


| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017095472 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2016-12-07
|
|
| US2016318941 | SUBSTITUTED [1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS |
2016-07-08
|
|
| US2017298070 | NOVEL CHIRAL SYNTHESIS OF N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-A]PYRAZINES |
2015-09-25
|
|
| US9422299 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2015-04-23
|
2015-08-20
|
| US2018111943 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2017-10-27
|
Home » Posts tagged 'APPROVALS 2023' (Page 3)
Tag Archives: APPROVALS 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
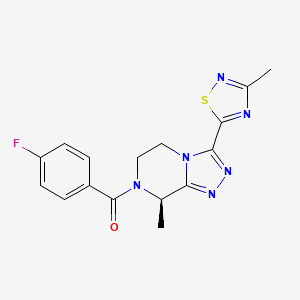
Zuranolone
Zuranolone
CAS 1632051-40-1
FDA APPROVED 8/4/2023, To treat postpartum depression
Press Release
WeightAverage: 409.574
Monoisotopic: 409.272927379Chemical FormulaC25H35N3O2
- SAGE 217
- SAGE-217
- SAGE217
Zuranolone, sold under the brand name Zurzuvae, is a medication used for the treatment of postpartum depression.[1][2] It is taken by mouth.[1]
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis, and urinary tract infection.[1][2] An orally active inhibitory pregnane neurosteroid, zuranolone acts as a positive allosteric modulator of the GABAA receptor.[6][7][8]
Zuranolone was approved for medical use in the United States for the treatment of postpartum depression in August 2023.[2] It was developed by Sage Therapeutics and Biogen.[9]
Medical uses
Zuranolone is indicated for the treatment of postpartum depression.[1][2]
Adverse effects
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis (cold-like symptoms), and urinary tract infection.[2]
The US FDA label contains a boxed warning noting that zuranolone can impact a person’s ability to drive and perform other potentially hazardous activities.[2] Use of zuranolone may cause suicidal thoughts and behavior.[2] Zuranolone may cause fetal harm.[2]
History
Zuranolone was developed as an improvement on the intravenously administered neurosteroid brexanolone, with high oral bioavailability and a biological half-life suitable for once-daily administration.[7][10] Its half-life is around 16 to 23 hours, compared to approximately 9 hours for brexanolone.[4][5]
The efficacy of zuranolone for the treatment of postpartum depression in adults was demonstrated in two randomized, double-blind, placebo-controlled, multicenter studies.[2] The trial participants were women with postpartum depression who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode and whose symptoms began in the third trimester or within four weeks of delivery.[2] In study 1, participants received 50 mg of zuranolone or placebo once daily in the evening for 14 days.[2] In study 2, participants received another zuranolone product that was approximately equal to 40 mg of zuranolone or placebo, also for 14 days.[2] Participants in both studies were monitored for at least four weeks after the 14-day treatment.[2] The primary endpoint of both studies was the change in depressive symptoms using the total score from the 17-item Hamilton depression rating scale (HAMD-17), measured at day 15.[2] Participants in the zuranolone groups showed significantly more improvement in their symptoms compared to those in the placebo groups.[2] The treatment effect was maintained at day 42—four weeks after the last dose of zuranolone.[2]
Society and culture
Zuranolone is the international nonproprietary name.[11]
Legal status
Zuranolone was approved by the US Food and Drug Administration (FDA) for the treatment of postpartum depression in August 2023.[2][12] The FDA granted the application for zuranolone priority review and fast track designations.[2] Approval of Zurzuvae was granted to Sage Therapeutics, Inc.[2]
Zuranolone has also been under development for the treatment of major depressive disorder, but the application for this use was given a Complete Response Letter (CRL) by the FDA due to insufficient evidence of effectiveness.[13]
Research
In a randomized, placebo-controlled phase III trial to assess its efficacy and safety for the treatment of major depressive disorder, subjects in the zuranolone group (50 mg oral zuranolone once daily for 14 days) experienced statistically significant and sustained improvements in depressive symptoms (as measured by HAM-D score) throughout the treatment and follow-up periods of the study.[14]
Other investigational applications include insomnia, bipolar depression, essential tremor, and Parkinson’s disease.[15][6][16]
syn
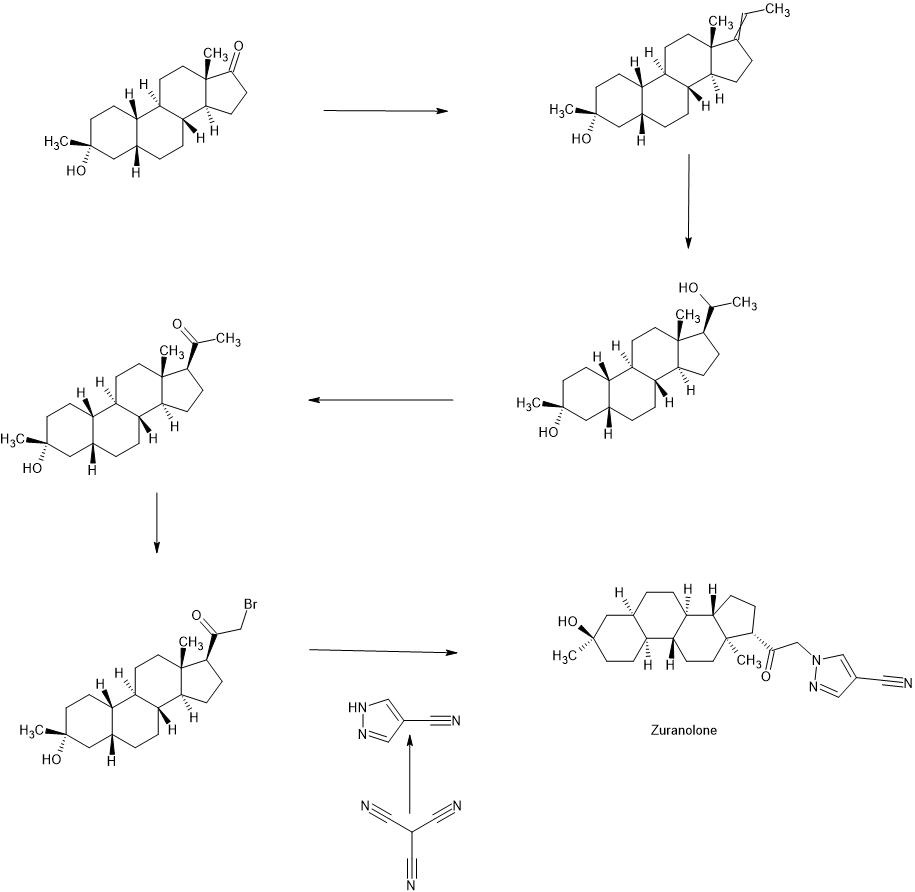

PATENT
WO2022020363
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022020363&_cid=P11-LLRZ9A-38538-1
Example 1. Synthesis of 1-(2-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl)-1H-pyrazole-4-carbonitrile (Compound 1).
[00488] To a suspension of K2CO3 (50 mg, 0.36 mmol) in THF (5 mL) was added 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol) and 2-bromo-1-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[ ^]phenanthren-17-yl)ethan-1-one (50 mg, 0.12 mmol). The mixture was stirred at room temperature for 15 hours. The reaction mixture was poured into 5 mL H2O and extracted with ethyl acetate (2×10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified by reverse-phase preparative HPLC to afford Compound 1 as a white solid (9 mg, 17.4% yield).1H NMR (500 MHZ, CDCl3) δ (ppm) 7.87 (1H, s), 7.82 (1H, s), 5.02 (1H, AB), 4.2 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dxt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt=2.24 min, m/z=410.1 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2017), 60(18), 7810-7819
https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b00846
Certain classes of neuroactive steroids (NASs) are positive allosteric modulators (PAM) of synaptic and extrasynaptic GABAA receptors. Herein, we report new SAR insights in a series of 5β-nor-19-pregnan-20-one analogues bearing substituted pyrazoles and triazoles at C-21, culminating in the discovery of 3α-hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217, 3), a potent GABAA receptor modulator at both synaptic and extrasynaptic receptor subtypes, with excellent oral DMPK properties. Compound 3 has completed a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial and is currently being studied in parallel phase 2 clinical trials for the treatment of postpartum depression (PPD), major depressive disorder (MDD), and essential tremor (ET).


3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19- nor-5β-pregnan-20-one (3). Yield: 28 g (49%) as an off-white solid. LC-MS: tR = 1.00 min, m/z = 410 (M + 1). 1 H NMR (400 MHz, CDCl3): δ 7.86 (s, 1H), 7.80 (s, 1H), 5.08−4.84 (m, 2H), 2.70−2.55 (m, 1H), 2.25−2.15 (m, 1H), 2.10−2.00 (m, 1H), 1.88−1.59 (m, 7H), 1.53−1.30 (m, 15H), 1.25−1.00 (m, 3H), 0.67 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 13.92 (CH3), 23.20, 24.44, 25.54, 25.78, 26.15 (5 × CH2), 26.69 (CH3), 31.43, 34.61 (2 × CH2), 34.77, 37.71 (2 × CH), 39.26 (CH2), 40.35 (CH), 41.21 (CH2), 41.75 (CH), 45.56 (C), 56.04, 61.24 (2 × CH), 61.78 (CH2), 72.14 (C), 93.25 (C), 113.35 (CN), 136.16, 142.49 (2 × CH), 202.23 (CO). HRMS m/z 410.2803 calcd for C25H36N3O2 + 410.2802
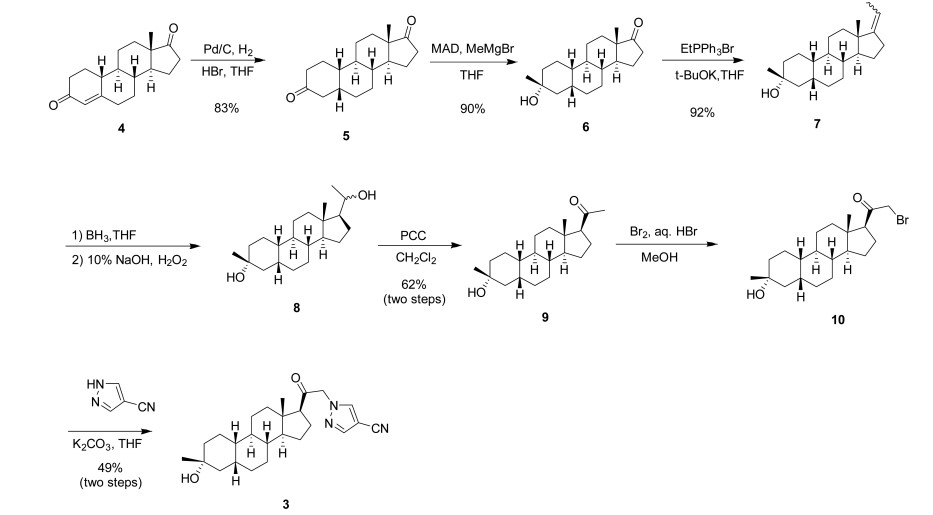
PATENT
WO2014169833
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014169833&_cid=P11-LLRZJ9-40598-1
Synthetic Procedures
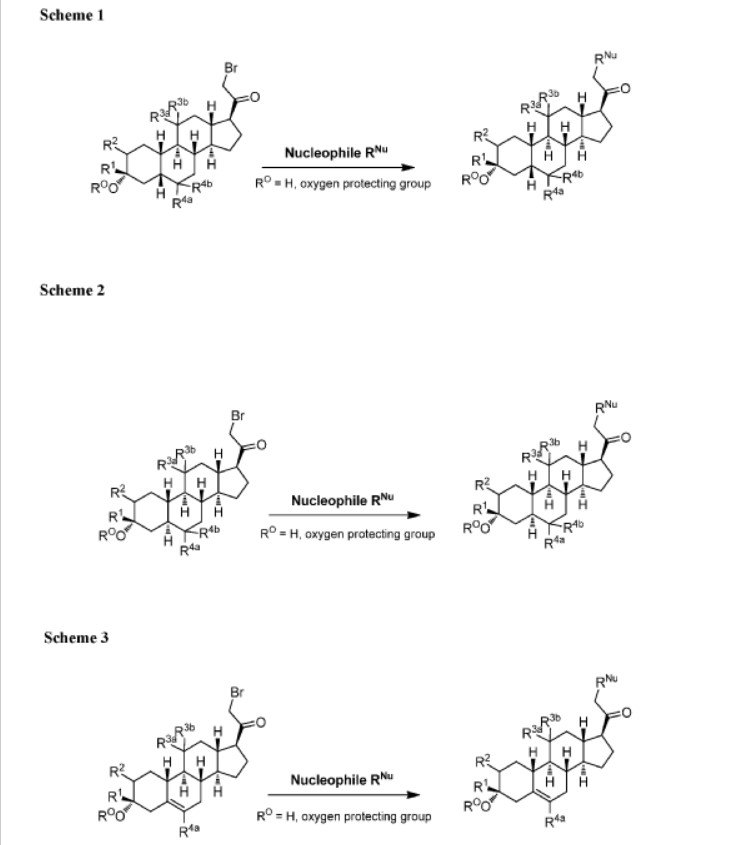
The compounds of the invention can be prepared in accordance with methods described in the art (Upasmi et al., J. Med. Chem. 1997, 40:73-84; and Hogenkamp et al., J. Med. Chem. 1997, 40:61- 72) and using the appropriate reagents, starting materials, and purification methods known to those skilled in the art. In some embodiments, compounds described herein can be prepared using methods shown in general Schemes 1-4, comprising a nucleophilic substitution of 19-nor pregnane bromide with a neucleophile. In certain embodiments, the nucleophile reacts with the 19-nor pregnane bromide in the presence of K2CO3 in THF.

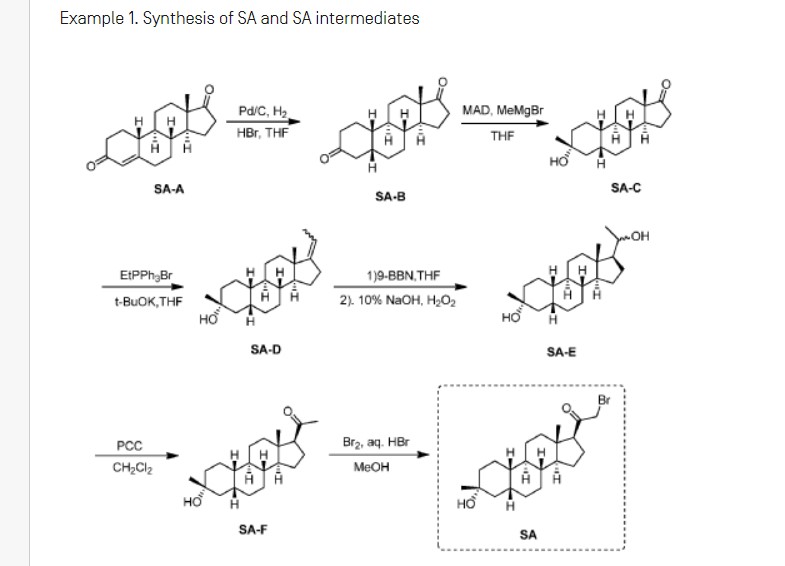

Synthesis of compound SA-B. Compound SA (50 g, 184 mmol) and palladium black (2.5 g) in tetrahydrofuran (300 mL) and concentrated hydrobromic acid (1.0 mL) was hydrogenated with 10 atm hydrogen. After stirring at room temperature for 24h, the mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to afford the crude compound. Recrystallization from acetone gave compound SA-B (42.0 g, yield: 83.4%) as white powder.
1H NMR: (400 MHz, CDCl3) δ 2.45-2.41 (m, 1H), 2.11-3.44 (m, 2H), 3.24 (s, 3H), 2.18-2.15 (m, 1H), 2.01-1.95 (m, 1H), 1.81-1.57 (m, 7H), 1.53-1.37 (m, 7H), 1.29-1.13 (m, 3H), 1.13-0.90 (m, 2H), 0.89 (s, 3H).
Synthesis of compound SA-C. A solution of SA-B (42.0 g, 153.06 mmol) in 600 mL anhydrous toluene was added dropwise to the methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD) (459.19 mmol, 3.0 eq, freshly prepared) solution under N2 at -78°C. After the addition was completed, the reaction mixture was stirred for 1 hr at -78°C. Then 3.0 M MeMgBr (153.06 mL, 459.19 mmol) was slowly added dropwise to the above mixture under N2 at -78°C. Then the reaction mixture was stirred for 3 hr at this temperature. TLC (Petroleum ether/ethyl acetate = 3:1) showed the reaction was completed. Then saturated aqueous NH4Cl was slowly added dropwise
to the above mixture at -78°C. After the addition was completed, the mixture was filtered, the filter cake was washed with EtOAc, the organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated, purified by flash Chromatography on silica gel (Petroleum ether/ ethyl acetate20:1 to 3:1) to afford compound SA-C (40.2 g, yield: 90.4%) as white powder. 1H NMR: (400 MHz, CDCl3) δ 2.47-2.41 (m, 1H), 2.13-2.03 (m, 1H), 1.96-1.74 (m, 6H), 1.70-1.62 (m, 1H), 1.54-1.47 (m, 3H), 1.45-1.37 (m, 4H), 1.35-1.23 (m, 8H), 1.22-1.10 (m, 2H), 1.10-1.01 (m, 1H), 0.87 (s, 3H).
Synthesis of compound SA-D. To a solution of PPh3EtBr (204.52 g, 550.89 mmol) in THF (500 mL) was added a solution of t-BuOK (61.82 g, 550.89 mmol) in THF (300 mL) at 0°C. After the addition was completed, the reaction mixture was stirred for 1 h 60 °C, then SA-C (40.0 g, 137.72 mmol) dissolved in THF (300 mL) was added dropwise at 60°C. The reaction mixture was heated to 60 °C for 18 h. The reaction mixture was cooled to room temperature and quenched with Sat. NH4Cl, extracted with EtOAc (3*500 mL). The combined organic layers were washed with brine, dried and concentrated to give the crude product, which was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 10:1) to afford compound SA-D (38.4 g, yield:92%) as a white powder. 1H NMR: (400 MHz, CDCl3) δ 5.17-5.06 (m, 1H), 2.42-2.30 (m, 1H), 2.27-2.13 (m, 2H), 1.89-1.80 (m, 3H), 1.76-1.61 (m, 6H), 1.55-1.43 (m, 4H), 1.42-1.34 (m, 3H), 1.33-1.26 (m, 6H), 1.22-1.05 (m, 5H), 0.87 (s, 3H).
Synthesis of compound SA-E. To a solution of SA-D (38.0 g, 125.62 mmol) in dry THF (800 mL) was added dropwise a solution of BH3.Me2S (126 mL, 1.26 mol) under ice-bath. After the addition was completed, the reaction mixture was stirred for 3 h at room temperature (14-20 °C). TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed. The mixture was cooled to 0 °C and 3.0 M aqueous NaOH solution (400 mL) followed by 30% aqueous H2O2 (30%, 300 mL) was added. The mixture was stirred for 2 h at room temperature (14-20 °C), and then filtered, extracted with EtOAc (3*500 mL). The combined organic layers were washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product (43 g , crude) as colorless oil. The crude product was used in the next step without further purification.
Synthesis of compound SA-F. To a solution of SA-E (43.0 g, 134.16 mmol) in dichloromethane (800 mL) at 0 °C and PCC (53.8 g, 268.32 mmol) was added portion wise. Then the reaction mixture was stirred at room temperature (16-22 °C) for 3 h. TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed, then the reaction mixture was filtered, washed with DCM. The organic phase was washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product. The crude product was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 8:1) to afford compound SA-F (25.0 g, yield:62.5%, over two steps) as a white powder. 1H NMR (SA-F): (400 MHz, CDCl3) δ 2.57-2.50 (m, 1H), 2.19-2.11 (m, 4H), 2.03-1.97 (m, 1H), 1.89-1.80 (m, 3H), 1.76-1.58 (m, 5H), 1.47-1.42 (m, 3H), 1.35-1.19 (m, 10H), 1.13-1.04 (m, 3H), 0.88-0.84 (m, 1H), 0.61 (s, 3H).
Synthesis of compound SA. To a solution of SA-F (10 g, 31.4 mmol) and aq. HBr (5 drops, 48% in water) in 200 mL of MeOH was added dropwise bromine (5.52 g, 34.54 mmol). The reaction mixture was stirred at 17 °C for 1.5 h. The resulting solution was quenched with saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (150 mLx2). The combined organic layers were dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (PE: EA=15:1 to 6:1) to afford compound SA (9.5 g, yield: 76.14%) as a white solid. LC/MS: rt 5.4 mm ; m/z 379.0, 381.1, 396.1.

To a suspension of K2CO3 (50 mg, 0.36mmol) in THF (5 mL) was added ethyl 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol ) and SA (50 mg,0.12 mmol). The mixture was stirred at rt for 15h. The reaction mixture was poured in to 5 mL H2O and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified with by reverse-phase prep-HPLC to afford the title compound as a white solid (9mg, 17.4%). 1H NMR (500 MHz, CDCl3), δ (ppm) 7.87 (1H, s),
7.82 (1H, s), 5.02 (1H, AB), 4.92 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dXt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt = 2.24 mm, m/z = 410.1 [M+H]+.
PATENT
WO2020150210


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
References
- ^ Jump up to:a b c d e “Zurzuvae (zuranolone) capsules, for oral use, [controlled substance schedule pending]” (PDF). Archived (PDF) from the original on 5 August 2023. Retrieved 5 August 2023.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Oral Treatment for Postpartum Depression”. U.S. Food and Drug Administration (FDA) (Press release). 4 August 2023. Retrieved 4 August 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Zuranolone”. DrugBank Online.
- ^ Jump up to:a b Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. (2022). “GABAkines – Advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors”. Pharmacology & Therapeutics. 234: 108035. doi:10.1016/j.pharmthera.2021.108035. PMC 9787737. PMID 34793859. S2CID 244280839.
- ^ Jump up to:a b Faden J, Citrome L (2020). “Intravenous brexanolone for postpartum depression: what it is, how well does it work, and will it be used?”. Therapeutic Advances in Psychopharmacology. 10: 2045125320968658. doi:10.1177/2045125320968658. PMC 7656877. PMID 33224470.
- ^ Jump up to:a b “SAGE 217”. AdisInsight. Archived from the original on 29 March 2019. Retrieved 10 February 2018.
- ^ Jump up to:a b Blanco MJ, La D, Coughlin Q, Newman CA, Griffin AM, Harrison BL, et al. (2018). “Breakthroughs in neuroactive steroid drug discovery”. Bioorganic & Medicinal Chemistry Letters. 28 (2): 61–70. doi:10.1016/j.bmcl.2017.11.043. PMID 29223589.
- ^ Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. (2017). “Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor”. Journal of Medicinal Chemistry. 60 (18): 7810–7819. doi:10.1021/acs.jmedchem.7b00846. PMID 28753313.
- ^ Saltzman J (4 August 2023). “FDA approves postpartum depression pill from two Cambridge drug firms”. The Boston Globe. Archived from the original on 6 August 2023. Retrieved 5 August 2023.
- ^ Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, et al. (2020). “Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator”. Neuropharmacology. 181: 108333. doi:10.1016/j.neuropharm.2020.108333. PMC 8265595. PMID 32976892.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
- ^ “FDA Approves Zurzuvae (zuranolone), the First and Only Oral Treatment Approved for Women with Postpartum Depression, and Issues a Complete Response Letter for Major Depressive Disorder” (Press release). Biogen Inc. 4 August 2023. Retrieved 4 August 2023 – via GlobeNewswire.
- ^ McKenzie H. “Sage Hints at Difficult Decisions After Zuranolone’s Rejection in MDD”.
- ^ Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. (May 2023). “Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial”. The American Journal of Psychiatry: appiajp20220459. doi:10.1176/appi.ajp.20220459. PMID 37132201. S2CID 258461851.
- ^ Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, et al. (2021). “Effect of Zuranolone vs Placebo in Postpartum Depression: A Randomized Clinical Trial”. JAMA Psychiatry. 78 (9): 951–959. doi:10.1001/jamapsychiatry.2021.1559. PMC 8246337. PMID 34190962.
- ^ Bullock A, Kaul I, Li S, Silber C, Doherty J, Kanes SJ (2021). “Zuranolone as an oral adjunct to treatment of Parkinsonian tremor: A phase 2, open-label study”. Journal of the Neurological Sciences. 421: 117277. doi:10.1016/j.jns.2020.117277. PMID 33387701. S2CID 229333842.
External links
- Clinical trial number NCT04442503 for “A Study to Evaluate the Efficacy and Safety of SAGE-217 in Participants With Severe Postpartum Depression (PPD)” at ClinicalTrials.gov
- Clinical trial number NCT02978326 for “A Study to Evaluate SAGE-217 in Participants With Severe Postpartum Depression” at ClinicalTrials.gov
/////////Zuranolone, FDA 2023, APPROVALS 2023, Zurzuvae, postpartum depression , SAGE 217, SAGE-217, SAGE217
[H][C@@]1(CC[C@@]2([H])[C@]3([H])CC[C@]4([H])C[C@](C)(O)CC[C@]4([H])[C@@]3([H])CC[C@]12C)C(=O)CN1C=C(C=N1)C#N
Palovarotene

Palovarotene
CAS 410528-02-8
4-[(E)-2-[5,5,8,8-tetramethyl-3-(pyrazol-1-ylmethyl)-6,7-dihydronaphthalen-2-yl]ethenyl]benzoic acid
FDA 8/16/2023
To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
- RG-667
- RO-3300074
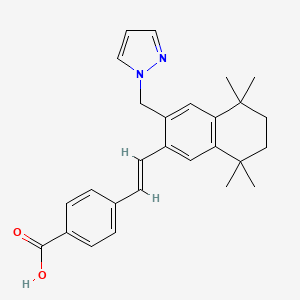
Palovarotene, sold under the brand name Sohonos, is a medication used for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva.[4][5] It is a highly selective retinoic acid receptor gamma (RARγ) agonist.[6]
It was approved for medical use in Canada in June 2022,[4] and in the United States in August 2023.[5]
Medical uses
Palovarotene is indicated for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva.[4][5]
History
Palovarotene is a retinoic acid receptor gamma (RARγ) agonist licensed to Clementia Pharmaceuticals from Roche Pharmaceuticals. At Roche, palovarotene was evaluated in more than 800 individuals including healthy volunteers and patients with chronic obstructive pulmonary disease (COPD).[7] A one-year trial did not demonstrate a significant benefit on lung density in moderate-to-severe emphysema secondary to severe α(1)-antitrypsin deficiency.[8]
In 2011, animal studies demonstrated that RARγ agonists, including palovarotene, blocked new bone formation in both an injury-induced mouse model of heterotopic ossification (HO) and a genetically modified biological mouse model of fibrodysplasia ossificans progressiva containing a continuously active ACVR1/ALK2 receptor in a dose-dependent manner.[9][10] A 2016 study demonstrated that palovarotene also inhibited spontaneous heterotopic ossification, maintained limb mobility and functioning, and restored skeletal growth in fibrodysplasia ossificans progressiva mouse models.[11]
Society and culture
Legal status
Palovarotene is being developed by Ipsen Biopharmaceuticals and was granted priority review and orphan drug designations by the United States Food and Drug Administration (FDA) for the treatment of fibrodysplasia ossificans progressiva[12][13] and orphan medicinal product designation by the European Medicines Agency (EMA) in 2014.[14][15][16][17] Phase II clinical studies failed to show a significant change in heterotopic bone volume, the main outcome measure, but prompted further investigation in a phase III clinical trial.[18] In December 2022, the FDA declined to approve palovarotene for the fibrodysplasia ossificans progressive without additional clinical trial data.[19] In January 2023, the European Medicines Agency (EMA) recommended the refusal of the marketing authorization for palovarotene for the treatment of fibrodysplasia ossificans progressiva.[20]
Research
Phase II
Clementia submitted a new drug application for palovarotene for the treatment of fibrodysplasia ossificans progressiva after observing positive phase II results.[21]
Phase III
In December 2019, Ipsen issued a partial clinical hold for people under the age of 14, due to reports of early fusion of growth plates.[22] Ipsen acquired Clementia in 2019.[23]
SYN
J. Med. Chem. 2025, 68, 2147−2182
Palovarotene (Sohonos). Palovarotene (7) is a selective retinoic acid receptor γ (RARγ) agonist that was
developed for the treatment of fibrodysplasia ossificans progressiva (FOP), a very rare autosomal dominant disorder, impacting ∼1 in2million individuals worldwide. 54,55 This orally bioavailable agonist reduces the incidence of heterotopic ossification in patients with FOP and was developed by the
French biopharmaceutical company Ipsen. 56 The small Molecule agonist was originally developed by Roche for a different indication, and was later licensed to Clementia Pharmaceuticals, which was ultimately acquired by Ipsen.
AlthoughapprovedbytheUSFDAinAugust2023,palovarotene was first approved by Health Canada in January 2022 for patients with FOP inadults andchildren aged 10 years and older for males and aged 8 years and older for females. With respect to pharmacodynamics, the agonist binds to RARγ and thus inhibits bone morphogenetic protein and Smad 1/5/8 signaling.57 This signaling inhibition permits normal muscle tissue repair and ultimately reduces the incidence of heterotopic ossification. A robust kilogram-scale synthesis of palovarotene has been disclosed in a patent by Roche and is depicted in Scheme 11.58
Starting from 2,5-dimethyl-2,5-hexanediol (7.1), the two tertiary alcohols were chlorinated with concentrated hydro chloric acid in toluene. Without isolation, the resulting

(54) Wentworth, K. L.; Masharani, U.; Hsiao, E. C. Therapeutic
advances for blocking heterotopic ossification in fibrodysplasia
ossificans progressiva. Br. J. Clin. Pharmacol. 2019, 85, 1180−1187.
(55) Semler, O.; Rehberg, M.; Mehdiani, N.; Jackels, M.; Hoyer
Kuhn, H. Current and emerging therapeutic options for the
management of rare skeletal diseases. Paediatr. Drugs 2019, 21, 95−
106.
(56) Hoy, S. M. Palovarotene: first approval. Drugs 2022, 82, 711−
716.
(57) Pignolo, R. J.; Pacifici, M. Retinoid agonists in the targeting of
heterotopic ossification. Cells 2021, 10, 3245.
(58) Martin, M. Process for preparing retinoid compounds. US
20070232810, 2007.

