chiralcel OJ-H (3 x 15 cm) 808041 column with 15% methanol(0.2% DEA)/C02, 100 bar eluent at 80 mL/min flow rate monitoring the wavelength of 220 nm with an injection volume: 0.5 mL, 20 mg/mL ethanol. 16.9 grams of the (R)-enantiomer and 17 grams of the titled compound were isolated with a chemical purity >99% and an enantiomeric excess (ee) >99% (measured using a Chiralcel OJ-H analytical column). NMR and mass were equivalent to the racemic material. The absolute configurationof the first eluting isomer was unambiguously assigned as the (R)-configuration via small-molecule X-ray using anomalous dispersion of the bis-p-bromobenzyl derivative: 4-bromo-benzoic acid 2-{4-[(R)-2-(4-bromo-
benzoylamino)- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl]-piperazin-l -yl} -ethyl ester. Thus, the second eluting enantiomer was determined to be (S)-configuration.
Step 7: 2-(2,5-Dichloro-pyrimidin-4-ylamino)-N-methyl-benzamide: 2-Amino-N-methyl-benzamide (24.4 g, 0.16 mol) in DMF (0.5 L) was added 2,4,5-Trichloro-pyrimidine (39 g, 1.3 eq) and Potassium carbonate (1.3 eq). Stired under argon at 75 °C for 5 hrs and then at RT overnight. Poured into 1 L water and precipitate isolated by filtration and washed 1 : 1 acetonitrile: water followed by drying in air stream and under vacuum to afford the title compound as a yellow solid (38 g, 78% yield). 11.70 (s, 1H), 8.74 (d, J = 8.2 Hz, 1H), 8.24 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.16 (t, J = 8.4 Hz, 1H), 6.28 (s, 1H), 3.06 (d, J = 4.7 Hz, 3H).
Step 8 : 2-(5-Chloro-2- {(S)-6-[4-(2-hydroxy-ethyl)-piperazin-l -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide: To a sealed vessel 2-[4-((S)-2-Amino-l-methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-6-yl)-piperazin-l-yl]-ethano (2.69 g, 8.41 mmol) and 2-(2,5-Dichloro-pyrimidin-4-ylamino)-N-methyl-benzamide (2.00 g, 6.73 mmol) were combined in 1-methoxy-2-propanol (120 mL, 1200 mmol) followed by the addition of Methanesulfonic acid (2.44 mL, 37.7 mmol). The reaction was then heated at 90°C for 18 hours.
The reaction mixture was added to a separatory funnel and diluted with sat. bicarb until a cloudy ppt formed. This was extracted with dichloromethane 3x. The organic
layer was then washed with brine, dried over MgS04, filtered and concentrated. The residue was pumped dry then chromatographed on ISCO flash column. It was injected in dichloromethane onto a normal phase column and eluted on a gradient of 0-10%
(dichloromethane: 10%NH4OH in MeOH). The desired product eluted around 9-10% and the 10% gradient was held until product eluted completely. Mixed fractions concentrated and were chromatographed on the Gilson reverse-phase HPLC gradient elution 0-40%
CH3CN. Chromatogrpahy was repeated using normal phase silica and reverse phase HPLC to effect further purification as desired. Following neutralization and concentration of all the material, the resulting solid was obtained by taking the foam up into EtOAc and concentrating to dryness several times to give the title compound (1.1 g, 28%>). 11.02 (s, 1H), 8.69 (d, J = 8.9 Hz, 1H), 8.13 (s, 1H), 8.08 (d, J = 8.4 Hz, 1Η),7.59-7.50 (m, 2H),
7.41 (s, 1H), 7.13 (t, J = 7.4 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.21 (s, 1H), 3.74 (m, 3H), 3.66-3.63 (m, 2H), 3.29-3.23 (m, 1H), 3.06 (d, J = 4.3 Hz, 3H), 2.92-2.72 (m, 5H), 2.66-2.55 (m, 8H), 2.48-2.39 (m, 2H), 2.16-2.10 (m, 2H), 1.87-1.77 (m, 1H), 1.42-1.32 (m, 1H).LC/MS (ESI+) m/z = 580 (M+H)+
CEP-37440 amorphous HC1 salt
2-(5-Chloro-2- {6-[4-(2-hydroxy-ethyl)-piperazin- 1 -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide hydrochloride: 2-(5-Chloro-2-{6-[4-(2-hydroxy-ethyl)-piperazin-l-yl]-l-methoxy-6, 7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino}-pyrimidin-4-ylamino)-N-methyl-benzamide (4.90 g, 8.45 mmol) and 2.5 M of HC1 in ethanol (13.5 niL, 33.8 mmol) were heated until they dissolved in ethanol (164 mL). The reaction was concentrated two times from ethanol, then warmed in a small amount of ethanol until completely dissolved. This solution was allowed to cool slowly with a stirring (<100 rpm). A solid preciptate formed quickly before the solution had cooled. This mixture was allowed to stir until ambient temperature was achieved and then filtered. The solid was washed with ethanol followed by ether then directly pumped dry under high vac to give 2-(5-chloro-2-{6-[4-(2-hydroxy-ethyl)-piperazin-l -yl]- 1 -methoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-2-ylamino} -pyrimidin-4-ylamino)-N-methyl-benzamide hydrochloride (5.3 grams, quantitative yield). 1H-NMR (MeOD, 400 MHz) δ 8.55 (s, 1H), 8.17 (s, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.55 (t, J = 6.8 Hz, 1H), 7.46 (broad s, 1H), 7.36 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.5 Hz, 1H), 4.00-3.95 (m, 4H), 3.83-3.72 (m, 5H), 3.73 (s, 3H), 3.65-3.59 (m, 2H), 3.47-3.38 (m, 5H), 2.95 (s, 3H), 2.72-2.65 (m, 1H), 2.44-2.38 (m, 1H), 2.29-2.28 (m, 1H), 2.19-2.12 (m, 1H), 1.59-1.49 (m, 1H). LC/MS (ESI+) m/z = 580 (M+H)+.
2-[[5-chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin-l-yl]-l-methoxy-6,7,8,9-tetrahydro-5Hbenzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methyl-benzamide (CEP-37440) is an orally available dual kinase inhibitor of the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) and focal adhesion kinase (FAK) with antineoplastic activity. See, e.g., WO 2013/134353.
H

