
Home » Posts tagged 'APPROVALS 2021' (Page 5)
Tag Archives: APPROVALS 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Dasiglucagon
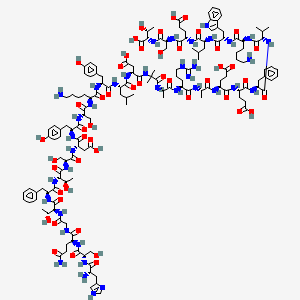



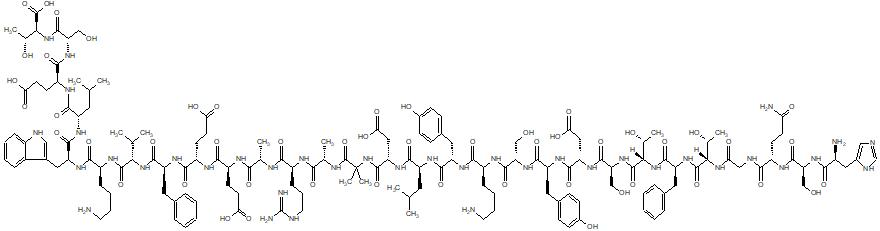

Dasiglucagon
Treatment of Hypoglycemia in Type 1 and Type 2 Diabetes Patients
| Formula | C152H222N38O50 |
|---|---|
| CAS | 1544300-84-6 |
| Mol weight | 3381.6137 |
FDA APPROVED, 2021/3/22, Zegalogue
Zealand Pharma A/S
UNIIAD4J2O47FQ
HypoPal rescue pen
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-2-methylpropanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-carboxy-1-[[(2S)-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-5-oxopentanoic acid
. [16-(2-methylalanine)(S>X),17-L-alanine(R>A),20-L-α-glutamyl(Q>E),21-L-αglutamyl(D>E),24-L-lysyl(Q>K),27-L-α-glutamyl(M>E),28-L-serine(N>S)]human glucagon
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L- phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L- lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L- arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L- valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-alpha-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-alpha-glutamyl-L-alphaC152 H222 N38 O50L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-Molecular Weight3381.61
Other Names
- L-Histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-L-threonine
- Developer Beta Bionics; Zealand Pharma
- ClassAntihyperglycaemics; Antihypoglycaemics; Peptides
- Mechanism of ActionGlucagon receptor agonists
- Orphan Drug StatusYes – Hypoglycaemia; Congenital hyperinsulinism
- RegisteredHypoglycaemia
- Phase IIICongenital hyperinsulinism
- Phase II/IIIType 1 diabetes mellitus
- 22 Mar 2021Registered for Hypoglycaemia (In children, In adolescents, In adults, In the elderly) in USA (SC) – First global approval
- 22 Mar 2021Zealand Pharma anticipates the launch of dasiglucagon in USA (SC, Injection) in June 2021
- 22 Mar 2021Pooled efficacy and safety data from three phase III trials in Hypoglycaemia released by Zealand Pharma
NEW DRUG APPROVALS
one time
$10.00
PATENTS
WO 2014016300
US 20150210744
PAPER
Pharmaceutical Research (2018), 35(12), 1-13
Dasiglucagon, sold under the brand name Zegalogue, is a medication used to treat severe hypoglycemia in people with diabetes.[1]
The most common side effects include nausea, vomiting, headache, diarrhea, and injection site pain.[1]
Dasiglucagon was approved for medical use in the United States in March 2021.[1][2][3] It was designated an orphan drug in August 2017.[4]
Dasiglucagon is under investigation in clinical trial NCT03735225 (Evaluation of the Safety, Tolerability and Bioavailability of Dasiglucagon Following Subcutaneous (SC) Compared to IV Administration).
Medical uses
Dasiglucagon is indicated for the treatment of severe hypoglycemia in people aged six years of age and older with diabetes.[1][2]
Contraindications
Dasiglucagon is contraindicated in people with pheochromocytoma or insulinoma.[1]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214231s000lbl.pdf
- ^ Jump up to:a b “Dasiglucagon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 March 2021.
- ^ “Zealand Pharma Announces FDA Approval of Zegalogue (dasiglucagon) injection, for the Treatment of Severe Hypoglycemia in People with Diabetes” (Press release). Zealand Pharma. 22 March 2021. Retrieved 22 March 2021 – via GlobeNewswire.
- ^ “Dasiglucagon Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 10 August 2017. Retrieved 22 March 2021.
External links
- “Dasiglucagon”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03378635 for “A Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Type 1 Diabetes Subjects” at ClinicalTrials.gov
- Clinical trial number NCT03688711 for “Trial to Confirm the Clinical Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Subjects With T1DM” at ClinicalTrials.gov
- Clinical trial number NCT03667053 for “Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in T1DM Children” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zegalogue |
| AHFS/Drugs.com | Zegalogue |
| License data | US DailyMed: Dasiglucagon |
| Routes of administration | Subcutaneous |
| Drug class | Glucagon receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1544300-84-6 |
| PubChem CID | 126961379 |
| DrugBank | DB15226 |
| UNII | AD4J2O47FQ |
| KEGG | D11359 |
| Chemical and physical data | |
| Formula | C152H222N38O50 |
| Molar mass | 3381.664 g·mol−1 |
| 3D model (JSmol) | Interactive image |
///////////Dasiglucagon, FDA 2021, APPROVALS 2021, Zegalogue, ダシグルカゴン, ZP 4207, ZP-GA-1, Hypoglycemia, Type 1, Type 2 , Diabetes Patients, Zealand Pharma A/S, Orphan Drug Status, Hypoglycaemia, Congenital hyperinsulinism, HypoPal rescue pen, DIABETES
#Dasiglucagon, #FDA 2021, #APPROVALS 2021, #Zegalogue, #ダシグルカゴン, #ZP 4207, ZP-GA-1, #Hypoglycemia, #Type 1, #Type 2 , #Diabetes Patients, #Zealand Pharma A/S, #Orphan Drug Status, #Hypoglycaemia, #Congenital hyperinsulinism, #HypoPal rescue pen, #DIABETESSMILES
- C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC3=CC=C(C=C3)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(=O)O)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC4=CC=CC=C4)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC5=CNC6=CC=CC=C65)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H](CC7=CNC=N7)N)O
Anamorelin hydrochloride

Anamorelin249921-19-5[RN]
3-{(2R)-3-{(3R)-3-Benzyl-3-[(trimethylhydrazino)carbonyl]-1-piperidinyl}-2-[(2-methylalanyl)amino]-3-oxopropyl}-1H-indole
3-Piperidinecarboxylic acid, 1-[(2R)-2-[(2-amino-2-methyl-1-oxopropyl)amino]-3-(1H-indol-3-yl)-1-oxopropyl]-3-(phenylmethyl)-, 1,2,2-trimethylhydrazide, (3R)-8846анаморелинأناموريلين阿那瑞林
| Formula | C31H42N6O3 |
|---|---|
| Molar mass | 546.716 g·mol−1 |
.HCL
Anamorelin hydrochloride
3-Piperidinecarboxylic acid, 1-[(2R)-2-[(2-amino-2-methyl-1-oxopropyl)amino]-3-(1H-indol-3-yl)-1-oxopropyl]-3-(phenylmethyl)-, 1,2,2- trimethylhydrazide, hydrochloride (1:1), (3R)-
| Formula | C31H42N6O3. HCl |
|---|---|
| CAS | 861998-00-7 |
| Mol weight | 583.1645 |
APPROVED JAPAN PMDA Adlumiz, 22/1/2021
アナモレリン塩酸塩
ONO-7643, RC-1291, ST-1291
Antineoplastic, Growth hormone secretagogue receptor (GHSR) agonist
Anamorelin is a non-peptidic ghrelin mimetic
Treatment of cancer anorexia and cancer cachexia
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Company:Novo Nordisk (Originator) , Helsinn,Ono
Anamorelin (INN) (developmental code names ONO-7643, RC-1291, ST-1291), also known as anamorelin hydrochloride (USAN, JAN), is a non-peptide, orally-active, centrally-penetrant, selective agonist of the ghrelin/growth hormone secretagogue receptor (GHSR) with appetite-enhancing and anabolic effects which is under development by Helsinn Healthcare SA for the treatment of cancer cachexia and anorexia.[2][3][4]
Anamorelin significantly increases plasma levels of growth hormone (GH), insulin-like growth factor 1 (IGF-1), and insulin-like growth factor-binding protein 3 (IGFBP-3) in humans, without affecting plasma levels of prolactin, cortisol, insulin, glucose, adrenocorticotropic hormone (ACTH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), or thyroid-stimulating hormone (TSH).[3][5] In addition, anamorelin significantly increases appetite, overall body weight, lean body mass, and muscle strength,[4][5] with increases in body weight correlating directly with increases in plasma IGF-1 levels.[3]
As of February 2016, anamorelin has completed phase III clinical trials for the treatment of cancer cachexia and anorexia associated with non-small-cell lung carcinoma.[6][7]
On 18 May 2017, the European Medicines Agency recommended the refusal of the marketing authorisation for the medicinal product, intended for the treatment of anorexia, cachexia or unintended weight loss in patients with non-small cell lung cancer. Helsinn requested a re-examination of the initial opinion. After considering the grounds for this request, the European Medicines Agency re-examined the opinion, and confirmed the refusal of the marketing authorisation on 14 September 2017.[8] The European Medicines Agency concluded that the studies show a marginal effect of anamorelin on lean body mass and no proven effect on hand grip strength or patients’ quality of life. In addition, following an inspection at clinical study sites, the agency considered that the safety data on the medicine had not been recorded adequately. Therefore, the agency was of the opinion that the benefits of anamorelin did not outweigh its risks.[9]
EMA
The chemical name of anamorelin hydrochloride is 2-Amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazine-1-carbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride corresponding to the molecular formula C31H42N6O3•HCl and has a relative molecular mass 583.16 g/mol and has the following structure:
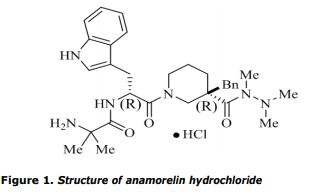
The structure of the active substance was elucidated by a combination of 1 H-NMR, 13C-NMR, elemental analysis, FT-IR, UV and and mass spectrometry. Anamorelin HCl appears as a white to off-white hygroscopic solid, freely soluble in water, methanol and ethanol, sparingly soluble in acetonitrile and practically insoluble in ethyl acetate, isopropyl acetate and n-heptane. Its pka was found to be 7.79 and the partition coefficient 2.98. It has two chiral centres with the R,R absolute configuration, which is controlled in the active substance specification by chiral HPLC. Based on the presented data, neither anamorelin hydrochloride, nor any of its salts have been previously authorised in medicinal products in the European Union. Anamorelin is therefore considered as a new active substance.
SYN
OPRD

PATENT
WO 9958501
PATENT
WO 2001034593
https://patents.google.com/patent/WO2001034593A1/enExample 1A procedure for the preparation of the compound which is either 2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

or2-Amino-N-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

Step aPiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester

A one-necked round-bottom flask (1 I) equipped with a magnetic stirrer and addition funnel was charged with NaOH-pellets (15,6 g), tetrahydrofuran (400 ml) and ethylnipecotate (50 ml, 324 mmol). To the stirred mixture at room temperature was added dropwise a solution of Boc2O (84,9 g, 389 mmol) dissolved in tetrahydrofuran (150 ml) (1 hour, precipitation of white solid, NaOH-pellets dissolved, exoterm). The mixture was stirred overnight at room temperature. The mixture was added to EtOAc (500 ml) and H2O (2000 ml), and the aqueous layer was re-extracted with EtOAc (2 X 500 ml) and the combined organic layers were washed with brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford piperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (82,5 g) as a thin yellow oil.1H-NMR (300 MHz, CDCI3): δ 1,25 (t, 3H, CH3); 1 ,45 (s, 9H, 3 X CH3); 2,05 (m, 1H); 2,45 (m, 1H); 2,85 (m, 1 H); 3,95 (d (broad), 1 H); 4,15 (q, 2H, CH2)Step b3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester (racemic mixture)

A three-necked round-bottom flask (2 I) equipped with a magnetic stirrer, thermometer, nitrogen bubbler and addition funnel was evacuated, flushed with nitrogen, charged with anhydrous tetrahydrofuran (500 ml) and cooled to -70 °C. Then lithium diisopropylamine (164 ml of a 2,0 M solution in tetrahydrofuran, 327 mmol) was added. To the stirred solution at -70 °C was added dropwise over 45 min. a solution of piperidine-1 ,3-dicarboxylic acid 1- tert-butyl ester 3-ethyl ester (80 g, 311 mmol) in anhydrous tetrahydrofuran (50 ml) (temperature between -70 °C and -60 °C, clear red solution). The mixture was stirred for 20 min. and followed by dropwise addition over 40 min. of a solution of benzylbromide (37 ml, 311 mmol) in anhydrous tetrahydrofuran (250 ml) (temperature between -70 °C and -60 °C). The mixture was stirred for 1 hour at -70 °C, and then left overnight at room temperature (pale orange).The reaction mixture was concentrated in vacuo to approx. 300 ml, transferred to a separating funnel, diluted with CH2CI2 (900 ml) and washed with H2O (900 ml). Due to poor separation the aqueous layer was re-extracted with CH2CI2 (200 ml), the combined organic layers were washed with aqueous NaHSO4 (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), H2O (200 ml), brine (100 ml), dried over MgSO4> filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The solids formed was removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-ter–butyl ester 3-ethyl ester (81 ,4 g). ■ HPLC (h8): Rt = 15,79 min.LC-MS: Rt = 7,67 min. (m+1) = 348,0Step c 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (racemic mixture)

3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (81 g, 233 mmol) was dissolved in EtOH (400 ml) and NaOH (400 ml, 16% aqueous solution) in a one neck round- bottom flask (1 L) equipped with a condenser and a magnetic stirrer. The mixture was refluxed for 10 h under nitrogen, and cooled to room temperature, concentrated in vacuo to approx. 600 ml (precipitation of a solid), diluted with H2O (400 ml), cooled in an icebath, and under vigorous stirring acidified with 4 M H2SO4 until pH = 3 (final temperature: 28 °C). The mixture was extracted with EtOAc (2 X 700 ml), and the combined organic layers were washed with brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals formed were removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester (66,0 g)HPLC (h8): Rt = 12,85 min.LC-MS: Rt = 5,97 min. (m+1) = 320,0Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)): Rt = 8,29 min. 46,5 % Rt = 13,69 min. 53,5 %Step d(3R)-3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-Benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester
(Resolution of 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester)

3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (76 g, 238 mmol) was dissolved in EtOAc (3,0 L) in a one neck flask (5L) equipped with magnetic stirring. Then H2O (30 ml), R(+)-1-phenethylamine (18,2 ml, 143 mmol) and Et3N (13,2 ml, 95 mmol) were added and the mixture was stirred overnight at room temperature resulting in precipitation of white crystals (41 ,9 g), which were removed by filtration, washed with EtOAc and dried in vacuo. The precipitate was dissolved in a mixture of aqueous NaHSO4 (300 ml, 10%) and EtOAc (600 ml), layers were separated and the aqueous layer re-extracted with EtOAc (100 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a colourless oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals that had been formed were removed by filtration, washed with heptane and dried in vacuo to give one compound which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine- 1,3-dicarboxylic acid 1-tert-butyl ester (27,8 g).Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)):Rt = 7,96 min. 95,8 % eeStep e(3R)-3-Benzyl-3-(N,N’1N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester

Trimethylhydrazine dihydrochloride (15,3 g, 104 mmol) was suspended in tetrahydrofuran (250 ml) in a one-neck round-bottom flask (1 I) equipped with a large magnetic stirrer, and an addition funnel/nitrogen bubbler. The flask was then placed in a water-bath (temp: 10- 20°C), bromo-rrts-pyrrolydino-phosphonium-hexafluorophosphate (40,4 g, 86,7 mmol) was added, and under vigorous stirring dropwise addition of diisopropylethylamine (59 ml, 347 mmol). The mixture (with heavy precipitation) was stirred for 5 min., and a solution of the product from step d which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester (27,7 g, 86,7 mmol) in tetrahydrofuran (250 ml) was added slowly over 1 ,5 hour. The mixture was stirred overnight at room temperature. The reaction was diluted with EtOAc (1000 ml), washed with H2O (500 ml), aqueous NaHSO4, (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford a thin orange oil. The mixture was dissolved in EtOAc (300 ml), added to SiO2 (150 g) and concentrated in vacuo to a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 I) and the desired compound was liberated with EtOAc (2,5 I). After concentration in vacuo, the product which is either (3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1- carboxylic acid tert-butyl ester or (3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)- piperidine-1-carboxylic acid tert-butyl ester (49 g) as an orange oil was obtained.HPLC (h8): Rt = 14,33 min.Ste f(3R)-3-Benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-Benzyl-piperidine-3- carboxylic acid trimethylhydrazide

The product from step e which is either (3R)-3-Benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck roundbottom flask (2L) equipped with magnetic stirring. The flask was then placed in a waterbath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust- like precipitation). After stirring for 1 hour (precipitation of large amount of white crystals), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration, washed with EtOAc (2 X 100 ml), and dried under vacuum at 40 °C overnight to give the product which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g).HPLC (h8): Rt = 7,84 min.Step q r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3- yl)methyl)-2-oxoethvncarbamic acid tert-butyl ester or .(1 R)-2-..3S)-3-Benzyl-3-(N,N’,N’- trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3-yl)methyl)-2-oxoethyllcarbamic acid tert-butyl ester

Boc-D-Trp-OH (32,3 g, 106 mmol) was dissolved in dimethylacetamide (250 ml) in a one- neck roundbottom flask (500 ml) equipped with a magnetic stirrer and a nitrogen bubbler. The solution was cooled to 0-5 °C and 1-hydroxy-7-azabenzotriazole (14,4 g, 106 mmol), 1- ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (20,3 g, 106 mmol), N- methylmorpholine (11 ,6 ml, 106 mmol) were added. After stirring for 20 min. at 0-5 °C the product from step f which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g, 106 mmol) and N-methylmorpholine (24,4 ml, 223 mmol) were added. The reaction was stirred overnight at room temperature. The mixture was then added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1H- indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3S)-3-benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1 H-indol-3-yl)methyl)-2- oxoethyljcarbamic acid tert-butyl ester (56,7g) as an orange oil.HPLC (h8): Rt = 14,61 min.LC-MS: Rt = 7,35 min. (m+1 ) = 562,6Step h1 -f(2R)-2-Amino-3-(1 H-indol-3-yl)propionylH3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-f(2R)-2-Amino-3-(1 H-indol-3-yl)propionvn-(3S)-3-benzylpiperidine-3- carboxylic acid trimethylhydrazide

The product from step g which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck round-bottom flask (2L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 10 min. (heavy precipitation of oil). The mixture was flushed with N2 to remove excess of HCI and then separated into an oil and an EtOAc-layer. The EtOAc-layer was discarded. The oil was dissolved in H2O (500 ml), CH2CI2 (1000 ml), and solid Na2CO3 was added until pH > 7. The layers were separated, and the organic layer was washed with H2O (100 ml), brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either 1-[(2R)-2-amino-3-(1 H-indol- 3-yl)propionyl]-(3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27 g) as an orange foam.HPLC (h8): Rt = 10,03 min.Step i(1-r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1 -methylethyl fcarbamic acid tert-butyl ester or1-r(1 R)-2-r(3S)-3-Benzyl-3-(N,N’.N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1-methylethyl)carbamic acid tert-butyl ester

Boc-Aib-OH (11 ,9 g, 58,4 mmol) was dissolved in dimethylacetamide (125 ml) in a one-neck roundbottom flask (500 ml) equipped with a magnetic stirrer and nitrogen bubbler. To the stirred solution at room temperature were added 1-hydroxy-7-azabenzotriazole (7,95 g, 58,4 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (11 ,2 g, 58,4 mmol), and diisopropylethylamine (13,0 ml, 75,8 mmol). After 20 min. (yellow with precipitation) a solution of the product from step h which is either 1-[(2R)-2-amino-3-(1 H-indol-3- yl)propionyl]-(3R)-3-benzylpiperidine-3:carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1 H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27,0 g, 58,4 mmol) in dimethylacetamide (125 ml) was added. The reaction was stirred at room temperature for 3 h. The mixture was added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to approx. 500 ml. Then SiO2 (150 g) was added and the remaining EtOAc removed in vacuo to give a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 L), and the desired compound was liberated with EtOAc (2,5 L). After concentration in vacuo, the product which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N, N’, N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester or {1-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester 33,9 g as an orange foam was obtained.HPLC (h8): Rt = 14,05 min.Step j2-Amino-N-r(1 R)-2-f(3R)-3-benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vπ-1- (1 H-indol-3-ylmethyl)-2-oxoethyll-2-methylpropionamide, fumarate or2-Amino-N-r(1 R)-2-r(3S)-3-benzyl-3-(N1N’1N’-trimethylhvdrazinocarbonyl)piperidin-1-yll-1- (1H-indol-3-ylmethyl)-2-oxoethvπ-2-methylpropionamide, fumarate

The product from step i which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1- methylethyl}carbamic acid tert-butyl ester or {1-[(1 R)-2-[(3S)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -(1 H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1 – methylethyljcarbamic acid tert-butyl ester (23,8 g, 36,8 mmol) was dissolved in of EtOAc (800 ml) (clear yellow solution) in a one neck round-bottom flask (1L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust-like precipitation). After stirring for 1 hour (precipitation of large amount of yellow powder), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration and dried under vacuum at 40 °C overnight.The non-crystallinic precipitate was dissolved in H2O (500 ml) and washed with EtOAc (100 ml). Then CH2CI2 (1000 ml) and solid Na2CO3 was added until pH > 7. The 2 layers were separated, and the aqueous layer was e-extracted with CH2CI2 (200 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was evaporated under reduced pressure and redissolved in EtOAc (500 ml) in a one neck round-bottom flask (1 L) equipped with magnetic stirring. A suspension of fumaric acid (3,67 g) in isopropanol (20 ml) and EtOAc (50 ml) was slowly added (5 min.), which resulted in precipitation of a white crystallinic salt. After 1 hour the precipitation was isolated by filtration and dried overnight in vacuum at 40 °C to give the fumarate salt of the compound which is either 2-amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-(1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide or 2-amino-N-[(1 R)-2-[(3S)-3- benzyl-3-(N,N,,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3-ylmethyl)-2- oxoethyl]-2-methylpropionamide (13,9 g) as a white powder.HPLC (A1): Rt = 33,61 min.HPLC (B1): Rt = 34,62 min. LC-MS: Rt = 5,09 min. (m+1) = 547,4
ClaimsHide Dependent
1. The compound obtainable by the procedure as described in example 1 , or a pharmaceutically acceptable salt thereof.2. The compound obtainable by the procedure as described in example 1 , and which compound is2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide

or a pharmaceutically acceptable salt thereof.3. A pharmaceutical composition comprising, as an active ingredient, a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof together with a pharmaceutically acceptable carrier or diluent.4. A pharmaceutical composition according to claim 3 for stimulating the release of growth hormone from the pituitary.5. A pharmaceutical composition according to claim 3 or claim 4 for administration to animals to increase their rate and extent of growth, to increase their milk and wool production, or for the treatment of ailments.6. A method of stimulating the release of growth hormone from the pituitary of a mammal, the method comprising administering to said mammal an effective amount of a compound according to any one of claims 1 or 2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3 – 5.7. A method of increasing the rate and extent of growth, the milk and wool production, or for the treatment of ailments, the method comprising administering to a subject in need thereof an effective amount of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3-5.8. Use of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof for the preparation of a medicament.9. Use according to claim 8 wherein the medicament is for stimulating the release of growth hormone from the pituitary of a mammal.
PATENT
CN 108239141
PATENT
US 20130281701
| Growth hormone is a major participant in the control of several complex physiologic processes, including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes, e.g., stimulation of protein synthesis and free fatty acid mobilization and to cause a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiency in growth hormone can result in a number of severe medical disorders, e.g., dwarfism. |
| The release of growth hormone from the pituitary is controlled, directly or indirectly, by number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin. In both cases the hormones are released from the hypothalamus but their action is mediated primarily via specific receptors located in the pituitary. Other compounds which stimulate the release of growth hormone from the pituitary have also been described. For example, arginine, L-3,4-dihydroxyphenylalanine (1-Dopa), glucagon, vasopressin, PACAP (pituitary adenylyl cyclase activating peptide), muscarinic receptor agonists and a synthetic hexapeptide, GHRP (growth hormone releasing peptide) release endogenous growth hormone either by a direct effect on the pituitary or by affecting the release of GHRH and/or somatostatin from the hypothalamus. |
| The use of certain compounds for increasing the levels of growth hormone in mammals has previously been proposed. For example, U.S. Pat. Nos. 6,303,620 and 6,576,648 (the entire contents of which are incorporated herein by reference), disclose a compound: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide, having the following chemical structure: |
(MOL) (CDX) which acts directly on the pituitary cells under normal experimental conditions in vitro to release growth hormone therefrom. This compound is also known under the generic name “anamorelin.” This growth hormone releasing compound can be utilized in vitro as a unique research tool for understanding, inter alia, how growth hormone secretion is regulated at the pituitary level. Moreover, this growth hormone releasing compound can also be administered in vivo to a mammal to increase endogenous growth hormone release.
Example 1
Crystallization of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide form A
| 0.0103 g of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide was dissolved in methanol (0.1 mL) in a glass vial. The glass vial was then covered with PARAFILM® (thermoplastic film) which was perforated with a single hole. The solvent was then allowed to evaporate under ambient conditions. An X-ray diffraction pattern showed the compound was crystalline ( FIG. 1). |
PATENT
WO 2017067438
https://patents.google.com/patent/WO2017067438A1/enAnamorelin, whose chemical name is: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2- Trimethylformylhydrazide is a compound that increases mammalian growth hormone levels and has a compound structure as shown in Formula I:

Cancer cachexia is a state of consumption in which patients lose a lot of weight and muscle mass. It is necessary for the treatment of cachexia because it weakens the patient, affects the quality of life and interferes with the patient’s treatment plan. The drug alamorelin produces the same effect as the so-called “starved hormone” ghrelin, which stimulates hunger. Alamolin is a mimetic of ghrelin, which is secreted by the stomach and is a ligand for growth hormone receptors. . Alamolin binds to this receptor, causing the release of growth hormone, causing a metabolic cascade that affects a variety of different factors, including fat-removing body weight, as well as blood sugar metabolism. Therefore, alamorelin can also enhance the appetite of patients and help patients stay healthy. The 2014 European Society of Medical Oncology (ESMO) in Madrid, Spain, announced that Alamolin is expected to be the first drug in history to effectively improve cancer cachexia.Alamolin is a drug developed by Helsinn Therapeutics (Switzerland) from Novo Nordisk for the development of a cachexia and anorexia for patients with cancer, including non-small cell lung cancer. It can also be used to treat hip fractures and preventive diseases. The strength of the elderly and the elderly has continued to decline. In two key, 12-week Phase III clinical trials (ROMANA 1, ROMANA 2), alamorelin can significantly increase the body fat loss, and is generally tolerated; the incidence of serious adverse drug reactions is less than 3%, mainly related to hyperglycemia and diabetes. Compared with the placebo group, alamorelin continued to increase body weight and improve cancer anorexia-cachexia-related symptoms and concerns; however, there was no significant difference in the improvement of grip strength between the alamolin group and the placebo group. Therefore, this product has excellent clinical value and market value.The polymorphic form of the drug free base and its preparation are reported as follows:Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl is disclosed in the patent ZL99806010.0 A method for synthesizing hydrazide, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonyl)piperidin-1-yl tert-Butyl ester of 1-((1H-indol-3-yl)methyl)-2-oxoethyl]carbamate is dissolved in dichloromethane, then trifluoroacetic acid is added to remove tert-butyl formate After the base, the mixture was concentrated to remove the solvent, and then the product was extracted with dichloromethane, and the obtained extract was concentrated to dryness to give (3R)-1-(2-methylalanyl-D-color ammonia as an amorphous powder. Acyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent ZL00815145.8 discloses the synthesis of alamorelin and its compounds as pharmaceutically acceptable salts, relating to novel diastereomeric compounds, pharmaceutically acceptable salts thereof, compositions containing them and their use in therapy Lack of use of medical conditions caused by growth hormone. Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformyl is disclosed in this patent. The synthesis method of hydrazine, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonylcarbonyl)piperidin-1-yl] 1-((1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was dissolved in ethyl acetate, and then hydrogen chloride gas was passed to remove the tert-butyl formate protection group. , the solid is dissolved in water, and then the pH is adjusted to about 7 with sodium carbonate, and the product is extracted with dichloromethane; the extract phase is concentrated to obtain (3R)-1-(2-methylalanyl-D-tryptophan). -3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent WO2006016995 discloses (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl as a medicament Crystalline polymorphs of hydrazides, methods of producing and separating these polymorphs, and pharmaceutical compositions and drug therapies containing these polymorphs, the crystalline polymorphs for direct application to the pituitary Gland cells release the growth hormone. This patent discloses (4R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone 4 Crystal form: Form A, Form B, Form C and Form D. The patent also provides the preparation of 3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone. The method of crystal form, especially the preparation method of Form C, in which the method of removing the tert-butyl formate protecting group of methanesulfonic acid in methanol is utilized without exception. As a well-known cause in the art, clinical studies have found that mesylate is genotoxic, and its DNA alkylation leads to mutagenic effects, in which methyl methanesulfonate and ethyl methanesulfonate have been reported. (eg document EMEA/44714/2008). The invention adopts hydrochloric acid or hydrogen chloride gas to remove the tert-butyl formate protecting group, avoids the method of removing methanesulfonic acid, thereby avoiding the risk of the genotoxic impurities in the process, and increasing the risk. The safety of the drug.(3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-three prepared by the patent ZL99806010.0 and the patent ZL00815145.8 Methyl formyl hydrazide, no data on the purity of its compounds, we found that (3R)-1-(2-methylalanyl-D-tryptophan)-3 was prepared by this method. -Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide does not help to remove the impurities produced, and the purity of the obtained product is not high, and it is difficult to meet the medicinal requirements. And (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethyl obtained by the preparation method of the present invention. The crystal form of the formyl hydrazide has a purity of 99.8% and a single impurity of less than 0.1%, which fully meets the requirements for medicinal purity. Moreover, the crystal form is stable to conditions such as pressure, temperature, humidity and illumination, and the preparation method is simple in operation and suitable for industrial production.Example 1:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, and then 4 L of dichloromethane was added to the reaction flask, and the raw material was completely dissolved by stirring.Then, the reaction system is cooled to 10 ° C or lower in an ice bath, hydrogen chloride gas is continuously supplied to the reaction liquid, and solids are gradually precipitated, and the reaction is further maintained at about 10 ° C for 3 to 5 hours, and the sample is detected. After the reaction of the raw materials is completed, the reaction system is completed. 1.5 L of water was added thereto, the solid was completely dissolved, and then the pH was adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the layers were separated; the aqueous phase was extracted once more with dichloromethane, and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 246 g, yield 97.2%. HPLC content (area normalization method) was 96.1%.Example 2:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, 36% concentrated hydrochloric acid was added to the reaction flask, and the reaction system was heated to 40 with stirring. The reaction was carried out at ° C to 50 for 3 hours.Then, the sample is detected. After the reaction of the raw material is completed, the reaction system is cooled to 10 or less, and 2.0 L of dichloromethane is added to the reaction system, and then the pH is adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the aqueous phase is further separated. It was extracted once with dichloromethane and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 248 g, yield 98%. HPLC content (area normalization method) was 96.2%.Preparation of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone E crystal formExample 3Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10g was added to the reaction flask and 30 ml of N- was added.Methylpyrrolidone, stirred and dissolved completely. Then, 60 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 20 ° C, filtered, and the filter cake was washed with a mixture of N-methylpyrrolidone / H 2 O; the cake was vacuum dried at about 55 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophan)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.5 g), HPLC content (area normalization) 99.72%. The XRD pattern is shown in Fig. 1, the DSC chart is shown in Fig. 2, and the TGA pattern is shown in Fig. 3, where the crystal form is defined as the E crystal form. The DSC of the crystal form has an endotherm at 120.05, the TGA is heated at 60A, and the crystal loss of 5 is about 3.1%. Combined with the Karl Fischer method, the moisture content of the product is determined. 3.1% and 3.2% indicate that the sample is present as a monohydrate.Example 4:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of N,N-dimethylformamide was added, stirred, and dissolved completely. Then, 30 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 50 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 h.Slowly cool to below 10 ° C, filter, filter cake washed with N, N-dimethylformamide / H 2 O mixture; vacuum cake dried at around 55 ° C to obtain (3R)-1-(2-A Alanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.5 g), HPLC content (area normalization) ) 99.87%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 5:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of dimethyl sulfoxide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C, the solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of dimethyl sulfoxide / H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.1 g), HPLC content (area normalization) 99.61%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 6Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of 1,4-dioxane was added, stirred, and dissolved completely. Then, 50 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of 1,4-dioxane/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methyl alanyl-D-tryptophan-3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.7 g), HPLC content (area normalization) 99.11%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of N,N-dimethylacetamide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and was slowly cooled to about 50 ° C. Seed crystals were added thereto, and cooling was continued to gradually precipitate a solid.The reaction system was cooled to about 10 ° C, filtered, and the filter cake was washed with a mixture of N,N-dimethylacetamide/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2- Methylalanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.1 g), HPLC content (area normalized) Law) 99.78%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 50 ml of acetone was added, stirred, and dissolved completely. Then, 70 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 45 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cool to below 10 ° C, filter, filter cake washed with acetone / H 2 O mixture; filter cake vacuum dried at around 50 ° C to obtain (3R)-1-(2-methylalanyl-D-color Aminoacyl-3-phenylmethyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.3 g), HPLC content (area normalization) 98.9%. Upon comparison, it was confirmed that the solid was in the E crystal form.
SYN
Reference:
1. Org. Process Res. Dev. 2006, 10, 339–345.