.
SYN
Desjardins, C., Grogan, D. R., Packman, J. N., & Harnett, M. (2017). Methods for treating heterotopic ossification (WO2017210792A1). World Intellectual Property Organization. https://patents.google.com/patent/WO2017210792A1
Chemical Communications (Cambridge, United Kingdom) (2019), 55(38), 5420-5422
WO2014105446
US20070232810
Patent
https://patents.google.com/patent/WO2002028810A3/en
WO2002028810

XAMPLE 12: PREPARATION OF 4-r(E)-2-(5,5.8.8-TETRAMETHYL-3-PYRAZOL-l-YLMETHYL -5.6.7.8-TETRAHYDRO-NAPHTHALEN-2-YL VINYLl BENZOIC ACID (6)
A mixture of 2.0 g (4.5 mmol) of (E)- methyl-4-[2-(3-bromomethyl-5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 0.65 g (9.5 mmol) of pyrazole in 15 mL of N-methyl pyrrolidine was heated at 100°. After 2 hours, the reaction mixture was cooled to room temperature, poured into brine and extracted with ethyl acetate. The organic extracts were washed with brine, dried over sodium sulfate and concentrated under reduced pressure. The residue was stirred with hexane and the product was filtered off, washed with hexane and dried to give 1.6 g (83%) of methyl-4-[2-(5,5,8,8-Tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate (M+ = 429).
A mixture of 27.6 g (64.4 mmol) of methyl-4-[2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 97 mL (193 mmol) of 2 N sodium hydroxide in 300 mL of ethyl alcohol was heated at reflux. After 1 hour, the reaction mixture was cooled to room temperature and diluted with 900 mL of water. The reaction mixture was acidified with 2 N HCl and the product was isolated by filtration, washed with water and pentane and dried to give 25.9 g (97%) of 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid (m.p. = 246.5-248 °C) 6.
Proceeding as described in the example above but substituting pyrazole with pyrrole, 4-methylpyrazole, 1,2,4-triazole, moφholine, 2-pyrrohdone, 3,5-dimethylpyrzole,
δ – valerolactone, 2-methyhmidazole and 4-methylimidzole gave 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrrol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 7, 4-{(E)-2-[5,5,8,8-Tetramemyl-3-(4-methylpyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 20, 4-[(E)-2-(5,5,8,8-Tetxamethyl-3-[l,2,4]triazol-l-ylmethyl-5,6,7,8Jetrahydro-naphthalen-2-yl]vinyl}benzoic acid 39, 4-[(E)-2-(5,5,8,8-tetramethyl-3-moφhohn-4-ylmethyl- 5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 138, 4-[(E)-2-(5,5,8,8-tetramethyl-3- (2-oxo-pyrrohdin-l-yl-methyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 139, 4-{(E)-2-[5,5,8,8-Tetramet yl-3-(3,5-mmemylpyτazol-l-yhnethyl-5,6,7,8-tetrahydro-napn^ 2-yl)vinyl]benzoic acid 143, 4-[(E)-2-(5,5,8,8-tetramethyl-3-(2-oxo-piperidin-l-yl-methyl-5,6,7,8-tetrahydro-naρhthalen-2-yl)vinyl]benzoic acid 146 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(2-methyhmidazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 149and 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(4-methyhmidazol-l-ylmethyl-5,6,7,8-tettahydro-naphthalen-2-yl)vinyl]benzoic acid 150 respectively.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Clinical data | |
|---|---|
| Trade names | Sohonos |
| Other names | R-667, RG-667 |
| License data | US DailyMed: Palovarotene |
| Routes of administration | By mouth |
| Drug class | Retinoic acid receptor gamma agonist |
| ATC code | M09AX11 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3][4]US: ℞-only[5] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 410528-02-8 |
| PubChem CID | 10295295 |
| DrugBank | DB05467 |
| ChemSpider | 8470763 |
| UNII | 28K6I5M16G |
| KEGG | D09365 |
| ChEBI | CHEBI:188559 |
| Chemical and physical data | |
| Formula | C27H30N2O2 |
| Molar mass | 414.549 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References[
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Summary Basis of Decision – Sohonos”. Health Canada. 23 October 2014. Archived from the original on 6 August 2022. Retrieved 6 August 2022.
- ^ “Sohonos product information”. Health Canada. 20 June 2022. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ Jump up to:a b c d “Sohonos Product Information”. Health Canada. 22 October 2009. Archived from the original on 18 August 2023. Retrieved 17 August 2023.
- ^ Jump up to:a b c d “Archived copy” (PDF). Archived (PDF) from the original on 18 August 2023. Retrieved 18 August 2023.
- ^ “Health Canada Approves Ipsen’s Sohonos (palovarotene capsules) as the First Approved Treatment for Fibrodysplasia Ossificans Progressiva” (Press release). Ipsen. 24 January 2022. Retrieved 28 May 2022 – via Business Wire.
- ^ Hind M, Stinchcombe S (November 2009). “Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema”. Current Opinion in Investigational Drugs. 10 (11): 1243–50. PMID 19876792.
- ^ Stolk J, Stockley RA, Stoel BC, Cooper BG, Piitulainen E, Seersholm N, et al. (August 2012). “Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor”. The European Respiratory Journal. 40 (2): 306–12. doi:10.1183/09031936.00161911. PMID 22282548.
- ^ Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, et al. (April 2011). “Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists”. Nature Medicine. 17 (4): 454–60. doi:10.1038/nm.2334. PMC 3073031. PMID 21460849.
- ^ Kaplan FS, Shore EM (April 2011). “Derailing heterotopic ossification and RARing to go”. Nature Medicine. 17 (4): 420–1. doi:10.1038/nm0411-420. PMC 4913781. PMID 21475232.
- ^ Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al. (September 2016). “Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation”. Journal of Bone and Mineral Research. 31 (9): 1666–75. doi:10.1002/jbmr.2820. PMC 4992469. PMID 26896819.
- ^ “Ipsen announces FDA Priority Review for NDA in patients with FOP”. Ipsen (Press release). 24 August 2022. Retrieved 28 January 2023.
- ^ “Palovarotene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 January 2013. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ “EU/3/14/1368”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 27 January 2023. Retrieved 28 January 2023.
- ^ “Public summary of opinion on orphan designation. Palovarotene for the treatment of fibrodysplasia ossificans progressiva” (PDF). European Medicines Agency (EMA). Archived (PDF) from the original on 22 April 2016. Retrieved 11 April 2016.
- ^ “Clementia Pharmaceuticals Receives Fast Track Designation for Palovarotene for Treatment of Fibrodysplasia Ossificans Progressiva (FOP)” (Press release). Clementia Pharmaceuticals. 1 December 2014. Retrieved 11 April 2016 – via PR Newswire.
- ^ “Clementia Pharmaceuticals Receives EMA Orphan Medicinal Product Designation for Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva” (Press release). Clementia Pharmaceuticals. 21 November 2014. Retrieved 11 April 2016 – via PR Newswire.
- ^ Pignolo RJ, Baujat G, Hsiao EC, Keen R, Wilson A, Packman J, et al. (October 2022). “Palovarotene for Fibrodysplasia Ossificans Progressiva (FOP): Results of a Randomized, Placebo-Controlled, Double-Blind Phase 2 Trial”. Journal of Bone and Mineral Research. 37 (10): 1891–1902. doi:10.1002/jbmr.4655. PMC 9804935. PMID 35854638. S2CID 250697248.
- ^ “FDA Tells Ipsen It Won’t Approve Palovarotene for FOP”. Global Genes. 27 December 2022. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ “Sohonos: Pending EC decision”. European Medicines Agency (EMA). 26 January 2023. Archived from the original on 27 January 2023. Retrieved 28 January 2023.
- ^ “Clementia Announces Plan to Submit a New Drug Application for Palovarotene for the Treatment of FOP Based on Positive Phase 2 Results”. 23 October 2018. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
- ^ “Ipsen Initiates Partial Clinical Hold for Palovarotene IND120181 and IND135403 Studies”. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
- ^ “Ipsen Completes Acquisition of Clementia Pharmaceuticals”. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
External links
Clinical trial number NCT03312634 for “An Efficacy and Safety Study of Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva. (MOVE)” at ClinicalTrials.gov
/////////FDA 2023, APPROVALS 2023, Palovarotene, Sohonos, RG-667, RO-3300074
syn
syn
European Journal of Medicinal Chemistry 265 (2024) 116124
Palovarotene (Sohonos)
On February 17, 2022, the FDA granted approval to Palovarotene for the treatment of heterotopic ossification (HO) linked to fibrodysplasia ossificans progressiva (FOP) [64]. FOP, or myositis ossificans pro
gressiva (MOP), is an uncommon hereditary condition marked by atypical bone growth in regions beyond the usual skeletal structure. It is commonly accompanied by recurring episodes of discomfort and abrupt
swelling of soft tissues. This disorder causes restricted mobility and fusion of joints, leading to deformities, limited movement, and premature mortality [65]. Palovarotene is an orally available retinoic acid receptor γ (RARγ) agonist [66]. Palovarotene specifically attaches to RARγ and hinders the phosphorylation process of mothers against decapentaplegic homolog (SMAD)1/5/8. This action results in the suppression of the bone morphogenetic protein (BMP)/ALK2 downstream signaling pathway, leading to a decrease in ALK2/SMAD-dependent chondrogenesis and osteoblast differentiation. Consequently, the over all effect is a reduction in endochondral ossification [67].
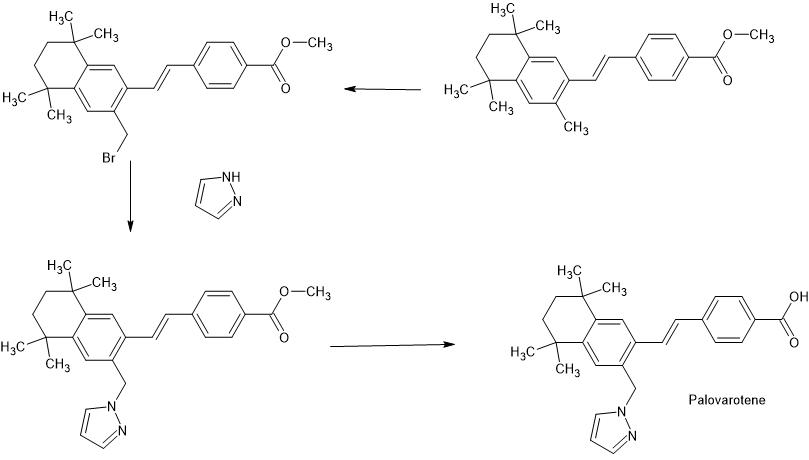
The preparation of Palovarotene is shown in Scheme 18 [68].Starting with 2,5-dimethylhexane-2,5-diol (PALO-001), a nucleophilic substitution reaction with HCl, followed by AlClpromoted Friedel-Crafts alkylation with 1-bromo-2-methylbenzene (PALO-003), gave PALO-004. PALO-005 was obtained by substitution with CuCN.The cyano group of PALO-005 was reduced to aldehyde by diisobutylalumium hydride (DIBAL-H) to obtain PALO-006. PALO-006 was subjected to Wittig-Horner reaction with methyl 4-((dimethoxyphosphoryl)methyl)benzoate PALO-007 to obtain olefin PALO-008.
PALO-008 was brominated with N-bromosuccinimide (NBS) to obtain PALO-009. PALO-009 was nucleophilic substituted with 1H-pyrazole (PALO-010) to obtain PALO-011, which was hydrolyzed under alkaline conditions to obtain the final product Palovarotene.
[64] S.M. Hoy, Palovarotene: first approval, Drugs 82 (2022) 711–716.
[65] R.J. Pignolo, E.M. Shore, F.S. Kaplan, Fibrodysplasia ossificans progressiva:
diagnosis, management, and therapeutic horizons, Pediatr. Endocrinol. Rev. 2
(2013) 437–448.
[66] G.J. Pavey, A.T. Qureshi, A.M. Tomasino, C.L. Honnold, D.K. Bishop, S. Agarwal,
S. Loder, B. Levi, M. Pacifici, M. Iwamoto, B.K. Potter, T.A. Davis, J.A. Forsberg,
Targeted stimulation of retinoic acid receptor-γ mitigates the formation of
heterotopic ossification in an established blast-related traumatic injury model,
Bone 90 (2016) 159–167.
[67] H. Kitoh, Clinical aspects and current therapeutic approaches for FOP,
Biomedicines 8 (2020) 325.
[68] J.-M. Lapierre, D.M. Rotstein, E.B. Sjogren, Preparation of New Retinoids for the
Treatment of Emphysema, Cancer and Dermatological Disorders, 2002.
WO2002028810.


NEW DRUG APPROVALS
ONE TIME
$10.00
Zavegepant

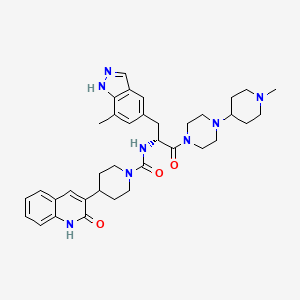
Zavegepant
ザベジェパント;
- 1337918-83-8
- as HCl: 1414976-20-7
C36H46N8O3 BASE
638.8 g/mol BASE
- Vazegepant
- BMS-742413
- BHV-3500
FDA APPR 3/9/2023Zavzpret
N-[(2R)-3-(7-methyl-1H-indazol-5-yl)-1-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]-1-oxopropan-2-yl]-4-(2-oxo-1H-quinolin-3-yl)piperidine-1-carboxamide
ZAVZPRET is indicated for the acute treatment of migraine with or without aura in adults.
The recommended dose of ZAVZPRET is 10 mg given as a single spray in one nostril, as needed. The maximum dose that may be given in a 24-hour period is 10 mg (one spray). The safety of treating more than 8 migraines in a 30-day period has not been established, Nasal spray: 10 mg of zavegepant per device. Each unit-dose nasal spray device delivers a single spray containing 10 mg of zavegepant.
ZAVZPRET (zavegepant) nasal spray contains zavegepant hydrochloride, a calcitonin generelated peptide receptor antagonist. Zavegepant hydrochloride is described chemically as (R)-N- (3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl) piperazin-1-yl)-1-oxopropan-2-yl)- 4-(2-oxo-1,2-dihydroquinolin-3-yl) piperidine-1-carboxamide hydrochloride and its structural formula is:
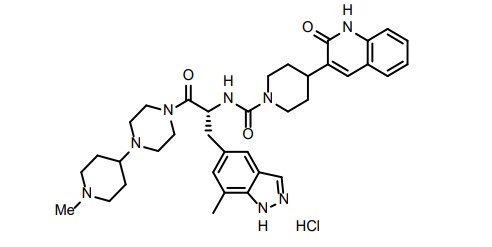
Its molecular formula is C36H46N8O3․HCl, representing a molecular weight of 675. 28 g/mol. Zavegepant free base has a molecular weight of 638.82 g/mol. Zavegepant hydrochloride is a white to off-white powder, freely soluble in water, and has pKa values of 4.8 and 8.8. Each unit-dose ZAVZPRET device for nasal administration delivers 10 mg of zavegepant (equivalent to 10.6 mg of zavegepant hydrochloride) in a buffered aqueous solution containing dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection. The solution has a pH of 5.3 to 6.7.
Active ingredients in ZAVZPRET: zavegepant Inactive ingredients in ZAVZPRET: dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection.
Zavegepant, sold under the brand name Zavzpret, is a medication used for the treatment of migraine.[1] Zavegepant is a calcitonin gene-related peptide receptor antagonist.[1] It is sprayed into the nose.[1] It is sold by Pfizer.[1]
The most common adverse reactions include taste disorders, nausea, nasal discomfort, and vomiting.[1]
Zavegepant was approved for medical use in the United States in March 2023.[1][2][3]
Medical usesZavegepant is a Calcitonin Gene-related Peptide Receptor Antagonist. The mechanism of action of zavegepant is as a Calcitonin Gene-related Peptide Receptor Antagonist.
Zavegepant is indicated for the acute treatment of migraine with or without aura in adults.[1]
Zavegepant is an antagonist of the calcitonin gene-related peptide (CGRP) receptor currently in phase 3 trials in an intranasal formulation for the treatment of migraine. If FDA approved, it will join other previously-approved “-gepant” drugs [rimegepant] and [ubrogepant] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. On April 15th, 2020, a phase 2 clinical trial (NCT04346615: Safety and Efficacy Trial of Vazegepant Intranasal for Hospitalized Patients With COVID-19 Requiring Supplemental Oxygen) began to investigate the use of intranasally administered zavegepant to combat the acute respiratory distress syndrome (ARDS) sometimes seen in patients with COVID-19. Acute lung injury activates the release of CGRP, which plays a role in the development of ARDS – CGRP antagonists, then, may help to blunt the significant inflammation associated with COVID-19. The clinical trial is expected to complete in September 2020.
Zavegepant is a highly soluble small molecule calcitonin gene related peptide (CGRP) receptor antagonist, with potential analgesic and immunomodulating activities. Upon administration, zavegepant targets, binds to and inhibits the activity of CGRP receptors located on mast cells in the brain. This may inhibit neurogenic inflammation caused by trigeminal nerve release of CGRP. In addition, by blocking the CGRP receptors located in smooth muscle cells within vessel walls, zavegepant inhibits the pathologic dilation of intracranial arteries. Zavegepant, by blocking the CGRP receptors, also suppresses the transmission of pain by inhibiting the central relay of pain signals from the trigeminal nerve to the caudal trigeminal nucleus. Altogether, this may relieve migraine. As CGRP receptors induce the release of pro-inflammatory mediators, such as interleukin-6 (IL-6), from inflammatory cells, zavegepant may prevent an IL-6-mediated inflammatory response. Zavegepant may also inhibit the CGRP-mediated induction of eosinophil migration and the stimulation of beta-integrin-mediated T cell adhesion to fibronectin at the site of inflammation, and may abrogate the CGRP-mediated polarization of the T cell response towards the pro-inflammatory state characterized by Th17 and IL-17. This may improve lung inflammation and oxygenation, prevent edema, and further lung injury. CGRP, a 37 amino-acid peptide expressed in and released from a subset of polymodal primary sensory neurons of the trigeminal ganglion and nerve fibers projecting to the airways and by pulmonary neuroendocrine cells, plays an important role in pain transmission, inflammation, and neurogenic vasodilatation. It is released upon acute lung injury and upregulation of transient receptor potential (TRP) channels.
SYN’
Synthesis of a CGRP Receptor Inhibitor
Publication Date: 2013
Publication Name: Synfacts
Azepino-indazoles as calcitonin gene-related peptide (CGRP) receptor antagonists
- PMID: 33096162Publication Date: 2021-01-01Journal: Bioorganic & medicinal chemistry lettersDiscovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)piperidine-1-carboxamide (BMS-742413): a potent human CGRP antagonist with superior safety profile for the treatment of migraine through intranasal delivery
PMID: 23632269Publication Date: 2013-06-01Journal: Bioorganic & medicinal chemistry letters
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
Patent
https://patents.google.com/patent/US20120245356A1/en
Patent
WO 2022165291
https://patents.google.com/patent/WO2022165291A1/en
Migraine is a chronic and debilitating disorder characterized by recurrent attacks lasting four to 72 hours with multiple symptoms, including typically one-sided, pulsating headaches of moderate to severe pain intensity that are associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia). Migraines are often preceded by transient neurological warning symptoms, known as auras, which typically involve visual disturbances such as flashing lights, but may also involve numbness or tingling in parts of the body. Migraine is both widespread and disabling. The Migraine Research Foundation ranks migraine as the world’s third most prevalent illness, and the Global Burden of Disease Study 2015 rates migraine as the seventh highest specific cause of disability worldwide. According to the Migraine Research Foundation, in the United States, approximately 36 million individuals suffer from migraine attacks. While most sufferers experience migraine attacks once or twice per month, more than 4 million people have chronic migraine, defined as experiencing at least 15 headache days per month, of which at least eight are migraine, for more than three months. Others have episodic migraine, which is characterized by experiencing less than 15 migraine days per month. People with episodic migraine may progress to chronic migraine over time. Migraine attacks can last four hours or up to three days. More than 90% of individuals suffering from migraine attacks are unable to work or function normally during a migraine attack, with many experiencing comorbid conditions such as depression, anxiety and insomnia. Also, those suffering from migraine often have accompanying nausea and have an aversion to consuming food or liquids during an attack.
CGRP (calcitonin gene-related peptide) is a 37 amino acid neuropeptide, which belongs to a family of peptides that includes calcitonin, adrenomedullin and amylin. In humans, two forms of CGRP (a-CGRP and 0-CGRP) exist and have similar activities. They vary by three amino acids and exhibit differential distribution. At least two CGRP receptor subtypes may also account for differential activities. The CGRP receptor is located within pain-signaling pathways, intracranial arteries and mast cells and its activation is thought to play a causal role in migraine pathophysiology. For example, research and clinical studies have shown: serum levels of CGRP are elevated during migraine attacks, infusion of intravenous CGRP produces persistent pain in migraine sufferers and non-migraine sufferers, and treatment with anti-migraine drugs normalizes CGRP activity.
Currently, clinicians use a number of pharmacologic agents for the acute treatment of migraine. A study published by the American Headache Society in 2015 concluded that the medications deemed effective for the acute treatment of migraine fell into the following classes: triptans, ergotamine derivatives, non-steroidal anti-inflammatory drugs (“NSAIDs”), opioids and combination medications. The current standard of care for the acute treatment of migraine is prescription of triptans, which are serotonin 5-HT IB/ID receptor agonists. Triptans have been developed and approved for the acute treatment of migraine over the past two decades. The initial introduction of triptans represented a shift toward drugs more selectively targeting the suspected pathophysiology of migraine. While triptans account for almost 80% of anti-migraine therapies prescribed at office visits by healthcare providers, issues such as an incomplete effect or headache recurrence remain important clinical limitations. In fact, only about 30% of patients from clinical trials are pain free at two hours after taking triptans. In addition, triptans are contraindicated in patients with cardiovascular disease, cerebrovascular disease, or significant risk factors for either because of potential systemic and cerebrovascular vasoconstriction from the 5-HT IB -mediated effects. Also, according to a January 2017 study published in the journal Headache, an estimated 2.6 million migraine sufferers in the United States have a cardiovascular event, condition or procedure that limits the potential of triptans as a treatment option.
Accordingly, there remains a significant unmet medical need for a novel migraine-specific medication that provides enhanced patient benefits compared to existing therapies.
Possible CGRP involvement in migraine has been the basis for the development and clinical testing of a number of compounds, including for example, advanced clinical candidates rimegepant (BHV-3000) and zavegepant (BHV-3500), which are developed by Biohaven Pharmaceutical Holding Company Ltd., New Haven, CT.
Zavegepant (also known as vazegepant) is a third generation, high affinity, selective and structurally unique small molecule CGRP receptor antagonist having the following formula I:

I
Zavegepant is described, for example, in WO 03/104236 published December 18, 2003 and US 8,481,546 issued July 9, 2013, which are incorporated herein in their entireties by reference.
While zavegepant is a highly soluble molecule, its bioavailability characteristics may render it challenging to prepare the drug in an oral dosage form. Enhancing the bioavailability of zavegepant and other CGRP inhibitors by different administration routes would therefore be desirable.
Calcitonin gene-related peptide (CGRP) is widely distributed in nociceptive pathways in human peripheral and central nervous system and its receptors are also expressed in pain pathways. While CGRP is involved in migraine pathophysiology, its role in non-headache pain has not been quite clear. There remains a need for new medicines to treat various pain disorders in patients in need thereof.
Scheme 1


Scheme 3

Scheme 4

tert-butyl 4-(2-methoxy-2-oxoethylidene)piperidine-l -carboxylate. Sodium hydride in mineral oil (60%, 7.92 g, 198.02 mmoles) was washed with hexanes then suspended in dimethylformamide (220 mL). The mixture was cooled to 0°C. Trimethyl phosphonoacetate (29.0 mL, 189.82 mmoles) was added dropwise to the stirred reaction mixture. After 20 min at 0°C, a solution of A-/c/7-butoxycarbonyl-4-pi peri done (30.41 g, 152.62 mmoles) in dimethylformamide (80 mL) was added to the mixture dropwise. The reaction was stirred at room temperature for 3 h and then diluted with diethyl ether (650 mL). The mixture was washed once with water and the aqueous layer was extracted once with diethyl ether. The combined organic layers were washed 4 times with water and the aqueous phase was discarded. The organic phase was washed with brine and dried over magnesium sulfate, filtered, and concentrated to dryness. The title compound was obtained as a white solid in 92% yield. 1 H- NMR (300 MHz, CDCh): 5 = 5.68 (s, 1 H), 3.66 (s, 3 H), 3.40-3.51 (m, 4 H), 2.90 (t, J= 5.49, 2 H), 2.25 (t, J= 5.49, 2 H), 1.44 (s, 9 H).

ed-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate. A solution of tert-butyl 4- (2-methoxy-2-oxoethylidene)piperidine-l -carboxylate (35.71 g, 140 mmoles) in a mixture of 1 : 1 ethyl acetate/methanol (220 mL) was carefully treated with 50% wet 10% palladium on carbon (3.3 g). The reaction vessel was charged with 55 psi of hydrogen gas and the mixture was shaken on a Parr apparatus at room temperature for 16 h. The reaction mixture was then filtered to remove the catalyst and the filtrate concentrated in vacuo. The title compound was obtained as a clear colorless oil in 97% yield. ‘H-NMR (300 MHz, CDCh): 5 = 4.04 (d, J= 10.25, 2 H), 3.64 (s, 3 H), 2.68 (t, J= 12.44, 2 H), 2.21 (d, J= 6.95, 2 H), 1.98-1.77 (m, 1 H), 1.64 (d, J= 13.54, 2 H), 1.41 (s, 9 H), 1.25-0.99 (m, 2 H).