CEP-37440
[0003] In view of the surprising and unexpected properties observed with CEP-37440, improved methods for its preparation in high enantiomeric purity are needed.

CEP-37825 CEP-38063-tartrate salt
Scheme 2

CEP-37440
[0059] Preferred methods for asymmetrically producing an intermediate for the formation of CEP-37440 according to the disclosure are depicted in Scheme 3.
Scheme 3

4
[0060] Preferred methods for asymmetrically producing an intermediate for the formation of CEP-37440 according to the disclosure are depicted in Scheme 4.

[0061] The following examples are provided to illustrate the compositions, processes, and properties of the present disclosure. The examples are merely illustrative and are not intended to limit the disclosure to the materials, conditions, or process parameters set forth therein.
EXAMPLES
Chiral HPLC method:
CHIRALCEL AD column
eluent: heptane/isopropanol (90:10)
flow: 1.2 mL/min
detection: 220 nm and 254 nm.
Example 1

5-methoxytetralone CEP-41158
[0063] To a 12L 4-neck round bottom flask was added methoxytetralone (500 g, 2830 mmol, 1 eq) and Pti3PCH3I (1320 g, 3264 mmol, 1.15 eq) as solids via a powder funnel. THF (5 L) was added and the mixture was stirred with overhead stirring at 19 °C. tBuO (525 g, 4683, 1.65 eq) was added portion-wise over 2 hours to maintain a maximum reaction temperature of 47 °C. A THF solution of tBuOK can also be used with similar results. The funnel was washed with additional THF as needed. The resulting slurry was stirred for an additional hour at 30 °C. HPLC analysis showed < 0.5% starting material.
[0064] The reaction was transferred to a single neck flask and concentrated by roto-evaporator and solvent switched to heptanes (approximately 2.5L, removing as much THF as possible). The slurry was filtered, washing with an additional 0.5 L heptanes. The combined filtrant and wash (approximately 3 L) was washed 2 times with water (2 x 250 mL). The water layers were back extracted 1 time with 0.5 L heptanes which was combined with the other organic layers (total volume was approximately 3.5L).
[0065] To remove the residual amounts of Pl^PO, the solution can be dried and cooled (overnight in the cold room) to precipitate out the triphenylphosphine oxide. Filtration results in a clean product stream that can be concentrated to an oil. Alternatively, the initial heptane product stream may be passed through a plug of S1O2 followed by concentration to an oil.
Example 2
![]()
[0066] CEP-41 158 (Limiting Reagent, see Example 1 ) was charged to a reaction vessel followed by methyl tertiary butyl ether (MTBE) (6.3 volumes), isopropanol (3.3 volumes),
Triton X (0.1 volumes), water (6.3 volumes), and 2-iodo-5-methylbenzenesulfonic acid (0.125 eq). While holding the batch at 20-25 °C, oxone (0.65 eq) was added portion-wise over
approximately 1.5 h and then continued to stir at room temperature. HPLC was used to monitor reaction conversion. A typical reaction time was 4 h but the reaction can be stirred overnight, as well.
[0067] The reaction was quenched by the slow addition of sodium sulfite (0.5 eq) over 10 minutes. Peroxide test strips were used to confirm no peroxides remained. NaOH (5N) was then added at <30 °C to obtain a pH of 8 (typically this was approximately 2 volumes of 5N NaOH). Celite was added and the solution was filtered. The aqueous layer was removed and the organics were washed with brine before being concentrated to an oil.
[0068] Bisulfite adduct formation: The above oil was taken up in isopropanol (7 volumes) before adding water (4 volumes). To this solution was slowly added a freshly prepared aqueous sodium bisulfite solution (6.4 M, 2 eq) at ambient temperature. The resulting slurry was stirred overnight then filtered. The solids were washed with isopropanol and then dried at 40 °C in a vacuum oven with a nitrogen bleed.
Example 3

