
Home » Posts tagged 'APPROVALS 2022' (Page 4)
Tag Archives: APPROVALS 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Somatrogon

>Somatrogon amino acid sequence SSSSKAPPPSLPSPSRLPGPSDTPILPQFPTIPLSRLFDNAMLRAHRLHQLAFDTYQEFE EAYIPKEQKYSFLQNPQTSLCFSESIPTPSNREETQQKSNLELLRISLLLIQSWLEPVQF LRSVFANSLVYGASDSNVYDLLKDLEEGIQTLMGRLEDGSPRTGQIFKQTYSKFDTNSHN DDALLKNYGLLYCFRKDMDKVETFLRIVQCRSVEGSCGFSSSSKAPPPSLPSPSRLPGPS DTPILPQSSSSKAPPPSLPSPSRLPGPSDTPILPQ
Somatrogon
CAS: 1663481-09-1
Protein Chemical FormulaC1359H2125N361O420S7
Protein Average Weight30465.1 Da (Aglycosylated)
NGENLA, JAPAN PMDA APPROVED 2022/1/20
ソマトロゴン;
- MOD-4023
Replenisher (somatotoropin)
- OriginatorModigene
- DeveloperOPKO Health; Pfizer
- ClassBiological proteins; Growth hormones; Hormonal replacements; Recombinant proteins
- Mechanism of ActionHuman growth hormone replacements
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 21 Jan 2022Pfizer and OPKO health receives complete response letter from the US FDA for somatrogon in Somatotropin deficiency (In children)
- 20 Jan 2022Registered for Somatotropin deficiency (In children) in Japan (SC)
- 01 Dec 2021CHMP issues a positive opinion and recommends approval of somatrogon for Somatotropin deficiency in the European Union
Somatrogon, sold under the brand name Ngenla, is a medication for the treatment of growth hormone deficiency.[1][2] Somatrogon is a glycosylated protein constructed from human growth hormone and a small part of human chorionic gonadotropin which is appended to both the N-terminal and C-terminal.[2]
Somatrogon is a long-acting recombinant human growth hormone used as the long-term treatment of pediatric patients who have growth failure due to growth hormone deficiency.
omatrogon is a long-acting recombinant human growth hormone. Growth hormone is a peptide hormone secreted by the pituitary gland that plays a crucial role in promoting longitudinal growth during childhood and adolescence and regulating metabolic function in adulthood.2 Recombinant growth hormone therapy for growth hormone deficiency and other conditions has been available since 1985, with daily administration being the standard treatment for many years. More recently, longer-acting forms of growth hormone were developed to improve patient adherence and thus, improve the therapeutic efficacy of treatment.1 Somatrogon was produced in Chinese Hamster Ovary (CHO) cells using recombinant DNA technology. It is a chimeric product generated by fusing three copies of the C-terminal peptide (CTP), or 28 carboxy-terminal residues, from the beta chain of human chorionic gonadotropin (hCG) to the N-terminus and C-terminus of human growth hormone.2,6 The glycosylation and the presence of CTPs in the protein sequence prolongs the half-life of somatrogon and allows its once-weekly dosing.6
In October 2021, Health Canada approved somatrogon under the market name NGENLA as the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone caused by growth hormone deficiency, marking Canada as the first country to approve this drug.4 It is available as a once-weekly subcutaneous injection.5
////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
About Somatrogon©
Somatrogon©, a long-acting human growth hormone (hGH) molecule, is a once-weekly injectable, created using recombinant technology, for the treatment of pediatric and adult growth hormone deficiency (GHD). The molecule consists of the natural peptide sequence of native growth hormone and the 28 amino acids of the C-Terminus Peptide (CTP) of the human chorionic gonadotropin hormone. This molecule, as compared to current GH replacement therapies, is intended to reduce the injection frequency from a daily to once a week in adults and children with GHD.
| Clinical data | |
|---|---|
| Trade names | Ngenla |
| Other names | MOD-4023 |
| Pregnancy category | AU: B1[1] |
| Routes of administration | Subcutaneous injection |
| ATC code | H01AC08 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1] |
| Identifiers | |
| CAS Number | 1663481-09-1 |
| DrugBank | DB14960 |
| UNII | 6D848RA61B |
Somatrogon© COMPETITIVE ADVANTAGES
In 2014, Pfizer and OPKO entered into a worldwide agreement for the development and commercialization of Somatrogon©. Under the agreement, OPKO is responsible for conducting the clinical program and Pfizer is responsible for registering and commercializing the product.
- New molecular entity (NME) that maintains natural native sequence of growth hormone
- Once weekly injection vs. current products requiring daily injections
- Human growth hormone is used for:
- Growth hormone deficient children and adults
- SGA, PWS, ISS
- Final presentation:
- Refrigerated, liquid, non-viscous formulation
- Disposable easy to handle pen injection device with thin needle and small injection volume
- Orphan drug designation in the U.S. and the EU for children and adults
Somatrogon© PROGRAM STATUS
Phase 3 Pediatric Somatrogon©
- Phase 3 study in naive growth hormone deficiency pediatric population was completed.
The study was conducted in over 20 countries. This study enrolled and treated 224 pre-pubertal, treatment-naive children with growth hormone deficiency.
- OPKO and Pfizer Announce Positive Phase 3 Top-Line Results for Somatrogon© during Oct 2019.
- Achieved Primary Endpoint
- Somatrogon© was proven non-inferior to daily Genotropin® (somatropin) with respect to height velocity after 12 months
- Height velocity at 12 months of treatment was higher in the Somatrogon© group (10.12 cm/year) than in the somatropin group (9.78 cm/year)
- Secondary Endpoints Achieved
- Change in height standard deviation scores at six and 12 months were higher with Somatrogon© in comparison to somatropin
- At six months, change in height velocity was higher with Somatrogon© in comparison to somatropin
- Somatrogon© was generally well tolerated in the study and comparable to that of somatropin dosed once-daily with respect to the types, numbers and severity of the adverse events observed between the treatment arms
- Children completing this study had the opportunity to enroll in a global, open-label, multicenter, long-term extension study, in which they were able to either continue receiving or switch to Somatrogon© Approximately 95% of the patients switched into the open-label extension study and received Somatrogon© treatment
Phase 3 adults Somatrogon© completed
- Primary endpoint of change in trunk fat mass from baseline to 26 weeks did not demonstrate a statistical significance between the Somatrogon© treated group and placebo
- Completed post hoc outlier analysis in June 2017 to assess the influence of outliers on the primary endpoint results
- Analyses which excluded outliers showed a statistically significant difference between Somatrogon© and placebo on the change in trunk fat mass: additional analyses that did not exclude outliers showed mixed results
- No safety concerns
- OPKO and Pfizer have agreed that OPKO may proceed with a pre-BLA meeting with FDA to discuss a submission plan
- OPKO plans to carry out an additional study in adults using a pen device
Pediatric Somatrogon© registration study in Japan- expected to be completed in Q1 2020
- 44 patients, comparison of weekly Somatrogon to daily growth hormone.
- Same pen device, dosage and formulation used in global study.
Somatrogon© Path to Approval
- BLA submission in US anticipated second half of 2020
- Completion of analysis of immunogenicity and safety data from pivotal Phase 3 study and open label extension study
- Two abstracts accepted for oral presentation of data set at the Endo Society’s Annual Meeting in March 2020
- “Somatrogon© Growth Hormone in the Treatment of Pediatric Growth Hormone Deficiency: Results of the Pivotal Phase 3”
- “Interpretation of Insulin-like Growth Factor (IGF-1) Levels Following Administration of Somatrogon© (a long acting Growth Hormone-hGH-CTP)”
- MAA submission in Europe to follow upon completion of open label study demonstrating benefit and compliance with reduced treatment burden
- Study expected to be completed in Q3 2020
References
Hershkovitz O, Bar-Ilan A, Guy R, et al. In vitro and in vivo characterization of MOD-4023, a long-acting carboxy-terminal peptide (CTP)-modified human growth hormone. Mol Pharm. 2016; 13:631–639 [PDF]
Strasburger CJ, Vanuga P, Payer J, et al. MOD-4023, a long-acting carboxy-terminal peptide-modified human growth hormone: results of a Phase 2 study in growth hormone-deficient adults. Eur J Endocrinol. 2017;176:283–294 [PDF]
Zelinska N, Iotova V, Skorodok J, et al. Long-acting CTP-modified hGH (MOD-4023): results of a safety and dose-finding study in GHD children. J Clin Endocrinol Metab. 2017;102:1578–1587 [PDF]
Fisher DM, Rosenfeld RG, Jaron-Mendelson M, et al. Pharmacokinetic and pharmacodynamic modeling of MOD-4023, a long-acting human growth hormone, in GHD Children. Horm Res Paediatr. 2017;87:324–332 [PDF]
Kramer W, Jaron-Mendelson M, Koren R, et al. Pharmacokinetics, Pharmacodynamics and Safety of a Long-Acting Human Growth Hormone (MOD-4023) in Healthy Japanese and Caucasian Adults. Clin Pharmacol Drug Dev. 2017 [in press]
Society and culture
Legal status
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Ngenla, intended for the treatment of growth hormone deficiency (GHD) in children and adolescents from 3 years of age.[3] The applicant for this medicinal product is Pfizer Europe MA EEIG.[3]
Somatrogon was approved for medical use in Australia in November 2021.[1]
References
- ^ Jump up to:a b c d “Ngenla”. Therapeutic Goods Administration (TGA). 13 December 2021. Retrieved 28 December 2021.
- ^ Jump up to:a b “Pfizer and OPKO Announce Extension of U.S. FDA Review of Biologics License Application of Somatrogon for Pediatric Growth Hormone Deficiency” (Press release). Opko Health. 24 September 2021. Retrieved 18 December 2021 – via GlobeNewswire.
- ^ Jump up to:a b “Ngenla: Pending EC decision”. European Medicines Agency (EMA). 16 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
- Fisher DM, Rosenfeld RG, Jaron-Mendelson M, Amitzi L, Koren R, Hart G (2017). “Pharmacokinetic and Pharmacodynamic Modeling of MOD-4023, a Long-Acting Human Growth Hormone, in Growth Hormone Deficiency Children”. Horm Res Paediatr. 87 (5): 324–32. doi:10.1159/000470842. PMC 5637306. PMID 28399519.
External links
- “Somatrogon”. Drug Information Portal. U.S. National Library of Medicine.
///////////Somatrogon, NGENLA, APPROVALS 2022, JAPAN 2022, ソマトロゴン , MOD-4023, Modigene, OPKO Health, Pfizer

NEW DRUG APPROVALS
one time
$10.00
Tebentafusp-tebn
Tebentafusp-tebn
- IMCGP100
UNIIN658GY6L3E
CAS number1874157-95-5
FDA APPROVED 1/25/2022, Kimmtrak, To treat unresectable or metastatic uveal melanoma
Immunocore Limited
- T cell receptor α chain (synthetic human) fusion protein with T cell receptor β chain (synthetic human) fusion protein with immunoglobulin, anti-(human CD3 antigen) (synthetic scFv fragment)
- Protein Sequence
- Sequence Length: 695, 500, 195
Sequence:
1AIQMTQSPSS LSASVGDRVT ITCRASQDIR NYLNWYQQKP GKAPKLLIYY51TSRLESGVPS RFSGSGSGTD YTLTISSLQP EDFATYYCQQ GNTLPWTFGQ101GTKVEIKGGG GSGGGGSGGG GSGGGGSGGG SEVQLVESGG GLVQPGGSLR151LSCAASGYSF TGYTMNWVRQ APGKGLEWVA LINPYKGVST YNQKFKDRFT201ISVDKSKNTA YLQMNSLRAE DTAVYYCARS GYYGDSDWYF DVWGQGTLVT251VSSGGGGSDG GITQSPKYLF RKEGQNVTLS CEQNLNHDAM YWYRQDPGQG301LRLIYYSWAQ GDFQKGDIAE GYSVSREKKE SFPLTVTSAQ KNPTAFYLCA351SSWGAPYEQY FGPGTRLTVT EDLKNVFPPE VAVFEPSEAE ISHTQKATLV401CLATGFYPDH VELSWWVNGK EVHSGVCTDP QPLKEQPALN DSRYALSSRL451RVSATFWQDP RNHFRCQVQF YGLSENDEWT QDRAKPVTQI VSAEAWGRAD
Sequence:
1AQQGEEDPQA LSIQEGENAT MNCSYKTSIN NLQWYRQNSG RGLVHLILIR51SNEREKHSGR LRVTLDTSKK SSSLLITASR AADTASYFCA TDGSTPMQFG101KGTRLSVIAN IQKPDPAVYQ LRDSKSSDKS VCLFTDFDSQ TNVSQSKDSD151VYITDKCVLD MRSMDFKSNS AVAWSNKSDF ACANAFNNSI IPEDT
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-23 – Cys-88 | disulfide bridge |
| bridge | Cys-153 – Cys-227 | disulfide bridge |
| bridge | Cys-281 – Cys-349 | disulfide bridge |
| bridge | Cys-401 – Cys-466 | disulfide bridge |
| bridge | Cys-427 – Cys-157′ | disulfide bridge |
| bridge | Cys-23′ – Cys-89′ | disulfide bridge |
| bridge | Cys-132′ – Cys-182′ | disulfide bridge |
Tebentafusp, sold under the brand name Kimmtrak, is an anti-cancer medication used to treat uveal melanoma (eye cancer).[1][2]
The most common side effects include cytokine release syndrome, rash, pyrexia (fever), pruritus (itching), fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager.[1][2] It was approved for medical use in the United States in January 2022.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager used to treat unresectable or metastatic uveal melanoma.
Tebentafusp is a gp100 peptide-HLA-directed CD3 T cell engager.5 It is a bispecific, fusion protein and first-in-class drug of immune-mobilizing monoclonal T cell receptors against cancer (ImmTACs), a recently developed cancer immunotherapy with a novel mechanism of action. ImmTACs bind to target cancer cells that express a specific antigen of interest and recruit cytotoxic T cells to lyse the cells, such as melanocytes.1,2
Uveal melanoma is a rare ocular tumour with often poor prognosis and limited treatment options. Even after surgical ablation or removal of the ocular tumour, almost 50% of patients with uveal melanoma develop metastatic disease.1 On January 26, 2022, tebentafusp was first approved by the FDA for the treatment of HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma. This approval marks the first bispecific T cell engager to be approved by the FDA to treat a solid tumour and being the first and only therapy for the treatment of unresectable or metastatic uveal melanoma to be approved by the FDA.5
FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma
On January 25, 2022, the Food and Drug Administration approved tebentafusp-tebn (Kimmtrak, Immunocore Limited), a bispecific gp100 peptide-HLA-directed CD3 T cell engager, for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 patients with metastatic uveal melanoma. Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if prior systemic therapy or localized liver-directed therapy were administered. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.
Patients were randomized (2:1) to receive tebentafusp-tebn (N=252) or investigator’s choice (N=126) of either pembrolizumab, ipilimumab, or dacarbazine. Tebentafusp-tebn was administered weekly by intravenous infusion at 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15 and every subsequent week until disease progression or unacceptable toxicity. The main efficacy outcome measure was overall survival (OS). An additional efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST 1.1. Median OS was 21.7 months (95% CI: 18.6, 28.6) for patients treated with tebentafusp-tebn and 16 months (95% CI: 9.7, 18.4) in the investigator’s choice arm (HR=0.51, 95% CI: 0.37, 0.71, p<0.0001) PFS was 3.3 months (95% CI: 3, 5) for those receiving tebentafusp-tebn and 2.9 months (95% CI: 2.8, 3) in the investigator’s choice arm (HR=0.73, 95% CI: 0.58, 0.94, p=0.0139).
The most common adverse reactions (≥30%) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common laboratory abnormalities (≥50%) were decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate.
The recommended tebentafusp-tebn dose administered intravenously is:
- 20 mcg on day 1,
- 30 mcg on day 8,
- 68 mcg on day 15, and
- 68 mcg once weekly thereafter.
View full prescribing information for Kimmtrak.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the United Kingdom’s Medicines and Healthcare product Regulatory Agency (MHRA). The application reviews may be ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Kimmtrak |
| Other names | IMCgp100, tebentafusp-tebn |
| License data | US DailyMed: Tebentafusp |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1874157-95-5 |
| DrugBank | DB15283 |
| UNII | N658GY6L3E |
Medical uses
Tebentafusp is indicated for HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma.[1][2]
History
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 participants with metastatic uveal melanoma.[2] Participants were required to be HLA-A*02:01 genotype positive identified by a central assay.[2] Participants were excluded if prior systemic therapy or localized liver-directed therapy were administered.[2] Prior surgical resection of oligometastatic disease was permitted.[2] Participants with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.[2]
The U.S. Food and Drug Administration (FDA) granted Immunocore‘s application for tebentafusp priority review, breakthrough therapy, and orphan drug designations.[2]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761228s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves tebentafusp-tebn for unresectable”. U.S. Food and Drug Administration (FDA). 25 January 2022. Retrieved 28 January 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
External links
- “Tebentafusp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03070392 for “Safety and Efficacy of IMCgp100 Versus Investigator Choice in Advanced Uveal Melanoma” at ClinicalTrials.gov
/////////////////Tebentafusp-tebn, Kimmtrak, priority review, breakthrough designation, orphan drug designation, Immunocore Limited, IMCGP100, APPROVALS 2022, FDA 2022

NEW DRUG APPROVALS
ONE TIME
$10.00
Gefapixant citrate

Gefapixant
- Molecular FormulaC14H19N5O4S
- Average mass353.397 Da
1015787-98-0[RN]
10642
AF 217
5-[(2,4-Diamino-5-pyrimidinyl)oxy]-4-isopropyl-2-methoxybenzenesulfonamide
5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamide
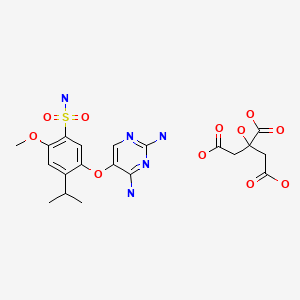
Gefapixant Citrate
| Formula | C14H19N5O4S. C6H8O7 |
|---|---|
| CAS | 2310299-91-1 |
| Mol weight | 545.5203 |
APPROVED JAPAN PMDA 2022/1/20, Lyfnua
ゲーファピキサントクエン酸塩
吉法匹生
| Efficacy | Analgesic, Anti-inflammatory, Antitussive, P2X3 receptor antagonist |
|---|---|
| Comment | Treatment of disorders associated with purinergic receptor activation |
Gefapixant (MK-7264) is a drug which acts as an antagonist of the P2RX3 receptor, and may be useful in the treatment of chronic cough.[1][2][3] It was named in honour of Geoff Burnstock.[4]
Gefapixant is under investigation in clinical trial NCT02397460 (Effect of Gefapixant (AF-219/MK-7264) on Cough Reflex Sensitivity).
PAPER
Organic Process Research & Development (2020), 24(11), 2445-2452.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00248
A robust, green, and sustainable manufacturing process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. The newly developed commercial process features low process mass intensity (PMI), short synthetic sequence, high overall yield, minimal environmental impact, and significantly reduced API costs. The key innovations are the implementation of a highly efficient two-step methoxyphenol synthesis, an innovative pyrimidine synthesis in flow, a simplified sulfonamide synthesis, and a novel salt metathesis approach to consistently deliver the correct active pharmaceutical ingredient (API) salt form in high purity.

SYN
Organic Process Research & Development (2020), 24(11), 2478-2490.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00252
Gefapixant citrate (MK-7264) is a P2X3 antagonist for the treatment of chronic cough. The second generation manufacturing route developed for the Step 3A/3B formylation–cyclization reaction to generate the key intermediate diaminopyrimidine (1) (AF-072) required a significant excess of ethyl formate (EF), potassium tert-butoxide (KOt-Bu), and guanidine•HCl (G•HCl) when both steps were run as batch processes. It was imperative to develop an alternative process that required less of each reagent and generated less carbon monoxide byproducts, as the annual production of the final active pharmaceutical ingredient (API) is expected to be over 50 MT. In addition, the second generation process was misaligned with our company’s strategy of having the best science in place at the first regulatory filing. The final flow–batch process described herein, which features a flow-based formylation combined with a batch cyclization, has been performed on a 500 kg scale and now requires 35% less EF (leading to a 70% reduction in waste carbon monoxide), 38% less KOt-Bu, and 50% less G•HCl. These improvements, along with a twofold increase in concentration, have resulted in a 54% reduction in the step process mass intensity (step-PMI) from the second generation two-step batch–batch process (PMI of 17.16) to the flow–batch process (PMI of 7.86), without sacrificing reaction performance.

SYN
H. REN*, K. M. MALONEY* ET AL. (MERCK & CO., INC., RAHWAY USA) Development of a Green and Sustainable Manufacturing Process for Gefapixant Citrate (MK-7264) Part 1: Introduction and Process Overview Org. Process Res. Dev. 2020, 24, 2445–2452, DOI: 10.1021/acs.oprd.0c00248.

Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
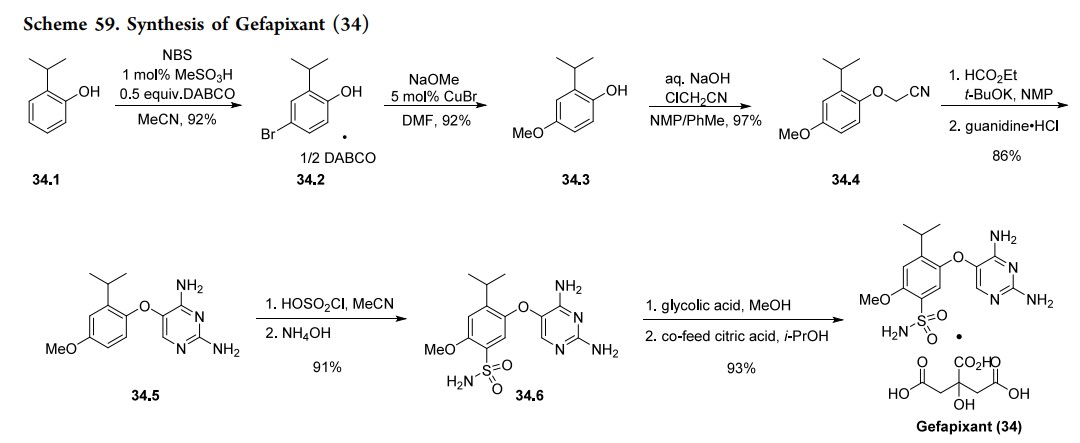
Gefapixant (Lyfnua). Gefapixant (34), also known as MK-7264, prior to that AF-219 and RO-4926219, is a P2 × 3antagonist for the treatment of chronic cough that was recently approved by the Japan Ministry of Health.243 Chronic cough is one of the most frequent reasons for patients to request medical consultation and is defined as cough ≥8 weeks in the past 12 months for those aged 18 years or older.244 The prevalence of chronic cough among US adults is 5% and can be associated with a deterioration of quality of life.244 The commercial manufacturing process of gefapixant has been described by Merck & Co., Inc., Rahway, NJ, USA, and is outlined in Scheme 59. Synthesis of 34 began with the regioselective bromination of isopropyl phenol 34.1. 245−247 The choice of polar MeCN solvent was found to play a critical role in the bromination regioselectivity providing the parabromophenol 34.2 in high yield. Interestingly, when toluene was used as the solvent the undesired ortho-substituted brominated phenol was the major product. In trial experiments it was discovered that a small amount of dibrominated product was formed which was alleviated using 1 mol % of methanesulfonic acid. Copper-mediated C−O bond formation proceeded with the use of NaOMe and CuBr in DMF to provide 34.3 in 92% yield. The authors describe in detail the screening conditions employed and the dimerization biproducts initially observed when obtaining 34.3. Ultimately, the use of DABCO in the first step allowed for the crystallization of the brominated phenol 34.2 as a DABCO
adduct. This enabled the Cu-catalyzed methoxylation to proceed without the need for phenol protection as well as the suppression of undesired dimerization products.247 Alkylation of phenol 34.3 with chloroacetonitrile in the presence of aqueous sodium hydroxide provided cyanomethyl intermediate 34.4.248 The diaminopyrimidine heterocycle was formed by formylation using ethyl formate and KOtBu
followed by reaction with guanidine HCl to complete the cyclization and obtain 34.5 in 81% yield.249 This was performed in a hybrid flow-batch telescoped process. Treat ment of 34.5 with chlorosulfonic acid in MeCN followed by ammonium hydroxide provided sulfonamide 34.6 in high yield.250,251 The final step in the manufacturing process was the isolation of gefapixant as a mono citrate salt.252,253 The free base of gefapixant was converted to a highly soluble glycolate salt which enabled complete dissolution in MeOH. Citric acid was added to crystallize final API as a mono citrate salt in 93%
yield.
(243) Merck & Co. Inc. Merck provides U.S. and Japan regulatory
update for gefapixant. https://www.merck.com/news/merck-providesu-s-and-japan-regulatory-update-for-gefapixant/ (accessed 2023-06).
(244) Yang, X.; Chung, K. F.; Huang, K. Worldwide prevalence, risk
factors and burden of chronic cough in the general population: a
narrative review. J. Thorac. Dis. 2023, 15, 2300−2313.
(245) Kocienski, P. Synthesis of gefapixant. Synfacts 2021, 17,
No. 0123.
(246) Ren, H.; Maloney, K. M.; Basu, K.; Di Maso, M. J.;
Humphrey, G. R.; Peng, F.; Desmond, R.; Otte, D. A. L.; Alwedi, E.;
Liu, W. J.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 1:
Introduction and process overview. Org. Process Res. Dev. 2020, 24,
2445−2452.
(247) Peng, F.; Humphrey, G. R.; Maloney, K. M.; Lehnherr, D.;
Weisel, M.; Levesque, F.; Naber, J. R.; Brunskill, A. P. J.; Larpent, P.;
Zhang, S. W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 2:
Development of a robust process for phenol synthesis. Org. Process
Res. Dev. 2020, 24, 2453−2461.
(248) Basu, K.; Lehnherr, D.; Martin, G. E.; Desmond, R. A.; Lam,
Y.-h.; Peng, F.; Chung, J. Y. L.; Arvary, R. A.; Zompa, M. A.; Zhang,
S.-W.; et al. Development of a green and sustainable manufacturing
process for gefapixant citrate (MK-7264). Part 3: development of a
one-pot formylation−cyclization sequence to the diaminopyrimidine
core. Org. Process Res. Dev. 2020, 24, 2462−2477.
(249) Otte, D. A. L.; Basu, K.; Jellett, L.; Whittington, M.; Spencer,
G.; Burris, M.; Corcoran, E. B.; Stone, K.; Nappi, J.; Arvary, R. A.;
et al. Development of a green and sustainable manufacturing process
for gefapixant citrate (MK-7264). Part 4: Formylation−cyclization as
a flow−batch process leads to significant improvements in process
mass intensity (PMI) and CO generated versus the batch−batch
process. Org. Process Res. Dev. 2020, 24, 2478−2490.
(250) Di Maso, M. J.; Ren, H.; Zhang, S.-W.; Liu, W.; Desmond, R.;
Alwedi, E.; Narsimhan, K.; Kalinin, A.; Larpent, P.; Lee, A. Y.; et al.
Development of a green and sustainable manufacturing process for
gefapixant citrate (MK-7264). Part 5: Completion of the API free
base via a direct chlorosulfonylation process. Org. Process Res. Dev.
2020, 24, 2491−2497.
(251) Rivera, N. R.; Cohen, R. D.; Zhang, S.-W.; Dance, Z. E. X.;
Halsey, H. M.; Song, S.; Bu, X.; Reibarkh, M.; Ren, H.; Lee, A. Y.;
et al. Gefapixant citrate (MK-7264) sulfonamide step speciation
study: Investigation into precipitation−dissolution events during
addition of chlorosulfonic acid. Org. Process Res. Dev. 2023, 27,
286−294.
(252) Maloney, K. M.; Zhang, S.-W.; Mohan, A. E.; Lee, A. Y.;
Larpent, P.; Ren, H.; Humphrey, G. R.; Desmond, R.; DiBenedetto,
M.; Liu, W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 6:
Development of an improved commercial salt formation process. Org.
Process Res. Dev. 2020, 24, 2498−2504.
(253) Mohan, A. E.; DiBenedetto, M.; Alwedi, E.; Ang, Y. S.; Asi
Sihombing, M. S. B.; Chang, H. Y. D.; Cote, A.; Desmond, R.; DiazSantana, A.; Khong, E.; et al. Development and Demonstration of a
Co-feed Process to Address Form and Physical Attribute Control of
the Gefapixant (MK-7264) Citrate Active Pharmaceutical Ingredient.
Org. Process Res. Dev. 2021, 25, 541−551.


AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
SYN
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00247

A scalable two-pot sulfonamidation through the process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. Direct conversion of the diaryl ether precursor to a sulfonyl chloride intermediate using chlorosulfonic acid, followed by treatment with aqueous ammonia hydroxide, provided the desired sulfonamide in high yield. A pH-swing crystallization allowed for the formation of a transient acetonitrile solvate that enables the rejection of two impurities. After drying, the desired anhydrous free base form was isolated in high yield and purity.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1566070221000898
Gefapixant is the approved generic name for a compound also known as MK-7264, and prior to that AF-219 and RO-4926219. It is the first-in-class clinically developed antagonist for the P2X3 subtype of trimeric ionotropic purinergic receptors, a family of ATP-gated excitatory ion channels, showing nanomolar potency for the human P2X3 homotrimeric channel and essentially no activity at related channels devoid of P2X3 subunits. As the first P2X3 antagonist to have progressed into clinical studies it has now progressed to the point of successful completion of Phase 3 investigations for the treatment of cough, and the NDA application is under review with US FDA for treatment of refractory chronic cough or unexplained chronic cough. The molecule was discovered in the laboratories of Roche Pharmaceuticals in Palo Alto, California, but clinical development then continued with the formation of Afferent Pharmaceuticals for the purpose of identifying the optimal therapeutic indication for this novel mechanism and establishing a clinical plan for development in the optimal patient populations selected. Geoff Burnstock was a close collaborator and advisor to the P2X3 program for close to two decades of discovery and development. Progression of gefapixant through later stage clinical studies has been conducted by the research laboratories of Merck & Co., Inc., Kenilworth, NJ, USA (MRL; following acquisition of Afferent in 2016), who may commercialize the product once authorization has been granted by regulatory authorities.
PATENT
WO 2008040652
https://patents.google.com/patent/WO2008040652A1/en

SCHEME AExample 1: 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamideThe synthetic procedure used in this Example is outlined in Scheme B.


not isolated


SCHEME BStep 1 2-Isopropyl-4-methoxy-phenolTo a cooled solution of l-(2-hydroxy-5-methoxy-phenyl)-ethanone (10.0 kg) in 79.0 kg of THF was gradually added 46.4 kg of 3M solution of MeMgCl in THF at a rate such that the reaction mixture temperature did not exceed 25°C. Following addition of the MeMgCl solution, the reaction mixture was stirred at ambient temperature for 18 hours, at which point HPLC (high pressure liquid chromatography) analysis showed more than 98% conversion of l-(2-hydroxy-5-methoxy-phenyl)-ethanone to 2- (1 -hydroxy- 1- methyl-ethyl)-4-methoxy-phenol (not shown in Scheme D). To the stirred solution was then added 10% palladium on carbon (1.02 kg, 50% water wet) suspended in 3.5 kg of THF. The reaction mixture was cooled and placed under a hydrogen atmosphere at 0.34 atmosphere pressure, and concentrated HCl (19.5 kg) was added while maintaining the reaction temperature at 25°C. The resultant mixture was stirred at ambient temperature for 18 hours, then treated with 44.4 kg water and filtered through a bed of Celite to remove suspended catalyst. The filter cake was rinsed with EtOAc and the combined filtrate was separated. The organic phase was washed with water, then concentrated by distillation to provide an oil. This oil was dissolved in 2-butanone (20.4 kg) and the crude solution was employed directly in the next step. A 161.8 g aliquot of the solution was concentrated under vacuum to provide 49.5 g of 2-isopropyl-4-methoxyphenol as an oil, projecting to 10.4 kg crude contained product in the bulk 2-butanone solution. 1H NMR (DMSO) delta: 1.14 (d, 6H, J = 6.9 Hz), 3.18 (septet, IH, J = 6.9 Hz), 3.65 (s, 3H), 6.56, (dd, IH, J = 8.6 Hz, 3.1 Hz), 6.67 (d, IH, J = 3.1 Hz), 6.69 (d, IH, 8.6 Hz).Step 2 (2-Isopropyl-4-methoxy-phenoxy)-acetonitrileA stirred slurry of toluene-4-sulfonic acid cyanomethyl ester (13.0 kg), potassium carbonate (13.0 kg) and 2-isopropyl-4-methoxyphenol (9.57 kg) in 79.7 kg of 2-butanone was heated to 55-600C for 4 days, then heated to reflux for 18 hours. The resultant slurry was cooled and filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was redissolved in toluene. The toluene solution was extracted with IN KOH, and the organic phase was concentrated by distillation to give 20.6 g of a 1:1 (by weight) solution of (2-isopropyl-4-methoxy-phenoxy)-acetonitrile in toluene, which was used directly in the next step. A aliquot (96.7 g) of this solution was concentrated to dryness to give 50.9 g of crude (2-isopropyl-4-methoxy-phenoxy)- acetonitrile, projecting to a yield of 10.9 kg in the bulk solution: MS (M+H) = 206; 1H NMR (CDCl3) delta: 1.25 (d, J = 6.9 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 4.76 (s, 2H), 6.73 (dd. IH, J = 8.8 Hz, 3.1 Hz), 6.87 (d, IH, J = 3.1 Hz), 6.91 (d, IH, J = 8.8 Hz).Step 3 5-(2-Isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine An approximately 1:1 (by weight) solution of 10.6 kg of (2-isopropyl-4-methoxy-phen- oxy) -acetonitrile in toluene was concentrated under reduced pressure and the residue was treated with 10.8 kg of tert-butoxybis(dimethylamino)methane (Brederick’s Reagent). The resulting mixture was dissolved in 20.2 kg of DMF and the solution was heated to 1100C for 2 hours, at which point HPLC analysis showed essentially complete conversion to 3,3-bis-dimethylamino-2-(2-isopropyl-4-methoxy-phenoxy)-propionitrile (not isolated, 1H NMR (CDCl3) delta: 1.21 (d, 3H, J = 7.2 Hz), 1.23 (d, 3H, J = 7.1 Hz), 2.46 (s, 6H), 2.48 (s, 6H), 3.43 (d, IH, J = 5.0 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.93 (d, IH, J = 5.0 Hz), 6.70 (dd, IH, J = 8.8 Hz, 3.0 Hz), 6.82 (d, IH, J = 3.0 Hz), 6.98 (d, IH, J = 8.8 Hz). The DMF solution was cooled and transferred onto 14.7 kg of aniline hydrochloride. The resulting mixture was heated to 1200C for 22 hours, at which point HPLC analysis showed greater than 97% conversion to 2-(2-isopropyl-4-methoxy-phenoxy)-3- phenylamino-acrylonitrile (not isolated, 1H nmr (CDCl3) delta: 1.31 (d, 6H, J = 6.9 Hz), 3.39 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 6.61 (d (br), IH, J = 12.7 Hz), 6.73 (dd, IH, J = 8.9 Hz, 3.1 Hz), 6.88 (d, IH, J = 3.0 Hz), 6.93 (m, 2H), 6.97 (d, IH, J = 8.9 Hz), 7.05 (m, IH), 7.17 (d, IH, J = 12.6 Hz), 7.35 (m. 2H)).The mixture was cooled, diluted with 21.5 kg toluene, then with 72.2 L of water. The organic layer was separated, washed with water, and concentrated by distillation. The concentrate was transferred into 23.8 kg DMF, and the DMF solution was transferred onto 6.01 kg of guanidine carbonate. The resulting mixture was heated to 1200C for 3 days, at which point HPLC analysis showed greater than 95% conversion of 2-(2- isopropyl-4-methoxy-phenoxy)-3-phenylamino-acrylonitrile into 5-(2-Isopropyl-4- methoxy-phenoxy)-pyrimidine-2,4-diamine. The reaction mixture was cooled, diluted with 7.8 kg of EtOAc, then reheated to 600C. Water (75.1 L) was added and the resultant mixture was allowed to cool to ambient temperature. The precipitated solid was collected by filtration, rinsed with isopropanol and dried under vacuum at 50 degrees to give 9.62 kg of 5-(2-isopropyl-4-methoxy- phenoxy)-pyrimidine-2,4-diamine: m.p. 170-171 degrees C; MS (M+H) = 275; H nmr (chloroform) delta: 1.25 (d, 6H, J = 6.9 Hz), 3.30 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.68 (br, 2H), 4.96 (br, 2H), 6.64 (dd, IH, J = 8.9 Hz, 3.0 Hz), 6.73, d, J = 8.9 Hz), 6.85 (d, IH, J = 3 Hz), 7.47 (s, IH).Step 4 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide, sulfolane solvate Chlorosulfonic acid (13.82 kg) was added to a slurry of 5-(2-isopropyl-4-methoxy-phen- oxy)-pyrimidine-2,4-diamine (10.07 kg) in sulfolane (50.0 kg) at a rate to maintain an internal pot temperature below 65°C. The reaction mixture was aged at 60-650C for 12 hours, at which point HPCL showed that all 5-(2-isopropyl-4-methoxy-phenoxy)- pyrimidine-2,4-diamine starting material had been converted to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonic acid. MS (M+H) = 355. Phosphorus oxychloride (3.41 kg) was then added to the reaction mixture at 600C. The reaction mixture was heated to 75°C and aged for 12 hours, at which point HPLC showed that approximately 99% of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonic acid had been converted to 5-(2,4-diamino-pyrimidin-5-yloxy)-4-iso- propyl-2-methoxy-benzenesulfonyl chloride. MS (M+H) = 373. The solution of 5-(2,4- diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride was then cooled to around 2°C).To a cooled (ca. 2°C) solution of ammonia (7N) in MeOH (74.1 kg) was added the cooled sulfolane solution of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride (a homogeneous syrup) at a rate such that the internal temperature did not exceed 23°C. The resultant slurry was stirred for 18 hours at ambient temperature, then filtered on a coarse porosity frit filter. The collected solids were rinsed with MeOH (15.9 kg), then dried under reduced pressure at 700C to a constant weight of 23.90 kg. HPLC showed 97.5% conversion of 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide sulfolane solvate. H nmr (DMSOd6) delta: 1.26 (d, 6H, J = 6.9 Hz), 2.07 (sym. m, 8H), 2.99 (sym. m, 8H), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 6.03 (s (br), 2H), 6.58 (s (br), 2H), 7.00 (s, IH), 7.04 (s (br), 2H), 7.08 (s, IH), 7.35 (s, IH).
Step 5 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamideA slurry of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide sulfolane solvate (23.86 kg) in a mixture of ethanol (74.3 kg) and 0.44 N HCl (109.4 kg) was heated to reflux to provide a homogeneous solution of the monohydrochloride salt of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamide. This solution was filterd while hot, then treated with concentrated ammonium hydroxide (3.4 L) to liberate the free base of 5-(2,4-diamino-pyrimidin-5- yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide. The resultant mixture was cooled slowly to 200C and the crystalline product isolated by filtration. The filter cake was washed with water (20.1 kg) and dried under reduced pressure at 700C to a constant weight of 8.17 kg (57.7% yield based on di-solvate of sulfolane).MP = 281-282 0C.1H nmr (DMSOd6) delta: 1.27 (d, 6H, J = 6.9 Hz), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 5.87 (s (br), 2H), 6.40 (s (br), 2H), 6.98 (s, IH), 7.01 (s (br), 2H), 7.07 (s, IH), 7.36 (s, IH).
PATENT
US 20080207655https://patents.google.com/patent/US20080207655
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016004358
xample 20
5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-N-methyl-benzenemethylsulfonamide Step 1. 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride
[211] A mixture of pyrimidine (0.400 g, 1.5 mmol) in 2 ml chlorosulfonic acid was allowed to stir 20 min. The mixture was poured over ice. The precipitate was filtered, washed by cold H2O and dried under vacuum to afford 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride (0.515 g, 95%) as a white solid; [MH]+= 373.
PATENT
WO 2017058645
https://patents.google.com/patent/WO2017058645A1/en
PATENTDisclosed herein is a novel process for preparing Compound A, a phenoxy diaminopyrimidine compound of the following formula, or a pharmaceutically acceptable salt thereof:

Compound A.Also disclosed herein are various salts and solvates of Compound A.
Scheme 1


Step 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalStep 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalThe following 4-bromo-2-isopropylphenol hemi-DABCO co-crystal is obtained in greater than 99% purity and at about 85-92% yield by the following process:

To a solution of 2-isopropyl phenol (75.0 g, 550 mmol) in acetonitrile (225 mL) was added MSA (0.520 g, 5.41 mmol). The mixture was cooled to -10 °C and NBS (98.01 g, 550 mmol) was added in portions while maintaining the internal temperature below 10 °C. The reaction was aged for 30 min to 1 h and then warmed to 20 °C, diluted with water (450 mL), and extracted with toluene (225 mL). The organic layer was sequentially washed with 9 wt% phosphoric acid (150 mL) and 5 wt% NaCl (150 mL). The organic layers were concentrated to roughly 150 mL and filtered into a clean reactor. The mixture was heated to 30-40 °C and n- heptane (28.5 mL) was added followed by DABCO (30.89 g, 275 mmol). The mixture was seeded (a seed can be synthesized from a previous batch of this procedure preformed without seeding) with 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (75 mg, 0.277 mmol), diluted with 52.5 mL of n-heptane, and stirred for 1 h. The slurry was cooled to 20 °C over 1 h and 370 mL of n-heptane is added over 2 h. The slurry was cooled to 5 °C over 2 h, aged for 2 h, filtered, and washed with n-heptane (2 x 75 mL). The solid was dried at 20-25 °C under vacuum to yield 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (134.8 g, 90 %) as a solid. 1H NMR (400 MHz, DMSO-76) d 7.20 (d, J= 2.5 Hz, 1H), 7.13 (dd, J= 8.5, 2.6 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.16 (hept, J= 6.9 Hz, 2H), 2.60 (s, 12H), 1.14 (d, J= 6.9 Hz, 12H).The crystallization of step 1 generates 4-bromo-2-isopropylphenol hemi-DABCO co-crystal, bromophenol mono-DABCO co-crystal, or a mixture of bromophenol hemi-DABCO co-crystal and bromophenol mono-DABCO co-crystal. An XRPD pattern of bromophenol hemi- DABCO co-crystal is shown in Figure 1.
The bromo-phenol mono-DABCO co-crystal can be generated in the following procedure:

bromophenol DABCO co-crystalTo a vial with a stir bar was charged DABCO (1.7 g, 15 mmol), phenol (2.5 g, 15 mmol), and 2 mL of n-heptane. The resulting slurry was stirred at 23 °C overnight. The slurry was then filtered and the resulting wet cake was washed with 2 mL of 5 °C n-heptane. The cake was dried under vacuum with nitrogen sweep to afford 4-bromo-2-isopropylphenol mono- DABCO co-crystal (2.9 g, 70% yield) as a solid. 1H NMR (500 MHz, DMSO-76) d 9.65 (s, 1H), 7.20 (s, 1H), 7.14 (d, J= 8.5 Hz, 1H), 6.74 (d, J= 8.5 Hz, 1H), 3.17 (hept, J= 6.8 Hz, 1H), 2.61(s, 12H), 1.15 (d, 7 = 6.9 Hz, 6H).An XRPD pattern of bromophenol mono-DABCO co-crystal is shown in Figure 2.Step 2a. Preparation of 2-Isopropyl-4-Methoxyphenol
The 2-isopropyl-4-Methoxyphenol shown below is obtained at about 92% yield by the following process:

bromophenol DABCO co-crystal methoxy phenolTo a solution of 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (120 g, 442 mmol) in 25 wt% sodium methoxide in methanol (430 g) was added 60 mL of DMF. The solution was pressure purged with nitrogen, copper (I) bromide (3.23 g, 22.5 mmol) was added to the mixture, and the reaction was heated to reflux for 12-16 h. The reaction is cooled to 0-5 °C and quenched with 6M HC1 until the pH of the solution is less than 5. The slurry is diluted with 492 mL of toluene and 720 mL of water to provide a homogeneous solution with a rag between the layers. The aqueous layer is cut to waste. The organic layer is filtered to remove the rag and washed with 240 mL of water to provide 2-isopropyl-4-methoxylphenol (491 g, 13.3 wt%, 89% assay yield) as a solution in toluene. 1H NMR (500 MHz, DMSO-76) d 8.73 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.66 (d, 7= 3.0 Hz, 1H), 6.55 (dd, 7= 8.6, 3.1 Hz, 1H), 3.65 (s, 3H), 3.17 (hept, j = 6.9 Hz, 1H), 1.14 (d, 7= 6.9 Hz, 6H).Step 2b. Preparation of 2-Isopropyl-4-Methoxyphenol
Alternatively, the methoxy phenol is obtained by the following process:

To a high-pressure vessel were charged 400 mL of anhydrous toluene, Re2(CO)io (3.16 g, 4.84 mmol) and mequinol (100 g, 806 mmol) at RT. The vessel was then degassed with propylene, and charged with propylene (85.0 g, 2.02 mol). The vessel was sealed and heated to 170 °C. Internal pressure was measured near 250 psi. The reaction was stirred at this condition for 72 h. The vessel was then allowed to cool down to 23 °C. The internal pressure was carefully released to 1 atmospheric pressure, and the toluene solution was assayed as 91% and used directly in the next step or isolated as a solid.Step 2a/2b results in anhydrous 2-isopropyl-4-methoxyphenol form 1. An XRPD pattern of the methoxy phenol form 1 is shown in Figure 3.In another embodiment, the product is isolated as a DMAP co-crystal:

To a vial with a stir bar was charged DMAP (3.67 g, 30.1 mmol), 2.5 ml of toluene, and 2-isopropyl-4-methoxylphenol (5.00 g, 30.1 mmol). The reaction mixture was stirred at RT for 5 min, and a homogeneous solution was formed. The reaction mixture was then cooled to 5 °C. Ten mL of n-heptane was slowly charged over 20 min. The resulting slurry was stirred at 5 °C overnight. The slurry was filtered and the resulting wet cake was washed with 3 mL of 5 °C n-heptane. The cake was dried under vacuum with a nitrogen sweep to provide 2- isopropyl-4-methoxylphenol DMAP co-crystal (7.01 g, 81%) as a solid. 1H NMR (500 MHz, DMSO-76) d 8.78 (s, 1H), 8.10 (d, J= 6.1 Hz, 2H), 6.71 – 6.65 (m, 2H), 6.57 (dd, J= 11.3, 6.0 Hz, 3H), 3.66 (s, 3H), 3.17 (hept, J= 6.8 Hz, 1H), 2.95 (s, 6H), 1.14 (d, J= 6.9 Hz, 6H).The crystallization generates anhydrous 2-isopropyl -4-methoxyphenol DMAP co crystal. An XRPD pattern of the 2-isopropyl-4-methoxyphenol DMAP co-crystal is shown in Figure 4.Step 3a. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
The cyanoether is obtained at about 95 % yield by the following process:

A 12-15 wt% solution of 2-isopropyl-4-methoxylphenol (314.3 g, 12 wt%, 226.8 mmol) was concentrated to greater than 50 wt% 2-isopropyl-4-methoxyphenol in toluene under vacuum at 40-50°C. To the solution was added 189 mL of NMP, and the mixture was cooled to 5 °C. Sodium hydroxide (27.2 g, 50 wt% in water, 340 mmol) and chloroacetonitrile (36 g, 340 mmol) were added sequentially to the mixture while maintaining the internal temperature below 10 °C. The reaction was aged for 2 h and then diluted with 150 mL of toluene and 226 mL of water while maintaining the temperature below 10 °C. The mixture was warmed to 20-25 °C, the layers were separated, and the organic layer was washed with 75 mL of 20 wt% NaCl (aq.). The organic layer was and filtered to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (56.8 g, 74.6 wt%) as a solution in toluene. The filter was washed with NMP to provide additional 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (27.1 g, 5.0 wt%) as a solution in NMP. The combined yield was about 94 %. 1H NMR (500 MHz, DMSO-i¾) d 7.05 (d, J= 8.8 Hz, 1H), 6.81 (d, 7= 3.0 Hz, 1H), 6.78 (dd, j= 8.8, 3.1 Hz, 1H), 5.11 (s, 2H), 3.73 (s, 3H), 3.20 (hept, j = 6.9 Hz, 1H), 1.17 (d, 7= 6.9 Hz, 6H).Step 3b. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
Alternatively, the cyanoether shown below is obtained at about 92% yield by the following process:

A solution of 2-isopropyl-4-methoxyphenol in toluene (491 g, 13.3 wt%, 393 mmol) was concentrated and solvent switched to acetonitrile under vacuum at 40-50 °C.Potassium carbonate (164.5 g, 1190 mmol) and tetrabutylammonium hydrogensulfate (1.5 g, 4.42 mmol) were added to a separate vessel, and the vessel was pressure purged with nitrogen gas.The solution of phenol in acetonitrile and chloroacetonitrile was added sequentially to the reaction vessel. The vessel was heated to 40 °C and aged for 4 h. The mixture was allowed to cool to 25 °C, and was diluted with 326 mL water. The layers were separated, and the organic layer was washed with 130 mL of 10 wt% NaCl. A solvent switch to toluene was performed under vacuum, and the organic layer was filtered through two 16D Cuno #5 cartridges. The organic layer was concentrated to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile in toluene (128.2 g, 58 wt%, 92% yield).Step 4 Preparation of the Dia inopyrimidine 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2.4-di amineThe diaminopyrimidine is obtained at about 90 % yield by the following process:

A solution of potassium tert-butoxide (44.8 g, 0399 mmol) in NMP (180 mL) was cooled to -10 °C. A solution of 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile, the cyanoether, (59.3 g, 61.4 wt%, 177 mmol) in toluene and ethyl formate (26.3 g, 355 mmol) was charged to the base solution while maintaining the internal temperature between -12 °C and -8 °C. After a 3 h age, guanidine hydrochloride (136 g, 1420 mmol) was added to the mixture and the reaction was heated to 115 °C for 6 h. The mixture was allowed to cool to 90 °C, diluted with 200 mL of water, and aged until the reaction mixture was homogeneous (about 30-45 min). After all solids dissolved, vacuum (400 mm Hg) was applied to the reactor to remove toluene. Vacuum was disconnected and the solution was allowed to cool to 85°C. 5-(2-Isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine seed (49.8 mg) (a seed can be synthesized by a route described in U.S. Patent 7,741,484) was charged, the solution was aged for 2 h, 200 mL of water was added, and the batch was allowed to cool to 20 °C over 6 h. The slurry was aged for 10 h at 20 °C, filtered, washed with 2: 1 water :NMP (3 x 100 mL) and water (3 x 100 mL), and dried under vacuum at 50 °C to provide the title compound (42.2 g, 88%) as a solid. 1H NMR (500 MHz, DMSO-r¾) d 7.23 (s, 1H), 6.83 (d, J= 3.0 Hz, 1H), 6.70 (dd, J= 8.9, 3.0 Hz, 1H), 6.63 (d, j= 8.8 Hz, 1H), 6.32 (s, 2H), 5.75 (s, 2H), 3.71 (s, 3H), 3.28 (hept, j= 6.9 Hz, 1H), 1.20 (d, j = 6.9 Hz, 6H); 13C NMR (126 MHz, DMSO-r¾) d 159.7, 157.2, 155.1, 148.4, 144.2, 139.0, 130.4,116.9, 112.5, 111.3, 55.4, 26.57, 22.83.The crystallization of step 4 generates an anhydrous 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1. An XRPD pattern of the 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 is shown in Figure 5.In one embodiment, 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2, 4-diamineNMP solvate 1 is obtained by adding excess amount of 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 into NMP in a closed vessel to form a suspension. The suspension is stirred at RT until the completion of form transition. The crystals of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of the 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 is shown in Figure 6.Step 5. Preparation of Compound A Free BaseCompound A free base is obtained at about 91% yield by a process comprising the steps:

To a suspension of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine, the diaminopyrimidine, (47.0 g, 171 mmol) in 141 mL of acetonitrile at -10 °C was added chlorosulfonic acid (63.1 mL, 942 mmol) while maintaining the internal temperature below 25 °C. The solution was aged for 1 h at 25 °C and then heated to 45 °C for 12 h. The solution was allowed to cool to 20 °C and added to a solution of 235 mL ammonium hydroxide and 71 mL of acetonitrile at -10 °C while maintaining the internal temperature below 15 °C. The slurry was aged at l0°C for 1 h, heated to 25 °C, and aged for 1 h. The slurry was diluted with 564 mL of water and 188 mL of 50 wt% sodium hydroxide to provide a homogeneous solution that was heated to 35 °C for 2 h. The solution was allowed to cool to 22 °C and the pH of the solution was adjusted to 12.9 with a 2M solution of citric acid. The solution was seeded with Compound A free base (470 mg, 1.19 mmol) (a seed can be synthesized by a route described in U.S. Patent 7,741,484), aged for 2 h, acidified to pH 10.5-11.3 with a 2M solution of citric acid over 5-10 h, and then aged for 2 h. The slurry was filtered, the resulting cake was washed with 90: 10 water: acetonitrile (2 x 118 mL) and water (2 x 235 mL), and dried at 55 °C under vacuum to provide Compound A free base (50.9 g, 91%) as a solid. 1H NMR (500 MHz, DMSO-i¾) d 7.36 (s, 1H), 7.07 (s, 1H), 7.05 – 6.89 (m, 3H), 6.37 (s, 2H), 5.85 (s, 2H), 3.89 (s, 3H), 3.41 (hept, J = 6.6 Hz, 1H), 1.27 (d, J= 6.8 Hz, 6H).The crystallization of step 5 generates anhydrous Compound A free base form 1. In one embodiment, Compound A free base acetonitrile solvate 1 can be prepared by adding excess amount of Compound A free base form 1 into acetonitrile in a closed vessel to form a suspension. The suspension is stirred at 50 °C until the completion of form transition.The crystals of Compound A free base acetonitrile solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of Compound A free base acetonitrile solvate 1 is shown in Figure 7.Step 6a. Preparation of Compound A Citrate SaltCompound A citrate salt is obtained by a process comprising the steps:

Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) were added to methanol (360 mL). The solution was heated to 60 °C, aged for 1 h, and filtered through a 0.6 pm filter into a clean vessel. A solution of citric acid (32.6 g, 170 mmol) in 2- propanol (180 mL) at RT was filtered through a 0.6 pm filter into the methanol solution over 30 min while the temperature of the methanol solution was maintained between 58-62 °C. The solution was seeded with Compound A citrate salt (450 mg, 0.825 mmol) (a seed can be synthesized by a route described in patent application number PCT/US17/66562), aged for 1 h, and diluted with 180 mL of 2-propanol over 3 h while the temperature was maintained between 58-62 °C. The slurry was cooled to 50 °C over 3 h. The slurry was filtered at 50 °C, washed with 1 : 1 methanol :2-propanol (120 mL) and 2-propanol (120 mL) at 50 °C, and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 97%) as a solid. 1H NMR (400 MHz, DMSO-76) d 10.89 (s, 3H), 7.33 (s, 1H), 7.10 (s, 1H), 7.07 (s, 3H), 7.04 (s, 2H), 6.44 (s, 2H), 3.91 (s, 3H), 3.34 (hept, J= 6.7 Hz, 1H), 2.69 (d, 7= 15.3 Hz, 2H), 2.60 (d, 7= 15.3 Hz, 2H), 1.26 (d, 7= 6.9 Hz, 6H). Step 6b. Alternative preparation of Compound A Citrate SaltAlternatively, Compound A citrate salt is obtained by a process comprising the steps:

To a suspension of Compound A citrate salt (4.5 g, 8.25 mmol) in methanol (72 mL) and 2-propanol (36 mL) at 50 °C were added simultaneously through separate 0.6 pm filters a solution of Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) in 360 mL of methanol at 50 °C and a solution of citric acid (19.5 g, 101 mmol) in 180 mL of 2- propanol at 25 °C over 8 h while maintaining the seed solution temperature of 60 °C. After the simultaneous addition is complete, citric acid (13.2 g, 68.7 mmol) in 180 mL of 2-propanol was added to the slurry over 8 h while the temperature was maintained at 60 °C. The slurry was allowed to cool to 50 °C and aged for 1 h, filtered at 50 °C, washed with 1 : 1 methanol :2- propanol (2 x 120 mL) and 2-propanol (120 mL), and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 88%) as a solid.The crystallization of step 6a/6b generates anhydrous Compound A citrate form 1. In another embodiment, Compound A citrate methanol solvate 1 can be prepared via a saturated solution of Compound A citrate form 1 in methanol at 50C. The solution is naturally cooled to ambient temperature or evaporated at ambient temperature until the crystals of Compound A citrate methanol solvate 1 can be acquired. An XRPD pattern of Compound A citrate methanol solvate 1 is shown in Figure 8.
PATENT
https://patents.google.com/patent/CN111635368B/enPreparation of the Compound Gefapixant of example 11Adding compound 7(16g) and dichloromethane (64mL) into a 250mL three-necked bottle, stirring for dissolving, cooling to below 5 ℃ in an ice bath, dropwise adding a mixed solution of chlorosulfonic acid (21.1g) and dichloromethane (16mL) into the reaction solution, and stirring for 1 hour at the temperature of not higher than 5 ℃; then heating to room temperature and continuing stirring for 10 hours, after the reaction is finished, pouring the reaction liquid into ice water, and quickly separating a water layer; the organic layer was washed once with ice water, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give a crude product. Dissolving the crude product with 30ml of acetonitrile, and cooling to below 5 ℃; 16ml of ammonia water (25-28%) is dripped into the solution, and after the dripping is finished, the solution is heated to room temperature and stirred for 20 hours. After the reaction is completed, concentrating the reaction solution under reduced pressure to remove acetonitrile, and separating out a white solid; and filtering again, and drying the filter cake at 70 ℃ under reduced pressure for 24h to obtain Gefapixant: white powder (19.50g), yield 94.6%, purity: 97.2 percent.Example 12 purification of the Compound GefapixantAdding a compound Gefapixant (20.77g) into a 500mL reaction bottle, adding 0.44N hydrochloric acid (95.4mL), absolute ethyl alcohol (64.4g) and nitrogen protection, heating to 75 ℃, stirring for dissolving, then carrying out heat preservation and reflux for 1 hour, filtering while hot, after filtering, heating the filtrate again to 60 ℃, dropwise adding ammonia water (25-28 percent and 2.96mL), closing and heating after dropwise adding, slowly cooling to room temperature, and gradually precipitating white solids. And continuously cooling the reaction solution to 20 ℃, keeping the temperature and stirring for 4h, filtering, washing a filter cake with 15ml of water, and performing vacuum drying on the obtained wet product at 60 ℃ for 24h to obtain Gefapixant: white powder (6.58g), yield 53.2%, purity: 99.5 percent.1H NMR(400MHz,DMSO)δ7.37(s,1H),7.08(s,1H),7.02(s,2H),7.00(s,1H),6.43(brs,2H),5.89(s,2H),3.90(s,3H),3.42(m,1H),1.28(d,J=8.0Hz,6H);LC-MS:m/z=354.1[M+H]+。
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Muccino D, Green S (June 2019). “Update on the clinical development of gefapixant, a P2X3 receptor antagonist for the treatment of refractory chronic cough”. Pulmonary Pharmacology & Therapeutics. 56: 75–78. doi:10.1016/j.pupt.2019.03.006. PMID 30880151.
- ^ Richards D, Gever JR, Ford AP, Fountain SJ (July 2019). “Action of MK-7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation”. British Journal of Pharmacology. 176 (13): 2279–2291. doi:10.1111/bph.14677. PMC 6555852. PMID 30927255.
- ^ Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (December 2019). “Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications”. Expert Opinion on Therapeutic Patents. 29 (12): 943–963. doi:10.1080/13543776.2019.1693542. hdl:11581/435751. PMID 31726893. S2CID 208037373.
- ^ Ford, Anthony P.; Dillon, Michael P.; Kitt, Michael M.; Gever, Joel R. (November 2021). “The discovery and development of gefapixant”. Autonomic Neuroscience. 235: 102859. doi:10.1016/j.autneu.2021.102859.
| Clinical data | |
|---|---|
| ATC code | R05DB29 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1015787-98-0 |
| PubChem CID | 24764487 |
| DrugBank | DB15097 |
| ChemSpider | 58828660 |
| UNII | 6K6L7E3F1L |
| KEGG | D11349 |
| ChEMBL | ChEMBL3716057 |
| Chemical and physical data | |
| Formula | C14H19N5O4S |
| Molar mass | 353.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Gefapixant, Lyfnua, JAPAN 2022, APPROVALS 2022, ゲーファピキサントクエン酸塩 , MK 7264, 吉法匹生 , AF 217