Abstract

The rapid process development of a scaleable synthesis of the pseudotripeptide RC-1291 for preclinical and clinical evaluation is described. By employing a nontraditional N-to-C coupling strategy, the peptide chain of RC-1291 was assembled in high yield, with minimal racemization and in an economical manner by introducing the most expensive component last. A one-pot deprotection/crystallization procedure was developed for the isolation of RC-1291 free base, which afforded the target compound in excellent yield and with a purity of >99.5% without chromatographic purification.
(R,R)-2-Amino-N-[2-[3-benzyl-3-(N,N′,N′-trimethyl-hydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)- 2-oxo-ethyl]-2-methyl-propionamide (1). Crude 7 (911 g; 1.28 mol theoretical)10 was dissolved in methanol (4.12 L) in a 22-L round-bottom flask equipped with a mechanical stirrer, a temperature probe, a reflux condenser, a gas (N2) inlet, and an addition funnel. The solution was heated to 55 °C; then methanesulfonic acid (269.5 g, 2.805 mol) was added over a period of 15 min. (Caution: gas evolution!) The solution was then heated to 60 °C for a period of 1 h, after which HPLC analysis showed that no 7 remained. The temperature of the reaction mixture was increased to reflux (68-72 °C) over a period of 35 min, while simultaneously adding a solution of KOH (85%, 210.4 g, 3.187 mol) in water (4.12 L). The clear, slightly yellow solution was then allowed to cool to 20 °C at a rate of 5 °C/h. The free base of RC1291 (1) crystallized as a pale-yellow solid, which was isolated by filtration. The filter cake was washed with two portions of 50% aqueous methanol (500 mL each) and then dried under high vacuum at 20 ( 5 °C to afford 1 as an off-white, crystalline solid (595 g, 85% yield for two steps, >99.5% AUC by HPLC).
HRMS (ESI) calcd for C31H43N6O3 [M + H]+ 547.3397, found 547.3432.
1H NMR (DMSO-d6; 413 K) δ 10.30 (s, 1H), 7.85 (bs, 1H), 7.50 (d, J ) 7.8 Hz, 1H), 7.27 (d, J ) 8.1 Hz, 1H), 7.1-7.2 (m, 3H), 6.95-7.0 (m, 5H), 5.07 (t, J ) 6.3 Hz, 1H), 3.54 (d, J ) 12.3 Hz, 1H), 3.36 (bs, 1H), 3.15-3.30 (m, 1H), 3.06 (dd, J ) 7.2, 14.4 Hz, 1H), 2.96 (dd, J ) 6.0, 14.3 Hz, 2H), 2.7-2.8 (m, 6H), 2.43 (m, 6H), 2.09 (bs, 1H), 1.73 (bs, 1H), 1.45-1.55 (m, 2H), 1.3-1.40 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H).
13C NMR (DMSO-d6; 413 K) δ 175.8, 173.4, 170.3, 137.0, 135.7, 129.0, 127.2, 127.1, 125.3, 122.9, 120.1, 117.6, 110.7, 109.4, 53.6, 49.0, 47.0, 42.7, 38.5, 30.7, 28.2, 28.0, 23.2, 21.1.

PAPER
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.8b00322
Cachexia and muscle wasting are very common among patients suffering from cancer, chronic obstructive pulmonary disease, and other chronic diseases. Ghrelin stimulates growth hormone secretion via the ghrelin receptor, which subsequently leads to increase of IGF-1 plasma levels. The activation of the GH/IGF-1 axis leads to an increase of muscle mass and functional capacity. Ghrelin further acts on inflammation, appetite, and adipogenesis and for this reason was considered an important target to address catabolic conditions. We report the synthesis and properties of an indane based series of ghrelin receptor full agonists; they have been shown to generate a sustained increase of IGF-1 levels in dog and have been thoroughly investigated with respect to their functional activity.

Patent
https://patents.google.com/patent/EP2838892A1/enGrowth hormone is a major participant in the control of several complex physiologic processes including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes such as stimulating protein synthesis and mobilizing free fatty acids, and causing a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiencies in growth hormone can result in dwarfism and other severe medical disorders.The release of growth hormone from the pituitary gland is controlled directly and indirectly by a number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin.The use of certain compounds to increase levels of growth hormone in mammals has previously been proposed. Anamorelin is one such compound. Anamorelin is a synthetic orally active compound originally synthesized in the 1990s as a growth hormone secretogogue for the treatment of cancer related cachexia. The free base of anamorelin is chemically defined as:® (3R) 1 -(2-methylaIanyl~D ryptophyl)~3-(phenylraethyl)~3~piperidineearboxylie acid 1 ,2,2trimethyihydrazide,* 3-{(2R)-3-{(3R)-3-benzyi-3-| (trimethylhydrazino)carbonyi]piperidin-l»yl}-2-[(2»met hylaianyl)amino]-3-ox.opropyi}-IH-indole, or• 2-Amino-N-[(lR)-2-[(3R)-3~benzyWcarbony piperidin- 1 -yl] – 1-( 1 H-indol-3 -yl^^and has the below chemical structure;
U.S. Patent No. 6,576,648 to Artkerson reports a process of preparing anamorelin as the fumarate salt, with the hydrochloride salt produced as an intermediate in Step (j) of Example 1 . U.S. Patent No. 7,825, 138 to Lorimer describes a process for preparing crystal forms of the free base of anamorelin.There is a need to develop anamorelin monohydrochloride as an active pharmaceutical ingredient with reduced impurities and improved stability over prior art forms of anamorelin hydrochloride, such as those described in U.S. Patent No, 6,576,648, having good solubility, bioavailability and processabi!ity. There is also a need to develop methods of producing pharmaceutically acceptable forms of anamorelin monohydrochloride thai have improved yield over prior art processes, reduced residual solvents, and controlled distribution of chloride content,it has unexpectedly been discovered that the process of making the hydrochloride salt of anamorelin described in Step (j) of U.S. Patent No. 6.576,648 can result in excessive levels of chloride in the final product, and that this excess chloride leads to the long-term instability of the final product due at least, partially to an increase in the amount of the less stable dihydrochloride salt of anamorelin. Conversely, because anamorelin free base is less soluble in water than the hydrochloride salt, deficient chloride content in the final product can lead to decreased solubility of the molecule. The process described in U.S. Patent No, 6,576,648 also yields a final product that contains more than 5000 ppm (0.5%) of residual solvents, which renders the product less desirable from a pharmaceutical standpoint, as described in CH Harmonized Tripartite Guideline. See Impurities; Guideline for residual solvents Q3C(R3). in order to overcome these problems, methods have been developed which, for the first time, allow for the efficient and precise control of the reaction between anarnorehn tree base and hydrochloric acid in situ, thereby increasing the yield of anarnorehn monohydrochioride from the reaction and reducing the incidence of unwanted anamorelin dihydroeh ride. According to the method, the free base of anamorelin is dissolved in an organic solvent and combined with water and hydrochloric acid, with the molar ratio of anarnorehn and chloride tightly controlled to prevent an excess of chloride in the final product. The water and hydrochloric acid can be added either sequentially or at the same time as long as two separate phases are formed. Without wishing to be bound by any theory, it is believed thai as the anamorelin free base in the organic phase is protonated by the hydrochloric acid it migrates into the aqueous phase. The controlled ratio of anamorelin free base and hydrochloric acid and homogenous distribution in the aqueous phase allows for the controlled formation of the monohydrochioride salt over the dihydrochloride, and the controlled distribution of the resulting chloride levels within individual batches and among multiple batches of anamorelin monohydrochioride.Thus, in a fust embodiment the invention provides methods for preparing anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride comprising: (a) dissolving anamorelin free base in an organic solvent to form a solution; (b) mixing said solution with water and hydrochloric acid for a time sufficient to: (i) react said anamorelin free base with said hydrochloric acid, and (ii) form an organic phase and an aqueous phase; (c) separating the aqueous phase from the organic phase; and (d) isolating anamorelin monohydrochioride from the aqueous phase.In a particularly preferred embodiment, the molar ratio of anamorelin to hydrochloric acid used in the process is less than or equal to 1 : 1 , so as to reduce the production of anamorelin dihydrochloride and other unwanted chemical species. Thus, for example, hydrochloric acid can be added at a molar ratio of from 0,90 to 1 ,0 relative to said anamorelin, from 0.90 to 0.99, or from 0.93 to 0.97.n another particularly preferred embodiment, the anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride is isolated from the aqueous phase via spray drying, preferably preceded by distillation. This technique has proven especially useful in the manufacture of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride because of the excellent reduction in solvent levels observed, and the production of a stable amorphous form of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride. In other embodiments, the invention relates to the various forms of anamorelin monohvdrochloride and compositions comprising anamorelin monohvdrochloride produced by the methods of the present invention. In a first embodiment, which derives from the controlled chloride content among batches accomplished by the present methods, the invention provides anamorelin monohvdrochloride or a composition comprising anamorelin monohydrochloride having an inter-batch chloride content of from 5.8 to 6.2%, preferably from 5.8 to less than 6.2%. Alternatively, the invention provides anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride having a molar ratio of chloride to anamorelin less than or equal to 1 : 1 , such as from 0.9 to 1.0 or 0.99, in yet another embodiment the invention provides an amorphous form of anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride. Further descriptions of the anamorelin monohydrochloride and compositions comprising the anamorelin monohydrochloride are given in the detailed description which follows.EXAMPLE 1 . PREPARATION OF ANAMOREUN HYDROCHLORIDEVarious methods have been developed to prepare the hydrochloric acid salt of anarnorelin, with differing results.In a first method, which is the preferred method of the present invention, anarnorelin free base was carefully measured and dissolved in isopropyl acetate. Anarnorelin free base was prepared according to known method (e.g., U.S. Patent No, 6,576,648). A fixed volume of HCl in water containing various molar ratios (0.80, 0,95, 1.00 or 1.05) of HCl relative to the anarnorelin free base was then combined with the anamorelin/isopropyl acetate solution, to form a mixture having an organic and an aqueous phase, The aqueous phase of the mixture was separated from the organic phase and the resulting aqueous phase was concentrated by spray drying to obtain the batches of anarnorelin monohydrochloride (or a composition comprising anarnorelin monohydrochloride ) shown in Table 1 A.Approximately 150mg of the resulting spray dried sample of anarnorelin monohydrochloride (or composition comprising anarnorelin monohydrochloride) was accurately weighed out and dissolved in methanol (50mL). Acetic acid (5mL) and distilled water (5mL) were added to the mixture. The resulting mixture was potentiometricaJ ly titrated using 0,0 IN silver nitrate and the e dpoint was determined. A blank determination was also performed and correction was made, if necessary. The chloride content in the sample was calculated by the following formula. This measurement method of chloride content was performed without any cations other than proton (! ! ‘ ).Chloride content (%) = VxNx35.453x l 00x l 00/{Wx[1 00-(water content (%))-(residual solvent (%))]}V: volume at the endpoint (ml.)N; actual normality of 0.01 mol/L silver nitrate35.453 : atomic weight of ChlorineW: weight of sample (mg)TABLE 1 AHCl Chloride ContentThis data showed that anamorelin monohydrochlonde produced by a fixed volume of HCl in water containing 0.80 or 1 .05 molar equivalents of HC1 relative to anamorelin free base had levels of chloride thai were undesirable, and associated with product instability as shown in Example 3.Alternatively, a fixed volume of HCl in water containing 0.95 moles of HCl relative to anamorelin free base was used to prepare anamorelin monohydrochlonde (or composition comprising anamorelin monohydrochloride) as follows. Anamorelin free base (18.8g, 34.4mmoi) and isopropyl acetate (341.8g) were mixed in a 1000 mL flask. The mixture was heated at 40±5°C to confirm dissolution of the crystals and then cooled at 25±5°C. Distilled water (22.3g) and 3.6% diluted hydrochloric acid (33. Ig, 32.7mmoL 0.95 equivalents) were added into the flask and washed with distilled water. After 30 minutes stirring, the reaction was static for more than 15 minutes and the lower layer (aqueous layer) was transferred into a separate 250mL flask. Distilled water was added to the flask and concentrated under pressure at 50i5cC. The resulting aqueous solution was then filtered and product isolated by spray drying to afford anamorelin monohydrochlonde A (the present invention).The physical properties of anamorelin monohydrochloride A were compared to anamorelin monohydrochloride produced by a traditional comparative method (“anamorelin monohydrochloride B”) (comparative example). Anamorelin mono hydrochloride B in the comparative example was produced by bubbling HCl gas into isopropyl acetate to produce a 2M solution of HCl, and reacting 0.95 molar equivalents of the 2M HCl in isopropyl acetate with anamorelin free base. The physical properties of anamorelin monohydrochloride B are reported in Table IB. This data shows that when 0.95 equivalents of HCl is added to anamorelin free base, the chloride content (or amount of anamorelin dihydrochloride) is increased, even when a stoichiometric ratio of hydrochloride to anamorelin of less than 1 ,0 is used, possibly due to uncontrolled precipitation. In addition, this data shows that the concentration of residual solvents in anamorelin monohydrochloride B was greater than the concentration in anamorelin monohydrochloride A, TABLE I B
A similar decrease in residual solvent concentration was observed when 2-methyltetrahydrofuran was used as the dissolving solvent for anamorelin free base instead of isopropvi acetate in the process for preparing spray dried anamorelin monohydrochloride A (data not reported).The residual solvent (organic volatile impurities) concentration (specifically isopropyl acetate) of anamorelin monohydrochloride in TABLE IB was measured using gas chromatography (GC-2010, Shimadzu Corporation) according to the conditions shown in TABLE 1 C,
References
- ^ Leese PT, Trang JM, Blum RA, de Groot E (March 2015). “An open-label clinical trial of the effects of age and gender on the pharmacodynamics, pharmacokinetics and safety of the ghrelin receptor agonist anamorelin”. Clinical Pharmacology in Drug Development. 4 (2): 112–120. doi:10.1002/cpdd.175. PMC 4657463. PMID 26640742.
- ^ Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802. doi:10.2217/fon.14.14. PMID 24472001.
- ^ Jump up to:a b c Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73. doi:10.1016/j.ghir.2008.12.003. PMID 19196529.
- ^ Jump up to:a b Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology. 16 (1): 108–16. doi:10.1016/S1470-2045(14)71154-4. PMID 25524795.
- ^ Jump up to:a b Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37. doi:10.1007/s00520-012-1500-1. PMID 22699302. S2CID 22853697.
- ^ Zhang H, Garcia JM (June 2015). “Anamorelin hydrochloride for the treatment of cancer-anorexia-cachexia in NSCLC”. Expert Opinion on Pharmacotherapy. 16 (8): 1245–53. doi:10.1517/14656566.2015.1041500. PMC 4677053. PMID 25945893.
- ^ Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531. doi:10.1016/S1470-2045(15)00558-6. PMID 26906526.
- ^ “Adlumiz”. European Medicines Agency.
- ^ “Refusal of the marketing authorisation for Adlumiz (anamorelin hydrochloride): Outcome of re-examination” (PDF). European Medicines Agency. 15 September 2017.
External links
| Clinical data | |
|---|---|
| Routes of administration | Oral |
| ATC code | None |
| Pharmacokinetic data | |
| Elimination half-life | 6–7 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 249921-19-5 |
| PubChem CID | 9828911 |
| ChemSpider | 8004650 |
| UNII | DD5RBA1NKF |
| CompTox Dashboard (EPA) | DTXSID20179702 |
| Chemical and physical data | |
| Formula | C31H42N6O3 |
| Molar mass | 546.716 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC(C)(C(=O)NC(CC1=CNC2=CC=CC=C21)C(=O)N3CCCC(C3)(CC4=CC=CC=C4)C(=O)N(C)N(C)C)N | |
| hideInChIInChI=1S/C31H42N6O3/c1-30(2,32)28(39)34-26(18-23-20-33-25-15-10-9-14-24(23)25)27(38)37-17-11-16-31(21-37,29(40)36(5)35(3)4)19-22-12-7-6-8-13-22/h6-10,12-15,20,26,33H,11,16-19,21,32H2,1-5H3,(H,34,39)/t26-,31-/m1/s1Key:VQPFSIRUEPQQPP-MXBOTTGLSA-N |
///////Anamorelin hydrochloride, Anamorelin, APPROVALS 2021, JAPAN 2021, PMDA, Adlumiz, 22/1/2021, アナモレリン塩酸塩, анаморелин , أناموريلين ,阿那瑞林 , ONO 7643, RC 1291, ST 1291,
#Anamorelin hydrochloride, #Anamorelin, #APPROVALS 2021, #JAPAN 2021, #PMDA, #Adlumiz, 22/1/2021, #アナモレリン塩酸塩, #анаморелин , #أناموريلين ,阿那瑞林 , #ONO 7643, #RC 1291, #ST 1291,
COVAXIN, BBV 152


COVAXIN
CAS 2501889-19-4
- Whole-Virion Inactivated SARS-CoV-2 Vaccine
- UNII76JZE5DSN6
- BBV 152
- A whole virion inactivated COVID-19 vaccine candidate derived from SARS-CoV-2 strain NIV-2020-770
REF
medRxiv (2020), 1-21.
bioRxiv (2020), 1-32.
BBV152 (also known as Covaxin) is an inactivated virus-based COVID-19 vaccine being developed by Bharat Biotech in collaboration with the Indian Council of Medical Research.
BBV152 is a vaccine candidate created by the Indian Council of Medical Research (ICMR). The candidate, a whole virion inactivated SARS-CoV-2 vaccine, was developed from a well-known SARS-CoV-2 strain and a vero cell platform (CCL-81) with adjuncts of either aluminum hydroxide gel (Algel) or a novel TLR7/8 agonist adsorbed gel. The components of the vaccine include BBV152A, BBV152B, and BBV152C. Animal studies in mice, rats, and rabbits reported BBV152 immunogenicity at two separate antigen concentrations with both types of adjuvants. The formulation with the TLR7/8 adjuvant specifically induced significant Th1 biased antibody responses and increased SARS-CoV-2 lymphocyte responses. Thus, as of July 2020, BBV152 is in Phase 1/2 clinical trials assessing safety and immunogenicity in humans (NCT04471519).
Clinical research
Phase I and II trials
In May 2020, Indian Council of Medical Research’s (ICMR‘s) National Institute of Virology approved and provided the virus strains for developing a fully indigenous COVID-19 vaccine.[1][2] In June 2020, the company got permission to conduct Phase I and Phase II human trials of a developmental COVID-19 vaccine named Covaxin, from the Drugs Controller General of India (DCGI), Government of India.[3] A total of 12 sites were selected by the Indian Council for Medical Research for Phase I and II randomised, double-blind and placebo-controlled clinical trials of vaccine candidate.[4][5][6]
In December 2020, the company announced the report for Phase I trials and presented the results through medRxiv preprint;[7][8] the report was later published in the The Lancet.[9]
On March 8, 2021, Phase II results were published in The Lancet. The study showed that Phase II trials had a higher immune response and induced T-cell response due to the difference in dosing regime from Phase I. The doses in Phase II were given at 4 weeks interval as opposed to 2 weeks in Phase I. Neutralization response of the vaccine were found significantly higher in Phase II.[10]
Phase III trials[edit]
In November 2020, Covaxin received the approval to conduct Phase III human trials[11] after completion of Phase I and II.[12] The trial involves a randomised, double-blinded, placebo-controlled study among volunteers of age group 18 and above and started on 25 November.[13] The Phase III trials involved around 26,000 volunteers from across India.[14] The phase III trials covered a total of 22 sites consisting several states in the country, including Delhi, Karnataka and West Bengal.[15] Refusal rate for Phase III trials was much higher than that for Phase I and Phase II. As a result only 13,000 volunteers had been recruited by 22 December with the number increasing to 23,000 by 5 January. [16][17]
As on March 2021, the stated interim efficacy rate for phase III trial is 81%.[18][10]
B.1.1.7 (United Kingdom) variant
In December 2020, a new SARS‑CoV‑2 variant, B.1.1.7, was identified in the UK.[19] A study on this variant was carried and preliminary results presented in biorxiv have shown Covaxin to be effective in neutralizing this strain.[20]
Manufacturing
The vaccine candidate is produced with Bharat Biotech’s in-house vero cell manufacturing platform[21] that has the capacity to deliver about 300 million doses.[22] The company is in the process of setting up a second plant at its Genome Valley facility in Hyderabad to make Covaxin. The firm is in talks with other state governments like Odisha[23] for another site in the country to make the vaccine. Beside this, they are also exploring global tie-ups for Covaxin manufacturing.[24]
In December 2020, Ocugen Inc entered a partnership with Bharat Biotech to co-develop Covaxin for the U.S. market.[25][26] In January 2021, Precisa Med entered an agreement with Bharat Biotech to supply Covaxin in Brazil[27]
Emergency use authorisation
See also: COVID-19 vaccine § Trial and authorization status
Bharat Biotech has applied to the Drugs Controller General of India (DCGI), Government of India seeking an emergency use authorisation (EUA).[31] It was the third firm after Serum Institute of India and Pfizer to apply for emergency use approval.[32]
On 2 January 2021, the Central Drugs Standard Control Organisation (CDSCO) recommended permission for EUA,[33] which was granted on 3 January.[34] The emergency approval was given before Phase III trial data was published. This was criticized in some sections of the media.[35][36]
The vaccine was also approved for Emergency Use in Iran and Zimbabwe.[30][29]
References
- ^ “ICMR teams up with Bharat Biotech to develop Covid-19 vaccine”. Livemint. 9 May 2020.
- ^ Chakrabarti A (10 May 2020). “India to develop ‘fully indigenous’ Covid vaccine as ICMR partners with Bharat Biotech”. ThePrint.
- ^ “India’s First COVID-19 Vaccine Candidate Approved for Human Trials”. The New York Times. 29 June 2020.
- ^ “Human clinical trials of potential Covid-19 vaccine ‘COVAXIN’ started at AIIMS”. DD News. Prasar Bharati, Ministry of I & B, Government of India. 25 July 2020.
- ^ Press, Associated (25 July 2020). “Asia Today: Amid new surge, India tests potential vaccine”. Washington Post. Retrieved 17 December 2020.
- ^ “Delhi: 30-year-old is first to get dose of trial drug Covaxin”. The Indian Express. 25 July 2020.
- ^ Perappadan, Bindu Shajan (16 December 2020). “Coronavirus | Covaxin phase-1 trial results show promising results”. The Hindu. Retrieved 17 December 2020.
- ^ Sabarwal, Harshit (16 December 2020). “Covaxin’s phase 1 trial result shows robust immune response, mild adverse events”. Hindustan Times. Retrieved 17 December 2020.
- ^ Ella, Raches; Vadrevu, Krishna Mohan; Jogdand, Harsh; Prasad, Sai; Reddy, Siddharth; Sarangi, Vamshi; Ganneru, Brunda; Sapkal, Gajanan; Yadav, Pragya; Abraham, Priya; Panda, Samiran; Gupta, Nivedita; Reddy, Prabhakar; Verma, Savita; Rai, Sanjay Kumar; Singh, Chandramani; Redkar, Sagar Vivek; Gillurkar, Chandra Sekhar; Kushwaha, Jitendra Singh; Mohapatra, Satyajit; Rao, Venkat; Guleria, Randeep; Ella, Krishna; Bhargava, Balram (21 January 2021). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: a double-blind, randomised, phase 1 trial”. The Lancet Infectious Diseases. doi:10.1016/S1473-3099(20)30942-7. PMC 7825810. PMID 33485468.
- ^ Jump up to:a b Ella, Raches; Reddy, Siddhart; Jogdand, Harsh; Sarangi, Vamsi; Ganneru, Brunda; Prasad, Sai; Das, Dipankar; Dugyala, Raju; Praturi, Usha; Sakpal, Gajanan; Yadav, Pragya; Reddy, Prabhakar; Verma, Savita; Singh, Chandramani; Redkar, Sagar Vivek; Singh, Chandramani; Gillurkar, Chandra Sekhar; Kushwaha, Jitendra Singh; Mohapatra, Satyajit; Mohapatra, Satyajit; Bhate, Amit; Rai, Sanjay; Panda, Samiran; Abraham, Priya; Gupta, Nivedita; Ella, Krishna; Bhargav, Balram; Vadrevu, Krishna Mohan (8 March 2021). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: interim results from a double-blind, randomised, multicentre, phase 2 trial, and 3-month follow-up of a double-blind, randomised phase 1 trial”. The Lancet Infectious Diseases. doi:10.1016/S1473-3099(21)00070-0.
- ^ “Coronavirus | Covaxin Phase III trial from November”. The Hindu. 23 October 2020.
- ^ Ganneru B, Jogdand H, Daram VK, Molugu NR, Prasad SD, Kannappa SV, et al. (9 September 2020). “Evaluation of Safety and Immunogenicity of an Adjuvanted, TH-1 Skewed, Whole Virion InactivatedSARS-CoV-2 Vaccine – BBV152”. doi:10.1101/2020.09.09.285445. S2CID 221635203.
- ^ “An Efficacy and Safety Clinical Trial of an Investigational COVID-19 Vaccine (BBV152) in Adult Volunteers”. clinicaltrials.gov(Registry). United States National Library of Medicine. NCT04641481. Retrieved 26 November 2020.
- ^ “Bharat Biotech begins Covaxin Phase III trials”. The Indian Express. 18 November 2020.
- ^ Sen M (2 December 2020). “List of states that have started phase 3 trials of India’s first Covid vaccine”. mint.
- ^ “70%-80% Drop In Participation For Phase 3 Trials Of Covaxin: Official”. NDTV. 17 December 2020.
- ^ “Bharat Biotech’s Covaxin given conditional nod based on incomplete Phase 3 trial results data”. The Print. 3 January 2021.
- ^ Kumar, N. Ravi (3 March 2021). “Bharat Biotech says COVID-19 vaccine Covaxin shows 81% efficacy in Phase 3 clinical trials”. The Hindu.
- ^ “Inside the B.1.1.7 Coronavirus Variant”. The New York Times. 18 January 2021. Retrieved 29 January 2021.
- ^ Sapkal, Gajanan N.; Yadav, Pragya D.; Ella, Raches; Deshpande, Gururaj R.; Sahay, Rima R.; Gupta, Nivedita; Mohan, V. Krishna; Abraham, Priya; Panda, Samiran; Bhargava, Balram (27 January 2021). “Neutralization of UK-variant VUI-202012/01 with COVAXIN vaccinated human serum”. bioRxiv: 2021.01.26.426986. doi:10.1101/2021.01.26.426986. S2CID 231777157.
- ^ Hoeksema F, Karpilow J, Luitjens A, Lagerwerf F, Havenga M, Groothuizen M, et al. (April 2018). “Enhancing viral vaccine production using engineered knockout vero cell lines – A second look”. Vaccine. 36 (16): 2093–2103. doi:10.1016/j.vaccine.2018.03.010. PMC 5890396. PMID 29555218.
- ^ “Coronavirus vaccine update: Bharat Biotech’s Covaxin launch likely in Q2 of 2021, no word on pricing yet”. http://www.businesstoday.in. India Today Group. Retrieved 13 December2020.
- ^ “Odisha fast tracks coronavirus vaccine manufacturing unit”. The New Indian Express. 7 November 2020.
- ^ Raghavan P (24 September 2020). “Bharat Biotech exploring global tie-ups for Covaxin manufacturing”. The Indian Express.
- ^ Reuters Staff (22 December 2020). “Ocugen to co-develop Bharat Biotech’s COVID-19 vaccine candidate for U.S.” Reuters. Retrieved 5 January 2021.
- ^ “Bharat Biotech, Ocugen to co-develop Covaxin for US market”. The Economic Times. Retrieved 5 January 2021.
- ^ “Bharat Biotech inks pact with Precisa Med to supply Covaxin to Brazil”. mint. 12 January 2021.
- ^ Schmall E, Yasir S (3 January 2021). “India Approves Oxford-AstraZeneca Covid-19 Vaccine and 1 Other”. The New York Times. Retrieved 3 January 2021.
- ^ Jump up to:a b “Iran issues permit for emergency use for three other COVID-19 vaccines: Official”. IRNA English. 17 February 2021.
- ^ Jump up to:a b Manral, Karan (4 March 2021). “Zimbabwe approves Covaxin, first in Africa to okay India-made Covid-19 vaccine”. Hindustan Times. Retrieved 6 March 2021.
- ^ Ghosh N (7 December 2020). “Bharat Biotech seeks emergency use authorization for Covid-19 vaccine”. Hindustan Times.
- ^ “Coronavirus | After SII, Bharat Biotech seeks DCGI approval for Covaxin”. The Hindu. 7 December 2020.
- ^ “Expert panel recommends granting approval for restricted emergency use of Bharat Biotech’s Covaxin”. The Indian Express. 2 January 2021.
- ^ “Coronavirus: India approves vaccines from Bharat Biotech and Oxford/AstraZeneca”. BBC News. 3 January 2021. Retrieved 3 January 2021.
- ^ “Disputes Mount, but Heedless Govt Intent on Rolling Vaccine Candidates Out”. The Wire. 12 January 2021.
- ^ “AIPSN urges govt to reconsider emergency approval for Covaxin till Phase 3 data is published – Health News , Firstpost”. Firstpost. 8 January 2021.
External links
| Scholia has a profile for Covaxin / BBV152 (Q98703813). |
COVAXIN®, India‘s indigenous COVID-19 vaccine by Bharat Biotech is developed in collaboration with the Indian Council of Medical Research (ICMR) – National Institute of Virology (NIV).
The indigenous, inactivated vaccine is developed and manufactured in Bharat Biotech’s BSL-3 (Bio-Safety Level 3) high containment facility.
The vaccine is developed using Whole-Virion Inactivated Vero Cell derived platform technology. Inactivated vaccines do not replicate and are therefore unlikely to revert and cause pathological effects. They contain dead virus, incapable of infecting people but still able to instruct the immune system to mount a defensive reaction against an infection.
Why develop Inactivated Vaccine? Conventionally, inactivated vaccines have been around for decades. Numerous vaccines for diseases such as Seasonal Influenza, Polio, Pertussis, Rabies, and Japanese Encephalitis use the same technology to develop inactivated vaccines with a safe track record of >300 million doses of supplies to date. It is the well-established, and time-tested platform in the world of vaccine technology.
Key Attributes:
- COVAXIN® is included along with immune-potentiators, also known as vaccine adjuvants, which are added to the vaccine to increase and boost its immunogenicity.
- It is a 2-dose vaccination regimen given 28 days apart.
- It is a vaccine with no sub-zero storage, no reconstitution requirement, and ready to use liquid presentation in multi-dose vials, stable at 2-8oC.
- Pre-clinical studies: Demonstrated strong immunogenicity and protective efficacy in animal challenge studies conducted in hamsters & non-human primates. For more information about our animal study, please visit our blog page on Non-Human Primates.
- The vaccine received DCGI approval for Phase I & II Human Clinical Trials in July, 2020.
- A total of 375 subjects have been enrolled in the Phase 1 study and generated excellent safety data without any reactogenicity. Vaccine-induced neutralizing antibody titers were observed with two divergent SARS-CoV-2 strains. Percentage of all the side-effects combined was only 15% in vaccine recipients. For further information, visit our blog page on phase 1 study.
- In Phase 2 study, 380 participants of 12-65 years were enrolled. COVAXIN® led to tolerable safety outcomes and enhanced humoral and cell-mediated immune responses. Know more about our phase 2 study.