CEP -41609 CEP-41159
[0069] Unless otherwise noted all volumes and equivalents are based on the wt% corrected charge of CEP-41609. To a nitrogen purged 2.0 L jacketed reactor equipped with an anchor overhead stirrer, and a thermocouple probe was charged 1 17.8 g of CEP-41609 (84.9 wt%, 100.0 g, 0.3398 mol). 500 mL of acetonitrile (5 volumes) was then introduced and the jacket was set to 15 °C. 300 mL of deionized H2O (3 volumes) was then charged and the slurry cooled from -17 °C to 10 °C with stirring at 130 RPM. HC1 (86.4 mL, 1 1.8M, 1.019 mol, 3 equiv.) was added in one portion with the slurry at 10 °C. The mixture exothermed to 17.3 °C and the jacket was adjusted to 22 °C. The slurry cooled to 13.8 °C before being warmed back up to 20 °C in 13 minutes. The stirring was turned up to 175 RPM to improve mixing while completing a 30 minute age. The solids had dissolved after 30 minutes. At 57 minutes the stirring was stopped and the biphasic solution was allowed to settle and sit overnight (15 h).
[0070] After sitting overnight, stirring (175 RPM) was reinitiated and the jacket was set to -40 °C. It took 28 minutes for the mixture to reach -15 °C and in that time the jacket was adjusted to -20 °C. With the jacket at -20 and the reaction at -15 °C the first charge of N-chlorosuccinimide (NCS) was done (28.4 g, 0.2127 mol, 0.626 equiv.). The reaction exothermed to -1.2 °C in 1.5 minutes. 4 minutes later with the reaction cooled to -6.9 °C and the jacket still at -20 °C the second charge of NCS was done (85.0 g, 0.6367 mol, 1.874 equiv.). The reaction exothermed to 2.2 °C in 1.5 minutes. The reaction was then cooled to 0 °C in 1 minute and held at 0 ± 2 °C. Acetonitrile (ACN) (25 mL, 0.25 volumes vs wt% corrected CEP-41609) was used to rinse residual NCS off the wall of the reactor and into the reaction just after the second charge. After 2 hours at 0 ± 2 °C an IPC was taken from the organic layer and the HPLC showed undetectable levels of CEP-41608.
[0071] At 2 hours 22 minutes the work up began with the addition of MTBE (650 mL, 6.5 volumes) over 6 minutes at 0 to 1.7 °C. The mixture was stirred at 175 RPM (complete mixing achieved) for 2.5 minutes then stirring was stopped and the layers settled in 2.5 minutes. 280 mL of an aqueous layer was then cut at -1.6 °C. To the remaining 1500 mL of organic solution was charged 650 mL NaCl solution (6.5 volumes, 24 wt% NaCl; 189.2 g NaCl mixed with 599.25 g D.I. H20) over 8 minutes at -1.3 to 1.7 °C. Stirring was increased to 325 RPM to achieve complete mixing. The solution was allowed to stir for 1.5 minutes before stopping the stirring and allowing the layers settle in 3 minutes. 1000 mL of aqueous layer was cut at -0.4 °C. To the remaining 1 100 mL of organic layer was charged 650 mL NaHCC solution (6.5 volumes, 7.5 wt% NaHC03; 53 g NaHC03 mixed with 655.5 g D.I. H20) over 9 minutes at -0.3 to 2.6 °C with stirring at 325 RPM to achieve complete mixing. The solution was allowed to stir for 5 minutes and then the stirring was stopped and the layers settled in 4 minutes. 800 mL of an aqueous layer (pH = 8) was cut at 0.2 °C.
[0072] The remaining organic layer (1000 mL) was checked by HPLC (70.2 A% CEP-41 159, 17.5 A% impurity 1, 5.9 A% impurity 2, 2.9 A% impurity 3). Thejacket was set to 10 °C while a solution of Na2S204 (33.4 g @ 85 wt%, 0.1631 mol, in 326 mL DI H20) was prepared in a capped imax jar. 50 minutes after cutting the NaHCC layer the organic layer was at 8.8 °C. With thejacket at 10 °C the Na2S204 solution was added in one portion with the stirring at 250 RPM. The mixture warms to 15.4 °C and the jacket was adjusted to maintain the reaction at 15 ± 1 °C for 15 minutes. Stirring was then stopped and an HPLC was taken from the organic layer while holding the biphasic solution at 15 °C. The HPLC showed no detectable impurity 1 (76.1 A% CEP-41 159, 6.4 A% impurity 2, 3.1 A% impurity 3, 0.54 A% CEP-41608). After a total of 39 minutes contact time with Na2S204 solution the aqueous layer was cut leaving 925 mL of organic layer. This solution was held overnight with the jacket at 20 °C.
[0073] After 16 hours 1 1 minutes the organic solution was drained into a round bottom flask, rinsing with 100 mL MTBE (1 volume). The solution was then concentrated at 120 mbar
with the bath temperature at 35 °C. Once 590 mL (5.9 volumes vs. wt% corrected CEP-41609) was removed the remaining solution was diluted with 550 mL AcOH (5.5 volumes vs. wt% corrected CEP-41609). This solution was then concentrated again at 65 mbar with the bath temperature at 45 °C. Once 320 mL (3.2 volumes vs. wt% corrected CEP-41609) was removed the distillation was stopped and the remaining solution was weighted (523.39 g) and checked by HPLC and Ή NMR. HPLC showed 66.31 g (87.0% yield) of CEP-41 159 in solution, and the NMR showed 96.0 wt% AcOH (3.1 wt% ACN, 0.9 wt% MTBE; 93 wt% AcOH desired). The desired AcOH solution mass was calculated from the HPLC assay of CEP-41 159 (9 x 66.3 lg = 596.79 g) and the amount of AcOH needed to dilute to this mass was also calculated (596.79-523.39 = 73.4 g x (lmL/1.049g) = 70 mL AcOH). The AcOH solution was then transferred back to the 2 L JLR (which had been rinsed with H2O and dried with an N2 sweep and 40 °C jacket) and 70 mL of AcOH was used to rinse out the round bottom flask and dilute the solution.
[0074] After sitting at 20 °C for 2.5 hours this solution was diluted with 199 mL DI H20 (3 volumes vs. CEP-41 159) while setting the stirring at 225 RPM and the jacket at -30 °C. The resulting homogeneous solution was cooled to -10 °C in 21 minutes. The stirring was set to 324 RPM and after 1 minute at -10 °C seed (249.4 mg, Lot # 3292-1 1 1-Pl ) was added to the solution. A thick slurry formed in <2 minutes (exotherms to -8.6 °C) but the stir speed allowed for good mixing. After 29 minutes added 332 mL DI H2O (5 volumes vs. CEP-41 159) over 12 minutes at -10 to -8 °C. Held the slurry for 28 minutes at -10 °C and then filtered a sample to check the mother liquor losses. Losses looked good at 7.7 mg/g (desired <9 mg g). After 50 minutes at -10 °C with all water added the slurry was filtered. Filtration was quick, taking <1 minute. The flask and cake were washed with 265.3 mL of ambient temperature 25 vol% ACOH H2O (4 volumes vs. CEP-41 159). The resulting mother liquors and wash were assayed and found to contain 6.7% losses (4.0 mg/g CEP-41 159). The solids were held on the filter with the vacuum pulling air across them and with aluminum foil keeping light from them.
[0075] After 67 hours and the solids were then left on the filter for another 48 hours. A total of 58.08 g of white cottony solids were recovered and were checked by HPLC and Ή NMR. HPLC indicates the solids were 100 A% and 102.7 wt% while the NMR showed only trace levels of AcOH. Based on this data the solids were deemed 100% pure and the yield was 76.2%.
Example 4