NEW DRUG APPROVALS
ONE TIME
$10.00
UPDATE
.WO/2022/060945SOLID STATE FORMS OF GEFAPIXANT AND PROCESS FOR PREPARATION THEREOF
TEVA
Gefapixant, 5-(2, 4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide, has the following chemical structure:
[0003] Gefapixant is a purinergic P2X3 receptor antagonist, and it is developed for the treatment of chronic cough. Gefapixant is also under clinical investigation as a treatment for asthma, interstitial cystitis, musculoskeletal pain, pelvic pain, and sleep apnea syndrome.
[0004] The compound is described in International Publication No. WO 2005/95359.
International Publication No. WO 2008/040652 disclosed a sulfonate solvate of Gefapixant. International Publication Nos. WO 2018/118668 and WO 2019/209607 disclose crystalline forms of Gefapixant as well as Gefapixant salts.
[0005] Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g., measured by thermogravimetric analysis (“TGA”), or differential scanning calorimetry (“DSC”)), X-ray diffraction (XRD) pattern, infrared absorption fingerprint, and solid state (13C) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
[0007] Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, including a different crystal habit, higher crystallinity, or polymorphic stability, which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemi cal/phy si cal stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of Gefapixant or salts or co-crystals thereof.
Daridorexant

Daridorexant
- Molecular FormulaC23H23ClN6O2
- Average mass450.921 Da
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1[RN]
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
ACT-541468, , Nemorexant
FDA APPROVED 2022, 1/7/2022, To treat insomnia,

Daridorexant HCl
CAS#: 1792993-84-0 (HCl)
Chemical Formula: C23H24Cl2N6O2
Molecular Weight: 487.39
Elemental Analysis: C, 56.68; H, 4.96; Cl, 14.55; N, 17.24; O, 6.57
Methanone, ((2S)-2-(6-chloro-7-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)-, hydrochloride (1:1)
Daridorexant HCl; Daridorexant hydrochloride; ACT541468A; ACT 541468A; ACT-541468A; ACT541468 hydrochloride; ACT 541468 hydrochloride; ACT-541468 hydrochloride
Daridorexant HCl is used in the treat of Insomnia Disorder in Adult Patients
Daridorexant, sold under the brand name Quviviq, is a medication used for the treatment of insomnia.[1] Daridorexant is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals.[3][4] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[3][4] The medication has a relatively short elimination half-life of 6 to 10 hours.[2] As of April 2020, daridorexant has passed its first phase III clinical trial for the treatment of insomnia.[3]Daridorexant was approved for medical use in the United States in January 2022.[1][5][6]
Daridorexant, formerly known as nemorexant, is a selective dual orexin receptor antagonist used to treat insomnia. Insomnia is characterized by difficulties with sleep onset and/or sleep maintenance and impairment of daytime functioning. It chronically affects the person’s daily functioning and long-term health effects, as insomnia is often associated with comorbidities such as hypertension, diabetes, and depression. Conventional treatments for insomnia include drugs targeting gamma-aminobutyric acid type-A (GABA-A), serotonin, histamine, or melatonin receptors; however, undesirable side effects are frequently reported, such as next-morning residual sleepiness, motor incoordination, falls, memory and cognitive impairment. Novel drugs that target orexin receptors gained increasing attention after discovering the role of orexin signalling pathway in wakefulness and almorexant, an orexin receptor antagonist that improved sleep. Daridorexant was designed via an intensive drug discovery program to improve the potency and maximize the duration of action while minimizing next-morning residual activity.1
Daridorexant works on orexin receptors OX1R and OX2R to block the binding of orexins, which are wake-promoting neuropeptides and endogenous ligands to these receptors. Daridorexant reduces overactive wakefulness: in the investigational trials, daridorexant reportedly improved sleep and daytime functioning in patients with insomnia.1 It was approved by the FDA on January 10, 2022, under the name QUVIVIQ.6 as the second orexin receptor antagonist approved to treat insomnia following suvorexant.2
QUVIVIQ
- Generic Name: daridorexant tablets
- Brand Name: Quviviq
QUVIVIQ contains daridorexant, an orexin receptor antagonist. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5- methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
 |
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains 27 mg or 54 mg of daridorexant hydrochloride equivalent to 25 mg or 50 mg of daridorexant, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
Dosage Forms And Strengths
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with “25” on one side and “i” (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with “50” on one side and “i” (Idorsia logo) on the other side, containing 50 mg daridorexant.
QUVIVIQ tablets are available as:
25 mg, light purple, arc-triangle shaped film-coated tablets debossed with “25” on one side, and “i” on the other side. NDC 80491-7825-3, bottle of 30 with child-resistant closure
50 mg: light orange, arc-triangle shaped film-coated tablets debossed with “50” on one side, and “i” on the other side. NDC 80491-7850-3, bottle of 30 with child-resistant closure
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202000453
Since its discovery in 1998, the orexin system has been of interest to the research community as a potential therapeutic target for the treatment of sleep/wake disorders. Herein we describe our efforts leading to the identification of daridorexant, which successfully finished two pivotal phase 3 clinical trials for the treatment of insomnia disorders.

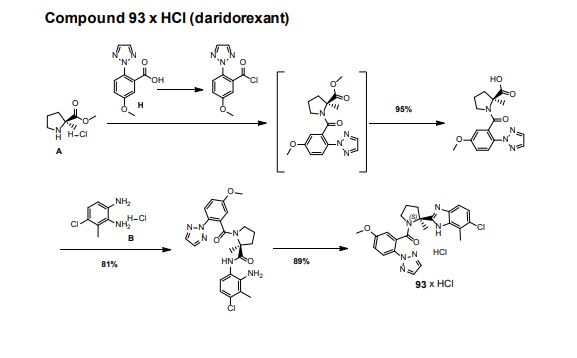
Step 3. Amide (S7) (1000 g, 2.13 mmol) was dissolved in EtOH (5 L) and 32% aqueous HCl (500 mL) was added at 23 °C. The solution was filtered through a Whatman filter (5 µm). The filtrate was heated to 75 °C for 4h. The resulting suspension was cooled to 0 °C and filtered. The product was dried under reduced pressure to yield 93 x HCl (922 g, 89%) as a white solid.
LC-MS B: tR = 0.78 min; [M+H]+ = 451.19, mp 280 °C.
1H NMR (500 MHz, D6-DMSO) δ: 15.05- 15.65 (m, 1 H), 8.06 (s, 2 H), 7.79 (s, 1 H), 7.75 (d, J = 8.9 Hz, 2 H), 7.66 (m, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.15 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 4.06-4.10 (m, 1 H), 3.92 (s, 3 H), 3.35 (s, 1 H), 2.78 (s, 3 H), 2.54-2.67 (m, 1 H), 2.23-2.31 (m, 1 H), 2.06-2.20 (m, 2 H), 1.97 (s, 3 H),
13C NMR (125 MHz, D6-DMSO) δ: 166.2, 159.3, 158.6, 136.5, 132.7, 131.9, 130.4, 130.3, 129.4, 126.8, 124.5, 123.4, 116.4, 113.7, 113.0, 61.6, 56.8, 49.7, 41.1, 23.9, 20.2, 15.7.

SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.201900618
Abstract
DORA explorers: The orexin system plays an important role in regulating the sleep-wake cycle. Herein we report our optimization efforts toward a novel dual orexin receptor antagonist (DORA) with improved properties over compound 6. Replacing the oxadiazole by a triazole resulted in compounds (e. g. compound 33) with improved properties, such as higher intrinsic metabolic stability, lower plasma protein binding, higher brain free fraction, and increased solubility. Further optimization was needed to decrease the compounds P-glycoprotein susceptibility. Our work led to the identification of compound 42, a potent, brain-penetrating DORA with improved in vivo efficacy in dogs compared with compound 6.

Abstract
The orexin system is responsible for regulating the sleep-wake cycle. Suvorexant, a dual orexin receptor antagonist (DORA) is approved by the FDA for the treatment of insomnia disorders. Herein, we report the optimization efforts toward a DORA, where our starting point was (5-methoxy-4-methyl-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-[5-(2-trifluoromethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-pyrrolidin-1-yl}methanone (6), a compound which emerged from our in-house research program. Compound 6 was shown to be a potent, brain-penetrating DORA with in vivo efficacy similar to suvorexant in rats. However, shortcomings from low metabolic stability, high plasma protein binding (PPB), low brain free fraction (fu brain), and low aqueous solubility, were identified and hence, compound 6 was not an ideal candidate for further development. Our optimization efforts addressing the above-mentioned shortcomings resulted in the identification of (4-chloro-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-methyl-2-[5-(2-trifluoromethoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-pyrrolidin-1-yl}l-methanone (42), a DORA with improved in vivo efficacy compared to 6.
PAT
WO 2015083071
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PAT
WO 2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689
Examples
Reference Example 1
Synthesis of 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
4,5-dibromo-2-(4-methoxy-2-nitrophenyl)-2H-1,2,3-triazole
4- Fluoro-3-nitroanisole (3.44 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (4.56 g, 1 eq.)1, K2C03 (2.78 g, 1 eq.) and DMF (30 mL) are heated to 1 10 °C for 32 h. The reaction mixture is cooled to 22 °C and treated with water (70 mL). The resulting suspension is filtered, washed with water (15 mL). The product is slurried in isopropanol (40 mL), filtered and dried under reduced pressure to yield a white solid. Yield: 6.42 g, 84%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, CDCI3) δ: 7.71 (d, J = 8.9 Hz, 1 H), 7.47 (d, J = 2.8 Hz, 1 H), 7.25 (dd, Ji = 2.8 Hz, J2 = 8.9 Hz, 1 H), 3.97 (s, 3 H).
1 X. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake, P. Wipf Organic Letters 2010 12 (20), 4632-4635.
5- methoxy-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(4-methoxy-2-nitrophenyl)-2/-/-1 ,2,3-triazole (2 g, 1 eq.), sodium acetate (1.3 g, 3 eq.), and 10% Pd/C 50% water wet (0.3 g) is suspended in EtOAc (10 mL). The mixture is heated to 50 °C and set under hydrogen until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with 1 N NaOH (10 mL) and water (15 mL). The organic layer is concentrated under reduced pressure to yield an oil. Yield: 0.95 g, 94%. Purity: 96% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.05 (s, 2 H), 7.53 (d, J = 8.9 Hz, 1 H), 6.49 (d, J = 2.7 Hz, 1 H), 6.30 (dd, Ji = 2.7 Hz, J2 = 8.9 Hz, 1 H), 5.94 (s, 2 H), 3.74 (s, 3 H).
5-methoxy-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline (455 g, 1 eq ) is dissolved in isopropanol (3 L). To the solution is added cone. H2SO4 (235 g, 1 eq.) below 40 °C. The suspension is cooled to
20 °C and filtered. The cake is washed with isopropanol (700 mL) and TBME (1.5 L). The product is dried to obtain a white solid. Yield: 627 g, 91 %. Purity: 100% a/a (LC-MS method 2).
2-(2-iodo-4-methoxyphenyl)-2H-1,2,3-triazole
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (200 g, 1 eq.) is dissolved in 2 M aq. H2SO4 soln. (1.4 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (62 g, 1.3 eq.) in water (600 mL) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of Kl (161 g, 1.4 eq.) in water (700 mL) at 65 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (27 g, 0.4 eq.) in water (120 mL). The mixture is extracted with isopropyl acetate (2 L). The organic layer is washed with a mixture of 2 N NaOH (500 mL) and 40% NaHS03 soln. (100 mL), and a mixture of 1 N HCI (50 mL) and water (500 mL). The organic layer is concentrated to dryness. The residue is dissolved in isopropanol (700 mL) and cooled to 0 °C. The resulting suspension is filtered. The solid is dried under reduced pressure. Yield: 164 g, 79%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.08 (s, 2 H), 7.57 (d, J = 2.8 Hz, 1 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.13 (dd, Ji = 2.8 Hz, J2 = 8.8 Hz, 1 H), 3.85 (s, 3 H).
5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-4-methoxyphenyl)-2/-/-1 ,2,3-triazole (200 g, 1 eq.) is dissolved in THF (2 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (350 mL, 1.05 eq.) is added at 0 °C. The mixture is cooled to -20 °C and C02 (gas) is bubbled into the solution over 30 min until the exothermicity is ceased. To the mixture is added 2 N HCI (600 mL) at 8 °C and concentrated under reduced pressure to remove 2.4 L solvent. The residue is extracted with TBME (1.6 L). The organic layer is washed with 1 N HCI (200 mL) and extracted with 1 N NaOH (600 mL and 200 mL). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (160 mL). The resulting suspension is filtered and washed with water (200 mL). Yield: 127 g, 87%. Purity: 100% a/a (LC-MS method 2); MP: 130 °C (DSC goldpan). The obtained product may be re-crystallized from toluene (MP: 130.9 °C) or water (MP: 130 °C).
Table Ref 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 2 (recrystallization from toluene)
Technique Data Summary Remarks
XRPD Crystalline see Fig. 8
Reference Example 2
Synthesis of 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
4,5-Dibromo-2-(5-methyl-2-nitrophenyl)-2H-1 ,2,3-triazole
3- Fluoro-4-nitrotoluene (1367 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (1999 g, 1 eq.), K2C03 (1340 g, 1.1 eq.) and DMF (1 1 L) is heated to 75 °C for 15 h. The reaction mixture is cooled to 22 °C and treated with water (18 L). The resulting suspension is filtered, washed with water (4 L). The product is washed with isopropanol (5 L), and dried under reduced pressure to yield a white solid. Yield: 281 1 g, 88%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.10 (d, J = 8.3 Hz, 1 H), 7.86 (d, J = 1.0 Hz, 1 H), 7.66 (dd, J1 = 0.9 Hz, J2 = 8.3 Hz, 1 H), 2.51 (s, 3 H).
4- Methyl-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(5-methyl-2-nitrophenyl)-2/-/-1 ,2,3-triazole (205 g, 1 eq.), sodium acetate (149 g, 3.2 eq.), and 5% Pd/C 50% water wet (37.8 g) is suspended in EtOAc (0.8 L). The mixture is heated to 40-50 °C and set under hydrogen (2 bar) until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with water (300 mL), 2N NaOH (300 ml_+250 mL) and water (300 mL). The organic layer is concentrated under reduced pressure to yield a yellow oil. Yield: 132 g, 90%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.09 (s, 2 H), 7.48 (d, J = 1.3 Hz, 1 H), 6.98 (dd, J1 = 1.8 Hz, J2 = 8.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 5.79 (s, 2 H), 2.23 (s, 3 H).
4-Methyl-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl) aniline (199 g, 1 eq ) is dissolved in isopropanol (1.7 L). To the solution is added cone. H2SO4 (118 g, 1.05 eq.) below 40 °C. The suspension is cooled to 20 °C and filtered. The cake is washed with isopropanol (500 mL). The product is dried to obtain a white solid. Yield: 278 g, 89%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.21 (s, 2 H), 7.70 (s, 1 H), 7.23 (s, 2 H), 2.35 (s, 3 H).
2-(2-iodo-5-methylphenyl)-2H-1 ,2,3-triazole
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (1553 g, 1 eq.) is dissolved in 1 M aq. H2S04 Soln. (1 1 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (433 g, 1.1 eq.) in water (4 L) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of potassium iodide (1325 g, 1.4 eq.) in water (4 L) at 55-70 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (220 g, 0.4 eq.) in water (900 mL). The mixture is extracted with isopropyl acetate (13 L). The organic layer is washed with a mixture of 2 N NaOH (3.5 L) and 40% NaHSOs soln. (330 g), and a mixture of 1 N HCI (280 mL) and water (3.5 L). The
organic layer is concentrated to dryness. Yield: 1580 g, 97%. Purity: 91 % a/a (LC-MS method 2). 1 H NMR (400 MHz, CDCI3) <5: 7.90 (s, 2 H), 7.87 (d, J = 8.1 Hz, 1 H), 7.34 (d, J = 1 .6 Hz, 1 H), 7.03-7.06 (m, 1 H), 2.40 (s, 3 H).
The crude product, together with a second batch (141 1 g) is purified by distillation on a short path distillation equipment at 120 °C jacket temperature, feeding tank (70 °C), cooling finger (20 °C) and at a pressure of 0.004 mbar. Yield: 2544 g (78%), Purity: 100 % a/a ()LC-MS method 2).
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-5-methylphenyl)-2/-/-1 ,2,3-triazole (1250 g, 1 eq.) is dissolved in THF (13 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (2.2 L, 1 eq.) is added at 0 °C. The mixture is cooled to -25 °C and CO2 (gas) is bubbled into the solution over 60 min until the exothermicity is ceased. To the mixture is added 2 N HCI (5 L) at 4 °C and concentrated under reduced pressure to remove 14.5 L solvent. The residue is extracted with TBME (10 L). The organic layer is extracted with 1 N NaOH (6 L and 3 L). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (1 .23 L). The resulting suspension is filtered and washed with water (5 L). Yield: 796 g, 89%. Purity: 100% a/a (LC-MS method 2); MP: 125 °C (DSC goldpan).
The following examples illustrate the invention.
Example 1 :
Example 1.1: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methoxybenzoic acid (21 .5 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (600 mL) and water (8.4 mL). To the mixture were added 1 H-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-/V,/V-dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 3.5 h. IPC showed full conversion. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 84: 16. The mixture was cooled to 40 °C and filtered. The cake was washed with dioxane (100 mL). The solid was dried to obtain 50.6 g of a blue solid. The ratio of N{2) to Λ/(1 ) isomer of was 98.6: 1 .4.
Table 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 1.2: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.1 was dissolved in water (300 mL). TBME (200 mL) and 32% aq. HCI (35 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (20 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 optionally seed crystals were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.4 g, 61 %. Purity: 100% a/a, tR 0.63 min. Seed crystals may be obtained by careful crystallization according to the above procedure.
MP: 80 °C (DSC).
1H NMR (400 MHz, DMSO) & 3.87 (s, 3 H), 7.26 (m, 2 H), 7.64 (d, J = 8.7 Hz, 1 H), 8.02 (s, 2 H), 13.01-13.22 (br, 1 H).
Table 2: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Example 1.3: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid, e.g. obtained according to the procedure of Reference Example 1 (5 g, 0.0228 mol) and KHCO3 (1.61 g, 0.7 eq) were suspended in dioxane (100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.56 g, 44%. 1H NMR (400 MHz, D20) & 3.80 (s, 3 H), 7.04 (m, 2 H), 7.46 (d, J = 8.7 Hz, 1 H), 7.82 (s, 2 H). MP: 279.5°C (DSC shows additionally a broad endothermic event at about 153 °C to 203 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
Table 3: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 2
Example 1.4: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
In an alternative procedure, 2-Bromo-5-methoxybenzoic acid (20 g, 0.086 mol, 1 eq.) copper (I) iodide (0.824 g, 0.05 eq.), and K2C03 powder (26.9 g, 2.25 eq.) were suspended in dioxane (494 mL). To the mixture was added 1 H-1 ,2,3-triazole (12 g, 2 eq.). The mixture was heated at reflux for 1 h. To the mixture was added water (12.5 g, 8 eq.). The mixture was heated at reflux for 2 h. Solvent (100 mL) was removed by distillation. The residue was cooled to 45 °C in 8 min, filtered and washed with dioxane (50 mL).
XRPD corresponds to crystalline form 1 (see Fig. 1 , Example 1.1 ).
Example 1.5: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.4 was dissolved in water (200 mL). The mixture was heated to 50 °C and 20% aq. H2SO4 (40 mL) was added to adjust the pH to 5. The mixture was filtered over Celite. The filtrate was treated at 45 °C with 20% aq. H2S04 (40 mL). At pH 3 seeds (obtained for example using the procedure of reference example 1 ) were added. The suspension was stirred at 45 °C and filtered. The product was washed with water (20 mL) and dried at 60 °C and 10 mbar to yield a white solid. Yield: 10.8 g, 57%. Purity: 100% a/a, tR 0.63 min.
Characterisation of 5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid obtained according to Example 1.5:
XRPD corresponds to crystalline form 1 (see Fig. 2, Example 1.2).
Example 2:
Example 2.1: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-4-methylbenzoic acid (20 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 A7-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-Λ/,ΛΑ-
dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of 98.5%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 75:25. The mixture was concentrated at normal pressure and external temperature of 130 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 48.8 g of a blue solid. The ratio of N(2) to Λ/(1 ) isomer was 98.7: 1 .3.
Table 4: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 2.2: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid
The solid of Example 2.1 was dissolved in water (300 mL) and filtered. To the filtrate were added TBME (200 mL) and 32% aq. HCI (30 mL). The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (10 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 1 1 .7 g, 62%. Purity: 100% a/a. tR 0.66 min.
MP: 125 °C (DSC).
1H NMR (400 MHz, DMSO) & 2.44 (s, 3 H), 7.41 (d, J = 7.9 Hz, 1 H), 7.56 (s, 1 H), 7.68 (d, J = 7.9 Hz, 1 H), 8.06 (s, 2 H), 12.53-13.26 (br, 1 H)
Table 5: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Technique Data Summary Remarks
XRPD Crystalline see Fig. 5
Example 2.3: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and KHC03 (1 .74 g , 0.7 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.47 g, 42% . MP: 277 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.28 (d, J = 7.9 Hz, 1 H), 7.39 (m, 2 H), 7.84 (s, 2 H).
MP: 276.8 °C (DSC shows additionally a broad endothermic event at about 140 °C to 208 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
XRPD corresponds to crystalline form 1 (see Fig. 4, Example 2.1 ).
Reference Example 3:
Reference Example 3.1: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid sodium salt (sodium 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), Na2CC>3 powder (24.6 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 5 h. IPC showed a conversion of >99%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 78:22. The mixture was concentrated at normal pressure and external temperature of 135 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 36.2 g of a yellow solid. The ratio of N(2) to Λ/( 1 ) isomer of was 99: 1 .
Table 6: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid sodium salt in crystalline form 1
Reference Example 3.2: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid obtaind in Reference Example 3.1 was dissolved in water (300 mL) and filtered. To the filtrate was added TBME (200 mL) and 32% aq. HCI (30 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with 1 N aq. HCI ( 100 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed
under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of Reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.1 g, 64%. Purity: 100% a/a. tR 0.67 min.
MP: 173 °C (DSC)
1 H NMR (400 MHz, DMSO) & 2.42 (s, 3 H), 7.50-7.52 (m, 1 H), 7.58 (s, 1 H), 7.63 (m, 1 H), 8.05 (s, 2 H), 13.01 (s, 1 H).
Table 7: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Reference Example 3.3: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid sodium salt
5-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and Na2C03 (1 .05 g, 0.4 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.79 g, 50%. MP: 341 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.30 (m, 2 H), 7.43 (m, 1 H), 7.83 (s, 2 H).
XRPD corresponds to crystalline form 1 (see Fig. 6, Reference Example 3.1 ).
Reference Example 3.4: 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.1 g, 2.5 eq.) were suspended in dioxane (600 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of >94%. The ratio of the desired N(2) to the regioisomeric Λ/( 1 ) isomer was 78:22. The mixture was cooled to 35 °C and filtered. The cake was washed with dioxane (100 mL). The products were dissolved in water and a LC-MS was recorded. The ratio of N(2) to Λ/(1 ) isomer of was 83: 17.
Reference Example 4.1: Methyl (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoic acid (100 g, 0.46 mol) was suspended in DCM (650 mL) and DMF (10 mL) at 20 °C. To this suspension was added oxalyl chloride (51 mL, 0.59 mol) over a period of 30 min. LC-MS showed 60% conversion to acid chloride intermediate. Oxalyl chloride (17.6 mL, 0.45 eq.) was added dropwise. LC-MS showed full conversion to acid chloride intermediate.
Methyl (S)-2-methylpyrrolidine-2-carboxylate hydrochloride (84 g, 0.47 mol) was suspended in DCM (800 mL) in a second flask. The suspension was cooled to 10 °C. Triethylamine (200 mL, 1.41 mol) was added over 15 min. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. The reaction mixture was washed with 1 M HCI (500 mL), 1 N NaOH (500 mL) and water (500 mL). The organic layer was concentrated to dryness to give a light-yellow solid as product. Yield: 157 g, 100%, 99% a/a (LC-MS), M+1 =345. 1H NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.79 (d, J = 8.9 Hz, 1 H), 7.21 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 6.85 (d, J = 1.9 Hz, 1 H), 3.89 (s, 3 H), 3.66 (s, 3 H), 3.29 (m, 1 H), 3.03 (m, 1 H), 2.08 (m, 1 H), 1.82 (m, 3 H), 1.50 (s, 3 H).
Reference Example 4.2: (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid
Methyl (S)-1-(5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate (157 g, 0.46 mol) was dissolved in MeOH (750 mL) at 20 °C. To this solution was added 16% NaOH (300 mL). The resulting solution was heated up to 80 °C and stirred for 60 min. Solvent was distilled off under reduced pressure (850 mL). The residue was taken up in DCM (1500 mL) and water (450 ml) at 20 °C. 32% HCI (200 mL) was added. Layers were separated and the organic layer was washed with water (450 mL). The organic layer was concentrated to the minimum stirring volume under reduced pressure. Toluene (750 mL) was added and solvent was further distilled under vacuum (150 mL distilled). The mixture was cooled to 20 °C and stirred for 15 min. The suspension was filtered at 20 °C. The cake was rinsed with toluene (150 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 128 g, 85%, 94% a/a (LC-MS), M+1 =331. Melting point: 178 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 12.3 (s, 1 H), 8.04 (s, 2 H), 7.79 (d, 1 H), 7.20 (dd, J1 = 2.8 Hz, J2 = 8.9 Hz, 1 H), 6.84 (m, 1 H), 3.88 (s, 3 H), 3.29 (m, 1 H), 2.99 (m, 1 H), 2.1 1 (m, 1 H), 1.81 (m, 3 H), 1.47 (s, 3 H).
Reference Example 4.3: (S)-N-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide
(S)-1-(5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid (128 g, 0.39 mol) was suspended in DCM (850 mL) and DMF (6 mL) at 20 °C. To this suspension was added oxalyl chloride (39 mL, 0.45 mol) over a period of 30 min. 4-Chloro-3-methylbenzene-1 ,2-diamine hydrochloride (75 g, 0.39 mol) was suspended in DCM (1300 mL) in a second flask. The suspension was cooled down to 10 °C. Triethylamine (180 mL, 1.27 mol) was added. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. Water (650 mL) was added to the reaction mixture. Layers were separated and the organic phase was concentrated under reduced pressure (1900 mL distilled out). TBME (1000 mL) was added and solvent was further distilled under vacuum (400 mL distilled). The mixture was finally cooled down to 20 °C and stirred for 15 min. The resulting suspension was filtered off at 20 °C. The cake was rinsed with TBME (250 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 145 g, 80%, 97% a/a (LC-MS), M+1=469. Melting point: 185 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 9.10-9.14 (m, 1 H), 7.88-8.12 (m, 2 H), 7.81-7.82 (m, 1 H), 7.38-7.44 (m, 1 H), 7.21 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 6.84 (d, J = 7.8 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 1 H), 5.01 (brs, 2 H), 3.88 (s, 3 H), 3.61-3.73 (m, 1 H), 3.14-3.26 (m, 1 H), 2.25-2.30 (m, 1 H), 2.13 (s, 3 H), 1.97 (m, 3 H), 1.47-1.61 (m, 3 H).
Reference Example 4.4: (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl) (5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride
(S)-/V-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1 ,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide (145 g, 0.31 mol) was dissolved in isopropanol (870 mL) at 20 °C. To this solution was added carefully 5-6 N HCI in isopropanol (260 mL) over 10 min. the reaction mixture was then heated up to 90 °C and stirred for 4 hours. Water (28 mL) was added and the reaction mixture was stirred for an additional one hour. The reaction mixture was cooled to 20 °C. A light brown suspension was obtained which was filtered. The cake was rinsed with isopropanol (220 mL). The solid was finally dried under reduced pressure at 60 °C to give a beige solid. Yield: 133 g, 88%, 100% a/a (LC-MS), M+1 =451. Melting point: 277 °C (DSC). Ή NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.76 (d, J = 8.9 Hz, 1 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.55 (m, 1 H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 3.98 (m, 1 H), 3.90 (s, 3 H), 3.33 (m, 2H), 3.32 (m, 1 H), 2.74 (s, 3 H), 2.55 (m, 1 H), 2.23 (m, 1 H), 2.10 (m, 2 H), 1.95 (s, 3 H).
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Quviviq |
| Other names | Nemorexant; ACT-541468 |
| License data | US DailyMed: Daridorexant |
| Routes of administration | By mouth |
| Drug class | Orexin antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Pharmacokinetic data | |
| Elimination half-life | 6–10 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1505484-82-1 |
| PubChem CID | 91801202 |
| DrugBank | DB15031 |
| ChemSpider | 64854514 |
| UNII | LMQ24G57E9 |
| KEGG | D11886 |
| PDB ligand | NS2 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.93 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
REF
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214985s000lbl.pdf
- ^ Jump up to:a b Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J (November 2020). “Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders”. Expert Opin Drug Metab Toxicol. 16 (11): 1063–1078. doi:10.1080/17425255.2020.1817380. PMID 32901578.
- ^ Jump up to:a b c “Daridorexant – Idorsia Pharmaceuticals – AdisInsight”.
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- ^ “Daridorexant: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 January 2022.
- ^ “Idorsia receives US FDA approval of Quviviq (daridorexant)” (Press release). Idorsia Pharmaceuticals. 10 January 2022. Retrieved 11 January 2022 – via GlobeNewswire.
External links
- “Daridorexant”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03545191 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects With Insomnia Disorder” at ClinicalTrials.gov
- Clinical trial number NCT03575104 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
- Clinical trial number NCT03679884 for “Study to Assess the Long Term Safety and Tolerability of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
///////////////Daridorexant, Quviviq, FDA 2022, APPROVALS 2022, INSOMNIA, ACT541468A, ACT 541468A. ACT-541468A, ACT541468, FDA 2022, APPROVALS 2022
O=C(N1[C@](C)(C2=NC3=CC=C(Cl)C(C)=C3N2)CCC1)C4=CC(OC)=CC=C4N5N=CC=N5.[H]Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
DEUCRAVACITINIB
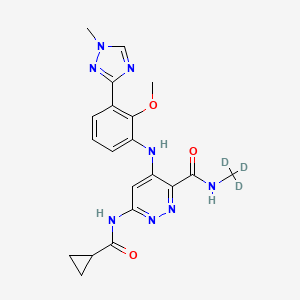
DEUCRAVACITINIB
BMS-986165
CAS 1609392-27-9, C20H22N8O3, 425.46
6-(cyclopropanecarbonylamino)-4-[2-methoxy-3-(1-methyl-1,2,4-triazol-3-yl)anilino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
- OriginatorBristol-Myers Squibb
- ClassAmides; Aniline compounds; Anti-inflammatories; Antipsoriatics; Antirheumatics; Cyclopropanes; Ethers; Hepatoprotectants; Organic deuterium compounds; Pyridazines; Skin disorder therapies; Small molecules; Triazoles
- Mechanism of ActionTYK2 kinase inhibitors
- Phase IIIPlaque psoriasis
- Phase IICrohn’s disease; Lupus nephritis; Psoriatic arthritis; Systemic lupus erythematosus; Ulcerative colitis
- Phase IAutoimmune disorders
- No development reportedInflammatory bowel diseases; Psoriasis
- 02 Jul 2021Bristol-Myers Squibb plans a phase I pharmacokinetics trial (In volunteers) in USA (PO, Tablet) in July 2021 (NCT04949269)
- 14 Jun 2021Bristol-Myers Squibb plans a phase III trial for Psoriatic arthritis (Treatment-naïve) in USA, Brazil, Colombia, Czech republic, Hungary, Italy, Mexico, Romania, Spain and Taiwan in July 2021 (NCT04908202) (EudraCT2020-005097-10)
- 02 Jun 2021Interim efficacy and adverse events data from the phase III POETYK-PSO-1 trial in Psoriatic psoriasis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
BMS , presumed to be in collaboration with Jinan University and Chinese Academy of Sciences , is developing deucravacitinib, a TYK2 inhibitor, for treating autoimmune diseases, primarily psoriasis. In July 2021, deucravacitinib was reported to be in phase 3 clinical development.
Deucravacitinib (BMS-986165) is a highly selective, orally bioavailable allosteric TYK2 inhibitor for the treatment of autoimmune diseases, which selectively binds to TYK2 pseudokinase (JH2) domain (IC50=1.0 nM) and blocks receptor-mediated Tyk2 activation by stabilizing the regulatory JH2 domain. Deucravacitinib inhibits IL-12/23 and type I IFN pathways.
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b00444