4-[2-Hydroxy-l-methoxycarbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l-carboxylic acid tert-butyl ester. A A-diisopropylamine (4.40 mL, 31.3 mmoles) was dissolved in tetrahydrofuran (50 mL). The mixture was cooled to -78°C. Butyllithium (2.5 M in hexanes, 12.4 mL, 31 mmoles) was added dropwise to the stirred solution. After stirring at -78°C for 30 min, a solution of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate (6.65 g, 25.8 mmoles) in tetrahydrofuran (15 mL) was added dropwise to the mixture. Stirring was continued at -78°C for 1 h. A solution of 2-nitrobenzaldehyde (3.90 g, 25.8 mmoles) in tetrahydrofuran (20 mL) was then added to the mixture dropwise, and then stirring was continued at -78°C for a further 2.5 h. The reaction was quenched with cold aqueous ammonium chloride and then diluted with water. The mixture was extracted twice with ethyl acetate and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the desired product in 94% yield as light yellow foam. MS m/e (M- C4H8+H)+= 353.1.

4-(4-Hydroxy-2-oxo-l , 2, 3, 4-tetrahydro-quinolin-3-yl)-piperidine-l -carboxylic acid tertbutyl ester. In a 3 neck flask fitted with a nitrogen inlet, thermometer, and a mechanical stirrer, 4-[2-hydroxy-l -methoxy carbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l -carboxylic acid tertbutyl ester (9.93 g, 24.3 mmoles) was dissolved in acetic acid (1.75 moles, 100 mL). Iron powder (8.90 g, 159 mmoles) was added to the vessel with stirring. The stirred mixture was slowly heated to 80°C for 30 min and then cooled to room temperature. It was then diluted with ethyl acetate and filtered through a pad of celite. Solids were washed with 20% methanol/ethyl acetate, and then with methanol. The filtrate was concentrated and the residue partitioned between ethyl acetate and aqueous sodium bicarbonate. The layers were separated. The resulting aqueous phase was extracted twice with ethyl acetate. The organic layers were combined. The mixture was washed twice with water and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the title compound as light yellow foam in 77% yield. MS m/e (M-H)’ = 345.1.

3-(Piperidin-4-yl)quinolin-2(lH) hydrochloride . A stirred solution of 4-(4-hydroxy-2- oxo-l,2,3,4-tetrahydro-quinolin-3-yl)-piperidine-l-carboxylic acid tert-butyl ester (5.60 g, 16.2 mmoles) in ethyl acetate (70 mL) was treated with HC1 in dioxane (4N, 40 mmoles, 10 mL). The mixture was stirred at room temperature for 45 min. More HC1 in dioxane (4N, 120 mmoles, 30 mL) was then added and stirring was continued at room temperature for 16 h. The resulting solid was collected by filtration and washed with ethyl acetate. It was then suspended in 5% water-isopropanol (100 mL) and the mixture was warmed to reflux and stirred for 20 min. The mixture was cooled to room temperature and stirred at room temperature for 16 h. The solid was collected by filtration, washed with isopropanol, and dried under high vacuum. The title compound was obtained as white solid in 75% yield. ‘H-NMR (DMSO-de) 5 11.85 (s, 1 H), 9.02 (bs, 1 H), 8.88 (bs, 1 H), 7.70 (t, J= 3.81 Hz, 2 H), 7.53 – 7.30 (d, J= 8.24 Hz, 1 H), 7.17 (t, J= 7.48 Hz, 2 H), 3.36 (d, J= 12.51 Hz, 2 H), 3.10 – 2.94 (m, 3 H), 2.01 (d, J= 13.43 Hz, 2 H), 1.87 – 1.73 (m, 2 H); MS m/e (M+H)+ = 229.0.

4-Iodo-2,6-dimethylbenzenamine hydrochloride . To a suspension of sodium bicarbonate (126 g, 1.5 moles) and 2,6-dimethylaniline (61.5 mL, 500 mmoles) in methanol (700 mL) was added iodine monochloride (1.0 M in dichloromethane, 550 mL, 550 mmoles) at room temperature over 1 h. After addition was complete, stirring was continued for 3 h. The reaction was filtered to remove excess sodium bicarbonate and the solvent removed in vacuo. The residue was re-dissolved in diethyl ether (1.5 L) and treated with hydrochloric acid (2M in ether, 375 mL, 750 mmoles). The resulting suspension was stored in the freezer (-15°C) overnight. The solid was filtered and washed with diethyl ether until it became colorless, to give 126.5 g (89%) as a grey-green powder. ‘H-NMR (DMSO-de) 5 2.33 (s, 6 H), 7.48 (s, 2 H), 9.05 (bs, 3 H); 13C-NMR (DMSO-de) 5 17.4, 91.5, 133.1, 131.2, 136.9.

Methyl 2 -(benzyloxy carbonyl) acrylate . To a flame dried three-neck round bottom flask, fitted with a mechanical stirrer, was added (S)-methyl 2-(benzyloxycarbonyl)-3- hydroxypropanoate (129 g, 509 mmoles), anhydrous dichloromethane (2 L), and methanesulfonyl chloride (49.3 mL, 636 mmoles). The mixture was cooled to -15°C, and treated with tri ethylamine (213 mL, 1527 mmoles), dropwise, to ensure the temperature of the reaction mixture did not exceed 0°C. The addition of the first equivalent of triethylamine was exothermic. After addition of tri ethylamine, the mixture was stirred at 0°C for 30 min. The cooling bath was removed and the mixture stirred at room temperature for 1.5 h. The reaction was quenched by addition of methanol (21 mL). The mixture was washed with 0.5% aqueous potassium bisulfate until the washings were pH 5, then saturated sodium bicarbonate, and brine, dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, 1 :9 ethyl acetate/hexanes) gave I l l g (92%) as a viscous colorless oil, which crystallized upon standing. ’H-NMR (DMSO-de) 5 3.71 (s, 3 H), 5.10 (s, 2 H), 5.60 (s, 1 H), 5.76 (s, 1 H), 7.39-7.35 (m, 5 H), 8.96 (s, 1 H); 13C-NMR (DMSO-de) 5 52.3, 65.9, 127.8, 128.1, 128.3, 128.8, 133.3, 136.3, 153.5, 163.7.

(Z)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl) acrylate. A 2 L round bottom flask was charged 4-iodo-2,6-dimethylbenzenamine hydrochloride salt (55 g, 194 mmoles), methyl 2-(benzyloxycarbonyl)acrylate (59.2 g, 252 mmoles), tetrabutylammonium chloride (59.2 g, 213 mmoles), palladium (II) acetate (4.34 g, 19.4 mmoles), and tetrahydrofuran (1.2 L, degassed by a flow of nitrogen for 30 min). The mixture was stirred so that a suspension was formed and then degassed by a flow of nitrogen for 30 min. Triethylamine (110 mL, 789 mmoles) was added and the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, the reaction mixture was filtered through a pad of celite, washed with tetrahydrofuran (2 x 100 mL), and concentrated. The residue was dissolved in di chloromethane, washed with water (3X) and brine (2X), dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, using 1 :9 ethyl acetate/dichloromethane) gave a tan solid. The solid was recrystallized from warm methanol (210 mL) and water (100 mL). The mixture was held at room temperature overnight, then at 0°C for 2 h, and finally at -15°C for 2 h. The resulting solid was filtered, washed with ice cold 1 : 1 methanol/water, and dried under high vacuum overnight to give 44.7 g (65%) as a light tan solid which was a mixture of ZZE isomers (73 :27). ’H-NMR (DMSO-de) 5, 2.05 (s, 6 H), 3.61 (s, 0.8 H), 3.68 (s, 2.2 H), 5.00 (s, 0.54 H), 5.13 (s, 1.46 H), 5.24 (s, 2 H), 7.40-7.21 (m, 8 H), 8.51 (s, 0.27 H), 8.79 (s, 0.73 H); 13C-NMR (DMSO-de) 5 17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7, 166.0.

(R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate. A flame- dried 2 L Parr hydrogenation bottle was charged with (Z)-methyl 3-(4-amino-3,5- dimethylphenyl)-2-(benzyloxycarbonyl)acrylate (84.5 g, 239 mmoles), di chloromethane (300 mL), and methanol (300 mL). The bottle was swirled so that a light brown suspension was formed. The mixture was degassed using a flow of nitrogen for 30 min. To this was quickly added (-)-l,2-bis((2A,5A)-2,5-diethylphospholano)-bezene(cyclooctadiene) rhodium (I) tetrafluoroborate ([(2A,5A)-Et-DuPhosRh]BF4) (2.11 g, 3.20 mmoles). The bottle was immediately attached to a Parr Hydrogenator. After 5 cycles of hydrogen (60 psi) and vacuum, the bottle was pressurized to 65 psi and the suspension was agitated at room temperature for 16 h. The reaction had become homogeneous. The reaction mixture was concentrated, and the resulting residue purified by flash chromatography (silica gel, 1 :9 ethyl acetate/dichloromethane) to give 82.9 g (98%). ‘H-NMR (DMSO-de) 5 2.04 (s, 6 H), 2.65 (dd, J= 13.4, 9.8 Hz, 1H), 2.82 (dd, J= 13.7, 5.2 Hz, 1 H), 3.62 (s, 3 H), 4.15-4.10 (m, 1H), 4.41 (s, 2 H), 5.00 (s, 2 H), 6.68 (s, 2 H), 7.37-7.28 (m, 5 H), 7.70 (d, J= 7.9 Hz, 1 H); 13C-NMR (DMSO-de) 5 17.7, 35.9, 51.7, 56.1, 65.3, 120.4, 124.0, 127.5, 127.7, 128.2, 128.3, 136.9, 142.6, 155.9, 172.5.

(R)-Methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5-yl)propanoate. (R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate (50.0 g, 140 mmoles) was weighed into a flame-dried 5 L three neck round bottom flask, followed by the addition of toluene (2.4 L) and glacial acetic acid (120 mL, 2.1 moles). The mixture was mechanically stirred to form a clear solution, and then potassium acetate (103 g, 1.05 moles) was added. To the resulting white suspension, z.w-amyl nitrite (20.7 mL, 154 mmoles) was added dropwise at room temperature, and the resulting mixture was stirred at room temperature for 16 h. Saturated sodium bicarbonate (I L) was added, followed by the careful addition of solid sodium bicarbonate to neutralize the acetic acid. The mixture was extracted with a mixture of di chloromethane (2 L) and brine (1.5 L). After separation, the aqueous layer was extracted with di chloromethane (500 mL). The combined organic layers were dried over anhydrous sodium sulfate and filtered. Solvents were removed to afford a tan solid, which was washed with hexanes (2 L) and toluene (150 mL). The solid was recrystallized from hot acetone (260 mL) and hexanes (700 mL). The slightly cloudy mixture was allowed to cool to room temperature slowly, then to 0°C for 1.5 h, and finally to -15°C for 1.5 h. The resulting solid was filtered and washed with ice-cold acetone/hexanes (1 : 1, 200 mL) to afford 39.1 g (76% yield). Analytical HPLC showed >98% UV purity. The enantiomeric excess (ee) was determined to be 99.8% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; A = ethanol, B = 0.05% diethylamine/heptane; 85%B @1.0 mL/min. for 55 min. The retention times for R was 44.6 min and for S was 28.8 min). ‘H-NMR (DMSO-de) 5 2.48 (s, 3 H), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s, 3H), 4.32-4.27 (m, 1 H), 4.97 (s, 2 H), 7.03 (s, 1 H), 7.24-7.22 (m, 2 H), 7.29 -7.27 (m, 3 H), 7.41 (s, 1 H), 7.83 (d, J= 8.2 Hz, 1H), 7.99 (s, 1H), 13.1 (s, 1 H); 13C-NMR (DMSO-de) 5 16.7, 36.5, 51.8, 56.0, 65.3, 117.6, 119.6, 122.7, 127.2, 127.4, 127.6, 128.2, 129.3, 133.4, 136.8, 139.2, 155.9, 172.4. Mass spec.: 368.16 (MH)+.

(R)-Methyl 2-amino-3-(7-methyl-lH-indazol-5-yl)propanoate. A Parr hydrogenation bottle was charged with (R)-methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5- yl)propanoate (11.0 g, 29.9 mmoles) and methanol (75 mL). The suspension was purged with nitrogen and treated with palladium (10% on charcoal, 700 mg). The bottle was shaken under hydrogen (15 psi) overnight. The mixture was filtered through a pad of celite to remove the catalyst. Concentration of the eluent gave 7.7 g (quant.) as an oil which was used without further purification. XH-NMR (CD3OD) 5 2.54 (s, 3 H), 2.98 (dd, J= 13.5, 7.0 Hz, 1 H), 3.09 (dd, J= 13.5, 5.9 Hz, 1 H), 3.68 (s, 3 H), 3.75 (dd, J= 7.0, 6.2 Hz, 1 H), 7.01 (s, 1 H), 7.39 (s, 1 H), 7.98 (s, 1 H). Mass spec.: 232.34 (M-H)’.

(R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoate. To a solution of (R)-methyl 2-amino-3-(7-methyl-lH- indazol-5-yl)propanoate hydrochloride (7.26 g, 27.0 mmoles) in dimethylformamide (50 mL) at room temperature was added N, A’-disuccinimidyl carbonate (7.60 g, 29.7 mmoles) followed by triethylamine (11.29 mL, 81 mmoles). The resulting mixture was stirred for 30 min and treated with 3-(piperidin-4-yl)quinolin-2(lH)-one (6.77 g, 29.9 mmoles) in portions. The reaction was allowed to stir for 24 h. The mixture was concentrated, dissolved in ethyl acetate, and washed sequentially with water, brine, and 0.5 N HC1 (2X). The organic phase was dried over magnesium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (silica gel, 20: 1 ethyl acetate/methanol) to give 11.9 g (78%). 1 H-NMR (CD3OD) 5 13.0 (s, 1 H), 11.8 (s, 1 H), 7.98 (s, 1 H), 7.63 (d, J= 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.45 – 7.41 (m, 2 H), 7.27 (d, J= 8.2Hz, 1 H), 7.16 (t, J= 7.9 Hz, 1 H), 7.03 (s, 1 H), 6.85 (d, J= 7.9 Hz, 1 H), 4.31 – 4.26 (m, 1 H), 4.10 – 4.08 (m, 2 H), 3.60 (s, 3 H), 3.07 – 3.01 (m, 2 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.67 (m, 2 H), 2.48 (s, 3 H), 1.78 – 1.72 (m, 2 H), 1.34 – 1.26 (m, 2 H). Mass spec.: 488.52 (MH)+.

(R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l- carboxamido)propanoic acid. A solution of (R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2- oxo-1, 2-dihydroquinolin-3-yl)piperidine-l-carboxamido)propanoate_(5.50 g, 11.3 mmoles) in tetrahydrofuran (50 mL) and methanol (10 mL) was cooled to 0°C. To this was added a cold (0°C) solution of lithium hydroxide monohydrate (0.95 g, 22.6 mmoles) in water (20 mL), dropwise over 15 min. The reaction was stirred at room temperature for additional 3 h. The mixture was concentrated to remove the organic solvents. The resulting residue was dissolved in a minimum amount of water, cooled to 0°C, and treated with cold (0°C) IN HC1 until pH 2 was attained. The resulting solid was collected by filtration, washed with cold water and ether, and then dried overnight under high vacuum to give 5.0 g (94%) as a white solid. ’H-NMR (DMSO- d6) 5 13.05 (bs, 1 H), 11.77 (s, 1 H), 7.98 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.55 (s, 1 H), 7.44 (d, J= 8.2Hz, 1 H), 7.42 (s, 1 H), 7.27 (d, J= 8.2 Hz, 1 H), 7.16 (t, J= 7.6 Hz, 1 H), 7.05 (s, 1 H), 6.65 (d, J= 7.9 Hz, 1 H), 4.27 – 4.22 (m, 1 H), 4.10 – 4.07 (m, 2 H), 3.12 – 3.07 (m, 1 H), 3.03 – 2.99 (m, 1 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.66 (m, 2 H), 2.47 (s, 3 H), 1.77 – 1.74 (m, 2 H), 1.34 – 1.27 (m, 2 H). Mass spec.: 474.30 (MH)+.

(R)-N-(3-(7-methyl-lH-indazol-5-yl)-l-(4-(l-methylpiperidin-4-yl)piperazin-l-yl)-l- oxopropan-2-yl)-4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l-carboxamide (I). A flask was charged with (R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoic acid (2.9 g, 6.11 mmoles), triethylamine (3.00 mL, 21.5 mmoles), l-(l-methylpiperidin-4-yl)piperazine (1.23 g, 6.72 mmoles), and dimethylformamide (10 mL). The resulting solution was treated with 2-(lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium tetrafluoroborate (2.26 g, 7.03 mmoles) in portions. The reaction was allowed to stir at room temperature overnight. The mixture was concentrated under vacuum to remove dimethylformamide. The crude product was dissolved in 7% methanol in di chloromethane and purified by flash chromatography using 7% methanol in di chloromethane containing 2% of aqueous ammonium hydroxide as eluent. The pure fractions were collected and solvent was removed under vacuum. The desired product was crystallized from hot acetone to give the compound having Formula I in 77% yield. Analytical HPLC showed 99.0 % UV purity at 230 nm. The enantiomeric excess (ee) was determined to be >99.9% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; eluent: 70% (0.05% diethylamine)/heptane/30%ethanol; @1.0 mL/min. for 45 min. The retention times were 18.7 min for R and 28.1 min for S). ‘H-NMR (500 MHz, DMSO-de) 5 ppm 13.01 (s, 1 H), 11.76 (s, 1 H), 7.96 (s, 1 H), 7.62 (d, J= 7.10 Hz, 1 H), 7.60 (s, 1 H), 7.42 (m, 1 H), 7.36 (s, 1 H), 7.26 (d, J = 8.25 Hz, 1 H), 7.14 (m, 1 H), 7.00 (s, 1 H), 6.69 (d, J= 8.25 Hz, 1 H), 4.78 (q, J= 7.79 Hz, 1 H), 4.14 (d, J= 12.37 Hz, 2 H), 3.54 (dd, J= 9.16, 4.58 Hz, 1 H), 3.24 (m, 1 H), 3.11 (m, 1 H), 2.97 (m, 1 H), 2.89 (m, 2 H), 2.69 (m, 4 H), 2.32 (m, 1 H), 2.21 (m, 1 H), 2.07 (m, 4 H), 1.95 (t, J= 8.25 Hz, 1 H), 1.87 (m, J= 11.28, 11.28, 3.55, 3.44 Hz, 1 H), 1.76 (t, J= 12.03 Hz, 2 H), 1.68 (t, J= 11.11 Hz, 2 H), 1.53 (t, J= 8.25 Hz, 1 H), 1.32 (m, 4 H), 1.16 (m, 2 H); 13C-NMR (DMSO-de) 5 16.80, 27.30, 30.51, 30.51, 30.67, 35.50, 38.04, 41.74, 44.00, 44.16, 45.35, 45.78, 48.14, 48.39, 51.45, 54.76, 54.76, 60.61, 114.53, 117.79, 119.29, 119.34, 121.57, 122.78, 127.46, 127.79, 129.29, 129.79, 133.31, 133.72, 136.98, 137.41, 139.12, 156.50, 161.50, 170.42.
Accurate mass analysis: m/z 639.3770, [MH]+, A = -0.2 ppm. Optical rotation: -27.36° @ 589 nm, concentration = 4.71 mg/mL in methanol. DESCRIPTION AND DOSAGE FORM
The physical and chemical properties of zavegepant (BHV-3500) drug substance mono-hydrochloride salt form are provided in Table 1.
Table 1 Physical and Chemical Properties
Biohaven number BHV-3500
Molecular formula C36H47CIN8O3
Molecular weight 675.26 (HO salt); 638.82 (free base)
Appearance White to off-white powder
Melting point ~178°C pH-solubility profile 105 mg/mL at pH = 8.2 and > 300 mg/mL at lower pH pKa 4.8 and 8.8 logD 1.21
Patent
US2022401439Bioorg Med Chem Lett
. 2021 Jan 1;31:127624.
doi: 10.1016/j.bmcl.2020.127624. Epub 2020 Oct 21.
References
- ^ Jump up to:a b c d e f g h https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216386s000lbl.pdf
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/216386Orig1s000ltr.pdf
- ^ “Pfizer’s Zavzpret (Zavegepant) Migraine Nasal Spray Receives FDA Approval” (Press release). 10 March 2023.
Further reading
- Croop R, Madonia J, Stock DA, Thiry A, Forshaw M, Murphy A, Coric V, Lipton RB (October 2022). “Zavegepant nasal spray for the acute treatment of migraine: A Phase 2/3 double-blind, randomized, placebo-controlled, dose-ranging trial”. Headache. 62 (9): 1153–1163. doi:10.1111/head.14389. PMC 9827820. PMID 36239038.
- Noor N, Angelette A, Lawson A, Patel A, Urits I, Viswanath O, et al. (2022). “A Comprehensive Review of Zavegepant as Abortive Treatment for Migraine”. Health Psychology Research. 10 (3): 35506. doi:10.52965/001c.35506. PMC 9239361. PMID 35774914.
- Scuteri D, Tarsitano A, Tonin P, Bagetta G, Corasaniti MT (November 2022). “Focus on zavegepant: the first intranasal third-generation gepant”. Pain Management. 12 (8): 879–885. doi:10.2217/pmt-2022-0054. PMID 36189708. S2CID 252681912.
External links
- Clinical trial number NCT04571060 for “Randomized Trial in Adult Subjects With Acute Migraines” at ClinicalTrials.gov
- Clinical trial number NCT03872453 for “Acute Treatment Trial in Adult Subjects With Migraines” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zavzpret |
| Other names | BHV-3500 |
| License data | US DailyMed: Zavegepant |
| Routes of administration | Nasal |
| Drug class | Calcitonin gene-related peptide receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1337918-83-8as HCl: 1414976-20-7 |
| PubChem CID | 53472683as HCl: 134819878 |
| DrugBank | DB15688 |
| ChemSpider | 30814207 |
| UNII | ODU3ZAZ94Jas HCl: 000QCM6HAL |
| KEGG | D11898as HCl: D11899 |
| ChEMBL | ChEMBL2397415as HCl: ChEMBL4650220 |
| Chemical and physical data | |
| Formula | C36H46N8O3 |
| Molar mass | 638.817 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////FDA 2023, APPROVALS 2023, Vazegepant, BMS-742413, BHV-3500, ザベジェパント , Zavegepant, ZAVZPRET, BMS
PFIZERCC1=CC(=CC2=C1NN=C2)CC(C(=O)N3CCN(CC3)C4CCN(CC4)C)NC(=O)N5CCC(CC5)C6=CC7=CC=CC=C7NC6=O

NEW DRUG APPROVALS
ONE TIME help to run this blog
$10.00
NIROGACESTAT
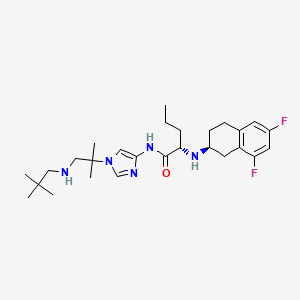

NIROGACESTAT
(2S)-2-[[(2S)-6,8-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl]amino]-N-[1-[1-(2,2-dimethylpropylamino)-2-methylpropan-2-yl]imidazol-4-yl]pentanamide
489.6 g/mol, C27H41F2N5O
CAS 1290543-63-3
FDA APPROVED 11/27/2023, To treat adults with progressing desmoid tumors who require systemic treatment, Ogsiveo
PF-03084014, 1290543-63-3, PF-3084014, 865773-15-5QZ62892OFJUNII:QZ62892OFJUNII-QZ62892OFJнирогацестат [Russian] [INN]نيروغاسيستات [Arabic] [INN]尼罗司他 [Chinese] [INN]ニロガセスタット;
orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation
Nirogacestat, also known as PF-03084014, is a potent and selective gamma secretase (GS) inhibitor with potential antitumor activity. PF-03084014 binds to GS, blocking proteolytic activation of Notch receptors. Nirogacestat enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Nirogacestat enhances docetaxel-mediated tumor response and provides a rationale to explore GSIs as adjunct therapy in conjunction with docetaxel for men with CRPC (castration-resistant prostate cancer).
Nirogacestat was disclosed to be a gamma-secretase inhibitor, which can inhibit Aβ-peptide production. SpringWorks Therapeutics (a spin-out of Pfizer ) is developing nirogacestat, as hydrobromide salt, a gamma-secretase inhibitor, for treating aggressive fibromatosis. In February 2021, nirogacestat was reported to be in phase 3 clinical development.
Nirogacestat is a selective gamma secretase (GS) inhibitor with potential antitumor activity. Nirogacestat binds to GS, blocking proteolytic activation of Notch receptors; Notch signaling pathway inhibition may follow, which may result in the induction of apoptosis in tumor cells that overexpress Notch. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains. Overexpression of the Notch signaling pathway has been correlated with increased tumor cell growth and survival.
Nirogacestat has been used in trials studying the treatment of Breast Cancer, HIV Infection, Desmoid Tumors, Advanced Solid Tumors, and Aggressive Fibromatosis, among others.
Nirogacestat (Gamma Secretase Inhibitor)
Nirogacestat is an oral, selective, small molecule, gamma secretase inhibitor (GSI) in Phase 3 clinical development for patients with desmoid tumors. Gamma secretase is a protease complex that cleaves, or divides, multiple transmembrane protein complexes, including Notch, which, when dysregulated, can play a role in activating pathways that contribute to desmoid tumor growth.
Gamma secretase has also been shown to directly cleave BCMA, a therapeutic target that is highly expressed on multiple myeloma cells. By inhibiting gamma secretase with nirogacestat, membrane-bound BCMA can be preserved, thereby increasing target density while simultaneously reducing levels of soluble BCMA, which may serve as decoy receptors for BCMA-directed therapies. Together, these mechanisms combine to potentially enhance the activity of BCMA therapies and improve outcomes for multiple myeloma patients. SpringWorks is seeking to advance nirogacestat as a cornerstone of multiple myeloma combination therapy in collaboration with industry leaders who are advancing BCMA therapies.
SpringWorks Therapeutics Announces Clinical Collaboration with Pfizer
By Satish October 05, 2020
SpringWorks Therapeutics today announced that the company has entered into a clinical trial collaboration agreement with Pfizer to evaluate SpringWorks Therapeutics’ investigational gamma secretase inhibitor (GSI), nirogacestat, in combination with Pfizer’s anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody, PF‐06863135, in patients with relapsed or refractory multiple myeloma.
Gamma secretase inhibition prevents the cleavage and shedding of BCMA from the surface of myeloma cells. In preclinical models, nirogacestat has been shown to increase the cell surface density of BCMA and reduce levels of soluble BCMA, thereby enhancing the activity of BCMA-targeted therapies, including CD3 bispecific antibodies.
Saqib Islam, Chief Executive Officer of SpringWorks Therapeutics Said: This collaboration is another important step in continuing to advance our goal of developing nirogacestat as a best-in-class BCMA potentiator, and we are pleased to work with Pfizer to study nirogacestat in combination with PF‐06863135, which has recently demonstrated promising monotherapy clinical data, We now have five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities. We look forward to generating clinical data with our collaborators to further evaluate the ability of nirogacestat to improve outcomes for patients with multiple myeloma.