CEP -41159 CEP-41160
All volumes and equivalents are based on the wt% corrected charge of CEP-41 159 unless other wise stated. Potassium nitrate (22.99 g, 0.2274 mol, 1.02 equivalents) was dissolved in trifluoroacetic acid (TFA) (125 mL, 2.5 volumes). This dissolution took ~5 minutes with vigorous stirring. CEP-41 159 (50.0 g, 0.2229 mol, 1.00 equivalents) was taken up in TFA (125 mL, 2.5 volumes) at 22 °C. This solution was cooled to -13 °C over 21 minutes and then the addition of the potassium nitrate/TFA solution was started. This solution was added in four equal portions. After each portion was added an HPLC was run on a sample of the reaction to evaluate if the reaction had progressed. The sample was diluted in ACN before it could warm up as a temperature rise would likely cause further reaction to occur. The HPLC after the first addition was completed (addition took 13 min at -13 to -6.2 °C) showed 20.9 % conversion. The batch was cooled to -13 °C and the second addition was done over 5 minutes at -13 to -1.2 °C. HPLC after the second addition showed 43.6% conversion. After the reaction was cooled back to -13 °C the third addition was started. This third addition was done over 5 minutes at -13 to -2.7 °C, and the HPLC showed 70.1% conversion. After the reaction was cooled back to -13 °C the final addition was started. This final addition was done over 3 minutes at -13 to -7.5 °C, and the HPLC showed 98.9% conversion. The reaction was held at -13 °C while sodium acetate (22.85 g, 0.2787 mol, 1.25 equivalents) was taken up in DI water (600 mL, 12.0 volumes). After 19 minutes at -14 to -13 °C the reaction was diluted with half of the sodium acetate solution over 3 minutes while allowing the reaction to warm from -14 °C to 14.5 °C. The resulting solution was warmed to 22 °C over 14 minutes and seeded with CEP-41 160 (50 mg, 0.001 x CEP-41 159). The resulting slurry was allowed to stir overnight. After 15 hours 51 minutes the second half of the sodium acetate solution was added over 12 minutes at 21.6-22.6 °C. The resulting slurry was stirred for 34 minutes and then assayed for losses. The losses were at 2.88mg/g. The slurry was then filtered 1 hour 07 minutes after final sodium acetate solution addition was done. The reaction vessel and cake were washed with Dl water (200 mL, 4.0 volumes). The mother liquors and wash were combined and assayed by HPLC to determine they contain 4.3% of the product. The solids were dried in the filter open to air with the vacuum pulling on the bottom of the filter. After 4 hours 52 minutes of drying 52.985g of CEP-41 160 was recovered. These were bright
yellow sandy solids which were 100 A% and 100.8 wt% (@ 238 nm). This is 88.3% yield of CEP-41 160.
Example 5