- A total of 25,800 subjects have been enrolled and randomized in a 1:1 ratio to receive the vaccine and control in a Event-Driven, randomized, double-blind, placebo-controlled, multicentre phase 3 study.
The purpose of this study is to evaluate the efficacy, safety, and immunogenicity of COVAXIN® in volunteers aged ≥18 years.
Of the 25,800 participants, >2400 volunteers were above 60 years of age and >4500 with comorbid conditions.
COVAXIN® demonstrated 81% interim efficacy in preventing COVID-19 in those without prior infection after the second dose.
COVAXIN® effective against UK variant strain:
Analysis from the National Institute of Virology indicates that vaccine-induced antibodies can neutralize the UK variant strains and other heterologous strains.
Global Acceptance of COVAXIN®:
Bharat biotech has been approached by several countries across the world for the procurement of COVAXIN®.
- Clinical trials in other countries to commence soon.
- Supplies from government to government in the following countries to take place: Mongolia, Myanmar, Sri Lanka, Philippines, Bahrain, Oman, Maldives and Mauritius.
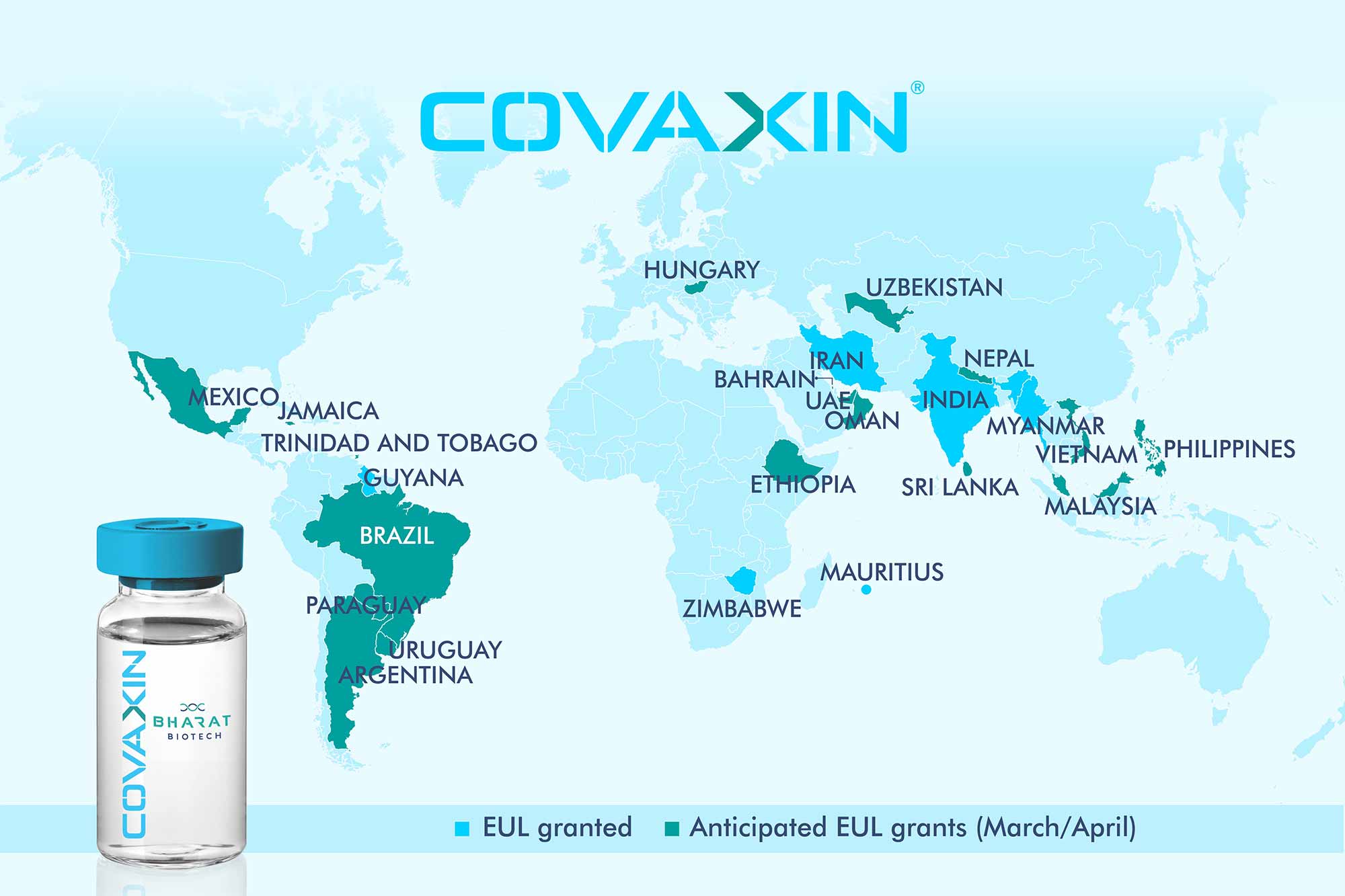
| A person holding a vial of the Covaxin vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Trade names | Covaxin |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | EUA : IND, IRN, ZBW |
| Identifiers | |
| DrugBank | DB15847 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////COVAXIN, BBV152, BBV 152, INDIA 2021, APPROVALS 2021, COVID 19, CORONA VIRUS, bharat biotech
#COVAXIN, #BBV152, #BBV 152, #INDIA 2021, #APPROVALS 2021, #COVID 19, #CORONA VIRUS, #bharat biotech
Lenalidomide hydrate,


Lenalidomide hydrate
レナリドミド水和物
An immunomodulator.
CC-5013 hemihydrate
2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-, hydrate (2:1)
(+/-)-2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-, hydrate (2:1)
| Formula | (C13H13N3O3)2. H2O |
|---|---|
| CAS | 847871-99-2 |
| Mol weight | 536.5365 |
EMA APPROVED 2021/2/11, Lenalidomide KRKA
Research Code:CDC-501; CC-5013
Trade Name:Revlimid®
MOA:Angiogenesis inhibitor
Indication:Myelodysplastic syndrome (MDS); Mantle cell lymphoma (MCL); Multiple myeloma (MM)
Status:Approved
Company:Celgene (Originator)
Sales:$5,801.1 Million (Y2015); 
$4,980 Million (Y2014);;
$4280 Million (Y2013);;
$3766.6 Million (Y2012);;
$3208.2 Million (Y2011);ATC Code:L04AX04
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2005-12-27 | Marketing approval | Revlimid | Multiple myeloma (MM),Myelodysplastic syndrome (MDS),Mantle cell lymphoma (MCL) | Capsule | 2.5 mg/5 mg/10 mg/15 mg/20 mg/25 mg | Celgene | Priority; Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2007-06-14 | Marketing approval | Revlimid | Multiple myeloma (MM),Myelodysplastic syndrome (MDS) | Capsule | 2.5 mg/5 mg/7.5 mg/10 mg/15 mg/20 mg/25 mg | Celgene | Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-08-20 | New indication | Revlimid | Myelodysplastic syndrome (MDS) | Capsule | 5 mg | Celgene | |
| 2010-06-25 | Marketing approval | Revlimid | Multiple myeloma (MM) | Capsule | 5 mg | Celgene |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 5 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 10 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 15 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 25 mg | Celgene |
| Molecular Weight | 259.26 |
| Formula | C13H13N3O3 |
| CAS No. | 191732-72-6 (Lenalidomide); |
| Chemical Name | 3(4-amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione |
Lenalidomide was first approved by the U.S. Food and Drug Administration (FDA) on Dec 27, 2005, then approved by European Medicine Agency (EMA) on June 14, 2007, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on June 25, 2010. It was developed and marketed as Revlimid® by Celgene.
Lenalidomide is an analogue of thalidomide with immunomodulatory, antiangiogenic, and antineoplastic properties. In multiple myeloma cells, the combination of lenalidomide and dexamethasone synergizes the inhibition of cell proliferation and the induction of apoptosis. Revlimid® is indicated for the treatment of multiple myeloma (MM), in combination with dexamethasone, in patients who have received at least one prior therapy, transfusion-dependent anemia due to low-or intermediate-1-risk myelodysplastic syndromes (MDS) associated with a deletion 5q abnormality with or without additional cytogenetic abnormalities and mantle cell lymphoma (MCL) whose disease has relapsed or progressed after two prior therapies, one of which included bortezomib.
Revlimid® is available as capsule for oral use, containing 2.5, 5, 10, 15, 20 or 25 mg of free Lenalidomide. The recommended dose is 25 mg once daily for multiple myeloma (MM), in combination with 40 mg dexamethasone once daily, 10 mg once daily for myelodysplastic syndromes (MDS) and 25 mg once daily for mantle cell lymphoma (MCL).
Lenalidomide, sold under the trade name Revlimid among others, is a medication used to treat multiple myeloma (MM) and myelodysplastic syndromes (MDS).[2] For MM it is used after at least one other treatment and generally together with dexamethasone.[2] It is taken by mouth.[2]
Common side effects include diarrhea, itchiness, joint pain, fever, headache, and trouble sleeping.[2] Severe side effects may include low blood platelets, low white blood cells, and blood clots.[2] Use during pregnancy may harm the baby.[2] The dose may need to be adjusted in people with kidney problems.[2] It has a chemical structure similar to thalidomide but has a different mechanism of action.[3][2] How it works is not entirely clear as of 2019.[2]
Lenalidomide was approved for medical use in the United States in 2005.[2] It is on the World Health Organization’s List of Essential Medicines.[4]
Medical uses
Multiple myeloma
Lenalidomide is used to treat multiple myeloma.[5] It is a more potent molecular analog of thalidomide, which inhibits tumor angiogenesis, tumor-secreted cytokines, and tumor proliferation through induction of apoptosis.[6][7][8]
Lenalidomide is effective at inducing a complete or “very good partial” response and improves progression-free survival. Adverse events more common in people receiving lenalidomide for myeloma include neutropenia, deep vein thrombosis, infections, and an increased risk of other hematological malignancies.[9] The risk of second primary hematological malignancies does not outweigh the benefit of using lenalidomide in relapsed or refractory multiple myeloma.[10] It may be more difficult to mobilize stem cells for autograft in people who have received lenalidomide.[6]
In 2006, lenalidomide received U.S. Food and Drug Administration (FDA) clearance for use in combination with dexamethasone in people with multiple myeloma who have received at least one prior therapy.[11] In 2017, the FDA approved lenalidomide as standalone maintenance therapy (without dexamethasone) for people with multiple myeloma following autologous stem cell transplant.[12]
In 2009, The National Institute for Health and Clinical Excellence issued a final appraisal determination approving lenalidomide in combination with dexamethasone as an option to treat people with multiple myeloma who have received two or more prior therapies in England and Wales.[13]
The use of lenalidomide combined with other drugs was evaluated. It was seen that the drug combinations of lenalidomide plus dexamethasone and continuous bortezomib plus lenalidomide plus dexamethasone probably result in an increase of the overall survival.[14]
Myelodysplastic syndromes
Lenalidomide was approved by the FDA on 27 December 2005 for patients with low- or intermediate-1-risk myelodysplastic syndromes who have chromosome 5q deletion syndrome (5q- syndrome) with or without additional cytogenetic abnormalities.[15][16][17] It was approved on 17 June 2013 by the European Medicines Agency for use in patients with low- or intermediate-1-risk myelodysplastic syndromes who have 5q- deletion syndrome but no other cytogenetic abnormalities and are dependent on red blood cell transfusions, for whom other treatment options have been found to be insufficient or inadequate.[18]
Mantle cell lymphoma
Lenalidomide is approved by FDA as a specialty drug requiring a specialty pharmacy distribution for mantle cell lymphoma in patients whose disease has relapsed or progressed after at least two prior therapies, one of which must have included the medicine bortezomib.[3]
Amyloidosis
Although not specifically approved by the FDA for use in treating amyloidosis, Lenalidomide is widely used in the treatment of that condition, often in combination with dexamethasone. [19]
Adverse effects
In addition to embryo-fetal toxicity, lenalidomide carries black box warnings for hematologic toxicity (including neutropenia and thrombocytopenia) and thromboembolism.[3] Serious potential side effects include thrombosis, pulmonary embolus, hepatotoxicity, and bone marrow toxicity resulting in neutropenia and thrombocytopenia. Myelosuppression is the major dose-limiting toxicity, which is not the case with thalidomide.[20]
Lenalidomide may be associated with such adverse effects as second primary malignancy, severe cutaneous reactions, hypersensitivity reactions, tumor lysis syndrome, tumor flare reaction, hypothyroidism, and hyperthyroidism.[3]
Teratogenicity
Lenalidomide is related to thalidomide, which is known to be teratogenic. Tests in monkeys suggest that lenalidomide is likewise teratogenic.[21] It cannot be prescribed for women who are pregnant or who may become pregnant during therapy.[1] For this reason, the drug is only available in the United States through a restricted distribution system in conjunction with a risk evaluation and mitigation strategy. Females who may become pregnant must use at least two forms of reliable contraception during treatment and for at least four weeks after discontinuing treatment with lenalidomide.[3][22]
Venous thromboembolism
Lenalidomide, like its parent compound thalidomide, may cause venous thromboembolism (VTE), a potentially serious complication with their use. High rates of VTE have been found in patients with multiple myeloma who received thalidomide or lenalidomide in conjunction with dexamethasone, melphalan, or doxorubicin.[23]
Stevens-Johnson syndrome
In March 2008, the U.S. Food and Drug Administration (FDA) included lenalidomide on a list of twenty prescription drugs under investigation for potential safety problems. The drug was investigated for possibly increasing the risk of developing Stevens–Johnson syndrome, a life-threatening skin condition.[24]
FDA ongoing safety review
In 2011, the FDA initiated an ongoing review of clinical trials that found an increased risk of developing cancers such as acute myelogenous leukemia and B-cell lymphoma,[25] though it did not advise patients to discontinue treatment with lenalidomide.[26]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past ten years.[when?] There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[27] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of angiogenesis, and immunomodulation. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
On a molecular level, lenalidomide has been shown to interact with the ubiquitin E3 ligase cereblon[28] and target this enzyme to degrade the Ikaros transcription factors IKZF1 and IKZF3.[29] This mechanism was unexpected as it suggests that the major action of lenalidomide is to re-target the activity of an enzyme rather than block the activity of an enzyme or signaling process, and thereby represents a novel mode of drug action. A more specific implication of this mechanism is that the teratogenic and anti-neoplastic properties of lenalidomide, and perhaps other thalidomide derivatives, could be disassociated.
History
See also: Development of analogs of thalidomide
Lenalidomide was approved for medical use in the United States in 2005.[2]
Society and culture
Economics
Lenalidomide costs US$163,381 per year for the average person in the United States as of 2012.[25] Lenalidomide made almost $9.7bn for Celgene in 2018.[30]
In 2013, the UK National Institute for Health and Care Excellence (NICE) rejected lenalidomide for “use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)” in England and Scotland, arguing that Celgene “did not provide enough evidence to justify the GB£3,780 per month (US$5,746.73) price-tag of lenalidomide for use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)”.[31]
Research
Lenalidomide is undergoing clinical trial as a treatment for Hodgkin’s lymphoma,[32] as well as non-Hodgkin’s lymphoma, chronic lymphocytic leukemia and solid tumor cancers, such as carcinoma of the pancreas.[33] One Phase III clinical trial being conducted by Celgene in elderly patients with B-cell chronic lymphocytic leukemia was halted in July 2013, when a disproportionate number of cancer deaths were observed during treatment with lenalidomide versus patients treated with chlorambucil.[34]
1. WO9803502A1 / US2002173658A1.
2. Bioorg. Med. Chem. Lett. 1999, 9, 1625-1630.Route 2
Reference:
1. WO2010139266A1 / US2012077982A1.Route 3
Reference:
1. CN103497175A.Route 4
Reference:
1. WO2010139266A1 / US2012077982A1.Route 5
Reference:
1. CN103554082A.
Clip
SYN

SCALABLE AND GREEN PROCESS FOR THE SYNTHESIS OF ANTICANCER DRUG LENALIDOMIDE
Yuri Ponomaryov, Valeria Krasikova, Anton Lebedev, Dmitri Chernyak, Larisa Varacheva, Alexandr Chernobroviy

Abstract
A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
How to Cite
Ponomaryov, Y.; Krasikova, V.; Lebedev, A.; Chernyak, D.; Varacheva, L.; Chernobroviy, A. Chem. Heterocycl. Compd. 2015, 51, 133. [Khim. Geterotsikl. Soedin. 2015, 51, 133.]
For this article in the English edition see DOI 10.1007/s10593-015-1670-0
SYN
https://link.springer.com/article/10.1007/s10593-015-1670-0

A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
SYN

SYN
EP 0925294; US 5635517; WO 9803502
Cyclization of N-(benzyloxycarbonyl)glutamine (I) by means of CDI in refluxing THF gives 3-(benzyloxycarbonylamino)piperidine-2,6-dione (II), which is deprotected with H2 over Pd/C in ethyl acetate/4N HCl to yield 3-aminopiperidine-2,6-dione hydrochloride (III). Bromination of 2-methyl-3-nitrobenzoic acid methyl ester (IV) with NBS in CCl4 provides 2-(bromomethyl)-3-nitrobenzoic acid methyl ester (V), which is cyclized with the aminopiperidine (III) by means of triethylamine in hot DMF to afford 3-(4-nitro-1-oxoisoindolin-2-yl)piperidine-2,6-dione (VI). Finally, the nitro group of compound (VI) is reduced with H2 over Pd/C in methanol (1, 2).
SYN
Bioorg Med Chem Lett 1999,9(11),1625
Treatment of 3-nitrophthalimide (I) with ethyl chloroformate and triethylamine produced 3-nitro-N-(ethoxycarbonyl)phthalimide (II), which was condensed with L-glutamine tert-butyl ester hydrochloride (III) to afford the phthaloyl glutamine derivative (IV). Acidic cleavage of the tert-butyl ester of (IV) provided the corresponding carboxylic acid (V). This was cyclized to the required glutarimide (VI) upon treatment with thionyl chloride and then with triethylamine. The nitro group of (VI) was finally reduced to amine by hydrogenation over Pd/C.
Lenalidomide
- Synonyms:CC-5013, CDC 501
- ATC:L04AX04
- MW:259.27 g/mol
- CAS-RN:191732-72-6
- InChI Key:GOTYRUGSSMKFNF-JTQLQIEISA-N
- InChI:InChI=1S/C13H13N3O3/c14-9-3-1-2-7-8(9)6-16(13(7)19)10-4-5-11(17)15-12(10)18/h1-3,10H,4-6,14H2,(H,15,17,18)/t10-/m0/s1
Synthesis
References
- ^ Jump up to:a b c “Lenalidomide (Revlimid) Use During Pregnancy”. Drugs.com. 13 March 2020. Retrieved 13 August 2020.
- ^ Jump up to:a b c d e f g h i j k “Lenalidomide Monograph for Professionals”. Drugs.com. Retrieved 27 October 2019.
- ^ Jump up to:a b c d e “DailyMed – Revlimid- lenalidomide capsule”. dailymed.nlm.nih.gov. Retrieved 27 October 2019.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Armoiry X, Aulagner G, Facon T (June 2008). “Lenalidomide in the treatment of multiple myeloma: a review”. Journal of Clinical Pharmacy and Therapeutics. 33 (3): 219–26. doi:10.1111/j.1365-2710.2008.00920.x. PMID 18452408. S2CID 1228171.
- ^ Jump up to:a b Li S, Gill N, Lentzsch S (November 2010). “Recent advances of IMiDs in cancer therapy”. Current Opinion in Oncology. 22 (6): 579–85. doi:10.1097/CCO.0b013e32833d752c. PMID 20689431. S2CID 205547603.
- ^ Tageja N (March 2011). “Lenalidomide – current understanding of mechanistic properties”. Anti-Cancer Agents in Medicinal Chemistry. 11 (3): 315–26. doi:10.2174/187152011795347487. PMID 21426296.
- ^ Kotla V, Goel S, Nischal S, Heuck C, Vivek K, Das B, Verma A (August 2009). “Mechanism of action of lenalidomide in hematological malignancies”. Journal of Hematology & Oncology. 2: 36. doi:10.1186/1756-8722-2-36. PMC 2736171. PMID 19674465.
- ^ Yang B, Yu RL, Chi XH, Lu XC (2013). “Lenalidomide treatment for multiple myeloma: systematic review and meta-analysis of randomized controlled trials”. PLOS ONE. 8 (5): e64354. Bibcode:2013PLoSO…864354Y. doi:10.1371/journal.pone.0064354. PMC 3653900. PMID 23691202.
- ^ Dimopoulos MA, Richardson PG, Brandenburg N, Yu Z, Weber DM, Niesvizky R, Morgan GJ (March 2012). “A review of second primary malignancy in patients with relapsed or refractory multiple myeloma treated with lenalidomide”. Blood. 119 (12): 2764–7. doi:10.1182/blood-2011-08-373514. PMID 22323483.
- ^ “FDA approves lenalidomide oral capsules (Revlimid) for use in combination with dexamethasone in patients with multiple myeloma”. Food and Drug Administration (FDA). 29 June 2006. Retrieved 15 October 2015.[dead link]
- ^ “Lenalidomide (Revlimid)”. Food and Drug Administration(FDA). 22 February 2017.
- ^ “REVLIMID Receives Positive Final Appraisal Determination from National Institute for Health and Clinical Excellence (NICE) for Use in the National Health Service (NHS) in England and Wales”. Reuters. 23 April 2009.
- ^ Piechotta V, Jakob T, Langer P, Monsef I, Scheid C, Estcourt LJ, et al. (Cochrane Haematology Group) (November 2019). “Multiple drug combinations of bortezomib, lenalidomide, and thalidomide for first-line treatment in adults with transplant-ineligible multiple myeloma: a network meta-analysis”. The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD013487. PMC 6876545. PMID 31765002.
- ^ List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, et al. (February 2005). “Efficacy of lenalidomide in myelodysplastic syndromes”. The New England Journal of Medicine. 352 (6): 549–57. doi:10.1056/NEJMoa041668. PMID 15703420.
- ^ List AF (August 2005). “Emerging data on IMiDs in the treatment of myelodysplastic syndromes (MDS)”. Seminars in Oncology. 32 (4 Suppl 5): S31-5. doi:10.1053/j.seminoncol.2005.06.020. PMID 16085015.
- ^ List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. (October 2006). “Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion”. The New England Journal of Medicine. 355 (14): 1456–65. doi:10.1056/NEJMoa061292. PMID 17021321.
- ^ “Revlimid Approved In Europe For Use In Myelodysplastic Syndromes”. The MDS Beacon. Retrieved 17 June 2013.
- ^ “Revlimid and Amyloidosis AL” (PDF). MyelomaUK. Retrieved 3 October 2020.
- ^ Rao KV (September 2007). “Lenalidomide in the treatment of multiple myeloma”. American Journal of Health-System Pharmacy. 64 (17): 1799–807. doi:10.2146/ajhp070029. PMID 17724360.
- ^ “Revlimid Summary of Product Characteristics. Annex I” (PDF). European Medicines Agency. 2012. p. 6.
- ^ Ness, Stacey (13 March 2014). “New Specialty Drugs”. Pharmacy Times. Retrieved 5 November 2015.
- ^ Bennett CL, Angelotta C, Yarnold PR, Evens AM, Zonder JA, Raisch DW, Richardson P (December 2006). “Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer”. JAMA. 296 (21): 2558–60. doi:10.1001/jama.296.21.2558-c. PMID 17148721.
- ^ “Potential Signals of Serious Risks/New Safety Information Identified from the Adverse Event Reporting System (AERS) between January – March 2008”. Food and Drug Administration(FDA). March 2008. Archived from the original on 19 April 2014. Retrieved 16 December 2019.
- ^ Jump up to:a b Badros AZ (May 2012). “Lenalidomide in myeloma–a high-maintenance friend”. The New England Journal of Medicine. 366(19): 1836–8. doi:10.1056/NEJMe1202819. PMID 22571206.
- ^ “FDA Drug Safety Communication: Ongoing safety review of Revlimid (lenalidomide) and possible increased risk of developing new malignancies”. Food and Drug Administration (FDA). April 2011.
- ^ Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma. 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080. S2CID 43350339.
- ^ Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. (November 2011). “Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide”. Blood. 118 (18): 4771–9. doi:10.1182/blood-2011-05-356063. PMC 3208291. PMID 21860026.
- ^ Stewart AK (January 2014). “Medicine. How thalidomide works against cancer”. Science. 343 (6168): 256–7. doi:10.1126/science.1249543. PMC 4084783. PMID 24436409.
- ^ “Top 10 Best-Selling Cancer Drugs of 2018”. Genetic Engineering and Biotechnology News. 22 April 2019. Retrieved 25 April 2019.
- ^ “Revlimid faces NICE rejection for use in rare blood cancer Watchdog’s draft guidance does not recommend Celgene’s drug for NHS use in England and Wales”. Pharma News. 11 July 2013. Retrieved 5 November 2015.
- ^ “Phase II Study of Lenalidomide for the Treatment of Relapsed or Refractory Hodgkin’s Lymphoma”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “276 current clinical trials world-wide, both recruiting and fully enrolled, as of 27 February 2009”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “Celgene Discontinues Phase 3 Revlimid Study after ‘Imbalance’ of Deaths”. Nasdaq. 18 July 2013.
External links[edit]
- “Lenalidomide”. Drug Information Portal. U.S. National Library of Medicine.
//////////Lenalidomide hydrate, Lenalidomide KRKA, EU 2021, APPROVALS 2021, レナリドミド水和物 , CC-5013 hemihydrate,
#Lenalidomide hydrate, #Lenalidomide KRKA, #EU 2021, #APPROVALS 2021, #レナリドミド水和物 , #CC-5013 hemihydrate,
O.Nc1cccc2C(=O)N(Cc12)C3CCC(=O)NC3=O.Nc4cccc5C(=O)N(Cc45)C6CCC(=O)NC6=O
Lisocabtagene maraleucel
 Lisocabtagene maraleucel (liso-cel; JCAR017; Anti-CD19 CAR T-Cells) is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, [1][2] which is a surface glycoprotein expressed during normal B-cell development and maintained following malignant transformation of B cells. [3][4][5] Liso-cel CAR T-cells aim to target and CD-19 expressing cells through a CAR construct that includes an anti-CD19 single-chain variable fragment (scFv) targeting domain for antigen specificity, a transmembrane domain, a 4-1BB costimulatory domain hypothesized to increase T-cell proliferation and persistence, and a CD3-zeta T-cell activation domain. [1][2][6][7][8][9] The defined composition of liso-cel may limit product variability; however, the clinical significance of defined composition is unknown. [1][10] Image Courtesy: 2019/2020 Celgene/Juno Therapeutics / Bristol Meyers Squibb.
Lisocabtagene maraleucel (liso-cel; JCAR017; Anti-CD19 CAR T-Cells) is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, [1][2] which is a surface glycoprotein expressed during normal B-cell development and maintained following malignant transformation of B cells. [3][4][5] Liso-cel CAR T-cells aim to target and CD-19 expressing cells through a CAR construct that includes an anti-CD19 single-chain variable fragment (scFv) targeting domain for antigen specificity, a transmembrane domain, a 4-1BB costimulatory domain hypothesized to increase T-cell proliferation and persistence, and a CD3-zeta T-cell activation domain. [1][2][6][7][8][9] The defined composition of liso-cel may limit product variability; however, the clinical significance of defined composition is unknown. [1][10] Image Courtesy: 2019/2020 Celgene/Juno Therapeutics / Bristol Meyers Squibb.
Lisocabtagene maraleucel
リソカブタゲンマラルユーセル;
JCAR 017
STN# BLA 125714
- Adoptive immunotherapy agent JCAR 017
- Autologous anti-CD19 scFv/4-1BB/CD3ζ/CD28 chimeric antigen receptor-expressing CD4+/CD8+ central memory T cell JCAR 017
- CAR T-cell JCAR 017
FDA 2021, 2021/2/24, BREYANZI
Juno Therapeutics
Antineoplastic, Anti-CD19 CAR-T cell
An immunotherapeutic autologous T cell preparation expressing a chimeric antigen receptor (CAR) specific to the CD19 antigen (Juno Therapeutics, Inc., Seattle, Washington, USA – FDA Clinical Trial Data)
- For the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B.
Lisocabtagene maraleucel, sold under the brand name Breyanzi, is a cell-based gene therapy used to treat large B-cell lymphoma.[1][3]
Side effects of lisocabtagene maraleucel include hypersensitivity reactions, serious infections, low blood cell counts and a weakened immune system.[3]
Lisocabtagene maraleucel, a chimeric antigen receptor (CAR) T cell therapy, is the third gene therapy approved by the U.S. Food and Drug Administration (FDA) for certain types of non-Hodgkin lymphoma, including diffuse large B-cell lymphoma (DLBCL).[3] Lisocabtagene maraleucel was approved for medical use in the United States in February 2021.[1][3]

