
Home » Posts tagged 'APPROVALS 2021' (Page 4)
Tag Archives: APPROVALS 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Upacicalcet sodium hydrate
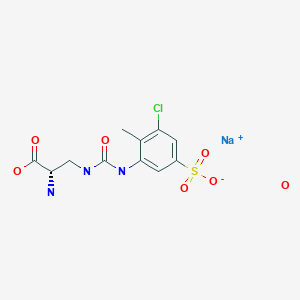
Upacicalcet sodium hydrate, ウパシカルセトナトリウム水和物
CAS 2052969-18-1
1333218-50-0 free
PMDA JAPAN APPROVED 2021/6/23, Upasita
Calcium sensing receptor agonist
(2S)-2-amino-3-[(3-chloro-2-methyl-5-sulfophenyl)carbamoylamino]propanoic acid
| Formula | C11H13ClN3O6S. Na. xH2O |
|---|
- OriginatorAjinomoto Pharma
- DeveloperSanwa Kagaku Kenkyusho
- ClassAmines; Chlorobenzenes; Propionic acids; Small molecules; Sulfonic acids; Toluenes
- Mechanism of ActionCalcium-sensing receptor agonists
- RegisteredSecondary hyperparathyroidism
- 25 Jun 2021Chemical structure information added
- 23 Jun 2021Sanwa Kagaku Kenkyusho and Kissei Pharmaceutical agree to co-promote upacicalcet in Japan for Secondary hyperparathyroidism
- 23 Jun 2021Registered for Secondary hyperparathyroidism in Japan (IV) – First global approval
| Upacicalcet Sodium HydrateMonosodium 3-({[(2S)-2-amino-2-carboxyethyl]carbamoyl}amino)-5-chloro-4-methylbenzenesulfonate hydrateC11H13ClN3NaO6S▪xH2O [2052969-18-1 , anhydride] |
Announcement of Marketing Authorization Approval in Japan and Co-promotion Agreement of UPASITA® IV Injection Syringe for the Treatment of Secondary Hyperparathyroidism in Dialysis Patients
SANWA KAGAKU KENKYUSHO Co., Ltd. (Head Office: Nagoya, President and CEO : Shusaku Isono, Suzuken Group, ; “SANWA KAGAKU”) has received Marketing Authorization approval today for UPASITA® IV Injection Syringes (generic name: Upacicalcet Sodium Hydrate; “UPASITA®”) for the treatment of secondary hyperparathyroidism in patients on hemodialysis.
UPASITA® was created by Ajinomoto Pharmaceuticals Co., Ltd. (currently EA Phama Co., Ltd.) and developed by SANWA KAGAKU for the treatment of secondary hyperparathyroidism under a licensing agreement with EA Pharma. UPASITA® acts on calcium sensing receptor in the parathyroid and suppresses excessive secretions of parathyroid hormones (PTH). UPASITA® is administered by intravenous injection to dialysis patients through dialysis circuit by physicians or medical staffs upon completion of dialysis and such administration is expected to reduce the burden of patients with many oral medications whose drinking water volume is severely restricted.
Regarding provision of medical and drug information, SANWA KAGAKU entered into a co-promotion agreement in Japan with Kissei Pharmaceutical Co., Ltd. (Head Office: Matsumoto, Nagano; Chairman and CEO: Mutsuo Kanzawa ; “Kissei”). SANWA KAGAKU will handle the production, marketing, and distribution of the Product while SANWA KAGAKU and Kissei collaboratively promote it to medical institutions in the field in accordance with the agreement. Through the co-promotion activity in the field, SANWA KAGAKU and Kissei will contribute to the treatment of dialysis patients suffering from secondary hyperparathyroidism.
《Reference》
About secondary hyperparathyroidism (SHPT)
SHTP is one of complications that occur as chronic kidney disease (chronic kidney failure) progresses and is a pathological condition where excessive PTH is secreted by the parathyroid gland. It has been reported that excessive secretion of parathyroid hormone promotes efflux of phosphorus and calcium from the bone into the blood, thereby increasing the risk of developing bone fractures and arteriosclerosis due to calcification of the cardiovascular system and affecting the vital prognosis.
Product Summary of UPASITA® IV Injection Syringe for Dialysis
Brand name:
UPASITA® IV Injection Syringe for Dialysis 25μg
UPASITA® IV Injection Syringe for Dialysis 50μg
UPASITA® IV Injection Syringe for Dialysis 100μg
UPASITA® IV Injection Syringe for Dialysis 150μg
UPASITA® IV Injection Syringe for Dialysis 200μg
UPASITA® IV Injection Syringe for Dialysis 250μg
UPASITA® IV Injection Syringe for Dialysis 300μg
Generic Name (JAN):
Upacicalcet Sodium Hydrate
Date of Marketing Approval:
June 23, 2021
Indications:
Secondary hyperparathyroidism in patients on hemodialysis
Dosage and Administration:
In adults, UPASITA® is usually administered into venous line of the dialysis circuit at the end of dialysis session during rinse back at a dose of 25 μg sodium upacicalcet 3 times a week as a starting dose.
The starting dose can be 50 μg depending on the concentration of serum calcium. Thereafter, the dose may be adjusted in a range from 25 to 300 μg while parathyroid hormone (PTH) and serum calcium level should be carefully monitored in patients.
SYN
WO 2020204117