CEP-37825 CEP-3B063-Tartrate Salt
(A-Ring)
[0076] A glass jar was charged with CEP-37825 (17.0 g physical, 16.7 g corrected, 52.3m mol, 1.00 equiv, Johnson Matthey 4239.A.13.2), MeOH (167 mL) and H20 (40.5 g). The mixture was stirred at ambient temperature until complete dissolution occurred. The resulting solution was filtered over a sintered glass funnel into a 500 mL jacketed reactor, rinsing with MeOH (85 mL). The reactor was evacuated and filled with N2. The solution was heated to 35 °C (internal temperature). A solution of L-tartaric acid (7.85 g, 52.3 mmol, 1.00 equiv) in MeOH (84 mL) was added via addition funnel over 1 1 minutes. Additional MeOH (30 mL) was used as a rinse. The solution was seeded with a slurry of CEP-38063 L-tartrate (50.0 mg) in MeOH (2.5 mL). The vial containing the slurry was rinsed with additional MeOH (1.2 mL). A slurry gradually formed, and the mixture was aged at 33-34 °C for 90 min. The internal temperature was decreased to 29 °C and agitated for 63 min after which the mixture was cooled to 20-25 °C and stirred overnight.
[0077] After stirring overnight at ambient temperature, the mixture was filtered, rinsing with a solution of H20 (2.6 mL) in MeOH (49 mL). The mixture was dried at room temperature overnight under vacuum. Crude CEP-38063 L-tartrate (10.07 g) was obtained in 87.6% de (93.8% dr). The CEP-38063 free base content was 64.2%. The yield was 36% from CEP-37825 racemate.
[0078] Recrystallization: A 1 L OptiMax vessel was charged with two lots of crude CEP-38063 L-tartrate (18.9 g, 89.3% dr, 9.47 g, 88.6% de, 57.1 mmol total). Methanol (346 mL) and H20 (38.8 g) were added and the reactor was placed under nitrogen. The slurry was heated to 66.5 °C during which time the solids dissolved. The solution was then cooled to 55 °C and seeded with a slurry of CEP-38063 L-tartrate (106.9 mg) in MeOH (5.4 mL). The vial containing the slurry was rinsed with additional MeOH (2.6 mL). The slurry was agitated at 53- 54 °C for 82 min. It was then cooled to 48.5 °C over 60 minutes. It was agitated for 1 h, then cooled to 20 °C over 60 minutes.
[0079] After stirring overnight at ambient temperature, the mixture was filtered, rinsing with a solution of H2O (5.7 mL) in MeOH (107 mL). The mixture was dried under vacuum at room temperature overnight. CEP-38063 L-tartrate (21.4 g) was obtained in >99 A%, 99.4% de (99.7% dr) . The CEP-38063 free base content was 65.3%.
Example 6: Procedure for CEP-19036 (amidation and coupling step)
[0084] Into a 20-L jacketed glass reactor were charged isatoic anhydride (500 g, 3.06 mol,) and ethanol (2500 mL). This was followed by the controlled addition of 30 wt% methylamine in ethanol (378.5 g, 3.98 mol) further diluted with ethanol (330 mL) over 70 minutes from an addition funnel at 20±5 °C. The resulting mixture was stirred at 20±5 °C for 60 min and checked by HPLC for reaction completion (<1 A% S ). Upon completion, 13% NaCl
(prepared by dissolving 390 g of NaCl in 2610 mL of DI water) was added over 20.0 min. The resulting mixture was agitated at 20±5 °C for 10 min then allowed to settle for 10 min. The bottom aqueous layer was removed. The organic layer was washed with 26% NaCl (prepared by dissolving 792 g of NaCl in 2210 mL of DI water). The combined aqueous washes were extracted with ethyl acetate (5100 mL). The ethyl acetate extract was combined with the batch and concentrated under reduced pressure (50-80 mmHg) at 30-40 °C in a 12-L round-bottomed flask until the batch volume was approximately 1.5 L (3x the weight of isatoic anhydride). To the residue was added ethyl acetate (5100 mL), which was then concentrated under reduced pressure (50-80 mmHg), a second time, to approximately 1.5L (3 x the weight of isatoic anhydride). To the concentrated batch was added 5.0 L of acetonitrile. The resulting mixture was stirred at room temperature for 60 min, and filtered through a pad of Celite in a sintered glass funnel with fine porosity. The pad was rinsed with acetonitrile (500 mL) and the rinse was combined with the batch. The clear filtrate was transferred to a 20-L jacketed glass reactor. Hunig’s base (712.6 g) and 2,4,5-trichloropyrimidine (657.3 g) were added. The resulting solution was heated to 73±3 °C and agitated at 73±3 °C until the reaction was complete as indicated by an in-process test. The reaction mixture was then cooled to 0±5 °C over 30 min and stirred at this temperature for 1 -2 hrs. The product was collected by vacuum filtration on a sintered glass funnel. The cake was rinsed with acetonitrile (1240 mL) and pulled dry under vacuum with a nitrogen bleed until the residual water and acetonitrile content were less than 1 wt% by NMR analysis and KF titration. A total of 746.3 g (81.9% overall yield) of CEP- 19036 was obtained as a light tan solid with the following quality attributes: 99.8 A%, 98.99 wt%, 0.1 wt% NaCl, 0.1 wt% H20, 0.1 wt% acetonitrile.
Example 7