Medical uses
Lisocabtagene maraleucel is indicated for the treatment of adults with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B.[1][3]
Lisocabtagene maraleucel is not indicated for the treatment of people with primary central nervous system lymphoma.[3]
Adverse effects
The labeling carries a boxed warning for cytokine release syndrome (CRS), which is a systemic response to the activation and proliferation of CAR T cells, causing high fever and flu-like symptoms and neurologic toxicities.[3]
History
The safety and efficacy of lisocabtagene maraleucel were established in a multicenter clinical trial of more than 250 adults with refractory or relapsed large B-cell lymphoma.[3] The complete remission rate after treatment with lisocabtagene maraleucel was 54%.[3]
The FDA granted lisocabtagene maraleucel orphan drug, regenerative medicine advanced therapy (RMAT) and breakthrough therapy designations.[3] Lisocabtagene maraleucel is the first regenerative medicine therapy with RMAT designation to be licensed by the FDA.[3] The FDA granted approval of Breyanzi to Juno Therapeutics Inc., a Bristol-Myers Squibb Company.[3]
SYN
WO 2018156680
WO 2018183366
Saishin Igaku (2018), 73(11), 1504-1512.
WO 2019148089
WO 2019220369
Leukemia & Lymphoma (2020), 61(11), 2561-2567.
WO 2020097350
WO 2020086943
Journal of Immunotherapy (2020), 43(4), 107-120.
CLIP
On February 5, 2021, the Food and Drug Administration approved lisocabtagene maraleucel (Breyanzi, Juno Therapeutics, Inc.) for the treatment of adult patients with relapsed or refractory (R/R) large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B.
Lisocabtagene maraleucel is a CD19-directed chimeric antigen receptor (CAR) T cell immunotherapy. It consists of autologous T cells that are genetically modified to produce a CAR protein, allowing the T cells to identify and eliminate CD19-expressing normal and malignant cells.
Efficacy was evaluated in TRANSCEND (NCT02631044), a single-arm, open label, multicenter trial that evaluated lisocabtagene maraleucel, preceded by lymphodepleting chemotherapy, in adults with R/R large B-cell lymphoma after at least two lines of therapy.
Of the 192 patients evaluable for response, the overall response rate (ORR) per independent review committee assessment was 73% (95% CI: 67, 80) with a complete response (CR) rate of 54% (95% CI: 47, 61). The median time to first response was one month. Of the 104 patients who achieved CR, 65% had remission lasting at least 6 months and 62% had remission lasting at least 9 months. The estimated median duration of response (DOR) was not reached (95% CI: 16.7 months, NR) in patients who achieved a CR. The estimated median DOR among patients with partial response was 1.4 months (95% CI: 1.1, 2.2).
Cytokine release syndrome (CRS) occurred in 46% of patients (Grade 3 or higher, 4%) and neurologic toxicity occurred in 35% (Grade 3 or higher, 12%). Three patients had fatal neurologic toxicity. Other Grade 3 or higher adverse reactions included infections (19%) and prolonged cytopenias (31%). FDA approved lisocabtagene maraleucel with a Risk Evaluation and Mitigation Strategy because of the risk of fatal or life-threatening CRS and neurologic toxicities.
The recommended regimen is a single dose containing 50 to 110 x 106 CAR-positive viable T cells with a 1:1 ratio of CD4 and CD8 components, administered by IV infusion and preceded by fludarabine and cyclophosphamide for lymphodepletion. Lisocabtagene maraleucel is not indicated for the treatment of patients with primary central nervous system lymphoma.
References
- ^ Jump up to:a b c d “Lisocabtagene maraleucel”. U.S. Food and Drug Administration (FDA). 5 February 2021. Retrieved 5 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ https://www.fda.gov/media/145711/download
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves New Treatment For Adults With Relapsed Or Refractory Large-B-Cell Lymphoma”. U.S. Food and Drug Administration (FDA) (Press release). 5 February 2021. Retrieved 5 February 2021. This article incorporates text from this source, which is in the public domain.
External links
- “Lisocabtagene maraleucel”. NCI Drug Dictionary. National Cancer Institute.
- Clinical trial number NCT02631044 for “Study Evaluating the Safety and Pharmacokinetics of JCAR017 in B-cell Non-Hodgkin Lymphoma (TRANSCEND-NHL-001)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Breyanzi |
| Other names | JCAR017 |
| License data | US DailyMed: Lisocabtagene_maraleucel |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| UNII | 7K2YOJ14X0 |
| KEGG | D11990 |
| ChEMBL | ChEMBL4297236 |
///////////Lisocabtagene maraleucel, BREYANZI, FDA 2021, APPROVALS 2021, リソカブタゲンマラルユーセル , Juno Therapeutics, JCAR 017, STN# BLA 125714
#Lisocabtagene maraleucel, #BREYANZI, #FDA 2021, #APPROVALS 2021, #リソカブタゲンマラルユーセル , #Juno Therapeutics, #JCAR 017, #STN# BLA 125714
Casimersen
Casimersen
カシメルセン;
RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(C-A-A-m5U-G-C-C-A-m5U-C-C-m5U-G-G-A-G-m5U-m5U-C-C-m5U-G), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N-dimethylphosphonamidate]
| Formula | C268H424N124O95P22 |
|---|---|
| CAS | 1422958-19-7 |
| Mol weight | 7584.4307 |
FDA 2021/2/25 , Amondys 45, Antisense oligonucleotide
Treatment of Duchenne muscular dystrophy
Nucleic Acid Sequence
Sequence Length: 224 a 7 c 5 g 6 umodified
- Exon-45: NG-12-0064
- SRP-4045
- WHO 10354
Casimersen, sold under the brand name Amondys 45, is an antisense oligonucleotide medication used for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the dystrophin gene that is amenable to exon 45 skipping.[1][2][3][4] It is an antisense oligonucleotide of phosphorodiamidate morpholino oligomer (PMO).[1]
The most common side effects include upper respiratory tract infections, cough, fever, headache, joint pain and throat pain.[2]
Casimersen was approved for medical use in the United States in February 2021,[1][2] and it is the first FDA-approved targeted treatment for people who have a confirmed mutation of the DMD gene that is amenable to skipping exon 45.[2]
Duchenne muscular dystrophy (DMD) is an X-linked recessive allelic disorder characterized by a lack of functional dystrophin protein, which leads to progressive impairment of ambulatory, pulmonary, and cardiac function and is invariably fatal. A related, albeit a less severe, form of muscular dystrophy known as Becker muscular dystrophy (BMD) is characterized by shortened and partially functional dystrophin protein production. Although corticosteroids effectively slow disease progression in both DMD and BMD patients, they do not address the underlying molecular pathogenesis.1,2,3
The application of antisense oligonucleotides in DMD patients with specific mutations allows for exon skipping to produce truncated BMD-like dystrophin proteins, which restore partial muscle function and slow disease progression.1,2,4,5,7 Casimersen is a phosphorodiamidate morpholino oligonucleotide (PMO); PMOs are oligonucleotides in which the five-membered ribofuranosyl ring is replaced with a six-membered morpholino ring, and the phosphodiester links between nucleotides are replaced with a phosphorodiamidate linkage.6,7 In this manner, PMOs are much less susceptible to endo- and exonucleases and exhibit drastically reduced metabolic degradation compared to traditional synthetic oligonucleotides.6 Casimersen is the most recent in a line of approved PMOs for treating DMD, including eteplirsen and viltolarsen. However, the specific mutations, and hence the precise exon skipping, targeted by each is different.
Casimersen was granted accelerated FDA approval on February 25, 2021, based on data showing an increase in dystrophin levels in skeletal muscle of patients treated with casimersen; this approval is contingent on further verification in confirmatory trials. Casimersen is currently marketed under the tradename AMONDYS 45™ by Sarepta Therapeutics, Inc.7
Casimersen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients confirmed to have a DMD gene mutation amenable to exon 45 skipping. This indication represents an accelerated approval based on observed efficacy; continued approval for this indication may be contingent on the verification of safety and efficacy in a confirmatory trial.7
Medical uses
Casimersen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[1][2]
History
Casimersen was evaluated in a double-blind, placebo-controlled study in which 43 participants were randomized 2:1 to receive either intravenous casimersen or placebo.[2] All participants were male, between 7 and 20 years of age, and had a genetically confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[2]
The U.S. Food and Drug Administration (FDA) granted the application for casimersen fast track, priority review, and orphan drug designations.[2][5] The FDA granted the approval of Amondys 45 to Sarepta Therapeutics, Inc.[2]
Pharmacodynamics
Casimersen is an antisense phosphorodiamidate morpholino oligonucleotide designed to bind to exon 45 of the DMD pre-mRNA, preventing its inclusion in mature mRNA and allowing the production of an internally truncated dystrophin protein in patients who would normally produce no functional dystrophin. Due to the need for continuous alteration of mRNA splicing and its relatively short half-life, casimersen is administered weekly.7 Although casimersen is associated with mostly mild adverse effects, animal studies suggest a potential for nephrotoxicity, which has also been observed after administration of some oligonucleotides.4,7 Measurement of glomerular filtration rate before starting casimersen is advised. Serum cystatin C, urine dipstick, and urine protein-to-creatinine ratio should be measured before starting therapy. They should be measured monthly (urine dipstick) or every three months (serum cystatin C and urine protein-to-creatinine ratio) during treatment. Creatinine levels are not reliable in muscular dystrophy patients and should not be used. Any persistent alteration in kidney function should be further investigated.7
Mechanism of action
Duchenne muscular dystrophy (DMD) is an X-linked recessive allelic disorder that results in the absence of functional dystrophin, a large protein comprising an N-terminal actin-binding domain, C-terminal β-dystroglycan-binding domain, and 24 internal spectrin-like repeats.1,2,3 Dystrophin is vital for normal muscle function; the absence of dystrophin leads to muscle membrane damage, extracellular leakage of creatinine kinase, calcium influx, and gradual replacement of normal muscle tissue with fibrous and adipose tissue over time.1,2 DMD shows a characteristic disease progression with early functional complaints related to abnormal gait, locomotion, and falls that remain relatively stable until around seven years of age. The disease then progresses rapidly to loss of independent ambulatory function, ventilatory insufficiency, and cardiomyopathy, with death typically occurring in the second or third decade of life.1,2,3
The human DMD gene contains 79 exons spread over approximately 2.4 million nucleotides on the X chromosome.1 DMD is associated with a variety of underlying mutations, including exon duplications or deletions, as well as point mutations leading to nonsense translation through direct production of an in-frame stop codon, frameshift production of an in-frame stop codon, or aberrant inclusion of an intronic pseudo-exon with the concomitant production of an in-frame stop codon.1,2 In all cases, no functional dystrophin protein is produced. Becker muscular dystrophy (BMD) is a related condition with in-frame mutations that result in the production of a truncated but partially functional dystrophin protein. BMD patients, therefore, have milder symptoms, delayed disease progression, and longer life expectancy compared to DMD patients.1,2,3
Casimersen is an antisense phosphorodiamidate morpholino oligonucleotide designed to bind to exon 45 of the DMD pre-mRNA and prevent its inclusion within the mature mRNA before translation.4,7 It is estimated that around 8% of DMD patients may benefit from exon 45 skipping, in which the exclusion of this exon results in the production of an internally truncated and at least partly functional dystrophin protein.4,7,5 Although fibrotic or fatty muscle tissue developed previously cannot be improved, this therapy aims to slow further disease progression through the production of partially functional dystrophin and alleviation of the pathogenic mechanism of muscle tissue necrosis.1,2
| TARGET | ACTIONS | ORGANISM |
|---|---|---|
| ADMD gene (exon 45 casimersen target site) | binder | Humans |
Absorption
DMD patients receiving IV doses of 4-30 mg/kg/week revealed exposure in proportion to dose with no accumulation of casimersen in plasma with once-weekly dosing. Following a single IV dose, casimersen Cmax was reached by the end of infusion. Inter-subject variability, as measured by the coefficient of variation, ranged from 12-34% for Cmax and 16-34% for AUC.7
Pre-clinical studies in nonhuman primates (cynomolgus monkeys) investigated the pharmacokinetics of once-weekly casimersen administered at doses of 5, 40, and 320 mg/kg. On days 1 and 78, the 5 mg/kg dose resulted in a Cmax of 19.5 ± 3.43 and 21.6 ± 5.60 μg/mL and an AUC0-t of 24.9 ± 5.17 and 26.9 ± 7.94 μg*hr/mL. The 40 mg/kg dose resulted in a Cmax of 208 ± 35.2 and 242 ± 71.1 μg/mL and an AUC0-t of 283 ± 68.5 and 320 ± 111 μg*hr/mL. Lastly, the 320 mg/kg dose resulted in a a Cmax of 1470 ± 88.1 and 1490 ± 221 μg/mL and an AUC0-t of 1960 ± 243 and 1930 ± 382 μg*hr/mL.4
Volume of distribution
Casimersen administered at 30 mg/kg had a mean steady-state volume of distribution (%CV) of 367 mL/kg (28.9%).7
Protein binding
Casimersen binding to human plasma proteins is not concentration-dependent, ranging from 8.4-31.6%.7
Metabolism
Casimersen incubated with human hepatic microsomal preparations is metabolically stables and no metabolites are detected in plasma or urine.7
Route of elimination
Casimersen is predominantly (more than 90%) excreted in the urine unchanged with negligible fecal excretion.7
Half-life
Casimersen has an elimination half-life of 3.5 ± 0.4 hours.7
Clearance
Casimersen administered at 30 mg/kg has a plasma clearance of 180 mL/hr/kg.7
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Amondys 45 | Injection | 50 mg/1mL | Intravenous | Sarepta Therapeutics, Inc. | 2021-02-25 | Not applicable |  |
Synthesis Reference
Diane Elizabeth Frank and Richard K. Bestwick, “Exon skipping oligomers for muscular dystrophy.” U.S. Patent US20190262375A1, issued August 29, 2019.
PATENT
https://patents.google.com/patent/WO2017205879A2/en
also
WO 2021025899
References
- ^ Jump up to:a b c d e “Amondys 45- casimersen injection”. DailyMed. Retrieved 1 March 2021.
- ^ Jump up to:a b c d e f g h i j “FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation”. U.S. Food and Drug Administration (FDA) (Press release). 25 February 2021. Retrieved 25 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ “Sarepta Therapeutics Announces FDA Approval of Amondys 45 (casimersen) Injection for the Treatment of Duchenne Muscular Dystrophy (DMD) in Patients Amenable to Skipping Exon 45” (Press release). Sarepta Therapeutics. 25 February 2021. Retrieved 25 February 2021 – via GlobeNewswire.
- ^ Rodrigues M, Yokota T (2018). “An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases”. Exon Skipping and Inclusion Therapies. Methods in Molecular Biology. 1828. Clifton, N.J. pp. 31–55. doi:10.1007/978-1-4939-8651-4_2. ISBN 978-1-4939-8650-7. PMID 30171533.
- ^ “Casimersen Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 4 June 2019. Retrieved 25 February 2021.
General References
- Wein N, Alfano L, Flanigan KM: Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr Clin North Am. 2015 Jun;62(3):723-42. doi: 10.1016/j.pcl.2015.03.008. Epub 2015 Apr 20. [PubMed:26022172]
- Verhaart IEC, Aartsma-Rus A: Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019 Jul;15(7):373-386. doi: 10.1038/s41582-019-0203-3. [PubMed:31147635]
- Mercuri E, Bonnemann CG, Muntoni F: Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-2038. doi: 10.1016/S0140-6736(19)32910-1. [PubMed:31789220]
- Carver MP, Charleston JS, Shanks C, Zhang J, Mense M, Sharma AK, Kaur H, Sazani P: Toxicological Characterization of Exon Skipping Phosphorodiamidate Morpholino Oligomers (PMOs) in Non-human Primates. J Neuromuscul Dis. 2016 Aug 30;3(3):381-393. doi: 10.3233/JND-160157. [PubMed:27854228]
- Rodrigues M, Yokota T: An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol Biol. 2018;1828:31-55. doi: 10.1007/978-1-4939-8651-4_2. [PubMed:30171533]
- Smith CIE, Zain R: Therapeutic Oligonucleotides: State of the Art. Annu Rev Pharmacol Toxicol. 2019 Jan 6;59:605-630. doi: 10.1146/annurev-pharmtox-010818-021050. Epub 2018 Oct 9. [PubMed:30285540]
- FDA Approved Drug Products: AMONDYS 45 (casimersen) injection [Link]
External links
- “Casimersen”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02500381 for “Study of SRP-4045 and SRP-4053 in DMD Patients (ESSENCE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Amondys 45 |
| Other names | SRP-4045 |
| License data | US DailyMed: Casimersen |
| Routes of administration | Intravenous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1422958-19-7 |
| DrugBank | DB14984 |
| UNII | X8UHF7SX0R |
| KEGG | D11988 |
| Chemical and physical data | |
| Formula | C268H424N124O95P22 |
| Molar mass | 7584.536 g·mol−1 |
////////////Casimersen, FDA 2021, APPROVALS 2021, カシメルセン , Exon-45: NG-12-0064, SRP-4045, WHO 10354, Amondys 45, Antisense oligonucleotide, Duchenne muscular dystrophy
#Casimersen, #FDA 2021, #APPROVALS 2021, #カシメルセン , #Exon-45: NG-12-0064, #SRP-4045, #WHO 10354, #Amondys 45, #Antisense oligonucleotide, #Duchenne muscular dystrophy
Sequence:
1caaugccauc cuggaguucc ug
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| modified base | c-1 | 5′-ester |
| modified base | c-1 | modified cytidine |
| modified base | a-2 | modified adenosine |
| modified base | a-3 | modified adenosine |
| modified base | u-4 | m5u |
| modified base | u-4 | modified uridine |
| modified base | g-5 | modified guanosine |
| modified base | c-6 | modified cytidine |
| modified base | c-7 | modified cytidine |
| modified base | a-8 | modified adenosine |
| modified base | u-9 | modified uridine |
| modified base | u-9 | m5u |
| modified base | c-10 | modified cytidine |
| modified base | c-11 | modified cytidine |
| modified base | u-12 | m5u |
| modified base | u-12 | modified uridine |
| modified base | g-13 | modified guanosine |
| modified base | g-14 | modified guanosine |
| modified base | a-15 | modified adenosine |
| modified base | g-16 | modified guanosine |
| modified base | u-17 | modified uridine |
| modified base | u-17 | m5u |
| modified base | u-18 | modified uridine |
| modified base | u-18 | m5u |
| modified base | c-19 | modified cytidine |
| modified base | c-20 | modified cytidine |
| modified base | u-21 | m5u |
| modified base | u-21 | modified uridine |
| modified base | g-22 | modified guanosine |
| uncommon link | c-1 – a-2 | unavailable |
| uncommon link | a-2 – a-3 | unavailable |
| uncommon link | a-3 – u-4 | unavailable |
| uncommon link | u-4 – g-5 | unavailable |
| uncommon link | g-5 – c-6 | unavailable |
| uncommon link | c-6 – c-7 | unavailable |
| uncommon link | c-7 – a-8 | unavailable |
| uncommon link | a-8 – u-9 | unavailable |
| uncommon link | u-9 – c-10 | unavailable |
| uncommon link | c-10 – c-11 | unavailable |
| uncommon link | c-11 – u-12 | unavailable |
| uncommon link | u-12 – g-13 | unavailable |
| uncommon link | g-13 – g-14 | unavailable |
| uncommon link | g-14 – a-15 | unavailable |
| uncommon link | a-15 – g-16 | unavailable |
| uncommon link | g-16 – u-17 | unavailable |
| uncommon link | u-17 – u-18 | unavailable |
| uncommon link | u-18 – c-19 | unavailable |
| uncommon link | c-19 – c-20 | unavailable |
| uncommon link | c-20 – u-21 | unavailable |
| uncommon link | u-21 – g-22 | unavailable |
Fosdenopterin hydrobromide

Fosdenopterin hydrobromide
FDA APPR 2021/2/26, NULIBRY
BBP-870/ORGN001
a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.
| ホスデノプテリン臭化水素酸塩水和物; |
| Formula | C10H14N5O8P. 2H2O. HBr |
|---|---|
| CAS | 2301083-34-9DIHYDRATE |
| Mol weight | 480.1631 |
2301083-34-9
(1R,10R,12S,17R)-5-amino-11,11,14-trihydroxy-14-oxo-13,15,18-trioxa-2,4,6,9-tetraza-14λ5-phosphatetracyclo[8.8.0.03,8.012,17]octadeca-3(8),4-dien-7-one;dihydrate;hydrobromide
1,3,2-DIOXAPHOSPHORINO(4′,5′:5,6)PYRANO(3,2-G)PTERIDIN-10(4H)-ONE, 8-AMINO-4A,5A,6,9,11,11A,12,12A-OCTAHYDRO-2,12,12-TRIHYDROXY-, 2-OXIDE, HYDROBROMIDE, HYDRATE (1:1:2), (4AR,5AR,11AR,12AS)-
| CYCLIC PYRANOPTERIN MONOPHOSPHATE MONOHYDROBROMIDE DIHYDRATE |
(4aR,5aR,11aR,12aS)-8-Amino-2,12,12-trihydroxy-4a,5a,6,7,11,11a,12,12aoctahydro-2H-2lambda5-(1,3,2)dioxaphosphinino(4′,5′:5,6)pyrano(3,2-g)pteridine-2,10(4H)-dione, hydrobromide (1:1:2)
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide, hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780



C10H14N5O8P, Average: 363.223
150829-29-1
- ALXN-1101
- WHO 11150
- Synthesis ReferenceClinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA: Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem. 2013 Feb 28;56(4):1730-8. doi: 10.1021/jm301855r. Epub 2013 Feb 19.
Fosdenopterin (or cyclic pyranopterin monophosphate, cPMP), sold under the brand name Nulibry, is a medication used to reduce the risk of death due to a rare genetic disease known as molybdenum cofactor deficiency type A (MoCD-A).[1]
Adverse effects
The most common side effects include complications related to the intravenous line, fever, respiratory infections, vomiting, gastroenteritis, and diarrhea.[1]
Mechanism of action
People with MoCD-A cannot produce cyclic pyranopterin monophosphate (cPMP) in their body.[1] Fosdenopterin is an intravenous medication that replaces the missing cPMP.[1][2] cPMP is a precursor to molybdopterin, which is required for the enzyme activity of sulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
History
Fosdenopterin was developed by José Santamaría-Araujo and Guenter Schwarz at the German universities TU Braunschweig and the University of Cologne.[4][5]
The effectiveness of fosdenopterin for the treatment of MoCD-A was demonstrated in thirteen treated participants compared to eighteen matched, untreated participants.[1][6] The participants treated with fosdenopterin had a survival rate of 84% at three years, compared to 55% for the untreated participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for fosdenopterin priority review, breakthrough therapy, and orphan drug designations along with a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Nulibry to Origin Biosciences, Inc., in February 2021.[1] It is the first medication approved for the treatment of MoCD-A.[1]
References
- ^ Jump up to:a b c d e f g h i j “FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A”. U.S. Food and Drug Administration (FDA) (Press release). 26 February 2021. Retrieved 26 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ DrugBank DB16628 . Accessed 2021-03-05.
- ^ Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (April 2004). “The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor”. The Journal of Biological Chemistry. 279 (16): 15994–9. doi:10.1074/jbc.M311815200. PMID 14761975.
- ^ Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ, et al. (June 2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli”. Human Molecular Genetics. 13 (12): 1249–55. doi:10.1093/hmg/ddh136. PMID 15115759.
- ^ Tedmanson S (5 November 2009). “Doctors risk untried drug to stop baby’s brain dissolving”. TimesOnline.
- ^ Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, et al. (November 2015). “Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study”. Lancet. 386 (10007): 1955–63. doi:10.1016/S0140-6736(15)00124-5. PMID 26343839. S2CID 21954888.
External links
- “Fosdenopterin”. Drug Information Portal. U.S. National Library of Medicine.
Molybdenum cofactor deficiency (MoCD) is an exceptionally rare autosomal recessive disorder resulting in a deficiency of three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde oxidase.1 Signs and symptoms begin shortly after birth and are caused by a build-up of toxic sulfites resulting from a lack of SOX activity.1,5 Patients with MoCD may present with metabolic acidosis, intracranial hemorrhage, feeding difficulties, and significant neurological symptoms such as muscle hyper- and hypotonia, intractable seizures, spastic paraplegia, myoclonus, and opisthotonus. In addition, patients with MoCD are often born with morphologic evidence of the disorder such as microcephaly, cerebral atrophy/hypodensity, dilated ventricles, and ocular abnormalities.1 MoCD is incurable and median survival in untreated patients is approximately 36 months1 – treatment, then, is focused on improving survival and maintaining neurological function.
The most common subtype of MoCD, type A, involves mutations in MOCS1 wherein the first step of molybdenum cofactor synthesis – the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP) – is interrupted.1,3 In the past, management strategies for this disorder involved symptomatic and supportive treatment,5 though efforts were made to develop a suitable exogenous replacement for the missing cPMP. In 2009 a recombinant, E. coli-produced cPMP was granted orphan drug designation by the FDA, becoming the first therapeutic option for patients with MoCD type A.1
Fosdenopterin was approved by the FDA on Februrary 26, 2021, for the reduction of mortality in patients with MoCD type A,5 becoming the first and only therapy approved for the treatment of MoCD. By improving the three-year survival rate from 55% to 84%,7 and considering the lack of alternative therapies available, fosdenopterin appears poised to become a standard of therapy in the management of this debilitating disorder.
Fosdenopterin replaces an intermediate substrate in the synthesis of molybdenum cofactor, a compound necessary for the activation of several molybdenum-dependent enzymes including sulfite oxidase (SOX).1 Given that SOX is responsible for detoxifying sulfur-containing acids and sulfites such as S-sulfocysteine (SSC), urinary levels of SSC can be used as a surrogate marker of efficacy for fosdenopterin.7 Long-term therapy with fosdenopterin has been shown to result in a sustained reduction in urinary SSC normalized to creatinine.7
Animal studies have identified a potential risk of phototoxicity in patients receiving fosdenopterin – these patients should avoid or minimize exposure to sunlight and/or artificial UV light.7 If sun exposure is necessary, use protective clothing, hats, and sunglasses,7 in addition to seeking shade whenever practical. Consider the use of a broad-spectrum sunscreen in patients 6 months of age or older.8
Molybdenum cofactor deficiency (MoCD) is a rare autosomal-recessive disorder in which patients are deficient in three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde dehydrogenase.1 The loss of SOX activity appears to be the main driver of MoCD morbidity and mortality, as the build-up of neurotoxic sulfites typically processed by SOX results in rapid and progressive neurological damage. In MoCD type A, the disorder results from a mutation in the MOCS1 gene leading to deficient production of MOCS1A/B,7 a protein that is responsible for the first step in the synthesis of molybdenum cofactor: the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP).1,4
Fosdenopterin is an exogenous form of cPMP, replacing endogenous production and allowing for the synthesis of molybdenum cofactor to proceed.7
- Mechler K, Mountford WK, Hoffmann GF, Ries M: Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015 Dec;17(12):965-70. doi: 10.1038/gim.2015.12. Epub 2015 Mar 12. [PubMed:25764214]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, Jameson E, Konig K, McGregor TL, Font-Montgomery E, Santamaria-Araujo JA, Santra S, Vaidya M, Vierzig A, Wassmer E, Weis I, Wong FY, Veldman A, Schwarz G: Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015 Nov 14;386(10007):1955-63. doi: 10.1016/S0140-6736(15)00124-5. Epub 2015 Sep 3. [PubMed:26343839]
- Iobbi-Nivol C, Leimkuhler S: Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim Biophys Acta. 2013 Aug-Sep;1827(8-9):1086-101. doi: 10.1016/j.bbabio.2012.11.007. Epub 2012 Nov 29. [PubMed:23201473]
- Mendel RR: The molybdenum cofactor. J Biol Chem. 2013 May 10;288(19):13165-72. doi: 10.1074/jbc.R113.455311. Epub 2013 Mar 28. [PubMed:23539623]
- FDA News Release: FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A [Link]
- OMIM: MOLYBDENUM COFACTOR DEFICIENCY, COMPLEMENTATION GROUP A (# 252150) [Link]
- FDA Approved Drug Products: Nulibry (fosdenopterin) for intravenous injection [Link]
- Health Canada: Sun safety tips for parents [Link]
SYN
Journal of Biological Chemistry (1995), 270(3), 1082-7.
https://linkinghub.elsevier.com/retrieve/pii/S0021925818829696
PATENT
WO 2005073387
PATENT
WO 2012112922
PAPER

Journal of Medicinal Chemistry (2013), 56(4), 1730-1738
https://pubs.acs.org/doi/10.1021/jm301855r

Cyclic pyranopterin monophosphate (1), isolated from bacterial culture, has previously been shown to be effective in restoring normal function of molybdenum enzymes in molybdenum cofactor (MoCo)-deficient mice and human patients. Described here is a synthesis of 1 hydrobromide (1·HBr) employing in the key step a Viscontini reaction between 2,5,6-triamino-3,4-dihydropyrimidin-4-one dihydrochloride and d-galactose phenylhydrazone to give the pyranopterin (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one (10) and establishing all four stereocenters found in 1. Compound 10, characterized spectroscopically and by X-ray crystallography, was transformed through a selectively protected tri-tert-butoxycarbonylamino intermediate into a highly crystalline tetracyclic phosphate ester (15). The latter underwent a Swern oxidation and then deprotection to give 1·HBr. Synthesized 1·HBr had in vitro efficacy comparable to that of 1 of bacterial origin as demonstrated by its enzymatic conversion into mature MoCo and subsequent reconstitution of MoCo-free human sulfite oxidase–molybdenum domain yielding a fully active enzyme. The described synthesis has the potential for scale up.





PAPER
European Journal of Organic Chemistry (2014), 2014(11), 2231-2241.
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/ejoc.201301784
Abstract
The first synthesis of an oxygen‐stable analogue of the natural product cyclic pyranopterin monophosphate (cPMP) is reported. In this approach, the hydropyranone ring is annelated to pyrazine by a sequence comprising ortho‐lithiation/acylation of a 2‐halopyrazine, followed by nucleophilic aromatic substitution. The tetrose substructure is introduced from the chiral pool, from D‐galactose or D‐arabitol.

Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation‐labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D‐galactose or D‐arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Patent
https://patents.google.com/patent/US9260462B2/en
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclic pyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, untreatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis (see U.S. Pat. No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
Scheme 3.

Scheme 4.

(I).
Scheme 6.

(I).

Scheme 8.

(I).

Scheme 10.

EXAMPLESExample 1Preparation of Precursor Z (cPMP)