PATENT
WO 2011108724
WO 2011108690
JP 2013063971
WO 2016194881
JP 6510136
PATENT
WO 2016194881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016194881&tab=FULLTEXT(Example 1) Synthesis of
(2S) -2-amino-3-{[(5-chloro-2-hydroxy-3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 1 )
[Chemical formula 14]CDI 150. 2 g (926.6Mmol, 1.1 eq. vs Boc-DAP-O t Bu) to and stirred at 5 ° C. acetone was added 750mL (3.0L / kg). 250 g (842.6 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, completion of the IC (imidazolylcarbonylation) reaction was confirmed by HPLC. 282.6 g (1263.8 mmol, 1.5 eq.) Of ACHB was added in 3 portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 124.5 mL (1432.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1000 mL (4.0 L / kg) of acetone. The filtrate was concentrated to 1018 g (4.1 kg / kg), the temperature was raised to 50 ° C., and 625.0 mL (7187 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 750 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1730 g (6.9 kg / kg) to precipitate a solid. After stirring at 20 ° C. for 14 hours, vacuum filtration was performed. The filtered solid was washed with 500 mL (2.0 L / kg) of acetone and then dried under reduced pressure at 60 ° C. for 6 hours to obtain 201.4 g of the target product (64.5%).
1H-NMR (400MHz, DMSO-d6): δ 8.3 (s, 1H), 8.2 (bs, 3H), 8.1 (d, 1H, J = 2.6Hz), 7.3 (t, 1H, J = 6.0Hz), 7.0 (d, 1H, J = 2.6Hz), 4.0-4.1 (m, 1H), 3.6-3.7 (m, 1H), 3.4-3.5 (m, 1H)[0026](Example 2) Synthesis of
(2S) -2-amino-3-{[(3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 2 )
[Chemicalformula 15] CDI 120.2 g (741.2 mmol, 1. 600 mL (3.0 L / kg) of acetone was added to 1 eq. Vs Boc-DAP-OtBu), and the mixture was stirred at 5 ° C. 200 g (673.9 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 100 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 175.0 g (1010.8 mmol, 1.5 eq.) Of ABS was added in 3 portions and washed with 100 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 99.6 mL (1145.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1400 mL (7.0 L / kg) of acetone. The filtrate was concentrated to 800.1 g (4.0 kg / kg), heated to 50 ° C., and then 500.0 mL (5750.0 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 600 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1653.7 g to precipitate a solid. After aging at 20 ° C. for 15 hours, vacuum filtration was performed. The filtered solid was washed with 400 mL (2.0 L / kg) of acetone and then dried under reduced pressure at room temperature for 6 hours to obtain 140.3 g of the desired product (net 132.2 g, 64.7%).
1H-NMR (400MHz, DMSO-d6): δ 8.8 (s, 1H), 8.2 (bs, 3H), 7.7 (s, 1H), 7.3-7.4 (m, 1H), 7.1-7.2 (m, 2H) , 6.3-6.4 (bs, 1H), 4.0-4.1 (bs, 1H), 3.6-3.7 (bs, 1H), 3.5-3.6 (bs, 1H)[0027](Example 3) Synthesis of
(2S) -2-amino-3-{[(3-chloro-2-methyl-5-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 3 )
[Chemical formula 16]CDI 14. To 4 g (88.8 mmol, 1.05 eq. Vs Boc-DAP-OtBu), 75 mL (3.0 L / kg vs DAP-OtBu) of acetone was added and stirred at 5 ° C. After adding 25 g (84.3 mmol) of Boc-DAP-OtBu in two portions and stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 26.1 g (118.0 mmol, 1.4 eq.) Of ACTS was added in 3 portions and washed with 25 mL (1.0 L / kg) of acetone. After the temperature was raised to 30 ° C., the mixture was stirred overnight, and the completion of the urea conversion reaction was confirmed by HPLC. After concentrating under reduced pressure at 10 kPa and 40 ° C. until the solvent was completely removed, 37.5 mL (1.5 L / kg) of water and 22.8 mL (257.6 mmol) of concentrated hydrochloric acid were added to perform deprotection for 2 hours. After confirming the completion of the reaction by HPLC, the mixture was cooled to 5 ° C., 60 mL (2.4 L / kg) of MeCN was added, and the mixture was stirred overnight. Further, when 120 mL (4.8 L / kg) of MeCN was added, stratification occurred, so 10 mL (0.4 L / kg) of water and 2.5 mL (0.1 L / kg) of MeCN were added. The precipitated solid was filtered under reduced pressure, washed with 60 mL of MeCN / water (1/2), and then dried under reduced pressure at 60 ° C. for 14 hours to obtain 20.1 g of the desired product as a white solid (net18.3 g, yield 61). 0.8%).
1H-NMR (400MHz, DMSO-d6): δ 14.70-13.30 (bs, 1H), 8.27 (bs, 3H), 8.15 (s, 1H), 7.98 (d, 1H, J = 1.6Hz), 7.27 (d , 1H, J = 1.6Hz), 6.82 (t, 1H, J = 6.0Hz), 4.04 (bs, 1H), 3.70-3.60 (m, 1H), 3.60-3.50 (m, 1H), 2.22 (s, 3H)[0028](Example 4) Synthesis of
compound 3 using phenylchloroformate as a carbonyl group-introducing reagent
(Step 1)
[Chemicalformula 17] MeCN 375 mL (7.5 L / kg vs ACTS), Py for 50 g (225.6 mmol) of ACTS. 38.1 mL (473.7 mmol, 2.1 eq.) Was added and stirred at 25 ° C. 29.9 mL (236.8 mmol, 1.05 eq.) Of ClCO 2 Ph (phenyl chloroformate) was added dropwise, and after stirring for 30 minutes, completion of the CM (carbamate) reaction was confirmed by HPLC. 68.9 g (232.4 mmol) of Boc-DAP-OtBu was added, 97.5 mL (699.3 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. The completion of the urea conversion reaction was confirmed by HPLC. Here, 103.5 g of the total amount of 517.43 g was used to move to the next step (down to ACTS 10 g scale).
30 mL of water was added and concentrated to 77.0 g at 40 ° C. and 5 kPa. After 100 mL (10 L / kg) of AcOEt was added and the liquid separation operation was performed, 30 mL of water was added to the organic layer and the liquid separation operation was performed again. The organic layer was concentrated to 47.6 g at 40 ° C. and 10 kPa, and then 15 mL (1.5 L / kg) of AcOEt and 100 mL (10 L / kg) of THF were added. Again, it was concentrated to 50.7 g and THF was added up to 146 g. When it was concentrated again to 35.5 g and added to AcOEt 30 mL (3 L / kg) and THF 100 mL (10 L / kg), a solid was precipitated. It was cooled to 5 ° C. and aged overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of THF, and then dried under reduced pressure at 40 ° C. for 3 hours overnight at 30 ° C. to obtain 24.9 g of the desired product as a white solid (net). 23.0 g, 83.6%).
1 H-NMR (400MHz, DMSO-d6): δ 8.86 (bs, 1H), 8.09 (s, 1H), 7.88 (s, 1H), 7.25 (d, 1H, J = 1.6Hz), 7.14 (d, 1H, J = 7.6Hz), 6.60 (t, 1H, J = 5.6Hz), 4.00-3.90 (m, 1H), 3.60-3.50 (m, 1H), 3.30-3.20 (m, 1H), 3.15-3.05 (m, 6H), 2.19 (s, 3H), 1.50-1.30 (m, 18H), 1.20-1.10 (m, 9H)
(Step 2)
[Chemical
formula 18] Compound 4 21.64 g (net. 20.0 g, 68 mL of water (3.4 L / kg vs. compound 4) vs. 32.8 mmol) ) Was added, the mixture was stirred at 50 ° C., and 12 mL (135.6 mmol, 4.1 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 1 hour, the temperature was raised to 70 ° C. to dissolve the precipitated solid. After confirming the completion of the reaction by HPLC, the mixture was cooled to 50 ° C. and aged for 1 hour, and then cooled to 5 ° C. over 4 hours. The precipitated solid was filtered under reduced pressure, washed with 40 mL (2.0 L / kg) of MeCN / water (2/1), and then dried under reduced pressure at 60 ° C. for 3 hours to obtain 11.2 g of the desired product as a white solid (11.2 g). net 10.5 g, 91.1%).[0029](Example 5)
[Chemicalformula 19] MeCN 10.0 mL (10.0 L / kg vs ACSS), Py 0.75 mL (9.25 mmol, 2.05 eq.) For 1.00 g (4.51 mmol) of ACTS. , And stirred at 8 ° C. After dropping 0.59 mL (4.74 mmol, 1.05 eq.) Of ClCO 2 Ph, raising the temperature to room temperature and stirring for 1 hour, completion of the CM conversion reaction was confirmed by HPLC. 1.33 g (4.51 mmol, 1.0 eq.) Of Boc-DAP-OtBu was added, 1.92 mL (13.76 mmol, 3.05 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 1 hour. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 1.0 mL of water and 2.0 mL of concentrated hydrochloric acid (22.6 mmol, 5.0 eq.) Were added, and the mixture was stirred at 50 ° C. for 4 hours. After confirming the completion of deprotection by HPLC, MeCN 7.5 mL (7.5 L / kg), 1 M HCl aq. After adding 4.5 mL, the mixture was stirred at 5 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 3.0 mL (3.0 L / kg) of MeCN, and then dried at 60 ° C. overnight to obtain 1.28 g of the desired product as a white solid (net 1.18 g, 77). .0%).[0030](Example 6)
(Step 1)
3-({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid ( Synthesis of Compound 5 )
[Chemical formula 20] To5 g (22.56 mmol) of ACTS, 37.5 mL (7.5 L / kg vs ACTS) of MeCN and 3.81 mL (47.38 mmol, 2.1 eq.) Of Py were added. The mixture was stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the CM reaction was confirmed by HPLC. 5.92 g (23.23 mmol, 1.03 eq.) Of Boc-DAP-OMe was added, 9.75 mL (69.93 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. 0.4 g (1.58 mmol, 0.07 eq.) Of Boc-DAP-OMe and 0.22 mL (1.58 mmol, 0.07 eq.) Of TEA were added, and the completion of the ureaization reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of MeCN and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / MeCN (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the target product as a white solid (772 g of the target product). net 7.20 g, 87.3%).
1H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) , 2.21 (s, 3H)
HRMS (FAB – ): calcd for m / z 364.0369 (MH), found The m / z 364.0395 (MH)
(step 2)
[Formula 21]
compound 5 10.64 g (net Non 10.0 g, To 27.34 mmol), 18 mL of water (1.8 L / kg vs. compound 5 ) was added and stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. The pH was adjusted to 5.8 by adding about 3.55 mL. After confirming the precipitation of the target product by dropping 65 mL (6.5 L / kg) of IPA, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of IPA was added dropwise and aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of IPA, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92. 6%).
1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)[0031](Example 7)
(Step 1)
[Chemicalformula 22] For 10.0 g (45.1 mmol) of ACTS, 50 mL (5.0 L / kg vs ACTS) of MeCN, 7.46 mL (92.5 mmol, 2.05 eq. ) Was added, and the mixture was stirred at 8 ° C. 5.98 mL (47.4 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, the temperature was raised to 25 ° C., and the mixture was stirred for 1 hour, and then the completion of the CM reaction was confirmed by HPLC. 100 ml of acetone (10.0 L / kg vs ACTS) was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 30 mL of acetone (3.0 L / kg vs ACTS), and then dried under reduced pressure at 60 ° C. for 2 hours to obtain 17.8 g of the target product (net 14.4 g as a free form). Quant).
1 H-NMR (400MHz, DMSO-d6): δ 9.76 (bs, 1H), 8.93-8.90 (m, 2H), 8.60-8.50 (m, 1H), 8.10-8.00 (m, 2H), 7.60 (s , 1H), 7.50-7.40 (m, 3H), 7.30-7.20 (m, 3H), 2.30 (s, 3H)
(Step 2)
[Chemical 23]
Compound 6 To 5.0 g (11.9 mmol), 50 ml of acetonitrile and 3.53 g (11.9 mmol) of Boc-DAP-OtBu were added, and the mixture was stirred at 8 ° C. 3.5 ml (25 mmol) of triethylamine was added dropwise, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and 25 ml of ethyl acetate and 5 ml of water were added for extraction. The organic layer was washed with 5 ml of water, the solvent was distilled off, 50 ml of tetrahydrofuran was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 10 ml of tetrahydrofuran, and dried under reduced pressure at 60 ° C. overnight to obtain 6.3 g of the desired product as a white solid.[0032](Example 8)
[Chemicalformula 24] For 1.08 g (4.89 mmol) of ACTS, 8.1 mL (7.5 L / kg vs ACTS) of MeCN and 827 μL (10.27 mmol, 2.1 eq.) Of Py were added. In addition, it was stirred at room temperature. ClCO 2 Ph 649 μL (5.14 mmol, 1.05 eq.) Was added dropwise, and the mixture was stirred for 30 minutes, and then the completion of the CM conversion reaction was confirmed by HPLC. 1.48 g (5.04 mmol, 1.03 eq.) Of Cbz-DAP-OMe HCl was added, 2.1 mL (15.17 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at room temperature for about 5 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 15.0 mL of 30% HBr / AcOH was added, and the mixture was stirred at room temperature for 70 minutes, and the completion of deprotection was confirmed by HPLC. After concentration to dryness, 10 mL of water and 4 mL of AcOEt were added to carry out an extraction operation, and then the aqueous layer was stirred at room temperature overnight. The precipitated solid was filtered under reduced pressure, washed with 15 mL of water and 10 mL of AcOEt, and then dried at 40 ° C. for 3 hours to obtain 1.45 g of the desired product as a white solid (58.8%).[0033](Example 9) Synthesis of compound 7 ( methyl ester of compound 1 )
using phenyl chloroformate as a carbonyl group introduction reagent [Chemical formula 25] MeCN 73 mL (14.6 L) with respect to 5.00 g (22.4 mmol) of ACHB. / Kg vs ACHB), Py 3.8 mL (47 mmol, 2.1 eq.), Was added and stirred at 40 ° C. After adding 3.0 mL (24 mmol, 1.05 eq.) Of ClCO 2 Ph and stirring for 30 minutes, the completion of the CM conversion reaction was confirmed by HPLC. 5.87 g (23 mmol, 1.0 eq.) Of Boc-DAP-OMe was added, washed with a small amount of MeCN, 9.7 mL (70 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 3 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (112 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 7 hours. Further, 1.5 mL (23 mmol, 1.0 eq.) Of MsOH was added, and the reaction was carried out at 50 ° C. overnight. After confirming the completion of deprotection by HPLC, 90 mL of acetone was added to the reaction solution, and the mixture was cooled to room temperature. The precipitated solid was obtained and dried under reduced pressure at 60 ° C. to obtain the desired product. 1 H-NMR (400MHz, DMSO-d6): δ 7.22 (m, 1H), 7.14 (m, 1H), 4.36 (m, 1H), 3.80 (s, 3H), 3.20-3.40 (m, 2H).[0034](Example 10) Synthesis of
compound 5 using 4-chlorophenylchloroformate as a carbonyl group-introducing reagent
[Chemical formula 26] For5.00 g (22.6 mmol) of ACTS, 73 mL (14.6 L / kg vs ACTS) of MeCN, 3.8 mL (47 mmol, 2.1 eq.) Of Py was added and stirred at 40 ° C. After adding 3.25 mL (23.7 mmol, 1.05 eq.) Of 4-chloroformic acid 4-chlorophenylate and stirring at 40 ° C. for 1.5 hours, completion of the CM conversion reaction was confirmed by HPLC. Add 5.92 g (23.2 mol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 6.94 g of the desired product as a white solid (84.1%).[0035](Example 11) Synthesis of
compound 5 using 4-nitrophenyl chloroformate as a carbonyl group-introducing reagent
[Chemical formula 27]73 mL (14.6 L / kg vs. ACTS) of MeCN with respect to 5.00 g (22.6 mmol) of ACTS. , Py 3.8 mL (47 mmol, 2.1 eq.), And stirred at 40 ° C. 4.77 mL (23.7 mmol, 1.05 eq.) Of 4-nitrophenyl chloroformate was added dropwise, and the mixture was stirred at 40 ° C. for 3.5 hours, and then the completion of the CM reaction was confirmed by HPLC. Add 5.92 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 5.96 g of the desired product as a white solid (72.2%).[0036](Example 12) Synthesis of
compound 3 using Boc-DAP-OH
[Chemical 28]MeCN 73 mL (14.6 L / kg vs ACTS), Py 3.8 mL, relative to 5.00 g (22.6 mmol) of ACTS. (47 mmol, 2.1 eq.) Was added and stirred at 40 ° C. After adding 3.00 mL (23.8 mmol, 1.05 eq.) Of phenylchloroformate and stirring at 40 ° C. for 0.5 hours, the completion of the CM conversion reaction was confirmed by HPLC (CM conversion reaction product: 4.37 minutes). , ACTS: N.D.). Add 4.75 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OH, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea-forming reaction by HPLC (urea-forming reaction product: 3.81 minutes, CM-forming reaction product: 0.02 area% vs. urea-forming reaction product), the mixture was cooled to room temperature. By adding 7.3 mL (113 mmol, 5.0 eq.) Of MsOH, raising the temperature to 50 ° C., stirring for 4.5 hours, and further adding 1.5 mL (23 mmol, 1.0 eq.) Of MsOH, stirring for 1 hour. , The formation of the target product was confirmed by HPLC (Compound 3: 2.49 minutes, urea conversion reaction product: 0.50 area vs. compound 3, area of compound 3 with respect to the total area excluding pyridine: 71.0 area).
PATENT
JP 6510136
PATENT
WO 2020204117
Reference Example 1
Synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate sodium (Compound A1)
(Step 1)
Synthesis of
3 -({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid 3-amino- 37.5 mL (7.5 L / kg vs ACTS) of acetonitrile and 3.81 mL (47.38 mmol, 2.1 eq.) Of pyridine against 5 g (22.56 mmol) of 5-chloro-4-methylbenzenesulfonic acid (ACTS). Was added and stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the carbamate reaction was confirmed by HPLC. Add 5.92 g (23.23 mmol, 1.03 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 9.75 mL (69.93 mmol, 3.1 eq.) Triethylamine. Was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. Add 0.4 g (1.58 mmol, 0.07 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 0.22 mL (1.58 mmol, 0.07 eq.) Of triethylamine. Then, the completion of the urea conversion reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of methanesulfonic acid was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of acetonitrile and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / acetonitrile (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the desired product as a white solid (. net 7.20 g, 87.3%).
1 H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) ), 2.21 (S, 3H)HRMS (FAB – ): Calcd For M / Z 364.0369 (MH & lt;), Found M / Z 364.0395 (MH & lt;)
(Step 2)
(2)
Compound obtained in step 1 of synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate . To 64 g (net 10.0 g, 27.34 mmol), 18 mL of water (1.8 L / kg vs. the compound of Step 1) was added, and the mixture was stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. About 3.55 mL was added to adjust the pH to 5.8. After confirming the precipitation of the desired product by dropping 65 mL (6.5 L / kg) of isopropyl alcohol, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of isopropyl alcohol was added dropwise and the mixture was aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of isopropyl alcohol, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92). .6%).
1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)
///////////Upacicalcet sodium hydrate, Upasita, ウパシカルセトナトリウム水和物 , APPROVALS 2021, JAPAN 2021, Upacicalcet
NEW DRUG APPROVALS
ONE TIME
$10.00
Meglimin hydrochloride
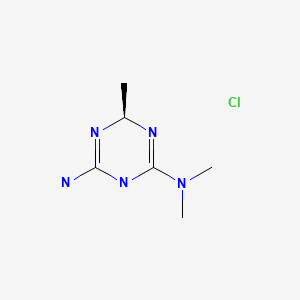

Meglimin hydrochloride
Imeglimin
hydrochloride
Twymeeg
| Formula | C6H13N5. HCl |
|---|---|
| CAS | 775351-61-6 (HCl). , C6H14ClN5 191.66CAS 775351-65-0, FREEFORM 155.20 |
| Mol weight | 191.6619 |
AntidiabeticAPPROVED PMDA JAPAN2021/6/23, イメグリミン塩酸塩
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
1,3,5-Triazine-2,4-diamine,1,6-dihydro-N,N,6-trimethyl-,(+)-(9CI)
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
JAPAN
Twymeeg Tablets 500 mg
(Sumitomo Dainippon Pharma Co., Ltd.)

Imeglimin is an experimental drug being developed as an oral anti-diabetic.[1][2] It is an oxidative phosphoryl
Imeglimin (brand name Twymeeg) is an oral anti-diabetic medication.[1][2] It was approved for use in Japan in June 2021.[3]
It is an oxidative phosphorylation blocker that acts to inhibit hepatic gluconeogenesis, increase muscle glucose uptake, and restore normal insulin secretion. It is the first approved drug of this class of anti-diabetic medication.


PATENT
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
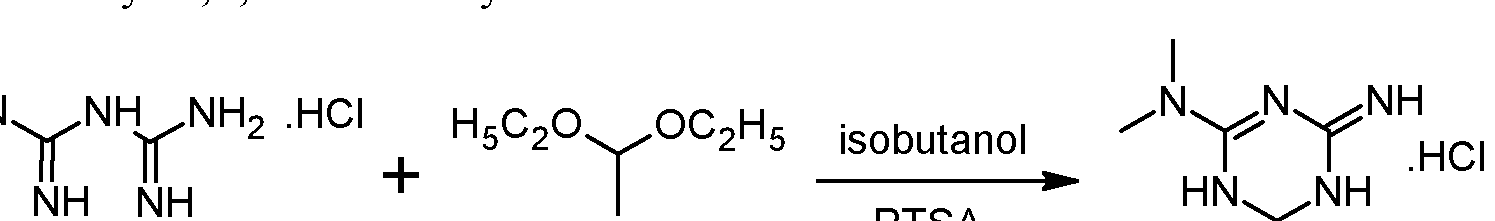
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer

Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt

γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
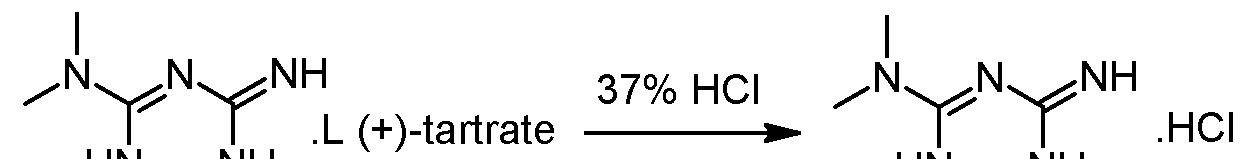
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
References
- ^ Vuylsteke V, Chastain LM, Maggu GA, Brown C (September 2015). “Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes”. Drugs in R&D. 15 (3): 227–32. doi:10.1007/s40268-015-0099-3. PMC 4561051. PMID 26254210.
- ^ Dubourg J, Fouqueray P, Thang C, Grouin JM, Ueki K (April 2021). “Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial”. Diabetes Care. 44 (4): 952–959. doi:10.2337/dc20-0763. PMID 33574125.
- ^ Poxel SA (June 23, 2021). “Poxel and Sumitomo Dainippon Pharma Announce the Approval of TWYMEEG® (Imeglimin hydrochloride) for the Treatment of Type 2 Diabetes in Japan” (Press release).
| Clinical data | |
|---|---|
| Trade names | Twymeeg |
| Legal status | |
| Legal status | Rx-only in Japan |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 775351-65-0 |
| PubChem CID | 24812808 |
| ChemSpider | 26232690 |
| UNII | UU226QGU97 |
| CompTox Dashboard (EPA) | DTXSID50228237 |
| Chemical and physical data | |
| Formula | C6H13N5 |
| Molar mass | 155.205 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Imeglimin hydrochloride, Twymeeg, JAPAN 2021, APPROVALS 2021, Antidiabetic, イメグリミン塩酸塩, ATI DIABETES, DIABETES, Imeglimin
CC1N=C(NC(=N1)N(C)C)N.Cl
NEW DRUG APPROVALS
ONE TIME
$10.00
Tucidinostat, Chidamide