[0085] CEP-38063-tartrate salt (30.1 g of salt, Limiting Reagent) was charged to a vessel along with 10 volumes of water and 10 volumes of dichloromethane. At room temperature NaOH (10 N aqueous solution, 2 equiv) was added and stirred for 15 minutes. The bottom organic layer was removed and the aqueous layer was washed a second time with 10 volumes of dichloromethane. The combined organics were washed with brine (5 volumes) then concentrated under vacuum by distillation. To the concentrate, l-methoxy-2-propanol (12.7 volumes) was added along with CEP- 19036 (1.25 equiv) and methanesulfonic acid (2.75 eq). The resulting mixture was heated to 70 °C for approximately 48 hours then cooled to room temperature. Water (14 volumes) and dichloromethane (14 volumes) was added and stirred for 15 minutes. The bottom organic layer was cut to waste prior to adding an additional 14 volumes of dichloromethane and NaOH (ION, 3.6 equiv). The bottom organic layer was removed and the aqueous was washed with an additional 14 volumes of dichloromethane. The organic layers were combined, washed with brine and concentrated under vacuum by distillation. Isopropanol (30 volumes) and water (0.6 volumes) was added and heated to 70 °C. The resulting solution was cooled to 50 °C before adding additional water (5.8 volumes) which results in
crystallization. The slurry was then cooled to room temperature, filtered and dried at 60 °C to afford a 74% yield of product with >99% purity.
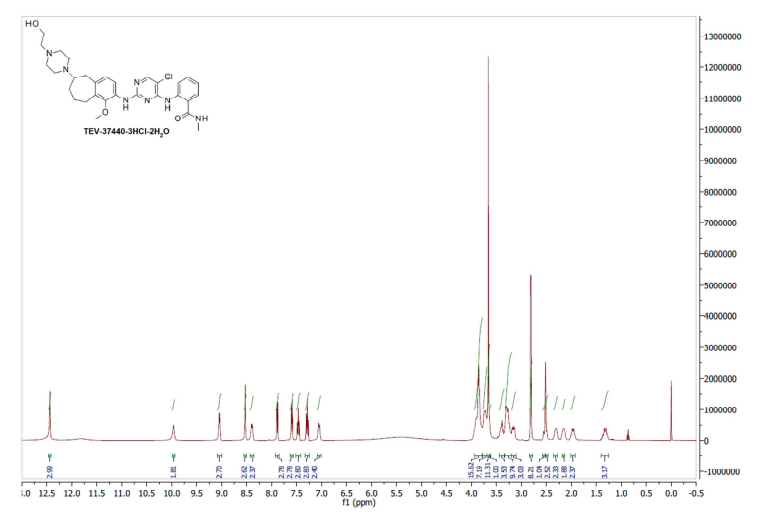
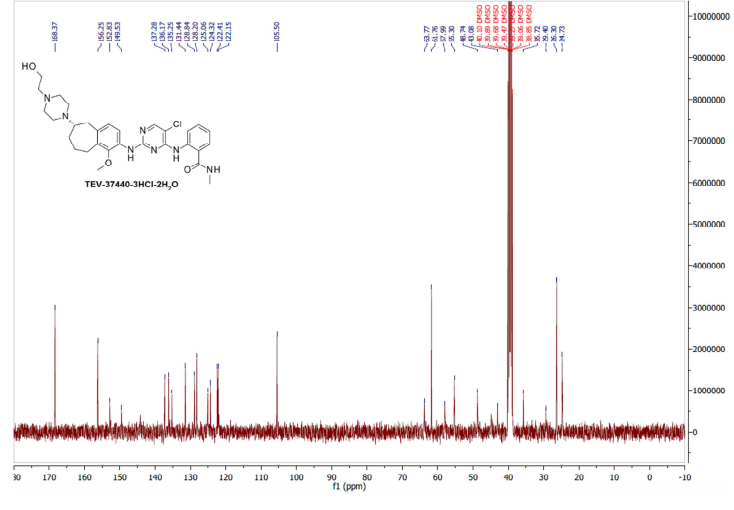
Example 8: Procedure for CEP-37440-3HCl-2H2O (salt formation)
[0086] Combined free base (145.83 g, 251.4 mmol) into a 5 L 3-neck round bottom flask equipped with overhead stirring and an addition funnel. To these solids was added nBuOH which resulted in a yellow cloudy solution (free of large solids) after stirring for 40 minutes. Precipitation occurred immediately and a slight exotherm to 23 °C was observed. The resulting slurry was heated to 85 °C over 45 minutes. Near 60 °C, the solids went into solution. Once the heating reached 80 °C, HC1 (2.5 M in 1 : 1 : 1 MeOH/EtOH/water, 307 mL, 767 mmol) was added via addition over 4 minutes and seed crystals (CEP-37440 H2A3, 1.7 g) were added. The seed held and more solids formed during a 1 h age at 85 °C. The solution was then cooled to room temperature over 1 h then further cooled to 2 °C over an additional 30 minutes. The slurry was stirred at 0-5 °C for lh then filtered, washing with 0 °C nBuOH (400 mL). Initial cake dimensions (cylinder) were 6.0 cm high with a diameter of 13.5 cm. After wash, compressed cake was 4.3 cm high. Losses to the mother liquor were approximately 0.5%. The resulting solids were dried in a vacuum oven at 60 °C for 24 h (with N2 purging after the first 16 h) to give 162 g of the desired product (98.8 HPLC purity, 88% isolated yield assuming 100 wt% starting material, 0.5% residual nBuOH by NMR, XRPD and m.p. confirmed H2A3 salt form).
Example 9: Enamine formation and hydrogenatlon