Under the terms of the agreement, Pfizer will sponsor and conduct the Phase 1b/2 study to evaluate the safety, tolerability and preliminary efficacy of the combination, and will assume all costs associated with the study, other than expenses related to the manufacturing of nirogacestat and certain expenses related to intellectual property rights. Pfizer and SpringWorks Therapeutics will also form a joint development committee to manage the clinical study, which is expected to commence in the first half of 2021.
Chris Boshoff, MD, PhD, Chief Development Officer for Pfizer Oncology at Pfizer Said: Entering into this clinical collaboration is a proud milestone in our strong relationship with SpringWorks,We believe that studying nirogacestat in combination with PF-06863135 could hold significant therapeutic promise for patients with relapsed or refractory multiple myeloma, and we look forward to working together to advance this important area of research.
In addition to its ongoing clinical collaborations with BCMA-directed therapies, SpringWorks is also currently conducting a global Phase 3, double-blind, randomized, placebo-controlled clinical trial (the DeFi Trial) to evaluate nirogacestat in adults with progressing desmoid tumors.
About Nirogacestat
Nirogacestat is an investigational, oral, selective, small molecule gamma secretase inhibitor in Phase 3 clinical development for desmoid tumors, which are rare and often debilitating and disfiguring soft-tissue tumors. Gamma secretase cleaves multiple transmembrane protein complexes, including Notch, which is believed to play a role in activating pathways that contribute to desmoid tumor growth.
In addition, gamma secretase has been shown to directly cleave membrane-bound BCMA, resulting in the release of the BCMA extracellular domain, or ECD, from the cell surface. By inhibiting gamma secretase, membrane-bound BCMA can be preserved, increasing target density while reducing levels of soluble BCMA ECD, which may serve as decoy receptors for BCMA-directed therapies. Nirogacestat’s ability to enhance the activity of BCMA-directed therapies has been observed in preclinical models of multiple myeloma. SpringWorks is evaluating nirogacestat as a BCMA potentiator and has five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities, including with an antibody-drug conjugate, two CAR T cell therapies and two bispecific antibodies. In addition, SpringWorks and Fred Hutchinson Cancer Research Center have entered into a sponsored research agreement to further characterize the ability of nirogacestat to modulate BCMA and potentiate BCMA directed therapies using a variety of preclinical and patient-derived multiple myeloma models developed by researchers at Fred Hutch.
Nirogacestat has received Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) for the treatment of desmoid tumors (June 2018) and from the European Commission for the treatment of soft tissue sarcoma (September 2019). The FDA also granted Fast Track and Breakthrough Therapy Designations for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis (November 2018 and August 2019).
About PF‐06863135
PF‐06863135 is an anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody being investigated in a Phase 1 clinical study to treat relapsed or refractory multiple myeloma. This bispecific antibody can be administered subcutaneously and has been optimized for binding affinity to both BCMA and CD3, enabling more potent T-cell-mediated tumor cell toxicity.
Source: SpringWorks Therapeutics
FDA Grants Breakthrough Designation to Nirogacestat for Desmoid Tumors
The FDA has granted nirogacestat, an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
The FDA has granted nirogacestat (PF-03084014), an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.1
The breakthrough designation was granted as a result of positive findings seen in phase I and II trials of nirogacestat monotherapy in patients with desmoid tumors. A phase III trial has also been initiated investigating nirogacestat in patients with desmoid tumors or aggressive fibromatosis (NCT03785964).
“We are committed to pursuing the rapid development of nirogacestat given the important need for new therapies for patients with desmoid tumors and are pleased to receive this breakthrough therapy designation,” Saqib Islam, CEO of SpringWorks, the company developing the small molecule inhibitor, said in a statement. “We are currently enrolling adult patients in our phase III DeFi trial and will continue to work closely with the FDA with the goal of bringing nirogacestat to patients as quickly as possible.”
The open-label, single-center phase II trial of nirogacestat enrolled 17 patients with desmoid tumors who were not eligible for surgical resection or definitive radiation therapy and who had experienced disease progression after at least 1 prior treatment regimen. Patients received 150 mg twice per day of continuous, oral nirogacestat in 21-day cycles.2
The median age of patients was 34 years (range, 19-69), 82% of the patients were female, and 53% of patients had aCTNNB1T41A somatic missense mutation. The median number of prior therapies was 4 (range, 1-9), which included cytotoxic chemotherapy in 71% and a tyrosine kinase inhibitor in 59%.
Sixteen patients were evaluable for response. After a median follow-up of more than 25 months, 5 patients (29%) achieved a partial response and 11 (65%) had stable disease, for a disease control rate of 100%. Ten patients (59%) remained on treatment with nirogacestat for more than 2 years.
Grade 1/2 adverse events were observed in all patients, with diarrhea (76%) and skin disorders (71%) being the most common toxicities. The only treatment-related grade 3 event was reversible hypophosphatemia, which was reported in 8 patients (47%) and was considered to be a class effect of gamma-secretase inhibitors. Four patients met the criteria for dose reduction.
Findings from the phase I study also showed a disease control rate of 100% with nirogacestat. However, the median progression-free survival was not reached in either study due to a lack of patients progressing on treatment. Only 1 patient discontinued treatment due to an adverse event between the 2 studies.1
The FDA had previously granted nirogacestat with an orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation in November 2018 for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
References
- SpringWorks Therapeutics Receives Breakthrough Therapy Designation for Nirogacestat for the Treatment of Adult Patients with Progressive, Unresectable, Recurrent or Refractory Desmoid Tumors [press release]. Stamford, CT: SpringWorks Therapeutics, Inc; August 29, 2019. https://bit.ly/30IV0Eb. Accessed September 3, 2019.
- Kummar S, O’Sullivan Coyne G, Do KT, et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis).J Clin Oncol.2017;35(14):1561-1569. doi: 10.1200/JCO.2016.71.1994.
PAPER

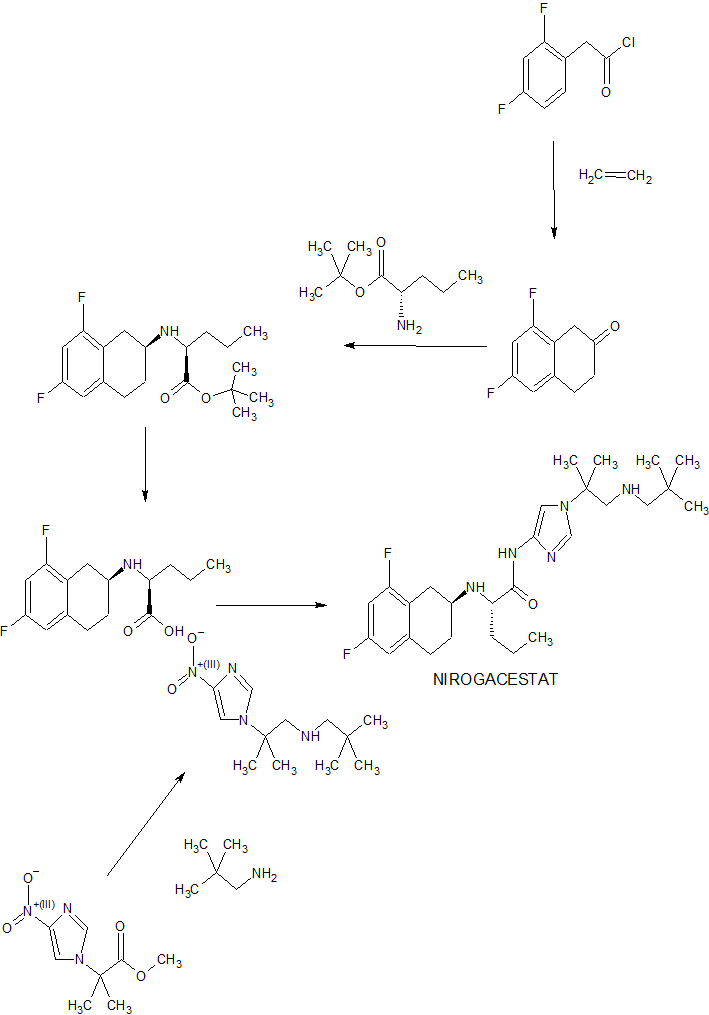
Bioorganic & medicinal chemistry letters (2011), 21(9), 2637-40.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X10018822



PATENT
WO 2016089208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016089208
PATENT
WO-2021029854
Novel, stable crystalline polymorphic (A to N) and amorphous forms of nirogacestat hydrobromide , useful for treating desmoid tumors such as multiple myeloma, a cancer having a mutation in a Notch pathway gene, adenoid cystic carcinoma and T-cell acute lymphoblastic leukemia.
(S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)-N-(l-(2- methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide (“Compound 1”) is a gamma-secretase inhibitor which can inhibit Ab-peptide production.
[0003] Not all compounds that are gamma-secretase inhibitors have characteristics affording the best potential to become useful therapeutics. Some of these characteristics include high affinity at the gamma-secretase, duration of gamma-secretase deactivation, oral bioavailability, tissue distribution, and stability (e.g., ability to formulate or crystallize, shelf life). Favorable characteristics can lead to improved safety, tolerability, efficacy, therapeutic index, patient compliance, cost efficiency, manufacturing ease, etc.
[0004] In addition, the isolation and commercial -scale preparation of a solid state form of hydrobromide salts of Compound 1 and corresponding pharmaceutical formulations having acceptable solid state properties (including chemical stability, thermal stability, solubility, hygroscopicity, and/or particle size), compound manufacturability (including yield, impurity rejection during crystallization, filtration properties, drying properties, and milling properties), and formulation feasibility (including stability with respect to pressure or compression forces during tableting) present a number of challenges.
[0005] Accordingly, there is a current need for one or more solid state forms of hydrobromide salts of Compound 1 that have an acceptable balance of these properties and can be used in the preparation of pharmaceutically acceptable solid dosage forms.
Crystalline Form A
[0147] In one aspect, the present disclosure relates to crystalline Form A of a hydrobromide salt of (S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)- N-(l -(2 -methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide having Formula (I),
[0148] In one embodiment, crystalline Form A is anhydrous.
[0149] In another embodiment, the melting point of crystalline Form A is about 254 °C.
[0150] In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, and 23.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, and 29.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 20.0 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, 29.3 ± 0.2, and 32.5 ± 0.2 degrees two theta when measured by Cu Ka radiation.
Patent
Product case, WO2005092864 ,
hold protection in the EU states until March 2025, and expire in the US in February 2026 with US154 extension.
PATENT
WO2020208572 , co-assigned to GSK and SpringWorks, claiming a combination of nirogacestat with anti-BCMA antibody (eg belantamab mafodotin ), for treating cancer.
PATENT
US10590087 , for a prior filing from Pfizer, claiming crystalline forms of nirogacestat hydrobromide.
////////////NIROGACESTAT, orphan drug designation, esmoid tumors, fast track designation, PF-03084014, PF 03084014, QZ62892OFJ , UNII:QZ62892OFJ ,UNII-QZ62892OFJ, ,нирогацестат , نيروغاسيستات , 尼罗司他 , ニロガセスタット, phase 3
CCCC(C(=O)NC1=CN(C=N1)C(C)(C)CNCC(C)(C)C)NC2CCC3=C(C2)C(=CC(=C3)F)F
TROFINETIDE

Trofinetide
- Molecular FormulaC13H21N3O6
- Average mass315.322 Da
Tofinetide , NNZ-256610076853400-76-7[RN]
glycyl-2-methyl-L-prolyl-L-glutamic acid
H-Gly-PMe-Glu-OHL-Glutamic acid, glycyl-2-methyl-L-prolyl-UNII-Z2ME8F52QLZ2ME8F52QLтрофинетид [Russian] [INN]تروفينيتيد [Arabic] [INN]曲非奈肽 [Chinese] [INN]
| IUPAC Condensed | H-Gly-aMePro-Glu-OH |
|---|---|
| Sequence | GXE |
| HELM | PEPTIDE1{G.[*C(=O)[C@@]1(CCCN1*)C |$_R2;;;;;;;;_R1;$|].E}$$$$ |
| IUPAC | glycyl-alpha-methyl-L-prolyl-L-glutamic acid |
An (1-3) IGF-1 analog with neuroprotective activity.
OPTICAL ROT; -52.4 ° Conc: 0.19 g/100mL; water ; 589.3 nm; Temp: 20 °C; Len: 1.0 dm…Tetrahedron 2005, V61(42), P10018-10035
EU Customs Code CN, 29339980
Harmonized Tariff Code, 293399
- L-Glutamic acid, glycyl-2-methyl-L-prolyl-
- glycyl-2-methyl-L-prolyl-L-glutamic acid
- Glycyl-L-2-methylprolyl-L-glutamic acid
FDA APPROVED 2023/3/10, Daybue
853400-76-7 CAS
トロフィネチド;
Trofinetide (NNZ-2566) is a drug developed by Neuren Pharmaceuticals that acts as an analogue of the neuropeptide (1-3) IGF-1, which is a simple tripeptide with sequence Gly–Pro–Glu formed by enzymatic cleavage of the growth factor IGF-1 within the brain. Trofinetide has anti-inflammatory properties and was originally developed as a potential treatment for stroke,[1][2] but has subsequently been developed for other applications and is now in Phase II clinical trials against Fragile X syndrome and Rett syndrome.[3][4][5]
Trofinetide (NNZ-2566), a neuroprotective analogue of glypromate, is a novel molecule that has a profile suitable for both intravenous infusion and chronic oral delivery. It is currently in development to treat traumatic brain injury.
In February 2021, Neuren is developing trofinetide (NNZ-2566, phase 2 clinical ), a small-molecule analog of the naturally occurring neuroprotectant and N-terminus IGF-1 tripeptide Glypromate (glycine-proline-glutamate), for intravenous infusion treatment of various neurological conditions, including moderate to severe traumatic brain injury (TBI), stroke, chronic neurodegenerative disorders and peripheral neuropathies. At the same time, Neuren is also investigating an oral formulation of trofinetide (phase 3 clinical) for similar neurological indications, including mild TBI.
Autism Spectrum Disorders and neurodevelopment disorders (NDDs) are becoming increasingly diagnosed. According to the fourth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-4), Autism spectrum disorders (ASD) are a collection of linked developmental disorders, characterized by abnormalities in social interaction and communication, restricted interests and repetitive behaviours. Current classification of ASD according to the DSM-4 recognises five distinct forms: classical autism or Autistic Disorder, Asperger syndrome, Rett syndrome, childhood disintegrative disorder and pervasive developmental disorder not otherwise specified (PDD-NOS). A sixth syndrome, pathological demand avoidance (PDA), is a further specific pervasive developmental disorder.
More recently, the fifth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-5) recognizes recognises Asperger syndrome, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDD-NOS) as ASDs.
This invention applies to treatment of disorders, regardless of their classification as either DSM-4 or DSM-5.
Neurodevelopment Disorders (NDDs) include Fragile X Syndrome (FXS), Angelman Syndrome, Tuberous Sclerosis Complex, Phelan McDermid Syndrome, Rett Syndrome, CDKL5 mutations (which also are associated with Rett Syndrome and X-Linked Infantile Spasm Disorder) and others. Many but not all NDDs are caused by genetic mutations and, as such, are sometimes referred to as monogenic disorders. Some patients with NDDs exhibit behaviors and symptoms of autism.
As an example of a NDD, Fragile X Syndrome is an X-linked genetic disorder in which affected individuals are intellectually handicapped to varying degrees and display a variety of associated psychiatric symptoms. Clinically, Fragile X Syndrome is characterized by intellectual handicap, hyperactivity and attentional problems, autism spectrum symptoms, emotional lability and epilepsy (Hagerman, 1997a). The epilepsy seen in Fragile X Syndrome is most commonly present in childhood, but then gradually remits towards adulthood. Hyperactivity is present in approximately 80 percent of affected males (Hagerman, 1997b). Physical features such as prominent ears and jaw and hyper-extensibility of joints are frequently present but are not diagnostic. Intellectual handicap is the most common feature defining the phenotype. Generally, males are more severely affected than females. Early impressions that females are unaffected have been replaced by an understanding of the presence of specific learning difficulties and other neuropsychiatric features in females. The learning disability present in males becomes more defined with age, although this longitudinal effect is more likely a reflection of a flattening of developmental trajectories rather than an explicit neurodegenerative process.
The compromise of brain function seen in Fragile X Syndrome is paralleled by changes in brain structure in humans. MRI scanning studies reveal that Fragile X Syndrome is associated with larger brain volumes than would be expected in matched controls and that this change correlates with trinucleotide expansion in the FMRP promoter region (Jakala et al, 1997). At the microscopic level, humans with Fragile X Syndrome show abnormalities of neuronal dendritic structure, in particular, an abnormally high number of immature dendritic spines (Irwin et al, , 2000).
Currently available treatments for NDDs are symptomatic – focusing on the management of symptoms – and supportive, requiring a multidisciplinary approach. Educational and social skills training and therapies are implemented early to address core issues of learning delay and social impairments. Special academic, social, vocational, and support services are often required. Medication, psychotherapy or behavioral therapy may be used for management of co-occurring anxiety, ADHD, depression, maladaptive behaviors (such as aggression) and sleep issues, Antiepileptic drugs may be used to control seizures.
Patent
WO 2014085480,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014085480

EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro-Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
WO95/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS to increase TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses certain GPE analogs having amino acid substitutions and certain other modification that are capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including injury or disease in the CNS.
EXAMPLES
The following examples are intended to illustrate embodiments of this invention, and are not intended to limit the scope to these specific examples. Persons of ordinary skill in the art can apply the disclosures and teachings presented herein to develop other embodiments without undue experimentation and with a likelihood of success. All such embodiments are considered part of this invention.
Example 1: Synthesis of N,N-Dimethylglycyl-L-prolyl)-L-glutamic acid
The following non-limiting example illustrates the synthesis of a compound of the invention, N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
All starting materials and other reagents were purchased from Aldrich; BOC=tert-butoxycarbonyl; Bn=benzyl.
BOC-L-proline-(P-benzyl)-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem. Soc: 79, 6810, 1994] (10 mmol) in dichloromethane (50 mi), cooled to 0°C, was added triethylamine (1 .39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl-L-glutamate (10 mmol) was then added and the mixture stirred at 0° C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol 1-1) then dried (MgSO4) and concentrated at reduced pressure to give BOC-L-proline-L-glutamic acid dibenzyl ester (5.0 g, 95%).
L-proline-L-glutamic acid dibenzyl ester
A solution of BOC-L-glutamyl-L-proline dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 h. at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give L-proline-L-glutamic acid dibenzyl ester.
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of L-proline-L-glutamic acid dibenzyl ester (10 mmol), N,N-dimethylglycine (10 mmol) and triethylamine ( 10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0°C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallised from ethyl acetate to yield the tripeptide derivative.
It can be appreciated that following the method of the Examples, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Eample 2: Synthesis of Glycyl-L-2-Methyl-L-Prolyl-L-Glutamate
L-2-Methylproline and L-glutamic acid dibenzyl ester p-toluenesulphonate were purchased from Bachem, N-benzyloxycarbonyl-glycine from Acros Organics and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BoPCl, 97%) from Aldrich Chem. Co.
Methyl L-2-methylprolinate hydrochloride 2
Thionyl chloride (5.84 cm3, 80.1 mmol) was cautiously added dropwise to a stirred solution of (L)-2-methylproline 1 (0.43 g, 3.33 mmol) in anhydrous methanol (30 cm3) at -5 °C under an atmosphere of nitrogen. The reaction mixture was heated under reflux for 24 h, and the resultant pale yellow-coloured solution was. concentrated to dryness in vacuo. The residue was dissolved in a 1 : 1 mixture of methanol and toluene (30 cm3) then concentrated to dryness to remove residual thionyl chloride. This procedure was repeated twice more, yielding hydrochloride 2 (0.62 g, 104%) as an hygroscopic, spectroscopically pure, off-white solid: mp 127- 131 °C; [α]D -59.8 (c 0.24 in CH2Cl2); vmax (film)/cm-1 3579, 3398 br, 2885, 2717, 2681 , 2623, 2507, 1743, 1584, 1447, 1432, 1374, 1317, 1294, 1237, 1212, 1172, 1123, 981 , 894, 861 and 764; δH (300 MHz; CDCl3; Me4Si) 1.88 (3H, s, Proα-CH3), 1 .70-2.30 (3H, br m, Proβ-HAΗΒ and Proγ-H2), 2.30-2.60 (1H, br m, Proβ-HAΗΒ), 3.40-3.84 (2H, br m, Proδ-H2), 3.87 (3H, s, CO2CH3), 9.43 (1H, br s, NH) and 10.49 ( 1H, br s, HCl); δC (75 MHz; CDCl3) 21.1 (CH3, Proα-CH3), 22.4 (CH2, Proγ-C), 35.6 (CH2, Proβ-C), 45.2 (CH2, Proδ-C), 53.7 (CH3, CO2CH3), 68.4 (quat., Proα-C) and 170.7 (quat, CO); m/z (FAB+) 323.1745 [M2.H35Cl.H+: (C7H13NO2)2. H35Cl.H requires 323.1738] and 325.1718 [M2.H37Cl.H+: (C7H13NOz)2. H37Cl.H requires 325.1708],
N-Benxyloxycarbonyl-glycyl-L-2-methylproline 5
Anhydrous triethylamine (0.45 cm3, 3.23 mmol) was added dropwise to a mixture of methyl L-2-methylprolinate hydrochloride 2 (0.42 g, 2.34 mmol) and N-benzyloxycarbonyl-glycine (98.5%) 3 (0.52 g, 2.45 mmol) in methylene chloride (16 cm3), at 0 °C, under an atmosphere of nitrogen. The resultant solution was stirred for 20 min and a solution of 1 ,3-dicyclohexylcarbodiimide (0.56 g, 2.71 mmol) in methylene chloride (8 cm3) at 0 °C was added dropwise and the reaction mixture was warmed to room temperature and stirred for a further 20 h. The resultant white mixture was filtered through a Celite™ pad to partially remove 1 ,3-dicyclohexylurea, and the pad was washed with methylene chloride (50 cm3). The filtrate was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and concentrated to dryness in vacuo. Further purification of the residue by flash column chromatography (35 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) afforded tentatively methyl N-benzyloxycarbonyl-glycyl-L-2-methylprolinate 4 (0.56 g), containing 1 ,3-dicyclohexylurea, as a white semi-solid: Rf 0.65 (EtOAc); m/z (ΕI+) 334.1534 (M+. C17H22N2O5 requires 334.1529) and 224 ( 1 ,3-dicyclohexylurea).
To a solution of impure prolinate 4 (0.56 g, ca. 1.67 mmol) in 1,4-dioxane (33 cm3) was added dropwise 1 M aqueous sodium hydroxide (10 cm3, 10 mmol) and the mixture was stirred for 19 h at room temperature. Methylene chloride ( 100 cm3) was then added and the organic layer extracted with saturated aqueous sodium hydrogen carbonate (2 x 100 cm3). The combined aqueous layers were carefully acidified with hydrochloric acid (32%), extracted with methylene chloride (2 x 100 cm3), and the combined organic layers dried (MgSO4), filtered, and
concentrated to dryness in vacuo. Purification of the ensuing residue (0.47 g) by flash column chromatography ( 17 g SiO2; 50% ethyl acetate – hexane to 30% methanol – dichloromethane; gradient elution) gave N-protected dipeptide 5 (0.45 g, 60%) as a white foam in two steps from hydrochloride 2. Dipeptide 5 was shown to be exclusively the frafw-orientated conformer by NMR analysis: Rf 0.50 (20% MeOH – CH2Cl2); [α]D -62.3 (c 0.20 in CH2Cl2); vmax (film)/cm-1 3583, 3324 br, 2980, 2942, 1722, 1649, 1529, 1454, 1432, 1373, 1337, 1251 , 1219, 1179, 1053, 1027, 965, 912, 735 and 698; δH (300 MHz; CDCl3; Me4Si) 1.59 (3H, s, Proα-CH3), 1 .89 (1H, 6 lines, J 18.8, 6.2 and 6.2, Proβ-HAHB), 2.01 (2H, dtt, J 18.7, 6.2 and 6.2, Proγ-H2), 2.25-2.40 (1H, m, Proβ-HAΗΒ), 3.54 (2H, t, J 6.6, Proδ-H2), 3.89 (1H, dd, J 17.1 and 3.9, Glyα-HAHB), 4.04 (1H, dd, J 17.2 and 5.3, Glyα-HAΗΒ), 5.11 (2H, s, OCH2Ph), 5.84 (I H, br t, J 4.2, N-H), 7.22-7.43 (5H, m, Ph) and 7.89 (1 H, br s, -COOH); δC (75 MHz; CDCl3) 21.3 (CH3, Proα-CH3), 23.8 (CH2, Proγ-C), 38.2 (CH2, Proβ-C), 43.6 (CH2, Glyα-C), 47.2 (CH2, Proδ-C), 66.7 (quat, Proα-C), 66.8 (CH2, OCH2Ph), 127.9 (CH, Ph), 127.9 (CH, Ph), 128.4, (CH, Ph), 136.4 (quat., Ph), 156.4 (quat., NCO2), 167.5 (quat., Gly-CON) and 176.7 (quat., CO); m/z (EI+) 320.1368 (M+. C16Η20Ν2Ο5 requires 320.1372).
Dibenzyl N-benzyloxycarbonyl-glycyl-L-2-methylprolyl-L-glutamate 7
Triethylamine (0.50 cm3, 3.59 mmol) was added dropwise to a solution of dipeptide 5 (0.36 g, 1.12 mmol) and L-glutamic acid dibenzyl ester /Moluenesulphonate 6 (0.73 g, 1.46 mmol) in methylene chloride (60 cm3) under nitrogen at room temperature, and the reaction mixture stirred for 10 min. Bis(2-oxo-3-oxazoIidinyl)phosphinic chloride (BoPCl, 97%) (0.37 g, 1.41 mmol) was added and the colourless solution stirred for 17 h. The methylene chloride solution was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and evaporated to dryness in vacuo. Purification of the resultant residue by repeated (2x) flash column chromatography (24 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) yielded ƒully protected tripeptide 7 (0.63 g, 89%) as a colourless oil. Tripeptide 7 was shown to be exclusively the trans-orientated conformer by NMR analysis: Rf 0.55 (EtOAc); [α]D -41.9 (c 0.29 in CH2Cl2); vmax (film)/cm-1 3583, 3353 br, 2950, 1734, 1660, 1521, 1499, 1454, 1429, 1257, 1214, 1188, 1166, 1051, 911, 737 and 697; δH (400 MHz; CDCl3; Me4Si) 1.64 (3H, s, Proot-CH3), 1.72 (1H, dt, J 12.8, 7.6 and 7.6, Proβ-HAHB), 1.92 (2H, 5 lines, J 6.7, Proγ-H2), 2.04 (1H, 6 lines, J 7.3 Gluβ-HAHB), 2.17-2.27 (1H, m, Gluβ-HAΗΒ), 2.35-2.51 (3H, m, Proβ-HAΗΒ and Gluγ-H2), 3.37-3.57 (2H, m, Proδ-H2), 3.90 (1 H, dd, J 17.0 and 3.6, Glyα-HAHB), 4.00 (1H, dd, J 17.1 and 5.1, Glyα-HAΗΒ), 4.56 (1H, td, J 7.7 and 4.9, Glyα-H), 5.05-5.20 (6H, m, 3 x OCH2Ph), 5.66-5.72 (1H, br m, Gly-NH), 7.26-7.37 (15H, m, 3 x Ph) and 7.44 (1H, d, J 7.2, Glu-NH); δC (100 MHz; CDCl3) 21.9 (CH3, Proα-CH3), 23.4 (CH2, Proγ-C), 26.6 (CH2, Gluβ-C), 30.1 (CH2, Gluγ-C), 38.3 (CH2, Proβ-C),
43.9 (CH2, Glyα-C), 47.6 (CH2, Proδ-C), 52.2 (CH, Glua-C), 66.4 (CH2, OCH2Ph), 66.8 (CH2, OCH2Ph), 67.1 (CH2, OCH2Ph), 68.2 (quat, Proα-C), 127.9 (CH, Ph), 128.0 (CH, Ph), 128.1, (CH, Ph), 128.2, (CH, Ph), 128.2, (CH, Ph), 128.3, (CH, Ph), 128.4, (CH, Ph), 128.5, (CH, Ph), 128.5, (CH, Ph), 135.2 (quat., Ph), 135.7 (quat., Ph), 136.4 (quat, Ph), 156.1 (quat, NCO2), 167.3 (quat., Gly-CO), 171.4 (quat., CO), 172.9 (quat., CO) and 173.4 (quat., CO); m/z (FAB+) 630.2809 (MH+. C35H40N3O8 requires 630.2815).
Glycyl-L-2-methylprolyl-L-glutamic acid (G-2-MePE)
A mixture of the protected tripeptide 7 (0.63 g, 1.00 mmol) and 10 wt % palladium on activated carbon (0.32 g, 0.30 mmol) in 91 :9 methanol – water (22 cm3) was stirred under an atmosphere of hydrogen at room temperature, protected from light, for 23 h. The reaction mixture was filtered through a Celite™ pad and the pad washed with 75 :25 methanol – water (200 cm3). The filtrate was concentrated to dryness under reduced pressure and the residue triturated with anhydrous diethyl ether to afford a 38: 1 mixture of G-2-MePE and tentatively methylamine 8 (0.27 g, 86%) as an extremely hygroscopic white solid. Analytical reverse-phase HPLC studies on the mixture [Altech Econosphere C 18 Si column, 150 x 4.6 mm, 5 ☐m; 5 min flush with H2O (0.05% TFA) then steady gradient over 25 min to MeCN as eluent at flow rate of 1 ml/min; detection using diode array] indicated it was a 38: 1 mixture of two eluting peaks with retention times of 13.64 and 14.44 min at 207 and 197 nm, respectively. G-2-MePE was shown to be a 73 :27 trans:cis mixture of conformers by 1H NMR analysis (the ratio was estimated from the relative intensities of the double doublet and triplet at δ 4.18 and 3.71 , assigned to the Gluα-H protons of the major and minor conformers, respectively):
mp 144 °Cɸ;
[ α]D -52.4 (c 0.19 in H2O);
δα (300 MHz; D2O; internal MeOH) 1.52 (3H, s, Proα-CH3), 1.81-2.21 (6H, m, Proβ-H2, Proγ-H, and Gluβ-H2), 2.34 (1.46H, t, J 7.2, Gluy-H2), 2.42* (0.54H, t, 77.3, Gluγ-H2), 3.50-3.66 (2H, m, Pro6-H2), 3.71 * (0.27H, t, J 6.2, Gluoc-H), 3.85 (1H, d, J 16.6, Glyα-HAHB), 3.92 (1H, d, J 16.6, Glyα-HAΗΒ) and 4.18 (0.73H, dd, J 8.4 and 4.7, Glua-H);
δC (75 MHz; D2O; internal MeOH) 21.8 (CH3, Proα-CH3), 25.0 (CH2, Proγ-C), 27.8* (CH2: Gluβ-C), 28.8 (CH2, Gluβ-C), 32.9 (CH2, Gluγ-C), 40.8 (CH2, Proβ-C), 42.7 (CH2, Glyα-C), 49.5 (CH2, Proδ-C), 56.0* (CH, Gluα-C), 56.4 (CH, Gluα-C), 69.8 (quat, Proα-C), 166.5 (quat., Gly-CO), 177.3 (quat., Pro-CON), 179.2 (quat., Gluα-CO), 180.2* (quat., Gluγ-CO) and 180.6 (quat., Gluγ-CO);
m/z (FAB+) 3 16.1508 (MH+. C13H22N3O6 requires 316.1509).
PATENT
WO02094856
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Testing; Material and Methods
The following experimental protocol followed guidelines approved by the
University of Auckland animal ethics committee.
Preparation of cortical astrocyte cultures for harvest of metabolised cell culture supernatant
One cortical hemisphere from a postnatal day 1 rat was used and collected into
4ml of DMEM. Trituration was done with a 5ml glass pipette and subsequently through an 18 gauge needle. Afterwards, the cell suspension was sieved through a lOOμm cell strainer and washed in 50ml DMEM (centrifugation for 5min at 250g). The sediment was resuspended into 20ml DMEM+10% fetal calf serum. 10 Milliliters of suspension was added into each of two 25cm3 flasks and cultivated at 37°C in the presence of 10% C02, with a medium change twice weekly. After cells reached confluence, they were washed three times with PBS and adjusted to Neurobasal/B27 and incubated for another 3 days. This supernatant was frozen for transient storage until usage at -80°C.
Preparation of striatal and cortical tissue from rat E18/E19 embryos
A dam was sacrificed by C02-treatment in a chamber for up to 4 minutes and was prepared then for cesarean section. After surgery, the embryos were removed from their amniotic sacs, decapitated and the heads put on ice in DMEM/F12 medium for striatum and PBS + 0.65% D(+)-glucose for cortex.
Striatal tissue extraction procedure and preparation of cells
Whole brain was removed from the skull with the ventral side facing upside in DMEM/F12 medium. The striatum was dissected out from both hemispheres under a stereomicroscope and the striatal tissue was placed into the Falcon tube on ice.
The collected striatal tissue was triturated by using a PI 000 pipettor in 1ml of volume. The tissue was triturated by gently pipetting the solution up and down into the pipette tip about 15 times, using shearing force on alternate outflows. The tissue pieces settled to the bottom of the Falcon tube within 30 seconds, subsequently the supernatant was transferred to a new sterile Falcon tube on ice. The supernatant contained a suspension of dissociated single cells. The tissue pieces underwent a second trituration to avoid excessively damaging cells already dissociated by over triturating them. 1 Milliliter of ice-cold DMEM/F12 medium was added to the tissue pieces in the first tube and triturated as before. The tissue pieces were allowed to settle and the supernatant was removed to a new sterile Falcon tube on ice. The cells were centrifuged at 250g for 5 minutes at 4°C. The resuspended cell pellet was ready for cell counting.
Plating and cultivation of striatal cells
Striatal cells were plated into Poly-L-Lysine (O.lmg/ml) coated 96-well plates (the inner 60 wells only) at a density of 200,000 cells /cm2 in Neurobasal/B27 medium (Invitrogen). The cells were cultivated in the presence of 5% C02 at 37°C under 100% humidity. Complete medium was changed on days 1, 3 and 6.
Cortical tissue extraction procedure and preparation of cells
The two cortical hemispheres were carefully removed by a spatula from the whole brain with the ventral side facing upside into a PBS +0.65% D(+)-glucose containing petri dish. Forcips were put into the rostral part (near B. olfactorius) of the cortex for fixing the tissue and two lateral – sagittal oriented cuttings were done to remove the paraform and entorhinal cortices. The next cut involved a frontal oriented cut at the posterior end to remove the hippocampal formation. A final frontal cut was done a few millimeters away from the last cut in order to get hold of area 17/18 of the visual cortex.
The collected cortices on ice in PBS+0.65% D(+)-glucose were centrifuged at 350g for 5min. The supernatant was removed and trypsin/EDTA (0.05%/0.53mM) was added for 8min at 37°C. The reaction was stopped by adding an equal amount of DMEM+10%) fetal calf serum. The supernatant was removed by centrifugation followed by two subsequent washes in Neurobasal/B27 medium.
The cells were triturated once with a glass Pasteur pipette in 1 ml of
Neurobasal/B27 medium and subsequently twice by using a 1ml insulin syringe with a 22 gauge needle. The cell suspension was passed through a lOOμm cell strainer and subsequently rinsed by 1ml of Neurobasal B27 medium. Cells were counted and adjusted to 50,000 cells per 60μl.
Plating and cultivation of cortical cells
96-well plates were coated with 0.2mg/ml Poly-L-Lysine and subsequently coated with 2μg/ml laminin in PBS, after which 60μl of cortical astrocyte-conditioned medium was added to each well. Subsequently, 60μl of cortical cell suspension was added. The cells were cultivated in the presence of 10% C02 at 37°C under 100%) humidity. At day 1, there was a complete medium change (1:1- Neurobasal/B27 and astrocyte-conditioned medium) with addition of lμM cytosine-β-D-arabino-furanoside (mitosis inhibitor). On the second day, 2/3 of medium was changed. On day 5, 2/3 of the medium was changed again.
Cerebellar microexplants from P8 animals: preparation, cultivation and fixation
The laminated cerebellar cortices of the two hemispheres were explanted from a P8 rat, cut into small pieces in PBS + 0.65% D(+)glucose solution and triturated by a 23gauge needle and subsequently pressed through a 125 μm pore size sieve. The microexplants that were obtained were centrifuged (60 g) twice (media exchange) into serum-free BSA-supplemented START V-medium (Biochrom). Finally, the
microexplants were reconstituted in 1500 μl STARTV-medium (Biochrom). For cultivation, 40μl of cell suspension was adhered for 3 hours on a Poly-D-Lysine
(O.lmg/ml) coated cover slip placed in 35mm sized 6-well plates in the presence of 5% C02 under 100% humidity at 34°C. Subsequently, 1ml of STARTV-medium was added together with the toxins and drugs. The cultures were monitored (evaluated) after 2-3 days of cultivation in the presence of 5% C02 under 100% humidity. For cell counting analysis, the cultures were fixed in rising concentrations of paraformaldehyde (0.4%, 1.2%, 3% and 4% for 3min each) followed by a wash in PBS.
Toxin and drug administration for cerebellar, cortical and striatal cells: analysis
All toxin and drug administration experiments were designed that 1/100 parts of okadaic acid (30nM and lOOnM concentration and 0.5mM 3-nitropropionic acid for cerebellar microexplants only), GPE (InM -ImM) and G-2Methyl-PE (InM-lmM) were used respectively at 8DIV for cortical cultures and 9DIV for striatal cultures. The incubation time was 24hrs. The survival rate was determined by a colorimetric end-point MTT-assay at 595nm in a multi-well plate reader. For the cerebellar microexplants four windows (field of 0.65 mm2) with highest cell density were chosen and cells displaying neurite outgrowth were counted.
Results
The GPE analogue G-2Methyl-PE exhibited comparable neuroprotective capabilities within all three tested in vitro systems (Figures 12-15).
The cortical cultures responded to higher concentrations of GPE (Figure 12) /or
G-2Methyl-PE (lOμM, Figure 13) with 64% and 59% neuroprotection, respectively.
Whereas the other 2 types of cultures demonstrated neuroprotection at lower doses of G-2Methyl-PE (Figures 14 and 15). The striatal cells demonstrated
neuroprotection within the range of InM to ImM of G-2Methyl-PE (Figure 15) while the postnatal cerebellar microexplants demonstrated neuroprotection with G-2Methyl-PE in the dose range between InM and lOOnM (Figure 14).
While this invention has been described in terms of certain preferred embodiments, it will be apparent to a person of ordinary skill in the art having regard to that knowledge and this disclosure that equivalents of the compounds of this invention may be prepared and administered for the conditions described in this application, and all such equivalents are intended to be included within the claims of this application.
PATENT
WO-2021026066
Composition and kits comprising trofinetide and other related substances. Also claims a process for preparing trofinetide and the dosage form comprising the same. Disclosed to be useful in treating neurodegenerative conditions, autism spectrum disorders and neurodevelopmental disorders.
Trofinetide is a synthetic compound, having a similar core structure to Glycyl-Prolyl-Glutamic acid (or “GPE”). Trofinetide has been found to be useful in treating neurodegenerative conditions and recently has been found to be effective in treating Autism Spectrum disorders and Neurodevelopmental disorders.
Formula (Ila),
Example 1: Trofinetide Manufacturing Process
In general, trofinetide and related compounds can be manufactured from a precursor peptide or amino acid reacted with a silylating or persilylating agent at one or more steps. In the present invention, one can use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group.
Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
Step 1: Preparation of Z-Gly-OSu
Several alternative procedures can be used for this step.
Procedure 1A
One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 27 eq of iPrOH and 4 eq of CH2Cl2 at 21 °C. The mixture was cooled and when the temperature reached -4 °C, 1.1 eq of EDC.HCl was added gradually, keeping the temperature below 10 °C. During the reaction a dense solid appeared. After addition of EDC.HCl, the mixture was allowed to warm to 20 °C. The suspension was cooled to 11 °C and filtered. The cake was washed with 4.9 eq of cold iPrOH and 11 eq of IPE before drying at 34 °C (Z-Gly-OSu dried product -Purity: 99.5%; NMR assay: 96%; Yield: 84%).
Procedure 1B
This Procedure is for a variant of Procedure 1A, and differs by replacing iPrOH with ACN. One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 22 eq of ACN at 35 °C. The mixture was cooled in an ice bath. When the temperature reached 1 °C, 0.9 eq of DCC in 5.5 eq of ACN was added gradually to keep the temperature below 5 °C. The coupling reaction took about 20 hrs. During the reaction, DCU precipitated and was removed by filtration at the end of the coupling. After filtration, DCU was washed with ACN to recover the product. The mixture of Z-Gly-OSu was then concentrated to reach 60% by weight. iPrOH (17 eq) was added to initiate the crystallization. Quickly after iPrOH addition a dense solid appeared. An additional 17 eq of iPrOH was needed to liquify the suspension. The suspension was cooled in an ice bath and filtered. The solid was washed with 9 eq of iPrOH before drying at 45 °C (Z-Gly-OSu dried product – Purity: 99.2%; HPLC assay: 99.6%; Yield: 71%).
Step 2: Preparation of Z-Gly-MePro-OH
Several alternative procedures can be used for this step.
Procedure 2A
One (1) eq of MePro.HCl was partially solubilized in 29 eq of CH2Cl2 at 35 °C with 1.04 eq of TEA and 1.6 eq of TMA. The mixture was heated at 35 °C for 2 hrs to perform the silylation. Then 1.02 eq of Z-Gly-OSu was added to the mixture. The mixture was kept at 35 °C for 3 hrs and then 0.075 eq of butylamine was added to quench the reaction. The mixture was allowed to return to room temperature and mixed for at least 15 min. The Z-Gly-MePro-OH was extracted once with 5% w/w NaHCO3 in 186 eq of water, then three times successively with 5% w/w NaHCO3 in 62 eq of water. The aqueous layers were pooled and the pH was brought to 2.2 by addition of 34 eq of HCl as 12N HCl at room temperature. At this pH, Z-Gly-MePro-OH formed a sticky solid that was solubilized at 45 °C with approximately 33 eq of EtOAc and 2.3 eq of iButOH. Z-Gly-MePro-OH was extracted into the organic layer and washed with 62 eq of demineralized water. The organic layer was then dried by azeotropic distillation with 11.5 eq of EtOAc until the peptide began to precipitate. Cyclohexane (12 eq) was added to the mixture to complete the precipitation. The suspension was cooled at 5 °C for 2 hrs and filtered. The solid was washed with 10 eq of cyclohexane before drying at 45 °C (Z-Gly-MePro-OH dried product – Purity: 100%; HPLC assay: 100%; Yield 79%).
Procedure 2B
This Procedure is for a variant of Procedure 2A. One (1) eq of MePro.HCl was partially solubilized in 36.6 eq of CH2Cl2 at 34 °C with 1.01 eq of TEA and 0.1 eq of TMA. Then 1.05 eq of Z-Gly-OSu was added to the mixture, followed by 1.0 eq of TEA. The mixture was maintained at 35 °C for approximately 1 hr, cooled to 25 to 30 °C and 0.075 eq of DMAPA was added to stop the reaction. One hundred (100) eq of water, 8.6 eq of HCl as 12N HCl and 0.3 eq of KHSO4 were added to the mixture (no precipitation was observed, pH=1.7). Z-Gly-MePro-OH was extracted into the organic layer and washed twice with 97 eq of demineralized water with 0.3 eq of KHSO4, then 100 eq of demineralized water, respectively. EtOAc (23 eq) was added to the mixture and CH2Cl2 was removed by distillation until the peptide began to precipitate. Cyclohexane (25 eq) was added to the mixture to complete the precipitation. The suspension was cooled at -2 °C overnight and filtered. The solid was washed with 21 eq of cyclohexane before drying at 39 °C (Z-Gly-MePro-OH dried product – Purity: 98.7%; NMR assay: 98%; Yield 86%).
Procedure 2C
In reactor 1, MePro.HCl (1 eq) was suspended in EtOAc (about 7 eq). DIPEA (1 eq) and TMA (2 eq) were added, and the mixture heated to dissolve solids. After dissolution, the solution was cooled to 0 °C. In reactor 2, Z-Gly-OH (1 eq) was suspended in EtOAc (about 15 eq). DIPEA (1 eq), and pyridine (1 eq) were added. After mixing, a solution was obtained, and cooled to -5 °C. Piv-Cl (1 eq) was added to reactor 2, and the contents of reactor 1 added to reactor 2. Upon completed addition, the contents of reactor 2 were taken to room temperature. The conversion from Z-Gly-OH to Z-Gly-MePro-OH was monitored by HPLC. When the reaction was complete, the reaction mixture was quenched with DMAPA (0.1 eq), and washed with an aqueous solution comprised of KHSO4, (about 2.5 wt%), NaCl (about 4 wt%), and conc. HCl (about 6 wt%) in 100 eq H2O. The aqueous layer was re-extracted with EtOAc, and the combined organic layers washed with an aqueous solution comprised of KHSO4 (about 2.5 wt%) and NaCl (about 2.5 wt%) in 100 eq H2O, and then with water (100 eq). Residual water was removed from the organic solution of Z-Gly-MePro-OH by vacuum distillation with EtOAc. The resulting suspension was diluted with heptane (about 15 eq) and cooled to 0 °C. The product was isolated by filtration, washed with cold heptane (about 7 eq), and dried under vacuum at 45 °C. Z-Gly-MePro-OH (85% yield) was obtained.
Step 3: Preparation of Z-Gly-MePro-Glu-OH
Several alternative procedures can be used in this step.
Procedure 3A
H-Glu-OH (1.05 eq) was silylated in 2 eq of CH2Cl2 with 3.5 eq of TMA at 65 °C. Silylation was completed after 2 hrs. While the silylation was ongoing, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 24 eq of CH2Cl2 and 1.0 eq of DMA at room temperature in another reactor. EDC.HCl (1.0 eq.) was added. The activation rate reached 97% after 15 min. The activated Oxyma Pure solution, was then added to silylated H-Glu-OH at 40 °C and cooled at room temperature. Coupling duration was approximately 15 min, with a coupling rate of 97%. Addition of 8.2% w/w NaHCO3 in 156 eq of water to the mixture at room temperature (with the emission of CO2) was performed to reach pH 8. Z-Gly-MePro-Glu-OH was extracted in water. The aqueous layer was washed twice with 29 eq of CH2Cl2. Residual CH2Cl2 was removed by concentration. The pH was brought to 2.5 with 2.5N HCl, followed by 1.4 eq of solid KHSO4 to precipitate Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 3 x 52 eq of water. The filtered solid was added to 311 eq of demineralized water and heated to 55-60 °C. iPrOH (29 eq) was added gradually until total solubilization of the product. The mixture was slowly cooled to 10 °C under moderate mixing during 40 min to initiate the crystallization. The peptide was filtered and washed with 2 x 52 eq of water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.5%; NMR assay: 96%; Yield 74%).
Procedure 3B
One (1) eq of Z-Gly-MePro-OH and 1.05 eq of Suc-OH were solubilized in 40 eq of ACN and 30 eq of CH2Cl2 at room temperature. The mixture was cooled in an ice bath, and when the temperature was near 0 °C, 1.05 eq of DCC dissolved in 8 eq of ACN was added gradually, keeping the temperature below 5 °C. After addition of DCC, the mixture was progressively heated from 0 °C to 5 °C over 1 hr, then to 20 °C between 1 to 2 hrs and then to 45 °C between 2 to 5 hrs. After 5 hrs, the mixture was cooled to 5 °C and maintained overnight. The activation rate reached 98% after approximately 24 hrs. DCU was removed by filtration and washed with 13.5 eq of ACN. During the activation step, 1.1 eq of H-Glu-OH was silylated in 30 eq of ACN with 2.64 eq of TMA at 65 °C. Silylation was completed after 2 hrs. Z-Gly-MePro-OSu was then added gradually to the silylated H-Glu-OH at room temperature, with 0.4 eq of TMA added to maintain the solubility of the H-Glu-OH. The mixture was heated to 45 °C and 0.7 eq of TMA was added if precipitation occurred. The coupling duration was about 24 hrs to achieve a coupling rate of approximately 91%. The reaction was quenched by addition of 0.15 eq of butylamine and 2.0 eq of TEA. Water (233 eq) was added and the mixture concentrated until gelation occurred. Z-Gly-MePro-Glu-OH was extracted in water by addition of 5% w/w NaHCO3 in 233 eq of water and 132 eq of CH2Cl2. The aqueous layer was washed twice with 44 eq of CH2Cl2. Residual CH2Cl2 was removed by distillation. The pH was brought to 2.0 with 24 eq of HCl as 12N HCl followed by 75 eq of HCl as 4N HCl. At this pH, Z-Gly-MePro-Glu-OH precipitated. The mixture was cooled in an ice bath over 1 hr and filtered. The solid was washed with 186 eq of cold water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – HPLC Purity: 98.4%; NMR assay: 100%; Yield 55%).
Procedure 3C
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq) was silylated in 3.7 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after approximately 1.5 to 2 hrs, as evidenced by solubilization. During the silylation step, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 31.5 eq of CH2Cl2 at 22 °C. One (1.06) eq of EDC.HCl was added to complete the activation. The silylated H-Glu-OH was then added to the activated Oxyma Pure solution. The temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 153 eq of water and 9 eq of iPrOH to reach a pH of 1.65. Residual CH2Cl2 was removed by concentration. The mixture was cooled to 12 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 90 eq of water before drying at 36 °C.
Procedure 3D
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq.) was silylated in 3.9 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after 2 hrs, as evidenced by Solubilization. During the silylation step, 1 eq of Z-Gly-MePro-OH and 1 eq of Oxyma Pure were solubilized in 25 eq of CH2Cl2 at 23 °C. One (1) eq of EDC.HCl was added. To complete the activation, an additional 0.07 eq of EDC. HCl was added. Silylated H-Glu-OH was then added to the activated Oxyma Pure solution. Temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 160 eq of water and 9.6 eq of iPrOH to reach pH 1.63.
Residual CH2Cl2 was removed by concentration. The mixture was cooled to 20 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 192 eq of water before drying at about 25 °C for 2.5 days. The solid was then solubilized at 64 °C by addition of 55 eq of water and 31 eq of iPrOH. After solubilization, the mixture was diluted with 275 eq of water and cooled to 10 °C for crystallization. The mixture was filtered and the solid was washed with 60 eq of water before drying at 27 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.6%; NMR assay: 98%; Yield 74%).
Procedure 3E
In reactor 1, H-Glu-OH (1.05 eq) was suspended in ACN (about 2.2 eq). TMA (about 3.5 eq) added, and the mixture was heated to dissolve solids. After dissolution, the solution was cooled to room temperature. In reactor 2, Z-Gly-MePro-OH (1 eq) was suspended in ACN (14 eq). Oxyma Pure (1 eq) and EDC.HCl (1 eq) were added. The mixture was stirred at room temperature until the solids dissolved. The contents of reactor 2 were added to reactor 1. The conversion from Z-Gly-MePro-OH to Z-Gly-MePro-Glu-OH was monitored by HPLC. Upon completion the reaction mixture was added to an aqueous solution comprised of KHSO4 (about 2.5 wt%) dissolved in about 100 eq H2O. ACN was removed from the aqueous suspension of Z-Gly-MePro-Glu-OH by vacuum distillation with H2O. After stirring at room temperature, the product in the resulting suspension was isolated by filtration and washed with water. The solid obtained was dissolved in an aqueous solution comprised of NaHCO3 (about 5 wt%) in 110 eq H2O, and recrystallized by addition of an aqueous solution comprised of KHSO4 (about 10 wt%) in 90 eq H2O. The product was isolated by filtration, washed with water, and dried under vacuum at 45 °C. Z-Gly-MePro-Glu-OH (75% yield) was obtained.
Step 4: Deprotection and Isolation of Trofinetide
Several alternative procedures can be used in this step.
Procedure 4A
Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 25 eq) and EtOAc (about 15 eq). Pd/C (0.025 eq by weight and containing 10% Pd by weight) was added, and the reaction mixture hydrogenated by bubbling hydrogen through the reaction mixture at room temperature. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration, and the layers separated. Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained. Alternatively, deprotection can be accomplished using MeOH only, or a combination of iPrOH and MeOH, or by use of ethyl acetate in water.
Procedure 4B
This Procedure is for a variant of Procedure 4A, excluding EtOAc. Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 50 eq). Pd/C (0.05 eq, 5% Pd by weight) was added, and the reaction mixture hydrogenated at room temperature with a pressure of 5 bar. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon
reaction completion the catalyst was removed by filtration, and the aqueous layer washed with EtOAc (about 5 eq). Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained.
Procedure 4C
This Procedure is for a variant of Procedure 4A, replacing EtOAc with MeOH. Z-Gly-MePro-Glu-OH (1 eq) was suspended in MeOH (100 eq) and water (12 eq). Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration and the layers were washed with MeOH and iPrOH. The solvents were concentrated under vacuum at 45 °C, and trofinetide precipitated. The precipitate was filtered and dried at 45 °C to provide trofinetide.
Procedure 4D
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 105 eq of MeOH and 12 eq of water. Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate approximately 99% after 1 hr), the catalyst was filtered off and washed with 20-30 eq of MeOH. iPrOH (93 eq) was added and MeOH was replaced by iPrOH by concentration at 45 °C under vacuum. The peptide was concentrated until it began to precipitate. The peptide was filtered and dried at 45 °C (H-Gly-MePro-Glu-OH dried product: Purity: 98.1%; NMR assay: 90%; Yield 81%).
Procedure 4E
This Procedure is for a variant of Procedure 4A, removing H2O and replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 44 eq of MeOH. Pd/Si type 340 (0.02 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 99.9%, after 3-3.5 hrs), the catalyst was filtered off and washed with 8 eq of MeOH. Deprotected peptide was then precipitated in 56 eq of iPrOH. After 30 min at 5 °C, the peptide was filtered and washed with three times with 11 eq of iPrOH before drying at 25 °C (H-Gly-MePro-Glu-OH dried product: Purity: 99.4%; HPLC assay: ~98%; Yield: 81%).
Procedure 4F
This Procedure is for a variant of Procedure 4A. One (1) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 14 eq of EtOAc and 25 eq of water. Pd/C (0.01 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 100%, after about 3.5 hrs), the catalyst was filtered off and washed with a mixture of 3.5 eq of EtOAc and 6 eq of water. The aqueous layer was then ready for spray-drying (Aqueous H-Gly-MePro-Glu-OH peptide solution: Purity: 98.6%; Yield: ~95%).
Procedure 4G
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si, EtOAc with MeOH, and removing H2O. Pd/Si type 340 (0.02 eq by weight) was added to 2.9 vols of MeOH for pre-reduction during 30 min. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 34 eq of MeOH. The reduced palladium was then transferred to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. Pd/C type 39 (0.007 eq by weight) was added to the mixture to increase reaction kinetics. At the end of the deprotection, the catalyst was filtered off and washed with 13.6 eq of MeOH. The deprotected peptide was then precipitated in 71 eq of iPrOH. After about 40 min, the peptide was filtered and washed with 35 eq of iPrOH. The peptide was dried below 20 °C and was then ready for solubilization in water and spray-drying.
Procedure 4H
This Procedure is for a variant of Procedure 4A. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 24.8 eq of water and 13.6 eq of EtOAc. Pd/C type 39 (0.025 eq by weight) was added to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (19 hrs), the catalyst was removed by filtration and washed with 5.3 eq of water and 2.9 eq of EtOAc. The biphasic mixture was then decanted to remove the upper organic layer. The aqueous layer was diluted with water to reach an H-Gly-MePro-Glu-OH concentration suitable for spray-drying the solution.
Example 2: Alternative Trofinetide Manufacturing Process
An alternative method for synthesis of Trofinetide is based on U.S. Patent No.
8,546,530 adapted for a tripeptide as follows.
The persilylated compounds used to synthesis Formula (Ia) (trofinetide) are obtained by silylating a corresponding peptide or amino acid by reaction with a silylating agent, optionally in an organic solvent. The persilylated peptide or amino acid can be isolated and purified if desired. One can use the persilylated peptide or amino acid in situ, e.g. by combining a solution containing persilylated peptide or amino acid with a solution containing, optionally activated, peptide or amino acid.
In step 2, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-MePro-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid can be isolated and purified if desired. One can use the persilylated amino acid in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated, amino acid (for example, Z-Gly-OH).
In step 3, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-Glu-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid or peptide can be isolated and purified if desired. It is however useful to use the persilylated amino acid or peptide in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated (for example, by using EDC.HCl and Oxyma Pure), peptide (for example, Z-Gly-MePro-OH).
In the present invention, it is useful to use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group. Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
The reaction of step 2 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 40 °C, and optionally from 15 °C to 30 °C.
The reaction of step 3 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 60 °C, optionally from 15 °C to 50 °C.
In the reaction of step 2, generally 0.5 to 5 equivalents, optionally 1 to 3 equivalents, optionally about 1.5 to 2.5 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2 to 4 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible. “Functional groups to be silylated” means particular groups having an active hydrogen atom that can react with the silylating agent such as amino, hydroxyl, mercapto or carboxyl groups.
In the reaction of step 3, generally 0.5 to 5 equivalents, optionally 2 to 4.5 equivalents, optionally about 3 to 4 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2.5 to 4.5 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible.
It is understood that “persilylated” means an amino acid or peptide or amino acid analogue or peptide analogue in which the groups having an active hydrogen atom that can react with the silylating agent are sufficiently silylated to ensure that a homogeneous reaction medium for a coupling step is obtained.
In the process according to the invention, the reaction between the amino acid or peptide and the persilylated amino acid or peptide is often carried out in the presence of a carboxyl group activating agent. In that case the carboxylic activating reagent is suitably selected from carbodiimides, acyl halides, phosphonium salts and uronium or guanidinium salts. More optionally, the carboxylic activating agent is an acyl halide, such as isobutyl chloroformate or pivaloyl chloride or a carbodiimide, such as EDC.HC1 or DCC.
Good results are often obtained when using additional carboxylic activating reagents which reduce side reactions and/or increase reaction efficiency. For example, phosphonium and uronium salts can, in the presence of a tertiary base, for example, N,N-diisopropylethylamine (DIPEA) and triethylamine (TEA), convert protected amino acids into activated species. Other reagents help prevent racemization by providing a protecting reagent. These reagents include carbodiimides (for example, DCC) with an added auxiliary nucleophile (for example, 1-hydroxy-benzo triazole (HOBt), 1-hydroxy-azabenzotriazole (HOAt), or Suc-OH) or derivatives thereof. Another reagent that can be utilized is TBTU. The mixed anhydride method, using isobutyl chloroformate, with or without an added auxiliary nucleophile, is also used, as is the azide method, due to the low racemization associated with it. These types of compounds can also increase the rate of carbodiimide-mediated couplings. Typical additional reagents include also bases such as N,N-diisopropylethylamine (DIPEA), triethylamine (TEA) or N-methylmorpholine (NMM).
When the silylation is carried out in the presence of a solvent, said solvent is optionally a polar organic solvent, more optionally a polar aprotic organic solvent. An amide type solvent such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAC)
can be used. In the present invention for step 2, one can use an alkyl acetate solvent, in particular ethyl acetate is more particularly optional.
In the present invention for step 3, one can use a chlorinated hydrocarbon solvent or alkyl cyanide solvent, in particular dichloromethane or acetonitrile are more particularly optional.
In another embodiment, silylation is carried out in a liquid silylation medium consisting essentially of silylating agent and amino acid or peptide.
In the present invention, amino acid or peptide is understood to denote in particular an amino acid or peptide or amino acid analogue or peptide analogue which is bonded at its N-terminus or optionally another position, to a carboxylic group of an amino protected amino acid or peptide.
Example 3: Specifications for Compositions Containing Compounds of Formula (I)
1 ICH guideline Q3C on impurities: guideline for residual solvents
Example 4: Alternative Manufacturing of Trofinetide Example 1, Step 4, Procedure 4B
This Procedure is for a variant of Step 4, Procedure 4B. Z-Gly-MePro-Glu-OH (1 eq) was added in portions to Pd/C (0.027 eq by weight and containing 5% Pd by weight) in about 50 eq of water. The reaction mixture was hydrogenated at 20 °C at a pressure of 5 bar for at least 4 cycles of 4 hrs each. Pd/C (0.0027 eq by weight) was charged between cycles, as needed, to speed up the reaction. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon reaction completion the catalyst was removed by filtration, washed with water (12.5 eq) and the aqueous layer washed with EtOAc (about 14 eq). After phase separation, residual EtOAc was removed from the aqueous solution containing
trofinetide by sparging with nitrogen under vacuum at 20 °C for about 3 hrs. The aqueous solution was filtered. The final concentration of trofinetide was about 25 wt% and the solution was then ready for spray-drying to isolate the product.
Example 5: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (I)
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (I) on an anhydrous basis.
Example 6: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (Ia) on an anhydrous basis.
Example 7: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, wherein the product comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101
wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 8: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, and additionally comprising one or more compounds selected from the group consisting of Formula (III), Formula (IIIa), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), and Formula (IX), wherein the composition comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101 wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 9: Analysis of Products and Compositions
The products and compositions disclosed herein may be analyzed by liquid chromatography, a suitable chromatographic method using UPLC, e.g. using materials and conditions such as Waters Acquity CSH C18, 1.7 µm, 150 x 2.1 mm column, water with 0.1 % TFA (mobile phase A), and water/ACN 70/30 + 0.1 % TFA (mobile phase B), ranging from (4% phase A/6% phase B to 100% phase B and flushed with 4% phase A/6% phase B).
Flow rate: 0.35 ml/min, Column temperature: 40 °C, autosampler temperature: 4 °C, injection volume: 4 ml (e.g. prepared by weighing about 10 mg of powder in a 10 ml volumetric flask and diluted to volume with water). Examples of detectors are UV (ultraviolet, UV 220 nm) and MS (mass spectrometry).
INDUSTRIAL APPLICABILITY
This invention finds use in the pharmaceutical, medical, and other health care fields.
PATENT
WO2014085480 ,
claiming use of trofinetide for treating autism spectrum disorders including autism, Fragile X Syndrome or Rett Syndrome.
EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro- Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
W095/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS for increasing TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses GPE analogs capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including but not limited to, injury or disease in the CNS.
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
PAPER
Tetrahedron (2005), 61(42), 10018-10035. (CLICK HERE)
The synthesis of ten proline-modified analogues of the neuroprotective tripeptide GPE is described. Five of the analogues incorporate a proline residue with a hydrophobic group at C-2 and two further analogues have this side chain locked into a spirolactam ring system. The pyrrolidine ring was also modified by replacing the γ-CH2 group with sulfur and/or incorporation of two methyl groups at C-5.
Graphical Abstract

PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(9), 2279-2283
A series of GPE analogues, including modifications at the Pro and/or Glu residues, was prepared and evaluated for their NMDA binding and neuroprotective effects. Main results suggest that the pyrrolidine ring puckering of the Pro residue plays a key role in the biological responses, while the preference for cis or trans rotamers around the Gly-Pro peptide bond is not important.
Graphical abstract
A series of Pro and/or Glu modified GPE analogues is described. Compounds incorporating PMe and dmP showed higher affinity for glutamate receptors than GPE and neuroprotective effects similar to those of this endogenous tripeptide in culture hippocampal neurons exposed to NMDA.

PATENT
US 20060251649
WO 2006127702
US 20070004641
US 20080145335
WO 2012102832
WO 2014085480
US 20140147491
References
- ^ Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. (March 2009). “NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke”. Journal of the Neurological Sciences. 278 (1–2): 85–90. doi:10.1016/j.jns.2008.12.003. PMID 19157421. S2CID 7789415.
- ^ Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, Schmid KE (September 2013). “Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3”. Neuromolecular Medicine. 15 (3): 504–14. doi:10.1007/s12017-013-8236-z. PMID 23765588. S2CID 12522580.
- ^ Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, Cogram P (March 2015). “NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome”. Neuromolecular Medicine. 17 (1): 71–82. doi:10.1007/s12017-015-8341-2. PMID 25613838. S2CID 11964380.
- ^ Study Details – Rett Syndrome Study
- ^ Neuren’s trofinetide successful in Phase 2 clinical trial in Fragile X
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Rett’s Syndrome | 1 |
| 3 | Recruiting | Treatment | Rett’s Syndrome | 1 |
| 2 | Completed | Supportive Care | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Fragile X Syndrome (FXS) | 1 |
| 2 | Completed | Treatment | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Rett’s Syndrome | 2 |
| 2 | Terminated | Treatment | Concussions | 1 |
| 1 | Completed | Treatment | Brain Injuries,Traumatic | 2 |
| Legal status | |
|---|---|
| Legal status | US: Investigational New Drug |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 853400-76-7 |
| PubChem CID | 11318905 |
| ChemSpider | 9493869 |
| UNII | Z2ME8F52QL |
| Chemical and physical data | |
| Formula | C13H21N3O6 |
| Molar mass | 315.322 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@]1(CCCN1C(=O)CN)C(=O)N[C@@H](CCC(=O)O)C(=O)O | |
| InChI[hide]InChI=1S/C13H21N3O6/c1-13(5-2-6-16(13)9(17)7-14)12(22)15-8(11(20)21)3-4-10(18)19/h8H,2-7,14H2,1H3,(H,15,22)(H,18,19)(H,20,21)/t8-,13-/m0/s1Key:BUSXWGRAOZQTEY-SDBXPKJASA-N |
////////////Tofinetide , NNZ 2566, PHASE 2, PHASE 3. NEUREN, Amino Acids, Peptides, Proteins,
CC1(CCCN1C(=O)CN)C(=O)NC(CCC(=O)O)C(=O)O
J. Med. Chem. 2025, 68, 2147−2182
Trofinetide (Daybue). Trofinetide (8) was developed by Neuren Pharmaceuticals and Acadia Pharmaceuticals for the treatment of rare childhood neurodevelopmental disorders and
was approved by the USFDA in March 2023 for adults and pediatric patients two years of age or older with Rett syndrome. 59 In most cases, Rett syndrome is caused by loss of-function mutations in the X-linked gene that encodes methyl CpG-binding protein 2 (MeCP2). 60,61 MeCP2 is a critical transcriptional regulator required for normal neurological development. In the past, treatment of Rett syndrome has
been limited to symptom management based on knowledge from treating other conditions, 62
but new research has focused on targeting the underlying genetic cause and finding agents to
restore MeCP2 function. Trofinetide is an orally available synthetic analog of glycine-proline-glutamate (GPE), the Nterminal tripeptide metabolite of insulin like growth factor-1 (IGF-1). GPE has been shown to partially reverse Rett-like symptoms in MeCP2 deficient mouse models and trofenitide was developed to have an improved pharmakokinetic profile to GPE. 64 Its use has shown significant improvement over placebo in clinical trials.
Anumberofaccountsrelated to the preparation of trofinetide have been reported with various protecting group strategies, but they are moreamenabletosmall-scaleproductionduetoreagent selection and challenging isolations requiring column chromatography.65−67 A commercially viable synthesis of the drug has been described by researchers at Neuren Pharmaceuticals and is depicted in Scheme 12.68
This synthesis takes advantage of in situ silyl protection/deprotection during its amidation steps to
avoid lengthy protecting group manipulations. Activation of Cbz protected glycine 8.1 with hydroxysuccinimide 8.2 using EDCI provided 8.3 in 84% yield as a direct drop crystallization
(isolated by crystallization directly from the reaction mixture). This activated ester was then coupled with commercially available methyl proline 8.4 69 by first silylation of the carboxylic acid of the proline analog in situ with 8.5, then adding 8.3 to couple with the amine. Deprotection of the silyl ester during the
workup provided amide 8.6 in 79% yield. In a similar sequence, 8.6 was activated with Oxyma Pure and EDCI while in a second vessel 8.7 was silyl protected using 8.5. Subsequent combination of these streams followed by workup and crystallization gave amide 8.8 in good yield. Finally, Cbz deprotection was
accomplished using Pd/C and hydrogen. Trofinetide (8) was extracted into the aqueous layer and isolated by spray drying in 90% yield.
(60) Kyle, S. M.; Vashi, N.; Justice, M. J. Rett syndrome: a
neurological disorder with metabolic components. Open Biol. 2018,
8, No. 170216.
(61) Collins, B. E.; Neul, J. L. Rett syndrome and MECP2 duplication
syndrome: disorders of MeCP2 dosage. Neuropsychiatr. Dis. Treat.
2022, 18, 2813−2835.
(62) Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt,
R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus
guidelines on managing Rett syndrome across the lifespan. BMJ
Paediatr. Open 2020, 4, No. e000717.
(63) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D.G.;Marsh,E.D.;Lin,T.;Stankovic,S.;Bishop,K. M.;Youakim,J.M.
Trofinetide for the treatment of Rett syndrome: a randomized phase 3
study. Nat. Med. 2023, 29, 1468−1475.
(64) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D. G.; Peters, S. U.; Jones, N. E.; Youakim, J. M. Design and outcome
measures of LAVENDER, a phase 3 study of trofinetide for Rett
syndrome. Contemp. Clin. Trials 2022, 114, No. 106704.
(65) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl-l-2-methylprolyl-l-glutamic acid. US
20140147491, 2014.
(66) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl L2012102832, 2012.-2-methylprolyl
L-glutamic acid. WO 2012102832, 2012
(67) Brimble, M.A.;Harris, P. W.R.;Sieg, F. Preparation of analogsof
glycyl-prolyl-glutamate as neuroprotective agents. US 20080145335,
2008
(68) Blower, C.; Peterson, M.; Shaw, J. M.; Bonnar, J. A.; Moniotte, E.
D. F. P.; Bousmanne, M. B. C.; Betti, C.; Decroos, K. W. L.; Ayoub, M.
Compositions of trofinetide. WO 2021026066, 2021.
(69) Beck, A. K.; Blank, S.; Job, K.; Seebach, D.; Sommerfeld, T.
Synthesis of (S)-2-methylproline: a general method for the preparation
of α-branched amino acids (L.-proline, 2-methyl-). Org. Synth. 1995, 72, 62−73

.
Ritlecitinib, PF 06651600

Ritlecitinib
PF-06651600
CAS 1792180-81-4
C₁₅H₁₉N₅O, 285.34, UNII-2OYE00PC25
Fda approved Litfulo, 6/23/2023, To treat severely patchy hair loss
Drug Trials Snapshot
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one

1-[(2S,5R)-2-Methyl-5-(7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-1-piperidinyl]-2-propen-1-one malonate
PF-06651600 malonate
CAS: 2140301-97-7 (malonate)
Chemical Formula: C18H23N5O5
Molecular Weight: 389.412
PHASE 2 alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis.
Ritlecitinib, sold under the brand name Litfulo, is a medication used for the treatment of severe alopecia areata (hair loss).[6] Ritlecitinib is a kinase inhibitor which inhibits Janus kinase 3 and tyrosine kinase.[6][9][10]
The most common side effects include headache, diarrhea, acne, rashes, eczema, fever, mouth ulcers, dizziness, shingles rash, and abnormal findings in some laboratory test results.[11]
Ritlecitinib was approved for medical use in the United States in June 2023,[6][11][12] in the European Union in September 2023,[7] and in Canada in November 2023.[4]
Pfizer is developing ritlecitinib, an irreversible, covalent and selective dual JAK3/TEC inhibitor, for treating AA, RA, vitiligo and inflammatory bowel diseases, including UC and CD. In July 2021, this drug was reported to be in phase 3 clinical development.
PF-06651600 is a potent and selective JAK3 inhibitor. PF-06651600 is a potent and low clearance compound with demonstrated in vivo efficacy. The favorable efficacy and safety profile of this JAK3-specific inhibitor PF-06651600 led to its evaluation in several human clinical studies. JAK3 was among the first of the JAKs targeted for therapeutic intervention due to the strong validation provided by human SCID patients displaying JAK3 deficiencies
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitors in development as potential medicines. PF-06651600 is a highly selective and orally bioavailable Janus Kinase 3 (JAK3) inhibitor that represents a potential immunomodulatory therapy. With the favorable efficacy, safety profile, and ADME properties, this JAK3-specific covalent inhibitor has been under clinical investigation for the treatment of alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis. Supported by positive results from a Phase 2 study, 1 was granted Breakthrough Therapy designation by the FDA on Sept. 5, 2018 for treatment of alopecia areata.
SYN

PAPER
J. Med. Chem. 2017, 60 (5), 1971–1993, DOI: 10.1021/acs.jmedchem.6b01694
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b01694
Paper
Process Development and Scale Up of a Selective JAK3 Covalent Inhibitor PF-06651600,
Yong Tao*