Tucidinostat, Chidamide
ツシジノスタット
2021/6/23 PMDA JAPAN APPROVED,
| Hiyasta |
| Formula | C22H19FN4O2 |
|---|---|
| CAS | 1616493-44-7 |
| Mol weight | 390.4103 |
Antineoplastic, Histone deacetylase inhibitor
Chidamide (Epidaza) is a histone deacetylase inhibitor (HDI) developed in China.[1] It was also known as HBI-8000.[2] It is a benzamide HDI and inhibits Class I HDAC1, HDAC2, HDAC3, as well as Class IIb HDAC10.[3]
Chidamide is approved by the Chinese FDA for relapsed or refractory peripheral T-cell lymphoma (PTCL), and has orphan drug status in Japan.[2][better source needed] As of April 2015 it is only approved in China.[1]
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
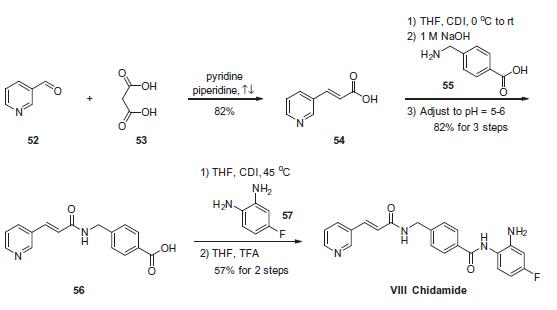
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
References
- ^ Jump up to:a b Lowe D (April 2015). “China’s First Homegrown Pharma”. Seeking Alpha.
- ^ Jump up to:a b “Chipscreen Biosciences Announces CFDA Approval of Chidamide (Epidaza) for PTCLs in China”. PR Newswire Association LLC.
- ^ “HUYA Bioscience International Grants An Exclusive License For HBI-8000 In Japan And Other Asian Countries To Eisai”. PR Newswire Association LLC. February 2016.
- ^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID 23541946.
- ^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID 25829268. S2CID 36758974.
- ^ Wang SS (2015-04-02). “A New Cancer Drug, Made in China”. The Wall Street Journal. Retrieved 13 April 2015.
| Clinical data | |
|---|---|
| Trade names | Epidaza |
| Other names | Tucidinostat |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1616493-44-7 |
| PubChem CID | 9800555 |
| ChemSpider | 7976319 |
| UNII | 87CIC980Y0 |
| Chemical and physical data | |
| Formula | C22H19FN4O2 |
| Molar mass | 390.418 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////Tucidinostat, Antineoplastic, Histone deacetylase inhibitor, ツシジノスタット , Epidaza, Chidamide, APPROVALS 2021, JAPAN 2021
NEW DRUG APPROVALS
one time
$10.00
Samidorphan
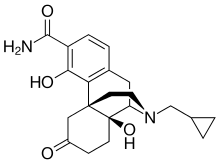
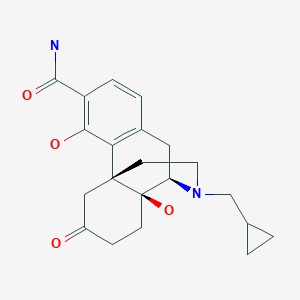
Samidorphan
サミドルファン;
| Formula | C21H26N2O4 |
|---|---|
| CAS | 852626-89-2 |
| Mol weight | 370.4421 |
FDA APPROVED 5/28/2021 Lybalvi
- ALKS 33
- ALKS-33
- RDC-0313
- RDC-0313-00
Product Ingredients

UNII0AJQ5N56E0
CAS Number1204592-75-5
WeightAverage: 504.536
Monoisotopic: 504.210780618
Chemical FormulaC25H32N2O9
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Samidorphan L-malate | 0AJQ5N56E0 | 1204592-75-5 | RARHXUAUPNYAJF-QSYGGRRVSA-N |
IUPAC Name(1R,9R,10S)-17-(cyclopropylmethyl)-3,10-dihydroxy-13-oxo-17-azatetracyclo[7.5.3.0^{1,10}.0^{2,7}]heptadeca-2,4,6-triene-4-carboxamide; (2S)-2-hydroxybutanedioic acid
MOA:mu-Opioid antagonist; delta-Opioid partial agonist; kappa-Opioid partial agonistsIndication:Alcohol dependence
New Drug Application (NDA): 213378
Company: ALKERMES INChttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213378s000lbl.pdfhttps://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/213378Orig1s000,%20Orig2s000ltr.pdf
To treat schizophrenia in adults and certain aspects of bipolar I disorder in adults
LYBALVI is a combination of olanzapine, an atypical antipsychotic, and samidorphan (as samidorphan L-malate), an opioid antagonist.
Olanzapine is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine. The molecular formula of olanzapine is: C17H20N4S and the molecular weight is 312.44 g/mol. It is a yellow crystalline powder and has pKa values of 7.80 and 5.44. The chemical structure is:
 |
Samidorphan L-malate is morphinan-3-carboxamide, 17-(cyclopropylmethyl)-4, 14-dihydroxy-6-oxo-, (2S)-2-hydroxybutanedioate. The molecular formula of samidorphan L-malate is C21H26N2O4 • C4H6O5 and the molecular weight is 504.54 g/mol. It is a white to off-white crystalline powder and has pKa values of 8.3 (amine) and 10.1 (phenol). The chemical structure is:
 |
LYBALVI is intended for oral administration and is available as film-coated, bilayer tablets in the following strengths: 5 mg/10 mg, 10 mg/10 mg, 15 mg/10 mg, and 20 mg/10 mg of olanzapine and samidorphan (equivalent to 13.6 mg of samidorphan L-malate).
Inactive ingredients include colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The film coating ingredients include hypromellose, titanium dioxide, triacetin, and color additives [iron oxide yellow (5 mg/10 mg); iron oxide yellow and iron oxide red (10 mg/10 mg); FD&C Blue No. 2/ indigo carmine aluminum lake (15 mg/10 mg); iron oxide red (20 mg/10 mg)].
- to treat schizophrenia
- alone for short-term (acute) or maintenance treatment of manic or mixed episodes that happen with bipolar I disorder
- in combination with valproate or lithium to treat manic or mixed episodes that happen with bipolar I disorder
Olanzapine is an effective atypical antipsychotic that, like other antipsychotics, is associated with weight gain, metabolic dysfunction, and increased risk of type II diabetes.5,6 Samidorphan is a novel opioid antagonist structurally related to naltrexone, with a higher affinity for opioid receptors, more potent μ-opioid receptor antagonism, higher oral bioavailability, and a longer half-life, making it an attractive candidate for oral dosing.1,5,11 Although antipsychotic-induced weight gain is incompletely understood, it is thought that the opioid system plays a key role in feeding and metabolism, such that opioid antagonism may be expected to ameliorate these negative effects. Samidorphan has been shown in animal models and clinical trials to ameliorate olanzapine-induced weight gain and metabolic dysfunction.5,6
Samidorphan was first approved as a variety of fixed-dose combination tablets with olanzapine by the FDA on May 28, 2021, and is currently marketed under the trademark LYBALVI™ by Alkermes Inc.11
Samidorphan (INN, USAN) (developmental code names ALKS-33, RDC-0313), also known as 3-carboxamido-4-hydroxynaltrexone,[2] is an opioid antagonist that preferentially acts as an antagonist of the μ-opioid receptor (MOR). It is under development by Alkermes for the treatment of major depressive disorder and possibly other psychiatric conditions.[3]
Development
Samidorphan has been investigated for the treatment of alcoholism and cocaine addiction by its developer, Alkermes,[4][5] showing similar efficacy to naltrexone but possibly with reduced side effects.
However, it has attracted much more attention as part of the combination product ALKS-5461 (buprenorphine/samidorphan), where samidorphan is combined with the mixed MOR weak partial agonist and κ-opioid receptor (KOR) antagonist buprenorphine, as an antidepressant. Buprenorphine has shown antidepressant effects in some human studies, thought to be because of its antagonist effects at the KOR, but has not been further developed for this application because of its MOR agonist effects and consequent abuse potential. By combining buprenorphine with samidorphan to block the MOR agonist effects, the combination acts more like a selective KOR antagonist, and produces only antidepressant effects, without typical MOR effects such as euphoria or substance dependence being evident.[6][7]
Samidorphan is also being studied in combination with olanzapine, as ALKS-3831 (olanzapine/samidorphan), for use in schizophrenia.[8] A Phase 3 study found that the addition of samidorphan to olanzapine significantly reduced weight gain compared to olanzapine alone.[9] The combination is now under review for approval by the US Food and Drug Administration.[10]
Pharmacology
Pharmacodynamics
The known activity profile of samidorphan at the opioid receptors is as follows:[11][12]
- μ-Opioid receptor (Ki = 0.052 nM; EC50 = N/A; Emax = 3.8%; IC50 = 0.88 nM; Imax = 92%)
- κ-Opioid receptor (Ki = 0.23 nM; EC50 = 3.3 nM; Emax = 36%; IC50 = 38 nM; Imax = 57%)
- δ-Opioid receptor (Ki = 2.6 nM; EC50 = 1.5 nM; Emax = 35%; IC50 = 6.9 nM; Imax = 56%)
As such, samidorphan is primarily an antagonist, or extremely weak partial agonist of the MOR.[11][12] In accordance with its in vitro profile, samidorphan has been observed to produce some side effects that are potentially consistent with activation of the KOR such as somnolence, sedation, dizziness, and hallucinations in some patients in clinical trials at the doses tested.[13]
SYNPATENT
WO2006052710A1.
https://patents.google.com/patent/WO2006052710A1/enExample 1 -Synthesis of 3-Carboxyamido-4-hvdroxy-naltrexone derivative 3

(A) Synthesis of 3-Carboxyamido-naltrexone 2[029] The triflate 11 of naltrexone was prepared according to the method of Wentland et al. (Bioorg. Med. Chem. Lett. 9, 183-187 (2000)), and the carboxamide 2 was prepared by the method described by Wentland et al. [(Bioorg. Med. Chem. Lett. ϋ, 623-626 (2001); and Bioorg. Med. Chem. Lett. 11, 1717-1721 (2001)] involving Pd-catalyzed carbonylation of the triflate 11 in the presence of ammonia and the Pd(O) ligand, DPPF ([l,l’-bis(diphenylρhosphino)ferrocene]) and DMSO.(B) Synthesis of 3-Carboxyamido-4-hydroxy-naltrexone derivative 3[030] Zinc dust (26 mg, 0.40 mmol) was added in portions to a solution of 2 (50 mg, 0.14 mmol) in HCl (37%, 0.2 mL) and AcOH (2 mL) at reflux. After heating at reflux for a further 15 min, the reaction was cooled by the addition of ice/water (10 mL) and basified (pH=9) with NH3/H2O, and the solution was extracted with EtOAc (3×10 mL). The organic extracts were washed with brine, dried, and concentrated. The residue was purified by column chromatography (SiO2, CH2Cl2, CH3OH : NH3/H2O = 15:1:0.01) to give compound 3 as a foam (25 mg, 50%). 1H NMR (CDC13) δl3.28(s, IH, 4-OH), 7.15(d, IH, J=8.1, H-2), 6.47(d, IH, J=8.4, H- 1), 6.10(br, IH, N-H), 4.35(br, IH, N-H), 4.04(dd,lH, J=I.8, 13.5, H-5), 3.11( d, IH, J=6), 2.99( d, IH, J=5.7), 2.94( s, IH), 2.86( d, IH, J= 6), 2.84-2.75(m, 2H), 2.65-2.61(m, 2H), 2.17-2.05(m, IH), 1.89-1.84(m, 2H), 0.85(m, IH), 0.56-0.50(m, 2H), 0.13-0.09(m, 2H). [α]D25= -98.4° (c=0.6, CH2Cl2). MS m/z (ESI) 371(MH+).
Paper
Bioorg. Med. Chem. Lett. 2000, 10, 183-187.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X99006708
Abstract
Opioid binding affinities were assessed for a series of cyclazocine analogues where the prototypic 8-OH substituent of cyclazocine was replaced by amino and substituted-amino groups. For μ and κ opioid receptors, secondary amine derivatives having the (2R,6R,11R)-configuration had the highest affinity. Most targets were efficiently synthesized from the triflate of cyclazocine or its enantiomers using Pd-catalyzed amination procedures.
PAPER
Bioorg. Med. Chem. Lett. 2001, 11, 1717-1721.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X01002785
Abstract
In response to the unexpectedly high affinity for opioid receptors observed in a novel series of cyclazocine analogues where the prototypic 8-OH was replaced by a carboxamido group, we have prepared the corresponding 3-CONH2 analogues of morphine and naltrexone. High affinity (Ki=34 and 1.7 nM) for μ opioid receptors was seen, however, the new targets were 39- and 11-fold less potent than morphine and naltrexone, respectively.
Abstract
High-affinity binding to μ opioid receptors has been identified in a series of novel 3-carboxamido analogues of morphine and naltrexone.