Small molecule JAK inhibitors have emerged as a major therapeutic advancement in treating autoimmune diseases. The discovery of isoform selective JAK inhibitors that traditionally target the catalytically active site of this kinase family has been a formidable challenge. Our strategy to achieve high selectivity for TYK2 relies on targeting the TYK2 pseudokinase (JH2) domain. Herein we report the late stage optimization efforts including a structure-guided design and water displacement strategy that led to the discovery of BMS-986165 (11) as a high affinity JH2 ligand and potent allosteric inhibitor of TYK2. In addition to unprecedented JAK isoform and kinome selectivity, 11 shows excellent pharmacokinetic properties with minimal profiling liabilities and is efficacious in several murine models of autoimmune disease. On the basis of these findings, 11 appears differentiated from all other reported JAK inhibitors and has been advanced as the first pseudokinase-directed therapeutic in clinical development as an oral treatment for autoimmune diseases.
Bristol Myers Squibb Presents Positive Data from Two Pivotal Phase 3 Psoriasis Studies Demonstrating Superiority of Deucravacitinib Compared to Placebo and Otezla® (apremilast)
Significantly more patients treated with deucravacitinib achieved PASI 75 and sPGA 0/1 compared to patients treated with placebo and Otezla at Week 16, with an increased benefit versus Otezla at Week 24 and maintained through Week 52
Deucravacitinib was well tolerated with a low rate of discontinuation due to adverse events
Deucravacitinib is a first-in-class, oral, selective tyrosine kinase 2 (TYK2) inhibitor with a unique mechanism of action
Results presented as late-breaking research at the 2021 American Academy of Dermatology Virtual Meeting Experience
PRINCETON, N.J.–(BUSINESS WIRE)– Bristol Myers Squibb (NYSE:BMY) today announced positive results from two pivotal Phase 3 trials evaluating deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, for the treatment of patients with moderate to severe plaque psoriasis. The POETYK PSO-1 and POETYK PSO-2 trials, which evaluated deucravacitinib 6 mg once daily, met both co-primary endpoints versus placebo, with significantly more patients achieving Psoriasis Area and Severity Index (PASI) 75 response and a static Physician’s Global Assessment score of clear or almost clear (sPGA 0/1) after 16 weeks of treatment with deucravacitinib. Deucravacitinib was well tolerated with a low rate of discontinuation due to adverse events (AEs).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20210423005134/en(Graphic: Business Wire)
Deucravacitinib demonstrated superior skin clearance compared with Otezla® (apremilast) for key secondary endpoints in both studies, as measured by PASI 75 and sPGA 0/1 responses at Week 16 and Week 24. Findings include:
PASI 75 Response in POETYK PSO-1 and POETYK PSO-2:
- At Week 16, 58.7% and 53.6% of patients receiving deucravacitinib achieved PASI 75 response, respectively, versus 12.7% and 9.4% receiving placebo and 35.1% and 40.2% receiving Otezla.
- At Week 24, 69.0% and 59.3% of patients receiving deucravacitinib achieved PASI 75 response, respectively, versus 38.1% and 37.8% receiving Otezla.
- Among patients who achieved PASI 75 response at Week 24 with deucravacitinib and continued treatment with deucravacitinib, 82.5% and 81.4%, respectively, maintained PASI 75 response at Week 52.
sPGA 0/1 Response in POETYK PSO-1 and POETYK PSO-2:
- At Week 16, 53.6% and 50.3% of patients receiving deucravacitinib achieved sPGA 0/1 response, respectively, versus 7.2% and 8.6% receiving placebo and 32.1% and 34.3% receiving Otezla.
- At Week 24, 58.4% and 50.4% of patients receiving deucravacitinib achieved sPGA 0/1 response, respectively, versus 31.0% and 29.5% receiving Otezla.
“In both pivotal studies, deucravacitinib was superior to Otezla across multiple endpoints, including measures of durability and maintenance of response, suggesting that deucravacitinib has the potential to become a new oral standard of care for patients who require systemic therapy and need a better oral option for their moderate to severe plaque psoriasis,” said April Armstrong, M.D., M.P.H., Associate Dean and Professor of Dermatology at the University of Southern California. “As many patients with moderate to severe plaque psoriasis remain undertreated or even untreated, it is also highly encouraging to see that deucravacitinib improved patient symptoms and outcomes to a greater extent than Otezla.”
Superiority of Deucravacitinib Versus Placebo and Otezla
Deucravacitinib demonstrated a robust efficacy profile, including superiority to placebo for the co-primary endpoints and to Otezla for key secondary endpoints. In addition to PASI 75 and sPGA 0/1 measures, deucravacitinib was superior to Otezla across both studies in multiple other secondary endpoints, demonstrating significant and clinically meaningful efficacy improvements in symptom burden and quality of life measures.
| POETYK PSO-1 and POETYK PSO-2 Results at Week 16 and Week 24 | ||||||
| Endpoint | POETYK PSO-1 (n=666) | POETYK PSO-2 (n=1,020) | ||||
| Deucravacitinib6 mg(n=332) | Otezla30 mg(n=168) | Placebo(n=166) | Deucravacitinib6 mg(n=511) | Otezla30 mg(n=254) | Placebo(n=255) | |
| PASI 75*a | ||||||
| Week 16 | 58.7%*† | 35.1% | 12.7% | 53.6%*‡ | 40.2% | 9.4% |
| Week 24 | 69.0%† | 38.1% | – | 59.3%† | 37.8% | – |
| sPGA 0/1*b | ||||||
| Week 16 | 53.6%*† | 32.1% | 7.2% | 50.3%*† | 34.3% | 8.6% |
| Week 24 | 58.4%† | 31.0% | – | 50.4%† | 29.5% | – |
| (Scalp) ss-PGA 0/1c | ||||||
| Week 16 | 70.8%*† | 39.1% | 17.4% | 60.3%*† | 37.3% | 17.3% |
| Week 24 | 71.8%† | 42.7% | – | 59.7%‡ | 41.6% | – |
| PSSD-Symptoms CFBd | ||||||
| Week 16 | -26.7*† | -17.8 | -3.6 | -28.3*† | -21.1 | -4.7 |
| Week 24 | -31.9† | -20.7 | – | -29.1† | -21.4 | – |
| DLQI 0/1e | ||||||
| Week 16 | 40.7%*† | 28.6% | 10.6% | 38.0%*† | 23.1% | 9.8% |
| Week 24 | 47.8%‡ | 24.2% | – | 41.8%† | 21.5% | – |
| *Co-primary endpoints for POETYK PSO-1 and POETYK PSO-2 were PASI 75 and sPGA 0/1 for deucravacitinib vs placebo at Week 16. |
| a. PASI 75 is defined as at least a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) scores. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. ‡p=0.0003 vs Otezla. |
| b. sPGA 0/1 is defined as a static Physician’s Global Assessment (sPGA) score of clear or almost clear. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
| c. ss-PGA 0/1 is defined as a scalp-specific Physician’s Global Assessment (ss-PGA) score of clear or almost clear in those with ss-PGA of at least 3 (moderate) at baseline. POETYK PSO-1: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. POETYK PSO-2: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. ‡p=0.0002 vs Otezla. |
| d. Change from baseline (CFB) in Psoriasis Symptoms and Signs Diary (PSSD) captures improvement in symptoms of itch, pain, stinging, burning and skin tightness in patient eDiaries. *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
| e. Dermatology Life Quality Index (DLQI) 0/1 scores reflect no effect at all on patient’s life in patients with a baseline DLQI score of ≥2. POETYK PSO-1: *p<0.0001 vs placebo. †p=0.0106 vs Otezla. ‡p<0.0001 vs Otezla. POETYK PSO-2: *p<0.0001 vs placebo. †p<0.0001 vs Otezla. |
Safety and Tolerability
Deucravacitinib was well-tolerated and had a similar safety profile in both trials. At Week 16, 2.9% of 419 patients on placebo, 1.8% of 842 patients on deucravacitinib and 1.2% of 422 patients on Otezla experienced serious adverse events (SAEs) across both studies. The most common AEs (≥5%) with deucravacitinib treatment at Week 16 were nasopharyngitis and upper respiratory tract infection with low rates of headache, diarrhea and nausea. At Week 16, 3.8% of patients on placebo, 2.4% of patients on deucravacitinib and 5.2% of patients on Otezla experienced AEs leading to discontinuation. Across POETYK PSO-1 and POETYK PSO-2 over 52 weeks, SAEs when adjusted for exposure (exposure adjusted incidence per 100 patient-years [EAIR]) were 5.7 with placebo, 5.7 with deucravacitinib and 4.0 with Otezla. In the same timeframe across both studies, EAIRs for AEs leading to discontinuation were 9.4 with placebo, 4.4 with deucravacitinib and 11.6 with Otezla. No new safety signals were observed during Weeks 16‒52.
Across both Phase 3 trials, rates of malignancy, major adverse cardiovascular events (MACE), venous thromboembolism (VTE) and serious infections were low and generally consistent across active treatment groups. No clinically meaningful changes were observed in multiple laboratory parameters (including anemia, blood cells, lipids and liver enzymes) over 52 weeks.
“The findings from both studies affirm that deucravacitinib – a first-in-class, oral, selective TYK2 inhibitor with a unique mechanism of action that inhibits the IL-12, IL-23 and Type 1 IFN pathways –may become an oral treatment of choice for people living with psoriasis. We believe deucravacitinib has significant potential across a broad range of immune-mediated diseases, and we are committed to further advancing our expansive clinical program with this agent,” said Mary Beth Harler, M.D., head of Immunology and Fibrosis Development, Bristol Myers Squibb. “We are in discussions with health authorities with the goal of bringing this new therapy to appropriate patients as soon as possible. At Bristol Myers Squibb, we are committed to building an immunology portfolio that addresses pressing unmet needs that exist for those impacted by serious dermatologic conditions and other immune-mediated diseases, to ultimately deliver the promise of living a better life.”
These results are available as a late-breaking research presentation (Session S033 – Late-Breaking Research Abstracts) as part of the 2021 American Academy of Dermatology (AAD) Virtual Meeting Experience (VMX). Full results of both studies will be submitted to a medical journal for peer review. In November 2020 and February 2021, respectively, Bristol Myers Squibb announced positive topline results from POETYK PSO-1 and POETYK PSO-2.
Visit www.bms.com/media/medical-meetings/bms-at-aad-vmx.html for more information on Bristol Myers Squibb’s scientific approach and resources on psoriasis and immune-mediated diseases.
About Deucravacitinib
Deucravacitinib (pronounced doo-krav-a-sih-ti-nib) is a first-in-class, oral, selective tyrosine kinase 2 (TYK2) inhibitor with a unique mechanism of action. Deucravacitinib is the first and only TYK2 inhibitor in clinical studies across multiple immune-mediated diseases. Bristol Myers Squibb scientists designed deucravacitinib to selectively target TYK2, thereby inhibiting signaling of interleukin (IL)-12, IL-23 and Type 1 interferon (IFN), key cytokines involved in psoriasis pathogenesis. Deucravacitinib achieves a high degree of selectivity by uniquely binding to the regulatory, rather than the active, domain of TYK2, which is structurally distinct from the regulatory domains of Janus kinase (JAK) 1, 2 and 3. At therapeutic doses, deucravacitinib does not inhibit JAK1, JAK2 or JAK3. Due to the innovative design of deucravacitinib, Bristol Myers Squibb earned recognition with the 2019 Thomas Alva Edison Patent Award for the science underpinning the clinical development of deucravacitinib.
Deucravacitinib is being studied in multiple immune-mediated diseases, including psoriasis, psoriatic arthritis, lupus and inflammatory bowel disease. In addition to POETYK PSO-1 and POETYK PSO-2, Bristol Myers Squibb is evaluating deucravacitinib in three other Phase 3 studies in psoriasis: POETYK PSO-3 (NCT04167462); POETYK PSO-4 (NCT03924427); POETYK PSO-LTE (NCT04036435). Deucravacitinib is not approved for any use in any country.
About the Phase 3 POETYK PSO-1 and POETYK PSO-2 Studies
PrOgram to Evaluate the efficacy and safety of deucravacitinib, a selective TYK2 inhibitor (POETYK) PSO-1 (NCT03624127) and POETYK PSO-2 (NCT03611751) are global Phase 3 studies designed to evaluate the safety and efficacy of deucravacitinib compared to placebo and Otezla® (apremilast) in patients with moderate to severe plaque psoriasis. Both POETYK PSO-1, which enrolled 666 patients, and POETYK PSO-2, which enrolled 1,020 patients, were multi-center, randomized, double-blind trials that evaluated deucravacitinib (6 mg once daily) compared with placebo and Otezla (30 mg twice daily). POETYK PSO-2 included a randomized withdrawal and retreatment period after Week 24.
The co-primary endpoints of both POETYK PSO-1 and POETYK PSO-2 were the percentage of patients who achieved Psoriasis Area and Severity Index (PASI) 75 response and those who achieved static Physician’s Global Assessment (sPGA) score of 0 or 1 at Week 16 versus placebo. Key secondary endpoints of the trials included the percentage of patients who achieved PASI 75 and sPGA 0/1 compared to Otezla at Week 16 and other measures.
About Psoriasis
Psoriasis is a widely prevalent, chronic, systemic immune-mediated disease that substantially impairs patients’ physical health, quality of life and work productivity. Psoriasis is a serious global problem, with at least 100 million people worldwide impacted by some form of the disease, including around 14 million people in Europe and approximately 7.5 million people in the United States. Up to 90 percent of patients with psoriasis have psoriasis vulgaris, or plaque psoriasis, which is characterized by distinct round or oval plaques typically covered by silvery-white scales. Despite the availability of effective systemic therapy, many patients with moderate to severe psoriasis remain undertreated or even untreated and are dissatisfied with current treatments. People with psoriasis report an impact on their emotional well-being, straining both personal and professional relationships and causing a reduced quality of life. Psoriasis is associated with multiple comorbidities that may impact patients’ well-being, including psoriatic arthritis, cardiovascular disease, metabolic syndrome, obesity, diabetes, inflammatory bowel disease and depression.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Celgene and Juno Therapeutics are wholly owned subsidiaries of Bristol-Myers Squibb Company. In certain countries outside the U.S., due to local laws, Celgene and Juno Therapeutics are referred to as, Celgene, a Bristol Myers Squibb company and Juno Therapeutics, a Bristol Myers Squibb company.
Otezla® (apremilast) is a registered trademark of Amgen Inc.
PATENT
WO-2021129467
Novel crystalline polymorphic forms (CSI and CSII) of deucravacitinib (also known as BMS-986165), useful a tyrosine kinase 2 pseudokinase domain (TYK2) inhibitor for treating psoriasis, systemic lupus erythematosus, and Crohn’s disease.Tyrosine kinase 2 (TYK2) is an intracellular signal transduction kinase that can mediate interleukin-23 (IL-23), interleukin-12 (IL-12) and type I interferon (IFN) These cytokines are involved in inflammation and immune response.
BMS-986165 is the first and only new oral selective TYK2 inhibitor, clinically used to treat autoimmune and autoinflammatory diseases (such as psoriasis, psoriatic arthritis, lupus and inflammatory bowel disease, Crowe Graciousness, etc.). The results of a phase III clinical study of the drug announced in November 2020 showed that BMS-986165 has shown positive clinical effects in the treatment of moderate to severe plaque psoriasis. In addition, BMS-986165 also shows good therapeutic effects in the treatment of systemic lupus erythematosus and Crohn’s disease.
The chemical name of BMS-986165 is 6-(cyclopropaneamido)-4-((2-methoxy-3-(1-methyl-1H-1,2,4-triazol-3-yl)benzene (Yl)amino)-N-(methyl-D3)pyridazine-3-carboxamide, the structural formula is shown below, and is hereinafter referred to as “compound I”:
The crystal form is a solid in which the compound molecules are arranged in a three-dimensional order in the microstructure to form a crystal lattice. The phenomenon of drug polymorphism refers to the existence of two or more different crystal forms of the drug. Because of different physical and chemical properties, different crystal forms of the drug may have different dissolution and absorption in the body, which in turn affects the clinical efficacy and safety of the drug to a certain extent. Especially for poorly soluble solid drugs, the crystal form will have a greater impact. Therefore, drug crystal form must be an important content of drug research and also an important content of drug quality control.
WO2018183656A1 discloses compound I crystal form A (hereinafter referred to as “crystal form A”) and a preparation method thereof. The crystalline form A disclosed in WO2018183656A1 is the only known free crystalline form of Compound I. The inventor of the present application repeated the preparation method disclosed in WO2018183656A1 to obtain and characterize the crystal form A. The results show that the crystal form A has poor compressibility and high adhesion. Therefore, there is still a need in the art to develop a compound I crystalline form with good stability, good compressibility, and low adhesion for the development of drugs containing compound I.
The inventor of the present application has paid a lot of creative work and unexpectedly discovered the crystalline form CSI of compound I and the crystalline form CSII of compound I provided by the present invention, which have advantages in physical and chemical properties, preparation processing performance and bioavailability, for example, There are advantages in at least one aspect of melting point, solubility, hygroscopicity, purification, stability, adhesion, compressibility, fluidity, dissolution in vivo and in vitro, and bioavailability, especially good physical and chemical stability and mechanical stability It has good performance, good compressibility, and low adhesion, which solves the problems existing in the prior art, and is of great significance to the development of drugs containing compound I.
PATENT
US9505748 , a family member of WO2014074661 .
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014074661
Preparation 1
Step l Int1
Step 2 Int2 Step 3 Int3 Step 4 Int4
Example 52
Step 1
[00219] To a solution of 2-methoxy-3-(l-methyl-lH-l ,2,4-triazol-3-yl)aniline (10.26 g, 50.2 mmol) and Int8 (10.5 g, 50.2 mmol) in THF (120 mL) was added lithium bis(trimethylsilyl)amide (LiHMDS, 1M in THF, 151 mL, 151 mmol) in a dropwise manner using a pressure equalized addition funnel. The reaction was run for 10 minutes after the completion of the addition and then quenched with HCl (1M aq., 126 mL, 126 mmol). The reaction was concentrated on a rotary evaporator until the majority of the THF was removed and a precipitate prevailed throughout the vessel. Water (-500 mL) was then added and the slurry sonicated for 5 minutes and stirred for 15 min. The solid was filtered off, rinsing with water and then air dried for 30 minutes. The powder was collected and dissolved in dichloromethane. The organic layer was washed with water and brine and then dried over sodium sulfate, filtered and concentrated to provide the product (12.5 g, 66% yield) (carried on as is). 1H NMR (400MHz, DMSO-d6) δ 11.11 (s, 1H), 9.36 (s, 1H), 8.56 (s, 1H), 7.72 (dd, J=7.8, 1.6 Hz, 1H), 7.60 (dd, J=7.9, 1.5 Hz, 1H), 7.29 (t, J=7.9 Hz, 1H), 7.19 (s, 1H), 3.95 (s, 3H), 3.72 (s, 3H). LC retention time 1.18 [E]. MS(E+) m/z: 377 (MH+).
Step 2
[00220] Intl3 (2.32 g, 6.16 mmol) and cyclopropanecarboxamide (1.048 g, 12.31 mmol) were dissolved in dioxane (62 mL) and Pd2(dba)3 (564 mg, 0.616 mmol), Xantphos (534 mg, 0.924 mmol) and cesium carbonate (4.01 g, 12.3 mmol) were added. The vessel was evacuated three times (backfilling with nitrogen) and then sealed and heated to 130 °C for 140 minutes. The reaction was filtered through CELITE® (eluting with ethyl acetate) and concentrated (on smaller scale this material could then be purified using preparative HPLC). The crude product was adsorbed onto CELITE® using dichloromethane, dried and purified using automated chromatography (100% EtOAc) to provide example 52 (1.22 g, 46% yield). 1H NMR (500MHz, chloroform-d) δ 10.99 (s, 1H), 8.63 (s, 1H), 8.18 (s, 1H), 8.10 (d, J=0.5 Hz, 2H), 7.81 (dd, J=7.9, 1.7 Hz, 1H), 7.51 (dd, J=7.9, 1.4 Hz, 1H), 7.33 – 7.20 (m, 7H), 4.01 (d, J=0.3 Hz, 3H), 3.82 (s, 3H), 1.73 -1.60 (m, 1H), 1.16 – 1.06 (m, 2H), 0.97 – 0.84 (m, 2H). LC retention time 6.84 [N]. MS(E+) m/z: 426 (MH+).
Example 53
[00221] To a homogeneous solution of Example 52 (50 mg, 0.12 mmol) in dichloromethane (3 mL) was added HCI (1M aq., 0.13 mL, 0.13 mmol) resulting in the solution turning yellow. The homogenous solution was concentrated down and then re-concentrated from dichloromethane twice to remove residual water, resulting in a white powder. The powder was suspended in dichloromethane and sonicated for 15 minutes, the powder was then collected via filtration, rinsing with dichloromethane to provide the corresponding HCI salt (38 mg, 70% yield). 1H NMR (500MHz, chloroform-d) δ 12.02 (s, 1H), 8.35 (s, 1H), 8.16 (s, 1H), 8.01 (dd, J=7.9, 1.5 Hz, 1H), 7.57 (br. s., 1H), 7.52 -7.46 (m, 1H), 7.36 (t, J=7.9 Hz, 1H), 4.03 (s, 3H), 3.83 (s, 3H), 2.05 – 1.95 (m, 1H), 1.16 – 1.09 (m, 2H), 1.03 (dd, J=7.4, 3.6 Hz, 2H). LC retention time 0.62 [j]. MS(E+) m/z: 426 (MH+).
[00222] Compare to NMR of parent free base: 1H NMR (500MHz, chloroform-d) δ 10.99 (s, 1H), 8.63 (s, 1H), 8.18 (s, 1H), 8.10 (d, J=0.5 Hz, 2H), 7.81 (dd, J=7.9, 1.7 Hz, 1H), 7.51 (dd, J=7.9, 1.4 Hz, 1H), 7.33 – 7.20 (m, 7H), 4.01 (d, J=0.3 Hz, 3H), 3.82 (s, 3H), 1.73 – 1.60 (m, 1H), 1.16 – 1.06 (m, 2H), 0.97 – 0.84 (m, 2H).
////////////DEUCRAVACITINIB, phase 3, BMS-986165, BMS 986165, psoriasis, systemic lupus erythematosus, Crohn’s disease,
CNC(=O)C1=NN=C(C=C1NC2=CC=CC(=C2OC)C3=NN(C=N3)C)NC(=O)C4CC4