[0087] To CEP-41 160 (10 g) was added MgS04 (2.5 g, 25 wt%) and dichloromethane (8 volumes wrt CEP-41 160) at room temperature and stirred for 4 days. The resulting slurry was filtered and the filtrant was concentrated to an oil. The oil was then taken up into MTBE (70 mL, 7 volumes) which resulted in crystallization. The slurry was cooled to 0 °C and the product was filtered washing with cold MTBE. The product was dried with nitrogen under vacuum to afford 10.5 g of the product (68%). Additional MTBE crystallizations could be applied to increase purity as desired.
[0088] Prior to the reaction, the NaBArF was dried by co-evaporation with dry toluene (3 times) to remove traces of water. Trifluoroethanol was dried over molecular sieves (Union Carbide, Type 13X). In a pre-dried Schlenk, the appropriate amount of [Ir(COE)2Cl]2 precursor and RD81 was dissolved in dry DCM. After stirring the solution for 30 minutes, the NaBArF was added and stirred for an additional 30 minutes. In a separate Schlenk, the appropriate amount of substrate was dissolved in dry trifluoroethanol and stirred for 30 minutes. To a pre-dried high-pressure autoclave both the catalyst solution and substrate solution were transferred under a gentle stream of dry nitrogen. The autoclave was closed and pressurized to 50 bars of hydrogen and stirred for the desired reaction time. Then, the autoclave was vented and the reaction mixture collected. Work-up of the samples: all volatiles were removed in vacuo.
[
Example 10

I II
[0090] Preparation of hydrogenation substrate was accomplished by condensation of the ketone (1 eq.) and amide (1.1 eq. – 1.3 eq) catalyzed by TsOH (0.05 eq. to 0.1 eq.) in toluene.
[0091] To a solution of I (4.7 g, 20 mmol) in toluene (30 mL, 7 vol) was added acetamide (1.54 g, 26 mmol, 1.3 equiv) and TsOH.H20 (0.02 g, 1 mmol, 0.05 equiv.). The mixture was refluxed under N2 equipped with a Dean-Stark apparatus to remove H20. The progress of the reaction was monitored by TLC. When the reaction was completed, the mixture was cooled down. The solution was washed with H20, dried over Na2S04 and concentrated. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (5/l→ 1/1, v/v) as eluent to give the desired product as a yellow solid (5.0 g, 90% yield).
Example 11

Π m
[0092] Asymetric hydrogenations were conducted using catalytic Ru(R-C3-TunePhos)(acac)2 in methanol with 0.25 eq. of H3P04 (20 mg/mL), at about 30, 50, and 70 °C using 10, 20, 35, 50, and 70 atm of H2. Reaction times were about 15 – 18 h.Conversions of >99% were achieved. Enantiomeric excesses (%) of 67% – 82% were observed.
Example 12

Π m
[0093] Asymetric hydrogenations were conducted using catalytic Ru(S-Cs-TunePhos)(acac)2 in methanol or ethanol with 0.063 eq., 0.125 eq. 0.25 eq., and 0.5 eq. of H3PO4 (20 mg/mL), at about 50, 70, and 90 °C using 20, 50, 70,and 75 atm of H2. Reaction times were about 15 – 18 h.Conversions of >99% were achieved. Enantiomeric excesses (%) of 79% – 86% were observed.
Example 13

π m
[0094] In a glove box, to a hydrogenator (with glass liner) were added the substrate (4.14 g, 15 mmol), Ru(S-C5-TunePhos)(acac)2 (13.8 mg, 0.015 mmol, TONI OOO), Η3ΡΟ» (9 mL, 20 mg/mL in CH3OH, 0.125 equiv), CH3OH (19 mL, 7 vol) and a magnetic stirring bar. The hydrogenator was sealed and taken out of the glove box. The hydrogenator was charged with hydrogen to 50 atm. and put in an oil bath. The reaction mixture was stirred under 50 atm. at 50 °C for 48 h. After hydrogen was released carefully, the reaction mixture was concentrated.
Recrystallization from CH3OH (55 mL) gave the desired product as an off-white solid (2.29 g, 55% yield).
References:
Allwein, S.P et al., Org. Process Res. Dev. 2012, 16, 148-155.
Purohit, V.C. Org. Lett. 2013, 15, 1650-1653.
WO2008/051547
Discovery of Clinical Candidate CEP-37440, a Selective Inhibitor of Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK)