References
- ^ Turncliff R, DiPetrillo L, Silverman B, Ehrich E (February 2015). “Single- and multiple-dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers”. Clinical Therapeutics. 37 (2): 338–48. doi:10.1016/j.clinthera.2014.10.001. PMID 25456560.
- ^ Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, Bidlack, JM (April 2005). “Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone”. Bioorganic & Medicinal Chemistry Letters. 15 (8): 2107–10. doi:10.1016/j.bmcl.2005.02.032. PMID 15808478.
- ^ “Samidorphan”. Adis Insight. Springer Nature Switzerland AG.
- ^ Hillemacher T, Heberlein A, Muschler MA, Bleich S, Frieling H (August 2011). “Opioid modulators for alcohol dependence”. Expert Opinion on Investigational Drugs. 20 (8): 1073–86. doi:10.1517/13543784.2011.592139. PMID 21651459.
- ^ Clinical trial number NCT01366001 for “ALK33BUP-101: Safety and Pharmacodynamic Effects of ALKS 33-BUP Administered Alone and When Co-administered With Cocaine” at ClinicalTrials.gov
- ^ “ALKS 5461 drug found to reduce depressive symptoms in Phase 1/2 study”.
- ^ “Investigational ALKS 5461 Channels ‘Opium Cure’ for Depression”.
- ^ LaMattina J (15 January 2013). “Will Alkermes’ Antipsychotic ALKS-3831 Become Another Tredaptive?”. Forbes.
- ^ Correll, Christoph U.; Newcomer, John W.; Silverman, Bernard; DiPetrillo, Lauren; Graham, Christine; Jiang, Ying; Du, Yangchun; Simmons, Adam; Hopkinson, Craig; McDonnell, David; Kahn, René S. (2020-08-14). “Effects of Olanzapine Combined With Samidorphan on Weight Gain in Schizophrenia: A 24-Week Phase 3 Study”. American Journal of Psychiatry. 177 (12): 1168–1178. doi:10.1176/appi.ajp.2020.19121279. ISSN 0002-953X.
- ^ “FDA Panel: Some Risk OK for Olanzapine Combo With Less Weight Gain”. http://www.medpagetoday.com. 2020-10-09. Retrieved 2021-01-23.
- ^ Jump up to:a b Linda P. Dwoskin (29 January 2014). Emerging Targets & Therapeutics in the Treatment of Psychostimulant Abuse. Elsevier Science. pp. 398–399, 402–403. ISBN 978-0-12-420177-4.
- ^ Jump up to:a b Wentland MP, Lou R, Lu Q, Bu Y, Denhardt C, Jin J, et al. (April 2009). “Syntheses of novel high affinity ligands for opioid receptors”. Bioorganic & Medicinal Chemistry Letters. 19 (8): 2289–94. doi:10.1016/j.bmcl.2009.02.078. PMC 2791460. PMID 19282177.
- ^ McElroy SL, Guerdjikova AI, Blom TJ, Crow SJ, Memisoglu A, Silverman BL, Ehrich EW (April 2013). “A placebo-controlled pilot study of the novel opioid receptor antagonist ALKS-33 in binge eating disorder”. The International Journal of Eating Disorders. 46(3): 239–45. doi:10.1002/eat.22114. PMID 23381803.
External links
| Clinical data | |
|---|---|
| Other names | ALKS-33, RDC-0313; 3-Carboxamido-4-hydroxynaltrexone |
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Elimination half-life | 7–9 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 852626-89-2 |
| PubChem CID | 11667832 |
| ChemSpider | 23259667 |
| UNII | 7W2581Z5L8 |
| KEGG | D10162 |
| Chemical and physical data | |
| Formula | C21H26N2O4 |
| Molar mass | 370.449 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////samidorphan, サミドルファン, ALKS 33, ALKS-33, RDC-0313, RDC-0313-00, APPROVALS 2021, FDA 2021, Lybalvi
SMILESO[C@@H](CC(O)=O)C(O)=O.NC(=O)C1=CC=C2C[C@H]3N(CC4CC4)CC[C@@]4(CC(=O)CC[C@@]34O)C2=C1O
NEW DRUG APPROVALS
one time
$10.00
Pegcetacoplan
Sequence:
1ICVWQDWGAH RCTXK
Sequence:
1ICVWQDWGAH RCTXK
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| terminal mod. | Lys-15 | C-terminal amide |
| terminal mod. | Lys-15′ | C-terminal amide |
| bridge | Cys-2 – Cys-12 | disulfide bridge, dimer |
| bridge | Lys-15 – Lys-15′ | covalent bridge, dimer |
| bridge | Cys-2′ – Cys-12′ | disulfide bridge, dimer |
| uncommon | Oaa-14 | – |
| uncommon | Oaa-14′ | – |
Pegcetacoplan
ペグセタコプラン;
FDA APPROVED Empaveli, 2021/5/14
Protein Sequence
Sequence Length: 30, 15, 15multichain; modifiedPoly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 15,15′-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide)Polymer
Poly(oxy-1,2-ethanediyl), alpha-hydro-omega-hydroxy-, 15,15′-diester with N-acetyl-Lisoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-N6-carboxy-L-lysinamide cyclic (2�->12)-(disulfide)
O,O’-bis((S2,S12-cyclo(N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-Ltryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-L-lysinamide))-N6.15-carbonyl)polyethylene glycol(n = 800-1100)
- APL-2
- WHO 10743
| Formula | C170H248N50O47S4. (C2H4O)n3872.40 g·mol−1 |
|---|---|
| EfficacyDisease | Complement inhibitorParoxysmal nocturnal hemoglobinuria |
| CAS | 2019171-69-6 |
| Comment | Treatment of paroxysmal nocturnal hemoglobinuria (PNH), complement-mediated nephropathies, and age-related macular degeneration (AMD) |
- OriginatorApellis Pharmaceuticals
- ClassAnti-inflammatories; Anti-ischaemics; Antianaemics; Cyclic peptides; Eye disorder therapies; Polyethylene glycols; Urologics
- Mechanism of ActionComplement C3 inhibitors
- Orphan Drug StatusYes – Paroxysmal nocturnal haemoglobinuria; Autoimmune haemolytic anaemia; Glomerulonephritis
- RegisteredParoxysmal nocturnal haemoglobinuria
- Phase IIIAge-related macular degeneration
- Phase IIAmyotrophic lateral sclerosis; Autoimmune haemolytic anaemia; Glomerulonephritis; IgA nephropathy; Lupus nephritis; Membranous glomerulonephritis
- Phase I/IIWet age-related macular degeneration
- DiscontinuedIschaemia
- 02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
- 25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
- 18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
Pegcetacoplan, sold under the brand name Empaveli, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
 |
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Empaveli received priority review, fast track and orphan drug designations for this indication.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
References
- ^ Jump up to:a b c d e f g h i “FDA approves new treatment for adults with serious rare blood disease”. U.S. Food and Drug Administration (FDA). 14 May 2021. Retrieved 14 May 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d https://pi.apellis.com/files/PI_Empaveli.pdf
- ^ “Apellis Announces U.S. Food and Drug Administration (FDA) Approval of Empaveli (pegcetacoplan) for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH)” (Press release). Apellis Pharmaceuticals. 14 May 2021. Retrieved 14 May 2021 – via GlobeNewswire.
External links
- “Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Empaveli |
| Other names | APL-2 |
| License data | US DailyMed: Pegcetacoplan |
| Routes of administration | Subcutaneous infusion |
| Drug class | Complement inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2019171-69-6 |
| UNII | TO3JYR3BOU |
| KEGG | D11613 |
| ChEMBL | ChEMBL4298211 |
| Chemical and physical data | |
| Formula | C170H248N50O47S4 |
| Molar mass | 3872.40 g·mol−1 |
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track, orphan drug
https://www.sec.gov/Archives/edgar/data/1492422/000156459020007350/apls-10k_20191231.htm
NEW DRUG APPROVALS
ONE TIME
$10.00
Idecabtagene vicleucel
Idecabtagene vicleucel
CAS 2306267-75-2
STN: BLA 125736
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
- Bb2121
| Name | Idecabtagene vicleucel (USAN); Abecma (TN) |
|---|---|
| Product | ABECMA (Celgene Corporation) |
| CAS | 2306267-75-2 |
| Efficacy | Antineoplastic, Anti-BCMA CAR-T cell |
| Disease | Multiple myeloma [DS:H00010] |
| Comment | Cellular therapy product |
USFDA 2021/4/21 APPROVED
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
NEW DRUG APPROVALS
ONE TIME
$10.00
DESCRIPTION
ABECMA is a BCMA-directed genetically modified autologous T cell immunotherapy product consisting of a patient’s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patient’s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
The recommended dose range for idecabtagene vicleucel is 300 to 460 × 106 CAR-positive T cells. View full prescribing information for Abecma.
This application was granted breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
Follow the Division of Drug Information on Twitter @FDA_Drug_InfoExternal Link Disclaimer and the Oncology Center of Excellence @FDAOncologyExternal Link Disclaimer. Send your feedback via email to FDAOncology@fda.hhs.gov. Thanks for tuning in today to the DISCO Burst Edition.
PAT
WO 2019148089
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
The 5G7 heavy chain is:
QAYLQQSGAELVRPGASVKMSCKASGYTFTSHNLHWVKQTPRQGLQWIGAIYPGNGNTAYNQKFKGKATLTVD
KSSSTAYMQLSSLTSDDSAVYFCARWGSVVGDWYFDVWGTGTTVTVSSASTKGPSVFPLAPCSRSTSESTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVE
RKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPREE
QFNSTFRVVSVLTWHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLV
KGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLS
LSPGK (SEQ ID NO: 223)
The 5G7 light chain is:
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
REF
New England Journal of Medicine (2021), 384(8), 705-716
https://www.rxlist.com/abecma-drug.htm#indications
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company
Indications:
- Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
Product Information
- Package Insert – ABECMA
- Demographic Subgroup Information – idecabtagene vicleucel [ABECMA]
Refer to Section 1.1 of the Clinical Review Memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Loncastuximab tesirine


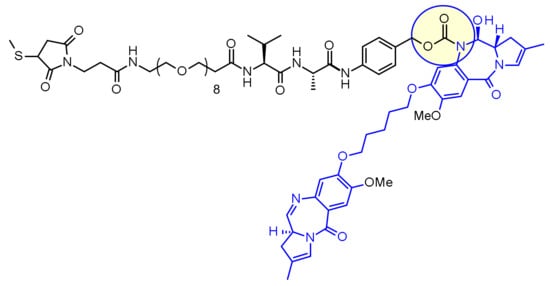
Loncastuximab tesirine
ZYNLONTA FDA APPROVED 2021/4/23
| Formula | C6544H10048N1718O2064S52 |
|---|---|
| Exact mass | 147387.9585 |
| CAS | 1879918-31-6 |
| Efficacy | Antineoplasitc, Anti-CD19 antibody |
| Disease | Diffuse large B-cell lymphoma not otherwise specified [DS:H02434] |
| Comment | Antibody-drug conjugate Treatment of hematological cancers |
ロンカスツキシマブテシリン; ADCT-402, ADCX 19
Immunoglobulin G1, anti-(human CD19 antigen) (human-Mus musculus monoclonal RB4v1.2 γ1-chain), disulfide with human-Mus musculus monoclonal RB4v1.2 κ-chain, dimer, bis(thioether) with N-[31-(3-mercapt-2,5-dioxo-1-pyrrolidinyl)-1,29-dioxo-4,7,10,13,16,19,22,25-octaoxa-28-azahentriacont-1-yl]-L-valyl-N-[4-[[[[(11S,11aS)-8-[[5-[[(11aS)-5,11a-dihydro-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-8-yl]oxy]pentyl]oxy]-11,11a-dihydro-11-hydroxy-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-10(5H)-yl]carbonyl]oxy]methyl]phenyl]-L-alaninamide
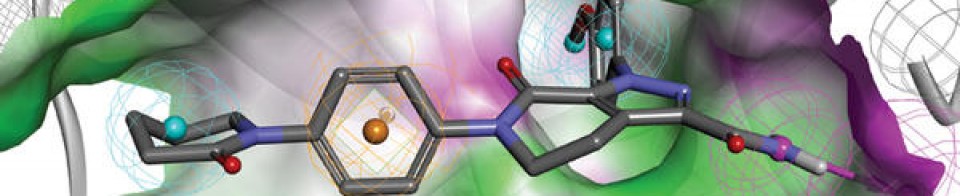
NEW DRUG APPROVALS
ONETIME
$10.00
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | CD19 |
| Clinical data | |
| Trade names | Zynlonta |
| Other names | ADCT-402, loncastuximab tesirine-lpyl |
| License data | US DailyMed: Loncastuximab_tesirine |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1879918-31-6 |
| DrugBank | DB16222 |
| ChemSpider | none |
| UNII | 7K5O7P6QIU |
| KEGG | D11338 |
| Chemical and physical data | |
| Formula | C6544H10048N1718O2064S52 |
| Molar mass | 147481.45 g·mol−1 |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Zynlonta | Injection, powder, lyophilized, for solution | 5 mg/1mL | Intravenous | ADC Therapeutics America, Inc. | 2021-04-30 | Not applicable |  |
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
|
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
References
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf
- ^ WHO Drug Information: International Nonproprietary Names for Pharmaceutical Substances
- ^ Wang K, Wei G, Liu D (November 2012). “CD19: a biomarker for B cell development, lymphoma diagnosis and therapy”. Experimental Hematology & Oncology. 1 (1): 36. doi:10.1186/2162-3619-1-36. PMC 3520838. PMID 23210908.
- ^ “Pyrrolobenzodiazepine”. ADC Review.
- ^ Clinical trial number NCT02669017 for “ADCT-402 in B-NHL” at ClinicalTrials.gov
- ^ Kahl B, Hamadani M, Caimi PF, Reid EG, Havenith K, He S, Feingold JM, O’Connor O (June 2017). “First clinical results of ADCT‐402, a novel pyrrolobenzodiazepine-based antibody drug conjugate (ADC), in relapsed/refractory B‐cell linage NHL” (PDF). Hematol Oncol. 35 (S2): 49–51. doi:10.1002/hon.2437_33.
- ^ “First clinical results of ADCT-402”. ADC Review.
- ^ Bainbridge K. “Grandfather fighting deadly cancer reveals scans of tumors after testing new drug”. Mirror.
- ^ “ADCT-402 Orphan Drug Designation” (PDF). ADC Therapeutics press release.
External links
- “Loncastuximab tesirine”. Drug Information Portal. U.S. National Library of Medicine.
/////////Loncastuximab tesirine, FDA 2021, APPROVALS 2021, ZYNLONTA, ロンカスツキシマブテシリン, ORPHAN DRUG, ADCT-402, priority review, ADCX 19
ZyCoV-D