NEW DRUG APPROVALS
one time
$10.00
NOVAWAX, NVX-CoV2373,
NOVAWAX
SARS-CoV-2 rS Nanoparticle Vaccine
MCDC OTA agreement number W15QKN-16-9-1002
Novavax COVID-19 vaccine, Coronavirus disease 19 infection
SARS-CoV-2 rS, TAK 019
Novavax, Inc. is an American vaccine development company headquartered in Gaithersburg, Maryland, with additional facilities in Rockville, Maryland and Uppsala, Sweden. As of 2020, it had an ongoing Phase III clinical trial in older adults for its candidate vaccine for seasonal influenza, NanoFlu and a candidate vaccine (NVX-CoV2373) for prevention of COVID-19.
NVX-CoV2373 is a SARS-CoV-2 rS vaccine candidate and was shown to have high immunogenicity in studies. The vaccine is created from the genetic sequence of COVID-19 and the antigen derived from the virus spike protein is generated using recombinant nanoparticle technology. The vaccine was developed and tested by Novavax. As of May 2020, the company is pursuing a Phase 1 clinical trial (NCT04368988) to test the vaccine.
History
Novavax was founded in 1987. It focused principally on experimental vaccine development, but did not achieve a successful launch up to 2021.[4]
In June 2013, Novavax acquired the Matrix-M adjuvant platform with the purchase of Swedish company Isconova AB and renamed its new subsidiary Novavax AB.[5]
In 2015, the company received an $89 million grant from the Bill & Melinda Gates Foundation to support the development of a vaccine against human respiratory syncytial virus for infants via maternal immunization.[6][7][8][9]
In March 2015 the company completed a Phase I trial for its Ebola vaccine candidate,[10] as well as a phase II study in adults for its RSV vaccine, which would become ResVax.[11] The ResVax trial was encouraging as it showed significant efficacy against RSV infection.[11]
2016 saw the company’s first phase III trial, the 12,000 adult Resolve trial,[11] for its respiratory syncytial virus vaccine, which would come to be known as ResVax, fail in September.[3] This triggered an eighty-five percent dive in the company’s stock price.[3] Phase II adult trial results also released in 2016 showed a stimulation of antigencity, but failure in efficacy.[11] Evaluation of these results suggested that an alternative dosing strategy might lead to success, leading to plans to run new phase II trials.[3] The company’s difficulties in 2016 led to a three part strategy for 2017: cost reduction through restructuring and the termination of 30% of their workforce; pouring more effort into getting ResVax to market; and beginning clinical trials on a Zika virus vaccine.[3]
Alongside the adult studies of ResVax, the vaccine was also in 2016 being tested against infant RSV infection through the route of maternal immunization.[11]
In 2019, late-stage clinical testing of ResVax, failed for a second time, which resulted in a major downturn in investor confidence and a seventy percent reduction in capital value for the firm.[12][13] As a secondary result, the company was forced to conduct a reverse stock split in order to maintain Nasdaq minimum qualification, meaning it was in risk of being delisted.[13]
The company positions NanoFlu for the unmet need for a more effective vaccine against influenza, particularly in the elderly who often experience serious and sometimes life-threatening complications. In January 2020, it was granted fast track status by the U.S. Food and Drug Administration (FDA) for NanoFlu.
External sponsorships
In 2018, Novavax received a US$89 million research grant from the Bill and Melinda Gates Foundation for development of vaccines for maternal immunization.[14]
In May 2020, Novavax received US$384 million from the Coalition for Epidemic Preparedness Innovations to fund early-stage evaluation in healthy adults of the company’s COVID-19 vaccine candidate NVX-CoV2373 and to develop resources in preparation for large-scale manufacturing, if the vaccine proves successful.[15] CEPI had already invested $4 million in March.[15]
Drugs in development
ResVax is a nanoparticle-based treatment using a recombinant F lipoprotein or saponin, “extracted from the Quillaja saponaria [or?] Molina bark together with cholesterol and phospholipid.”[16] It is aimed at stimulating resistance to respiratory syncytial virus infection, targeting both adult and infant populations.[11]
In January 2020, Novavax was given Fast Track status by the FDA to expedite the review process for NanoFlu, a candidate influenze vaccine undergoing a Phase III clinical trial scheduled for completion by mid-2020.[17]
COVID-19 vaccine candidate
See also: NVX-CoV2373 and COVID-19 vaccine
In January 2020, Novavax announced development of a vaccine candidate, named NVX-CoV2373, to establish immunity to SARS-CoV-2.[18] NVX-CoV2373 is a protein subunit vaccine that contains the spike protein of the SARS-CoV-2 virus.[19] Novavax’s work is in competition for vaccine development among dozens of other companies.
In January 2021, the company released phase 3 trials showing that it has 89% efficacy against Covid-19, and also provides strong immunity against new variants.[20] It has applied for emergency use in the US and UK but will be distributed in the UK first.Novavax COVID-19 Vaccine Demonstrates 89.3% Efficacy in UK Phase 3 TrialJan 28, 2021 at 4:05 PM ESTDownload PDF
First to Demonstrate Clinical Efficacy Against COVID-19 and Both UK and South Africa Variants
- Strong efficacy in Phase 3 UK trial with over 50% of cases attributable to the now-predominant UK variant and the remainder attributable to COVID-19 virus
- Clinical efficacy demonstrated in Phase 2b South Africa trial with over 90% of sequenced cases attributable to prevalent South Africa escape variant
- Company to host investor conference call today at 4:30pm ET
GAITHERSBURG, Md., Jan. 28, 2021 (GLOBE NEWSWIRE) — Novavax, Inc. (Nasdaq: NVAX), a biotechnology company developing next-generation vaccines for serious infectious diseases, today announced that NVX-CoV2373, its protein-based COVID-19 vaccine candidate, met the primary endpoint, with a vaccine efficacy of 89.3%, in its Phase 3 clinical trial conducted in the United Kingdom (UK). The study assessed efficacy during a period with high transmission and with a new UK variant strain of the virus emerging and circulating widely. It was conducted in partnership with the UK Government’s Vaccines Taskforce. Novavax also announced successful results of its Phase 2b study conducted in South Africa.
“With today’s results from our UK Phase 3 and South Africa Phase 2b clinical trials, we have now reported data on our COVID-19 vaccine from Phase 1, 2 and 3 trials involving over 20,000 participants. In addition, our PREVENT-19 US and Mexico clinical trial has randomized over 16,000 participants toward our enrollment goal of 30,000. NVX-CoV2373 is the first vaccine to demonstrate not only high clinical efficacy against COVID-19 but also significant clinical efficacy against both the rapidly emerging UK and South Africa variants,” said Stanley C. Erck, President and Chief Executive Officer, Novavax. “NVX-CoV2373 has the potential to play an important role in solving this global public health crisis. We look forward to continuing to work with our partners, collaborators, investigators and regulators around the world to make the vaccine available as quickly as possible.”
NVX-CoV2373 contains a full-length, prefusion spike protein made using Novavax’ recombinant nanoparticle technology and the company’s proprietary saponin-based Matrix-M™ adjuvant. The purified protein is encoded by the genetic sequence of the SARS-CoV-2 spike (S) protein and is produced in insect cells. It can neither cause COVID-19 nor can it replicate, is stable at 2°C to 8°C (refrigerated) and is shipped in a ready-to-use liquid formulation that permits distribution using existing vaccine supply chain channels.
UK Phase 3 Results: 89.3% Efficacy
The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65. The primary endpoint of the UK Phase 3 clinical trial is based on the first occurrence of PCR-confirmed symptomatic (mild, moderate or severe) COVID-19 with onset at least 7 days after the second study vaccination in serologically negative (to SARS-CoV-2) adult participants at baseline.
The first interim analysis is based on 62 cases, of which 56 cases of COVID-19 were observed in the placebo group versus 6 cases observed in the NVX-CoV2373 group, resulting in a point estimate of vaccine efficacy of 89.3% (95% CI: 75.2 – 95.4). Of the 62 cases, 61 were mild or moderate, and 1 was severe (in placebo group).
Preliminary analysis indicates that the UK variant strain that was increasingly prevalent was detected in over 50% of the PCR-confirmed symptomatic cases (32 UK variant, 24 non-variant, 6 unknown). Based on PCR performed on strains from 56 of the 62 cases, efficacy by strain was calculated to be 95.6% against the original COVID-19 strain and 85.6% against the UK variant strain [post hoc].
The interim analysis included a preliminary review of the safety database, which showed that severe, serious, and medically attended adverse events occurred at low levels and were balanced between vaccine and placebo groups.
“These are spectacular results, and we are very pleased to have helped Novavax with the development of this vaccine. The efficacy shown against the emerging variants is also extremely encouraging. This is an incredible achievement that will ensure we can protect individuals in the UK and the rest of the world from this virus,” said Clive Dix, Chair, UK Vaccine Taskforce.
Novavax expects to share further details of the UK trial results as additional data become available. Additional analysis on both trials is ongoing and will be shared via prepublication servers as well as submitted to a peer-reviewed journal for publication. The company initiated a rolling submission to the United Kingdom’s regulatory agency, the MHRA, in mid-January.
South Africa Results: Approximately 90% of COVID-19 cases attributed to South Africa escape variant
In the South Africa Phase 2b clinical trial, 60% efficacy (95% CI: 19.9 – 80.1) for the prevention of mild, moderate and severe COVID-19 disease was observed in the 94% of the study population that was HIV-negative. Twenty-nine cases were observed in the placebo group and 15 in the vaccine group. One severe case occurred in the placebo group and all other cases were mild or moderate. The clinical trial also achieved its primary efficacy endpoint in the overall trial population, including HIV-positive and HIV-negative subjects (efficacy of 49.4%; 95% CI: 6.1 – 72.8).
This study enrolled over 4,400 patients beginning in August 2020, with COVID-19 cases counted from September through mid-January. During this time, the triple mutant variant, which contains three critical mutations in the receptor binding domain (RBD) and multiple mutations outside the RBD, was widely circulating in South Africa. Preliminary sequencing data is available for 27 of 44 COVID-19 events; of these, 92.6% (25 out of 27 cases) were the South Africa escape variant.
Importantly in this trial, approximately 1/3 of the patients enrolled (but not included in the primary analyses described above) were seropositive, demonstrating prior COVID-19 infection at baseline. Based on temporal epidemiology data in the region, the pre-trial infections are thought to have been caused by the original COVID-19 strain (i.e., non-variant), while the subsequent infections during the study were largely variant virus. These data suggest that prior infection with COVID-19 may not completely protect against subsequent infection by the South Africa escape variant, however, vaccination with NVX-CoV2373 provided significant protection.
“The 60% reduced risk against COVID-19 illness in vaccinated individuals in South Africans underscores the value of this vaccine to prevent illness from the highly worrisome variant currently circulating in South Africa, and which is spreading globally. This is the first COVID-19 vaccine for which we now have objective evidence that it protects against the variant dominating in South Africa,” says Professor Shabir Maddi, Executive Director of the Vaccines and Infectious Diseases Analytics Research Unit (VIDA) at Wits, and principal investigator in the Novavax COVID-19 vaccine trial in South Africa. “I am encouraged to see that Novavax plans to immediately begin clinical development on a vaccine specifically targeted to the variant, which together with the current vaccine is likely to form the cornerstone of the fight against COVID-19.”
Novavax initiated development of new constructs against the emerging strains in early January and expects to select ideal candidates for a booster and/or combination bivalent vaccine for the new strains in the coming days. The company plans to initiate clinical testing of these new vaccines in the second quarter of this year.
“A primary benefit of our adjuvanted platform is that it uses a very small amount of antigen, enabling the rapid creation and large-scale production of combination vaccine candidates that could potentially address multiple circulating strains of COVID-19,” said Gregory M. Glenn, M.D., President of Research and Development, Novavax. “Combined with the safety profile that has been observed in our studies to-date with our COVID-19 vaccine, as well as prior studies in influenza, we are optimistic about our ability to rapidly adapt to evolving conditions.”
The Coalition for Epidemic Preparedness Innovations (CEPI) funded the manufacturing of doses of NVX-CoV2373 for this Phase 2b clinical trial, which was supported in part by a $15 million grant from the Bill & Melinda Gates Foundation.
Significant progress on PREVENT-19 Clinical Trial in US and Mexico
To date, PREVENT-19 has randomized over 16,000 participants and expects to complete our targeted enrollment of 30,000 patients in the first half of February. PREVENT-19 is being conducted with support from the U.S. government partnership formerly known as Operation Warp Speed, which includes the Department of Defense, the Biomedical Advanced Research and Development Authority (BARDA), part of the U.S. Department of Health and Human Services (HHS) Office of the Assistant Secretary for Preparedness and Response, and the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH) at HHS. BARDA is also providing up to $1.75 billion under a Department of Defense agreement.
PREVENT-19 (the PRE-fusion protein subunit Vaccine Efficacy Novavax Trial | COVID-19) is a Phase 3, randomized, placebo-controlled, observer-blinded study in the US and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 30,000 subjects 18 years of age and older compared with placebo. The trial design has been harmonized to align with other Phase 3 trials conducted under the auspices of Operation Warp Speed, including the use of a single external independent Data and Safety Monitoring Board to evaluate safety and conduct an unblinded review when predetermined interim analysis events are reached.
The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Conference Call
Novavax will host a conference call today at 4:30pm ET. The dial-in numbers for the conference call are (877) 212-6076 (Domestic) or (707) 287-9331 (International), passcode 7470222. A replay of the conference call will be available starting at 7:30 p.m. ET on January 28, 2021 until 7:30 p.m. ET on February 4, 2021. To access the replay by telephone, dial (855) 859-2056 (Domestic) or (404) 537-3406 (International) and use passcode 7470222.
A webcast of the conference call can also be accessed on the Novavax website at novavax.com/events. A replay of the webcast will be available on the Novavax website until April 28, 2021.
About NVX-CoV2373
NVX-CoV2373 is a protein-based vaccine candidate engineered from the genetic sequence of SARS-CoV-2, the virus that causes COVID-19 disease. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike (S) protein and is adjuvanted with Novavax’ patented saponin-based Matrix-M™ to enhance the immune response and stimulate high levels of neutralizing antibodies. NVX-CoV2373 contains purified protein antigen and can neither replicate, nor can it cause COVID-19. Over 37,000 participants have participated to date across four different clinical studies in five countries. NVX-CoV2373 is currently being evaluated in two pivotal Phase 3 trials: a trial in the U.K that completed enrollment in November and the PREVENT-19 trial in the U.S. and Mexico that began in December.
About Matrix-M™
Novavax’ patented saponin-based Matrix-M™ adjuvant has demonstrated a potent and well-tolerated effect by stimulating the entry of antigen presenting cells into the injection site and enhancing antigen presentation in local lymph nodes, boosting immune response.
About Novavax
Novavax, Inc. (Nasdaq: NVAX) is a biotechnology company that promotes improved health globally through the discovery, development and commercialization of innovative vaccines to prevent serious infectious diseases. The company’s proprietary recombinant technology platform combines the power and speed of genetic engineering to efficiently produce highly immunogenic nanoparticles designed to address urgent global health needs. Novavax is conducting late-stage clinical trials for NVX-CoV2373, its vaccine candidate against SARS-CoV-2, the virus that causes COVID-19. NanoFlu™, its quadrivalent influenza nanoparticle vaccine, met all primary objectives in its pivotal Phase 3 clinical trial in older adults and will be advanced for regulatory submission. Both vaccine candidates incorporate Novavax’ proprietary saponin-based Matrix-M™ adjuvant to enhance the immune response and stimulate high levels of neutralizing antibodies.
For more information, visit www.novavax.com and connect with us on Twitter and LinkedIn.
Candidate: NVX-CoV2373
Category: VAX
Type: Stable, prefusion protein made using Novavax’ proprietary nanoparticle technology, and incorporating its proprietary saponin-based Matrix-M™ adjuvant.
2021 Status: Novavax on March 11 announced final efficacy of 96.4% against mild, moderate and severe disease caused by the original COVID-19 strain in a pivotal Phase III trial in the U.K. of NVX–CoV2373. The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65.
The company also announced the complete analysis of its Phase IIb trial in South Africa, showing the vaccine had an efficacy of 55.4% among a cohort of HIV-negative trial participants, and an overall efficacy of 48.6% against predominantly variant strains of SARS-CoV-2 among 147 PCR-positive cases (51 cases in the vaccine group and 96 in the placebo group). Across both trials, NVX-CoV2373 demonstrated 100% protection against severe disease, including all hospitalization and death.
Philippines officials said March 10 that they secured 30 million doses of NVX-CoV2373 through an agreement with the Serum Institute of India, the second vaccine deal signed by the national government, according to Agence France-Presse. The first was with AstraZeneca for 2.6 million doses of its vaccine, developed with Oxford University.
The Novavax vaccine will be available from the third quarter, at a price that has yet to be finalized. The government hopes to secure 148 million doses this year from seven companies—enough for around 70% of its population.
In announcing fourth quarter and full-year 2020 results on March 1, Novavax said it could file for an emergency use authorization with the FDA in the second quarter of 2021. Novavax hopes it can use data from its Phase III U.K. clinical trial in its FDA submission, and expects the FDA to examine data in May, a month after they are reviewed by regulators in the U.K., President and CEO Stanley C. Erck said on CNBC. Should the FDA insist on waiting for U.S. data, the agency may push the review timeline by one or two months, he added.
The company also said that NVX-CoV2373 showed 95.6% efficacy against the original strain of COVID-19 and 85.6% against the UK variant strain, and re-stated an earlier finding that its vaccine met the Phase III trial’s primary endpoint met with an efficacy rate of 89.3%.
Novavax said February 26 that it signed an exclusive license agreement with Takeda Pharmaceutical for Takeda to develop, manufacture, and commercialize NVX-CoV2373 in Japan.
Novavax agreed to transfer the technology for manufacturing of the vaccine antigen and will supply its Matrix-M™ adjuvant to Takeda. Takeda anticipated the capacity to manufacture over 250 million doses of the COVID-19 vaccine per year. Takeda agreed in return to pay Novavax undisclosed payments tied to achieving development and commercial milestones, plus a portion of proceeds from the vaccine.
Takeda also disclosed that it dosed the first participants in a Phase II clinical trial to test the immunogenicity and safety of Novavax’ vaccine candidate in Japanese participants.
Novavax on February 18 announced a memorandum of understanding with Gavi, the Vaccine Alliance (Gavi), to provide 1.1 billion cumulative doses of NVX-CoV2373 for the COVAX Facility. Gavi leads the design and implementation of the COVAX Facility, created to supply vaccines globally, and has committed to working with Novavax to finalize an advance purchase agreement for vaccine supply and global distribution allocation via the COVAX Facility and its partners.
The doses will be manufactured and distributed globally by Novavax and Serum Institute of India (SII), the latter under an existing agreement between Gavi and SII.
Novavax and SK Bioscience said February 15 that they expanded their collaboration and license agreement, with SK finalizing an agreement to supply 40 million doses of NVX-CoV2373 to the government of South Korea beginning in 2021, for an undisclosed price. SK also obtained a license to manufacture and commercialize NVX-CoV2373 for sale to South Korea, as a result of which SK said it will add significant production capacity.
The agreement also calls on Novavax to facilitate technology transfer related to the manufacturing of its protein antigen, its Matrix M adjuvant, and support to SK Bioscience as needed to secure regulatory approval.
Rolling review begins—On February 4, Novavax announced it had begun a rolling review process for authorization of NVX-CoV2373 with several regulatory agencies worldwide, including the FDA, the European Medicines Agency, the U.K. Medicines and Healthcare products Regulatory Agency (MHRA), and Health Canada. The reviews will continue while the company completes its pivotal Phase III trials in the U.S. and U.K., and through initial authorization for emergency use granted under country-specific regulations, and through initial authorization for emergency use.
A day earlier, Novavax executed a binding Heads of Terms agreement with the government of Switzerland to supply 6 million doses of NVX-CoV2373, to the country. Novavax and Switzerland plan to negotiate a final agreement, with initial delivery of vaccine doses slated to ship following successful clinical development and regulatory review.
On January 28, Novavax electrified investors by announcing that its COVID-19 vaccine NVX-CoV2373 showed efficacy of 89.3% in the company’s first analysis of data from a Phase III trial in the U.K., where a variant strain (B.1.1.7) accounted for about half of all positive cases.
However, NVX-CoV2373 achieved only 60% efficacy in a Phase IIb trial in South Africa, where that country’s escape variant of the virus (B.1.351, also known as 20H/501Y.V2) was seen in 90% of cases, Novavax said.
Novavax said January 7 it executed an Advance Purchase Agreement with the Commonwealth of Australia for 51 million doses of NVX-CoV2373 for an undisclosed price, with an option to purchase an additional 10 million doses—finalizing an agreement in principle announced in November 2020. Novavax said it will work with Australia’s Therapeutics Goods Administration (TGA), to obtain approvals upon showing efficacy in clinical studies. The company aims to deliver initial doses by mid-2021.
2020 Status: Phase III trial launched—Novavax said December 28 that it launched the pivotal Phase III PREVENT-19 trial (NCT04611802) in the U.S. and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373. The randomized, placebo-controlled, observer-blinded study will assess the efficacy, safety and immunogenicity of NVX-CoV2373 in up to 30,000 participants 18 years of age and older compared with placebo. The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Two thirds of the participants will be assigned to randomly receive two intramuscular injections of the vaccine, administered 21 days apart, while one third of the trial participants will receive placebo. Trial sites were selected in locations where transmission rates are currently high, to accelerate the accumulation of positive cases that could show efficacy. Participants will be followed for 24 months following the second injection
PREVENT-19 is being conducted with support from federal agencies involved in Operation Warp Speed, the Trump administration’s effort to promote development and distribution of COVID-19 vaccines and drugs. Those agencies include the Department of Defense (DoD), the NIH’s National Institute of Allergy and Infectious Diseases (NIAID), and the Biomedical Advanced Research and Development Authority (BARDA)—which has committed up to $1.6 billion to Novavax under a DoD agreement (identifier MCDC OTA agreement number W15QKN-16-9-1002).
Novavax is also conducting a pivotal Phase III study in the United Kingdom, a Phase IIb safety and efficacy study in South Africa, and an ongoing Phase I/II trial in the U.S. and Australia. Data from these trials are expected as soon as early first quarter 2021, though timing will depend on transmission rates in the regions, the company said.
Novavax said November 9 that the FDA granted its Fast Track designation for NVX-CoV2373. By the end of November, the company expected to finish enrollment in its Phase III U.K. trial, with interim data in that study expected as soon as early first quarter 2021.
Five days earlier, Novavax signed a non-binding Heads of Terms document with the Australian government to supply 40 million doses of NVX-CoV2373 to Australia starting as early as the first half of 2021, subject to the successful completion of Phase III clinical development and approval of the vaccine by Australia’s Therapeutic Goods Administration (TGA). The vaccine regimen is expected to require two doses per individual, administered 21 days apart.
Australia joins the U.S., the U.K., and Canada in signing direct supply agreements with Novavax. The company is supplying doses in Japan, South Korea, and India through partnerships. Australian clinical researchers led the global Phase I clinical trial in August, which involved 131 Australians across two trial sites (Melbourne and Brisbane). Also, approximately 690 Australians have participated in the Phase II arm of the clinical trial, which has been conducted across up to 40 sites in Australia and the U.S.
Novavax joined officials in its headquarters city of Gaithersburg, MD, on November 2 to announce expansion plans. The company plans to take 122,000 square feet of space at 700 Quince Orchard Road, and has committed to adding at least 400 local jobs, nearly doubling its current workforce of 450 worldwide. Most of the new jobs are expected to be added b March 2021.
Maryland’s Department of Commerce—which has prioritized assistance to life sciences companies—approved a $2 million conditional loan tied to job creation and capital investment. The state has also approved a $200,000 Partnership for Workforce Quality training grant, and the company is eligible for several tax credits, including the Job Creation Tax Credit and More Jobs for Marylanders.
Additionally, Montgomery County has approved a $500,000 grant tied to job creation and capital investment, while the City of Gaithersburg said it will approve a grant of up to $50,000 from its Economic Development Opportunity Fund. The city accelerated its planning approval process to accommodate Novavax’ timeline, given the company’s role in fighting COVID-19 and resulting assistance from Operation Warp Speed, the Trump administration’s effort to accelerate development of COVID-19 vaccines.
On October 27, Novavax said that it had enrolled 5,500 volunteers in the Phase III U.K. trial, which has been expanded from 10,000 to 15,000 volunteers. The increased enrollment “is likely to facilitate assessment of safety and efficacy in a shorter time period,” according to the company.
The trial, which is being conducted with the U.K. Government’s Vaccines Taskforce, was launched in September and is expected to be fully enrolled by the end of November, with interim data expected by early first quarter 2021, depending on the overall COVID-19 attack rate. Novavax has posted the protocol for the Phase III U.K. trial online. The protocol calls for unblinding of data once 152 participants have achieved mild, moderate or severe endpoints. Two interim analyses are planned upon occurrence of 66 and 110 endpoints.
Novavax also said it expects to launch a second Phase III trial designed to enroll up to 30,000 participants in the U.S. and Mexico by the end of November—a study funded through the U.S. government’s Operation Warp Speed program. The patient population will reflect proportional representation of diverse populations most vulnerable to COVID-19, across race/ethnicity, age, and co-morbidities.
The company cited progress toward large-scale manufacturing while acknowledging delays from original timeframe estimates. Novavax said it will use its contract manufacturing site at FUJIFILM Diosynth Biotechnologies’ Morrisville, NC facility to produce material for the U.S. trial.
On September 25, Novavax entered into a non-exclusive agreement with Endo International subsidiary Par Sterile Products to provide fill-finish manufacturing services at its plant in Rochester, MI, for NVX-CoV2373. Under the agreement, whose value was not disclosed, the Rochester facility has begun production of NVX-CoV2373 final drug product, with initial batches to be used in Novavax’ Phase III clinical trial in the U.S. Par Sterile will also fill-finish NVX-CoV2373 vaccine intended for commercial distribution in the U.S.
A day earlier, Novavax launched the U.K. trial. The randomized, placebo-controlled, observer-blinded study to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 10,000 subjects 18-84 years of age, with and without “relevant” comorbidities, over the following four to six weeks, Novavax said. Half the participants will receive two intramuscular injections of vaccine comprising 5 µg of protein antigen with 50 µg Matrix‑M adjuvant, 21 days apart, while half of the trial participants will receive placebo. At least 25% of the study population will be over age 65.
The trial’s first primary endpoint is first occurrence of PCR-confirmed symptomatic COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2. The second primary endpoint is first occurrence of PCR-confirmed symptomatic moderate or severe COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2
“The data from this trial is expected to support regulatory submissions for licensure in the UK, EU and other countries,” stated Gregory M. Glenn, M.D., President, Research and Development at Novavax.
Maryland Gov. Larry Hogan joined state Secretary of Commerce Kelly M. Schulz and local officials in marking the launch of Phase III studies with a tour of the company’s facilities in Gaithersburg: “The coronavirus vaccine candidate that’s been developed by Novavax is one of the most promising in the country, if not the world.”
On August 31, Novavax reached an agreement in principle with the government of Canada to supply up to 76 million doses of NVX-CoV2373. The value was not disclosed. Novavax and Canada did say that they expect to finalize an advance purchase agreement under which Novavax will agree to supply doses of NVX-CoV2373 to Canada beginning as early as the second quarter of 2021.
The purchase arrangement will be subject to licensure of the NVX-CoV2373 by Health Canada, Novavax said. The vaccine is in multiple Phase II clinical trials: On August 24, Novavax said the first volunteers had been enrolled in the Phase II portion of its ongoing Phase I/II clinical trial (NCT04368988), designed to evaluate the immunogenicity and safety of two doses of of NVX-CoV2373 (5 and 25 µg) with and without 50 µg of Matrix‑M™ adjuvant in up to 1,500 volunteers ages 18-84.
The randomized, placebo-controlled, observer-blinded study is designed to assess two dose sizes (5 and 25 µg) of NVX-CoV2373, each with 50 µg of Matrix‑M. Unlike the Phase I portion, the Phase II portion will include older adults 60-84 years of age as approximately half of the trial’s population. Secondary objectives include preliminary evaluation of efficacy. The trial will be conducted at up to 40 sites in the U.S. and Australia, Novovax said.
NVX-CoV2373 is in a pair of Phase II trials launched in August—including a Phase IIb study in South Africa to assess efficacy, and a Phase II safety and immunogenicity study in the U.S. and Australia.
On August 14, the U.K. government agreed to purchase 60 million doses of NVX-CoV2373 from the company, and support its planned Phase III clinical trial in the U.K., through an agreement whose value was not disclosed. The doses are set to be manufactured as early as the first quarter of 2021.
The trial will be designed to evaluate the ability of NVX-CoV2373 to protect against symptomatic COVID-19 disease as well as evaluate antibody and T-cell responses. The randomized, double-blind, placebo-controlled efficacy study will enroll approximately 9,000 adults 18-85 years of age in the U.K., and is expected to start in the third quarter.
Novavax also said it will expand its collaboration with FUJIFILM Diosynth Biotechnologies (FDB), which will manufacture the antigen component of NVX-CoV2373 from its Billingham, Stockton-on-Tees site in the U.K., as well as at U.S. sites in Morrisville, NC, and College Station, TX. FDB’s U.K. sitevis expected to produce up to 180 million doses annually.
On August 13, Novavax said it signed a development and supply agreement for the antigen component of NVX-CoV2373 with Seoul-based SK bioscience, a vaccine business subsidiary of SK Group. The agreement calls for supply to global markets that include the COVAX Facility, co-led by Gavi, the Coalition for Epidemic Preparedness Innovations (CEPI) and the World Health Organization.
Novavax and SK signed a letter of intent with South Korea’s Ministry of Health and Welfare to work toward broad and equitable access to NVX-CoV2373 worldwide, as well as to make the vaccine available in South Korea. SK bioscience agreed to manufacture the vaccine antigen component for use in the final drug product globally during the pandemic, at its vaccine facility in Andong L-house, South Korea, beginning in August. The value of the agreement was not disclosed.
On August 7, Novavax licensed its COVID-19 vaccine technology to Takeda Pharmaceutical through a partnership by which Takeda will develop, manufacture, and commercialize NVX‑CoV2373 in Japan, using Matrix-M adjuvant to be supplied by Novavax. Takeda will also be responsible for regulatory submission to Japan’s Ministry of Health, Labour and Welfare (MHLW).
MHLW agreed to provide funding to Takeda—the amount was not disclosed in the companies’ announcement—for technology transfer, establishment of infrastructure, and scale-up of manufacturing. Takeda said it anticipated the capacity to manufacture over 250 million doses of NVX‑CoV2373 per year.
Five days earlier, Serum Institute of India agreed to license rights from Novavax to NVX‑CoV2373 for development and commercialization in India as well as low- and middle-income countries (LMIC), through an agreement whose value was not disclosed. Novavax retains rights to NVX-CoV2373 elsewhere in the world.
Novavax and Serum Institute of India agreed to partner on clinical development, co-formulation, filling and finishing and commercialization of NVX-CoV2373. Serum Institute will oversee regulatory submissions and marketing authorizations in regions covered by the collaboration. Novavax agreed to provide both vaccine antigen and Matrix‑M adjuvant, while the partners said they were in talks to have the Serum Institute manufacture vaccine antigen in India. Novavax and Seerum Institute plan to split the revenue from the sale of product, net of agreed costs.
A day earlier, Novavax announced positive results from the Phase I portion of its Phase I/II clinical trial (NCT04368988), designed to evaluate two doses of NVX-CoV2373 (5 and 25 µg) with and without Matrix‑M™ adjuvant in 131 healthy adults ages 18-59. NVX-CoV2373, adjuvanted with Matrix-M, elicited robust antibody responses numerically superior to human convalescent sera, according to data submitted for peer-review to a scientific journal.
All participants developed anti-spike IgG antibodies after a single dose of vaccine, Novavax said, many also developing wild-type virus neutralizing antibody responses. After the second dose, all participants developed wild-type virus neutralizing antibody responses. Both anti-spike IgG and viral neutralization responses compared favorably to responses from patients with clinically significant COVID‑19 disease, the company said—adding that IgG antibody response was highly correlated with neutralization titers, showing that a significant proportion of antibodies were functional.
For both dosages of NVX‑CoV2373 with adjuvant, the 5 µg dose performed “comparably” with the 25 µg dose, Novavax said. NVX‑CoV2373 also induced antigen-specific polyfunctional CD4+ T cell responses with a strong bias toward the Th1 phenotype (IFN-g, IL-2, and TNF-a).
Based on an interim analysis of Phase I safety and immunogenicity data, the trial was expanded to Phase II clinical trials in multiple countries, including the U.S. The trial—which began in Australia in May—is being funded by up-to $388 million in funding from the Coalition for Epidemic Preparedness Innovations (CEPI). If the Phase I/II trial is successful, CEPI said, it anticipates supporting further clinical development that would advance NVX-CoV2373 through to licensure.
On July 23, Novavax joined FDB to announce that FDB will manufacture bulk drug substance for NVX-CoV2373, under an agreement whose value was not disclosed. FDB’s site in Morrisville, NC has begun production of the first batch of NVX-CoV2373. Batches produced at FDB’s Morrisville site will be used in Novavax’s planned pivotal Phase III clinical trial, designed to assess NVX-CoV2373 in up to 30,000 participants, and set to start this fall.
The Phase III trial is among R&D efforts to be funded through the $1.6 billion awarded in July to Novavax through President Donald Trump’s “Operation Warp Speed” program toward late-stage clinical trials and large-scale manufacturing to produce 100 million doses of its COVID-19 vaccine by year’s end. Novavax said the funding will enable it to complete late-stage clinical studies aimed at evaluating the safety and efficacy of NVX-CoV2373.
In June, Novavax said biotech investor and executive David Mott was joining its board as an independent director, after recently acquiring nearly 65,000 shares of the company’s common stock. Also, Novavax was awarded a $60 million contract by the U.S. Department of Defense (DoD) for the manufacturing of NVX‑CoV2373. Through the Defense Health Program, the Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense Enabling Biotechnologies (JPEO-CBRND-EB) agreed to support production of several vaccine components to be manufactured in the U.S. Novavax plans to deliver this year for DoD 10 million doses of NVX‑CoV2373 that could be used in Phase II/III trials, or under an Emergency Use Authorization (EUA) if approved by the FDA.
Also in June, AGC Biologics said it will partner with Novavax on large-scale GMP production of Matrix-M– significantly increasing Novavax’ capacity to deliver doses in 2020 and 2021—through an agreement whose value was not disclosed. And Novavax joined The PolyPeptide Group to announce large-scale GMP production by the global CDMO of two unspecified key intermediate components used in the production of Matrix-M.
In May, Novavax acquired Praha Vaccines from the India-based Cyrus Poonawalla Group for $167 million cash, in a deal designed to ramp up Novavax’s manufacturing capacity for NVX-CoV2373. Praha Vaccines’ assets include a 150,000-square foot vaccine and biologics manufacturing facility and other support buildings in Bohumil, Czech Republic. Novavax said the Bohumil facility is expected to deliver an annual capacity of over 1 billion doses of antigen starting in 2021 for the COVID-19 vaccine.
The Bohumil facility is completing renovations that include the addition of Biosafety Level-3 (BSL-3) capabilities. The site’s approximately 150 employees with “significant experience” in vaccine manufacturing and support have joined Novavax, the company said.
On May 11, Novavax joined CEPI in announcing up to $384 million in additional funding for the company toward clinical development and large-scale manufacturing of NVX-CoV2373. CEPI agreed to fund preclinical as well as Phase I and Phase II studies of NVX-CoV2373. The funding multiplied CEPI’s initial $4 million investment in the vaccine candidate, made two months earlier. Novavax’s total $388 million in CEPI funding accounted for 87% of the total $446 million awarded by the Coalition toward COVID-19 vaccine R&D as of that date.
Novavax identified its COVID-19 vaccine candidate in April. The company said NVX-CoV2373 was shown to be highly immunogenic in animal models measuring spike protein-specific antibodies, antibodies that block the binding of the spike protein to the receptor, and wild-type virus neutralizing antibodies. High levels of spike protein-specific antibodies with ACE-2 human receptor binding domain blocking activity and SARS-CoV-2 wild-type virus neutralizing antibodies were also seen after a single immunization.
In March, Emergent Biosolutions disclosed it retained an option to allocate manufacturing capacity for an expanded COVID-19 program under an agreement with Novavax to provide “molecule-to-market” contract development and manufacturing (CDMO) services to produce Novavax’s NanoFlu™, its recombinant quadrivalent seasonal influenza vaccine candidate.
Earlier in March, Emergent announced similar services to support clinical development of Novavax’s COVID-19 vaccine candidate, saying March 10 it agreed to produce the vaccine candidate and had initiated work, anticipating the vaccine candidate will be used in a Phase I study within the next four months. In February, Novavax said it had produced and was assessing multiple nanoparticle vaccine candidates in animal models prior to identifying an optimal candidate for human testing.
References
- ^ “Company Overview of Novavax, Inc”. Bloomberg.com. Archived from the original on 24 February 2017. Retrieved 2 June2019.
- ^ https://www.globenewswire.com/news-release/2021/03/01/2184674/0/en/Novavax-Reports-Fourth-Quarter-and-Full-Year-2020-Financial-Results-and-Operational-Highlights.html
- ^ Jump up to:a b c d e Bell, Jacob (November 14, 2016). “Novavax aims to rebound with restructuring, more trials”. BioPharma Dive. Washington, D.C.: Industry Dive. Archived from the original on 2017-03-29. Retrieved 2017-03-28.
- ^ Thomas, Katie; Twohey, Megan (2020-07-16). “How a Struggling Company Won $1.6 Billion to Make a Coronavirus Vaccine”. The New York Times. ISSN 0362-4331. Retrieved 2021-01-29.
- ^ Taylor, Nick Paul (3 June 2013). “Novavax makes $30M bid for adjuvant business”. FiercePharma. Archived from the original on 14 September 2016. Retrieved 9 September 2016.
- ^ “Gaithersburg Biotech Receives Grant Worth up to $89 million”. Bizjournals.com. Archived from the original on 2017-04-01. Retrieved 2017-03-28.
- ^ “With promising RSV data in hand, Novavax wins $89M Gates grant for PhIII | FierceBiotech”. Fiercebiotech.com. Archivedfrom the original on 2017-04-14. Retrieved 2017-03-28.
- ^ “Novavax RSV vaccine found safe for pregnant women, fetus”. Reuters. 2016-09-29. Archived from the original on 2016-10-07. Retrieved 2017-03-28.
- ^ Herper, Matthew. “Gates Foundation Backs New Shot To Prevent Babies From Dying Of Pneumonia”. Forbes. Archived from the original on 2016-09-21. Retrieved 2017-03-28.
- ^ “Novavax’s Ebola vaccine shows promise in early-stage trial”. Reuters. 2017-07-21. Archived from the original on 2016-10-02. Retrieved 2017-03-28.
- ^ Jump up to:a b c d e f Adams, Ben (September 16, 2016). “Novavax craters after Phase III RSV F vaccine failure; seeks path forward”. FierceBiotech. Questex. Archived from the original on 18 August 2020. Retrieved 25 Jan 2020.
- ^ Shtrubel, Marty (December 12, 2019). “3 Biotech Stocks That Offer the Highest Upside on Wall Street”. Biotech. Nasdaq. Archived from the original on 2020-01-26. Retrieved 25 Jan 2020.
- ^ Jump up to:a b Budwell, George (January 20, 2020). “3 Top Biotech Picks for 2020”. Markets. Nasdaq. Novavax: A catalyst awaits. Archivedfrom the original on 2020-01-25. Retrieved 25 Jan 2020.
- ^ Mark Terry (February 16, 2018). “Why Novavax Could be a Millionaire-Maker Stock”. BioSpace. Archived from the original on 22 November 2020. Retrieved 6 March 2020.
- ^ Jump up to:a b Eric Sagonowsky (2020-05-11). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 2020-05-16. Retrieved 2020-05-12.
- ^ “Novavax addresses urgent global public health needs with innovative technology”. novavax.com. Archived from the original on 10 September 2020. Retrieved 30 August 2020.
- ^ Sara Gilgore (January 15, 2020). “Novavax earns key FDA status for its flu vaccine. Wall Street took it well”. Washington Business Journal. Archived from the original on 10 November 2020. Retrieved 6 March 2020.
- ^ Sara Gilgore (February 26, 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on March 16, 2020. Retrieved March 6, 2020.
- ^ Nidhi Parekh (July 24, 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on November 22, 2020. Retrieved July 24, 2020.
- ^ “Covid-19: Novavax vaccine shows 89% efficacy in UK trials”. BBC news. Retrieved 1 February 2021.
Further reading
- “Novavax, Inc. Common Stock (NVAX) News Headlines”. Market Activity. Nasdaq. Retrieved 25 Jan 2020. Continuously updated listing of Nasdaq publications related to Novavax, newest items first.
External links
- Official website
- Business data for Novavax, Inc.:
General References
| Type | Public |
|---|---|
| Traded as | Nasdaq: NVAX Russell 2000 Component |
| Industry | Biotechnology |
| Founded | 1987; 34 years ago [1] |
| Headquarters | Gaithersburg, Maryland,United States |
| Area served | Worldwide |
| Key people | Stanley Erck (CEO) |
| Products | Vaccines |
| Revenue | |
| Number of employees | 500+[3] |
| Website | www.novavax.com |
The Novavax COVID-19 vaccine, codenamed NVX-CoV2373, and also called SARS-CoV-2 rS (recombinant spike) protein nanoparticle with Matrix-M1 adjuvant, is a COVID-19 vaccine candidate developed by Novavax and Coalition for Epidemic Preparedness Innovations (CEPI). It requires two doses[1] and is stable at 2 to 8 °C (36 to 46 °F) (refrigerated).[2]
Description
NVX-CoV2373 has been described as both a protein subunit vaccine[3][4][5] and a virus-like particle vaccine,[6][7] though the producers call it a “recombinant nanoparticle vaccine”.[8]
The vaccine is produced by creating an engineered baculovirus containing a gene for a modified SARS-CoV-2 spike protein. The baculovirus then infects a culture of Sf9 moth cells, which create the spike protein and display it on their cell membranes. The spike proteins are then harvested and assembled onto a synthetic lipid nanoparticle about 50 nanometers across, each displaying up to 14 spike proteins.[3][4][8]
The formulation includes a saponin-based adjuvant.[3][4][8]
Development
In January 2020, Novavax announced development of a vaccine candidate, codenamed NVX-CoV2373, to establish immunity to SARS-CoV-2.[9] Novavax’s work is in competition for vaccine development among dozens of other companies.[10]
In March 2020, Novavax announced a collaboration with Emergent BioSolutions for preclinical and early-stage human research on the vaccine candidate.[11] Under the partnership, Emergent BioSolutions will manufacture the vaccine at large scale at their Baltimore facility.[12] Trials have also taken place in the United Kingdom, and subject to regulatory approval, at least 60 million doses will be manufactured by Fujifilm Diosynth Biotechnologies in Billingham for purchase by the UK government.[13][14] They also signed an agreement with Serum Institute of India for mass scale production for developing and low-income countries.[15] It has also been reported, that the vaccine will be manufactured in Spain.[16] The first human safety studies of the candidate, codenamed NVX-CoV2373, started in May 2020 in Australia.[17][18]
In July, the company announced it might receive $1.6 billion from Operation Warp Speed to expedite development of its coronavirus vaccine candidate by 2021—if clinical trials show the vaccine to be effective.[19][20] A spokesperson for Novavax stated that the $1.6 billion was coming from a “collaboration” between the Department of Health and Human Services and Department of Defense,[19][20] where Gen. Gustave F. Perna has been selected as COO for Warp Speed. In late September, Novavax entered the final stages of testing its coronavirus vaccine in the UK. Another large trial was announced to start by October in the US.[21]
In December 2020, Novavax started the PREVENT-19 (NCT04611802) Phase III trial in the US and Mexico.[22][full citation needed][23]
On 28 January 2021, Novavax reported that preliminary results from the United Kingdom trial showed that its vaccine candidate was more than 89% effective.[24][2] However, interim results from a trial in South Africa showed a lower effectiveness rate against the 501.V2 variant of the virus, at around 50-60%.[1][25]
On 12 March 2021, they announced their vaccine candidate was 96.4% effective in preventing the original strain of COVID-19 and 86% effective against the U.K variant. It proved 55% effective against the South African variant in people without HIV/AIDS. It was also 100% effective at preventing severe illness.[citation needed]
Deployment
On 2 February 2021, the Canadian Prime Minister Justin Trudeau announced that Canada has signed a tentative agreement for Novavax to produce millions of doses of its COVID-19 vaccine in Montreal, Canada, once it’s approved for use by Health Canada, making it the first COVID-19 vaccine to be produced domestically.[26]
References
- ^ Jump up to:a b Wadman M, Jon C (28 January 2021). “Novavax vaccine delivers 89% efficacy against COVID-19 in UK—but is less potent in South Africa”. Science. doi:10.1126/science.abg8101.
- ^ Jump up to:a b “New Covid vaccine shows 89% efficacy in UK trials”. BBC News. 28 January 2021. Retrieved 28 January 2021.
- ^ Jump up to:a b c Wadman M (November 2020). “The long shot”. Science. 370 (6517): 649–653. Bibcode:2020Sci…370..649W. doi:10.1126/science.370.6517.649. PMID 33154120.
- ^ Jump up to:a b c Wadman M (28 December 2020). “Novavax launches pivotal U.S. trial of dark horse COVID-19 vaccine after manufacturing delays”. Science. doi:10.1126/science.abg3441.
- ^ Parekh N (24 July 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on 22 November 2020. Retrieved 24 July 2020.
- ^ Chung YH, Beiss V, Fiering SN, Steinmetz NF (October 2020). “COVID-19 Vaccine Frontrunners and Their Nanotechnology Design”. ACS Nano. 14 (10): 12522–12537. doi:10.1021/acsnano.0c07197. PMC 7553041. PMID 33034449.
- ^ Medhi R, Srinoi P, Ngo N, Tran HV, Lee TR (25 September 2020). “Nanoparticle-Based Strategies to Combat COVID-19”. ACS Applied Nano Materials. 3 (9): 8557–8580. doi:10.1021/acsanm.0c01978. PMC 7482545.
- ^ Jump up to:a b c “Urgent global health needs addressed by Novavax”. Novavax. Retrieved 30 January 2021.
- ^ Gilgore S (26 February 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on 16 March 2020. Retrieved 6 March 2020.
- ^ “COVID-19 vaccine tracker (click on ‘Vaccines’ tab)”. Milken Institute. 11 May 2020. Archived from the original on 6 June 2020. Retrieved 12 May 2020. Lay summary.
- ^ Gilgore S (10 March 2020). “Novavax’s coronavirus vaccine program is getting some help from Emergent BioSolutions”. Washington Business Journal. Archived from the original on 9 April 2020. Retrieved 10 March 2020.
- ^ McCartney R. “Maryland plays an outsized role in worldwide hunt for a coronavirus vaccine”. Washington Post. Archived from the original on 7 May 2020. Retrieved 8 May 2020.
- ^ Boseley S, Davis N (28 January 2021). “Novavax Covid vaccine shown to be nearly 90% effective in UK trial”. The Guardian. Retrieved 29 January 2021.
- ^ Brown M (14 August 2020). “60m doses of new covid-19 vaccine could be made in Billingham – and be ready for mid-2021”. TeesideLive. Reach. Retrieved 29 January 2021.
- ^ “Novavax signs COVID-19 vaccine supply deal with India’s Serum Institute”. Reuters. 5 August 2020.
- ^ “Spain, again chosen to produce the vaccine to combat COVID-19”. This is the Real Spain. 18 September 2020.
- ^ Sagonowsky E (11 May 2020). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 16 May 2020. Retrieved 12 May 2020.
- ^ “Novavax starts clinical trial of its coronavirus vaccine candidate”. CNBC. 25 May 2020. Archived from the original on 26 May 2020. Retrieved 26 May 2020.
- ^ Jump up to:a b Thomas K (7 July 2020). “U.S. Will Pay $1.6 Billion to Novavax for Coronavirus Vaccine”. The New York Times. Archived from the original on 7 July 2020. Retrieved 7 July 2020.
- ^ Jump up to:a b Steenhuysen J (7 July 2020). “U.S. government awards Novavax $1.6 billion for coronavirus vaccine”. Reuters. Archived from the original on 14 September 2020. Retrieved 15 September 2020.
- ^ Thomas K, Zimmer C (24 September 2020). “Novavax Enters Final Stage of Coronavirus Vaccine Trials”. The New York Times. ISSN 0362-4331. Archived from the original on 28 September 2020. Retrieved 28 September 2020.
- ^ Clinical trial number NCT04611802 for “A Study Looking at the Efficacy, Immune Response, and Safety of a COVID-19 Vaccine in Adults at Risk for SARS-CoV-2” at ClinicalTrials.gov
- ^ “Phase 3 trial of Novavax investigational COVID-19 vaccine opens”. National Institutes of Health (NIH). 28 December 2020. Retrieved 28 December 2020.
- ^ Lovelace B (28 January 2020). “Novavax says Covid vaccine is more than 89% effective”. CNBC.
- ^ Facher L, Joseph A (28 January 2021). “Novavax says its Covid-19 vaccine is 90% effective in late-stage trial”. Stat. Retrieved 29 January 2021.
- ^ “Canada signs deal to produce Novavax COVID-19 vaccine at Montreal plant”. CP24. 2 February 2021. Retrieved 2 February2021.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Subunit |
| Clinical data | |
| Other names | NVX-CoV2373 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15810 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////////// Novavax, COVID-19, vaccine, CORONA VIRUS, NVX-CoV2373, SARS-CoV-2 rS, TAK 019
#Novavax, #COVID-19, #vaccine, #CORONA VIRUS, #NVX-CoV2373, #SARS-CoV-2 rS, #TAK 019
UPDATE
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373
SARS-CoV-2 rS;
Novavax Covid-19 vaccine (TN);
Nuvaxovid (TN)
SARS-CoV-2 rS;
組換えコロナウイルス (SARS-CoV-2) ワクチン;
コロナウイルス(SARS-CoV-2)スパイク糖タンパク質抗原nvx-cov2373ワクチン;
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373;
SARS-CoV-2 rS
APPROVED JAPAN Nuvaxovid, 2022/4/19
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373, JAPAN 2022, APPROVALS 2022, VACCINE, COVID 19. CORONA VACCINE, SARS-CoV-2
Desidustat