A scalable process for PF-06651600 (1) has been developed through successful enabling of the first generation syntheis. The synthesis highlights include the following: (1) replacement of costly PtO2 with a less expensive 5% Rh/C catalyst for a pyridine hydrogenation, (2) identification of a diasteroemeric salt crystallization to isolate the enantiomerically pure cis-isomer directly from a racemic mixture of cis/trans isomers, (3) a high yielding amidation via Schotten–Baumann conditions, and (4) critical development of a reproducible crystallization procedure for a stable crystalline salt (1·TsOH), which is suitable for long-term storage and tablet formulation. All chromatographic purifications, including two chiral SFC chromatographic separations, were eliminated. Combined with other improvements in each step of the synthesis, the overall yield was increased from 5% to 14%. Several multikilogram batches of the API have been delivered to support clinical studies.
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00198
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one p-Toluenesulfonate (1·TsOH)
1·TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. Mp 199 °C. Rotomers observed for NMR spectroscopies. 1H NMR (400 MHz, DMSO-d6): δ ppm 12.68 (brs, 1H), 9.22 (brs, 1H), 8.40 (s, 1H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1.2 Hz, 1H), 6.84 (m, 1H), 6.13 (m, 1H), 5.70 (m, 1H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1.91–1.72 (m, 4H), 1.24–1.17 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51, 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41, 25.22, 21.28, 16.97, 15.51. HRMS (ESI) m/z: calculated for C15H20N5O [M + H]+286.1668; observed 286.1692.
PAPER
PATENT
WO2015083028
PATENT
WO 2015083028
https://patents.google.com/patent/WO2015083028A1
PATENT
WO 2020084435
1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one has the structural formula:
The synthesis of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is described in WO2015/083028, commonly assigned to the assignee of the present invention and which is incorporated herein by reference in its entirety. 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is useful as an inhibitor of protein kinases, such as the enzyme Janus Kinase (JAK) and as such is useful therapy as an immunosuppressive agent for organ transplants, xeno transplantation, lupus, multiple sclerosis, rheumatoid arthritis, psoriasis, Type I diabetes and complications from diabetes, cancer, asthma, atopic dermatitis, autoimmune thyroid disorders, ulcerative colitis, Crohn’s disease, alopecia, vitiligo, Alzheimer’s disease, leukemia and other indications where immunosuppression would be desirable. See ACS Chem. Biol. , 2016, 11 (12), pp 3442-3451 . The present invention relates to a novel p-toluenesulfonic acid salt and crystalline solid form of the said salt of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one that demonstrate improved properties for use in a pharmaceutical dosage form, particularly for oral dosage forms.
Preparations
Scheme 1. Synthesis of 1
Scheme 2. Alternate Synthesis of Intermediates 7 and 10
1. K2C03, MIBK/water 1. H2, H2O
2. EtOAc, aq. NaCI Pd(OH)2/C (wet)
3. MeOH, H20 2. NaOH, MeOH
7 + 8 – ► 9 – – 10 . H2O
89% 89%
Scheme 3. First Alternate Preparation of 1
Scheme 4. Second Alternate Preparation of 1
Preparation 1
ferf-Butyl (6-methylpyridin-3-yl)carbamate (3). To a 3000L reactor was charged 2 (72.00 kg, 665.8 mol) and THF (660 kg). A solution of NH4CI (1 .07 kg , 20 mol) in water (72 kg, 4000 mol) was added. The mixture was heated to 57 °C and Di-f-butyl dicarbonate (220.0 kg, 1003 mol) was added slowly with rinse of THF (45 kg) while maintaining the temperature between 55 – 60 °C. The mixture was stirred at 55 – 60 °C for 10 h. Upon reaction completion, the slurry was cooled to 20 °C and ethyl acetate (654 kg) and water (367 kg) were added. The organic phase was separated, washed by water (2 x 360 kg) and stirred with active carbon (22 kg) for 5 h. The mixture was filtered through a layer of diatomaceous earth (22 kg) with THF rinse and the filtrates were concentrated under vacuum at <40 °C to a residual volume of ~370 L. n-Heptane (500 kg) was added slowly over 1 h and the resulting slurry was cooled to 20 °C and stirred for 2 h. The solid was collected by centrifuge with an n-heptane wash (420 kg), then dried at 45 °C under vacuum for 20 h to give 3 (131 .15 kg, 629.7 mol) as a white powder in 94.5% yield. HPLC purity: 99.9%. 1H NMR (400 MHz, DMSO-c/6): d ppm 9.42 (brs, 1 H), 8.48 (d, J = 1 .9 Hz, 1 H), 7.75 (d, J = 8.6 Hz, 1 H), 7.13 (d, J = 8.6 Hz, 1 H), 2.38 (s, 3H), 1 .49 (s, 9H). 13C NMR (100 MHz, DMSO-d6y d ppm 153.34, 151 .56, 139.75, 134.13, 126.10, 123.09, 79.87, 28.56, 23.70. HRMS (ESI) m/z: calculated for C11H17N2O2 [M + H]+ 209.1290; observed 209.1285.
Preparation 2
ferf-Butyl (6-methylpiperidin-3-yl)carbamate (rac-4). To a 3000L reactor was charged 3 (137.0 kg, 667.8 mol), ethanol (988 kg) and acetic acid (139 kg). The reactor was purged with nitrogen three times and 5 wt% Rhodium on carbon (wet, 27.4 kg, 20 wt% loading relative to 3) was added. The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 47 °C. The mixture was stirred at 45 – 60 °C under hydrogen pressure at 0.34 – 0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and flushed with nitrogen. The mixture was filtered through a layer of diatomaceous earth (20 kg) with an ethanol rinse (1320 kg) and the filtrates were concentrated under vacuum at <50 °C to a residual volume of ~350 L. n-Heptane (571 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of~350 L. This operation was repeated twice until the residual acetic acid <8.0%. Ethanol (672 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of ~350 L. This operation was repeated twice until the residual n-heptane was <0.2% and water was <0.2%. Ethanol (889 kg) was added and the solution (1254 kg) was transferred to drums for use in the subsequent classical resolution step. Achiral HPLC assay indicated that the solution contained 10.8 wt% of the total reduced product (rac-4) in 96% mass recovery and chiral SFC showed that the solution contained 36.3% of the desired stereoisomer cis-4.
Preparation 3
ferf-Butyl ((3R,6S)-6-methylpiperidin-3-yl)carbamate (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid salt (15). To a 2000L reactor (R1 ) was charged rac-4 as a 10.8 wt% solution in ethanol (620.5 kg, ~312.7 mol. of all 4 isomers). The solution was concentrated under vacuum at <45 °C to a residual volume of ~210 L and then cooled to 20 °C. To a 3000 L reactor (R2) was charged (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid 14 (47.0 kg, 136.1 mol) and ethanol (1 125 kg). With high speed agitation, reactor R2 was heated to 70 °C, stirred at 68 – 70 °C for ~2 h to dissolve all solid 14, and then seeded with crystalline 15 (1 1 g). The solution containing 4 in reactor R1 was slowly transferred to reactor R2 over 30 min with ethanol rinse (160 kg). Reactor R2 was stirred at ~74 °C for 3 h and then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 16 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg). The wet cake (with 97.1 % e.e.) was charged back to reactor R2. The slurry was heated to 74 °C and the mixture was stirred for 17 h. The mixture was then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 4 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg) and dried at 35 °C under vacuum for 25 h to give 15 (56.05 kg, 100.2 mol) as a white powder in 30.7% yield over 2 steps. Chiral HPLC purity: 99.1 %. 1H NMR (400 MHz, DMSO-d6): d ppm 9.46 (d, J = 7.0 Hz, 1 H), 9.07 (d, J = 2.2 Hz, 2H), 8.96 (t, J = 2.2 Hz, 1 H), 7.49 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.3 Hz, 2H), 7.23 (t, J = 7.3, 1 H), 7.1 1 (m, 1 H), 5.31 (d, J = 7.0 Hz, 1 H), 3.66 (m, 1 H), 2.98 (m, 3H), 1 .63 (m, 2H), 1 .45 (m, 2H), 1 .40 (s, 9H), 1 .1 1 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 172.71 , 161 .71 , 155.42, 148.51 , 141 .27, 137.70, 128.29, 128.25, 128.02, 127.05, 121 .12, 78.49, 59.74, 50.66, 46.29, 43.34, 28.66, 26.88, 26.1 1 , 18.60.
Preparation 4
Benzyl (2S,5R)-5-amino-2-methylpiperidine-1 -carboxylate hydrochloride (7»HCI) -telescoped process. To a 2000L reactor was charged 15 (70.0 kg, 125 mol) and MTBE (500 kg). The mixture was cooled to 12 °C and 6.9 wt% aqueous NaOH solution (378 kg, 652 mol) was added slowly while maintaining the temperature between 10 – 25 °C. The mixture was stirred at 18 °C for 1 h . The organic phase was separated and washed with 3.8 wt% aqueous NaOH solution (2 x 221 kg) and then 25 wt% aqueous NaCI solution (2 x 220 kg). The organic layer (containing the free base cis-4) was concentrated under vacuum at <40 °C to a residual volume of ~300 L and then cooled to 20 °C. NaHCOs (53 kg, 632 mol) and water (200 kg) were added and the mixture was cooled to 7 °C. Benzyl chloroformate (32.30 kg, 189.3 mol) was added slowly while maintaining the temperature between 5 – 20 °C. The mixture was stirred at 17 °C for 20 h. Upon reaction completion, the mixture was cooled to 12 °C, 25 wt% aqueous ammonium hydroxide solution (79 kg, 1 160 mol) was added slowly while maintaining the temperature between 10 – 20 °C, and the mixture was stirred at 15 °C for 1 h. The organic phase was separated and washed with 25 wt% aqueous NaCI solution (3 x 90 kg). The organic layer (containing 5) was concentrated under vacuum at <45 °C to a residual volume of ~150 L. Isopropyl acetate (310 kg) was added and the mixture was concentrated under vacuum at <45 °C to a residual volume of ~150 L. This operation was repeated twice to meetthe criteria of water <0.1 % (by KF). Isopropyl acetate (130 kg) was then added and the mixture was cooled to -3 °C. 4-5N HCI in methanol (181 kg, ~730 mol) was added slowly while maintaining the temperature between -5 to 5 °C, and the mixture was stirred at 3 °C for 12 h. Upon reaction completion, the mixture was cooled to -3 °C and MTBE (940 kg) was added slowly while maintaining the temperature between -5 to 5 °C. The resulting slurry was stirred at 3 °C for 3 h. The solid was collected by centrifuge with MTBE washes (4 x 70 kg), and then dried at 45 °C under vacuum for 20 h to give 7»HCI (28.60 kg, 100.4 mol) as a white powder in 80.3% yield. Achiral HPLC purity: 100%. Chiral SFC purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 8.36 (brs, 3H), 7.37 (m, 5H), 5.09 (s, 2H), 4.31 (m, 1 H), 4.16 (d, J = 8.2 Hz, 1 H), 3.00 (m, 2H), 1 .82 (m, 2H), 1 .59 (m, 2H), 1 .1 1 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 154.71 , 137.24, 128.92, 128.34, 128.00, 66.89, 47.20, 45.66, 40.68, 28.16, 23.02, 15.67. HRMS (ESI) m/z. calculated for C H N O [M + H]+ 249.1603; observed 249.1598.
Preparation 5
Benzyl (2S, 5R)-5-((2-chloro-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2 -methyl-piperidine-1 -carboxylate (9). To a 2000L reactor was charged 7»HCI (88.6 kg, 31 1 .12 mol), 8 (56.0 kg, 298 mol), K2C03 (133.0 kg, 962.3 mol), water (570 kg) and MIBK (101 kg). The mixture was heated to 90 °C and stirred at this temperature for 22 h. Upon reaction completion, the mixture was cooled to 56 °C and ethyl acetate (531 kg) was added. After cooling the mixture to 22 °C, the organic phase was separated, washed with water (570 kg) and concentrated under vacuum at <40 °C to a residual volume of ~220 L. Methanol (360 kg) was added slowly over a period of 1 h and the mixture concentrated under vacuum at <50 °C to a residual volume of ~220 L. This operation was repeated three times until residual MIBK reached <5 wt%. Methanol (270 kg) was added, followed by seeding with 9 (120 g). The mixture was stirred at 22 °C for >4 h and water (286 kg) was added slowly over 4 h. The slurry was stirred for 10 h and the solid was then collected by centrifuge. The wet cake (165.6 kg) was charged back to a clean reactor and water (896 kg) was added. The slurry was heated to 55 °C and stirred at this temperature for 7 h; and then cooled to 22 °C and stirred at this temperature for 2 h. The solid was collected by centrifuge with water wash (3 x 170 kg) and dried at 55 °C under vacuum for 20 h to give 9 (106.62 kg, 266.6 mol) as a white powder in 89.5% yield. Achiral HPLC purity: 99.7%. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .71 (brs, 1 H), 7.72 (d, J = 7.9 Hz, 1 H), 7.38 (m, 5H), 7.10 (s, 1 H), 6.57 (d, J = 2.7 Hz, 1 H), 5.1 1 (m, 2H), 4.39 (m, 1 H), 4.17 (m, 1 H), 4.01 (m, 1 H), 3.36 (s, 2H), 2.77 (m, 1 H), 1 .73-1 .81 (m, 4H), 1 .16 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 156.65, 154.74, 153.04, 151 .31 , 137.43, 128.89, 128.27, 127.96, 122.13, 101 .65, 99.51 , 66.75, 49.10, 47.32, 45.64, 42.98, 29.05, 25.08. HRMS (ESI) m/z. calculated for C20H22CIN5O2 [M + H]+ 400.1540; observed 400.1535.
Preparation 6
N-((3R,6S)-6-methylpiperidin-3-yl)-7H-pyrrolo[2,3-dlpyrimidin-4-amine monohydrate (10·H2O) To a 1600L reactor was charged water (570 kg). The reactor was purged with nitrogen three times. 10% Pd(OH)2/C (wet, 3.2 kg) and 9 (53.34 kg, 133.2 mol) were added with water rinses (2 x 55 kg). The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 77 °C. The mixture was stirred at 75 – 80 °C under a hydrogen pressure of 0.34 -0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and purged with nitrogen. The mixture was filtered through a layer of diatomaceous earth (8 kg) with a water rinse (460 kg), and the filtrates were transferred to a 3000L reactor. Methanol (260 kg) was added, followed by slow addition of 50 wt% aqueous sodium hydroxide (12.0 kg , 150 mol) while maintaining the temperature between 15 – 25 °C. The slurry was heated to 55 °C and stirred for 2 h; then cooled to 22 °C and stirred for 10 h. The solid was collected by centrifuge with a 10:1 water/methanol wash (3 x 1 10 kg) and then dried at 55 °C under vacuum for 20 h to give 10·H2O (30.90 kg, 266.6 mol) as a white powder in 89.1 % yield. Achiral HPLC purity: 99.7%. Chiral SFC
purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .48 (brs, 1 H), 8.08 (s, 1 H), 7.07 (s, 1 H), 6.85 (d, J = 7.3 Hz, 1 H), 6.64 (s, 1 H), 4.16 (m, 1 H), 3.35 (brs, 2H), 2.96 (d, J = 12.7 Hz, 1 H), 2.82 (d, J = 12.7 Hz, 1 H), 2.67 (m, 1 H), 2.04 (brs, 1 H), 1 .92 (m, 1 H), 1 .63 (m, 1 H), 1 .44 (m, 1 H), 1 .33 (m, 1 H), 1 .03 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 155.95, 151 .87, 150.74, 121 .20, 102.97, 99.20, 51 .27, 49.94, 44.78, 29.97, 28.69, 22.35. HRMS (ESI) m/z\ calculated for C12H17N5 [M + H]+ 232.1562; observed 232.1558.
Preparation 7
1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (1 ). To a 100L reactor was charged water (18.0 L), 10·H2q (3.60 kg, 14.4 mol) and THF (36.0 L). The mixture was heated to 53 °C and stirred for 15 min to dissolve all the solids. The solution was then cooled to 18 °C and K3PO4 (6.38 kg, 30.1 mol) was added. The mixture was stirred at 18 °C for 10 min to dissolve all the solids, and then cooled to 10 °C. 3-Chloropropionyl chloride (2.20 kg, 17.3 mol) was added while maintaining the temperature <20 °C. The mixture was then stirred at 20 °C for 2 h. Upon reaction completion, 2 N aqueous NaOH solution (23.50 kg, 43.76 mol) was added while maintaining the temperature <25 °C. The mixture was stirred at 22 °C for >12 h until the elimination reaction was complete (11 <0.2%). KH2PO4 (10.32 kg, 75.8 mol) was added and the mixture was stirred at 20 °C for 10 min. The organic phase was separated and then washed with 23.5 wt% aqueous NaCI solution (2 x 8.5 kg). The isolated organic phase was concentrated under vacuum at <30 °C to a residual volume of ~10 L, whereupon MEK (39.6 L) was added. This operation was repeated once or twice until residual THF was <1 % and water was <2%. MgS04 (0.96 kg), Silica gel (4.90 kg) and Darco™ G-60 (0.48 kg) were added to the MEK solution, and the mixture was stirred at 20 °C for 1 h, then filtered through a layer of Diatomaceous Earth with a MEK rinse (76 L). The combined filtrates were concentrated under vacuum at <30 °C to a residual volume of ~8 L. The concentration of the residual solution was measured by qNMR, and the solution was transferred to a container with a rinse using the calculated amount of MEK to adjust the final concentration to 30 wt%. Thus, a 30 wt% solution of 1 in MEK (1 1 .09 kg, 1 1 .66 mol of 1) with 98.7% purity was obtained in 81 % yield, which was stored in a cold room (2 – 8 °C) for the next step.
Preparation 8
1 -((2S,5R)-5-((7H-pyrrolo[2,3-cdpyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one p-toluenesulfonate (1»TsOH). To a 20L reactor was charged a 30 wt% solution of 1 in MEK (9.80 kg, 10.30 mol of 1) and silica gel (0.74 kg). The mixture was stirred at 22 °C for 15 min and filtered through a 0.45 micron Teflon cartridge filter with a MEK rinse (7.89 kg, 9.8 L), collecting in a 100L reactor. Water (1 .27 L) was added, followed by a solution of p-toluenesulfonic acid monohydrate (2.18 kg, 1 1 .3 mol) in MEK (4.75 kg, 5.9 L) with a MEK rinse (3.14 kg, 3.9 L), followed by the addition of 1 »TsOH seed (188 g, 0.41 mol). The mixture was stirred at 22 °C for
4 h to form a slurry and MEK (31 .56 kg, 39.2 L) was added slowly over a period of 3 h. The slurry was stirred at 22 °C for an additional 2 h and then filtered. The cake was washed with MEK (4.02 kg, 5 L) and then dried at 50 °C under vacuum for 10 h to give 1 »TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. m.p. 199 °C. Rotomers observed for NMR spectroscopies. Ή NMR (400 MHz, DMSO-d6): d ppm 12.68 (brs, 1 H), 9.22 (brs, 1 H), 8.40 (s, 1 H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1 H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1 .2 Hz, 1 H), 6.84 (m, 1 H), 6.13 (m, 1 H), 5.70 (m, 1 H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1 H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1 .91 -1 .72 (m, 4H), 1 .24-1 .17 (m, 3H). 13C NMR (100 MHz, DMSO-c/6): d ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51 , 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41 , 25.22, 21 .28, 16.97, 15.51 . HRMS (ESI) m/z: calculated for Ci5H2oN50 [M + H]+ 286.1668; observed 286.1692.
Comparative Example
Preparation of 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methyl-piperidin-1 -yl)prop-2-en-1 -one Malonic Acid Salt (Form 1 )
A 250 ml_ round bottom flask was charged with 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (4.10 g, 14.4 mmol), MEK (Methyl Ethyl Ketone (15.0 ml_/g, 687 mmol, 49.5 g, 61 .5 ml_)). To the solution, malonic acid (0.950 equiv. 13.7 mmol, 1 .42 g) was added in one portion. The mixture was heated to 50 °C and stirred at 50 °C for 15min. The heating was turned off and the slurry was stirred for 16 hours. The resulting white slurry was filtered. The filter cake was washed with MEK (2 X 5 ml_) and dried in a vacuum oven (40 °C) for 2 hours give 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one malonic acid salt (Form 1) (4.48 g, 1 1 .5 mmol, 4.48 g, 80.1 % Yield) as white powder.
PATENT
WO-2021136482
Improved process for preparing ritlecitinib (PF06651600), useful for treating alopecia areata (AA), rheumatoid arthritis (RA), Crohn’s disease (CD) and ulcerative colitis (UC). Also claims novel intermediates of ritlecitinib and their preparation method.
PATENT
CN111732591
crystalline polymorphic form of ritlecitinib L-tartrate. Jiangsu Alicorn focuses on developing generic, innovative and first-line drugs, whose website lists an undisclosed JAK inhibitor under its product’s list.
REFERENCES
1: D’Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018 Jul;27(7):595-599. doi: 10.1080/13543784.2018.1492547. Epub 2018 Jul 6. Review. PubMed PMID: 29938545.
2: Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, Gilfillan S, Lin LL, Notarangelo LD, Colonna M. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. 2018 Jan;11(1):50-60. doi: 10.1038/mi.2017.38. Epub 2017 May 17. PubMed PMID: 28513593; PubMed Central PMCID: PMC5693788.
3: Thorarensen A, Dowty ME, Banker ME, Juba B, Jussif J, Lin T, Vincent F, Czerwinski RM, Casimiro-Garcia A, Unwalla R, Trujillo JI, Liang S, Balbo P, Che Y, Gilbert AM, Brown MF, Hayward M, Montgomery J, Leung L, Yang X, Soucy S, Hegen M, Coe J, Langille J, Vajdos F, Chrencik J, Telliez JB. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop -2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J Med Chem. 2017 Mar 9;60(5):1971-1993. doi: 10.1021/acs.jmedchem.6b01694. Epub 2017 Feb 16. PubMed PMID: 28139931.
4: Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, Moy E, Balbo P, Li W, Zhao Y, Crouse K, Dickinson C, Symanowicz P, Hegen M, Banker ME, Vincent F, Unwalla R, Liang S, Gilbert AM, Brown MF, Hayward M, Montgomery J, Yang X, Bauman J, Trujillo JI, Casimiro-Garcia A, Vajdos FF, Leung L, Geoghegan KF, Quazi A, Xuan D, Jones L, Hett E, Wright K, Clark JD, Thorarensen A. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem Biol. 2016 Dec 16;11(12):3442-3451. Epub 2016 Nov 10. PubMed PMID: 27791347.
5: Walker G, Croasdell G. The European League Against Rheumatism (EULAR) – 17th Annual European Congress of Rheumatology (June 8-11, 2016 – London, UK). Drugs Today (Barc). 2016 Jun;52(6):355-60. doi: 10.1358/dot.2016.52.6.2516435. PubMed PMID: 27458612.
References
- ^ Jump up to:a b “Litfulo (ritlecitinib)”. Therapeutic Goods Administration (TGA). 30 July 2024. Retrieved 12 October 2024.
- ^ https://www.tga.gov.au/resources/prescription-medicines-registrations/litfulo-pfizer-australia-pty-ltd
- ^ “Litfulo Product information”. Health Canada. 13 February 2024. Retrieved 3 March 2024.
- ^ Jump up to:a b c “Summary Basis of Decision for Litfulo”. Health Canada. 18 July 2024. Retrieved 28 August 2024.
- ^ “Regulatory Decision Summary for Litfulo”. Health Canada. 29 November 2023. Retrieved 2 April 2024.
- ^ Jump up to:a b c d e f “Litfulo- ritlecitinib capsule”. DailyMed. U.S. National Library of Medicine. 23 June 2023. Archived from the original on 29 August 2023. Retrieved 28 August 2023.
- ^ Jump up to:a b c d “Litfulo EPAR”. European Medicines Agency. 18 September 2023. Archived from the original on 19 September 2023. Retrieved 20 September 2023.
- ^ “Litfulo Product information”. Union Register of medicinal products. 18 September 2023. Archived from the original on 1 October 2023. Retrieved 1 October 2023.
- ^ “Ritlecitinib”. Inxight Drugs. Archived from the original on 25 June 2023. Retrieved 24 June 2023.
- ^ Ramírez-Marín HA, Tosti A (February 2022). “Evaluating the Therapeutic Potential of Ritlecitinib for the Treatment of Alopecia Areata”. Drug Design, Development and Therapy. 16: 363–374. doi:10.2147/DDDT.S334727. PMC 8860347. PMID 35210753.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Litfulo”. U.S. Food and Drug Administration. 23 June 2023. Retrieved 29 April 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “FDA Approves Pfizer’s Litfulo (Ritlecitinib) for Adults and Adolescents With Severe Alopecia Areata” (Press release). Pfizer. 23 June 2023. Archived from the original on 25 June 2023. Retrieved 24 June 2023 – via Business Wire.
- ^ Kansteiner F (26 June 2023). “Pfizer’s Litfulo enters the scene in alopecia with adolescent nod to rival Lilly’s Olumiant”. Fierce Pharma. Archived from the original on 8 July 2023. Retrieved 18 September 2023.
Further reading
- Guttman-Yassky E, Pavel AB, Diaz A, Zhang N, Del Duca E, Estrada Y, et al. (April 2022). “Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers”. The Journal of Allergy and Clinical Immunology. 149 (4): 1318–1328. doi:10.1016/j.jaci.2021.10.036. PMID 34863853. S2CID 244824663.
- King B, Zhang X, Harcha WG, Szepietowski JC, Shapiro J, Lynde C, et al. (May 2023). “Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b-3 trial”. Lancet. 401 (10387). London, England: 1518–1529. doi:10.1016/S0140-6736(23)00222-2. PMID 37062298. S2CID 258114404.
External links
- Clinical trial number NCT03732807 for “PF-06651600 for the Treatment of Alopecia Areata (ALLEGRO-2b/3)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Litfulo |
| Other names | PF-06651600 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624015 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
|
show
|
|
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H19N5O |
| Molar mass | 285.351 g·mol−1 |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
////////////PF-06651600, PF 06651600, PF06651600, Breakthrough Therapy designation, PHASE 2, alopecia areata, rheumatoid arthritis, Crohn’s disease, ulcerative colitis, Ritlecitinib, Litfulo
C=CC(N1[C@@H](C)CC[C@@H](NC2=C3C(NC=C3)=NC=N2)C1)=O
Quizartinib dihydrochloride, キザルチニブ塩酸塩
Quizartinib dihydrochloride
キザルチニブ塩酸塩
| Formula |
C29H32N6O4S. 2HCl
|
|---|---|
| CAS |
1132827-21-4
|
| Mol weight |
633.5891
|
JAPAN, PMDA APPROVED,. 2019/6/18, Vanflyta, To use as part of a treatment regimen for newly diagnosed acute myeloid leukemia that meets certain criteria
Drug Trials Snapshot
fda 2023, 7/20/2023
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor, originated Ambit Biosciences, and acquired by Daiichi Sankyo, that is currently under development for the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It reported good results in 2012 from a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
As of 2017 it has completed 5 clinical trials and another 7 are active.[3]
SYN

References
- ^ Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry. 52 (23): 7808–7816. doi:10.1021/jm9007533.
- ^ Drug Tames Refractory AML. ASH Dec 2012
- ^ Quizartinib studies
 |
|
| Names | |
|---|---|
| IUPAC name
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
|
|
| Other names
AC220
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C29H32N6O4S | |
| Molar mass | 560.67 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Quizartinib dihydrochloride, キザルチニブ塩酸塩, JAPAN 2019
OLD POST

QUIZARTINIB
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
FOR acute myeloidLeukemia,
950769-58-1 CAS
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |
PATENTS
U.S. Provisional Patent App. No. 60/743,543, filed March 17, 2006, U.S. Patent App. No. 11/724,992, filed March 16, 2007, and U.S. Patent App. Publication No. 2007/0232604, published October 4, 2007,

BioNews TexasAmbit initiates QUANTUM-R Phase 3 clinical trial of quizartinib in FLT3-ITD …News-Medical.net… the treatment of both newly diagnosed and relapsed FLT3-ITD positive and negative AML patients.
Both the U.S. Food and Drug Administration (FDA) and European Commission have granted orphan drug designation to quizartinib for the treatment of AML.AML, High Risk MDS Therapy
see

Quizartinib
Ambit Biosciences
13 MAY 2013
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]

AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.

Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
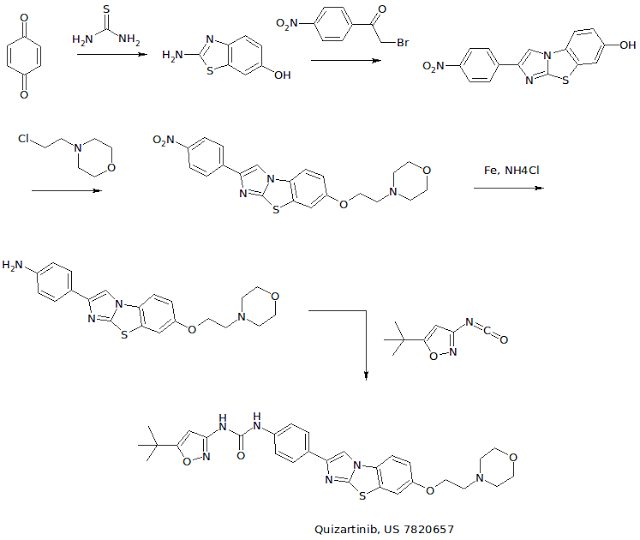

………………………..
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);

(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole

1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.