ZyCoV-D
CAS 2541524-47-2
DNA vaccine construct encoding a spike protein antigen of SARS-CoV-2 virus (Zydus-Cadila)
UPDATE. APPROVED IN INDIA AUG 2021
http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=51254&EncHid=&userName=ZyCoV-D
bioRxiv (2021), 1-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7423510/
| ZyCoV-D | (CTRI/2020/07/026352, 2020, CTRI/2020/07/026352, 2020; Myupchar, 2020) | ZYDUS CADILA |
ZyCoV-D is a genetically engineered DNA plasmid based vaccine encoding for the membrane proteins of the virus. The clinical trials to study the immunogenicity, and safety of the vaccine, will administer three doses at an interval of 28 days in 1048 individuals.
Phase 1/2: CTRI/2020/07/026352
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | DNA |
| Clinical data | |
| Routes of administration |
Intradermal |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15892 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
ZyCoV-D is a DNA plasmid based COVID-19 vaccine being developed by Cadila Healthcare with support from the Biotechnology Industry Research Assistance Council.
The ZYCOV-D vaccine candidate was developed by Cadila Healthcare Ltd. based in India1. The vaccine was developed using a DNA vaccine platform with a non-replicating and non-integrating plasmid carrying the gene of interest3. Once the plasmid DNA is introduced into host cells and the viral protein is translated, it elicits a strong immune response, stimulating the humoral and cellular components of the immune system3. The DNA vaccine platform offers minimal biosafety requirements, more improved vaccine stability, and lower cold chain requirements3. Phase I clinical trials of this vaccine candidate were completed in July 2020, with the company reporting successful dosing and tolerance1,2. As of August, 2020 the candidate is in Phase II clinical trials1.
NEW DRUG APPROVALS
ONE TIME
$10.00
Clinical research
Phase I and II trials
In February 2020, Cadila Healthcare decided to develop a DNA plasmid based COVID-19 vaccine at their Vaccine Technology Centre (VTC) in Ahmedabad.[1] The vaccine candidate was able to pass the pre-clinical trials on animal models successfully. A report of the study was made available via bioRxiv.[2] Thereafter, human trials for Phase I and II were approved by the regulator.[3]
The Phase II trials of the vaccine candidate were conducted in over 1,000 volunteers as part of the adaptive Phase I/II multi-centric, dose escalation, randomised, double-blind placebo controlled method.[4][5]
Phase III trials
In November 2020, the company announced it would test the vaccine candidate on 30,000 patients in Phase III trials.[6] The vaccine would be given out in three doses at five sites across four cities of India.[7] In January 2021, the Drugs Controller General of India (DCGI) granted permission to conduct the Phase III clinical trials for 28,216 Indian participants.[8][9]
In April 2021, the company reported that they expected to have initial data for the Phase III trials by May 2021.[10]
Production
On 23 April 2021, production of the ZyCoV-D vaccine was started, with a yearly capacity of 240 million doses. It is expected to get emergency use authorization in May or June.[11]
References
- ^ “Zydus Cadila launches a fast tracked programme to develop vaccine for the novel coronavirus, 2019-nCoV (COVID-19)”(PDF). http://www.zyduscadila.com. Cadila Healthcare.
- ^ Dey A, Rajanathan C, Chandra H, Pericherla HP, Kumar S, Choonia HS, et al. (26 January 2021). “Immunogenic Potential of DNA Vaccine candidate, ZyCoV-D against SARS-CoV-2 in Animal Models”. bioRxiv: 2021.01.26.428240. doi:10.1101/2021.01.26.428240. S2CID 231777527.
- ^ “A prospective, randomized, adaptive, phase I/II clinical study to evaluate the safety and immunogenicity of Novel Corona Virus −2019-nCov vaccine candidate of M/s Cadila Healthcare Limited by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry India. 15 December 2020. CTRI/2020/07/026352. Archived from the original on 22 November 2020.
- ^ “Zydus Cadila’s ZyCov-D vaccine found to be ‘safe and immunogenic'”. @businessline. The Hindu. 24 December 2020.
- ^ Rawat K, Kumari P, Saha L (February 2021). “COVID-19 vaccine: A recent update in pipeline vaccines, their design and development strategies”. European Journal of Pharmacology. 892: 173751. doi:10.1016/j.ejphar.2020.173751. PMC 7685956. PMID 33245898.
- ^ Thacker T (7 November 2020). “Zydus Cadila to test ZyCoV-D on 30,000 patients in Phase-3 trials”. The Economic Times.
- ^ “Covid 19 vaccine in India: Zydus Cadila begins enrolment for Phase 3 trial of ZyCoV-D in 4 cities”. The Financial Express. 22 January 2021.
- ^ “DBT-BIRAC supported indigenously developed DNA Vaccine Candidate by Zydus Cadila, approved for Phase III clinical trials”. pib.gov.in. Press Information Bureau. 3 January 2021.
- ^ “Novel Corona Virus-2019-nCov vaccine by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry – India. Retrieved 10 April 2021.
- ^ Das, Sohini (22 April 2021). “Cadila Healthcare testing two-shot regimen for ZyCoV-D, data likely by May”. Business Standard India.
- ^ Writer, Staff (24 April 2021). “Cadila Healthcare starts production of Covid vaccine candidate”. mint. Retrieved 27 April 2021.
Zydus Cadila Covid vaccine close to getting approved in India, says MD Sharvil Patel
In an exclusive interview with India Today TV, Managing Director of Zydus Cadila Dr Sharvil Patel said the company’s Covid vaccine candidate ZyCoV-D against the Covid-19 infection is very close to getting approved in India. They are likely to apply for emergency use authorisation this month.
Ahmedabad-based pharmaceutical company Zydus Cadila is likely to submit the application for emergency use authorisation of its Covid-19 vaccine candidate ‘ZyCoV-D’ in India this month. The company is confident that the vaccine will be approved in May itself. The company plants to produce one crore doses of its ‘painless’ Covid-19 vaccine per month.
If approved, ZyCoV-D will be the fourth vaccine to be used in India’s Covid-19 vaccination drive. Made in India, the company plans to ramp up the vaccine’s production to 3-4 crore doses per month and is already in talks with two other manufacturing companies for the same
Although the vaccine should ideally be stored between 2 and 8 degrees Celsius, it remains stable even at room temperature conditions at 25 degrees Celsius. It is easy to administer, the developers said, and will be administered via intradermal injection.
If approved for emergency use, ZyCoV-D could help India fill the vacuum of vaccine doses currently being experienced in the country’s immunisation drive.
Earlier in April, Zydus Cadila announced that its drug Virafin had received restricted emergency use approval from the Drug Controller General of India for the treatment of mild cases of Covid-19.
In an exclusive interview with India Today TV, Sharvil Patel sheds details on all aspects of the Covid-19 vaccine ZyCoV-D.
When asked the status of Covid vaccine candidate ZyCoV-D and when exactly Zydus Cadila would apply for emergency use authorisation in India, Dr Sharvil Patel said the vaccine was getting very close to getting approved in the country.
“I am very happy to say that India’s first indigenously developed DNA vaccine candidate against Covid, which is our ZyCoV-D, is getting very close to approval,” he said.
“We have almost completed all our recruitment for the clinical trials. We have, by far, recruited the largest number of patients for a Covid vaccine trial in India. The number of volunteers who have been vaccinated as a part of the trial is 28,000,” Sharvil Patel said.
Sharvil Patel also said that his company has also included children in the 12-17 age group for the vaccine trials.
He said, “The recruitment holds very important milestones in terms of cohorts because not only have we included the elderly and those with co-morbidities, but also children in the age group of 12 to 17 years.”
Sharvil Patel said as soon as the efficacy data is obtained, Sydus Cadila will file for emergency use authorisation. As soon as the approval is granted, Zydus Cadila will start production of Covid-19 vaccines from July, he said.
“We hope to see our efficacy data in the middle of May. As soon as we see strong efficacy which correlates to the vaccine’s strong immunogenicity in Phase 2, we will file for emergency use authorization. We hope to produce a good quantity of the vaccine from July onwards to make sure it is available to the people. That is the need of the hour right now,” Sharvil Patel said.
He said by May the company will be in a position to talk to the regulators about the restricted use of the Covid-19 vaccine. “The regulatory process is a rolling one. I believe the regulators look at the data in a short period of time,” Sharvil Patel said.
“We have submitted a lot of data already so that it will aid the regulators once we provide them with the efficacy results. We are, hence, expecting to get the approval in May itself,” Sharvil Patel said.
///////////ZyCoV-D, COVID 19, CORONA VIRUS, VACCINE, INDIA 2021, APPROVALS 2021, SARS-CoV-2
Pegylated Interferon alpha-2b, (PegIFN), Virafin
DB00022 sequence CDLPQTHSLGSRRTLMLLAQMRRISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMI QQIFNLFSTKDSSAAWDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVR KYFQRITLYLKEKKYSPCAWEVVRAEIMRSFSLSTNLQESLRSKE
CDLPQTHSLG SRRTLMLLAQ MRRISLFSCL KDRHDFGFPQ EEFGNQFQKA ETIPVLHEMI
QQIFNLFSTK DSSAAWDETL LDKFYTELYQ QLNDLEACVI QGVGVTETPL MKEDSILAVR
KYFQRITLYL KEKKYSPCAW EVVRAEIMRS FSLSTNLQES LRSKE

Chemical structure of peginterferon α-2a and α-2b. Abbreviations: PeG-IFN, peginterferon; IFN, interferon; Lys, lysine; His, histidine; Cys, cysteine; Ser, serine.
Pegylated Interferon alpha-2b
(PegIFN), Virafin

