
Home » Posts tagged 'anxiolytic'
Tag Archives: anxiolytic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Buspirone
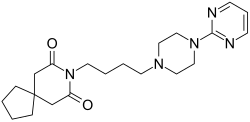
Buspirone
- Molecular FormulaC21H31N5O2
- Average mass385.503 Da
- буспиронبوسبيرون丁螺酮
251-489-4[EINECS]253-072-2[EINECS]36505-84-7[RN]8-[4-(4-Pyrimidin-2-yl-piperazin-1-yl)-butyl]-8-aza-spiro[4.5]decane-7,9-dione8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- Buspin
- Buspirone
- Spitomin
BuspironeCAS Registry Number: 36505-84-7CAS Name: 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dioneMolecular Formula: C21H31N5O2Molecular Weight: 385.50Percent Composition: C 65.43%, H 8.11%, N 18.17%, O 8.30%Literature References: Non-benzodiazepine anxiolytic; 5-hydroxytryptamine (5-HT1) receptor agonist. Prepn: Y. H. Wu et al.,J. Med. Chem.15, 477 (1972); Y. H. Wu, J. W. Rayburn, DE2057845 (1971 to Bristol-Myers); eidem,US3717634 (1973 to Mead-Johnson). Pharmacology: L. E. Allen et al.,Arzneim.-Forsch.24, 917 (1974). Comparison with diazepam in treatment of anxiety: H. L. Goldberg, R. J. Finnerty, Am. J. Psychiatry136, 1184 (1979); A. F. Jacobson et al.,Pharmacotherapy5, 290 (1985). Nonsynergistic effect with alcohol: T. Seppala et al.,Clin. Pharmacol. Ther.32, 201 (1982). Disposition and metabolism: S. Caccia et al.,Xenobiotica13, 147 (1983). Series of articles on chemistry, pharmacology, addictive potential, and clinical trials: J. Clin. Psychiatry43, pp 1-116 (1982); on pharmacology, safety and clinical comparison with clorazepate: Am. J. Med.80, Suppl. 3B, 1-51 (1986). Review of pharmacology and therapeutic efficacy: K. L. Goa, A. Ward, Drugs32, 114-129 (1986). Review: M. W. Jann, Pharmacotherapy8, 100-116 (1988); D. P. Taylor, FASEB J.2, 2445-2452 (1988).
Derivative Type: HydrochlorideCAS Registry Number: 33386-08-2Trademarks: Ansial (Vita); Ansiced (Abello); Axoren (Glaxo Wellcome); Bespar (BMS); Buspar (BMS); Buspimen (Menarini); Buspinol (Zdravlje); Buspisal (Lesvi); Narol (Almirall)Molecular Formula: C21H31N5O2.HClMolecular Weight: 421.96Percent Composition: C 59.77%, H 7.64%, N 16.60%, O 7.58%, Cl 8.40%Properties: Crystals from abs ethanol, mp 201.5-202.5°. LD50 i.p. in rats: 136 mg/kg (Allen).Melting point: mp 201.5-202.5°Toxicity data: LD50 i.p. in rats: 136 mg/kg (Allen)
Therap-Cat: Anxiolytic.Keywords: Anxiolytic; Arylpiperazines; Serotonin Receptor Agonist.