………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
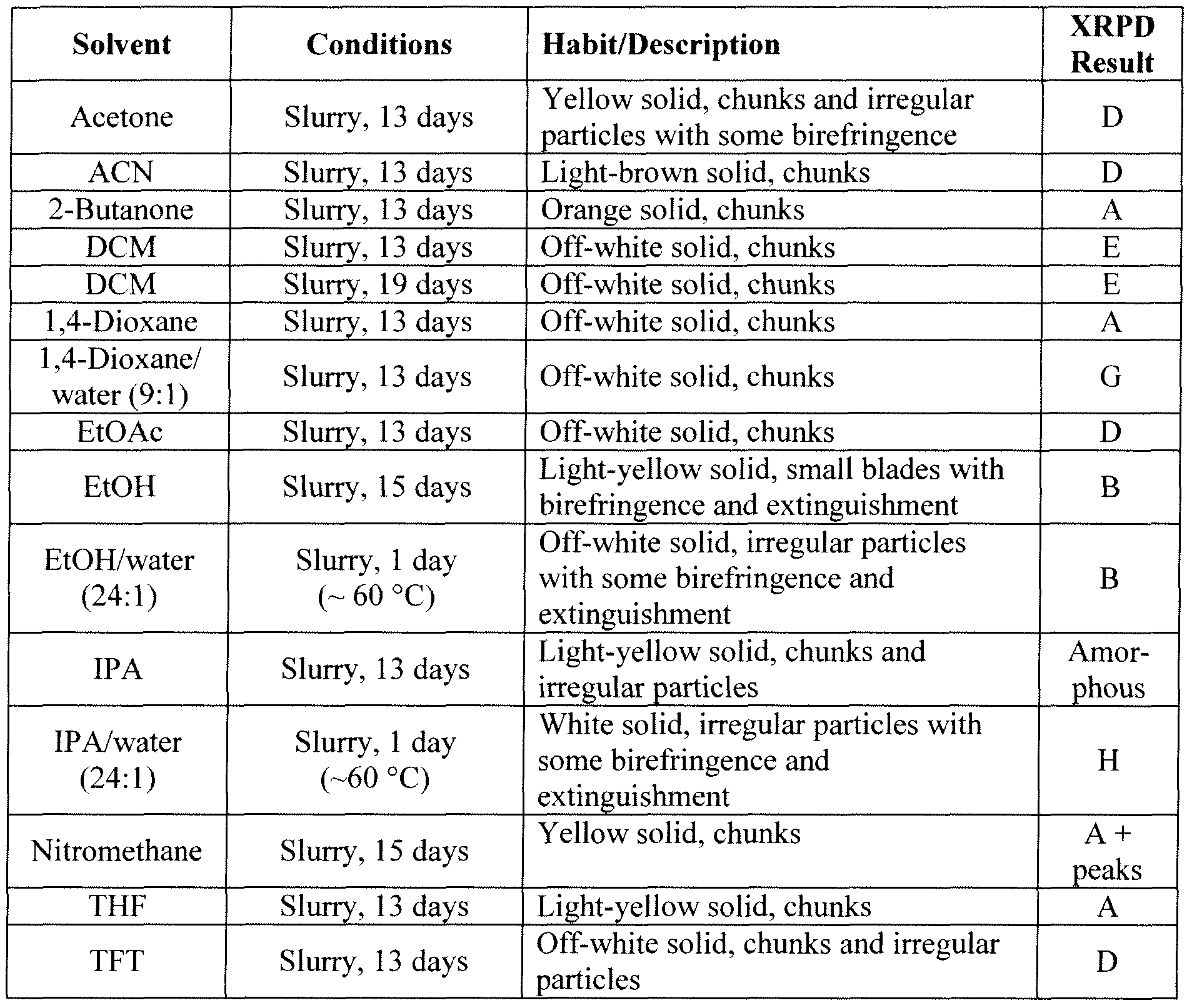
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8

“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
http://pubs.acs.org/doi/full/10.1021/jm9007533

N-(5-tert-Butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (7): General Procedure D
A suspension of 2-(4-aminophenyl)-7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazole (19c) (4.06 g, 10.3 mmol) and 5-tert-butyl-isoxazole-3-isocyanate (5) (1.994 g, 12 mmol) in toluene (60 mL) was heated at 120 °C overnight. The reaction was quenched with a mixture of dichloromethane and water containing a little methanol, and the mixture was neutralized with saturated aqueous NaHCO3. The aqueous phase was extracted twice with dichloromethane, and the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to a volume of about 20 mL and ethyl ether was added, resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of 7 (2.342 g, 41%).
1H NMR (DMSO-d6) δ 9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 1.3 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.18 (dd, J = 1.3 and 8.9 Hz, 1H), 6.52 (s, 1H), 4.16 (t, J = 5.7 Hz, 2H), 3.59 (t, J = 4.2 Hz, 4H), 3.36 (overlapping, 4H), 2.72 (t, J = 5.7 Hz, 2H), 1.30 (s, 9H).
General Procedure E for Preparation of Hydrochloride Salt
The free base was dissolved in a mixture of dichloromethane (20 mL) and methanol (1 mL). A solution of 1.0 M HCl in ethyl ether (1.1 equiv for all compounds except 7, for which 2.5 equiv were used) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration to give
N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (7) (2.441 g, 98%).
1H NMR (DMSO-d6) δ 11.0 (br, 1H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.78 (m, 3H), 7.53 (d, J = 8.7 Hz, 2H), 7.26 (dd, J = 2.4 and 8.9 Hz, 1H), 6.53 (s, 1H), 4.50 (t, J = 4.1 Hz, 2H), 3.97 (m, 2H), 3.81 (t, J = 12.1 Hz, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). LC-MS (ESI) m/z 561 (M + H)+.
Anal. (C29H32N6O4S·2HCl) C, H, N. C: calcd 54.97; found 54.54. H: calcd 5.22; found 5.87. N: calcd 13.26; found 13.16.
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf


Fezolinetant, фезолинетант , فيزولينيتانت , 非唑奈坦 ,
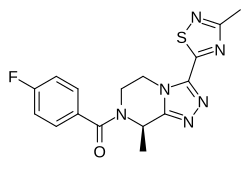
Fezolinetant ESN-364
- Molecular FormulaC16H15FN6OS
- Average mass358.393 Da
-
Methanone, [(8R)-5,6-dihydro-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-1,2,4-triazolo[4,3-a]pyrazin-7(8H)-yl](4-fluorophenyl)-UNII:83VNE45KXXфезолинетант [Russian] [INN]فيزولينيتانت [Arabic] [INN]非唑奈坦 [Chinese] [INN]
(4-Fluorophenyl)[(8R)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]methanone
10205
1629229-37-3 [RN]
83VNE45KXX
FDA APPROVED 5/12/2023, Veozah
- Originator Euroscreen
- Developer Ogeda
- Class Pyrazines; Small molecules; Triazoles
- Mechanism of Action Gonadal steroid hormone modulators; Neurokinin 3 receptor antagonists
- Phase II Hot flashes; Polycystic ovary syndrome; Uterine leiomyoma
- Preclinical Weight gain
- DiscontinuedBenign prostatic hyperplasia; Endometriosis
- 14 Sep 2018 Ogeda completes a phase II trial in Hot flashes (In the elderly, In adults) in USA (PO) (NCT03192176)
- 23 May 2018 Astellas Pharma completes a phase I trial in Polycystic ovary syndrome (In volunteers) in Japan (PO) (NCT03436849)
- 22 Feb 2018 Phase-I clinical trials in Polycystic ovary syndrome (In volunteers) in Japan (PO) (NCT03436849)
Fezolinetant (INN; former developmental code name ESN-364) is a small-molecule, orally active, selective neurokinin-3 (NK3) receptorantagonist which is under development by Ogeda (formerly Euroscreen) for the treatment of sex hormone-related disorders.[1][2] As of May 2017, it has completed phase I and phase IIa clinical trials for hot flashes in postmenopausal women.[1] Phase IIa trials in polycystic ovary syndrome patients are ongoing.[1] In April 2017, it was announced that Ogeda would be acquired by Astellas Pharma.[3]
Ogeda (formerly Euroscreen ) is developing fezolinetant, an NK3 antagonist, for treating endometriosis, benign prostate hyperplasia, polycystic ovary syndrome, uterine fibroids and hot flashes. In November 2018, drug was listed under phase II development for PCOS, uterine fibroids and hot flashes in company’s pipeline. In October 2018, the company was proceeding to phase III study preparation, and regulatory filings were expected in 2021 or later .
Fezolinetant shows high affinity for and potent inhibition of the NK3 receptor in vitro (Ki = 25 nM, IC50 = 20 nM).[2] Loss-of-function mutations in TACR and TACR3, the genes respectively encoding neurokinin B and its receptor, the NK3 receptor, have been found in patients with idiopathic hypogonadotropic hypogonadism.[2] In accordance, NK3 receptor antagonists like fezolinetant have been found to dose-dependently suppress luteinizing hormone (LH) secretion, though not that of follicle-stimulating hormone (FSH), and consequently to dose-dependently decrease estradiol and progesterone levels in women and testosterone levels in men.[4] As such, they are similar to GnRH modulators, and present as a potential clinical alternative to them for use in the same kinds of indications.[5]However, the inhibition of sex hormone production by NK3 receptor inactivation tends to be less complete and “non-castrating” relative to that of GnRH modulators, and so they may have a reduced incidence of menopausal-like side effects such as loss of bone mineral density.[4][5]
Unlike GnRH modulators, but similarly to estrogens, NK3 receptor antagonists including fezolinetant and MLE-4901 (also known as AZD-4901, formerly AZD-2624) have been found to alleviate hot flashes in menopausal women.[6][7] This would seem to be independent of their actions on the hypothalamic–pituitary–gonadal axis and hence on sex hormone production.[6][7] NK3 receptor antagonists are anticipated as a useful clinical alternative to estrogens for management of hot flashes, but with potentially reduced risks and side effects.[6][7]
PATENT
WO2011121137
hold protection in most of the EU states until 2031 and expire in the US in 2031.
PATENT
US 20170095472

PATENT
WO2016146712
PATENT
WO-2019012033
Novel deuterated analogs of fezolinetant , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, convulsion, obesity, inflammatory disease including irritable bowel syndrome, emesis, asthma, cough, urinary incontinence, reproduction disorders, testicular cancer and breast cancer. Further claims are processes for the preparation of fezolinetant. claiming use of NK3R antagonist eg fezolinetant, for treating pathological excess body fat or prevention of obesity.
Fezolinetant was developed as selective antagonist of NK-3 receptor and is useful as therapeutic compound, particularly in the treatment and/or prevention of sex-hormone dependent diseases. Fezolinetant corresponds to (R)-(4-fluorophenyl)-(8-methyl-3-(3-memyl-l,2,4-miacMazol-5-yl)-5,6-dmy(ko-[l,2,4]trizolo[4,3-a]pyrazin-7(8H)-yl)methanone and is described in WO2014/154895.
Drug-drug interactions are the most common type of drug interactions. They can decrease how well the medications works, may cause serious unexpected side effects, or even increase the blood level and possible toxicity of a certain drug.
Drug interaction may occur by pharmacokinetic interaction, during which one drug affects another drug’s absorption, distribution, metabolism, or excretion. Regarding metabolism, it should be noted that drugs are usually eliminated from the body as either the unchanged drug or as a metabolite. Enzymes in the liver, usually the cytochrome P450s (CYPs) enzymes, are often responsible for metabolizing drugs. Therefore, determining the CYP profile of a drug is of high relevancy to determine if it will affect the activity of CYPs and thus if it may lead to drug-drug interactions.The five most relevant CYPs for drug-drug interaction are CYP3A4, 2C9, 2C19, 1A2 and 2D6, among which isoforms 3A4, 2C9 and 2C19 are the major ones. The less a drug inhibits these CYPs, the less drug-drug interactions would be expected.
Therefore, it is important to provide drugs that present the safest CYP profile in order to minimize as much as possible the potential risks of drug-drug interactions.Even if fezolinetant possesses a good CYP profile, providing analogs of fezolinetant with a further improved CYP profile would be valuable for patients.
In a completely unexpected way, the Applicant evidenced that deuteration of fezolinetant provides a further improved CYP profile, especially on isoforms CYP 2C9 and 2C19. This was evidenced for the deuterated form (R)-(4-fluorophenyl)-(8-methyl-3-(3-(memyl-d.?)-l,2,4-miacttazol-5-y ^yl)methanone, hereafter referred to as “deuterated fezolinetant”.
Importantly, deuterated fezolinetant retains the biological activity of fezolinetant as well as its lipophilic efficiency.
Deuterated fezolinetant also presents the advantage to enable improvement of the in vivo half -life of the drug. For example, half -life is increased by a factor 2 in castrated monkeys, compared to fezolinetant.
Synthetic scheme
Deuterated fezolinetant may be synthesized using the methodology described following schemes (Part A and Part B):
Part A: Preparation of deuterated key intermediate (ii)
Part B: Synthesis of deuterated fezolinetant using intermediate (ii)
Synthesis of deuterated fezolinetant was performed through key intermediate (ii). Part A corresponds to the synthesis of intermediate (ii). Part B leads to deuterated fezolinetant (d3-fezolinetant), using intermediate (ii), using procedures adapted from WO2014/154895.
Experimental details
Part A – Step 1): Formation of d3-acetamide (b)
To i¾-acetic acid (a) (10 g, 1 equiv.) in DCM (100 mL) CDI (25.3 g, 1 equiv.) was added and the resultant mixture stirred at RT for 30 min, thereupon ammonia gas was bubbled through the reaction mixture for 40 min at 0-5 °C. Thereafter the bubbling was stopped, the mixture was filtered and the filtrate was evaporated under reduced pressure to give 30.95 g crude product that was purified using flash chromatography on silica to furnish 6.65 g (yield: 73 %) deuterated acetamide (b) was obtained (GC (column RTX-1301 30 m x 0.32 mm x 0.5 μπι) Rt 7.4 min, 98 %).
Part A – Step 2): Ring closure leading to compound (c)
<¾-Acetamide (b) (3.3 g, 1 equiv.) and chlorocarbonylsulfenyl chloride (CCSC) (8.4 g, 1.2 equiv.) were combined in 1,2-dichloroethane (63 mL), and refluxed for 4.5 h. CCSC can be prepared as per the procedure described in Adeppa et al. (Synth. Commun., 2012, Vol. 42, pp. 714-721). The volatiles were then removed to obtain 6.60 g (102 % yield) oxathiazolone (c) product as a yellow oil. The product was analyzed by GC (Rt= 7.8 min, 97 ). 13C NMR (CDC13): 16.0, 158.7, 174.4 ppm.
Part A – Step 3): formation of compound (d)
To oxathiazolone (c) (6.6 g, 1 equiv) in rn-xylene (231 mL) methyl cyanoformate (14.70 g, 3.2 equiv.) was added. The mixture was stirred at 130 °C for 19 h and thereafter the volatiles removed under reduced pressure at 50 °C to obtain 4.53 g brown oil (yield: 51 %). The product (d) was analyzed by GC (Rt = 11.8 min, 81 %) and mass spectrometry (M+H = 162).
Part A – Step 4): formation of intermediate (ii)
The ester (d) obtained above (3.65 g, lequiv.) was dissolved in ethanol (45 mL). The undissolved material was filtered off then hydrazine hydrate (2.3 mL, 1.15 equiv. 55w/w in H20) was added to the stirred solution. Thick suspension formed in minutes, the suspension was stirred for 45 min, filtered and washed with EtOH (3 mL) to furnish intermediate (ii) a pale yellow solid (2.43 g, 55 % yield). Mass spectrometry (M+H = 162, M+Na = 184); ¾ NMR (cfe-DMSO): 4.79 ppm (br s, 2H), 10.55 ppm (br s, 1H); 13C NMR (fife-DMSO): 17.4 ppm, 155.6 ppm, 173.4 ppm, 183.0 ppm.
Part B – Step a): formation of compound (iii)
Intermediate (i) was prepared as described in WO2014/154895.
Intermediate (ii) (490 mg, 3.04 mmol) and compound (i) (1.0 g (87 mol 1.3 content), 2.97 mmol) were taken up in MeOH and the reaction mixture was stirred at a temperature ranging from 55°C to 70°C for a period of time ranging from 6 hours to 8 hours. The reaction was deemed complete by TLC. The reaction mixture was evaporated and the crude product was purified by flash chromatography on silica in DCM : MeOH eluent to afford 1.13 g (97 % yield) of compound (iii) as a yellow oil. JH NMR (CDC13): δ (ppm) 7.26 (d, 1H), 6.48-6.49 (2H), 4.50 (m, 1H), 4.30 (m, 1H), 4.09 (m, 1H), 3.94 (d, 1H), 3.80 (s, 6H), 3.61 (d, 1H), 3.22 (m, 1H), 2.75 (m, 1H), 1.72 (d, 3H); Mass spectrometry (M+H = 390, 2M+Na = 801). Chiral LC (column: Chiralpak IC, 250 x 4.6 mm – eluent: MTBE MeOH DEA 98/2/0.1) 99.84 .
Part B – Step b): deprotection leading to compound (iv)
Intermediate (iii) prepared above (1.05 g, 2.7 mmol) was dissolved in DCM and washed with aq. NaOH. The organic phase was dried, then TFA (1.56 mL, 2.3 g, 7.5 equiv.) was added at RT. The resulting solution was stirred at RT for 2 h. The reaction was monitored by TLC. After completion of the reaction water was added to the reaction mixture, and the precipitate filtered and washed with water. The phases were separated, the pH of the aq. phase was adjusted to pH 13 by addition of 20 % aq. NaOH. NaCl was then added to the aqueous solution that was then extracted with DCM. The organic phase was evaporated under reduced pressure to give 504 mg of compound (iv) (78 % yield). ¾ NMR (cfe-DMSO): δ (ppm) 4.42 (m, 1H), 4.10 (m, 2H), 3.0 (m, 1H), 2.82 (m, 1H), 1.46 (d, 3H). 13C NMR (rf6-DMSO): δ (ppm) 174.8, 173.4, 156.2, 145.0, 48.1, 45.7, 40.7, 19.1. Mass spectrometry (M+H = 240, 2M+Na = 501).
Part B – Step c): acylation and recrystallization to form deuterated fezolinetant
Intermediate (iv) (450 mg, 1.88 mmol) was dissolved in DCM, then sat. aq. NaHC03 was added and the mixture was stirred for 30 min. To this mixture 4-fluorobenzoyl chloride (v) (220 1 equiv.) was added dropwise at RT. The reaction was stirred for a period of time ranging from about 20 min to overnight at RT and reaction progress monitored by TLC. After completion the phases were separated, the organic phase was washed with water, dried over MgS04, filtered and evaporated under reduced pressure to give 745 mg crude <i3-fezolinetant (110 % yield). The crude product was purified by flash chromatography using MeOH : DCM together with a second batch, then
crystallized (EtOH H20) before final analysis. ¾ NMR (d6-DMSO): δ (ppm) 7.60 (m, 2H), 7.33 (m, 2H), 5.73 (m, 1H), 4.68 (dd, 1H), 4.31 (m, 1H), 4.06 (m, 1H), 3.65 (m, 1H), 1.61 (d, 3H). 13C NMR (d6-DMSO): δ (ppm) 174.4, 173.5, 168.7, 163.7, 161.8, 154.1, 144.9, 131.6, 129.5, 115.5, 44.7, 18.7. Isotopic purity based on an intense molecular ion observed at m/z = 362.2 Da is estimated as approximately 100 % isotopic purity. Chiral purity (LC) (column: Chiralpak IC, 250 x 4.6 mm – eluent: n-hexane/EtOH DEA 80/20/0.1) >99.9 %. A single crystal X-ray structure of the deuterated fezolinetant final product was obtained (Figure 1) that confirmed the structure of the compound as well as the stereochemistry.
References
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800039455
- ^ Jump up to:a b c Hoveyda, Hamid R.; Fraser, Graeme L.; Dutheuil, Guillaume; El Bousmaqui, Mohamed; Korac, Julien; Lenoir, François; Lapin, Alexey; Noël, Sophie (2015). “Optimization of Novel Antagonists to the Neurokinin‑3 Receptor for the Treatment of Sex-Hormone Disorders (Part II)”. ACS Medicinal Chemistry Letters (6): 736-740. doi:10.1021/acsmedchemlett.5b00117.
- ^ http://www.prnewswire.com/news-releases/astellas-to-acquire-ogeda-sa-300433141.html
- ^ Jump up to:a b Fraser GL, Ramael S, Hoveyda HR, Gheyle L, Combalbert J (2016). “The NK3 Receptor Antagonist ESN364 Suppresses Sex Hormones in Men and Women”. J. Clin. Endocrinol. Metab. 101 (2): 417–26. doi:10.1210/jc.2015-3621. PMID 26653113.
- ^ Jump up to:a b Fraser GL, Hoveyda HR, Clarke IJ, Ramaswamy S, Plant TM, Rose C, Millar RP (2015). “The NK3 Receptor Antagonist ESN364 Interrupts Pulsatile LH Secretion and Moderates Levels of Ovarian Hormones Throughout the Menstrual Cycle”. Endocrinology. 156 (11): 4214–25. doi:10.1210/en.2015-1409. PMID 26305889.
- ^ Jump up to:a b c http://www.medscape.com/viewarticle/878262
- ^ Jump up to:a b c https://www.clinicalleader.com/doc/ogeda-announces-positive-fezolinetant-treatment-menopausal-flashes-0001
External links
 |
|
| Clinical data | |
|---|---|
| Synonyms | ESN-364 |
| Routes of administration |
By mouth |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H15FN6OS |
| Molar mass | 358.40 g·mol−1 |
| 3D model (JSmol) | |
////////////////Fezolinetant, ESN-364, фезолинетант , فيزولينيتانت , 非唑奈坦 , Phase II, Hot flashes, Polycystic ovary syndrome, Uterine leiomyoma, Euroscreen, Ogeda, FDA 2023, APPROVALS 2023, Veozah
Smiles
C[C@H]1N(CCn2c1nnc2c3nc(C)ns3)C(=O)c4ccc(F)cc4
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

READ
ANTHONY MELVIN CRASTO

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
CALL +919323115463 INDIA
//////////////
GDC 0853, Fenebrutinib
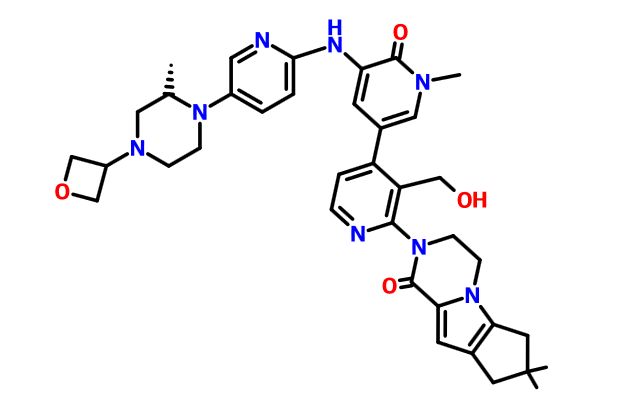
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
Haiming Zhang*†  , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† †Department of Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡Department of Process Chemistry and Catalysis and ¶Department of Drug Substance Scale-up and Supply, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, 4070 Basel, Switzerland
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. https://pubs.acs.org/doi/10.1021/acs.oprd.8b00134

Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
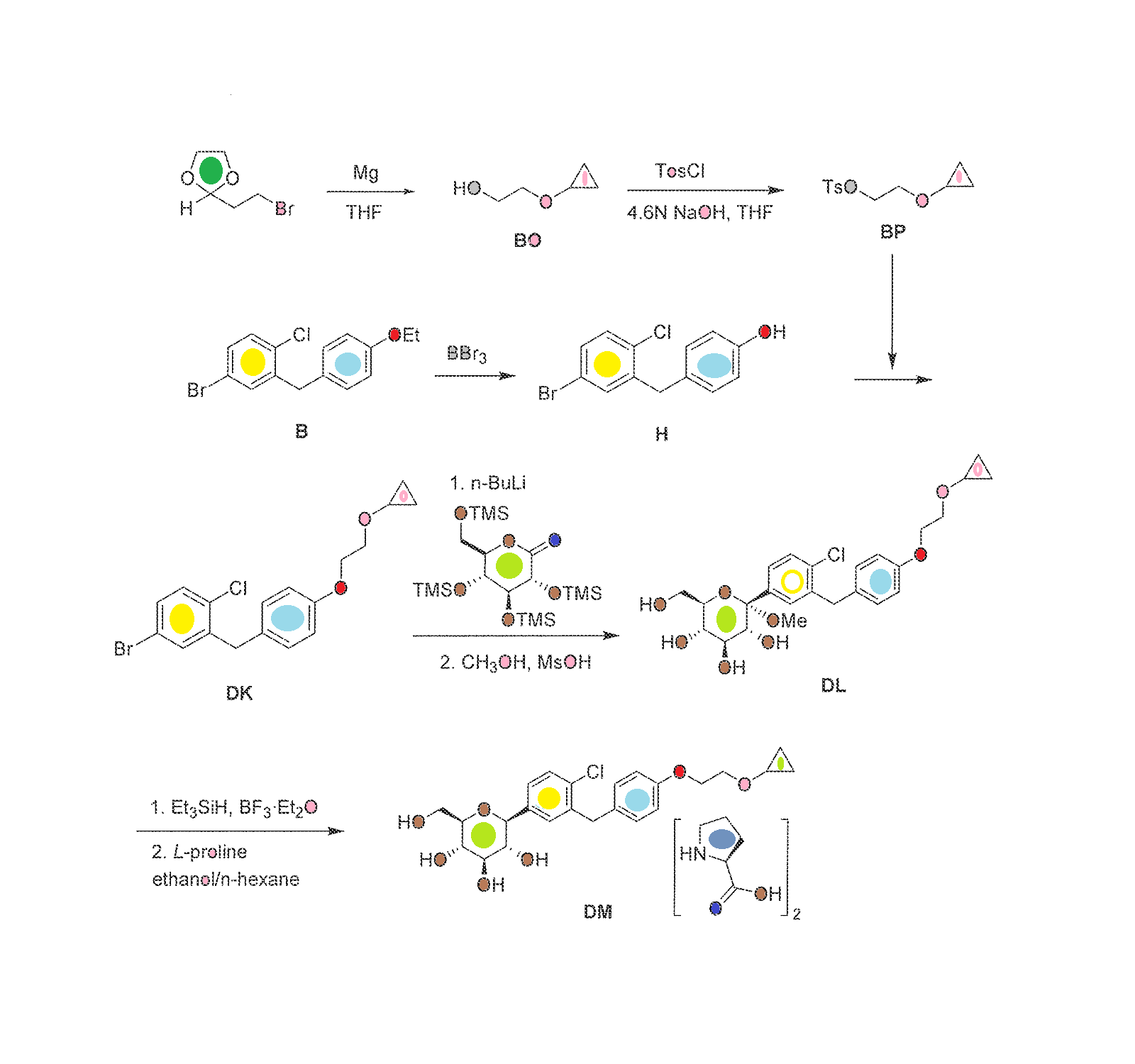

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……

{kind=link}
{kind=link}
{kind=link}