| Formula | C860H1353N229O255S9 |
|---|---|
| CAS | 99210-65-8, 98530-12-2, 215647-85-1 |
| Mol weight | 19268.9111 |
- Interferon α2b, pegylated
- PegIFN a-2b
- PegIFN a-2b (biologics)
- PegIFN α-2b
- PegIntron
- Pegaferon
- PegiHep
- Peginterferon alfa-2b
- Peginterferon α-2b
- Pegylated interferon alfa-2b
- Pegylated interferon α-2b
- Pegylated interferons, PegIFN a-2b
- Proteinaceous biopharmaceuticals, PegIFN a-2b
- Sch 54031
- Sylatron
- ViraferonPeg
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Interferon alfa-2b | unknown | 43K1W2T1M6 | 98530-12-2 | Not applicable |
| Clinical data | |
|---|---|
| Trade names | PegIntron, Sylatron, ViraferonPeg, others |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a605030 |
| License data | EU EMA: by INN |
| Routes of administration | Subcutaneous injection |
| ATC code | L03AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only |
| Pharmacokinetic data | |
| Elimination half-life | 22–60 hrs |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 215647-85-1 |
| IUPHAR/BPS | 7462 |
| DrugBank | DB00022 |
| ChemSpider | none |
| UNII | G8RGG88B68 |
| KEGG | D02745 |
| ChEMBL | ChEMBL1201561 |
| ECHA InfoCard | 100.208.164 |
| Chemical and physical data | |
| Formula | C860H1353N229O255S9 |
| Molar mass | 19269.17 g·mol−1 |
NEW DRUG APPROVALS
ONE TIME
$10.00
New Delhi: ,,,,,,https://www.ndtv.com/india-news/zydus-virafin-gets-emergency-use-approval-for-treating-moderate-covid-19-cases-2420358
Zydus Cadila received emergency use approval from the Drugs Controller General of India (DGCI) on Friday for the use of “Virafin”, Pegylated Interferon alpha-2b (PegIFN) in treating moderate COVID-19 infection in adults.
A single-dose subcutaneous regimen of the antiviral Virafin will make the treatment more convenient for the patients. When administered early on during COVID-19, Virafin will help patients recover faster and avoid much of the complications, the company said.
In a release, Cadila Health highlighted that “the drug has also shown efficacy against other viral infections.”
Speaking on the development, Dr Sharvil Patel, Managing Director, Cadila Healthcare Limited said, “The fact that we are able to offer a therapy which significantly reduces the viral load when given early on can help in better disease management. It comes at a much-needed time for patients and we will continue to provide them access to critical therapies in this battle against COVID-19.”
In its Phase III clinical trials, the therapy had shown better clinical improvement in the patients suffering from COVID-19. During the trials, a higher proportion of patients administered with PegIFN arm were RT-PCR negative by day 7. The drug ensures faster viral clearance and has several add-on advantages compared to other anti-viral agents, the release further reads.
The development and the nod from DGCI come at a time when India is combating the second wave of coronavirus.
The central government in one of its major announcements decided to administer COVID-19 vaccines to all age above 18 years.
India recorded 3,32,730 new COVID-19 cases in the last 24 hours, the highest single-day spike since the pandemic broke out last year. India has crossed the mark of 3 lakh COVID-19 cases for two consecutive days now. This has taken the cumulative count of the COVID infection in the country to 1,62,63,695.
2CommentsThe country has recorded 2,263 new deaths due to COVID-19 in the last 24 hours. As many as 1,86,920 people have succumbed to the viral infection in India so far. There are 24,28,616 active COVID-19 cases in the country now.
PATENT
https://patents.google.com/patent/EP1562634B1/en
- Interferon alpha-2a plays an important role for the treatment of chronic hepatitis C, but it is limited in its efficacy by the short in vivo half-life. To improve the half-life and efficacy, interferon alpha-2a was conjugated with a polyethylene glycol moiety. Pegylation changes physicochemical and biological properties of the protein. One effect is the decrease of the proteolytic degradation and the renal clearance. This increases the half-life of the pegylated protein in blood. Another effect is the altered distribution in the body, depending on the size of the PEG moiety of the protein. Interferon alpha 2a pegylated with a large polyethylene glycol moiety (PEG moiety) such as a 40 kDa branched polyethylene moietywherein R and R’ are independently lower alkyl; n and n’ are integers having a sum of from 600 to 1500; and the average molecular weight of the polyethylene glycol units in said conjugate is from about 26,000 daltons to about 66,000 daltons;
has an improved biological activity and exhibits sustained adsorption and reduced renal clearance, resulting in a strong antiviral pressure throughout a once-weekly dosing schedule, see Perry M. C., et al. Drugs, 2001,15,2263-2288 and Lamb M. W., et al. The Annals of Pharmacotherapy, 2002, 36, 933-938. - [0003]See also Monkarsh et al. Analytical Biochemistry, 1997, 247, 434- 440 (Positional Isomers of Mono-pegylated Interferon α-2a) and Bailon et al. Bioconjugate Chemistry, 2001, 12, 195-202 (Rational Design of a Potent, Long-Lasting Form of interferon).
- [0004]The method for the pegylation of interferon alpha-2a is described in EP A 809 996. Since this pegylation is performed by reaction of PEG2-NHS of formulawith primary amino groups on for example lysine or to the N-terminus of the interferon alpha.one or more PEG moieties may be attached and form a mixture of unpegylated, mono- and multiple-pegylated interferon. Monopegylated interferon alpha can be isolated from the mixture by methods known in the art. Furthermore, since interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus) it is a mixture of positional isomers. From these possible twelve isomers, nine were isolated and characterized, each of these being conjugated to the branched polyethylene glycol chain at a specific lysine, namely,
at Lys(31) to form interferon alpha 2a pegylated at Lys(31) [referred to as PEG-Lys(31)],
at Lys(49) to form interferon.alpha 2a pegylated at Lys(49) [referred to as PEG-Lys(49)],
at Lys(70) to form interferon alpha 2a pegylated at Lys(70) [referred to as PEG-Lys(70)],
at Lys(83) to form interferon alpha 2a pegylated at Lys(83) [referred to as PEG-Lys(83)],
at Lys(112) to form interferon alpha 2a pegylated at Lys(112) [referred to as PEG-Lys(112)],
at Lys(121) to form interferon alpha 2a pegylated at Lys(121) [referred to as PEG-Lys(121)],
at Lys(131) to form interferon alpha 2a pegylated at Lys(131) [referred to as PEG-Lys(131)],
at Lys(134) to form interferon alpha 2a pegylated at Lys(134) [referred to as PEG-Lys(134)],
at Lys(164) to form interferon alpha 2a pegylated at Lys(164) [referred to as PEG-Lys(164)]. - [0005]It has been found that PEG-Lys(31) and PEG-Lys(134) have higher activities in an antiviral assay than the mixture, the activity of PEG-Lys(164) was equal to the mixture, whereas the activities of PEG-Lys(49), PEG-Lys(70), PEG-Lys(83), PEG-Lys(112), PEG-Lys(121) and PEG-Lys(131) were lower.
- The following examples will further illustrate the invention
Example 1A Separation of the positional isomers
- [0035]A two-step isolation and purification scheme was used to prepare the monopegylated isoforms of PEG-interferon alpha 2a.
- a) The first step was a separation of the positional isomers on a preparative low pressure liquid chromatography column with a weak-cation exchange matrix (TOSOH-BIOSEP, Toyopearl CM-650S, e.g. Resin Batch no. 82A the diameter of the column being 16 mm, the length 120 cm). A linear pH-gradient of increasing sodium acetate concentration (25 mM, pH 4.0 up 75 mM to pH 7.8) was applied at a flow rate of 0.7 mL/min. Detection was at 280 nm. With this chromatographic step species 1, 2, 5,6 and a mixture of 3, 4, 4a, 7 and 8 could be collected, see Table 1.
- b) The fractions were further separated and purified in the second preparation step. A preparative column with the same matrix as the analytical strong-cation exchange column (Resin Batch no. 82A having a ion exchange capacity of 123 mEq/ml) as described above but larger dimensions (30 mm i.d. and 70 mm length), further a higher flow rate and an extended run time was used. As for the analytical method the column was pre-equilibrated with 3.4 mM sodium acetate, 10% ethanol and 1% diethylene glycol, adjusted to pH 4.4 (buffer A). After loading the PEG-IFN samples, the column was washed with buffer A, followed by an ascending linear gradient to 10 mM dibasic potassium phosphate, 10% ethanol and 1% diethylene glycol, adjusted to pH 6.6 (buffer B). The flow rate was 1.0 mL/min and the detection at 218 nm.
- [0036]The protein concentration of the PEG-IFN alpha 2a isomer was determined by spectrophotometry, based on the 280 nm absorption of the.protein moiety of the PEG-IFN alpha 2a.
- [0037]An analytical elution profile of 180 µg of PEG-IFN alpha 2a is shown in Figure 1. The result of this method is a separation into 8 peaks, 2 peaks with baseline separation and 6 with partial separation. The decrease of the baseline absorption towards the end of the chromatogram suggests that there were no other monopegylated species of IFN alpha 2a eluting at higher retention time.
- [0038]In addition, looking carefully at the IEC-chromatogram a further peak close to the detection limit is visible between peaks 2 and 3 indicating the presence of additional positional isomers that should also contribute to the specific activity of the PEG-IFN alpha 2a mixture. Additional species were expected as the interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus). However, given the low abundance of the these species, they were not isolated and characterised.
- [0039]Isomer samples derived from IEC optimisation runs were investigated directly after the isolation (t = 0) and after 2 of weeks of storage at 5°C (data not shown). No significant differences were observed for the protein derived from IEC-peaks with regard to the protein content as determined by spectrometric methods; nor were any changes to be detected in the monopegylation site, the content of oligo-PEG-IFN alpha 2a, the amount of aggregates and the bioassay activity. Taking into account the relative abundance of the individual isomers – as determined by the IEC method – as well as the specific activities – as determined in the anti-viral assay – almost the total specific bioactivity of the PEG-IFN alpha 2a mixture used for their isolation is recovered (approximately 93%).
- [0040]The analytical IE-HPLC was used to check the purity of the individual isomers with respect to contamination with other positional isomers in the IEC fractions. The peaks 2, 3, 4, 4a, 5 and 7 had more than 98%, the peaks 1 and 8 had 93% and peak 6 had 88 % purity. Table 1:PEG-peptides identified by comparison of the Lys-C digest spectra of the isomers and the reference standard.Identified PEG Sites in the separated PEG-IFN SpeciesPeakmissing peaks in peptide mapPEG-IFNPEG siteMr (DA)SequencePeak 1K31A,E24-49Peak 2K134I, I’134-164Peak 3K131C122-131aPeak 4K121B, C113-131Peak 4aK164b134-164a,bPeak 5K70D, F50-83Peak 6K83D, H71-112Peak 7K49E, F32-70Peak 8K112B, H84-121a132-133 too small to detect.a,b RP-HPLC.
- [0041]The fractions were characterised by the methods described in examples 2 to 6.
Example 1B Analytical separation of positional isomers of mono-pegylated interferon alpha 2a
- [0042]HPLC Equipment:HP1100Column:SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous, Order#: 13076Injection:5-10 µg monopegylated IFNmobile Phase:Buffer A: 10% v/vEthanol 1% v/vDiethylenglycol 2.3 mMNa-Acetat 5.2 mMAcetic acid, in purified water, no pH adjustment Buffer B: 10% v/vEthanol 1% v/vDiethylenglycol 16.4 mMKH2PO4 4.4 mMK2HPO4, in purified water, no pH adjustmentGradient:0 Min40 %B 2 Min40 %B 2.1 Min48 %B 25 Min68 %B 27 Min75 %B 30 Min75 %B 34 Min40 %B 40 Min40 %BFlow:1.0 ml/min Column Temperature:25°C Detection:218 nm a typical Chromatogram is given in Figure 8.
Example 2 Analysis of the fractions by mass spectrometry peptide mapping
- [0043]Mass spectra were recorded on a MALDI-TOF MS instrument (PerSeptive Biosystems Voyager-DE STR with delayed extraction). Each IEC fraction (Ion Exchange Chromatography) was desalted by dialysis, reduced with 0.02 M 1,4-dithio-DL-threitol (DTT) and alkylated with 0.2 M 4-vinyl pyridine. Then the proteins were digested with endoproteinase Lys-C (Wako Biochemicals) in 0.25 M Tris (tris(hydroxymethyl)-aminoethane) at pH 8.5 with an approximate enzyme to protein ratio of 1:30. The reaction was carried out over night at 37 °C.
- [0044]A solution of 20 mg/ml α-cyano-4-hydroxycinnamic acid and 12 mg/ml nitrocellulose in acetone/isopropanol 40/60 (v/v) was used as matrix (thick-layer application). First, 0.5 µL of matrix was placed on the target and allowed to dry. Then, 1.0 µL of sample was added. The spectra were obtained in linear positive ionisation mode with an accelerating voltage of 20.000 V and a grid voltage of 95 %. At least 190 laser shots covering the complete spot were accumulated for each spectrum. Des-Arg1-bradykinin and bovine insulin were used for internal calibration.
Example 3 high-performance liquid chromatography (RP-HPLC) Peptide Mapping
- [0045]The peptides were characterized by reverse-phase high-performance liquid chromatography (RP-HPLC) Peptide Mapping. The IEC fractions were reduced, alkylated and digested with endoproteinase Lys-C as described for the MALDI-TOF MS peptide mapping. The analysis of the digested isomers was carried out on a Waters Alliance HPLC system with a Vydac RP-C18 analytical column (5 µm, 2.1 × 250 mm) and a precolumn with the same packing material. Elution was performed with an acetonitrile gradient from 1 % to 95 % for 105 min in water with a flow rate of 0.2 mL/min. Both solvents contained 0.1 % (v/v) TFA. 100 µL of each digested sample were injected and monitored at 215 nm.
Example 4 MALDI-TOF spectra of undigested protein
- [0046]An 18 mg/ml solution of trans-3-indoleacrylic acid in acetonitrile/0.1 % trifluoroacetic acid 70/30 (v/v) was premixed with the same volume of sample solution. Then 1.0 µL of the mixture was applied to the target surface. Typically 150 – 200 laser shots were averaged in linear positive ionisation mode. The accelerating voltage was set to 25.000 V and the grid voltage to 90 %. Bovine albumin M+ and M2+ were used for external calibration.
Example 5 SE-HPLC (size exclusion HPLC)
- [0047]SE-HPLC was performed with a Waters Alliance 2690 HPLC system equipped with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm). Proteins were eluted using a mobile phase containing 0.02 M NaH2PO4, 0.15 M NaCl, 1% (v/v) diethylene glycol and 10 % (v/v) ethanol (pH 6.8) at a flow rate of 0.4 mL/min and detected at 210 nm. The injection amounts were 20 µg of each isomers.
- [0048]Size Exclusion HPLC and SDS-PAGE were used to determine the amount of oligo-PEG-IFN alpha 2a forms and aggregates in the different IEC fractions. The reference material contains 2.3 % aggregates and 2.2 % oligomers (Figure 4).
- [0049]Peaks 1, 4, 4a, 5, 6 and 8 contain < 0.7 % of the oligopegylated IFN alpha 2a forms, whereas in,peaks 2, 3, and 7 the percentage of the oligopegylated IFN alpha 2a forms are under the detection limit (< 0.2 %). In the case of the aggregates a different trend could be seen. In all peaks the amount of aggregates is below 0.9 %.
Example 6 SDS-PAGE
- [0050]SDS-PAGE was carried out both under non-reducing and under reducing conditions using Tris-Glycine gels of 16 % (1.5 mm, 10 well). Novex Mark 12 molecular weight markers with a mass range from 2.5 to 200 kDa were used for calibration, bovine serum albumin (BSA) was used as sensitivity standard (2 ng). Approximately 1 µg of all the samples and 0.5 µg of standard were applied to the gel. The running conditions were 125 V and 6 W for 120 min. The proteins were fixed and stained using the silver staining kit SilverXpress from Novex.
- [0051]The gels that were recorded under non-reducing conditions for the IEC fractions 1- 8 (Figure 2) show a pattern that is comparable to that of the PEG-IFN alpha 2a reference standard.
- [0052]Under reducing conditions, the gels show an increase in intensity of the minor bands at about 90 kDa as compared to the standard. Between 6 and 10 kDa protein fragments appear for peaks 6, 7 and 8 (Figure 3). Both bands together correspond to approximately 1 % of clipped material. In the lanes of isomer 1, 5, 6, 7, 8 additional bands with more than 100 kDa can be seen which are also present in the standard. These can be assigned to oligomers. Thus SDS-PAGE confirms the results of the SE-HPLC analysis.
- [0053]Overall, RP-HPLC and SDS-PAGE experiments indicate that the purity of the IEC fractions can be considered comparable to the PEG-IFN alpha 2a reference standard.
- [0054]The structure of the PEG-IFN alpha 2a species derived from the 9 IEC-fractions were identified based on the results of the methods described above using the strategy mentioned above.
Example 7 The antiviral activity (AVA)
- [0055]The antiviral activity was estimated by its protective effect on Madin-Darby bovine kidney (MDBK) cells against the infection by vesticular stomatitis virus (VSV) and compared with a PEG-IFN alpha 2a standard. Samples and reference standard were diluted in Eagle’s Minimum Essential Medium (MEM) containing 10 % fetal bovine serum to a final concentration of 10 ng/mL (assay starting concentration). Each sample was assayed in quadruplicate.
- [0056]The antiviral protection of Madin-Darby bovine kidney cells (MDBK) with vesicular stomatitis virus was tested according to the method described in Virol. 1981, 37, 755-758. All isomers induced an activity in the anti-viral assay as presented in Table 2. The activities range between 1061 and 339 U/µg, indicating that the difference in specific activities of the protein in the positional isomers is significant. The know-how and the results generated so far will allow the initiation of further investigations to establish this structure-function relationship between the positional isomers and the IFN alpha receptors. Table 2:In Vitro Antiviral Activities of PEG-IFN alpha 2a and individual PEG-IFN alpha 2a isomers. The Antiviral activity was determined in MDBK cells infected with vesicular stomatitis virus. The results present the averages of three assays performed independently.Antiviral Assay of PEG-IFNPeakU/µgPEG-IFN1061 ± 50Peak 11818 ± 127Peak 21358 ± 46Peak 3761197Peak 4339 ± 33Peak 4a966 ± 107Peak 5600 ± 27Peak 6463 ± 25Peak7513 ± 20Peak 8468 ± 23
- [0057]The results are further illustrated by the following figures
- Figure 1: Analytical IEC-HPLC of 180µg of PEG-IFN alpha 2a. An analytical strong-cation exchange column was used to check the purity of the separated positional isomers from each purification step (TOSOH-BIOSEP, SP-SPW,10 µm particle size, 7.5 mm diameter, 7.5 cm length).
- Figure 2: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under non-reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/10, Peak 8/ 11, Ix PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 3: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/ 10, Peak 8/ 11, 1x PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 4: Size Exclusion (SE-) HPLC was used to determine the amount of oligo PEG-IFN forms and aggregates in the different IEC fractions. SE-HPLC was performed with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm).
- Figure 5: MALDI-TOF spectrometry was used to determine the molecular weight of each isomer in order to ensure that the PEG-IFN molecules were still intact after IEC chromatography and to confirm the monopegylation.
- Figure 6: MALDI-TOF Lys-C peptide maps of the PEG-IFN reference standard and the peaks 1, 2, 3, 4, 4a, 5, 6, 7, 8. Missing peaks compared to the standard are indicated by arrows.
- Figure 7: RP-HPLC chromatograms of the Lys-C digests of the PEG-IFN reference and peak 4a
- Figure 8: Analytical HPLC of 5-10µg of PEG-IFN alpha 2a mixture of positional isomers on a column charged with SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous as described in Example 1B..
- Figure 9: Ribbon structure of interferon alpha-2a showing the pegylation sites. This is the high resolution structure of human interferon alpha-2a determined with NMR spectroscopy see J. Mol. Biol. 1997, 274, 661-675. The pegylation sites of pegylated interferon alpha-2a are coloured red and labelled with residue type and residue number.
Pegylated interferon alfa-2b, sold under the brand name PegIntron among others, is a medication used to treat hepatitis C and melanoma.[3] For hepatitis C it is typically used with ribavirin and cure rates are between 33 and 82%.[3][4] For melanoma it is used in addition to surgery.[3] It is given by injection under the skin.[3]
Side effects are common.[5] They may include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever.[3] Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3] Pegylated interferon alfa-2b is in the alpha interferon family of medications.[3] It is pegylated to protect the molecule from breakdown.[5]
Pegylated interferon alfa-2b was approved for medical use in the United States in 2001.[3] It is on the World Health Organization’s List of Essential Medicines.[6]
Peginterferon alfa-2b is a form of recombinant interferon used as part of combination therapy to treat chronic Hepatitis C, an infectious liver disease caused by infection with Hepatitis C Virus (HCV). HCV is a single-stranded RNA virus that is categorized into nine distinct genotypes, with genotype 1 being the most common in the United States, and affecting 72% of all chronic HCV patients 3. Treatment options for chronic Hepatitis C have advanced significantly since 2011, with the development of Direct Acting Antivirals (DAAs) resulting in less use of Peginterferon alfa-2b. Peginterferon alfa-2b is derived from the alfa-2b moeity of recombinant human interferon and acts by binding to human type 1 interferon receptors. Activation and dimerization of this receptor induces the body’s innate antiviral response by activating the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Use of Peginterferon alfa-2b is associated with a wide range of severe adverse effects including the aggravation and development of endocrine and autoimmune disorders, retinopathies, cardiovascular and neuropsychiatric complications, and increased risk of hepatic decompensation in patients with cirrhosis. The use of Peginterferon alfa-2b has largely declined since newer interferon-free antiviral therapies have been developed.
In a joint recommendation published in 2016, the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) no longer recommend Peginterferon alfa-2b for the treatment of Hepatitis C 2. Peginterferon alfa-2b was used alongside Ribavirin(https://go.drugbank.com/drugs/DB00811) with the intent to cure, or achieve a sustained virologic response (SVR), after 48 weeks of therapy. SVR and eradication of HCV infection is associated with significant long-term health benefits including reduced liver-related damage, improved quality of life, reduced incidence of Hepatocellular Carcinoma, and reduced all-cause mortality 1.
Peginterferon alfa-2b is available as a variable dose injectable product (tradename Pegintron) used for the treatment of chronic Hepatitis C. Approved in 2001 by the FDA, Pegintron is indicated for the treatment of HCV with Ribavirin or other antiviral drugs Label. When combined together, Peginterferon alfa-2b and Ribavirin have been shown to achieve a SVR between 41% for genotype 1 and 75% for genotypes 2-6 after 48 weeks of treatment.
Medical uses
It is used to treat hepatitis C and melanoma. For hepatitis C it is typically used with ribavirin. For melanoma it is used in addition to surgery.[3]
For hepatitis C it may also be used with boceprevir, telaprevir, simeprevir, or sofosbuvir.[5]
In India, in 2021, DGCI approved emergency use of Zydus Cadila‘s Virafin in treating moderate COVID-19 infection.[7]
Host genetic factors
For genotype 1 hepatitis C treated with pegylated interferon-alfa-2a or pegylated interferon-alfa-2b combined with ribavirin, it has been shown that genetic polymorphisms near the human IL28B gene, encoding interferon lambda 3, are associated with significant differences in response to the treatment. This finding, originally reported in Nature,[8] showed that genotype 1 hepatitis C patients carrying certain genetic variant alleles near the IL28B gene are more likely to achieve sustained virological response after the treatment than others. A later report from Nature[9] demonstrated that the same genetic variants are also associated with the natural clearance of the genotype 1 hepatitis C virus.
Side effects
Common side effects include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever. Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3]
Mechanism of action
One of the major mechanisms of PEG-interferon alpha-2b utilizes the JAK-STAT signaling pathway. The basic mechanism works such that PEG-interferon alpha-2b will bind to its receptor, interferon-alpha receptor 1 and 2 (IFNAR1/2). Upon ligand binding the Tyk2 protein associated with IFNAR1 is phosphorylated which in turn phosphorylates Jak1 associated with IFNAR2. This kinase continues its signal transduction by phosphorylation of signal transducer and activator of transcription (STAT) 1 and 2 via Jak 1 and Tyk2 respectively. The phosphorylated STATs then dissociate from the receptor heterodimer and form an interferon transcription factor with p48 and IRF9 to form the interferon stimulate transcription factor-3 (ISGF3). This transcription factor then translocates to the nucleus where it will transcribe several genes involved in cell cycle control, cell differentiation, apoptosis, and immune response.[10][11]
PEG-interferon alpha-2b acts as a multifunctional immunoregulatory cytokine by transcribing several genes, including interleukin 4 (IL4). This cytokine is responsible for inducing T helper cells to become type 2 helper T cells. This ultimately results in the stimulation of B cells to proliferate and increase their antibody production. This ultimately allows for an immune response, as the B cells will help to signal the immune system that a foreign antigen is present.[12]
Another major mechanism of type I interferon alpha (IFNα) is to stimulate apoptosis in malignant cell lines. Previous studies have shown that IFNα can cause cell cycle arrest in U266, Daudi, and Rhek-1 cell lines.[13]
A follow-up study researched to determine if the caspases were involved in the apoptosis seen in the previous study as well as to determine the role of mitochondrial cytochrome c release. The study confirmed that there was cleavage of caspase-3, -8, and -9. All three of these cysteine proteases play an important role in the initiation and activation of the apoptotic cascade. Furthermore, it was shown that IFNα induced a loss in the mitochondrial membrane potential which resulted in the release of cytochrome c from the mitochondria. Follow-up research is currently being conducted to determine the upstream activators of the apoptotic pathway that are induced by IFNα.[14]
History
It was developed by Schering-Plough. Merck studied it for melanoma under the brand name Sylatron. It was approved for this use in April 2011.
References
- ^ “PegIntron- peginterferon alfa-2b injection, powder, lyophilized, for solution PegIntron- peginterferon alfa-2b kit”. DailyMed. Retrieved 28 September 2020.
- ^ “Sylatron- peginterferon alfa-2b kit”. DailyMed. 28 August 2019. Retrieved 28 September 2020.
- ^ Jump up to:a b c d e f g h i j k l “Peginterferon Alfa-2b (Professional Patient Advice) – Drugs.com”. http://www.drugs.com. Archived from the original on 16 January 2017. Retrieved 12 January 2017.
- ^ “ViraferonPeg Pen 50, 80, 100, 120 or 150 micrograms powder and solvent for solution for injection in pre-filled pen CLEAR CLICK – Summary of Product Characteristics (SPC) – (eMC)”. http://www.medicines.org.uk. Archived from the original on 13 January 2017. Retrieved 12 January 2017.
- ^ Jump up to:a b c “Peginterferon alfa-2b (PegIntron)”. Hepatitis C Online. Archived from the original on 23 December 2016. Retrieved 12 January 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ https://www.aninews.in/news/national/general-news/dgci-approves-emergency-use-of-zyduss-virafin-in-treating-moderate-covid-19-infection20210423163622/
- ^ Ge D, Fellay J, Thompson AJ, et al. (2009). “Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance”. Nature. 461 (7262): 399–401. Bibcode:2009Natur.461..399G. doi:10.1038/nature08309. PMID 19684573. S2CID 1707096.
- ^ Thomas DL, Thio CL, Martin MP, et al. (2009). “Genetic variation in IL28B and spontaneous clearance of hepatitis C virus”. Nature. 461 (7265): 798–801. Bibcode:2009Natur.461..798T. doi:10.1038/nature08463. PMC 3172006. PMID 19759533.
- ^ Ward AC, Touw I, Yoshimura A (January 2000). “The JAK-STAT pathway in normal and perturbed hematopoiesis”. Blood. 95 (1): 19–29. doi:10.1182/blood.V95.1.19. PMID 10607680. Archived from the original on 2014-04-26.
- ^ PATHWAYS :: IFN alpha[permanent dead link]
- ^ Thomas H, Foster G, Platis D (February 2004). “Corrigendum toMechanisms of action of interferon and nucleoside analogues J Hepatol 39 (2003) S93–8″. J Hepatol. 40 (2): 364. doi:10.1016/j.jhep.2003.12.003.
- ^ Sangfelt O, Erickson S, Castro J, Heiden T, Einhorn S, Grandér D (March 1997). “Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines”. Cell Growth Differ. 8 (3): 343–52. PMID 9056677. Archived from the original on 2014-04-26.
- ^ Thyrell L, Erickson S, Zhivotovsky B, et al. (February 2002). “Mechanisms of Interferon-alpha induced apoptosis in malignant cells”. Oncogene. 21 (8): 1251–62. doi:10.1038/sj.onc.1205179. PMID 11850845.
External links
- Peginterferon alfa-2b in the U.S. National Library of Medicine’s Drug Information Portal
- Medicines patent loophole ‘found’ at the BBC, 2007
- PEG-Intron (Peginterferon Alfa-2B) — Platelet Count Decreased from DrugLib.com
///////////Pegylated Interferon alpha-2b, PegIFN, Virafin, COVID 19, CORONA VIRUS, INDIA 2021, APPROVALS 2021
Sacituzumab govitecan-hziy