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder.[9][10] Benefits support its short term use.[11] It has not been found to be effective in treating psychosis.[9] It is taken by mouth, and it may take up to four weeks to have an effect.[9][10]
Common side effects of buspirone include nausea, headaches, dizziness, and difficulty concentrating.[9][11] Serious side effects may include hallucinations, serotonin syndrome, and seizures.[11] Its use in pregnancy appears to be safe but has not been well studied, while use during breastfeeding is not recommended.[11][12] It is a serotonin 5-HT1A receptor agonist.[2]
Buspirone was first made in 1968 and approved for medical use in the United States in 1986.[9][10] It is available as a generic medication.[11] In 2018, it was the 92nd most-commonly prescribed medication in the United States, with more than 8 million prescriptions.[13][14]
Medical uses
Anxiety
Buspirone is used for the short-term treatment of anxiety disorders or symptoms of anxiety.[15][16][17][18][19] It is generally less preferred than selective serotonin reuptake inhibitors (SSRIs).[10]
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself.[20] The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate.[2] Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD,[21] although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).[2][22]
Other uses
Sexual dysfunction
There is some evidence that buspirone on its own may be useful in the treatment of hypoactive sexual desire disorder (HSDD) in women.[23]
Miscellaneous
Buspirone is not effective as a treatment for benzodiazepine withdrawal, barbiturate withdrawal, or alcohol withdrawal/delirium tremens.[24]
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.[25][26]
Contraindications
Buspirone has these contraindications:[27][28]
- Hypersensitivity to buspirone
- Metabolic acidosis, as in diabetes
- Should not be used with MAO inhibitors
- Severely compromised liver and/or kidney function
Side effects
Main article: List of side effects of buspirone
Known side effects associated with buspirone include dizziness, headaches, nausea, nervousness, and paresthesia.[2] Buspirone is relatively well tolerated, and is not associated with sedation, cognitive and psychomotor impairment, muscle relaxation, physical dependence, or anticonvulsant effects.[2] In addition, buspirone does not produce euphoria[20] and is not a drug of abuse.[16]
It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.[9]
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available.[29] In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress.[15][16][18] In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed.[30] Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals.[30] One death has been reported in association with 450 mg buspirone together with alprazolam, diltiazem, alcohol, cocaine.[30]
Interactions
Buspirone has been shown in vitro to be metabolized by the enzyme CYP3A4.[8] This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:[27]
- Itraconazole: Increased plasma level of buspirone
- Rifampicin: Decreased plasma levels of buspirone
- Nefazodone: Increased plasma levels of buspirone
- Haloperidol: Increased plasma levels of haloperidol
- Carbamazepine: Decreased plasma levels of buspirone
- Grapefruit: Significantly increases the plasma levels of buspirone.[31] See grapefruit–drug interactions.
- Fluvoxamine: Moderately increase plasma levels of buspirone.[32]
Elevated blood pressure has been reported when buspirone has been administered to patients taking monoamine oxidase inhibitors (MAOIs).[27]
Pharmacology
Pharmacodynamics
| Site | Ki (nM) | Species | Ref |
|---|---|---|---|
| 5-HT1A | 3.98–214 21 (median) | Human | [33][34] |
| 5-HT1B | >100,000 | Rat | [35] |
| 5-HT1D | 22,000–42,700 | Human | [36][37] |
| 5-HT2A | 138 759–1,300 | Human Rat | [38] [35][38] |
| 5-HT2B | 214 | Human | [38] |
| 5-HT2C | 490 1,100–6,026 | Human Rat/pig | [38] [35][38] |
| 5-HT3 | >10,000 | Rat | [39][40] |
| 5-HT4 | >10,000 | Rat | [40] |
| 5-HT6 | 398 | Mouse | [41] |
| 5-HT7 | 375–381 | Rat | [42][43] |
| α1 | 1,000 | Rat | [35] |
| α2 | 6,000 | Rat | [44] |
| α2A | 7.3 (1-PP) | Human | [35] |
| β | 8,800 | Rat | [35] |
| D1 | 33,000 | Rat | [35] |
| D2 | 484 240 | Human Rat | [45] [35] |
| D3 | 98 | Human | [45] |
| D4 | 29 | Human | [45] |
| mACh | 38,000 | Rat | [35] |
| GABAA (BDZ) | >100,000 | Rat | [35] |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. |
Buspirone acts as an agonist of the serotonin 5-HT1A receptor with high affinity.[2][35] It is a partial agonist of both presynaptic 5-HT1A receptors, which are inhibitory autoreceptors, and postsynaptic 5-HT1A receptors.[2] It is thought that the main effects of buspirone are mediated via its interaction with the presynaptic 5-HT1A receptor, thus reducing the firing of serotonin-producing neurons.[2] Buspirone also has lower affinities for the serotonin 5-HT2A, 5-HT2B, 5-HT2C, 5-HT6, and 5-HT7 receptors.[33]