Ranjit Desai
DESIDUSTAT
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
 |
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022

Benvitimod, Tapinarof, тапинароф , تابيناروف , 他匹那罗 ,
Benvitimod, Tapinarof
- Molecular FormulaC17H18O2
- Average mass254.324 Da
3,5-dihydroxy-4-isopropyl-trans-stilbene
Launched – 2019 CHINA, Psoriasis, Tianji Pharma
тапинароф [Russian] [INN]WBI-1001
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
Benvitimod is in phase III clinical trials, Dermavant Sciences for the treatment of atopic dermatitis and psoriasis.
The compound was co-developed by Welichem Biotech and Stiefel Laboratories (subsidiary of GSK). However, Shenzhen Celestial Pharmaceuticals acquired the developement rights in China, Taiwan, Macao and Hong Kong.
Benvitimod (also known as Tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes.It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters. It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis.
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters .[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Tapinarof is a non-steroidal anti-inflammatory drug originated by Welichem Biotech. Dermavant Sciences is developing the product outside China in phase III clinical trials for the treatment of plaque psoriasis. The company is also conducting phase II clinical trials for the treatment of atopic dermatitis. Phase II studies had also been conducted by Welichem Biotech and Stiefel (subsidiary of GlaxoSmithKline) for these indications.
Tapinarof was originated at Welichem Biotech, from which Tianji Pharma and Shenzen Celestial Pharmaceuticals obtained rights to the product in the Greater China region in 2005. In 2012, Welichem licensed development and commercialization rights in all other regions to Stiefel. In 2013, Welichem entered into an asset purchase agreement to regain Greater China rights to the product from Tianji Pharma and Celestial; however, this agreement was terminated in 2014. In 2018, Stiefel transferred its product license to Dermavant Sciences.
_Poinar,_1975.jpg)
Entomopathogenic nematodesemerging from a wax moth cadaver
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
PATENTS

1. US2003171429A1.
2. US2005059733A1.

Reference:1. CN103265412A.


Patent
https://patents.google.com/patent/CN103992212A/en
phenalkenyl Maude (Benvitimod) is a new generation of anti-inflammatory drugs, are useful for treating a variety of major autoimmune diseases, such as psoriasis, eczema, hair and more concentrated colitis allergic diseases.Phenalkenyl Maud stilbene compound, comprising cis and trans isomers, the trans alkenyl benzene Maude has a strong physiological activity, stability and physical and chemical properties, and cis alkenyl benzene Modesto predominantly trans phenalkenyl Maud byproducts during synthesis, conventional methods such as benzene alkenyl Maude Wittig reaction of cis-isomer impurity is inevitable.

[0004] benzyl trans-alkenyl Maude as main impurities in the synthesis, whether a drug is detected, or monitored during the reaction, the synthesis and analysis methods established cis alkenyl benzene Maude has very important significance.Phenalkenyl Maud conventional synthetic methods the impurity content is very low, and the properties of the cis compound is extremely unstable, easily converted to trans-structure, the synthetic method according to the preceding, the cis compound difficult to separate. The synthesis method has not been reported before in the literature. Thus, to find a synthesis route of cis-alkenyl benzene Maude critical.
[0005] The synthesis of compounds of cis-stilbene, in the prior art, there have been many reports, however, the prior art method of synthesizing a reaction product of the cis starting materials and reagents difficult source, the catalyst used is expensive higher costs, operational difficulties, is not conducive to large-scale production, such as:
① Gaukroger K, John A.Hadfield.Novel syntheses of cis and trans isomers ofcombretastatin A-4 [J] .J.0rg.Chemj 2001, (66): 8135-8138, instead of styrene and substituted phenyl bromide boric acid as the raw material, the Suzuki coupling reaction is a palladium catalyst, to give the cis compound, the reaction follows the formula:

Yield and selectivity of the process the structure is good, but the reaction is difficult source of raw materials, catalyst more expensive, limiting the use of this method.
[0006] ② Felix N, Ngassaj Erick A, Lindsey, Brandon Ej Haines.The first Cu- and
amine-free Sonogashira-type cross-coupling in the C_6 -alkynylation of protected
2, -deoxyadenosine [J] .Tetrahedron Letters, 2009, (65): 4085-4091, with a substituted phenethyl m
Alkynyl easily catalyst Pd / CaC03, Fe2 (CO) 9, Pd (OAc) 2 and the like produce cis compound to catalytic reduction. The reaction follows the formula:

Advantage of this method is stereospecific reduction of alkynes in the catalyst, to overcome the phenomenon of cis-trans isomerization of the Wittig reaction, but the reaction requires at _78 ° C, is not conducive to the operation, and the reagent sources difficult, expensive than high cost increase is not conducive to mass production.
[0007] ③ Belluci G, Chiappe C, Moro G L0.Crown ether catalyzed stereospecificsynthesis of Z_and E-stilbenes by Wittig reaction in a solid-liquid two-phasessystem [J] .Tetrahedron Letters, 1996, (37): 4225-4228 using Pd (PPh3) 4 as catalyst, an organic zinc reagent with a halide compound of cis-coupling reaction formula as follows:

The advantage of this method is that selective, high yield to give cis; deficiency is difficult to handle, the catalyst is expensive.
[0008] ④ new Wang, Zhangxue Jing, Zhou Yue, Zouyong Shun, trans-3,4 ‘, 5-trihydroxy-stilbene China Pharmaceutical Synthesis, 2005, 14 (4);. 204-208, reported that the trans compound of formula was dissolved in DMSO solution at a concentration dubbed, ultraviolet irradiation was reacted at 365nm, converted into cis compounds, see the following reaction formula:

However, the concentration of the solution preparation method, the reaction time is more stringent requirements.

The synthesis of cis-alkenyl benzene Maude application embodiments Example 1 A synthesis of cis-alkenyl Maude benzene and benzene-cis-ene prepared Maude, the reaction was carried out according to the following scheme:

Specific preparation process steps performed in the following order:
(O methylation reaction
The 195.12g (Imol) of 3, 5-hydroxy-4-isopropyl benzoic acid, 414.57g (3mol) in DMF was added 5000ml anhydrous potassium carbonate, mixing, stirred at room temperature, then cooled in an ice-salt bath next, slowly added dropwise 425.85g (3mol) of iodomethane, warmed to room temperature after the addition was complete, the reaction 2h, after completion of the reaction was stirred with water, extracted with ethyl acetate, and concentrated to give 3,5-dimethoxy-4- isopropyl benzoate; yield 93%, purity of 99%.
[0033] (2) a reduction reaction
3000ml tetrahydrofuran and 240g (Imol) 3,5-dimethoxy-4-isopropyl benzoate, 151.40g (4mol) mixing at room temperature sodium borohydride was stirred and heated to reflux was slowly added dropwise 400ml methanol, reaction 4h, was added 3L of water was stirred, extracted with ethyl acetate, washed with water, the solvent was removed by rotary evaporation to give a white solid, to give 3,5-dimethoxy-4-isopropylbenzene methanol; 96% yield purity was 99%.
[0034] (3) the oxidation reaction
The 212g (ImoI) of 3,5-dimethoxy-4-isopropylbenzene methanol, DMSO 800ml and 500ml of acetic anhydride were mixed and stirred at rt After 2h, stirred with water, extracted with ethyl acetate, washed with water, dried , and concentrated to give 3,5-dimethoxy-4-isopropyl-benzaldehyde; 94% yield, 99% purity.
[0035] (4) a condensation reaction
The mixture was 209.18g (lmol) of 3,5-dimethoxy-4-isopropyl-benzoic awake and 136.15g (Imol) phenylacetic acid was added 5000ml of acetic anhydride, stirred to dissolve, sodium acetate was added 246.09g , heating to 135 ° C, the reaction after 6h, cooled to room temperature after adjusting the dilute acid 2 was added, extracted with ethyl acetate, the pH was concentrated, added saturated sodium bicarbonate solution adjusted to pH 7, stirred 2h, and extracted with dichloromethane , adding dilute aqueous hydrochloric acid pH 2, the yellow solid was filtered, to obtain 3,5-dimethoxy-4-isopropyl-stilbene acid; 96% yield, 80% purity.
[0036] (5) decarboxylation reaction
The 327g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene acid and 384g (6mol) of copper powder were added to 5000ml of quinoline, 180 ° C reaction 3h, cooled to room temperature ethyl acetate was added with stirring, filtered, and the filtrate was washed with dilute hydrochloric acid to the aqueous layer was colorless and the aqueous phase was extracted with ethyl acetate inverted, the organic layers were combined, washed with water and saturated brine until neutral, i.e., spin-dried to give 3,5 – dimethoxy-4-isopropyl-stilbene; 92% yield, 77% purity.
[0037] (6) Demethylation
The 282.32g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene 4000ml toluene was placed in an ice bath and stirring, was cooled to 0 ° C, and dissolved slowly added 605.9g (5mol after) in N, N- dimethylaniline, was added 666.7g (5mol) of anhydrous aluminum chloride. after stirring for 0.5h, warmed to room temperature, the reaction was heated to 100 ° C 2h, cooled to 60 ° C , hot toluene layer was separated, diluted hydrochloric acid was added to the aqueous phase with stirring to adjust the PH value of 2, extracted with ethyl acetate, washed with water, and concentrated to give the cis-alkenyl benzene Modesto; crude yield 95%, purity 74 %.After separation by column chromatography using 300-400 mesh silica gel, benzene-cis-ene was isolated Maude pure, 68% yield, 98.5% purity. The resulting cis-alkenyl benzene Maud NMR shown in Figure 1, NMR data are as follows:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.255 (m, 5H), 6.558 (d, 1H), 6.402 (d, 1H), 6.218 (s, 2H), 4.872 (s, 2H), 3.423 (m , 1H), 1.359 (q, 6H). Coupling constants / = 12.
[0038] trans-alkenyl benzene Maud NMR shown in Figure 2, the following NMR data:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.477 (d, 2H), 7.360 (t, 2H), 6.969 (q, 2H), 6.501 (s, 1H), 4.722 (s, 2H), 3.486 (m , 1H), 1.380 (t, 6H). Coupling constants / = 16.
[0039] HPLC conditions a cis alkenyl benzene Maude pure product: column was Nucleosil 5 C18; column temperature was 20 ° C; detection wavelength 318nm; mobile phase consisting of 50:50 by volume of acetonitrile and water; flow rate It was 0.6mL / min, injection volume of 5 μ L; cis phenalkenyl Maude 18.423min retention time of a peak in an amount of 96.39%, see Figure 3. Trans phenalkenyl Maude 17.630min retention time of a peak, the content was 99.8%, see Figure 4.After mixing the two, trans-alkenyl benzene Maude 17.664min retention time of the peak, cis-alkenyl benzene Maude 18.458min retention time of the peak, see Figure 5.
PATENT
https://patents.google.com/patent/CN103172497A/en

phenalkenyl Maude is a natural product, a metabolite as to be symbionts.Phenalkenyl Maud Escherichia coli, Staphylococcus aureus has a very significant inhibitory effect, in addition, there is a styrenic Maude suppression of inflammation and its reactive derivative with immunomodulating activity. Alkenyl benzene Modesto topical ointment as an active ingredient, as a class of drugs has been completed two clinical treatment of psoriasis and eczema, the results of ongoing clinical phase III clinical studies, it has been shown to be completed in both psoriasis and eczema clearly effect, together with a styrenic Maude is a non-hormonal natural small molecule compounds, can be prepared synthetically prepared, therefore, it exhibits good market prospect.
[0004] a styrenic Maude initial synthesis route is as follows:
[0005]