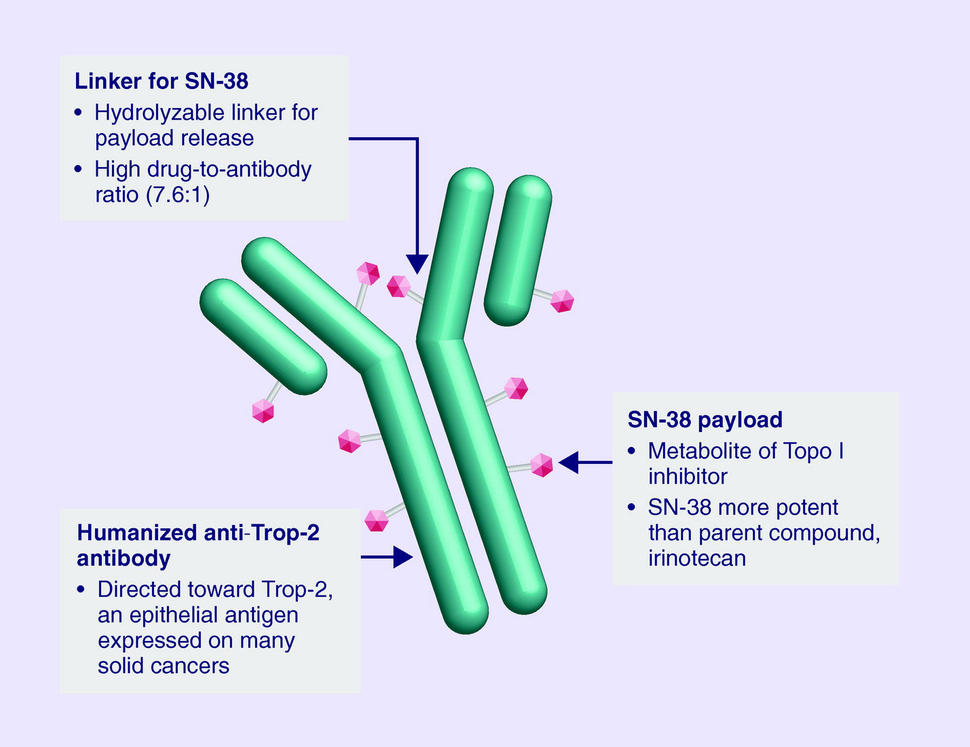
Sacituzumab govitecan-hziy
1601.8 g/mol
(2R)-2-amino-3-[1-[[4-[[1-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[2-[2-[[(2S)-6-amino-1-[4-[[(19S)-10,19-diethyl-7-hydroxy-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaen-19-yl]oxycarbonyloxymethyl]anilino]-1-oxohexan-2-yl]amino]-2-oxoethoxy]acetyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl]triazol-4-yl]methylcarbamoyl]cyclohexyl]methyl]-2,5-dioxopyrrolidin-3-yl]sulfanylpropanoic acid
Trodelvy
- hRS 7SN38
- hRS7-SN38
- IMMU 132
- IMMU-132
CAS: 1491917-83-9
UNII-DA64T2C2IO component ULRUOUDIQPERIJ-PQURJYPBSA-N
UNII-SZB83O1W42 component ULRUOUDIQPERIJ-PQURJYPBSA-N
| Efficacy | Antineoplastic, Topoisomerase I inhibitor |
|---|---|
| Disease | Breast cancer (triple negative) |
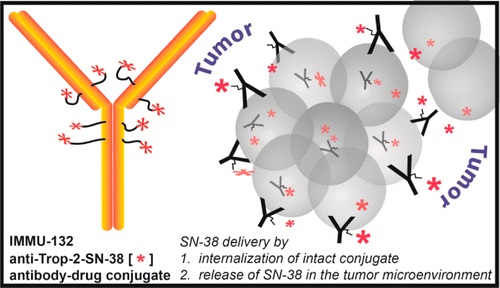