Experimental
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially (anhydrous grade solvent from Sigma-Aldrich Fine Chemicals) and used without any further purification. Thin layer chromatography (t.l.c.) was performed on glass or aluminum sheets coated with 60 F254 silica gel. Organic compounds were visualized under UV light or with use of a dip of ammonium molybdate (5 wt %) and cerium(IV) sulfate 4H2O (0.2 wt %) in aq. H2SO4 (2M), one of I2 (0.2%) and KI (7%) in H2SO4 (1M), or 0.1% ninhydrin in EtOH. Chromatography (flash column) was performed on silica gel (40-63 μm) or on an automated system with continuous gradient facility. Optical rotations were recorded at a path length of 1 dm and are in units of 10−1 deg cm2 g−1; concentrations are in g/100 mL. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0 ppm) or D2O(HOD, δ 4.79 ppm), and 13C NMR spectra in CDCl3 (center line, δ 77.0 ppm), CD3OD (center line, δ 49.0 ppm) or DMSO d6 (center line δ 39.7 ppm), D2O (no internal reference or internal CH3CN, δ 1.47 ppm where stated). Assignments of 1H and 13C resonances were based on 2D (1H—1H DQF-COSY, 1H—13C HSQC, HMBC) and DEPT experiments. 31P NMR were run at 202.3 MHz and are reported without reference. High resolution electrospray mass spectra (ESI-HRMS) were recorded on a Q-TOF Tandem Mass
Spectrometer. Microanalyses were performed by the Campbell Microanalytical Department, University of Otago, Dunedin, New Zealand.
A. Preparation of (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one mono hydrate (1)
2,5,6-Triamino-3,4-dihydropyrimidin-4-one dihydrochloride (Pfleiderer, W.; Chem. Ber. 1957, 90, 2272; Org. Synth. 1952, 32, 45; Org. Synth. 1963, Coll. Vol. 4, 245, 10.0 g, 46.7 mmol), D-galactose phenylhydrazone (Goswami, S.; Adak, A. K. Tetrahedron Lett. 2005, 46, 221-224, 15.78 g, 58.4 mmol) and 2-mercaptoethanol (1 mL) were stirred and heated to reflux (bath temp 110° C.) in a 1:1 mixture of MeOH—H2O (400 mL) for 2 h. After cooling to ambient temperature, diethyl ether (500 mL) was added, the flask was shaken and the diethyl ether layer decanted off and discarded. The process was repeated with two further portions of diethyl ether (500 mL) and then the remaining volatiles were evaporated. Methanol (40 mL), H2O (40 mL) and triethylamine (39.4 mL, 280 mmol) were successively added and the mixture seeded with a few milligrams of 1. After 5 min a yellow solid was filtered off, washed with a little MeOH and dried to give 1 as a monohydrate (5.05 g, 36%) of suitable purity for further use. An analytical portion was recrystallized from DMSO-EtOH or boiling H2O. MPt 226 dec. [α]D 20 +135.6 (c1.13, DMSO). 1H NMR (DMSO d6): δ 10.19 (bs, exchanged D2O, 1H), 7.29 (d, J=5.0 Hz, slowly exchanged D2O, 1H), 5.90 (s, exchanged D2O, 2H), 5.33 (d, J=5.4 Hz, exchanged D2O, 1H), 4.66 (ddd, J˜5.0, ˜1.3, ˜1.3 Hz, 1H), 4.59 (t, J=5.6 Hz, exchanged D2O, 1H), 4.39 (d, J=10.3 Hz, exchanged D2O, 1H), 3.80 (bt, J˜1.8 Hz, exchanged D2O, 1H), 3.70 (m, 1H), 3.58 (dd, J=10.3, 3.0 Hz, 1H), 3.53 (dt, J=10.7, 6.4 Hz, 1H), 3.43 (ddd, J=11.2, 5.9, 5.9 Hz, 1H), 3.35 (t, J=6.4 Hz, 1H), 3.04 (br m, 1H). 13C NMR (DMSO d6 center line 6 39.7): δ 156.3 (C), 150.4 (C), 148.4 (C), 99.0 (C), 79.4 (CH), 76.5 (CH), 68.9 (CH), 68.6 (CH), 60.6 (CH2), 53.9 (CH). Anal. calcd. for C10H15N5O5H2O 39.60; C, 5.65; H, 23.09; N. found 39.64; C, 5.71; H, 22.83; N.
B. Preparation of Compounds 2 (a or b) and 3 (a, b or c)
Di-tert-butyl dicarbonate (10.33 g, 47.3 mmol) and DMAP (0.321 g, 2.63 mmol) were added to a stirred suspension of 1 (1.5 g, 5.26 mmol) in anhydrous THF (90 mL) at 50° C. under Ar. After 20 h a clear solution resulted. The solvent was evaporated and the residue chromatographed on silica gel (gradient of 0 to 40% EtOAc in hexanes) to give two product fractions. The first product to elute was a yellow foam (1.46 g). The product was observed to be a mixture of two compounds by 1H NMR containing mainly a product with seven Boc groups (2a or 2b). A sample was crystallized from EtOAc-hexanes to give 2a or 2b as a fine crystalline solid. MPt 189-191° C. [α]D 20 −43.6 (c 0.99, MeOH). 1H NMR (500 MHz, CDCl3): δ 5.71 (t, J=1.7 Hz, 1H), 5.15 (dt, J=3.5, ˜1.0, 1H), 4.97 (t, J=3.8, 1H), 4.35 (br t, J=˜1.7, 1H), 4.09-3.97 (m, 3H), 3.91 (m, 1H), 1.55, 1.52, 1.51, 1.50, 1.45 (5s, 45H), 1.40 (s, 18H). 13C NMR (125.7 MHz, CDCl3): δ 152.84 (C), 152.78 (C), 151.5 (C), 150.9 (C), 150.7 (2×C), 150.3 (C), 149.1 (C), 144.8 (C), 144.7 (C), 118.0 (C), 84.6 (C), 83.6 (C), 83.5 (C), 82.7 (3×C), 82.6 (C), 76.3 (CH), 73.0 (CH), 71.4 (CH), 67.2 (CH), 64.0 (CH2), 51.4 (CH), 28.1 (CH3), 27.8 (2×CH3), 27.7 (CH3), 27.6 (3×CH3). MS-ESI+ for C45H72N5O19 +, (M+H)+, Calcd. 986.4817. found 986.4818. Anal. calcd. for C45H71N5O19H2O 54.39; C, 7.39; H, 6.34; N. found 54.66; C, 7.17; H, 7.05; N. A second fraction was obtained as a yellow foam (2.68 g) which by 1H NMR was a product with six Boc groups present (3a, 3b or 3c). A small amount was crystallized from EtOAc-hexanes to give colorless crystals. [α]D 2O −47.6 (c, 1.17, CHCl3). 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, exchanged D2O, 1H), 5.58 (t, J=1.8 Hz, 1H), 5.17 (d, J=3.4 Hz, 1H), 4.97 (t, J=3.9 Hz, 1H), 4.62 (s, exchanged D2O, 1H), 4.16 (dd, J=11.3, 5.9 Hz, 1H), 4.12 (dd, J=11.3, 6.4 Hz, 1H), 3.95 (dt, J=6.1, 1.1 Hz, 1H), 3.76 (m, 1H), 1.51, 1.50, 1.49, 1.48, 1.46 (5s, 54H). 13C NMR (125.7 MHz, CDCl3): δ 156.6 (C), 153.0 (C), 152.9 (C), 151.9 (C), 150.6 (C), 149.4 (2×C), 136.2 (C), 131.8 (C), 116.9 (C), 85.0 (2×C), 83.3 (C), 82.8 (C), 82.49 (C), 82.46 (C), 73.3 (CH), 71.5 (CH), 67.2 (CH), 64.5 (CH2), 51.3 (CH), 28.0, 27.72, 27.68, 27.6 (4×CH3). MS-ESI+ for C40H64N5O17 +, (M+H)+calcd. 886.4287. found 886.4289.
C. Preparation of Compound 4a, 4b or 4c
Step 1—The first fraction from B above containing mainly compounds 2a or 2b (1.46 g, 1.481 mmol) was dissolved in MeOH (29 mL) and sodium methoxide in MeOH (1M, 8.14 mL, 8.14 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
Step 2—The second fraction from B above containing mainly 3a, 3b or 3c (2.68 g, 3.02 mmol) was dissolved in MeOH (54 mL) and sodium methoxide in MeOH (1M, 12.10 mL, 12.10 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
The products from step 1 and step 2 above were combined and chromatographed on silica gel (gradient of 0 to 15% MeOH in CHCl3) to give 4a, 4b or 4c as a cream colored solid (1.97 g). 1H NMR (500 MHz, DMSO d6): δ 12.67 (br s, exchanged D2O, 1H), 5.48 (d, J=5.2 Hz, exchanged D2O, 1H), 5.43 (t, J=˜1.9 Hz, after D2O exchange became a d, J=1.9 Hz, 1H), 5.00 (br s, exchanged D2O, 1H), 4.62 (d, J=5.7 Hz, exchanged D2O, 1H), 4.27 (d, J=6.0 Hz, exchanged D2O, 1H), 3.89 (dt, J=5.2, 3.8 Hz, after D2O became a t, J=3.9 Hz, 1H), 3.62 (dd, J=6.0, 3.7 Hz, after D2O exchange became a d, J=3.7 Hz, 1H), 3.52-3.39 (m, 4H), 1.42 (s, 9H), 1.41 (s, 18H). 13C NMR (125.7 MHz, DMSO d6): δ 157.9 (C), 151.1, (C), 149.8 (2×C), 134.6 (C), 131.4 (C), 118.8 (C), 83.5 (2×C), 81.3 (C), 78.2 (CH), 76.5 (CH), 68.1 (CH), 66.8 (CH), 60.6 (CH2), 54.4 (CH), 27.9 (CH3), 27.6 (2×CH3). MS-ESI+ for C25H40N5O11 +, (M+H)+ calcd. 586.2719. found 586.2717.
D. Preparation of Compound 5a, 5b or 5c
Compound 4a, 4b or 4c (992 mg, 1.69 mmol) was dissolved in anhydrous pyridine and concentrated. The residue was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (5 mL) under a nitrogen atmosphere and the solution was cooled to −42° C. in an acetonitrile/dry ice bath. Methyl dichlorophosphate (187 μL, 1.86 mmol) was added dropwise and the mixture was stirred for 2 h 20 min. Water (10 mL) was added to the cold solution which was then removed from the cold bath and diluted with ethyl acetate (50 mL) and saturated NaCl solution (30 mL). The organic portion was separated and washed with saturated NaCl solution. The combined aqueous portions were extracted twice further with ethyl acetate and the combined organic portions were dried over MgSO4 and concentrated. Purification by silica gel flash column chromatography (eluting with 2-20% methanol in ethyl acetate) gave the cyclic methyl phosphate 5a, 5b or 5c (731 mg, 65%). 1H NMR (500 MHz, CDCl3,): δ 11.72 (bs, exchanged D2O, 1H), 5.63 (t, J=1.8 Hz, 1H), 5.41 (s, exchanged D2O, 1H), 4.95 (d, J=3.2 Hz, 1H), 4.70 (dt, J=12.4, 1.8 Hz, 1H), 4.42 (dd, J=22.1, 12.1 Hz, 1H). 4.15 (q, J=3.7 Hz, 1H), 3.82 (s, 1H), 3.75 (s, 1H), 3.58 (d, J=11.7 Hz, 3H), 2.10 (bs, exchanged D20, 1H+H2O), 1.50 (s, 9H), 1.46 (s, 18H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0): δ 157.5 (C), 151.2 (C), 149.6 (2×C), 134.5 (C), 132.3 (C), 117.6 (C), 84.7 (2×C), 82.8 (C), 77.3 (CH), 74.8 (d, J=4.1 Hz, CH), 69.7 (CH2), 68.8 (d, J=4.1 Hz, CH), 68.6 (d, J=5.9 Hz, CH), 56.0 (d, J=7.4 Hz, CH3), 51.8 (CH), 28.1 (CH3), 27.8 (CH3). MS-ESI+ for C26H40N5NaO13P+ (M+Na)+, calcd. 684.2252. found 684.2251.
E. Preparation of Compound 6a, 6b or 6c
Compound 5a, 5b or 5c (223 mg, 0.34 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) under a nitrogen atmosphere. Anhydrous DMSO (104 μL, 1.46 mmol) was added and the solution was cooled to −78° C. Trifluoroacetic anhydride (104 μL, 0.74 mmol) was added dropwise and the mixture was stirred for 40 min. N,N-diisopropylethylamine (513 μL, 2.94 mmol) was added and the stirring was continued for 50 min at −78° C. Saturated NaCl solution (20 mL) was added and the mixture removed from the cold bath and diluted with CH2Cl2 (30 mL). Glacial acetic acid (170 μL, 8.75 mmol) was added and the mixture was stirred for 10 min. The layers were separated and the aqueous phase was washed with CH2Cl2 (10 mL). The combined organic phases were washed with 5% aqueous HCl, 3:1 saturated NaCl solution:10% NaHCO3 solution and saturated NaCl solution successively, dried over MgSO4, and concentrated to give compound 6a, 6b or 6c (228 mg, quant.) of suitable purity for further use. 1H NMR (500 MHz, CDCl3): δ 5.86 (m, 1 H), 5.07 (m, 1 H), 4.70-4.64 (m, 2 H), 4.49-4.40 (m, 1 H), 4.27 (m, 1 H), 3.56, m, 4 H), 1.49 (s, 9 H), 1.46 (s, 18 H) ppm. 13C NMR (500 MHz, CDCl3): δ 157.5 (C), 151.1 (C), 150.6 (2 C), 134.6 (C), 132.7 (C), 116.6 (C), 92.0 (C), 84.6 (2 C), 83.6 (C), 78.0 (CH), 76.0 (CH), 70.4 (CH2), 67.9 (CH), 56.2 (CH3) δ6.0 (CH), 28.2 (3CH3), 26.8 (6 CH3) ppm. 31P NMR (500 MHz, CDCl3): δ−6.3 ppm.
F. Preparation of compound 7: (4aR,5aR,11aR,12aS)-1,3,2-Dioxaphosphorino[4′,5′:5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4-a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-2-oxide
Compound 6a, 6b or 6c (10 mg, 14.8 μmol was dissolved in dry acetonitrile (0.2 mL) and cooled to 0° C. Bromotrimethylsilane (19.2 μL, 148 μmol) was added dropwise and the mixture was allowed to warm to ambient temperature and stirred for 5 h during which time a precipitate formed. HCl(aq) (10 μl, 37%) was added and the mixture was stirred for a further 15 min. The mixture was centrifuged for 15 min (3000 g) and the resulting precipitate collected. Acetonitrile (0.5 mL) was added and the mixture was centrifuged for a further 15 min. The acetonitrile wash and centrifugation was repeated a further two times and the resulting solid was dried under high vacuum to give compound 7 (4 mg, 75%). 1H NMR (500 MHz, D2O): δ 5.22 (d, J=1.6 Hz, 1H), 4.34 (dt, J=13, 1.6 Hz, 1H), 4.29-4.27 (m, 1H), 4.24-4.18 (m, 1H), 3.94 (br m, 1H), 3.44 (t, J=1.4 Hz, 1H). 31P NMR (500 MHz, D2O): δ −4.8 MS-ESI+ for C10H15N5O8P+, (M+H)+calcd. 364.0653. found 364.0652.
Example 2Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. Coli in the In vitro Synthesis of Moco
In vitro synthesis of Moco was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. Moco synthesis also involved the use of the purified components E. coli MPT synthase, gephyrin, molybdate, ATP, and apo-sulfite oxidase. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. The assay is based on the conversion of cPMP into MPT, the subsequent molybdate insertion using recombinant gephyrin and ATP, and finally the reconstitution of human apo-sulfite oxidase.
As shown in FIG. 1, Moco synthesis from synthetic cPMP was confirmed, and no differences in Moco conversion were found in comparison to E. coli purified cPMP.
Example 3Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. coli in the In vitro Synthesis of MPT
In vitro synthesis of MPT was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. MPT synthesis also involved the use of in vitro assembled MPT synthase from E. coli. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. Three repetitions of each experiment were performed and are shown in FIGS. 2 and 3.
As shown in FIGS. 2 and 3, MPT synthesis from synthetic cPMP confirmed, and no apparent differences in MPT conversion were found when compared to E. coli purified cPMP. A linear conversion of cPMP into MPT is seen in all samples confirming the identity of synthetic cPMP (see FIG. 2). Slight differences between the repetitions are believed to be due to an inaccurate concentration determination of synthetic cPMP given the presence of interfering chromophores.
Example 4Preparation of Precursor Z (cPMP)
A. Preparation of Starting Materials

B. Introduction of the protected Phosphate

The formation of the cyclic phosphate using intermediate [10] (630 mg) gave the desired product [11] as a 1:1 mixture of diastereoisomers (494 mg, 69%).

C. Oxidation and Overall Deprotection of the Molecule
Oxidation of the secondary alcohol to the gem-diol did prove successful on intermediate [12], but the oxidized product [13] did show significant instability and could not be purified. For this reason, deprotection of the phosphate was attempted before the oxidation. However, the reaction of intermediate [11] with TMSBr led to complete deprotection of the molecule giving intermediate [14]. An attempt to oxidize the alcohol to the gem-diol using Dess-Martin periodinane gave the aromatized pteridine [15].
Oxidation of intermediate [11] with Dess-Martin periodinane gave a mixture of starting material, oxidized product and several by-products. Finally, intermediate [11] was oxidized using the method described Example 1. Upon treatment, only partial oxidation was observed, leaving a 2:1 mixture of [11]/[16]. The crude mixture was submitted to the final deprotection. An off white solid was obtained and analyzed by 1H-NMR and HPLC-MS. These analyses suggest that cPMP has been produced along with the deprotected precursor [11].
Because the analytical HPLC conditions gave a good separation of cPMP from the major impurities, this method will be repeated on a prep-HPLC in order to isolate the final material.
CLIP
BridgeBio Pharma And Affiliate Origin Biosciences Announces FDA Acceptance Of Its New Drug Application For Fosdenopterin For The Treatment Of MoCD Type A
Application accepted under Priority Review designation with Breakthrough Therapy Designation and Rare Pediatric Disease Designation previously grantedThere are currently no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury for infants and children.This is BridgeBio’s first NDA acceptanceSAN FRANCISCO, September 29, 2020 – BridgeBio Pharma, Inc. (Nasdaq: BBIO) and affiliate Origin Biosciences today announced the US Food and Drug Administration (FDA) has accepted its New Drug Application (NDA) for fosdenopterin (previously BBP-870/ORGN001), a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.The NDA has been granted Priority Review designation. Fosdenopterin has previously been granted Breakthrough Therapy Designation and Rare Pediatric Disease Designation in the US and may be eligible for a priority review voucher if approved. It received Orphan Drug Designation in the US and Europe. This is BridgeBio’s first NDA acceptance.“We want to thank the patients, families, scientists, physicians and all others involved who helped us reach this critical milestone,” said BridgeBio CEO and founder Neil Kumar, Ph.D. “MoCD Type A is a devastating disease with a median survival of less than four years and we are eager for our investigational therapy to be available to patients, who currently have no approved treatment options. BridgeBio exists to help as many patients as possible afflicted with genetic diseases, no matter how rare. We are grateful that the FDA has accepted our first NDA for priority review and we look forward to submitting our second NDA later this year for infigratinib for second line treatment of cholangiocarcinoma.”About Fosdenopterin
Fosdenopterin is being developed for the treatment of patients with MoCD Type A. Currently, there are no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury with a median survival between 3 to 4 years. Fosdenopterin is a first-in-class cPMP hydrobromide dihydrate and is designed to treat MoCD Type A by replacing cPMP and permitting the two remaining MoCo synthesis steps to proceed, with activation of MoCo-dependent enzymes and elimination of sulfites.About Molybdenum Cofactor Deficiency (MoCD) Type A
MoCD Type A is an ultra-rare, autosomal recessive, inborn error of metabolism caused by disruption in molybdenum cofactor (MoCo) synthesis which is vital to prevent buildup of s-sulfocysteine, a neurotoxic metabolite of sulfite. Patients are often infants with severe encephalopathy and intractable seizures. Disease progression is rapid with a high infant mortality rate.Those who survive beyond the first few month’s experience profuse developmental delays and suffer the effects of irreversible neurological damage, including brain atrophy with white matter necrosis, dysmorphic facial features, and spastic paraplegia. Clinical presentation that can be similar to hypoxic-ischemic encephalopathy (HIE) or other neonatal seizure disorders may lead to misdiagnosis and underdiagnosis. Immediate testing for elevated sulfite levels and S-sulfocysteine in the urine and very low serum uric acid may help with suspicion of MoCD.About Origin Biosciences
Origin Biosciences, an affiliate of BridgeBio Pharma, is a biotechnology company focused on developing and commercializing a treatment for Molybdenum Cofactor Deficiency (MoCD) Type A. Origin is led by a team of veteran biotechnology executives. Together with patients and physicians, the company aims to bring a safe, effective treatment for MoCD Type A to market as quickly as possible. For more information on Origin Biosciences, please visit the company’s website at www.origintx.com.
About BridgeBio Pharma
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio’s pipeline of over 20 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.
| Clinical data | |
|---|---|
| Trade names | Nulibry |
| Other names | Precursor Z, ALXN1101 |
| License data | US DailyMed: Fosdenopterin |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 150829-29-1 |
| PubChem CID | 135894389 |
| DrugBank | DB16628 |
| ChemSpider | 17221217 |
| UNII | 4X7K2681Y7 |
| KEGG | D11779 |
| ChEMBL | ChEMBL2338675 |
| CompTox Dashboard (EPA) | DTXSID90934067 |
| Chemical and physical data | |
| Formula | C10H14N5O8P |
| Molar mass | 363.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESNC1=NC(=O)C2=C(N[C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]4C(O)(O)[C@@H]3N2)N1 | |
| hideInChIInChI=1S/C10H14N5O8P/c11-9-14-6-3(7(16)15-9)12-4-8(13-6)22-2-1-21-24(19,20)23-5(2)10(4,17)18/h2,4-5,8,12,17-18H,1H2,(H,19,20)(H4,11,13,14,15,16)/t2-,4-,5+,8-/m1/s1Key:CZAKJJUNKNPTTO-AJFJRRQVSA-N |
//////////Fosdenopterin hydrobromide, ホスデノプテリン臭化水素酸塩水和物 , ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780, BBP-870/ORGN001, Priority Review designation, Breakthrough Therapy Designation, Rare Pediatric Disease Designation, Orphan Drug Designation, molybdenum cofactor deficiency, ALXN-1101, WHO 11150, FDA 2021, APPROVALS 2021
#Fosdenopterin hydrobromide, #ホスデノプテリン臭化水素酸塩水和物 , #ALXN1101 HBr, #UNII-X41B5W735T, X41B5W735T, #D11780, #BBP-870/ORGN001, #Priority Review designation, #Breakthrough Therapy Designation, #Rare Pediatric Disease Designation, #Orphan Drug Designation, #molybdenum cofactor deficiency, #ALXN-1101, #WHO 11150, #FDA 2021, #APPROVALS 2021
C1C2C(C(C3C(O2)NC4=C(N3)C(=O)NC(=N4)N)(O)O)OP(=O)(O1)O.O.O.Br
Melphalan flufenamide hydrochloride
 .HCl
.HCl
Melphalan flufenamide hydrochloride
メルファランフルフェナミド塩酸塩;
L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride
| Formula | C24H30Cl2FN3O3. HCl |
|---|---|
| CAS | 380449-54-7 |
| Mol weight | 534.8786 |
FDA APPROVED PEPAXTO, 2021/2/26
| Efficacy | Antineoplastic, Alkylating agent |
|---|---|
| Disease | Multiple myeloma |
- Ethyl (2S)-2-[(2S)-2-amino-3-{4-[bis(2-chloroethyl)amino]phenyl}propanamido]-3-(4-fluorophenyl)propanoate
- J 1
- J 1 (prodrug)
- L-Melphalanyl-L-p-fluorophenylalanine ethyl ester
- Melflufen
- Melphalan flufenamide
- Pepaxto
- Prodrug J 1
Melflufen
- Molecular FormulaC24H30Cl2FN3O3
- Average mass498.418 Da
- SP ROT +33.0 ° Conc: 1.3 g/100mL; chloroform ; 589.3 nm, Oncology Research 2003, V14(3), P113-132
мелфалана флуфенамид [Russian] [INN]ميلفالان فلوفيناميد [Arabic] [INN]氟美法仑 [Chinese] [INN]380449-51-4[RN]
9493Ethyl 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-L-phenylalaninate
F70C5K4786L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester
Melphalan flufenamide, sold under the brand name Pepaxto, is an anticancer medication used to treat multiple myeloma.[3][4]
The most common adverse reactions include fatigue, nausea, diarrhea, pyrexia and respiratory tract infection.[3]
Melphalan flufenamide is a peptidase enhanced cytotoxic (PEnC) that exerts a targeted delivery of melphalan in cells with high expression of aminopeptidases, such as aminopeptidase N, which has been described as over-expressed in human malignancies.Aminopeptidase N plays a functional role in malignant angiogenesis.
Melphalan flufenamide was approved for medical use in the United States in February 2021.[4][5]
Medical uses
Melphalan flufenamide is indicated in combination with dexamethasone for the treatment of adults with relapsed or refractory multiple myeloma, with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy and whose disease is refractory to at least one proteasome inhibitor, one immunomodulatory agent, and one CD-38 directed monoclonal antibody.[3][4]
Metabolism
Melphalan flufenamide is metabolized by aminopeptidase hydrolysis and by spontaneous hydrolysis on N-mustard.[6] Its biological half-life is 10 minutes in vitro.
Origin and development
Melphalan flufenamide is a peptidase enhanced cytotoxic (PEnC) with a targeted delivery within tumor cells of melphalan, a widely used classical chemotherapeutic belonging to a group of alkylating agents developed more than 50 years ago. Substantial clinical experience has been accumulated about melphalan since then. Numerous derivatives of melphalan, designed to increase the activity or selectivity, have been developed and investigated in vitro or in animal models.[7] Melphalan flufenamide was synthesized, partly due to previous experience of an alkylating peptide cocktail named Peptichemio[8] and its anti-tumor activity is being investigated.
Pharmacology
Compared to melphalan, melphalan flufenamide exhibits significantly higher in vitro and in vivo activity in several models of human cancer.[9][10][11][12][13][14][15][16] A preclinical study, performed at Dana–Farber Cancer Institute, demonstrated that melphalan flufenamide induced apoptosis in multiple myeloma cell lines, even those resistant to conventional treatment (including melphalan).[17] In vivo effects in xenografted animals were also observed, and the results confirmed by M Chesi and co-workers – in a unique genetically engineered mouse model of multiple myeloma – are believed to be predictive of clinical efficacy.[18]
Structure
Chemically, the drug is best described as the ethyl ester of a dipeptide consisting of melphalan and the amino acid derivative para-fluoro-L-phenylalanine.
Pharmacokinetics
Pharmacokinetic analysis of plasma samples showed a rapid formation of melphalan; concentrations generally exceeded those of melphalan flufenamide during ongoing infusion. Melphalan flufenamide rapidly disappeared from plasma after infusion, while melphalan typically peaked a few minutes after the end of infusion. This suggests that melphalan flufenamide is rapidly and widely distributed to extravasal tissues, in which melphalan is formed and thereafter redistributed to plasma.[19]
This rapid disappearance from plasma is likely due to hydrolytic enzymes.[20] The Zn(2+) dependent ectopeptidase (also known as alanine aminopeptidase), degrades proteins and peptides with a N-terminal neutral amino acid. Aminopeptidase N is frequently overexpressed in tumors and has been associated with the growth of different human cancers suggesting it as a suitable target for anti-cancerous therapy.[21]
Adverse effects
In a human Phase 1 trial, no dose-limiting toxicities (DLTs) were observed at lower doses. At doses above 50 mg, reversible neutropenias and thrombocytopenias were observed, and particularly evident in heavily pretreated patients.[22] These side-effects are shared by most chemotherapies, including alkylating agents in general.
Drug interactions
No drug interaction studies have been reported. Several in vitro studies indicate that melphalan flufenamide may be successfully combined with standard chemotherapy or targeted agents.[23][24]
Therapeutic efficacy
In a Phase 1/2 trial, in solid tumor patients refractory to standard therapy, response evaluation showed disease stabilization in a majority of patients.[25][26] In relapsed and refractory multiple-myeloma (RRMM) patients, promising activity was seen in heavily pre-treated RRMM patients where conventional therapies had failed; the median Progression-Free Survival was 9.4 months and the Duration of Response was 9.6 months.[27] An overall response rate of 41% and a clinical benefit rate of 56% were also shown, with similar results seen across patient populations regardless of their refractory status. Hematologic toxicity was common, but manageable with cycle prolongations, dose modifications and supportive therapy, and non-hematologic treatment-related adverse events were infrequent.
History
Efficacy was evaluated in HORIZON (NCT02963493), a multicenter, single-arm trial.[3] Eligible patients were required to have relapsed refractory multiple myeloma.[3] Patients received melphalan flufenamide 40 mg intravenously on day 1 and dexamethasone 40 mg orally (20 mg for patients ≥75 years of age) on day 1, 8, 15 and 22 of each 28-day cycle until disease progression or unacceptable toxicity.[3] Efficacy was evaluated in a subpopulation of 97 patients who received four or more prior lines of therapy and were refractory to at least one proteasome inhibitor, one immunomodulatory agent, and a CD38-directed antibody.[3]
The application for melphalan flufenamide was granted priority review and orphan drug designations.[3]
Society and culture
Names
Melphalan flufenamide is the International nonproprietary name (INN).[28]
PAPER
Organic Process Research & Development (2019), 23(6), 1191-1196.
https://pubs.acs.org/doi/pdf/10.1021/bk-2020-1369.ch005
Ethyl (2S)-2-[(2S)-2-amino-3-[bis-(2-chloroethyl)amino]phenyl]propaneamido]-3-(4-fluorophenyl)propanoate hydrochloride, (melphalan flufenamide or Melflufen), is an alkylating agent intended for the treatment of multiple myeloma. Initially only milligram quantities were synthesized, following a route starting from pharmaceutical-grade melphalan. Along with the pharmaceutical development, adjustments were made to the original medicinal chemistry route. This resulted in material for early clinical trials, but it became obvious that further development was necessary. Development resulted in a route in which two phenyl alanine derivatives were coupled to give a dipeptide. This intermediate was further manipulated to give an aniline which could be converted into the desired compound melflufen. The aniline derivative was converted to the corresponding N,N–bis-chloroethylaniline using chloroacetic acid and borane. Deprotection and conversion to the hydrochloride gave melflufen in good yield and excellent purity. Production was performed without chromatography at multi-kilogram scale to supply the API for Phase III studies and commercial validation batches.
PAPER
Antineoplastics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Melphalan
Melphalan, l-3-[p-[bis-(2-chloroethyl)amino]phenyl]alanine (30.2.1.13), is a structural analog of chlorambucil in which the butyric acid fragment is replaced with an aminoacid fragment, alanine. This drug is synthesized from l-phenylalanine, the nitration of which with nitric acid gives 4-nitro-l-phenylalanine (30.2.1.8). Reacting this with an ethanol in the presence of hydrogen chloride gives the hydrochloride of 4-nitro-l-phenylalanine ethyl ester (30.2.1.9), the amino group of which is protected by changing it to phthalamide by a reaction with succinic anhydride to give 30.2.1.10. The nitro group in this molecule is reduced to an amino group using palladium on calcium carbonate as a catalyst. The resulting aromatic amine (30.2.1.11) is then reacted with ethylene oxide, which forms a bis-(2-hydroxyethyl)-amino derivative (30.2.1.12). The hydroxy groups in this molecule are replaced with chlorine atoms upon reaction with thionyl chloride, after which treatment with hydrochloric acid removes the phthalamide protection, giving melphalan (30.2.13) [47–50].