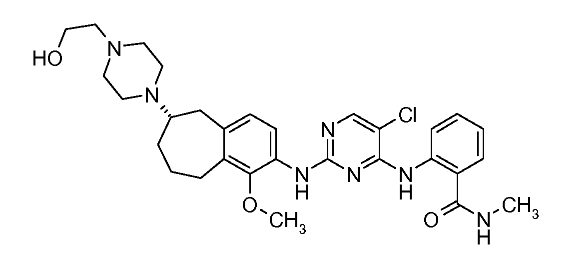
(27b) 2-[[5-Chloro-2-[[(6S)-6-[4-(2-hydroxyethyl)piperazin-1-yl]-1-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-N-methylbenzamide
Development of a Process Route to the FAK/ALK Dual Inhibitor TEV-37440
 , Dale R. Mowrey†, Daniel E. Petrillo†, James J. Reif†, Vikram C. Purohit†, Karen L. Milkiewicz†, Roger P. Bakale†, Michael A. Christie†, Mark A. Olsen‡, Christopher J. Neville‡, and Gregory J. Gilmartin‡
, Dale R. Mowrey†, Daniel E. Petrillo†, James J. Reif†, Vikram C. Purohit†, Karen L. Milkiewicz†, Roger P. Bakale†, Michael A. Christie†, Mark A. Olsen‡, Christopher J. Neville‡, and Gregory J. Gilmartin‡
The development of a scalable route to TEV-37440, a dual inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK), is presented. The medicinal chemistry route used to support this target through nomination is reviewed, along with the early process chemistry route to support IND (inversigational new drug) enabling activities within CMC (Chemistry, Manufacturing, and Controls). The identification and development of an improved route that was performed in the pilot plant to supply early phase clinical supplies are discussed. Details surrounding the use of a novel ring expansion, a selective nitration through a para-blocking group strategy, a single-pot amination–hydrogenation, a diastereomeric salt resolution, a through-process step to avoid a hazardous intermediate, and a practical formation of a trihydrochloride dihydrate salt are disclosed.
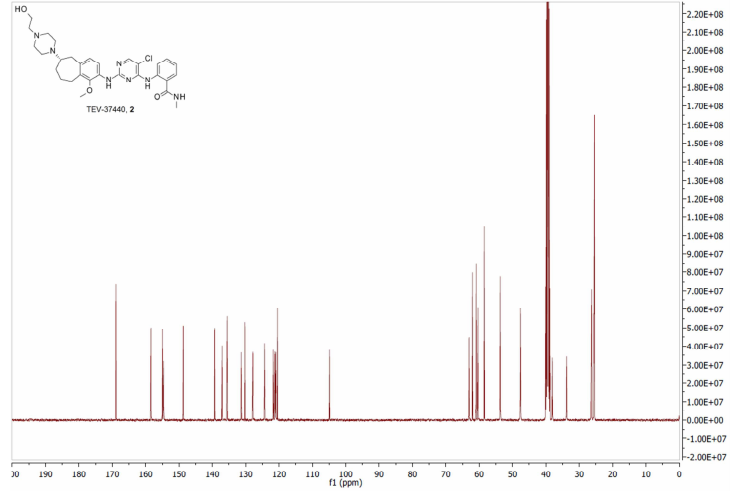
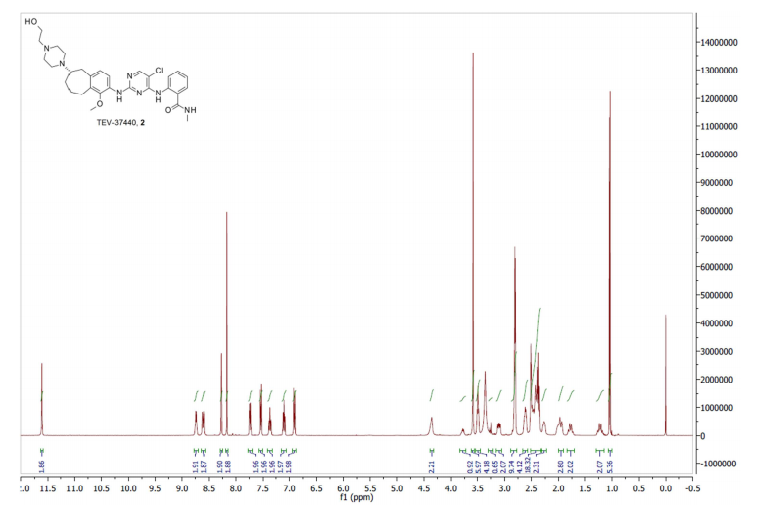
2-[(5-Chloropyrimidin-4-yl)amino]-N-methyl-benzamide; 2-[4-[(6S)-1-Methoxy-2-(methylamino)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl]piperazin-1-yl]ethanol (2, Free Base)
2-[(5-Chloropyrimidin-4-yl)amino]-N-methyl-benzamide; 2-[4-[(6S)-1-Methoxy-2-(methylamino)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl]piperazin-1-yl]ethanol Trihydrochloride Dihydrate Salt (2, Salt)
-
Allwein, S. P.; Roemmele, R. C.; Haley, J. J.; Mowrey, D. R.; Petrillo, D. E.; Reif, J. J.; Gingrich, D. E.;Bakale, R. P. Org. Process Res. Dev. 2012, 16, 148– 155, DOI: 10.1021/op200313v
-
2.Ott, G. R.; Cheng, M.; Learn, K. S.; Wagner, J.; Gingrich, D. E.; Lisko, J. G.; Curry, M.; Mesaros, E. F.;Ghose, A. K.; Quail, M. R.; Wan, W.; Lu, L.; Dobrzanski, P.; Albom, M. S.; Angeles, T. S.; Wells-Knecht, K.;Huang, Z.; Aimone, L. D.; Bruckheimer, E.; Anderson, N.; Friedman, J.; Fernandez, S. V.; Ator, M. A.;Ruggeri, B. A.; Dorsey, B. D. J. Med. Chem. 2016, 59, 7478– 7496, DOI: 10.1021/acs.jmedchem.6b00487
///////////////////TEV-37440, TEV 37440, CEP-37440, CEP 37440
CNC(=O)c5ccccc5Nc1nc(ncc1Cl)Nc3ccc2C[C@H](CCCc2c3OC)N4CCN(CC4)CCO