[0006] The reaction conditions for each step: 1) isopropanol, 80% sulfuric acid, 60 ° C, 65% .2) sodium borohydride, boron trifluoride, tetrahydrofuran, 0 ° C, 90% .3). of thionyl chloride, heated under reflux, 85% .4). triethyl phosphate, 120 ° C, 80% .5). benzaldehyde, sodium hydride, 85% .6) pyridine hydrochloride, 190 ° C, 60 %.
[0007] The chemical synthesis route, although ultimately obtained a styrenic Maude, but the overall yield is low, part of the reaction step is not suitable for industrial production, due to process conditions result in the synthesis of certain byproducts produced is difficult to remove impurities, difficult to achieve the quality standard APIs.
Preparation of 4-isopropyl-dimethoxy-benzoic acid [0011] 1,3,5_
[0012] 1000 l reactor 200 liters of 80% sulfuric acid formulation (V / V), the temperature was lowered to room temperature, put 80 kg 3,5_-dimethoxybenzoate ,, stirring gradually warmed to 60 ° C, in was added dropwise within 25 kg of isopropanol I hour, the reaction was complete after 5 hours, 500 liters of hot water, filtered, the filter cake was washed with a small amount of hot water I th, crushed cake was removed and dried. The dried powder was recrystallized from toluene, the product was filtered to give 78 kg `, yield 86%. Preparation 2,3,5_ dimethoxy-4-isopropylbenzene methanol
[0013] 1000 l reactor was added 50 kg 3,5_ _4_ isopropyl dimethoxy benzoic acid, 24 kg of potassium borohydride, 400 l of THF, at room temperature was slowly added dropwise 65 kg BF3.Et2O was stirred 12 hours, the reaction was complete, pure water was added dropwise to destroy excess BF3, filtered, concentrated to dryness, methanol – water to give an off-white recrystallized 40.3 kg, yield 90.1%.
[0014] Preparation of 3,3,5-_ ■ methoxy _4- isopropyl group gas section
[0015] 1000 l autoclave, 100 kg of 3,5-dimethoxy-4-isopropylbenzene methanol, 220 l of DMF, 0 ° C and added dropwise with stirring and 50 l of thionyl chloride, 24 hours after the reaction was complete, 300 liters of water and 300 liters of ethyl acetate, the aqueous phase was stirred layered discharged, and then washed with 200 liters of water was added 3 times, until complete removal of DMF, was added concentrated crystallized from petroleum ether to give 98 kg of white solid was filtered and dried a yield of 91%.
Preparation of methyl-dimethoxy-4-isopropylbenzene of diethyl [0016] 4,3,5_
[0017] 500 l autoclave, 98 kg 3,5_ _4_ isopropyl dimethoxy benzyl chloride and 120 l of triethyl phosphite, the reaction at 120 ° C 5h, fear distilled off under reduced pressure, the collection 145-155 ° C / 4mmHg fear minutes, cured at room temperature to give a colorless light solid was 118 kg, yield 81.6%.
, 3- [0018] 5, E-1 _ ■ methoxy-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene
[0019] 500 l autoclave, 33 kg 3,5_-dimethoxy-4-isopropylbenzene acid diethyl ester, 10.8 kg of benzaldehyde, and 120 l of tetrahydrofuran, at 40 ° C, and nitrogen with stirring, was added dropwise a solution of 11.8 kg potassium tert-butoxide in 50 liters of tetrahydrofuran, the temperature dropping control not to exceed 50 ° C. after the dropwise addition stirring was continued for I h, the reaction was complete, 150 liters of ethyl acetate and extracted , washed twice with 150 liters of water, 100 l I washed with brine, and the organic phase was dried and concentrated, methanol – water (I: D as a white crystalline solid 25.3 kg, yield 91%.
[0020] 6> 1, 3 ~ _ ■ Light-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene (I), (De Dae dilute benzene)
[0021] 100 l autoclave, 10 kg 1,3_-dimethoxy-2-isopropyl-5- (2-styryl) benzene _ pyridine hydrochloride and 25 kg nitrogen atmosphere was heated to 180 -190 ° C, stirred for 3 hours after the reaction was completed, 20 l HCl (2N) cooling to 100 ° C, and 20 liters of ethyl acetate the product was extracted, dried and concentrated to give the product 7.3 kg, 83% yield.
[0022] The method for purifying:
[0023] 100 l added to the reaction vessel 15.5 kg of crude product and 39 liters of toluene, heated to the solid all dissolved completely, filtered hot and left to crystallize, after crystallization, filtration, the crystals with cold toluene 10 washed liter at 60 ° C, protected from light vacuo dried for 24 hours, to obtain 14 kg of white needle crystals, yield 90%.
CLIP
https://www.eosmedchem.com/article/237.html
Design new synthesis of Route of Benvitimod
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)
(2)
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
- https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fanie.201814016&file=anie201814016-sup-0001-misc_information.pdf
///Benvitimod, Tapinarof, WBI-1001, тапинароф , تابيناروف , 他匹那罗 , Welichem Biotech, Stiefel Laboratories, Shenzhen Celestial Pharmaceuticals,CHINA 2019 , Psoriasis, Tianji Pharma, Dermavant Sciences, PHASE 3, fda 2022, approvals 2022, vtama, tapinarof
update….
5/23/2022 fda approved, To treat plaque psoriasis, vtama, tapinarof
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
5-[(E)-2-Phenylethen-1-yl]-2-(propan-2-yl)benzene-1,3-diol
|
|
| Other names
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C17H18O2 | |
| Molar mass | 254.329 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoid biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters.[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
See also
- Pinosylvin, a molecule produced in pines that does not bear the isopropyl alkylation.
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. CiteSeerX 10.1.1.603.247. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
PF 04965842, Abrocitinib
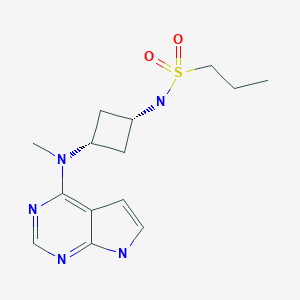

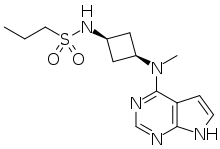
PF-04965842
PF 04965842, Abrocitinib
UNII: 73SM5SF3OR
CAS Number 1622902-68-4, Empirical Formula C14H21N5O2S, Molecular Weight 323.41
N-[cis-3-(Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)cyclobutyl]-1-propanesulfonamide,
N-((1s,3s)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)cyclobutyl)propane-1-sulfonamide
1-Propanesulfonamide, N-(cis-3-(methyl-7H-pyrrolo(2,3-d)pyrimidin-4-ylamino)cyclobutyl)-
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide
PHASE 3, for the potential oral treatment of moderate-to-severe atopic dermatitis (AD)
Jak1 tyrosine kinase inhibitor
UPDATE…… JAPAN APPROVED, 2021, 2021/9/27, CIBINQO
ALSO
fda 2022, APPROVALS 2022, 1/14/2022
THE US
In February 2018, the FDA granted Breakthrough Therapy designation for the treatment of patients with moderate-to-severe AD
PHASEIII
In December 2017, a randomized, double-blind, placebo-controlled, parallel-group, phase III trial (NCT03349060; JADE Mono-1; JADE; B7451012; 2017-003651-29) of PF-04965842 began in patients aged 12 years and older (expected n = 375) with moderate-to-severe AD
PRODUCT PATENT
| Pub. No.: | WO/2014/128591 | International Application No.: | PCT/IB2014/058889 | |||
| Publication Date: | 28.08.2014 | International Filing Date: | 11.02.2014 |
EXPIRY Roughly 2034
| form | powder |
| color | white to beige |
| solubility | DMSO: 10 mg/mL, clear |
| storage temp. | room temp |
- Biochem/physiol Actions
-
- PF-04965842 is a Janus Kinase (JAK) inhibitor selective for JAK1 with an IC50value of 29 nM for JAK1 compared to 803 nM for JAK2, >10000 nM for JAK3 and 1250 nM for Tyk2. JAKs mediate cytokine signaling, and are involved in cell proliferation and differentiation. PF-04965842 has been investigated as a possible treatment for psoriasis.
- Originator Pfizer
- Class Skin disorder therapies; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors
Highest Development Phases
- Phase IIIAtopic dermatitis
- DiscontinuedLupus vulgaris; Plaque psoriasis
Most Recent Events
- 08 Mar 2018Phase-III clinical trials in Atopic dermatitis (In children, In adults, In adolescents) in USA (PO) (NCT03422822)
- 14 Feb 2018PF 4965842 receives Breakthrough Therapy status for Atopic dermatitis in USA
- 06 Feb 2018Pfizer plans the phase III JADE EXTEND trial for Atopic Dermatitis (In children, In adults, In adolescents) in March 2018 (PO) (NCT03422822)
This compound was developed by Pfizer for Kinase Phosphatase Biology research. To learn more about Sigma′s partnership with Pfizer and view other authentic, high-quality Pfizer compounds,
PF-04965842 is an oral Janus Kinase 1 inhibitor being investigated for treatment of plaque psoriasis.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified into tyrosine and serine/threonine kinases. Inappropriate kinase activity, arising from mutation, over-expression, or inappropriate regulation, dys-regulation or de-regulation, as well as over- or under-production of growth factors or cytokines has been i mplicated in many diseases, including but not limited to cancer, cardiovascular diseases, allergies, asthma and other respiratory diseases, autoimmune d iseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative disorders such as Alzheimer’s disease. Inappropriate kinase activity triggers a variety of biological cellular responses relating to cell growth, cell differentiation , survival, apoptosis, mitogenesis, cell cycle control, and cel l mobility implicated in the aforementioned and related diseases.
Thus, protein kinases have emerged as an important class of enzymes as targets for therapeutic intervention. In particular, the JAK family of cellular protein tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2) play a central role in cytoki ne signaling (Kisseleva et al., Gene, 2002, 285 , 1; Yamaoka et al. Genome Biology 2004, 5, 253)). Upon binding to their receptors, cytokines activate JAK which then phosphorylate the cytokine receptor, thereby creating docking sites for signaling molecules, notably, members of the signal transducer and activator of transcription (STAT) family that ultimately lead to gene expression. Numerous cytokines are known to activate the JAK family. These cytokines include, the IFN family (IFN-alpha, IFN-beta, IFN-omega, Limitin, IFN-gamma, IL- 10, IL- 19, IL-20, IL-22), the gp 130 family (IL-6, IL- 11, OSM, LIF, CNTF, NNT- 1//SF-3, G-CSF, CT- 1, Leptin, IL- 12 , I L-23), gamma C family (IL-2 , I L-7, TSLP, IL-9, IL- 15 , IL-21, IL-4, I L- 13), IL-3 family (IL-3 , IL-5 , GM-CSF), single chain family (EPO, GH, PRL, TPO), receptor tyrosine kinases (EGF, PDGF, CSF- 1, HGF), and G-protein coupled receptors (ATI).
Abrocitinib, sold under the brand name Cibinqo, is a Janus kinase inhibitor medication used for the treatment of atopic dermatitis (eczema).[2] It was developed by Pfizer.[2]
Medical uses
Abrocitinib is indicated for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy.[2]
Side effects
The most common adverse effects in studies were upper respiratory tract infection, headache, nausea, and diarrhea.[3]
Pharmacology
Mechanism of action
It is a selective inhibitor of the enzyme janus kinase 1 (JAK1).[3]
Pharmacokinetics
Abrocitinib is quickly absorbed from the gut and generally reaches highest blood plasma concentrations within one hour. Only 1.0 to 4.4% of the dose are found unmetabolized in the urine.[4]
History
- April 2016: initiation of Phase 2b trial
- December 2017: initiation of JADE Mono-1 Phase 3 trial[5]
- May 2018: Results of Phase 2b trial posted
- October 2019: Results of Phase 3 trial presented[6]
- June 2020: Results of second Phase 3 trial published[7]
Society and culture
Legal status
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Cibinqo, intended for the treatment of atopic dermatitis.[8] The applicant for this medicinal product is Pfizer Europe MA EEIG.[8] In December 2021, the European Commission approved abrocitinib for the treatment of atopic dermatitis.[2][9]
In January 2022, the United States Food and Drug Administration (FDA) approved abrocitinib for adults with moderate-to-severe atopic dermatitis.[10]
////////////////////////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
EU
Click to access cibinqo-epar-public-assessment-report_en.pdf
Introduction
The finished product is presented as immediate release film-coated tablets containing 50 mg, 100 mg
or 200 mg of abrocitinib as active substance.
Other ingredients are:
Tablet core: microcrystalline cellulose (E460i), anhydrous dibasic calcium phosphate (E341ii), sodium
starch glycolate and magnesium stearate (E470b).
Film-coat: hypromellose (E464), titanium dioxide (E171), lactose monohydrate, macrogol (E1521),
triacetin (E1518) and red iron oxide (E172).
The product is available in high-density polyethylene (HDPE) bottles with polypropylene closure or
polyvinylidene chloride (PVDC) blisters with aluminium foil lidding film, as described in section 6.5 of
the SmPC.
The chemical name of abrocitinib is N-((1S,3S)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-
yl)amino)cyclobutyl)propane-1-sulfonamide corresponding to the molecular formula C14H21N5O2S. It
has a relative molecular mass of 323.42 Daltons and the following structure depicted in Figure 1:

The chemical structure of abrocitinib was elucidated by a combination of UV/VIS and IR spectroscopy,
mass spectrometry, NMR spectroscopy and X-ray diffraction.
The active substance is a white to pale-purple or pale pink crystalline powder. It is non-hygroscopic
and its solubility is pH dependent. Abrocitinib is classified as BCS Class II. The impact of particle size
on finished product uniformity of dosage units and dissolution has been studied (see finished product
section). Based on the abrocitinib finished product biopharmaceutics performance, stability, and
manufacturing experience, the active substance particle size specification was established.
Abrocitinib is an achiral molecule, but with 2 stereocentres.
Only one crystalline anhydrous form (Form 1) of abrocitinib has been identified. This form has been the
only form used in all toxicology and clinical studies. Extensive polymorph and hydrate screening have
been conducted to investigate if additional solid forms of abrocitinib could be discovered. Abrocitinib,
Form 1 was the only anhydrous crystalline form identified from these studies. No new anhydrous
polymorphs, hydrates or amorphous solids of abrocitinib were isolated from these screens.
Experiments with 1,4 dioxane and dimethyl sulfoxide yielded solvated forms of abrocitinib. When these
solvated structures were subjected to high temperature, these materials desolvated and converted to
Form 1, free base anhydrous form of abrocitinib. However, these are not relevant since the commercial
crystallisation step does not utilise either of these solvent systems.
It has been confirmed that the manufacturing process consistently yields polymorphic form I. This form
is physically and chemically stable under normal manufacturing and storage conditions as well as
under accelerated conditions. Hence the absence of control of form I is justified.
FDA
U.S. FDA Approves Pfizer’s CIBINQO® (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Additional Details on the CIBINQO Clinical Trial Program
Five clinical trials in the CIBINQO JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the New Drug Application (NDA) to support the FDA approval.
The safety and efficacy of CIBINQO was evaluated in three Phase 3, randomized, placebo-controlled clinical trials. The trials evaluated measures of improvements in skin clearance, itch, disease extent, and severity, including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Ratings Scale (PP-NRS). In each of the trials, over 40% of patients had prior exposure to a systemic therapy:
- JADE MONO-1 and JADE MONO-2: A pair of randomized, double-blind, placebo-controlled trials designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO monotherapy in 778 patients 12 years of age and older with moderate-to-severe AD. The trials assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
- JADE COMPARE: A randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO in 837 adult patients with moderate-to-severe AD on background topical medicated therapy. The trial also included an active control arm with dupilumab, a biologic treatment administered by subcutaneous injection, compared with placebo. The trial assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
Select findings for CIBINQO 100 mg, 200 mg, and placebo follow (*p<0.01 or **p<0.001):
- JADE MONO-1:
- IGA Response Rate (Week 12): 24%*, 44%**, and 8%, respectively
- EASI-75 Response Rate (Week 12): 40%**, 62%**, and 12%, respectively
- JADE MONO-2
- IGA Response Rate (Week 12): 28%**, 38%**, and 9%, respectively
- EASI-75 Response Rate (Week 12): 44%**, 61%**, and 10%, respectively
- JADE COMPARE
- IGA Response Rate (Week 12): 36%**, 47%**, and 14%, respectively
- EASI-75 Response Rate (Week 12): 58%**, 68%**, and 27%, respectively
Safety was additionally evaluated through a randomized dose-ranging trial and a long-term, open-label, extension trial (JADE EXTEND).
U.S. IMPORTANT SAFETY INFORMATION
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, AND THROMBOSIS
Serious Infections
Patients treated with CIBINQO may be at increased risk for developing serious infections that may lead to hospitalization or death. The most frequent serious infections reported with CIBINQO were herpes simplex, herpes zoster, and pneumonia.
If a serious or opportunistic infection develops, discontinue CIBINQO and control the infection.
Reported infections from Janus kinase (JAK) inhibitors used to treat inflammatory conditions:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral (including herpes zoster), and other infections due to opportunistic pathogens.
Avoid use of CIBINQO in patients with an active, serious infection, including localized infections. The risks and benefits of treatment with CIBINQO should be carefully considered prior to initiating therapy in patients with chronic or recurrent infections or those who have resided or traveled in areas of endemic tuberculosis or endemic mycoses.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIBINQO, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Consider yearly screening for patients in highly endemic areas for TB. CIBINQO is not recommended for use in patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, or for patients with a negative test for latent TB but who are at high risk for TB infection, start preventive therapy for latent TB prior to initiation of CIBINQO.
Viral reactivation, including herpes virus reactivation (eg, herpes zoster, herpes simplex), was reported in clinical studies with CIBINQO. If a patient develops herpes zoster, consider interrupting CIBINQO until the episode resolves. Hepatitis B virus reactivation has been reported in patients receiving JAK inhibitors. Perform viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with CIBINQO. CIBINQO is not recommended for use in patients with active hepatitis B or hepatitis C.
Mortality
In a large, randomized postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another JAK inhibitor to TNF blocker treatment, a higher rate of all-cause mortality (including sudden cardiovascular death) was observed with the JAK inhibitor. CIBINQO is not approved for use in RA patients.
Malignancies
Malignancies, including non-melanoma skin cancer (NMSC), were reported in patients treated with CIBINQO. Lymphoma and other malignancies have been observed in patients receiving JAK inhibitors used to treat inflammatory conditions. Perform periodic skin examination for patients who are at increased risk for skin cancer. Exposure to sunlight and UV light should be limited by wearing protective clothing and using broad-spectrum sunscreen.
In a large, randomized postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. CIBINQO is not approved for use in RA patients. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Major Adverse Cardiovascular Events
Major adverse cardiovascular events were reported in patients treated with CIBINQO. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients. Patients who are current or past smokers are at additional increased risk. Discontinue CIBINQO in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients treated with CIBINQO. Thrombosis, including PE, DVT, and arterial thrombosis have been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. Many of these adverse reactions were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of overall thrombosis, DVT, and PE were observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients.
Avoid CIBINQO in patients that may be at increased risk of thrombosis. If symptoms of thrombosis occur, discontinue CIBINQO and treat patients appropriately.
Contraindication
CIBINQO is contraindicated in patients taking antiplatelet therapies, except for low-dose aspirin (≤81 mg daily), during the first 3 months of treatment.
Laboratory Abnormalities
Hematologic Abnormalities: Treatment with CIBINQO was associated with an increased incidence of thrombocytopenia and lymphopenia. Prior to CIBINQO initiation, perform a complete blood count (CBC). CBC evaluations are recommended at 4 weeks after initiation and 4 weeks after dose increase of CIBINQO. Discontinuation of CIBINQO therapy is required for certain laboratory abnormalities.
Lipid Elevations: Dose-dependent increase in blood lipid parameters were reported in patients treated with CIBINQO. Lipid parameters should be assessed approximately 4 weeks following initiation of CIBINQO therapy, and thereafter patients should be managed according to clinical guidelines for hyperlipidemia. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Immunizations
Prior to initiating CIBINQO, complete all age-appropriate vaccinations as recommended by current immunization guidelines, including prophylactic herpes zoster vaccinations. Avoid vaccination with live vaccines immediately prior to, during, and immediately after CIBINQO therapy.
Renal Impairment
Avoid use in patients with severe renal impairment or end stage renal disease, including those on renal replacement therapy.
Hepatic Impairment
Avoid use in patients with severe hepatic impairment.
Adverse Reactions
Most common adverse reactions (≥1%) in subjects receiving 100 mg and 200 mg include: nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase, dizziness, urinary tract infection, fatigue, acne, vomiting, oropharyngeal pain, influenza, gastroenteritis.
Most common adverse reactions (≥1%) in subjects receiving either 100 mg or 200 mg also include: impetigo, hypertension, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia.
Use in Pregnancy
Available data from pregnancies reported in clinical trials with CIBINQO are not sufficient to establish a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise females of reproductive potential that CIBINQO may impair fertility.
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIBINQO during pregnancy. Pregnant women exposed to CIBINQO and health care providers are encouraged to call 1-877-311-3770.
Lactation
Advise women not to breastfeed during treatment with CIBINQO and for one day after the last dose.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
About Atopic Dermatitis
AD is a chronic skin disease characterized by inflammation of the skin and skin barrier defects.i,ii Most people know AD is a skin condition. But many don’t realize it can be caused in part by an abnormal immune response beneath the skin. This dysregulated immune response is thought to contribute to inflammation within the skin and the signs of AD on the surface. Lesions of AD are characterized by erythema (red/pink or discolored skin patches, depending on normal skin color), itching, lichenification (thick/leathery skin), induration (hardening)/papulation (formulation of papules), and oozing/crusting.i,ii
AD is one of the most common inflammatory skin diseases, affecting approximately 5-10% of adults in the U.S.iii,iv Approximately 1 in 3 adults with AD have moderate-to-severe disease.v,vi
About Pfizer Inflammation & Immunology
At Pfizer Inflammation & Immunology, we strive to deliver breakthroughs that enable freedom from day-to-day suffering for people living with autoimmune and chronic inflammatory diseases, which can be debilitating, disfiguring and distressing, dramatically affecting what they can do. With a focus on immuno-inflammatory conditions in Rheumatology, Gastroenterology and Medical Dermatology, our current portfolio of approved medicines and investigational molecules spans multiple action and delivery mechanisms, from topicals to small molecules, biologics and biosimilars. The root cause of many immunological diseases is immuno-inflammation, which requires specifically designed agents. Our differentiated R&D approach resulted in one of the broadest pipelines in the industry, where we purposefully match molecules to diseases where we believe they can make the biggest difference. Building on our decades-long commitment and pioneering science, we continue to advance the standard of care for patients living with immuno-inflammatory diseases and are working hand-in-hand with patients, caregivers and the broader healthcare community on healthcare solutions for the many challenges of managing chronic inflammatory diseases, allowing patients to live their best lives.
Pfizer Inc.: Breakthroughs that Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety, and value in the discovery, development, and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments, and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments, and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
There remains a need for new compounds that effectively and selectively inhibit specific JAK enzymes, and JAK1 in particular, vs. JAK2. JAK1 is a member of the Janus family of protein kinases composed of JAK1, JAK2, JAK3 and TYK2. JAK1 is expressed to various levels in all tissues. Many cytokine receptors signal through pairs of JAK kinases in the following combinations: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2 , JAK2/TYK2 or JAK2/JAK2. JAK1 is the most broadly
paired JAK kinase in this context and is required for signaling by γ-common (IL-2Rγ) cytokine receptors, IL—6 receptor family, Type I, II and III receptor families and IL- 10 receptor family. Animal studies have shown that JAK1 is required for the development, function and homeostasis of the immune system. Modulation of immune activity through inhibition of JAK1 kinase activity can prove useful in the treatment of various immune disorders (Murray, P.J.
J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285 , 1-24 (2002); O’Shea, J . J., et al., Ceil , 109, (suppl .) S121-S131 (2002)) while avoiding JAK2 dependent erythropoietin (EPO) and thrombopoietin (TPO) signaling (Neubauer H., et al., Cell, 93(3), 397-409 (1998);
Parganas E., et al., Cell, 93(3), 385-95 (1998)).

Tofacitinib (1), baricitinib (2), and ruxolitinib (3)
SYNTHESIS 5+1 =6 steps
Main synthesis
Journal of Medicinal Chemistry, 61(3), 1130-1152; 2018

INTERMEDIATE
CN 105732637
ONE STEP

CAS 479633-63-1, 7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-7-[(4- methylphenyl)sulfonyl]-
![]()
Pfizer Receives Breakthrough Therapy Designation from FDA for PF-04965842, an oral JAK1 Inhibitor, for the Treatment of Patients with Moderate-to-Severe Atopic Dermatitis
Dateline:
Public Company Information:
NEW YORK–(BUSINESS WIRE)–Pfizer Inc. (NYSE:PFE) today announced its once-daily oral Janus kinase 1 (JAK1) inhibitor PF-04965842 received Breakthrough Therapy designation from the U.S. Food and Drug Administration (FDA) for the treatment of patients with moderate-to-severe atopic dermatitis (AD). The Phase 3 program for PF-04965842 initiated in December and is the first trial in the J AK1 A topic D ermatitis E fficacy and Safety (JADE) global development program.
“Achieving Breakthrough Therapy Designation is an important milestone not only for Pfizer but also for patients living with the often devastating impact of moderate-to-severe atopic dermatitis, their providers and caregivers,” said Michael Corbo, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
Breakthrough Therapy Designation was initiated as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) signed in 2012. As defined by the FDA, a breakthrough therapy is a drug intended to be used alone or in combination with one or more other drugs to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as a breakthrough therapy, the FDA will expedite the development and review of such drug.1
About PF-04965842 and Pfizer’s Kinase Inhibitor Leadership
PF-04965842 is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD including interleukin (IL)-4, IL-13, IL-31 and interferon gamma.
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitor therapies in development. As a pioneer in JAK science, the Company is advancing several investigational programs with novel selectivity profiles, which, if successful, could potentially deliver transformative therapies for patients. Pfizer has three additional kinase inhibitors in Phase 2 development across multiple indications:
- PF-06651600: A JAK3 inhibitor under investigation for the treatment of rheumatoid arthritis, ulcerative colitis and alopecia areata
- PF-06700841: A tyrosine kinase 2 (TYK2)/JAK1 inhibitor under investigation for the treatment of psoriasis, ulcerative colitis and alopecia areata
- PF-06650833: An interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor under investigation for the treatment of rheumatoid arthritis
Working together for a healthier world®
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products. Our global portfolio includes medicines and vaccines as well as many of the world’s best-known consumer health care products. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 150 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
DISCLOSURE NOTICE: The information contained in this release is as of February 14, 2018. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about PF-04965842 and Pfizer’s ongoing investigational programs in kinase inhibitor therapies, including their potential benefits, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, the uncertainties inherent in research and development, including the ability to meet anticipated clinical trial commencement and completion dates and regulatory submission dates, as well as the possibility of unfavorable clinical trial results, including unfavorable new clinical data and additional analyses of existing data; risks associated with preliminary data; the risk that clinical trial data are subject to differing interpretations, and, even when we view data as sufficient to support the safety and/or effectiveness of a product candidate, regulatory authorities may not share our views and may require additional data or may deny approval altogether; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications may be filed in any jurisdictions for any potential indication for PF-04965842 or any other investigational kinase inhibitor therapies; whether and when any such applications may be approved by regulatory authorities, which will depend on the assessment by such regulatory authorities of the benefit-risk profile suggested by the totality of the efficacy and safety information submitted, and, if approved, whether PF-04965842 or any such other investigational kinase inhibitor therapies will be commercially successful; decisions by regulatory authorities regarding labeling, safety and other matters that could affect the availability or commercial potential of PF-04965842 or any other investigational kinase inhibitor therapies; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com .