Melaphalan is used intravenously and orally to treat multiple myeloma and cancers of the breast, neck, and ovaries. A synonym of this drug is alkeran.
The racemic form of this drug, d,l-3-[p-[bis-(2-chloroethyl)amino]phenyl]alanine, is also widely used under the name sarcolysine or racemelfalan.
PATENT WO 2001096367PAPEROncology Research (2003), 14(3), 113-132PATENTWO 2016180740https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016180740
Alkylating agents, such as drugs derived from nitrogen mustard, that is bis(2-chloroethyl)amine derivatives, are used as chemotherapeutic drugs in the treatment of a wide variety of cancers. Melphalan, or p-bis-(2-chloroethyl)-amino-L-phenylalanine (compound (Id), CAS No. 148-82-3), is an alkylating agent which is a conjugate of nitrogen mustard and the amino acid phenylalanine (US 3,032,584). Melphalan is used clinically in the treatment of metastatic melanomas, but has limited efficacy, dose-limiting toxicities and resistance can develop.
Melphalan flufenamide ethyl ester (L-melphalanyl-L-p-fluorophenylalanine ethyl ester, melflufen, compound (Ib)) is a derivative of melphalan conjugated to the amino acid phenylalanine, creating a dipeptide (WO 01/96367):
The monohydrochloride salt of melflufen (L-melphalanyl-L-p-fluorophenylalanine ethyl ester monohydrochloride; hydrochloride salt of (Ib); CAS No. 380449-54-7) is referred to as melflufen hydrochloride.
When studied in cultures of human tumor cells representing approximately 20 different diagnoses of human cancers, including myeloma, melflufen showed 50- to 100-fold higher potency compared with that of melphalan (http://www.oncopeptides.se/products/melflufen/ accessed 26 March 2015). Data disclosed in Arghya, et al, abstract 2086 “A Novel Alkylating Agent Melphalan Flufenamide Ethyl Ester Induces an Irreversible DNA Damage in Multiple Myeloma Cells” (2014) 5th ASH Annual Meeting and Exposition, suggest that melflufen triggers a rapid, robust and irreversible DNA damage, which may account for its ability to overcome melphalan-resistance in multiple myeloma cells. Melflufen is currently undergoing phase I/IIa clinical trials in multiple myeloma.
A process for preparing melflufen in hydrochloride salt form is described in WO 01/96367, and is illustrated in Scheme 1, below. In that process N-tert-butoxycarbonyl-L-melphalan is reacted with p-fluorophenylalanine ethyl ester to give N-tert-butoxycarbonyl-L-melphalanyl-L-p-fluorophenylalanine ethyl ester. After purification by gradient column chromatography the yield of that step is 43%.
Scheme 1. Current route to melflufen (in hydrochloride salt form)
As shown in Scheme 1, the known process for preparing melflufen (in hydrochloride salt form) uses the cytotoxic agent melphalan as a starting material, and melflufen is synthesised in a multistep sequence. Melphalan is highly toxic, thus the staring materials and all of the intermediates, and also the waste stream generated, are extremely toxic. That is a major disadvantage in terms of safety, environmental impact and cost when using the process on a large scale. Therefore, an improved and safer method is highly desired, especially for production of melflufen on a large scale. Further, the purity of commercially available melphalan is poor due to its poor stability, the yield in each step of the process is poor, and purity of the final product made by the known process is not high.
A process for preparing melphalan is described in WO 2014/141294. In WO 2014/141294 the step to introduce the bis(2-chloroethyl) group into the molecule comprises conversion of a primary phenyl amine to a tertiary phenyl amine diol, by reaction with ethylene oxide gas. This gives a 52.6% yield. The amine diol is then converted to a bis(2-chloroethyl) phenylamine by reaction with phosphoryl chloride. Using ethylene oxide, or chloroethanol, to convert an aromatic amine to the corresponding bis-(2-hydroxy ethyl) amine, followed by
chlorination of that intermediate, is a common technique for producing aromatic bis-(2-chloroethyl) amines. It is also known to start from a chloroarene and let it undergo a SNAr-reaction with diethanolamine. The present inventors have applied those methods to produce melflufen (in its salt form), shown in Scheme 2 below.
Scheme 2. Alternative pathways to melflufen
The inventors have found that using ethylene oxide in THF (route (a) of Scheme 2), no alkylation occurs at 55 °C; increasing the temperature to 60 °C lead to the dialkylated intermediate being formed, but the reaction was very slow. To increase yield and reaction rate the reaction would require high temperatures, but this would cause increased pressure so that the reaction would need be performed in a pressure reactor. Such conditions are likely lead to formation of side products. Similar reaction conditions but using a 50:50 mixture of ethylene oxide and acetic acid (route (b) of Scheme 2) lead to faster reaction times but formation of side products. Using potassium carbonate and chloroethanol (route (c) of Scheme 2) also lead to formation of side product, possibly due to the chloroethanol undergoing partial trans-esterification with the ethyl ester.
The inventors also attempted chlorination of the di-alkylated compound. Chlorination of the bis-(2-hydroxyethyl) compound (4) of Scheme 2 using thionyl chloride in dichloromethane led to significant de-protected side product formation. Chlorination of the bis-(2-hydroxyethyl) compound (4) of Scheme 2 using POCl3 required high temperature and long
reaction times. In addition, both thionyl chloride and POCl3 are challenging to handle at large scale due to safety concerns. The inventors also converted the bis-(2-hydroxyethyl) compound (4) of Scheme 2 to the corresponding dimesylate by treatment with methanesulfonyl chloride and triethylamine. The dimesylate was treated then with sodium chloride in DMF at 120 °C. However, the crude product of this reaction contained significant side products making this route unsuitable to be used economically at scale.
In summary, none of these routes were found to be suitable for large scale production of high purity melflufen. They do not work well for the synthesis of melflufen, resulting in poor yields and are inefficient. Further, the routes shown in Scheme 2 require multiple steps to form the N, N-bis-chloroethyl amine and use toxic reagents.
Example 1 – Synthesis of compound (VIc)
To a reactor with overhead stirring, equipped with nitrogen inlet and reflux condenser, was charged Boc-nitrophenylalanine (compound (IVc)) (35.0 g, 112.8 mmol, 1 eq.), followed by acetone (420 mL), N-methylmorpholine (43.4 mL, 394.8 mmol, 3.5 eq.), fluoro-L-phenylalanine ethyl ester hydrochloride (compound (V)) (28.5 g, 115 mmol, 1.02 eq.), EDC (23.8 g, 124.1 mmol, 1.1 eq.) and HOBt·H2O (1.7 g, 11.3 mmol, 0.1 eq.). The slurry was stirred at room temperature for 18.5 h which led to full consumption of compound (IVc) according to HPLC. Water (180 mL) and 2-MeTHF (965 mL) were charged. Approximately 640 g solvent was then removed by evaporation (TJ: 35 °C) from the clear two phase orange mixture. 360 mL 2-MeTHF was then added and evaporated off twice. The water phase was acidified to pH 3 via addition of 58 mL 2 M sulfuric acid. The organic layer was heated to 35-40 °C and was then sequentially washed with water (90 mL), twice with saturated aqueous NaHCO3 solution (90 mL) and then brine (90 mL) and finally water (90 mL). To the 2-MeTHF dissolved product was added heptane (270 mL) drop wise at 35-40 °C before the mixture was allowed to reach room temperature overnight with stirring. Another 135 mL heptane was added drop wise before the beige slurry was cooled to 10 °C. The product was isolated and was rinsed with 100 mL cold 2-MeTHF/heptane 6/4. Product compound (VIc) was stored moist (82.5 g). A small sample of the product was analyzed by limit of detection (LOD) which revealed the solid to contain 43.8% solvent residues. Based on this, the purified product was obtained in a yield of 82 %. The purity was determined by HPLC to be: 99.4 area%.
1 H-NMR (300 MHz, DMSO-D6) δ 8.48 (broad d, 1H, J=7.5 Hz), 8.16 (2H, d, J=8.7 Hz), 7.55 (2H, d, J=9 Hz), 7.28 (2H, dd, J=8,7, 8.1 Hz), 7.12-7.02 (3H, m), 4.49 (1H, dd, J=14.4, 7.2 Hz), 4.32-4.24 (1 H, m), 4.04 (2H, dd, J=14.4, 7.2 Hz), 3.08-2.95 (3H,m), 2.84 (1H, dd, J=13.2, 10.8 Hz), 1.27 (s, 9H), 1.11 (3H, t, J=7.2Hz)
13C-NMR (75 MHz, DMSO-D6) δ 171.4 (C=O), 171.2 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.2 (C=O), 146.6 (C), 146.2 (C), 133.1 (C), 131.1 (2 carbon, CH, d, J=8.3 Hz), 130.6 (2 carbon, CH), 123.1 (C), 114.9 (2 carbon, CH, J=20.4 Hz), 78.1 (C), 60.6 (CH2), 55.1 (CH), 53.6 (CH), 37.3 (CH2), 35.9 (CH2), 28.0 (3 carbons, CH3), 14.0 (CH3)
Example 2 – Synthesis of compound (IIc)
To a hydrogenation autoclave was added wet solid product compound (VIc) (approximately 4.9 g dry weight, 9.7 mmol, 1 eq.), 2-MeTHF (75 mL) and 3 w/w% of a 5% Pd/C-catalyst (147 mg, 50% moist). The reaction mixture was degased with nitrogen and then 1 barg hydrogen gas was charged. Stirring was set to 600 rpm and TJ to 36 °C. The reaction was completed in four hours, The hydrogenation autoclave was rinsed with 10 mL 2-MeTHF and the rinsing portion was added to the reaction solution in the E-flask. Charcoal (250 mg, 5 wt%) was then added and the resulting mixture was stirred for 15 minutes at room temperature before it was filtered. The filter was rinsed with 10 mL 2-MeTHF and the rinsing portion was added to the filter. The light yellow/pink filtrate contained white precipitated product. The slurry was heated to approximately 40 °C to dissolve the solid before heptane (42 mL) was added drop wise during one hour. The heating was turned off and the mixture was allowed to reach room temperature with overnight stirring. Additional 21 mL heptane was the added before the mixture was cooled to approximately 7 °C (ice/water bath). The solid was isolated and was washed through with 10 mL cold 2-MeTHF/heptane 6/4. The moist solid (5.7 g) was vacuum dried at 35 °C overnight which gave a dry weight of
compound (IIc) of 4.2 g which corresponds to a yield of 91 %. The purity was determined by HPLC to be 99.1 area%.
1H-NMR (300 MHz, DMSO-D6) δ 8.26 (1H, d, J=7.5Hz), 7.26 (dd, 2H, J=8.1, 5.7 Hz), 7.09 (2H, t, J=8.7 Hz), 6.86 (2H, d, J=8.1 Hz), 6.71 (1H, d, J=8.7 Hz), 6.45 (1H, d, J=8.1 Hz), 4.87 (2H, s), 4.45 (1H, dd, J=14.4, 7.5 Hz), 4.07-4.00 (3H, m), 3.06-2.91 (2H, m), 2.71 (1H, dd, J=13.8, 3.9 Hz), 2.54-2.46 (1H, m), 1.31 (s, 9H), 1.11 (3H, t, J=6.9 Hz).
13C-NMR (75 MHz, DMSO-D6) δ 171.4 (C=O), 171.2 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.1 (C=O), 146.9 (C), 133.2 (C, d, J=3.0 Hz), 131.1 (2 carbon, CH, d, J=8.3 Hz), 129.5 (2 carbon, CH), 124.8 (C), 114.8 (2 carbon, CH, J=21.1 Hz), 113.6 (2 carbon, CH), 77.9 (C), 60.5 (CH2), 56.0 (CH), 53.5 (CH), 36.7 (CH2), 35.9 (CH2), 28.1 (3 carbons, CH3), 13.9 (CH3)
The present inventors have repeated Example 2 several times using crude compound (VIc) or recrystallised compound (VIc) (purity: 99.1 area%) as starting material and varying various reaction conditions, e.g. pressure of H2, w/w% of Pd/C, solvent and temperature. The crude purity (97.2 area%) was a slightly higher when recrystallized compound (VIc) was used as starting material than when using crude compound (VIc), in which case the crude purity is generally 95-96 area%. Final yield and purity is also slightly higher than when starting from crude compound (VIc) (98-98.5 area%).
The present inventors have also repeated Example 2 several times varying the Pd/C w/w%, temperature, pressure of H2 and concentration using 2-MeTHF as the solvent. A high conversion of Compound (VIc) (>99.5 area%) was achieved for Pd/C w/w% from 3 to 6 bar; temperature ranges from 30 to 40 °C, H2 pressure from 1 to 6 barg, and for varying reaction concentrations. The resulting crude purity was similar in all attempts (95.3-96.2 area%), as was the purity of the isolated product after crystallization from 2-MeTHF/heptane (98.0-98.5 area%).
Example 3 – Preparation of compound (IIIc)
(i) carried out using BH3SMe2 in the presence of chloroacetic acid salt
In a 0.5 L dried reactor with overhead stirrer, compound (IIc) (6.99 g, 14.76 mmol) was added, followed by anhydrous tetrahydrofuran (46 mL), chloroacetic acid (36.3 g, 383.8 mmol), chloroacetic acid sodium salt (17.2 g, 147.6 mmol) at TI=5-13°C. A solution of
BH3SMe2 (14.6 g, 191.9 mmol, 18.2 mL) was then added over 45 minutes. After the addition, the reaction temperature was adjusted to TI=25-30°C and kept for 2 hr after reaching this temperature. The reaction was slowly quenched with ethanol (17.7 g, 383.8 mmol, 22.4 mL) and was stirred overnight at TJ=5°C and then slowly diluted with distilled water (138 mL) to precipitate the product, compound (IIIc). The temperature was adjusted to TI=15°C and the stirring rate was increased before addition of a solution of aqueous K2CO3 (8.0 M, 27 mL) to pH = 7.0-7.5. The reaction slurry was collected on a filter and reaction vessel and filter-cake were washed with water (2×40 mL). The filter-cake was re-slurred in water (200 mL) for 1 hr at TJ=20°C and then filtered again. Washing with water (50 mL), followed by drying at TJ=35°C under high vacuum, produced the crude white product, compound (IIIc), in 7.85 g (88.8%) uncorrected yield. HPLC purity 97.5 area %.
Crude compound (IIIc) (7.5 gram) prepared according to the described procedure was charged to a reactor and washed down with 2-MeTHF (80 mL). Heating at TJ=50°C dissolved the substance. Heptane (80 mL) was added with stirring at TI=45-50°C and then stirred before adjusting the temperature to TJ=10°C. The precipitated solid was collected by filtration and dried at TJ=35°C under high vacuum which produced white product, compound (IIIc), in 6.86 g (91.5%). HPLC purity 99.1 area %.
1H-NMR (300 MHz, DMSO-D6) δ 8.30 (1H, d, J=7.8 Hz), 7.26 (2H, dd, J=8.1, 6 Hz), 7.09-7.05 (3H, m), 6.79 (1H, d, J=8.9 Hz), 6.63 (2H, d, J=8.4 Hz), 4.49-4.42 (1H, dd, J=14.7, 7.5 Hz), 4.07-3.99 (3H, m), 3.68 (8H, s), 3.06-2.91 (2H, m), 2.76 (1H, dd, J=13.8, 4.2 Hz), 2.56 (1H, m), 1.29 (9H, s), 1.1 (3H, t, J=6.6 Hz)
13C-NMR (75 MHz, DMSO-D6) δ 172.1 (C=O), 171.3 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.2 (C=O), 144.7 (C), 133.2 (C, d, J=3.0 Hz), 131.1 (2 carbon, CH, d, J=7.5 Hz), 130.2 (2 carbon, CH), 126.1 (C), 114.9 (2 carbon, CH, J=21.1 Hz), 111.6 (2 carbon, CH), 78.0 (C), 60.6 (CH2), 55.9 (CH), 53.5 (CH), 52.2 (CH2), 41.2 (CH2), 36.4 (CH2), 35.9 (CH2), 28.1 (3 carbons, CH3), 14.0 (CH3)
(ii) Carried out using BH3SMe2 in the presence of chloroacetic acid salt
In a 0.5 L dried reactor with overhead stirrer, compound (IIe) (7.5 g, 15.84 mmol) was added, followed by 2-MeTHF (150 mL). The mixture was heated to 45 °C to form a clear solution. The solution was cooled to 4 °C and chloroacetic acid (38.9 g, 411.8 mmol), followed by chloroacetic acid sodium salt (18.4 g, 158.4 mmol) was added at TI=5-13°C. A solution of BH3SMe2 (15.6 g, 205.9 mmol, 19.5 mL) was then added over 90 minutes. After the addition, the reaction temperature was adjusted to TI=20-25°C and kept for 5 hr after reaching this temperature. The reaction was slowly quenched with water at TI=15-25 °C (150 g, 8333 mmol, 150 mL), pH=3.5 in water phase, and left overnight without stirring at TI=6 °C.
Product, compound (IIIc), had precipitated out in the organic phase and the temperature was adjusted to TI=35 °C while stirring, and two clear phases formed. The phases were allowed to separate and the water phase was removed. The organic phase was washed three times with 20% NaCl(aq). pH in the three water phases were: 1.7, 1.1, and 1.1. After the removal of the third water phase, the organic phase was transferred to a round bottom flask and concentrated to half its volume on an evaporator. Product, compound (IIIc), started to precipitate out and the product slurry was allowed to mature at 6 °C for 19 hr. The slurry was collected on a filter and round bottom flask and filter-cake were washed with 2-MeTHF:n-heptane (2×40 mL), followed by drying at TJ=35 °C under high vacuum, to produce the crude white product, compound (IIIc), in 8.3 g (87.6%) uncorrected yield. HPLC purity 99.4 area % .
(iii) Carried out using borane-tetrahydrofuran in the presence of chloroacetic acid salt
In a 100 mL dried round bottom flask with magnet stirrer bar, compound (IIc) (0.75 g, 1.58 mmol) was added under a slow nitrogen flow followed by anhydrous tetrahydrofuran (6 mL), chloroacetic acid (3.89 g, 41.2 mmol), and chloroacetic acid sodium salt (1.84 g, 15.8 mmol). At TI=5-13°C °C a 1 M solution of BH3THF (20.6 mmol, 20.6 mL) was added over 30
minutes. After the addition the reaction temperature was adjusted between TI=23-28 °C and kept for 2 hr after reaching this temperature. In process control sample (HPLC) indicated in-complete reaction and the jacket temperature was set to TJ=40°C and when the internal temperature reached TI=40°C the reaction was kept at this temperature for 2 hr when in-process sample (HPLC) showed 6.7 area% starting material, 7.1% acylation adduct
(impurity) and 84.1% compound (IIIc). The reaction was progressed at TI=23°C and left for 4 days before slowly quenched with ethanol (2.4 g, 3 mL). Water (100 mL) was added and the pH adjusted with 1 M aqueous K2CO3 to pH 7. The reaction slurry was collected on a filter and reaction vessel and filter-cake were washed with water (2×20 mL) followed by drying at TJ=35°C under high vacuum produced the crude colorless product in 0.85 g (89.6%) uncorrected yield. HPLC purity was 94.3 area %, with one major impurity attributed to a chloroacylation adduct of the starting material in 3.8 area %.
(iv) Carried out using BH3SMe2 without addition of chloroacetic acid salt
In a 100 mL dried round bottom flask with magnet stirrer bar, compound (IIc) (0.75 gram, 1.58 mmol) was added under a slow nitrogen flow followed by anhydrous tetrahydrofuran (6 mL) and chloroacetic acid (3.89 g, 41.2 mmol). At TI=5-16°C a solution of BH3SMe2 (1.56 g, 20.6 mmol, 2.0 mL) was added over 30. After the addition the reaction temperature was adjusted between TI=25°C and kept for 2.5 h after reaching this temperature. A process control sample (HPLC) indicated melflufen (Compound (Ib)), the Boc-deprotected form of Compound (IIIc), in 66 area %. The reaction was slowly quenched with ethanol (2.9 g, 3.7 mL). The pH of the reaction was adjusted with 1 M aqueous K2CO3 solution to pH=8, followed by addition of EtOAc (40 mL). Layers were separated and the aqueous layer re-extracted with EtOAc (50 mL). The organic layers were combined and reduced at <30 mbar / 35°C to an oil. The oil was re-distilled from EtOAc (30 mL) twice and the residue was dried at TJ=23°C / 5 mbar to leave 1.6 g brownish oil. HPLC purity of Compound (Ib) was 66.1 area %.
Example 4 – Preparation of compound (Ib) as hydrochloride salt
Boc-melflufen (compound (IIIc)) (5.0 g, 8.3 mmol) was charged to a round bottomed flask, equipped with magnet stirrer bar, and nitrogen inlet. 1.3 M HCl (anhydrous) in ethanol (64 mL, 83.5 mmol, 10 eq.) was added. After 19 h the conversion was 99.4%. The solvents were partially distilled at TJ=33°C on a rotary evaporator, followed by the addition of ethanol (18 mL). This was repeated twice. Seed crystals were added and after 30 minutes product had precipitated. The slurry was stirred for 21 h and was then concentrated. Methyl tert-butyl ether (MTBE) (108 mL) was added at room temperature with an even rate over 30 minutes. After 100 minutes of stirring at room temperature the precipitate was collected by vacuum filtration and washed with 2×25 mL ethanol: MTBE (1:6). Drying was performed overnight at TJ=35°C / 5 mbar in vacuum oven. Yield of compound (Ib) in the form of its hydrochloride salt, 4.0 g (90%). HPLC-purity 98.7 area%.
1H-NMR (300 MHz, MeOH-D4) δ 7.26 (2H, dd, J=8.4, 8.1 Hz), 7.17 (2H, d, J=8.4 Hz), 7.02 (2H, dd, J=9, 8.4 Hz), 6.74 (2H, d, J=8.4 Hz), 4.69 (1H, dd, J=7.8, 6.3 Hz), 4.15 (2H, dd, J=14.1, 7.2 Hz), 4.04 (1H, dd, J=8.4, 5.4 Hz), 3.76 (4H, dd, J=6.3, 6 Hz), 3.67 (4H, dd, 6.6, 5.7 Hz), 3.17 (2H, dd, J=14.4, 6 Hz), 3.06-2.88 (2H, m), 1.22 (3H, t, J=7.2 Hz)
13C-NMR (75 MHz, MeOH-D4) δ 172.2 (C=O), 169.8 (C=O), 163.4 (C-F, d, J=244.5 Hz), 147.4 (C), 133.9 (C, d, J=3 Hz), 132.1 (2 carbon, CH, d, J=7.5 Hz), 131.8 (2 carbon, CH), 123.4 (C), 116.2 (2 carbon, CH, d, J=21.9 Hz), 113.7 (2 carbon, CH), 62.6 (CH2), 55.6 (CH), 55.5 (CH), 54.3 (CH2), 41.6 (CH2), 37.6 (CH2), 37.6 (CH2), 14.5 (CH3)
Example 4 was repeated successfully in the presence ethyl acetate and with varying concentrations of HCl from 1.3 M to 2.5 M and at varying temperatures from 6 °C to room temperature.PAPERhttps://pubs.acs.org/doi/10.1021/acs.oprd.9b00116 Organic Process Research & Development (2019), 23(6), 1191-1196.Melflufen is a novel cytostatic currently in phase III clinical trials for treatment of multiple myeloma. Development of a process suitable for production is described. The two key features of the novel method are late introduction of the alkylating pharmacophore and an improved method for formation of the bis-chloroethyl group.


1H NMR spectrum of L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride (1) (in D4–MeOH).

13C NMR spectrum of L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride (1) (in D4–MeOH).

References
- ^ Berglund, Åke; Ullén, Anders; Lisyanskaya, Alla; Orlov, Sergey; Hagberg, Hans; Tholander, Bengt; Lewensohn, Rolf; Nygren, Peter; Spira, Jack; Harmenberg, Johan; Jerling, Markus; Alvfors, Carina; Ringbom, Magnus; Nordström, Eva; Söderlind, Karin; Gullbo, Joachim (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Strese, Sara; Wickström, Malin; Fuchs, Peder Fredlund; Fryknäs, Mårten; Gerwins, Pär; Dale, Tim; Larsson, Rolf; Gullbo, Joachim (2013). “The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo”. Biochemical Pharmacology. 86(7): 888–95. doi:10.1016/j.bcp.2013.07.026. PMID 23933387.
- ^ Jump up to:a b c d e f g h i “FDA grants accelerated approval to melphalan flufenamide for relapsed”. U.S. Food and Drug Administration(FDA). 26 February 2021. Retrieved 1 March 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “FDA Approves Oncopeptides’ Pepaxto (melphalan flufenamide) for Patients with Triple-Class Refractory Multiple Myeloma” (Press release). Oncopeptides AB. 1 March 2021. Retrieved 1 March 2021 – via PR Newswire.
- ^ “Pepaxto: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 1 March 2021.
- ^ Gullbo, J; Tullberg, M; Våbenø, J; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R; Luthman, K (2003). “Structure-activity relationship for alkylating dipeptide nitrogen mustard derivatives”. Oncology Research. 14 (3): 113–32. doi:10.3727/000000003771013071. PMID 14760861.
- ^ Wickstrom, M.; Lovborg, H.; Gullbo, J. (2006). “Future Prospects for Old Chemotherapeutic Drugs in the Target-Specific Era; Pharmaceutics, Combinations, Co-Drugs and Prodrugs with Melphalan as an Example”. Letters in Drug Design & Discovery. 3(10): 695. doi:10.2174/157018006778631893.
- ^ Gullbo, J; Dhar, S; Luthman, K; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R (2003). “Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): Comparison with melphalan”. Anti-Cancer Drugs. 14 (8): 617–24. doi:10.1097/00001813-200309000-00006. PMID 14501383. S2CID 10282399.
- ^ Berglund, Åke; Ullén, Anders; Lisyanskaya, Alla; Orlov, Sergey; Hagberg, Hans; Tholander, Bengt; Lewensohn, Rolf; Nygren, Peter; Spira, Jack; Harmenberg, Johan; Jerling, Markus; Alvfors, Carina; Ringbom, Magnus; Nordström, Eva; Söderlind, Karin; Gullbo, Joachim (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Strese, Sara; Wickström, Malin; Fuchs, Peder Fredlund; Fryknäs, Mårten; Gerwins, Pär; Dale, Tim; Larsson, Rolf; Gullbo, Joachim (2013). “The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo”. Biochemical Pharmacology. 86(7): 888–95. doi:10.1016/j.bcp.2013.07.026. PMID 23933387.
- ^ Wickström, M; Johnsen, J. I.; Ponthan, F; Segerström, L; Sveinbjörnsson, B; Lindskog, M; Lövborg, H; Viktorsson, K; Lewensohn, R; Kogner, P; Larsson, R; Gullbo, J (2007). “The novel melphalan prodrug J1 inhibits neuroblastoma growth in vitro and in vivo”. Molecular Cancer Therapeutics. 6 (9): 2409–17. doi:10.1158/1535-7163.MCT-07-0156. PMID 17876040.
- ^ Gullbo, J; Lindhagen, E; Bashir-Hassan, S; Tullberg, M; Ehrsson, H; Lewensohn, R; Nygren, P; de la Torre, M; Luthman, K; Larsson, R (2004). “Antitumor efficacy and acute toxicity of the novel dipeptide melphalanyl-p-L-fluorophenylalanine ethyl ester (J1) in vivo”. Investigational New Drugs. 22 (4): 411–20. doi:10.1023/B:DRUG.0000036683.10945.bb. PMID 15292711. S2CID 31613292.
- ^ Gullbo, J; Wickström, M; Tullberg, M; Ehrsson, H; Lewensohn, R; Nygren, P; Luthman, K; Larsson, R (2003). “Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1”. Journal of Drug Targeting. 11(6): 355–63. doi:10.1080/10611860310001647140. PMID 14668056. S2CID 25203458.
- ^ Gullbo, J; Dhar, S; Luthman, K; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R (2003). “Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): Comparison with melphalan”. Anti-Cancer Drugs. 14 (8): 617–24. doi:10.1097/00001813-200309000-00006. PMID 14501383. S2CID 10282399.
- ^ Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K. C. (2013). “In Vitro and in Vivo Antitumor Activity of a Novel Alkylating Agent, Melphalan-Flufenamide, against Multiple Myeloma Cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Viktorsson, K; Shah, C. H.; Juntti, T; Hååg, P; Zielinska-Chomej, K; Sierakowiak, A; Holmsten, K; Tu, J; Spira, J; Kanter, L; Lewensohn, R; Ullén, A (2016). “Melphalan-flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma”. Molecular Oncology. 10 (5): 719–34. doi:10.1016/j.molonc.2015.12.013. PMC 5423156. PMID 26827254.
- ^ Chauhan, D; Ray, A; Viktorsson, K; Spira, J; Paba-Prada, C; Munshi, N; Richardson, P; Lewensohn, R; Anderson, K. C. (2013). “In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Chesi, M; Matthews, G. M.; Garbitt, V. M.; Palmer, S. E.; Shortt, J; Lefebure, M; Stewart, A. K.; Johnstone, R. W.; Bergsagel, P. L. (2012). “Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy”. Blood. 120 (2): 376–85. doi:10.1182/blood-2012-02-412783. PMC 3398763. PMID 22451422.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Wickström, M; Viktorsson, K; Lundholm, L; Aesoy, R; Nygren, H; Sooman, L; Fryknäs, M; Vogel, L. K.; Lewensohn, R; Larsson, R; Gullbo, J (2010). “The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan”. Biochemical Pharmacology. 79 (9): 1281–90. doi:10.1016/j.bcp.2009.12.022. PMID 20067771.
- ^ Wickström, M; Larsson, R; Nygren, P; Gullbo, J (2011). “Aminopeptidase N (CD13) as a target for cancer chemotherapy”. Cancer Science. 102 (3): 501–8. doi:10.1111/j.1349-7006.2010.01826.x. PMC 7188354. PMID 21205077.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Wickström, M; Haglund, C; Lindman, H; Nygren, P; Larsson, R; Gullbo, J (2008). “The novel alkylating prodrug J1: Diagnosis directed activity profile ex vivo and combination analyses in vitro”. Investigational New Drugs. 26 (3): 195–204. doi:10.1007/s10637-007-9092-1. PMID 17922077. S2CID 19915448.
- ^ Chauhan, D; Ray, A; Viktorsson, K; Spira, J; Paba-Prada, C; Munshi, N; Richardson, P; Lewensohn, R; Anderson, K. C. (2013). “In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Viktorsson, K; Shah, C. H.; Juntti, T; Hååg, P; Zielinska-Chomej, K; Sierakowiak, A; Holmsten, K; Tu, J; Spira, J; Kanter, L; Lewensohn, R; Ullén, A (2016). “Melphalan-flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma”. Molecular Oncology. 10 (5): 719–34. doi:10.1016/j.molonc.2015.12.013. PMC 5423156. PMID 26827254.
- ^ https://ash.confex.com/ash/2015/webprogram/Paper85666.html
- ^ World Health Organization (2012). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 67”. WHO Drug Information. 26 (1): 72. hdl:10665/109416.
External links
- “Melphalan flufenamide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02963493 for “A Study of Melphalan Flufenamide (Melflufen) in Combination With Dexamethasone in Relapsed Refractory Multiple Myeloma Patients (HORIZON)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pepaxto |
| Other names | Melflufen, 4-[Bis-(2-chloroethyl)amino]-L-phenylalanine-4-fluoro-L-phenylalanine ethyl ester, J1[1][2] |
| License data | US DailyMed: Melphalan_flufenamide |
| Legal status | |
| Legal status | US: ℞-only [3] |
| Pharmacokinetic data | |
| Metabolism | Aminopeptidase hydrolysis, Spontaneous hydrolyisis on N-mustard |
| Elimination half-life | 10 min in vitro[medical citation needed] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 380449-51-4 |
| PubChem CID | 9935639 |
| DrugBank | DB16627 |
| ChemSpider | 8111267 |
| UNII | F70C5K4786 |
| ChEMBL | ChEMBL4303060 |
| Chemical and physical data | |
| Formula | C24H30Cl2FN3O3 |
| Molar mass | 498.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCCOC(=O)[C@H](CC1=CC=C(C=C1)F)NC(=O)[C@H](CC2=CC=C(C=C2)N(CCCl)CCCl)N | |
| hideInChIInChI=1S/C24H30Cl2FN3O3/c1-2-33-24(32)22(16-18-3-7-19(27)8-4-18)29-23(31)21(28)15-17-5-9-20(10-6-17)30(13-11-25)14-12-26/h3-10,21-22H,2,11-16,28H2,1H3,(H,29,31)/t21-,22-/m0/s1Key:YQZNKYXGZSVEHI-VXKWHMMOSA-N |
//////////Melphalan flufenamide hydrochloride, Melphalan flufenamide, FDA 2021, APPROVALS 2021, PEPAXTO, メルファランフルフェナミド塩酸塩 , J 1
#Melphalan flufenamide hydrochloride, #Melphalan flufenamide, #FDA 2021, #APPROVALS 2021, #PEPAXTO, メルファランフルフェナミド塩酸塩 , #J 1
Evinacumab
(Heavy chain)
EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA ISGDGGSTYY
ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL RNTIFGVVIP DAFDIWGQGT
MVTVSSASTK GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF
LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE
QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS
QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK
SRWQEGNVFS CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK ASSLESGVPS
RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H140-L214, H153-H209, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’140-L’214, H’153-H’209, H’267-H’327, H’373-H’431, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Evinacumab
エビナクマブ (遺伝子組換え)
Immunoglobulin G4, anti-(human protein ANGPTL3 (angiopoietin-like 3)) (human monoclonal REGN1500 heavy chain), disulfide with human monoclonal REGN1500 light chain, dimer
| Formula | C6480H9992N1716O2042S46 |
|---|---|
| CAS | 1446419-85-7 |
| Mol weight | 146081.9345 |
Protein Sequence
Sequence Length: 1334, 453, 453, 214, 214multichain; modified (modifications unspecified)
FDA APPROVED, 2021/2/11, EVKEEZA
Antihyperlipidemic, Anti-angiopietin like 3
Monoclonal antibody
Treatment of dyslipidemia
- REGN 1500
- REGN-1500
- REGN1500
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-22 – Cys-96 | disulfide bridge |
| bridge | Cys-140 – Cys-214” | disulfide bridge |
| bridge | Cys-153 – Cys-209 | disulfide bridge |
| bridge | Cys-232 – Cys-232′ | disulfide bridge |
| bridge | Cys-235 – Cys-235′ | disulfide bridge |
| bridge | Cys-267 – Cys-327 | disulfide bridge |
| bridge | Cys-373 – Cys-431 | disulfide bridge |
| bridge | Cys-22′ – Cys-96′ | disulfide bridge |
| bridge | Cys-140′ – Cys-214”’ | disulfide bridge |
| bridge | Cys-153′ – Cys-209′ | disulfide bridge |
| bridge | Cys-267′ – Cys-327′ | disulfide bridge |
| bridge | Cys-373′ – Cys-431′ | disulfide bridge |
| bridge | Cys-23” – Cys-88” | disulfide bridge |
| bridge | Cys-134” – Cys-194” | disulfide bridge |
| bridge | Cys-23”’ – Cys-88”’ | disulfide bridge |
| bridge | Cys-134”’ – Cys-194”’ | disulfide bridge |
PATENTS
WO 2017024062
US 20170305999
Evinacumab, sold under the brand name Evkeeza, is a monoclonal antibody medication for the treatment of homozygous familial hypercholesterolemia (HoFH).[1][2]
Evinacumab is a recombinant human IgG4 monoclonal antibody targeted against angiopoietin-like protein 3 (ANGPTL3) and the first drug of its kind. The ANGPTL family of proteins serve a number of physiologic functions – including involvement in the regulation of lipid metabolism – which have made them desirable therapeutic targets in recent years.2 Loss-of-function mutations in ANGPTL3 have been noted to result in hypolipidemia and subsequent reductions in cardiovascular risk, whereas increases in function appear to be associated with cardiovascular risk, and it was these observations that provided a rationale for the development of a therapy targeted against ANGPTL3.3
In February 2021, evinacumab became the first-and-only inhibitor of ANGPTL3 to receive FDA approval after it was granted approval for the adjunctive treatment of homozygous familial hypercholesterolemia (HoFH) under the brand name “Evkeeza”.8 Evinacumab is novel in its mechanism of action compared with other lipid-lowering therapies and therefore provides a unique and synergistic therapeutic option in the treatment of HoFH.
Common side effects include nasopharyngitis (cold), influenza-like illness, dizziness, rhinorrhea (runny nose), and nausea. Serious hypersensitivity (allergic) reactions have occurred in the Evkeeza clinical trials.[2]
Evinacumab binds to the angiopoietin-like protein 3 (ANGPTL3).[2] ANGPTL3 slows the function of certain enzymes that break down fats in the body.[2] Evinacumab blocks ANGPTL3, allowing faster break down of fats that lead to high cholesterol.[2] Evinacumab was approved for medical use in the United States in February 2021.[2][3]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | | |
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | |
History
The effectiveness and safety of evinacumab were evaluated in a double-blind, randomized, placebo-controlled, 24-week trial enrolling 65 participants with homozygous familial hypercholesterolemia (HoFH).[2] In the trial, 43 participants received 15 mg/kg of evinacumab every four weeks and 22 participants received the placebo.[2] Participants were taking other lipid-lowering therapies as well.[2]
The primary measure of effectiveness was the percent change in low-density lipoprotein (LDL-C) from the beginning of treatment to week 24.[2] At week 24, participants receiving evinacumab had an average 47% decrease in LDL-C while participants on the placebo had an average 2% increase.[2]
The U.S. Food and Drug Administration (FDA) granted the application for evinacumab orphan drug, breakthrough therapy, and priority review designations.[2] The FDA granted approval of Evkeeza to Regeneron Pharmaceuticals, Inc.[2]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves add-on therapy for patients with genetic form of severely”. U.S. Food and Drug Administration (FDA). 11 February 2021. Retrieved 12 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Approves First-in-class Evkeeza (evinacumab-dgnb) for Patients with Ultra-rare Inherited Form of High Cholesterol” (Press release). Regeneron Pharmaceuticals. 11 February 2021. Retrieved 12 February 2021 – via PR Newswire.
Further reading
- Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. (July 2017). “Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease”. N Engl J Med. 377 (3): 211–221. doi:10.1056/NEJMoa1612790. PMC 5800308. PMID 28538136.
External links
- “Evinacumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Angiopoietin-like 3 (ANGPTL3) |
| Clinical data | |
| Trade names | Evkeeza |
| Other names | REGN1500, evinacumab-dgnb |
| License data | US DailyMed: Evinacumab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1446419-85-7 |
| DrugBank | DB15354 |
| ChemSpider | none |
| UNII | T8B2ORP1DW |
| KEGG | D11753 |
| Chemical and physical data | |
| Formula | C6480H9992N1716O2042S46 |
| Molar mass | 146083.95 g·mol−1 |
//////////////
#Evinacumab, #Peptide, #APPROVALS 2021, #FDA 2021, #Monoclonal antibody, #dyslipidemia, #エビナクマブ (遺伝子組換え) , #REGN 1500, #REGN-1500, #REGN1500, #Anthony melvin crasto, #world drug tracker. # new drug approvals, #pharma
Tozinameran, Pfizer–BioNTech COVID‑19 vaccine