Sacituzumab Govitecan is an antibody drug conjugate containing the humanized monoclonal antibody, hRS7, against tumor-associated calcium signal transducer 2 (TACSTD2 or TROP2) and linked to the active metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), with potential antineoplastic activity. The antibody moiety of sacituzumab govitecan selectively binds to TROP2. After internalization and proteolytic cleavage, SN-38 selectively stabilizes topoisomerase I-DNA covalent complexes, resulting in DNA breaks that inhibit DNA replication and trigger apoptosis. TROP2, also known as epithelial glycoprotein-1 (EGP-1), is a transmembrane calcium signal transducer that is overexpressed by a variety of human epithelial carcinomas; this antigen is involved in the regulation of cell-cell adhesion and its expression is associated with increased cancer growth, aggressiveness and metastasis.
NEW DRUG APPROVALS
one time
$10.00
CLIP
FDA Approves Trodelvy®, the First Treatment for Metastatic Triple-Negative Breast Cancer Shown to Improve Progression-Free Survival and Overall Survival
– Trodelvy Significantly Reduced the Risk of Death by 49% Compared with Single-Agent Chemotherapy in the Phase 3 ASCENT Study –
– Trodelvy is Under Regulatory Review in the EU and in the United Kingdom, Canada, Switzerland and Australia as Part of Project Orbis –April 07, 2021 07:53 PM Eastern Daylight Time
FOSTER CITY, Calif.–(BUSINESS WIRE)–Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted full approval to Trodelvy® (sacituzumab govitecan-hziy) for adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also extended median overall survival (OS) to 11.8 months vs. 6.9 months (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death.
Trodelvy is directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including TNBC, where high expression is associated with poor survival and relapse. Prior to the FDA approval of Trodelvy, patients with previously treated metastatic TNBC had few treatment options in this high unmet-need setting. The FDA granted accelerated approval to Trodelvy in April 2020 based on objective response rate and duration of response results in a Phase 1/2 study. Today’s approval expands the previous Trodelvy indication to include treatment in adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“Women with triple-negative breast cancer have historically had very few effective treatment options and faced a poor prognosis,” said Aditya Bardia, MD, MPH, Director of Breast Cancer Research Program, Mass General Cancer Center and Assistant Professor of Medicine at Harvard Medical School, and global principal investigator of the ASCENT study. “Today’s FDA approval reflects the statistically significant survival benefit seen in the landmark ASCENT study and positions sacituzumab govitecan-hziy as a potential standard of care for pre-treated TNBC.”
“A metastatic TNBC diagnosis is frightening. As an aggressive and difficult-to-treat disease, it’s a significant advance to have an FDA-approved treatment option with a proven survival benefit for patients with metastatic disease that continues to progress,” said Ricki Fairley, Founder and CEO of Touch, the Black Breast Cancer Alliance. “For far too long, people with metastatic TNBC had very few treatment options. Today’s news continues the progress of bringing more options to treat this devastating disease.”
Among all patients evaluable for safety in the ASCENT study (n=482), Trodelvy had a safety profile consistent with the previously approved FDA label. The most frequent Grade ≥3 adverse reactions for Trodelvy compared to single-agent chemotherapy were neutropenia (52% vs. 34%), diarrhea (11% vs. 1%), leukopenia (11% vs. 6%) and anemia (9% vs. 6%). Adverse reactions leading to treatment discontinuation occurred in 5% of patients receiving Trodelvy.
“Today’s approval is the culmination of a multi-year development program and validates the clinical benefit of this important treatment in metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Building upon this milestone, we are committed to advancing Trodelvy with worldwide regulatory authorities so that, pending their decision, Trodelvy may become available to many more people around the world who are facing this difficult-to-treat cancer.”
Regulatory submissions for Trodelvy in metastatic TNBC have been filed in the United Kingdom, Canada, Switzerland and Australia as part of Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE) that provides a framework for concurrent submission and review of oncology products among international partners, as well as in Singapore through our partner Everest Medicines.The European Medicines Agency has also validated a Marketing Authorization Application for Trodelvy in the European Union. All filings are based on data from the Phase 3 ASCENT study.
Trodelvy Boxed Warning
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including metastatic triple-negative breast cancer (TNBC), where high expression is associated with poor survival and relapse.
Trodelvy is also being developed as an investigational treatment for metastatic urothelial cancer, hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER 2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
About Triple-Negative Breast Cancer (TNBC)
TNBC is an aggressive type of breast cancer, accounting for approximately 15% of all breast cancers. The disease is diagnosed more frequently in younger and premenopausal women and is more prevalent in African American and Hispanic women. TNBC cells do not have estrogen and progesterone receptors and have limited HER 2. Medicines targeting these receptors therefore are not typically effective in treating TNBC.
About the ASCENT Study
The Phase 3 ASCENT study, an open-label, active-controlled, randomized confirmatory trial, enrolled more than 500 patients with relapsed/refractory metastatic triple-negative breast cancer (TNBC) who had received two or more prior systemic therapies (including a taxane), at least one of them for metastatic disease. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline, as measured by a blinded, independent, centralized review using RECIST v1.1 criteria. Additional efficacy measures included PFS for the full population (all patients with and without brain metastases) and overall survival (OS). More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Important Safety Information for Trodelvy
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- Severe, life-threatening, or fatal neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity to TRODELVY
WARNINGS AND PRECAUTIONS
Neutropenia: Dose modifications may be required due to neutropenia. Neutropenia occurred in 62% of patients treated with TRODELVY, leading to permanent discontinuation in 0.5% of patients. Grade 3-4 neutropenia occurred in 47% of patients. Febrile neutropenia occurred in 6%.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3 diarrhea occurred in 12% of patients. Neutropenic colitis occurred in 0.5% of patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤ Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause severe and life-threatening hypersensitivity and infusion-related reactions, including anaphylactic reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 37% of patients. Grade 3-4 hypersensitivity occurred in 1% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. Pre-infusion medication is recommended. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
Nausea and Vomiting: Nausea occurred in 67% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 5% of patients. Vomiting occurred in 40% of patients and Grade 3-4 vomiting occurred in 3% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤ 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia in genotyped patients was 69% in patients homozygous for the UGT1A1*28, 48% in patients heterozygous for the UGT1A1*28 allele and 46% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia in genotyped patients was 24% in patients homozygous for the UGT1A1*28 allele, 8% in patients heterozygous for the UGT1A1*28 allele, and 10% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the ASCENT study (IMMU-132-05), the most common adverse reactions (incidence ≥25%) were nausea, neutropenia, diarrhea, fatigue, alopecia, anemia, vomiting, constipation, rash, decreased appetite, and abdominal pain. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced hemoglobin, lymphocytes, leukocytes, and neutrophils.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY
Please see full Prescribing Information, including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Sacituzumab govitecan, sold under the brand name Trodelvy, is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies.[1][2]
The most common side effects are nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia (hair loss), constipation, decreased appetite, rash and abdominal pain.[1][2] Sacituzumab govitecan has a boxed warning about the risk of severe neutropenia (abnormally low levels of white blood cells) and severe diarrhea.[1][2] Sacituzumab govitecan may cause harm to a developing fetus or newborn baby.[1] Women are advised not to breastfeed while on sacituzumab govitecan and 1 month after the last dose is administered.[3]
The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
Mechanism
Sacituzumab govitecan is a conjugate of the humanized anti-Trop-2 monoclonal antibody linked with SN-38, the active metabolite of irinotecan.[5] Each antibody having on average 7.6 molecules of SN-38 attached.[6] SN-38 is too toxic to administer directly to patients, but linkage to an antibody allows the drug to specifically target cells containing Trop-2.
Sacituzumab govitecan is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate, meaning that the drug targets the Trop-2 receptor that helps the cancer grow, divide and spread, and is linked to topoisomerase inhibitor, which is a chemical compound that is toxic to cancer cells.[1] Approximately two of every ten breast cancer diagnoses worldwide are triple-negative.[1] Triple-negative breast cancer is a type of breast cancer that tests negative for estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2) protein.[1] Therefore, triple-negative breast cancer does not respond to hormonal therapy medicines or medicines that target HER2.[1]
Development
Immunomedics announced in 2013, that it had received fast track designation from the US Food and Drug Administration (FDA) for the compound as a potential treatment for non-small cell lung cancer, small cell lung cancer, and metastatic triple-negative breast cancer. Orphan drug status was granted for small cell lung cancer and pancreatic cancer.[7][8] In February 2016, Immunomedics announced that sacituzumab govitecan had received an FDA breakthrough therapy designation (a classification designed to expedite the development and review of drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition) for the treatment of patients with triple-negative breast cancer who have failed at least two other prior therapies for metastatic disease.[9][10]
History
Sacituzumab govitecan was added to the proposed INN list in 2015,[11] and to the recommended list in 2016.[12]
Sacituzumab govitecan-hziy was approved for use in the United States in April 2020.[1][13][14][2]
Sacituzumab govitecan-hziy was approved based on the results of IMMU-132-01, a multicenter, single-arm clinical trial (NCT01631552) of 108 subjects with metastatic triple-negative breast cancer who had received at least two prior treatments for metastatic disease.[1][14][2] Of the 108 patients involved within the study, 107 were female and 1 was male.[15] Subjects received sacituzumab govitecan-hziy at a dose of 10 milligrams per kilogram of body weight intravenously on days one and eight every 21 days.[14][15] Treatment with sacituzumab govitecan-hziy was continued until disease progression or unacceptable toxicity.[15] Tumor imaging was obtained every eight weeks.[14][2] The efficacy of sacituzumab govitecan-hziy was based on the overall response rate (ORR) – which reflects the percentage of subjects that had a certain amount of tumor shrinkage.[1][14] The ORR was 33.3% (95% confidence interval [CI], 24.6 to 43.1). [1][14][15] Additionally, with the 33.3% of study participants who achieved a response, 2.8% of patients experienced complete responses.[15] The median time to response in patients was 2.0 months (range, 1.6 to 13.5), the median duration of response was 7.7 months (95% confidence interval [CI], 4.9 to 10.8), the median progression free survival was 5.5 months, and the median overall survival was 13.0 months.[15] Of the subjects that achieved an objective response to sacituzumab govitecan-hziy, 55.6% maintained their response for six or more months and 16.7% maintained their response for twelve or more months.[1][14]
Sacituzumab govitecan-hziy was granted accelerated approval along with priority review, breakthrough therapy, and fast track designations.[1][14] The U.S. Food and Drug Administration (FDA) granted approval of Trodelvy to Immunomedics, Inc.[1]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Therapy for Triple Negative Breast Cancer That Has Spread, Not Responded to Other Treatments”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 22 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Drug Trial Snapshot: Trodelvy”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ (PDF)https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf. Missing or empty
|title=(help) - ^ “New Drug Therapy Approvals 2020”. U.S. Food and Drug Administration (FDA). 31 December 2020. Retrieved 17 January2021. This article incorporates text from this source, which is in the public domain.
- ^ Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. 2015
- ^ “Novel Agents are Targeting Drivers of TNBC”. http://www.medpagetoday.com. 28 June 2016.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “New Therapy Shows Early Promise, Continues to Progress in Triple-Negative Breast Cancer”. Cure Today.
- ^ “U.S. Food and Drug Administration (FDA) Grants Breakthrough Therapy Designation to Immunomedics for Sacituzumab Govitecan for the Treatment of Patients With Triple-Negative Breast Cancer”(Press release). Immunomedics. 5 February 2016. Retrieved 25 April 2020 – via GlobeNewswire.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 113”. WHO Drug Information. 29 (2): 260–1. hdl:10665/331080.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 151–3. hdl:10665/331046.
- ^ “Trodelvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 April 2020.
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 23 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. The New England Journal of Medicine.
Further reading
- Bardia A, Mayer IA, Vahdat LT, et al. (February 2019). “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. N. Engl. J. Med. 380 (8): 741–751. doi:10.1056/NEJMoa1814213. PMID 30786188.
- Weiss J, Glode A, Messersmith WA, et al. (August 2019). “Sacituzumab govitecan: breakthrough targeted therapy for triple-negative breast cancer”. Expert Rev Anticancer Ther. 19 (8): 673–679. doi:10.1080/14737140.2019.1654378. PMID 31398063. S2CID 199518147.
External links
- “Sacituzumab govitecan”. Drug Information Portal. U.S. National Library of Medicine.
- “Sacituzumab govitecan”. ADC Review.
- “Sacituzumab govitecan”. National Cancer Institute.
- Clinical trial number NCT01631552 for “Phase I/II Study of IMMU-132 in Patients With Epithelial Cancers” at ClinicalTrials.gov
- Sacituzumab govitecan at the US National Library of Medicine Medical Subject Headings (MeSH)
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | Trop-2 |
| Clinical data | |
| Trade names | Trodelvy |
| Other names | IMMU-132, hRS7-SN-38, sacituzumab govitecan-hziy |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620034 |
| License data | US DailyMed: Sacituzumab_govitecan |
| Pregnancy category | Contraindicated |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| CAS Number | 1491917-83-9 |
| PubChem CID | 91668186 |
| DrugBank | DB12893 |
| ChemSpider | none |
| UNII | M9BYU8XDQ6 |
| KEGG | D10985 |
| Chemical and physical data | |
| Formula | C76H104N12O24S |
| Molar mass | 1601.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
//////////sacituzumab govitecan-hziy, fda 2021, approvals 2021, Trodelvy , hRS 7SN38, hRS7-SN38, IMMU 132, IMMU-132, MONOCLONAL ANTIBODY, Sacituzumab govitecan, sacituzumab govitecan-hziy, CANCER, MONOCLONAL ANTIBODIES
#sacituzumab govitecan-hziy, #fda 2021, #approvals 2021, #Trodelvy , #hRS 7SN38, #hRS7-SN38, #IMMU 132, #IMMU-132, #MONOCLONAL ANTIBODY, #Sacituzumab govitecan, #sacituzumab govitecan-hziy, #CANCER, #MONOCLONAL ANTIBODIES
CCC1=C2CN3C(=CC4=C(C3=O)COC(=O)C4(CC)OC(=O)OCC5=CC=C(C=C5)NC(=O)C(CCCCN)NC(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN6C=C(N=N6)CNC(=O)C7CCC(CC7)CN8C(=O)CC(C8=O)SCC(C(=O)O)N)C2=NC9=C1C=C(C=C9)O
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}