# # # # #
1 Food and Drug Administration Fact Sheet Breakthrough Therapies at https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm329491.htmaccessed on January 25, 2018
PATENT
CA 2899888
PATENT
WO 2014128591
PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US)
BROWN, Matthew Frank; (US).
FENWICK, Ashley Edward; (US).
FLANAGAN, Mark Edward; (US).
GONZALES, Andrea; (US).
JOHNSON, Timothy Allan; (US).
KAILA, Neelu; (US).
MITTON-FRY, Mark J.; (US).
STROHBACH, Joseph Walter; (US).
TENBRINK, Ruth E.; (US).
TRZUPEK, John David; (US).
UNWALLA, Rayomand Jal; (US).
VAZQUEZ, Michael L.; (US).
PARIKH, Mihir, D.; (US)
COMPD 2

Example 2 : N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane- l -sulƒonamide
This compound was prepared using 1-propanesulfonyl chloride. The crude compound was purified by chromatography on silica gel eluting with a mixture of dichloromethane and methanol (93 : 7) to afford the title compound as a tan sol id (78% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (br s, 1 H), 8.08 (s, 1 H), 7.46 (d, 1 H), 7.12 (d, 1 H), 6.61 (d, 1 H), 4.81-4.94 (m, 1 H), 3.47-3.62 (m, 1 H), 3.23 (s, 3 H), 2.87-2.96 (m, 2 H), 2.52-2.63 (m, 2 H), 2.14-2.27 (m, 2 H) 1.60- 1.73 (m, 2 H) 0.96 (t, 3 H). LC/MS (exact mass) calculated for C14H21N5O2S;
323.142, found (M + H+); 324.1.
PAPER
Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.

https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b01598
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (25)
Schmieder, G.; Draelos, Z.; Pariser, D.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; Page, K.; Peeva, E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study Br. J. Dermatol. 2017, DOI: 10.1111/bjd.16004
Compound 25, N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide is available through MilliporeSigma (cat. no. PZ0304).
CLIP
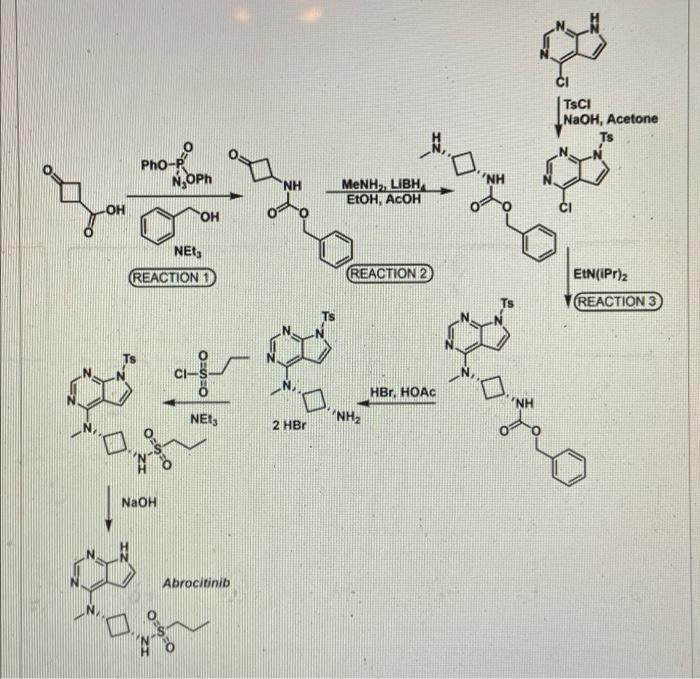
REFERENCES
1: Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, Kieras E, Parsons-Rich D, Menon S, Salganik M, Page K, Peeva E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2017 Sep 26. doi: 10.1111/bjd.16004. [Epub ahead of print] PubMed PMID: 28949012
2 Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.
- Originator Pfizer
- Class Anti-inflammatories; Antipsoriatics; Pyrimidines; Pyrroles; Skin disorder therapies; Small molecules; Sulfonamides
- Mechanism of Action Janus kinase 1 inhibitors
- Phase III Atopic dermatitis
- Discontinued Lupus vulgaris; Plaque psoriasis
- 21 May 2019Pfizer initiates enrolment in a phase I trial in Healthy volunteers in USA (PO) (NCT03937258)
- 09 May 2019 Pfizer plans a phase I pharmacokinetic and drug-drug interaction trial in healthy volunteers in May 2019 (NCT03937258)
- 30 Apr 2019 Pfizer completes a phase I trial (In volunteers) in USA (PO) (NCT03626415)
References[
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf
- ^ Jump up to:a b c d e “Cibinqo EPAR”. European Medicines Agency (EMA). 11 October 2021. Retrieved 17 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. (October 2019). “Efficacy and Safety of Oral Janus Kinase 1 Inhibitor Abrocitinib for Patients With Atopic Dermatitis: A Phase 2 Randomized Clinical Trial”. JAMA Dermatology. 155 (12): 1371–1379. doi:10.1001/jamadermatol.2019.2855. PMC 6777226. PMID 31577341.
- ^ Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. (August 2018). “Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: A phase 1, randomized, placebo-controlled, dose-escalation study”. British Journal of Clinical Pharmacology. 84 (8): 1776–1788. doi:10.1111/bcp.13612. PMC 6046510. PMID 29672897.
- ^ Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- ^ “Pfizer Presents Positive Phase 3 Data at the 28th Congress of the European Academy of Dermatology and Venereology for Abrocitinib in Moderate to Severe Atopic Dermatitis”. Drugs.com. 12 October 2019.
- ^ Silverberg, J. I.; Simpson, E. L.; Thyssen, J. P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; Dibonaventura, M.; Nduaka, C.; Rojo, R. (3 June 2020). “Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial”. JAMA Dermatology. 156 (8): 863–873. doi:10.1001/jamadermatol.2020.1406. PMC 7271424. PMID 32492087.
- ^ Jump up to:a b “Cibinqo: Pending EC decision”. European Medicines Agency. 15 October 2021. Retrieved 15 October 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “European Commission Approves Pfizer’s Cibinqo (abrocitinib) for the Treatment of Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 10 December 2021. Retrieved 17 December 2021.
- ^ “U.S. FDA Approves Pfizer’s Cibinqo (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 14 January 2022. Retrieved 16 January 2022.
External links
- “Abrocitinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- Clinical trial number NCT03575871 for “Study Evaluating Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-2)” at ClinicalTrials.gov
- {{ClinicalTrialsGov|NCT03720470|Study Evaluating Efficacy and Safety of PF-04965842 and Dupilumab in Adult Subjects With Moderate to Severe Atopic Dermatitis on Background Topical Therapy (JADE Compare)}
 |
|
| Clinical data | |
|---|---|
| Trade names | Cibinqo |
| Other names | PF-04965842 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Elimination half-life | 2.8–5.2 h |
| Excretion | 1.0–4.4% unchanged in urine |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.251.498 |
| Chemical and physical data | |
| Formula | C14H21N5O2S |
| Molar mass | 323.42 g·mol−1 |
| 3D model (JSmol) | |
/////////PF 04965842, Abrocitinib, Phase III, Atopic dermatitis, pfizer, fda 2022, APPROVALS 2022
CCCS(=O)(N[C@H]1C[C@@H](N(C)C2=C3C(NC=C3)=NC=N2)C1)=O
CCCS(=O)(=O)N[C@@H]1C[C@@H](C1)N(C)c2ncnc3[nH]ccc23
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
MITAPIVAT
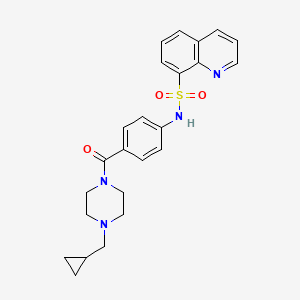

MITAPIVAT
CAS 1260075-17-9
MF C24H26N4O3S
MW 450.55
UPDATE…. FDA APPROVE 2/17/2022 To treat hemolytic anemia in pyruvate kinase deficiency, Pyrukynd
8-Quinolinesulfonamide, N-[4-[[4-(cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-
N-[4-[[4-(Cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-8-quinolinesulfonamide
- AG-348
- Originator Agios Pharmaceuticals
- Class Antianaemics; Piperazines; Quinolines; Small molecules; Sulfonamides
- Mechanism of Action Pyruvate kinase stimulants
- Orphan Drug Status Yes – Inborn error metabolic disorders
- New Molecular Entity Yes
- Phase III Inborn error metabolic disorders
- Phase II Thalassaemia
- 27 Feb 2019 Agios Pharmaceuticals plans a phase III trial for Inborn error metabolic disorders (Pyruvate kinase deficiency) (Treatment-experienced) in the US, Brazil, Canada, Czech Republic, Denmark, France, Germany, Ireland, Italy, Japan, South Korea, Netherlands, Portugal, Spain, Switzerland, Thailand, Turkey and United Kingdom in March 2019 (NCT03853798) (EudraCT2018-003459-39)
- 11 Dec 2018 Phase-II clinical trials in Thalassaemia in Canada (PO) (NCT03692052)
- 29 Aug 2018 Chemical structure information added
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Mitapivat sulfate | N4JTA67V3O | Not Available | Not applicable |
Mitapivat sulfate
CAS#: 2151847-10-6 (sulfate hydrate)
Chemical Formula: C48H60N8O13S3
Exact Mass: 1052.3442
Molecular Weight: 1053.23
Elemental Analysis: C, 54.74; H, 5.74; N, 10.64; O, 19.75; S, 9.13
N-(4-(4-(cyclopropylmethyl)piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide hemisulfate trihydrate
Related CAS #: 2151847-10-6 (sulfate hydrate) 1260075-17-9 (free) 2329710-91-8 (sulfate)
Mitapivat, sold under the brand name Pyrukynd, is a medication used to treat hemolytic anemia.[1] It is taken as the sulfate hydrate salt by mouth.[1]
Mitapivat is a pyruvate kinase activator.[1]
Mitapivat was approved for medical use in the United States in February 2022.[1][2][3]
Activator of pyruvate kinase isoenzyme M2 (PKM2), an enzyme involved in glycolysis. Since all tumor cells exclusively express the embryonic M2 isoform of PK, it is hypothesized that PKM2 is a potential target for cancer therapy. Modulation of PKM2 might also be effective in the treatment of obesity, diabetes, autoimmune conditions, and antiproliferation-dependent diseases.
Agios Pharmaceuticals is developing AG-348 (in phase 3 , in June 2019), an oral small-molecule allosteric activator of the red blood cell-specific form of pyruvate kinase (PK-R), for treating PK deficiency and non-transfusion-dependent thalassemia.
Mitapivat is a novel, first-in-class pyruvate kinase activator. It works to increase the activity of erythrocyte pyruvate kinase, a key enzyme involved in the survival of red blood cells. Defects in the pyruvate kinase enzyme in various red blood cells disorders lead to the lack of energy production for red blood cells, leading to lifelong premature destruction of red blood cells or chronic hemolytic anemia.1
On February 17, 2022, the FDA approved mitapivat as the first disease-modifying treatment for hemolytic anemia in adults with pyruvate kinase (PK) deficiency, a rare, inherited disorder leading to lifelong hemolytic anemia.6 Mitapivat has also been investigated in other hereditary red blood cell disorders associated with hemolytic anemia, such as sickle cell disease and alpha- and beta-thalassemia.1
SYN
WO 20100331307

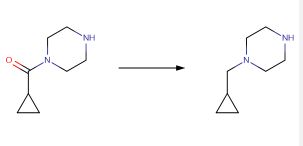
CAS 59878-57-8 TO CAS 57184-25-5
Eisai Co., Ltd., EP1508570, Lithium aluminium hydride (770 mg, 20.3 mmol) was suspended in tetrahydrofuran (150 mL), 1-(cyclopropylcarbonyl)piperazine (1.56 g, 10.1 mmol) was gradually added thereto, and the reaction mixture was heated under reflux for 30 minutes. The reaction mixture was cooled to room temperature, and 0.8 mL of water, 0.8 mL of a 15percent aqueous solution of sodium hydroxide and 2.3 mL of water were seque ntially gradually added thereto. The precipitated insoluble matter was removed by filtration through Celite, and the filtrate was evaporated to give the title compound (1.40g) as a colorless oil. The product was used for the synthesis of (8E,12E,14E)-7-((4-cyclopropylmethylpiperazin-1-yl)carbonyl)oxy-3,6,16,21-tetrahydroxy-6,10,12,16,20-pentamethyl-18,19-epoxytricosa-8,12,14-trien-11-olide (the co mpound of Example 27) without further purification.1H-NMR Spectrum (CDCl3,400MHz) delta(ppm): 0.09-0.15(2H,m), 0.48-0.56(2H,m),0.82-0.93(1H,m),2.25(2H,d,J=7.2Hz) 2.48-2.65(4H,m),2.90-2.99(4H,m).
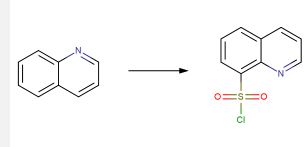
CAS 91-22-5 TO CAD 18704-37-5
chlorosulfonic acid;
Russian Journal of Organic Chemistry, vol. 36, 6, (2000), p. 851 – 853
NMR
US2010/331307
dimethylsulfoxide-d6, 1H
1H NMR (400 MHz, DMSO-d6) δ: 1.2 (t, 2H), 1.3 (t, 2H), 1.31-1.35 (m, 1H), 2.40 (s, 2H), 3.68 (br s, 4H), 3.4-3.6 (m, 4H), 7.06 (m, 6H), 7.25-7.42 (m, 3H), 9.18 (s, 1H) 10.4 (s, 1H)
1H NMR (400 MHz, DMSO-d6) δ: 0.04-0.45 (m, 2H), 0.61-0.66 (m, 2H), 1.4-1.6 (m, 1H), 2.21-2.38 (m, 4H), 2.61 (d, 2H), 3.31-3.61 (br s, 4H), 6.94-7.06 (m, 4H), 7.40 (d, 2H), 7.56-7.63 (m, 2H), 8.28 (d, 1H), 9.18 (s, 1H), 10.4 (s, 1H)
SYN
SYN
J. Med. Chem. 2024, 67, 4376−4418
Mitapivat (Pyrukynd). Mitapivat (9), developed by Agios Pharmaceuticals, Inc., was approved in the EU and US as an oral treatment of hemolytic anemia and pyruvate kinase deficiency (PKD), a rare, inherited condition associated with chronic hemolytic anemia. 66 Previous clinical management of PKD has been limited to only supportive therapies associated with short- and long-term risks, including red cell transfusions and splenectomy. 67 Mitapivat is a first-in-class activator of erythrocyte pyruvate kinase and has been shown to increase hemoglobin levels in patients who were not receiving regular red cell transfusions. In addition to its approved treatment of PKD, it is also currently in clinical studies for the treatment of sickle cell disease and thalassemia. 66 68
As compared to the linear medicinal chemistry route utilized for analog synthesis, Agios Pharmaceuticals, Inc. developed a convergent, high-yielding route to mitapivat depicted in Scheme 17 and Scheme 18.
69 Piperazine 9.3 was synthesized in two steps from tert-butyl piperazine-1-carboxylate (9.1) in
92% yield (Scheme 17) beginning with a reductive amination in the presence of excess cyclopropanecarbaldehyde (9.2) to give the desired tertiary amine. This was carried forward as a
solution into the N-Boc deprotection, precipitating the piperazine as bis-HCl salt 9.3. The second module of mitapivat, sulfonamide 9.7, was prepared in two steps beginning with the reaction of sulfonyl chloride 9.4 with aniline 9.5 to afford ester 9.6 in 99% yield (Scheme 18). Saponification of ester 9.6 under basic
conditions provided carboxylic acid 9.7 in 91% yield. Efficient coupling was demonstrated on the kilogram scale by activation of carboxylic acid 9.7 with CDI, followed by addition of piperazine 9.3. Finally, addition of water to the reaction mixture induced crystallization of the product, providing mitapivat in 90% isolated yield.
(66) Pilo, F.; Angelucci, E. Mitapivat for sickle cell disease and
thalassemia. Drugs Today (Barc) 2023, 59, 125−134.
(67) Al-Samkari, H.; Galacteros, F.; Glenthoej, A.; Rothman, J. A.;
Andres, O.; Grace, R. F.; Morado-Arias, M.; Layton, D. M.; Onodera,
K.; Verhovsek, M.; et al. Mitapivat versus placebo for pyruvate kinase
deficiency. N. Engl. J. Med. 2022, 386, 1432−1442.
(68) Salituro, F. G.; Saunders, J. O.; Yan, S. Preparation of
aroylpiperazines and related compounds as pyruvate kinase M2
modulators useful in treatment of cancer. U.S. Patent US
20100331307, 2010.
(69) Sizemore, J. P.; Guo, L.; Mirmehrabi, M.; Su, Y. Preparation of
amorphous and crystalline forms of N-(4-(4-(cyclopropylmethyl)
piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide useful for
treatment of pyruvate kinase associated disorders. WO 2019104134,
2019.

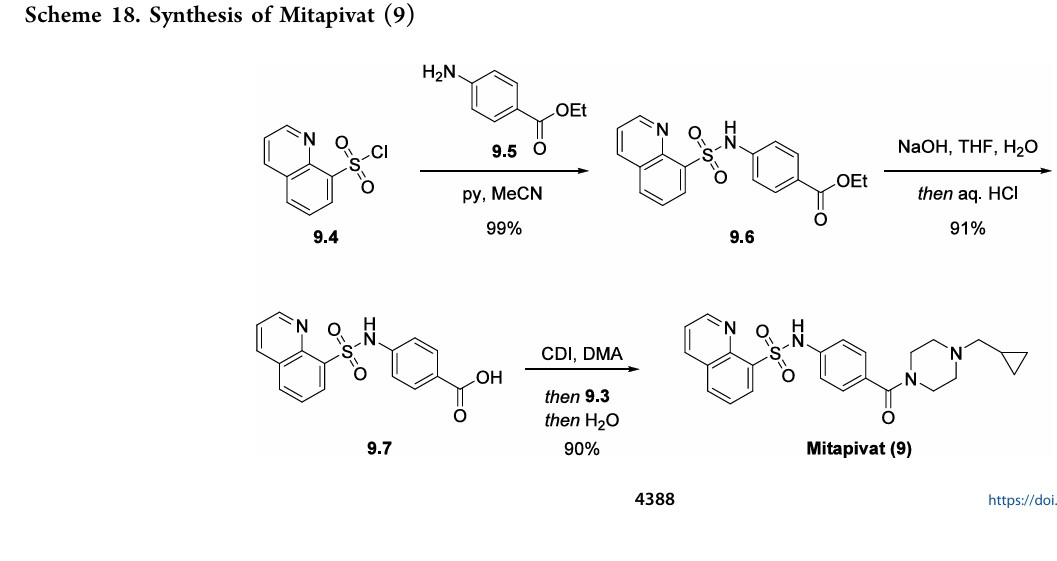
Development Overview
Introduction
Mitapivat (designated AG 348), an orally available, first-in-class, small molecule stimulator of pyruvate kinase (PK), is being developed by Agios Pharmaceuticals for the treatment of pyruvate kinase deficiency (Inborn error metabolic disorders in development table) and thalassemia. Mitapivat is designed to activate the wild-type (normal) and mutated PK-R (the isoform of pyruvate kinase that is present in erythrocytes), in order to correct the defects in red cell glycolysis found within mutant cells. Clinical development is underway for inborn error metabolic disorders in the US, Spain and Denmark and for Thalassaemia in Canada.
Mitapivat emerged from Agios’ research programme focussed on the discovery of small molecule therapeutics for inborn metabolic disorders [see Adis Insight Drug Profile 800036791].
Key Development Milestones
In June 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE trial to evaluate the efficacy and safety of orally administered mitapivat as compared with placebo in participants with pyruvate kinase deficiency (PKD), who are not regularly receiving blood transfusions (NCT03548220; AG348-C-006). The randomised, double-blind, placebo-controlled global trial intends to enrol 80 patients in the US, Canada, Denmark, France, Germany, Italy, Japan, South Korea, Netherlands, Poland, Portugal, Spain, Switzerland, Thailand and United Kingdom. The study design has two parts. Part 1 is a dose optimisation period where patients start at 5mg of mitapivat or placebo twice daily, with the flexibility to titrate up to 20mg or 50mg twice daily over a three month period to establish their individual optimal dose, as measured by maximum increase in hemoglobin levels. After the dose optimisation period, patients will receive their optimal dose for an additional three months in part 2. The primary endpoint of the study is the proportion of patients who achieve at least a 1.5 g/dL increase in haemoglobin sustained over multiple visits in part 2 of the trial
In February 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE-T trial to assess the efficacy and safety of mitapivat in regularly transfused adult subjects with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (EudraCT2017-003803-22; AG348-C-007). The open label trial will enrol approximately 20 patients in Denmark and Spain and will expand to Canada, France, Italy, Japan, the Netherlands, the UK and the US
In December 2018, Agios Pharmaceuticals initiated a phase II study to assess the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat (50mg and 100mg) for the treatment of patients with non-transfusion-dependent thalassemia (AG348-C-010; EudraCT2018-002217-35; NCT03692052). This study will include a 24-week core period followed by a 2-year extension period for eligible participants. The open-label trial intends to enrol approximately 17 patients. Enrolment has been initiated in Canada and may expand to the US and the UK
Agios Pharmaceuticals, in June 2015 initiated the phase II DRIVE PK trial to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat in adult transfusion-independent patients with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (AG348-C-003; NCT02476916). The trial will include two arms with 25 patients each. The patients in the first arm will receive 50mg twice daily, and the patients in the second arm will receive 300mg twice daily. The study will include a six-month dosing period with the opportunity for continued treatment beyond six months based on safety and clinical activity. The open-label, randomised trial completed enrolment of targeted 52 patients in the US, in November 2016. Preliminary data from the trial was presented at the 21st Congress of the European Haematology Association (EHA-2016). Updated results were presented by Agios at the 58th Annual Meeting and Exposition of the American Society of Haematology in December 2016. Based on results of the DRIVE PK trial, Agios plans to develop a registration path for mitapivat. Updated data from the trial was presented at the 22nd Congress of the European Haematology Association (EHA-2017)
In June 2018, Agios Pharmaceuticals completed a phase I trial in healthy male volunteers to assess the absorption, distribution, metabolism, excretion and absolute bioavailability of AG 348 (AG348-C-009; NCT03703505). Radiolabelled analytes of AG 348 ([14C]AG 348 and [13C6]AG 348) were administered in a single oral and intravenous dose on day 1. The open label trial was initiated in May 2018 and enrolled 8 volunteers in the US
In November 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the relative bioavailability and safety of the mitapivat tablet and capsule formulations after single-dose administration in healthy adults (AG348-C-005; NCT03397329). The open-label trial enrolled 26 subjects in the US and was initiated in October 2017
In October 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the pharmacokinetics, safety and effect on QTc interval of mitapivat in healthy volunteers (AG348-C-004; NCT03250598). This single-dose, open-label trial was initiated in August 2017 and enrolled 60 volunteers in the US
In November 2014, Agios completed a randomised, double-blind, placebo-controlled phase I trial that assessed the safety, pharmacokinetics and pharmacodynamics of multiple escalating doses of mitapivat in healthy volunteers (MAD; AG-348MAD; AG348-C-002; NCT02149966). Mitapivat was dosed daily for 14 days. The trial recruited 48 subjects in the US. In June 2015, positive results from the trial were presented at the 20th congress of the European Haematology Association (EHA-2015). Mitapivat showed a favourable pharmacokinetic profile with rapid absorption, low to moderate variability and a dose-proportional increase in exposure following multiple doses and serum hormone changes consistent with reversible aromatase inhibition were also observed
Agios Pharmaceuticals completed a randomised, double-blind, placebo-controlled phase I clinical trial of mitapivat in August 2014 (AG-348 SAD; AG348-C-001; NCT02108106). The study evaluated the safety, pharmacokinetics and pharmacodynamics of single escalating doses of the agent in healthy volunteers. Potential metabolic biomarkers were also explored. The trial enrolled 48 participants in the US
Patent Information
As of January 2018, Agios Pharmaceuticals owned approximately six issued US patents, 65 issued foreign patents, five pending US patent applications and 55 pending foreign patent applications in a number of jurisdictions directed to PK deficiency programme, including mitapivat (AG 348). The patents are valid till at least 2030
- Route of administrationPO
- FormulationTablet, unspecified
- ClassAntianaemics, Piperazines, Quinolines, Small molecules, Sulfonamides
- Mechanism of ActionPyruvate kinase stimulants
- WHO ATC codeA16A-X (Various alimentary tract and metabolism products)B03 (Antianemic Preparations)B06A (Other Hematological Agents)
- EPhMRA codeA16A (Other Alimentary Tract and Metabolism Products)B3 (Anti-Anaemic Preparations)B6 (All Other Haematological Agents)
- Chemical nameN-[4-[4-(cyclopropylmethyl)piperazine-1-carbonyl]phenyl]quinoline-8-sulfonamide
- Molecular formulaC24 H26 N4 O3 S
References
-
Agios Reports First Quarter 2017 Financial Results.
Media Release -
Agios Announces Initiation of Global Phase 3 Trial (ACTIVATE) of AG-348 in Adults with Pyruvate Kinase Deficiency Who Are Not Regularly Transfused.
Media Release -
A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Subjects With Pyruvate Kinase Deficiency
ctiprofile -
Agios Provides Business Update on Discovery Research Strategy and Pipeline, Progress on Clinical Programs, Commercial Launch Preparations and Reports First Quarter 2018 Financial Results at Investor Day.
Media Release -
An Open-Label Study To Evaluate the Efficacy and Safety of AG-348 in Regularly Transfused Adult Subjects With Pyruvate Kinase (PK) Deficiency
ctiprofile -
A Phase 2, Open-label, Multicenter Study to Determine the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of AG-348 in Adult Subjects With Non-transfusion-dependent Thalassemia
ctiprofile -
Agios Announces Key Upcoming Milestones to Support Evolution to a Commercial Stage Biopharmaceutical Company in 2017.
Media Release -
Agios to Present Clinical and Preclinical Data at the 20th Congress of the European Hematology Association.
Media Release -
Agios Announces Updated Data from Fully Enrolled DRIVE PK Study Demonstrating AG-348s Potential as the First Disease-modifying Treatment for Patients with Pyruvate Kinase Deficiency.
Media Release -
Agios Announces New Data from AG-348 and AG-519 Demonstrating Potential for First Disease-modifying Treatment for Patients with PK Deficiency.
Media Release -
Agios Provides Update on PKR Program.
Media Release -
AG-348 Achieves Proof-of-Concept in Ongoing Phase 2 DRIVE-PK Study and Demonstrates Rapid and Sustained Hemoglobin Increases in Adults with Pyruvate Kinase Deficiency.
Media Release -
Agios Reports New, Final Data from Phase 1 Multiple Ascending Dose (MAD) Study in Healthy Volunteers for AG-348, an Investigational Medicine for Pyruvate Kinase (PK) Deficiency.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Results Update from the DRIVE PK Study: Effects of AG-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency. ASH-Hem-2017 2017; abstr. 2194.
Available from: URL: https://ash.confex.com/ash/2017/webprogram/Paper102236.html -
A Phase 2, Open Label, Randomized, Dose Ranging, Safety, Efficacy, Pharmacokinetic and Pharmacodynamic Study of AG-348 in Adult Patients With Pyruvate Kinase Deficiency
ctiprofile -
A Phase I, Open-label Study to Evaluate the Absorption, Distribution, Metabolism, and Excretion and to Assess the Absolute Bioavailability of AG-348 in Healthy Male Subjects Following Administration of a Single Oral Dose of [14C]AG-348 and Concomitant Single Intravenous Microdose of [13C6]AG-348
ctiprofile -
A Phase 1, Randomized, Open-Label, Two-Period Crossover Study Evaluating the Relative Bioavailability and Safety of the AG-348 Tablet and Capsule Formulations After Single-Dose Administration in Healthy Adults
ctiprofile -
A Phase 1, Single-Dose, Open-Label Study to Characterize and Compare the Pharmacokinetics, Safety, and Effect on QTc Interval of AG-348 in Healthy Subjects of Japanese Origin and Healthy Subjects of Non-Asian Origin
ctiprofile -
Agios Pharmaceuticals Initiates Multiple Ascending Dose Trial in Healthy Volunteers of AG-348 for the Potential Treatment of PK Deficiency, a Rare, Hemolytic Anemia.
Media Release -
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose, Safety, Pharmacokinetic, and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Initiates Phase 1 Study of AG-348, a First-in-class PKR Activator, for Pyruvate Kinase Deficiency.
Media Release -
A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose, Safety, Pharmacokinetic and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Pharmaceuticals Reports First Quarter 2014 Financial Results.
Media Release -
Agios Pharmaceuticals Reports Third Quarter 2013 Financial Results.
Media Release -
Agios Pharmaceuticals to Present Preclinical Research at the 2013 American Society of Hematology Annual Meeting.
Media Release -
Agios Presents Preclinical Data from Lead Programs at American Society of Hematology Annual Meeting.
Media Release -
Agios Pharmaceuticals Form 10-K, February 2018. Internet-Doc 2018;.
Available from: URL: https://www.sec.gov/Archives/edgar/data/1439222/000143922218000004/agio-123117x10k.htm -
Agios Outlines Key 2018 Priorities Expanding Clinical and Research Programs to Drive Long Term Value.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Effects of Ag-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency: Updated Results from the Drive Pk Study. EHA-2017 2017; abstr. S451.
Available from: URL: https://learningcenter.ehaweb.org/eha/2017/22nd/181738/rachael.f.grace.effects.of.ag-348.a.pyruvate.kinase.activator.in.patients.with.html?f=m3e1181l15534 -
Agios Presents Updated Data from DRIVE PK Study Demonstrating AG-348 is Well-Tolerated and Results in Clinically Relevant, Rapid and Sustained Hemoglobin Increases in Patients with Pyruvate Kinase Deficiency.
Media Release
Medical uses
Mitapivat is indicated for the treatment of hemolytic anemia in adults with pyruvate kinase deficiency.[1][3]Pharmacology
Mechanism of action
Mitapivat binds to and activates pyruvate kinase, thereby enhancing glycolytic pathway activity, improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels.[4] Mutations in pyruvate kinase cause deficiency in pyruvate kinase which prevents adequate red blood cell (RBC) glycolysis, leading to a buildup of the upstream glycolytic intermediate 2,3-DPG and deficiency in the pyruvate kinase product ATP.[4][5]Society and culture
Names
Mitapivat is the international nonproprietary name (INN).[6]References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216196s000lbl.pdf
- ^ “Agios Announces FDA Approval of Pyrukynd (mitapivat) as First Disease-Modifying Therapy for Hemolytic Anemia in Adults with Pyruvate Kinase Deficiency” (Press release). Agios Pharmaceuticals. 17 February 2022. Retrieved 19 February 2022 – via GlobeNewswire.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/216196Orig1s000ltr.pdf
- ^ Jump up to:a b “Mitapivat (Code C157039)”. NCI Thesaurus. 31 January 2022. Retrieved 19 February 2022. This article incorporates text from this source, which is in the public domain.
- ^ “PK-R allosteric activator AG-348”. NCI Drug Dictionary. National Cancer Institute. Retrieved 19 February 2022. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 78”. WHO Drug Information. 31 (3): 539. hdl:10665/330961.
Further reading
- Kung C, Hixon J, Kosinski PA, Cianchetta G, Histen G, Chen Y, et al. (September 2017). “AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency”. Blood. 130 (11): 1347–1356. doi:10.1182/blood-2016-11-753525. PMC 5609468. PMID 28760888.
- Rab MA, Van Oirschot BA, Kosinski PA, Hixon J, Johnson K, Chubukov V, et al. (January 2021). “AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes”. Haematologica. 106 (1): 238–249. doi:10.3324/haematol.2019.238865. PMC 7776327. PMID 31974203.
External links
- “Mitapivat sulfate”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03548220 for “A Study to Evaluate Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
- Clinical trial number NCT03559699 for “A Study Evaluating the Efficacy and Safety of AG-348 in Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pyrukynd |
| Other names | AG-348, Mitapivat sulfate (USAN US) |
| License data | |
| Routes of administration | By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEMBL |
|
| Chemical and physical data | |
| Formula | C24H26N4O3S |
| Molar mass | 450.56 g·mol−1 |
| 3D model (JSmol) | |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}