SEQUENCE1
gagaauaaac uaguauucuu cuggucccca cagacucaga gagaacccgc51caccauguuc guguuccugg ugcugcugcc ucuggugucc agccagugug101ugaaccugac caccagaaca cagcugccuc cagccuacac caacagcuuu151accagaggcg uguacuaccc cgacaaggug uucagaucca gcgugcugca201cucuacccag gaccuguucc ugccuuucuu cagcaacgug accugguucc251acgccaucca cguguccggc accaauggca ccaagagauu cgacaacccc301gugcugcccu ucaacgacgg gguguacuuu gccagcaccg agaaguccaa351caucaucaga ggcuggaucu ucggcaccac acuggacagc aagacccaga401gccugcugau cgugaacaac gccaccaacg uggucaucaa agugugcgag451uuccaguucu gcaacgaccc cuuccugggc gucuacuacc acaagaacaa501caagagcugg auggaaagcg aguuccgggu guacagcagc gccaacaacu551gcaccuucga guacgugucc cagccuuucc ugauggaccu ggaaggcaag601cagggcaacu ucaagaaccu gcgcgaguuc guguuuaaga acaucgacgg651cuacuucaag aucuacagca agcacacccc uaucaaccuc gugcgggauc701ugccucaggg cuucucugcu cuggaacccc ugguggaucu gcccaucggc751aucaacauca cccgguuuca gacacugcug gcccugcaca gaagcuaccu801gacaccuggc gauagcagca gcggauggac agcuggugcc gccgcuuacu851augugggcua ccugcagccu agaaccuucc ugcugaagua caacgagaac901ggcaccauca ccgacgccgu ggauugugcu cuggauccuc ugagcgagac951aaagugcacc cugaaguccu ucaccgugga aaagggcauc uaccagacca1001gcaacuuccg ggugcagccc accgaaucca ucgugcgguu ccccaauauc1051accaaucugu gccccuucgg cgagguguuc aaugccacca gauucgccuc1101uguguacgcc uggaaccgga agcggaucag caauugcgug gccgacuacu1151ccgugcugua caacuccgcc agcuucagca ccuucaagug cuacggcgug1201uccccuacca agcugaacga ccugugcuuc acaaacgugu acgccgacag1251cuucgugauc cggggagaug aagugcggca gauugccccu ggacagacag1301gcaagaucgc cgacuacaac uacaagcugc ccgacgacuu caccggcugu1351gugauugccu ggaacagcaa caaccuggac uccaaagucg gcggcaacua1401caauuaccug uaccggcugu uccggaaguc caaucugaag cccuucgagc1451gggacaucuc caccgagauc uaucaggccg gcagcacccc uuguaacggc1501guggaaggcu ucaacugcua cuucccacug caguccuacg gcuuucagcc1551cacaaauggc gugggcuauc agcccuacag agugguggug cugagcuucg1601aacugcugca ugccccugcc acagugugcg gcccuaagaa aagcaccaau1651cucgugaaga acaaaugcgu gaacuucaac uucaacggcc ugaccggcac1701cggcgugcug acagagagca acaagaaguu ccugccauuc cagcaguuug1751gccgggauau cgccgauacc acagacgccg uuagagaucc ccagacacug1801gaaauccugg acaucacccc uugcagcuuc ggcggagugu cugugaucac1851cccuggcacc aacaccagca aucagguggc agugcuguac caggacguga1901acuguaccga agugcccgug gccauucacg ccgaucagcu gacaccuaca1951uggcgggugu acuccaccgg cagcaaugug uuucagacca gagccggcug2001ucugaucgga gccgagcacg ugaacaauag cuacgagugc gacaucccca2051ucggcgcugg aaucugcgcc agcuaccaga cacagacaaa cagcccucgg2101agagccagaa gcguggccag ccagagcauc auugccuaca caaugucucu2151gggcgccgag aacagcgugg ccuacuccaa caacucuauc gcuaucccca2201ccaacuucac caucagcgug accacagaga uccugccugu guccaugacc2251aagaccagcg uggacugcac cauguacauc ugcggcgauu ccaccgagug2301cuccaaccug cugcugcagu acggcagcuu cugcacccag cugaauagag2351cccugacagg gaucgccgug gaacaggaca agaacaccca agagguguuc2401gcccaaguga agcagaucua caagaccccu ccuaucaagg acuucggcgg2451cuucaauuuc agccagauuc ugcccgaucc uagcaagccc agcaagcgga2501gcuucaucga ggaccugcug uucaacaaag ugacacuggc cgacgccggc2551uucaucaagc aguauggcga uugucugggc gacauugccg ccagggaucu2601gauuugcgcc cagaaguuua acggacugac agugcugccu ccucugcuga2651ccgaugagau gaucgcccag uacacaucug cccugcuggc cggcacaauc2701acaagcggcu ggacauuugg agcaggcgcc gcucugcaga uccccuuugc2751uaugcagaug gccuaccggu ucaacggcau cggagugacc cagaaugugc2801uguacgagaa ccagaagcug aucgccaacc aguucaacag cgccaucggc2851aagauccagg acagccugag cagcacagca agcgcccugg gaaagcugca2901ggacgugguc aaccagaaug cccaggcacu gaacacccug gucaagcagc2951uguccuccaa cuucggcgcc aucagcucug ugcugaacga uauccugagc3001agacuggacc cuccugaggc cgaggugcag aucgacagac ugaucacagg3051cagacugcag agccuccaga cauacgugac ccagcagcug aucagagccg3101ccgagauuag agccucugcc aaucuggccg ccaccaagau gucugagugu3151gugcugggcc agagcaagag aguggacuuu ugcggcaagg gcuaccaccu3201gaugagcuuc ccucagucug ccccucacgg cgugguguuu cugcacguga3251cauaugugcc cgcucaagag aagaauuuca ccaccgcucc agccaucugc3301cacgacggca aagcccacuu uccuagagaa ggcguguucg uguccaacgg3351cacccauugg uucgugacac agcggaacuu cuacgagccc cagaucauca3401ccaccgacaa caccuucgug ucuggcaacu gcgacgucgu gaucggcauu3451gugaacaaua ccguguacga cccucugcag cccgagcugg acagcuucaa3501agaggaacug gacaaguacu uuaagaacca cacaagcccc gacguggacc3551ugggcgauau cagcggaauc aaugccagcg ucgugaacau ccagaaagag3601aucgaccggc ugaacgaggu ggccaagaau cugaacgaga gccugaucga3651ccugcaagaa cuggggaagu acgagcagua caucaagugg cccugguaca3701ucuggcuggg cuuuaucgcc ggacugauug ccaucgugau ggucacaauc3751augcuguguu gcaugaccag cugcuguagc ugccugaagg gcuguuguag3801cuguggcagc ugcugcaagu ucgacgagga cgauucugag cccgugcuga3851agggcgugaa acugcacuac acaugaugac ucgagcuggu acugcaugca3901cgcaaugcua gcugccccuu ucccguccug gguaccccga gucucccccg3951accucggguc ccagguaugc ucccaccucc accugcccca cucaccaccu4001cugcuaguuc cagacaccuc ccaagcacgc agcaaugcag cucaaaacgc4051uuagccuagc cacaccccca cgggaaacag cagugauuaa ccuuuagcaa4101uaaacgaaag uuuaacuaag cuauacuaac cccaggguug gucaauuucg4151ugccagccac acccuggagc uagcaaaaaa aaaaaaaaaa aaaaaaaaaa4201aaaagcauau gacuaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa4251aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaa
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| modified base | g-1 | m7g |
| modified base | g-1 | 3′-me |
| modified base | a-2 | am |
| uncommon link | g-1 – a-2 | 5′->5′ triphosphate |
Tozinameran
Pfizer–BioNTech COVID-19 vaccine
トジナメラン (JAN);
コロナウイルス修飾ウリジンRNAワクチン;
RNA (recombinant 5′-[1,2-[(3′-O-methyl)m7G-(5’→5′)-ppp-Am]]-capped all uridine→N1-methylpseudouridine-substituted severe acute respiratory syndrome coronavirus 2 secretory signal peptide contg. spike glycoprotein S1S2-specifying plus 5′- and 3′-untranslated flanking region-contg. poly(A)-tailed messenger BNT162b2), inner salt
Nucleic Acid Sequence
Sequence Length: 42841106 a 1315 c 1062 g 801 umodified
APPROVED JAPAN Comirnaty, 2021/2/14
CAS 2417899-77-3
| Active immunization (SARS-CoV-2) |
Tozinameran is mRNA encoding full length of spike protein analog of SARS-CoV-2
Target Severe acute respiratory syndrome coronavirus 2 spike glycoprotein
Coronavirus disease – COVID-19
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Injection, suspension | Intramuscular | 0.23 mg/1.8mL |
| Suspension | Intramuscular | 30 mcg |
| NAME | INGREDIENTS | DOSAGE | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Pfizer-BioNTech Covid-19 Vaccine | Pfizer-BioNTech Covid-19 Vaccine (0.23 mg/1.8mL) | Injection, suspension | Intramuscular | Pfizer Manufacturing Belgium NV | 2020-12-12 | Not applicable | |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Comirnaty | 30 mcg | Intramuscular | Bio N Tech Manufacturing Gmb H | 2021-01-06 | Not applicable |  | ||
| Pfizer-BioNTech Covid-19 Vaccine | Suspension | 30 mcg | Intramuscular | Biontech Manufacturing Gmbh | 2020-12-14 | Not applicable |  | |
| Pfizer-BioNTech Covid-19 Vaccine | Injection, suspension | 0.23 mg/1.8mL | Intramuscular | Pfizer Manufacturing Belgium NV | 2020-12-12 | Not applicable | |
The Pfizer–BioNTech COVID‑19 vaccine (pINN: tozinameran), sold under the brand name Comirnaty,[13] is a COVID-19 vaccine developed by the German company BioNTech in cooperation with Pfizer. It is both the first COVID-19 vaccine to be authorized by a stringent regulatory authority for emergency use[14][15] and the first cleared for regular use.[16]
It is given by intramuscular injection. It is an RNA vaccine composed of nucleoside-modified mRNA (modRNA) encoding a mutated form of the spike protein of SARS-CoV-2, which is encapsulated in lipid nanoparticles.[17] The vaccination requires two doses given three weeks apart.[18][19][20] Its ability to prevent severe infection in children, pregnant women, or immunocompromised people is unknown, as is the duration of the immune effect it confers.[20][21][22] As of February 2021, it is one of two RNA vaccines being deployed against COVID‑19, the other being the Moderna COVID‑19 vaccine. A third mRNA-based COVID-19 vaccine, CVnCoV, is in late-stage testing.[23]
Trials began in April 2020; by November, the vaccine had been tested on more than 40,000 people.[24] An interim analysis of study data showed a potential efficacy of over 90% in preventing infection within seven days of a second dose.[19][20] The most common side effects include mild to moderate pain at the injection site, fatigue, and headache.[25][26] As of December 2020, reports of serious side effects, such as allergic reactions, have been very rare,[a] and no long-term complications have been reported.[28] Phase III clinical trials are ongoing: monitoring of the primary outcomes will continue until August 2021, while monitoring of the secondary outcomes will continue until January 2023.[18]
In December 2020, the United Kingdom was the first country to authorize the vaccine on an emergency basis,[28] soon followed by the United States, the European Union and several other countries globally.[29][30][6][31][32]
BioNTech is the initial developer of the vaccine, and partnered with Pfizer for development, clinical research, overseeing the clinical trials, logistics, finances and for manufacturing worldwide with the exception of China.[33] The license to distribute and manufacture in China was purchased by Fosun, alongside its investment in BioNTech.[34][35] Distribution in Germany and Turkey is by BioNTech itself.[36] Pfizer indicated in November 2020, that 50 million doses could be available globally by the end of 2020, with about 1.3 billion doses in 2021.[20]
Pfizer has advanced purchase agreements of about US$3 billion for providing a licensed vaccine in the United States, the European Union, the United Kingdom, Japan, Canada, Peru, Singapore, and Mexico.[37][38] Distribution and storage of the vaccine is a logistics challenge because it needs to be stored at temperatures between −80 and −60 °C (−112 and −76 °F),[39] until five days before vaccination[38][39] when it can be stored at 2 to 8 °C (36 to 46 °F), and up to two hours at temperatures up to 25 °C (77 °F)[40][11] or 30 °C (86 °F).[41][42] In February 2021, Pfizer and BioNTech asked the U.S. Food and Drug Administration (FDA) to update the emergency use authorization (EUA) to permit the vaccine to be stored at between −25 and −15 °C (−13 and 5 °F) for up to two weeks before use.[43]
Development and funding
Before COVID-19 vaccines, a vaccine for an infectious disease had never before been produced in less than several years, and no vaccine existed for preventing a coronavirus infection in humans.[44] After the COVID-19 virus was detected in December 2019,[45] the development of BNT162b2 was initiated on 10 January 2020, when the SARS-CoV-2 genetic sequences were released by the Chinese Center for Disease Control and Prevention via GISAID,[46][47][48] triggering an urgent international response to prepare for an outbreak and hasten development of preventive vaccines.[49][50]
In January 2020, German biotech-company BioNTech started its program ‘Project Lightspeed’ to develop a vaccine against the new COVID‑19 virus based on its already established mRNA-technology.[24] Several variants of the vaccine were created in their laboratories in Mainz, and 20 of those were presented to experts of the Paul-Ehrlich-Institute in Langen.[51] Phase I / II Trials were started in Germany on 23 April 2020, and in the U.S. on 4 May 2020, with four vaccine candidates entering clinical testing. The Initial Pivotal Phase II / III Trial with the lead vaccine candidate ‘BNT162b2’ began in July. The Phase III results indicating a 95% effectiveness of the developed vaccine were published on 18 November 2020.[24]
BioNTech received a US$135 million investment from Fosun in March 2020, in exchange for 1.58 million shares in BioNTech and the future development and marketing rights of BNT162b2 in China,[35] Hong Kong, Macau and Taiwan.[52]
In June 2020, BioNTech received €100 million (US$119 million) in financing from the European Commission and European Investment Bank.[53] In September 2020, the German government granted BioNTech €375 million (US$445 million) for its COVID‑19 vaccine development program.[54]
Pfizer CEO Albert Bourla stated that he decided against taking funding from the US government’s Operation Warp Speed for the development of the vaccine “because I wanted to liberate our scientists [from] any bureaucracy that comes with having to give reports and agree how we are going to spend the money in parallel or together, etc.” Pfizer did enter into an agreement with the US for the eventual distribution of the vaccine, as with other countries.[55]
Clinical trials
See also: COVID-19 vaccine § Clinical trials started in 2020
Preliminary results from Phase I–II clinical trials on BNT162b2, published in October 2020, indicated potential for its efficacy and safety.[17][56] During the same month, the European Medicines Agency (EMA) began a periodic review of BNT162b2.[57]
The study of BNT162b2 is a continuous-phase trial in Phase III as of November 2020.[18] It is a “randomized, placebo-controlled, observer-blind, dose-finding, vaccine candidate-selection, and efficacy study in healthy individuals”.[18] The early-stage research determined the safety and dose level for two vaccine candidates, with the trial expanding during mid-2020 to assess efficacy and safety of BNT162b2 in greater numbers of participants, reaching tens of thousands of people receiving test vaccinations in multiple countries in collaboration with Pfizer and Fosun.[20][35]
The Phase III trial assesses the safety, efficacy, tolerability, and immunogenicity of BNT162b2 at a mid-dose level (two injections separated by 21 days) in three age groups: 12–15 years, 16–55 years or above 55 years.[18] For approval in the EU, an overall vaccine efficacy of 95% was confirmed by the EMA.[58] The EMA clarified that the second dose should be administered three weeks after the first dose.[59]
| Efficacy endpoint | Vaccine efficacy (95% confidence interval) [%] |
|---|---|
| After dose 1 to before dose 2 | 52.4 (29.5, 68.4) |
| ≥10 days after dose 1 to before dose 2 | 86.7 (68.6, 95.4) |
| Dose 2 to 7 days after dose 2 | 90.5 (61.0, 98.9) |
| ≥7 days after dose 2 (subjects without evidence of infection prior to 7 days after dose 2) | |
| Overall | 95.0 (90.0, 97.9) |
| 16–55 years | 95.6 (89.4, 98.6) |
| ≥55 years | 93.7 (80.6, 98.8) |
| ≥65 years | 94.7 (66.7, 99.9) |
The ongoing Phase III trial, which is scheduled to run from 2020 to 2022, is designed to assess the ability of BNT162b2 to prevent severe infection, as well as the duration of immune effect.[20][21][22]
Pfizer and BioNTech started a Phase II/III randomized control trial in healthy pregnant women 18 years of age and older (NCT04754594).[60] The study will evaluate 30 µg of BNT162b2 or placebo administered via intramuscular injection in 2 doses, 21 days apart. The Phase II portion of the study will include approximately 350 pregnant women randomized 1:1 to receive BNT162b2 or placebo at 27 to 34 weeks’ gestation. The Phase III portion of this study will assess the safety, tolerability, and immunogenicity of BNT162b2 or placebo among pregnant women enrolled at 24 to 34 weeks’ gestation. Pfizer and BioNTech announced on 18 February 2021 that the first participants received their first dose in this trial.[61]
Vaccine technology
See also: RNA vaccine and COVID-19 vaccine § Technology platforms
The BioNTech technology for the BNT162b2 vaccine is based on use of nucleoside-modified mRNA (modRNA) which encodes part of the spike protein found on the surface of the SARS-CoV-2 coronavirus (COVID‑19), triggering an immune response against infection by the virus protein.[62]
The vaccine candidate BNT162b2 was chosen as the most promising among three others with similar technology developed by BioNTech.[18][62][56] Prior to choosing BNT162b2, BioNTech and Pfizer had conducted Phase I trials on BNT162b1 in Germany and the United States, while Fosun performed a Phase I trial in China.[17][63] In these Phase I studies, BNT162b2 was shown to have a better safety profile than the other three BioNTech candidates.[63]
Sequence
The modRNA sequence of the vaccine is 4,284 nucleotides long.[64] It consists of a five-prime cap; a five prime untranslated region derived from the sequence of human alpha globin; a signal peptide (bases 55–102) and two proline substitutions (K986P and V987P, designated “2P”) that cause the spike to adopt a prefusion-stabilized conformation reducing the membrane fusion ability, increasing expression and stimulating neutralizing antibodies;[17][65] a codon-optimized gene of the full-length spike protein of SARS-CoV-2 (bases 103–3879); followed by a three prime untranslated region (bases 3880–4174) combined from AES and mtRNR1 selected for increased protein expression and mRNA stability[66] and a poly(A) tail comprising 30 adenosine residues, a 10-nucleotide linker sequence, and 70 other adenosine residues (bases 4175–4284).[64] The sequence contains no uridine residues; they are replaced by 1-methyl-3′-pseudouridylyl.[64]
Composition
In addition to the mRNA molecule, the vaccine contains the following inactive ingredients (excipients):[67][68][8]
- ALC-0315, ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate)
- ALC-0159, 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide
- 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
- cholesterol
- dibasic sodium phosphate dihydrate
- monobasic potassium phosphate
- potassium chloride
- sodium chloride
- sucrose
- water for injection
The first four of these are lipids. The lipids and modRNA together form nanoparticles. ALC-0159 is a polyethylene glycol conjugate (that is, a PEGylated lipid).[69]
The vaccine is supplied in a multidose vial as “a white to off-white, sterile, preservative-free, frozen suspension for intramuscular injection“.[11][12] It must be thawed to room temperature and diluted with normal saline before administration.[12]
Authorizations
Expedited
The United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA) gave the vaccine “rapid temporary regulatory approval to address significant public health issues such as a pandemic” on 2 December 2020, which it is permitted to do under the Medicines Act 1968.[70] It was the first COVID‑19 vaccine to be approved for national use after undergoing large scale trials,[71] and the first mRNA vaccine to be authorized for use in humans.[14][72] The United Kingdom thus became the first Western country to approve a COVID‑19 vaccine for national use,[73] although the decision to fast-track the vaccine was criticised by some experts.[74]
On 8 December 2020, Margaret “Maggie” Keenan, 90, from Fermanagh, became the first person to receive the vaccine.[75] In a notable example of museums documenting the pandemic, the vial and syringe used for that first dose were saved acquired by The Science Museum in London for its permanent collection.[76] By 20 December, 521,594 UK residents had received the vaccine as part of the national vaccination programme. 70% had been to people aged 80 or over.[77]
After the United Kingdom, the following countries expedited processes to approve the Pfizer–BioNTech COVID‑19 vaccine for use: Argentina,[78] Australia,[79] Bahrain,[80] Canada,[7][81] Chile,[82] Costa Rica,[83] Ecuador,[82] Hong Kong,[84] Iraq,[85] Israel,[86] Jordan,[87] Kuwait,[88] Mexico,[89] Oman,[90] Panama,[91] the Philippines,[92] Qatar,[93] Saudi Arabia,[32][94] Singapore,[95][96] the United Arab Emirates,[97] and the United States.[10]
The World Health Organization (WHO) authorized it for emergency use.[98]
In the United States, an emergency use authorization (EUA) is “a mechanism to facilitate the availability and use of medical countermeasures, including vaccines, during public health emergencies, such as the current COVID‑19 pandemic”, according to the FDA.[99] Following an EUA issuance, BioNTech and Pfizer are expected to continue the Phase III clinical trial to finalize safety and efficacy data, leading to application for licensure (approval) of the vaccine in the United States.[99][100][101] The United States Centers for Disease Control and Prevention (CDC) Advisory Committee on Immunization Practices (ACIP) approved recommendations for vaccination of those aged 16 years or older.[102][103]
Standard
On 19 December 2020, the Swiss Agency for Therapeutic Products (Swissmedic) approved the Pfizer–BioNTech COVID‑19 vaccine for regular use, two months after receiving the application, stating that the vaccine fully complied with the requirements of safety, efficacy and quality. This is the first authorization under a standard procedure.[1][104] On 23 December, a Lucerne resident, a 90-year-old woman, became the first person to receive the vaccine in Switzerland.[105] This marked the beginning of mass vaccination in continental Europe.[106]
On 21 December 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting conditional marketing authorization for the Pfizer–BioNTech COVID‑19 vaccine under the brand name Comirnaty.[2][107][108] The recommendation was accepted by the European Commission the same day.[107][109]
On February 23, 2021, the Brazilian Health Regulatory Agency approved the Pfizer–BioNTech COVID-19 vaccine under its standard marketing authorization procedure. It became the first COVID-19 vaccine to receive definitive registration rather than emergency use authorization in the country.[110]
Adverse effects
The adverse effect profile of the Pfizer–BioNTech COVID‑19 vaccine is similar to that of other adult vaccines.[20] During clinical trials, the side effects deemed very common[a] are (in order of frequency): pain and swelling at the injection site, tiredness, headache, muscle aches, chills, joint pain, and fever.[68] Fever is more common after the second dose.[68] These effects are predictable and to be expected, and it is particularly important that people be aware of this to prevent vaccine hesitancy.[111]
Severe allergic reaction has been observed in approximately 11 cases per million doses of vaccine administered.[112][113] According to a report by the US Centers for Disease Control and Prevention 71% of those allergic reactions happened within 15 minutes of vaccination and mostly (81%) among people with a documented history of allergies or allergic reactions.[112] The UK’s Medicines and Healthcare products Regulatory Agency (MHRA) advised on 9 December 2020, that people who have a history of “significant” allergic reaction should not receive the Pfizer–BioNTech COVID‑19 vaccine.[114][115][116] On 12 December, the Canadian regulator followed suit, noting that: “Both individuals in the U.K. had a history of severe allergic reactions and carried adrenaline auto injectors. They both were treated and have recovered.”[67]
On 28 January 2021, the European Union published a COVID-19 vaccine safety update which found that “the benefits of Comirnaty in preventing COVID‑19 continue to outweigh any risks, and there are no recommended changes regarding the use the vaccine.”[113][117] No new side effects were identified.[113]
Manufacturing
_E.jpg)
A doctor holding the Pfizer vaccine
Pfizer and BioNTech are manufacturing the vaccine in their own facilities in the United States and in Europe in a three-stage process. The first stage involves the molecular cloning of DNA plasmids that code for the spike protein by infusing them into Escherichia coli bacteria. In the United States, this stage is conducted at a small pilot plant in Chesterfield, Missouri[118] (near St. Louis). After four days of growth, the bacteria are killed and broken open, and the contents of their cells are purified over a week and a half to recover the desired DNA product. The DNA is stored in tiny bottles and frozen for shipment. Safely and quickly transporting the DNA at this stage is so important that Pfizer has used its company jet and helicopter to assist.[119]
The second stage is being conducted at plants in Andover, Massachusetts[120] in the United States, and in Germany. The DNA is used as a template to build the desired mRNA strands. Once the mRNA has been created and purified, it is frozen in plastic bags about the size of a large shopping bag, of which each can hold up to 5 to 10 million doses. The bags are placed on special racks on trucks which take them to the next plant.[119]
The third stage is being conducted at plants in Portage, Michigan[121] (near Kalamazoo) in the United States, and Puurs in Belgium. This stage involves combining the mRNA with lipid nanoparticles, then filling vials, boxing vials, and freezing them.[119] Croda International subsidiary Avanti Polar Lipids is providing the requisite lipids.[122] As of November 2020, the major bottleneck in the manufacturing process was combining mRNA with lipid nanoparticles.[119]
In February 2021, Pfizer revealed this entire sequence initially took about 110 days on average from start to finish, and that the company was making progress on reducing that number to 60 days.[123] Vaccine manufacturers normally take several years to optimize the process of making a particular vaccine for speed and cost-effectiveness before attempting large-scale production.[123] Due to the urgency presented by the COVID-19 pandemic, Pfizer began production immediately with the process by which the vaccine had been originally formulated in the laboratory, then started to identify ways to safely speed up and scale up that process.[123]
BioNTech announced in September 2020 that it had signed an agreement to acquire from Novartis a manufacturing facility in Marburg, Germany, to expand their vaccine production capacity.[124] Once fully operational, the facility would produce up to 750 million doses per year, or over 60 million doses per month. The site will be the third BioNTech facility in Europe which currently produces the vaccine, while Pfizer operates at least four production sites in the United States and Europe.
Advance orders and logistics
Pfizer indicated in its 9 November press release that 50 million doses could be available by the end of 2020, with about 1.3 billion doses provided globally by 2021.[20] In February 2021, BioNTech announced it would increase production by more than 50% to manufacture two billion doses in 2021.[125]
In July 2020, the vaccine development program Operation Warp Speed placed an advance order of US$1.95 billion with Pfizer to manufacture 100 million doses of a COVID‑19 vaccine for use in the United States if the vaccine was shown to be safe and effective.[34][126][127][128] By mid-December 2020, Pfizer had agreements to supply 300 million doses to the European Union,[129] 120 million doses to Japan,[130] 40 million doses (10 million before 2021) to the United Kingdom,[22] 20 million doses to Canada,[131] an unspecified number of doses to Singapore,[132] and 34.4 million doses to Mexico.[133] Fosun also has agreements to supply 10 million doses to Hong Kong and Macau.[134] The Hong Kong government said it would receive its first batch of one million doses by the first quarter of 2021.[135]
BioNTech and Fosun agreed to supply Mainland China with a batch of 100 million doses in 2021, subject to regulatory approval. The initial supply will be delivered from BioNTech’s production facilities in Germany.[136]
The vaccine is being delivered in vials that, once diluted, contain 2.25 ml of vaccine (0.45 ml frozen plus 1.8ml diluent).[101] According to the vial labels, each vial contains five 0.3 ml doses, however excess vaccine may be used for one, or possibly two, additional doses.[101][137] The use of low dead space syringes to obtain the additional doses is preferable, and partial doses within a vial should be discarded.[101][138] The Italian Medicines Agency officially authorized the use of excess doses remaining within single vials.[139] As of 8 January 2021, each vial contains six doses.[68][140][141][138] In the United States, vials will be counted as five doses when accompanied by regular syringes and as six doses when accompanied by low dead space syringes.[142]
.jpg)
Temperature the Pfizer vaccine must be kept at to ensure effectiveness, roughly between −80 and −60 °C (−112 and −76 °F)
Logistics in developing countries which have preorder agreements with Pfizer—such as Ecuador and Peru—remain unclear.[38] Even high-income countries have limited cold chain capacity for ultracold transport and storage of a vaccine that degrades within five days when thawed, and requires two shots three weeks apart.[38] The vaccine needs to be stored and transported at ultracold temperatures between −80 and −60 °C (−112 and −76 °F),[39][22][38][143][144] much lower than for the similar Moderna vaccine. The head of Indonesia‘s Bio Farma Honesti Basyir stated that purchasing the vaccine is out of the question for the world’s fourth-most populous country, given that it did not have the necessary cold chain capability. Similarly, India’s existing cold chain network can only handle temperatures between 2 and 8 °C (36 and 46 °F), far above the requirements of the vaccine.[145][146]
In January 2021, Pfizer and BioNTech offered to supply 50 million doses of COVID‑19 vaccine for health workers across Africa between March and the end of 2021, at a discounted price of US$10 per dose.[147]
Name
BNT162b2 was the code name during development and testing,[17][148] tozinameran is the proposed international nonproprietary name (pINN),[149] and Comirnaty is the brand name.[1][2] According to BioNTech, the name Comirnaty “represents a combination of the terms COVID‑19, mRNA, community, and immunity.”[150][151]
The vaccine also has the common name “COVID‑19 mRNA vaccine (nucleoside-modified)”[2] and may be distributed in packaging with the name Pfizer–BioNTech COVID‑19 Vaccine.”[152]
How the Pfizer-BioNTech Vaccine Works
By Jonathan Corum and Carl ZimmerUpdated Jan. 21, 2021Leer en español

The German company BioNTech partnered with Pfizer to develop and test a coronavirus vaccine known as BNT162b2, the generic name tozinameran or the brand name Comirnaty. A clinical trial demonstrated that the vaccine has an efficacy rate of 95 percent in preventing Covid-19.
A Piece of the Coronavirus
The SARS-CoV-2 virus is studded with proteins that it uses to enter human cells. These so-called spike proteins make a tempting target for potential vaccines and treatments.

Spikes
Spike
protein
gene
CORONAVIRUS
Like the Moderna vaccine, the Pfizer-BioNTech vaccine is based on the virus’s genetic instructions for building the spike protein.
mRNA Inside an Oily Shell
The vaccine uses messenger RNA, genetic material that our cells read to make proteins. The molecule — called mRNA for short — is fragile and would be chopped to pieces by our natural enzymes if it were injected directly into the body. To protect their vaccine, Pfizer and BioNTech wrap the mRNA in oily bubbles made of lipid nanoparticles.

Lipid nanoparticles
surrounding mRNA
Because of their fragility, the mRNA molecules will quickly fall apart at room temperature. Pfizer is building special containers with dry ice, thermal sensors and GPS trackers to ensure the vaccines can be transported at –94°F (–70°C) to stay viable.
Entering a Cell
After injection, the vaccine particles bump into cells and fuse to them, releasing mRNA. The cell’s molecules read its sequence and build spike proteins. The mRNA from the vaccine is eventually destroyed by the cell, leaving no permanent trace.

VACCINE
PARTICLES
VACCINATED
CELL
Spike
protein
mRNA
Translating mRNA
Three spike
proteins combine
Spike
Cell
nucleus
Spikes
and protein
fragments
Displaying
spike protein
fragments
Protruding
spikes
Some of the spike proteins form spikes that migrate to the surface of the cell and stick out their tips. The vaccinated cells also break up some of the proteins into fragments, which they present on their surface. These protruding spikes and spike protein fragments can then be recognized by the immune system.
Spotting the Intruder
When a vaccinated cell dies, the debris will contain many spike proteins and protein fragments, which can then be taken up by a type of immune cell called an antigen-presenting cell.

Debris from
a dead cell
Engulfing
a spike
ANTIGEN-
PRESENTING
CELL
Digesting
the proteins
Presenting a
spike protein
fragment
HELPER
T CELL
The cell presents fragments of the spike protein on its surface. When other cells called helper T cells detect these fragments, the helper T cells can raise the alarm and help marshal other immune cells to fight the infection.
Making Antibodies
Other immune cells, called B cells, may bump into the coronavirus spikes on the surface of vaccinated cells, or free-floating spike protein fragments. A few of the B cells may be able to lock onto the spike proteins. If these B cells are then activated by helper T cells, they will start to proliferate and pour out antibodies that target the spike protein.

HELPER
T CELL
Activating
the B cell
Matching
surface proteins
VACCINATED
CELL
B CELL
SECRETED
ANTIBODIES
Stopping the Virus
The antibodies can latch onto coronavirus spikes, mark the virus for destruction and prevent infection by blocking the spikes from attaching to other cells.

ANTIBODIES
VIRUS
Killing Infected Cells
The antigen-presenting cells can also activate another type of immune cell called a killer T cell to seek out and destroy any coronavirus-infected cells that display the spike protein fragments on their surfaces.

ANTIGEN-PRESENTING CELL Presenting a spike protein fragment ACTIVATED KILLER T CELL INFECTED CELL Beginning to kill the infected cell
Remembering the Virus
The Pfizer-BioNTech vaccine requires two injections, given 21 days apart, to prime the immune system well enough to fight off the coronavirus. But because the vaccine is so new, researchers don’t know how long its protection might last.

First dose, 0.3ml
Second dose, 21 days later
A preliminary study found that the vaccine seems to offer strong protection about 10 days after the first dose, compared with people taking a placebo:

Cumulative incidence of Covid-19 among clinical trial participants 2.5% 2.0 People taking a placebo
1.5 1.0 Second dose First dose People taking the
Pfizer-BioNTech vaccine
0.5
0
1
2
3
4
8
12
16
Weeks after the first dose
It’s possible that in the months after vaccination, the number of antibodies and killer T cells will drop. But the immune system also contains special cells called memory B cells and memory T cells that might retain information about the coronavirus for years or even decades.
For more about the vaccine, see Pfizer’s Covid Vaccine: 11 Things You Need to Know.
Preparation and Injection
Each vial of the vaccine contains 5 doses of 0.3 milliliters. The vaccine must be thawed before injection and diluted with saline. After dilution the vial must be used within six hours.

A diluted vial of the vaccine at Royal Free Hospital in London.Jack Hill/Agence France-Presse
References
- ^ Jump up to:a b According to the British National Formulary and MedDRA conventions, side effects are “very common” when they occur in more than 1 in 10 instances; “common”, 1 in 100 to 1 in 10; “uncommon”, 1 in 1,000 to 1 in 100; “rare”, 1 in 10,000 to 1 in 1,000; and “very rare” when they occur in less than 1 in 10,000 instances.[27]
- ^ Jump up to:a b c d “Swissmedic grants authorisation for the first COVID-19 vaccine in Switzerland”(Press release). Swiss Agency for Therapeutic Products (Swissmedic). 19 December 2020. Retrieved 19 December 2020.
- ^ Jump up to:a b c d e “Comirnaty EPAR”. European Medicines Agency (EMA). Retrieved 23 December 2020.
- ^ “Comirnaty”. Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ “Comirnaty (BNT162b2 [mRNA]) COVID‑19 Vaccine Product Information” (PDF). Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ Australian Public Assessment Report for BNT162b2 (mRNA) (PDF) (Report). Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ Jump up to:a b “Regulatory Decision Summary – Pfizer-BioNTech COVID-19 Vaccine”. Health Canada. 9 December 2020. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Jump up to:a b “Pfizer-BioNTech COVID-19 Vaccine (tozinameran)”. Health Canada. Retrieved 15 December 2020.
- ^ Jump up to:a b “Information for Healthcare Professionals on Pfizer/BioNTech COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 10 December 2020. Retrieved 21 December 2020.
- ^ “Conditions of Authorisation for Pfizer/BioNTech COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 31 December 2020. Retrieved 8 January2021.
- ^ Jump up to:a b “FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine” (Press release). U.S. Food and Drug Administration (FDA). 11 December 2020. Retrieved 11 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Pfizer-BioNTech COVID-19 Vaccine- rna ingredient bnt-162b2 injection, suspension”. DailyMed. Retrieved 14 December 2020.
- ^ Jump up to:a b c Pfizer-BioNTech COVID-19 Vaccine Emergency Use Authorization Review Memorandum (PDF). U.S. Food and Drug Administration (FDA) (Report). 14 December 2020. Retrieved 14 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Comirnaty EPAR”. European Medicines Agency (EMA). Retrieved 23 December 2020.
- ^ Jump up to:a b “UK medicines regulator gives approval for first UK COVID-19 vaccine” (Press release). Medicines and Healthcare products Regulatory Agency (MHRA). 2 December 2020. Retrieved 2 December 2020.
- ^ Boseley S, Halliday J (2 December 2020). “UK approves Pfizer/BioNTech Covid vaccine for rollout next week”. The Guardian. Retrieved 14 December 2020.
- ^ “Swissmedic grants authorisation for the first COVID-19 vaccine in Switzerland” (Press release). Swiss Agency for Therapeutic Products (Swissmedic). 19 December 2020. Retrieved 19 December 2020.
- ^ Jump up to:a b c d e Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, et al. (October 2020). “Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates”. The New England Journal of Medicine. 383 (25): 2439–50. doi:10.1056/NEJMoa2027906. PMC 7583697. PMID 33053279.
- ^ Jump up to:a b c d e f Clinical trial number NCT04368728 for “NCT04368728: Study to Describe the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals” at ClinicalTrials.gov
- ^ Jump up to:a b Palca J (9 November 2020). “Pfizer says experimental COVID-19 vaccine is more than 90% effective”. NPR. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b c d e f g h Herper M (9 November 2020). “Covid-19 vaccine from Pfizer and BioNTech is strongly effective, early data from large trial indicate”. STAT. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b Edwards E (9 November 2020). “Pfizer’s Covid-19 vaccine promising, but many questions remain”. NBC News. Archived from the original on 22 November 2020. Retrieved 12 November 2020.
- ^ Jump up to:a b c d Gallagher J (9 November 2020). “Covid vaccine: First ‘milestone’ vaccine offers 90% protection”. BBC News. Archived from the original on 26 November 2020. Retrieved 9 November 2020.
- ^ “CureVac Initiates Rolling Submission With European Medicines Agency for COVID-19 Vaccine Candidate, CVnCoV”. CureVac (Press release).
- ^ Jump up to:a b c “Update on our COVID-19 vaccine development program with BNT162b2” (PDF)(Press release). BioNTech. 2 December 2020. Retrieved 12 December 2020.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. N Engl J Med. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
- ^ “Questions and Answers About Pfizer-BioNTech COVID-19 Vaccine”. Pfizer. Retrieved 16 December 2020.
- ^ “Adverse reactions to drugs”. British National Formulary. Retrieved 19 December 2020.
- ^ Jump up to:a b “Coronavirus vaccine”. National Health Service. 7 December 2020. Archived from the original on 7 December 2020. Retrieved 7 December 2020.
- ^ Commissioner, Office of the (3 February 2021). “Pfizer-BioNTech COVID-19 Vaccine”. FDA.
- ^ “EMA recommends first COVID-19 vaccine for authorisation in the EU”. European Medicines Agency.
- ^ “Bahrain becomes second country to approve Pfizer COVID-19 vaccine”. Al Jazeera. Archived from the original on 4 December 2020. Retrieved 5 December 2020.
* “Coronavirus: Saudi Arabia approves Pfizer COVID-19 vaccine for use”. Al Arabiya English. 10 December 2020. Archived from the original on 11 December 2020. Retrieved 10 December 2020.
* Solomon DB, Torres N (11 December 2020). “Mexico approves emergency use of Pfizer’s COVID-19 vaccine”. Reuters. Retrieved 12 December 2020.
* Thomas K (20 November 2020). “F.D.A. Clears Pfizer Vaccine, and Millions of Doses Will Be Shipped Right Away”. The New York Times. Archived from the original on 12 December 2020. Retrieved 11 December 2020.
* “First shipments of Pfizer-BioNTech vaccine in Singapore by end-Dec; enough vaccines for all by Q3 2021”. The Straits Times. 14 December 2020. Retrieved 14 December 2020. - ^ Jump up to:a b Al Mulla Y (13 December 2020). “Kuwait approves emergency use of Pfizer vaccine”. Gulf News. Retrieved 14 December 2020.
- ^ Browne R (11 November 2020). “What you need to know about BioNTech – the European company behind Pfizer’s Covid-19 vaccine”. CNBC. Retrieved 14 January 2021.
- ^ Jump up to:a b Thomas K, Gelles D, Zimmer C (9 November 2020). “Pfizer’s early data shows vaccine is more than 90% effective”. The New York Times. Archived from the original on 23 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b c Burger L (15 March 2020). “BioNTech in China alliance with Fosun over coronavirus vaccine candidate”. Reuters. Archived from the original on 14 November 2020. Retrieved 10 November 2020.
- ^ “Pfizer and BioNTech Celebrate Historic First Authorization in the U.S. of Vaccine to Prevent COVID-19”. Pfizer Inc. and BioNTech SE.
- ^ “Securing Singapore’s access to COVID-19 vaccines”. gov.sg. Government of Singapore. 14 December 2020. Retrieved 1 February 2021.
- ^ Jump up to:a b c d e “Deep-freeze hurdle makes Pfizer’s vaccine one for the rich”. Bloomberg. 10 November 2020. Archived from the original on 22 November 2020. Retrieved 12 November 2020.
Vaccine goes bad five days after thawing, requires two shots; Many nations face costly ramp up of cold-chain infrastructure
- ^ Jump up to:a b c “Pfizer-BioNTech COVID-19 Vaccine Vaccination Storage & Dry Ice Safety Handling”. Pfizer. Retrieved 17 December 2020.
- ^ “Information for Healthcare Professionals on Pfizer/BioNTech COVID-19 vaccine”. Government of the United Kingdom. Retrieved 29 January 2021.
- ^ “Recommendation for an Emergency Use Listing of Tozinameran (Covid-19 Mrna Vaccine (Nucleoside Modified)) Submitted by Biontech Manufacturing Gmbh” (PDF). World Health Organization. 26 January 2021.
- ^ “Australian Product Information – Comirnaty (BNT162b2 [mRNA]) COVID-19 Vaccine”(PDF). Therapeutic Goods Administration. Australian Government.
- ^ “Pfizer and BioNTech Submit COVID-19 Vaccine Stability Data at Standard Freezer Temperature to the U.S. FDA”. Pfizer (Press release). 19 February 2021. Retrieved 19 February 2021.
- ^ Gates B (30 April 2020). “The vaccine race explained: What you need to know about the COVID-19 vaccine”. The Gates Notes. Archived from the original on 14 May 2020. Retrieved 2 May 2020.
- ^ “World Health Organization timeline – COVID-19”. World Health Organization. 27 April 2020. Archived from the original on 29 April 2020. Retrieved 2 May 2020.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. The New England Journal of Medicine. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
development of BNT162b2 was initiated on January 10, 2020, when the SARS-CoV-2 genetic sequence was released by the Chinese Center for Disease Control and Prevention and disseminated globally by the GISAID (Global Initiative on Sharing All Influenza Data) initiative
- ^ Bohn MK, Mancini N, Loh TP, Wang CB, Grimmler M, Gramegna M, et al. (October 2020). “IFCC Interim Guidelines on Molecular Testing of SARS-CoV-2 Infection”. Clinical Chemistry and Laboratory Medicine. 58 (12): 1993–2000. doi:10.1515/cclm-2020-1412. PMID 33027042.
- ^ “CEPI’s collaborative task force to assess COVID-19 vaccines on emerging viral strains”. BioSpectrum – Asia Edition. 23 November 2020.
the first SARS-CoV-2 viral genomes were shared via GISAID on 10 January 2020
- ^ Thanh Le T, Andreadakis Z, Kumar A, Gómez Román R, Tollefsen S, Saville M, Mayhew S (May 2020). “The COVID-19 vaccine development landscape”. Nature Reviews. Drug Discovery. 19 (5): 305–306. doi:10.1038/d41573-020-00073-5. PMID 32273591.
- ^ Fauci AS, Lane HC, Redfield RR (March 2020). “Covid-19 – Navigating the Uncharted”. The New England Journal of Medicine. 382 (13): 1268–1269. doi:10.1056/nejme2002387. PMC 7121221. PMID 32109011.
- ^ Papadopoulos C (14 December 2020). “Chronologie – So entstand der Corona-Impfstoff von Biontech” [Chronology – That’s how the Covid-vaccine of Biontech was being developed] (in German). Südwestrundfunk. Retrieved 20 December 2020.
- ^ 《Fosun Pharma and BioNTech form COVID‑19 vaccine strategic alliance in China》(Fosun Phrama News Content , 15 March 2020) Archived 15 August 2020 at the Wayback Machine
- ^ “Germany: Investment Plan for Europe – EIB to provide BioNTech with up to €100 million in debt financing for COVID-19 vaccine development and manufacturing”. European Investment Bank. 11 June 2020. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “BioNTech gets $445 million in German funding for vaccine”. Bloomberg L.P. 15 September 2020. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “Pfizer CEO says he would’ve released vaccine data before election if possible”. Axios. 9 November 2020. Archived from the original on 10 November 2020. Retrieved 11 November 2020.
- ^ Jump up to:a b Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (October 2020). “Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults”. Nature. 586(7830): 589–593. Bibcode:2020Natur.586..589M. doi:10.1038/s41586-020-2639-4. PMID 32785213. S2CID 221126922.
- ^ Hannah B (7 October 2020). “EMA begins rolling review of BNT162b2 COVID-19 vaccine”. European Pharmaceutical Review. Archived from the original on 11 November 2020. Retrieved 11 November 2020.
- ^ Jump up to:a b “EMA Assessment Report” (PDF). Europa (web portal). 21 December 2020. Retrieved 29 December 2020.
- ^ “Clarification of Comirnaty dosage interval”. European Medicines Agency (EMA). 28 January 2021. Retrieved 28 January 2021.
- ^ “Study to Evaluate the Safety, Tolerability, and Immunogenicity of SARS CoV-2 RNA Vaccine Candidate (BNT162b2) Against COVID-19 in Healthy Pregnant Women 18 Years of Age and Older”. ClinicalTrials.gov. Retrieved 21 February 2021.
- ^ “Pfizer and BioNTech Commence Global Clinical Trial to Evaluate COVID-19 Vaccine in Pregnant Women”. pfizer.com (Press release). 18 February 2021. Retrieved 21 February2021.
- ^ Jump up to:a b Gaebler C, Nussenzweig MC (October 2020). “All eyes on a hurdle race for a SARS-CoV-2 vaccine”. Nature. 586 (7830): 501–2. Bibcode:2020Natur.586..501G. doi:10.1038/d41586-020-02926-w. PMID 33077943. S2CID 224808629.
- ^ Jump up to:a b “China’s Fosun to end BioNTech’s COVID-19 vaccine trial, seek approval for another”. Reuters. 3 November 2020. Archived from the original on 12 December 2020. Retrieved 21 November 2020.
- ^ Jump up to:a b c World Health Organization. “Messenger RNA encoding the full-length SARS-CoV-2 spike glycoprotein” (DOC). WHO MedNet. Retrieved 16 December 2020.
- ^ Pallesen J, Wang N, Corbett KS, Wrapp D, Kirchdoerfer RN, Turner HL, et al. (August 2017). “Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen”. Proceedings of the National Academy of Sciences of the United States of America. 114 (35): E7348–E7357. doi:10.1073/pnas.1707304114. PMC 5584442. PMID 28807998.
- ^ Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, et al. (April 2019). “Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening”. Molecular Therapy. 27 (4): 824–836. doi:10.1016/j.ymthe.2018.12.011. PMC 6453560. PMID 30638957.
- ^ Jump up to:a b “Pfizer-BioNTech COVID-19 vaccine: Health Canada recommendations for people with serious allergies”. Health Canada. 12 December 2020.
- ^ Jump up to:a b c d Comirnaty: Product Information (PDF) (Report). European Medicines Agency(EMA). Retrieved 23 December 2020.
- ^ Public Assessment Report Authorisation for Temporary Supply COVID-19 mRNA Vaccine BNT162b2 (BNT162b2 RNA) concentrate for solution for injection (PDF). Regulation 174(Report). Medicines and Healthcare products Regulatory Agency (MHRA). 15 December 2020.
- ^ “UK medicines regulator gives approval for first UK COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 2 December 2020. Archived from the original on 2 December 2020. Retrieved 2 December 2020.
- ^ Neergaard L, Kirka D (2 December 2020). “Britain OKs Pfizer vaccine and will begin shots within days”. Associated Press. Archived from the original on 6 December 2020. Retrieved 6 December 2020.
- ^ Mueller B (2 December 2020). “U.K. Approves Pfizer Coronavirus Vaccine, a First in the West”. The New York Times. Retrieved 2 December 2020.
- ^ Roberts M (2 December 2020). “Covid Pfizer vaccine approved for use next week in UK”. BBC News. Archived from the original on 2 December 2020. Retrieved 2 December 2020.
- ^ Henley J, Connolly, Jones S (3 December 2020). “European and US experts question UK’s fast-track of Covid vaccine”. The Guardian. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ “First patient receives Pfizer Covid-19 vaccine”. BBC. 8 December 2020. Archivedfrom the original on 8 December 2020. Retrieved 8 December 2020.
- ^ “Vaccine vials and a virtual hug: a history of coronavirus in 15 objects”. The Guardian. 21 February 2021. Retrieved 22 February 2021.
- ^ “COVID-19 Vaccination Statistics –Week ending Sunday 20th December 2020” (PDF). NHS. 24 December 2020.
- ^ “Coronavirus en la Argentina: La ANMAT aprobo el uso de emergencia de la vacuna Pfizer”. La Nación (in Spanish). Retrieved 25 December 2020.
- ^ “TGA provisionally approves Pfizer COVID-19 vaccine”. Therapeutic Goods Administration (Press release). 25 January 2021. Retrieved 26 January 2021.
- ^ “Bahrain becomes second country to approve Pfizer COVID-19 vaccine”. Al Jazeera. Retrieved 5 December 2020.
- ^ “Drug and vaccine authorizations for COVID-19: List of applications received”. Health Canada. 9 December 2020. Retrieved 9 December 2020.
- ^ Jump up to:a b “Chile y Ecuador se adelantan en Sudamérica y autorizan la vacuna de Pfizer”. El Pais. Retrieved 17 December 2020.
- ^ “First Pfizer COVID-19 vaccines set to reach Costa Rica on Wednesday – president”. Reuters. 23 December 2020. Retrieved 24 December 2020.
- ^ “SFH authorises COVID-19 vaccine by Fosun Pharma/BioNTech for emergency use in Hong Kong”. The Government of Hong Kong (Press release). 25 January 2021. Retrieved 26 January 2021.
- ^ “Iraq grants emergency approval for Pfizer COVID-19 vaccine”. MSN. Retrieved 27 December 2020.
- ^ “Israeli Health Minister ‘pleased’ as FDA approves Pfizer COVID-19 vaccine”. The Jerusalem Post. Retrieved 28 December 2020.
- ^ “Jordan approves Pfizer-BioNTech Covid vaccine”. France 24. 15 December 2020. Retrieved 15 December 2020.
- ^ “Kuwait authorizes emergency use of Pfizer-BioNTech COVID-19 vaccine”. Arab News. 13 December 2020. Retrieved 15 December 2020.
- ^ “Mexico Approves Pfizer Vaccine for Emergency Use as Covid Surges”. Bloomberg. 12 December 2020. Retrieved 12 December 2020.
- ^ “Oman issues licence to import Pfizer BioNTech Covid vaccine – TV”. Reuters. 15 December 2020. Retrieved 16 December 2020.
- ^ “Panama approves Pfizer’s COVID-19 vaccine – health ministry”. Yahoo! Finance. Retrieved 16 December 2020.
- ^ “PH authorizes Pfizer’s COVID-19 vaccine for emergency use”. CNN Philippines. 14 January 2021.
- ^ “Qatar, Oman to receive Pfizer-BioNTech COVID-19 vaccine this week”. Reuters. Retrieved 24 December 2020.
- ^ “Saudi Arabia to Launch Its Coronavirus Vaccination Program” (in Spanish). Boomberg. Retrieved 17 December 2020.
- ^ Abdullah Z (14 December 2020). “Pfizer-BioNTech COVID-19 vaccine approved by Singapore, first shipment expected by end-December”. CNA. Retrieved 16 January 2021.
- ^ “Singapore approves use of Pfizer’s COVID-19 vaccine”. AP News. 14 December 2020. Retrieved 15 December 2020.
- ^ “Dubai approves the Pfizer-BioNTech vaccine which will be free of charge”. Emirates Woman. 23 December 2020. Retrieved 28 December 2020.
- ^ “WHO issues its first emergency use validation for a COVID-19 vaccine and emphasizes need for equitable global access”. World Health Organization (WHO) (Press release). 31 December 2020. Retrieved 6 January 2021.
- ^ Jump up to:a b “Emergency Use Authorization for vaccines explained”. U.S. Food and Drug Administration (FDA). 20 November 2020. Archived from the original on 20 November 2020. Retrieved 20 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Pfizer-BioNTech COVID-19 Vaccine EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA). 11 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Pfizer-BioNTech COVID-19 Vaccine EUA Fact Sheet for Healthcare Providers”(PDF). Pfizer. 11 December 2020.
- ^ Sun LH, Stanley-Becker I. “CDC greenlights advisory group’s decision to recommend Pfizer vaccine for use”. The Washington Post. Retrieved 14 December 2020.
- ^ Oliver SE, Gargano JW, Marin M, Wallace M, Curran KG, Chamberland M, et al. (December 2020). “The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Pfizer-BioNTech COVID-19 Vaccine — United States, December 2020” (PDF). MMWR. Morbidity and Mortality Weekly Report. 69 (50): 1922–24. doi:10.15585/mmwr.mm6950e2. PMC 7745957. PMID 33332292.
- ^ “COVID-19: Switzerland can start vaccinating vulnerable groups already in December”(Press release). Federal Office of Public Health. 19 December 2020. Retrieved 19 December 2020.
- ^ Erni S (23 December 2020). “90-jährige Luzernerin als erste Person in der Schweiz gegen Corona geimpft”. Neue Luzerner Zeitung. Retrieved 23 December 2020.
- ^ Pralong J (23 December 2020). “La piqûre de l’espoir pratiquée à Lucerne”. Heidi.news. Retrieved 23 December 2020.
- ^ Jump up to:a b “EMA recommends first COVID-19 vaccine for authorisation in the EU”. European Medicines Agency (EMA) (Press release). 21 December 2020. Retrieved 21 December2020.
- ^ “Comirnaty”. Union Register of medicinal products. Retrieved 8 January 2021.
- ^ “Statement by President von der Leyen on the marketing authorisation of the BioNTech-Pfizer vaccine against COVID-19”. European Commission. Retrieved 21 December 2020.
- ^ Cancian, Natália (23 February 2021). “Anvisa aprova registro da vacina da Pfizer contra Covid”. Folha de S. Paulo (in Portuguese). Retrieved 23 February 2021.
- ^ McKenna M (17 December 2020). “Vaccines Are Here. We Have to Talk About Side Effects”. Wired. Retrieved 23 December 2020.
- ^ Jump up to:a b CDC COVID-19 Response Team, Food and Drug Administration (January 2021). “Allergic Reactions Including Anaphylaxis After Receipt of the First Dose of Pfizer-BioNTech COVID-19 Vaccine — United States, December 14–23, 2020” (PDF). MMWR. Morbidity and Mortality Weekly Report. 70 (2): 46–51. doi:10.15585/mmwr.mm7002e1. PMC 7808711. PMID 33444297.
- ^ Jump up to:a b c “COVID-19 vaccine safety update: COMIRNATY” (PDF). European Medicines Agency. 28 January 2021.
- ^ Bostock N (9 December 2020). “MHRA warning after allergic reactions in NHS staff given COVID-19 vaccine”. GP. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Booth W, Cunningham E (9 December 2020). “Britain warns against Pfizer vaccine for people with history of ‘significant’ allergic reactions”. The Washington Post. Archivedfrom the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Cabanillas B, Akdis C, Novak N (December 2020). “Allergic reactions to the first COVID-19 vaccine: a potential role of Polyethylene glycol?”. Allergy. doi:10.1111/all.14711. PMID 33320974. S2CID 229284320.
- ^ “First COVID-19 vaccine safety update published”. European Medicines Agency (EMA)(Press release). 28 January 2021. Retrieved 29 January 2021.
- ^ Gray B (23 November 2020). “Pfizer’s Chesterfield workforce playing a key role in coronavirus vaccine development”. St. Louis Post-Dispatch.
- ^ Jump up to:a b c d Johnson CY (17 November 2020). “A vial, a vaccine and hopes for slowing a pandemic — how a shot comes to be”. The Washington Post. Retrieved 21 December2020.
- ^ Hughes M (20 December 2020). “Andover’s piece of the vaccine: Pfizer”. The Eagle-Tribune.
- ^ Shamus KJ (13 December 2020). “Historic journey: Pfizer prepares to deliver 6.4 million doses of COVID-19 vaccines”. Detroit Free Press.
- ^ Mullin R (25 November 2020). “Pfizer, Moderna ready vaccine manufacturing networks”. Chemical & Engineering News. Washington, D.C.: American Chemical Society. Retrieved 21 December 2020.
- ^ Jump up to:a b c Weise, Elizabeth (7 February 2021). “Pfizer expects to cut COVID-19 vaccine production time by close to 50% as production ramps up, efficiencies increase”. USA Today.
- ^ “BioNTech to Acquire GMP Manufacturing Site to Expand COVID-19 Vaccine Production Capacity in First Half 2021 | BioNTech”. investors.biontech.de. Retrieved 5 February2021.
- ^ “Statement on Manufacturing | BioNTech”. investors.biontech.de. Retrieved 5 February2021.
- ^ Erman M, Ankur B (22 July 2020). “U.S. to pay Pfizer, BioNTech $1.95 bln for millions of COVID-19 vaccine doses”. Reuters. Archived from the original on 22 July 2020. Retrieved 22 July 2020.
- ^ “U.S. Government Engages Pfizer to Produce Millions of Doses of COVID-19 Vaccine”. US Department of Health and Human Services. 22 July 2020. Archived from the original on 22 July 2020. Retrieved 23 July 2020.
- ^ Nazaryan A (9 November 2020). “So is Pfizer part of Operation Warp Speed or not? Yes and no”. Yahoo!. Archived from the original on 10 November 2020. Retrieved 9 November 2020.
- ^ Pleitgen F (11 November 2020). “EU agrees to buy 300 million doses of the Pfizer/BioNTech Covid-19 vaccine”. CNN. Archived from the original on 24 November 2020. Retrieved 26 November 2020.
- ^ “Japan and Pfizer reach COVID-19 vaccine deal to treat 60 million people”. The Japan Times. 1 August 2020. Archived from the original on 10 November 2020. Retrieved 21 November 2020.
- ^ Tasker JP (9 November 2020). “Trudeau says promising new Pfizer vaccine could be ‘light at the end of the tunnel'”. CBC News. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ “Pfizer and BioNTech to Supply Singapore with their BNT162b2 mRNA-based Vaccine Candidate to Combat COVID-19”. pfizer.com.sg. Pfizer Singapore. 14 December 2020. Retrieved 1 February 2021.
- ^ de Salud S. “233. Firma secretario de Salud convenio con Pfizer para fabricación y suministro de vacuna COVID-19”. gob.mx (in Spanish). Retrieved 17 December 2020.
- ^ Ng E (27 August 2020). “Fosun Pharma to supply Covid-19 vaccine to Hong Kong, Macau once approved”. South China Morning Post. Archived from the original on 20 November 2020. Retrieved 21 November 2020.
- ^ Ting V, Lau C, Wong O (11 December 2020). “Hong Kong buys 15 million Covid-19 vaccine doses from Sinovac, Pfizer”. South China Morning Post. Retrieved 18 December2020.
- ^ “BioNTech and Fosun Pharma to Supply China with mRNA-based COVID-19 Vaccine”(Press release). BioNTech. 16 December 2020. Retrieved 16 December 2020.
- ^ “Pfizer-BioNTech COVID-19 Vaccine Frequently Asked Questions”. U.S. Food and Drug Administration. 11 December 2020. Retrieved 29 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Extra dose from vials of Comirnaty COVID-19 vaccine”. European Medicines Agency (EMA). 8 January 2021. Retrieved 8 January 2021.
- ^ “AIFA, possibile ottenere almeno 6 dosi da ogni flaconcino del vaccino BioNTech/Pfizer”. aifa.gov.it (in Italian). Retrieved 29 December 2020.
- ^ “Global information about Comirnaty”. Comirnaty IE. 8 January 2021. Retrieved 16 January 2021.
- ^ “Comirnaty Package Insert” (PDF). BioNTech Manufacturing GmbH.
- ^ Rowland C (22 January 2021). “Biden wants to squeeze an extra shot of vaccine out of every Pfizer vial. It won’t be easy”. The Washington Post. Retrieved 29 January 2021.
- ^ Kollewe J. “Pfizer and BioNTech’s vaccine poses global logistics challenge”. The Guardian. Archived from the original on 10 November 2020. Retrieved 10 November2020.
- ^ Newey S (8 September 2020). “Daunting task of distribution exposed as it emerges some vaccines must be ‘deep frozen’ at −70C”. The Telegraph. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “How China’s COVID-19 could fill the gaps left by Pfizer, Moderna, AstraZeneca”. Fortune. 5 December 2020. Archived from the original on 12 December 2020. Retrieved 5 December 2020.
- ^ “Pfizer’s Vaccine Is Out of the Question as Indonesia Lacks Refrigerators: State Pharma Boss”. Jakarta Globe. 22 November 2020. Archived from the original on 7 December 2020. Retrieved 5 December 2020.
- ^ “Pfizer Has Offered South Africa Discounted Covid-19 Vaccines”. Bloomberg. 4 January 2021. Retrieved 5 January 2021.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. N Engl J Med. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)”(PDF). WHO Drug Information. 34 (3): 666. Archived (PDF) from the original on 27 November 2020. Retrieved 23 November 2020.
- ^ “Pfizer and BioNTech Receive Authorization in the European Union for COVID-19 Vaccine” (Press release). BioNTech. 21 December 2020. Retrieved 26 December 2020 – via GlobeNewswire.
- ^ Bulik BS (23 December 2020). “The inside story behind Pfizer and BioNTech’s new vaccine brand name, Comirnaty”. FiercePharma. Retrieved 25 December 2020.
- ^ “Comirnaty COVID-19 mRNA Vaccine”. Comirnaty Global. Retrieved 31 December2020.
External links
“Tozinameran”. Drug Information Portal. U.S. National Library of Medicine.
- Global Information About Pfizer–BioNTech COVID‑19 Vaccine (also known as BNT162b2) Pfizer
- Comirnaty assessment report European Medicines Agency Committee for Medicinal Products for Human Use
- A Phase 1/2/3 Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID‑19 in Healthy Individuals Pfizer clinical protocol
- Pfizer Vaccince News, updates and tracking of Israel’s vaccinaion campaign
- “How the Pfizer-BioNTech Covid-19 Vaccine Works”. The New York Times.
| A vial of the Pfizer–BioNTech COVID‑19 vaccine | |
| Vaccine description | |
|---|---|
| Target disease | COVID‑19 |
| Type | mRNA |
| Clinical data | |
| Trade names | Comirnaty[1][2] |
| Other names | BNT162b2, COVID-19 mRNA vaccine (nucleoside-modified) |
| License data | EU EMA: by INNUS DailyMed: Pfizer-BioNTech_COVID-19_Vaccine |
| Pregnancy category | AU: B1[3] |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [4][5]CA: Authorized by interim order [6][7]UK: Conditional and temporary authorization to supply [8][9]US: Unapproved (Emergency Use Authorization)[10][11][12]EU: Conditional marketing authorization granted [2]CH: Rx-only[further explanation needed][1] |
| Identifiers | |
| CAS Number | 2417899-77-3 |
| PubChem SID | 434370509 |
| DrugBank | DB15696 |
| UNII | 5085ZFP6SJ |
| KEGG | D11971 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
/////////
#Tozinameran, #APPROVALS 2021, #JAPAN 2021, Comirnaty, #Coronavirus disease, #COVID-19, #BNT162b2 , #BNT162b2, #SARS-CoV-2 Vaccine, #RNA ingredient BNT-162B2, #corona
The Pfizer-BioNTech COVID-19 vaccine (Tozinameran, INN), also known as BNT162b2, is one of four advanced mRNA-based vaccines developed through “Project Lightspeed,” a joint program between Pfizer and BioNTech.2,3 Tozinameran is a nucleoside modified mRNA (modRNA) vaccine encoding an optimized full-length version of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) protein. It is designed to induce immunity against SARS-CoV-2, the virus responsible for causing COVID-19.2 The modRNA is formulated in lipid nanoparticles for administration via intramuscular injection in two doses, three weeks apart.1,3
Tozinameran is undergoing evaluation in clinical trials in both the USA (NCT04368728) and Germany (NCT04380701).4,5 Tozinameran received fast track designation by the U.S. FDA on July 13, 2020.6 On December 11, 2020, the FDA issued an Emergency Use Authorization (EUA) based on 95% efficacy in clinical trials and a similar safety profile to other viral vaccines over a span of approximately 2 months.1 Tozinameran was granted an EUA in the UK on December 2, 2020,8 and in Canada on December 9, 20207 for active immunization against SARS-CoV-2.12
Currently, sufficient data are not available to determine the longevity of protection against COVID-19, nor direct evidence that the vaccine prevents the transmission of the SARS-CoV-2 virus from one individual to another.9 Fact sheets for caregivers, recipients, and healthcare providers are now available.10,11
Tozinameran has not yet been fully approved by any country. In both the UK and Canada, Tozinameran is indicated under an interim authorization for active immunization to prevent COVID-19 caused by SARS-CoV-2 in individuals aged 16 years and older.7,8
On December 11, 2020, the U.S. Food and Drug Administration granted emergency use authorization (EUA) for Tozinameran to prevent COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in patients aged 16 years and above.9 Safety and immune response information for adolescents 12-15 years of age will follow, and studies to further explore the administration of Tozinameran in pregnant women, children under 12 years of age, and those in special risk groups will be evaluated in the future.1
This vaccine should only be administered where appropriate medical treatment for immediate allergic reactions are immediately available in the case of an acute anaphylactic reaction after vaccine administration.12 Tozinameran administration should be postponed in any individual suffering from an acute febrile illness. Its use should be carefully considered in immunocompromised individuals and individuals with a bleeding disorder or on anticoagulant therapy. Appropriate medical treatment should be readily available in case of an anaphylactic reaction following vaccine administration.7,8
Tozinameran contains nucleoside modified mRNA (modRNA) encapsulated in lipid nanoparticles that deliver the modRNA into host cells. The lipid nanoparticle formulation facilitates the delivery of the RNA into human cells.12 Once inside these cells, the modRNA is translated by host machinery to produce the SARS-CoV-2 spike (S) protein antigen, which is subsequently recognized by the host immune system. Tozinameran has been shown to elicit both neutralizing antibody and cellular immune responses to the S protein, which helps protect against subsequent SARS-CoV-2 infection.7,8
Tozinameran is a nucleoside modified mRNA (modRNA) vaccine encoding an optimized full-length version of the SARS-CoV-2 spike (S) protein, translated and expressed in cells in vaccinated individuals to produce the S protein antigen against which an immune response is mounted. As with all vaccines, protection cannot be guaranteed in all recipients, and full protection may not occur until at least seven days following the second dose.7,8
In U.S. clinical trials, the vaccine was 95% effective in preventing COVID-19; eight COVID-19 cases occurred in the vaccine group and 162 cases occurred in the placebo group. Of the total 170 COVID-19 cases, one case in the vaccine group and three cases in the placebo group were considered to be severe infections.1,9
- Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Perez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW Jr, Hammitt LL, Tureci O, Nell H, Schaefer A, Unal S, Tresnan DB, Mather S, Dormitzer PR, Sahin U, Jansen KU, Gruber WC: Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020 Dec 10. doi: 10.1056/NEJMoa2034577. [PubMed:33301246]
- Gen Eng News: BNT162 vaccine candidates [Link]
- BioNTech BNT162 Update [Link]
- Clinical Trial NCT04368728 [Link]
- Clinical Trial NCT04380701 [Link]
- FDA fast track designation: BNT162b1 and BNT162b2 [Link]
- Health Canada Interim Product Monograph: BNT162b2 SARS-CoV-2 Vaccine [Link]
- MHRA Interim Product Monograph: BNT162b2 SARS-CoV-2 Vaccine [Link]
- FDA News Release: FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine [Link]
- Pfizer: Fact Sheet for Healthcare Providers Administering Vaccine, Pfizer-BioNtech COVID-19 vaccine [Link]
- Pfizer: Fact Sheet for Recipients and Caregivers, Pfizer BioNTech COVID-19 vaccine [Link]
- FDA Emergency Use Authorization: Full EUA Prescribing information, Pfizer-BioNTech COVID-19 vaccine [Link]
-
PHASESTATUSPURPOSECONDITIONSCOUNT2Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)12, 3Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)11, 2Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)11, 2RecruitingTreatmentCoronavirus Disease 2019 (COVID‑19) / Protection Against COVID-19 and Infections With SARS CoV 2 / Respiratory Tract Infections (RTI) / RNA Virus Infections / Vaccine Adverse Reaction / Viral Infections / Virus Diseases1

{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}