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamine D2 receptor with weak affinity.[2][35] It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses.[2] In accordance, buspirone has been found to increase dopaminergic neurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals.[2] Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.[45]
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist.[44][46][47] This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals.[46][48] In addition, 1-PP may play an important role in the antidepressant effects of buspirone.[48] Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor.[35][49] However, buspirone has been reported to have shown “significant and selective intrinsic efficacy” at the α1-adrenergic receptor expressed in a “tissue- and species-dependent manner”.[49]
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.[2][50]
Pharmacokinetics
Buspirone has a low oral bioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism.[2] The time to peak plasma levels following ingestion is 0.9 to 1.5 hours.[2] It is reported to have an elimination half-life of 2.8 hours,[2] although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours.[4] Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed.[7][8] Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP.[4][5][6] 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans.[5] The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo.[5] As such, it is likely to play an important role in the therapeutic effects of buspirone.[5] 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.[46][48]

Phase I Metabolism of buspirone in humans[51][52][8]
History
Buspirone was first synthesized, by a team at Mead Johnson, in 1968,[21] but was not patented until 1975.[54][55] It was initially developed as an antipsychotic drug acting on the D2 receptor, but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead.[2] In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD.[21][56] The patent placed on buspirone expired in 2001 and it is now available as a generic drug.
Society and culture

Buspar (buspirone) 10-mg tablets
Generic names
Buspirone is the INN, BAN, DCF, and DCIT of buspirone, while buspirone hydrochloride is its USAN, BANM, and JAN.[1][57][58][59]
Brand name
Buspirone was primarily sold under the brand name Buspar.[57][59] Buspar is currently listed as discontinued by the US Federal Drug Administration.[60] In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn for sale because of reasons of safety or effectiveness.[61]
2019 shortage
Due to interrupted production at a Mylan Pharmaceuticals plant in Morgantown, West Virginia, the United States experienced a shortage of buspirone in 2019.[62]
Research
Some tentative research supports other uses such as the treatment of depression and behavioral problems following brain damage.[2]
Chemistry
Buspirone is a member of the azapirone chemical class, and consists of azaspirodecanedione and pyrimidinylpiperazine components linked together by a butyl chain.
Analogues
Structural analogues of buspirone include other azapirones like gepirone, ipsapirone, perospirone, and tandospirone.[53]
Synthesis

Buspirone synthesis:[54] DE 2057845 U.S. Patent 3,717,634 U.S. Patent 3,907,801 U.S. Patent 3,976,776
Alkylation of 1-(2-pyrimidyl)piperazine (1) with 3-chloro-1-cyanopropane (2, 4-chlorobutyronitrile) gives 3, which is reduced either by hydrogenation over Raney nickel catalyst, or with LAH. The resulting 1° amine (4) from the previous step is then reacted with 3,3-tetramethyleneglutaric anhydride (5, 8-Oxaspiro[4.5]decane-7,9-dione) in order to yield buspirone (6).
PAPERS
- Koziol, Anna E.; Acta Crystallographica, Section E: Structure Reports Online 2006, V62(12), Po5616-o5618
- Mou, Jie; Organic Preparations and Procedures International 2008, V40(4), P391-394
- Kairisalo, Pekka Juhani; FI 72975 B 1987
- Journal of medicinal chemistry (1983), 26(2), 194-203
- Journal of medicinal chemistry (1986), 29(8), 1476-82.
- Medicinal research reviews (1990), 10(3), 283-326.
- Heterocycles (1993), 36(7), 1463-9
- Journal of medicinal chemistry (1996), 39(5), 1125-9.
- Journal of medicinal chemistry (1996), 39(16), 3195-202.
- Nature Catalysis, 3(10), 843-850; 2020
PAPER
https://pubs.rsc.org/en/content/articlelanding/2019/GC/C8GC03328E#!divAbstract
- Green Chemistry, 21(1), 59-63; 2019
Abstract
A continuous flow method for the direct conversion of alcohols to amines via a hydrogen borrowing approach is reported. The method utilises a low loading (0.5%) of a commercial catalyst system ([Ru(p-cymene)Cl2]2 and DPEPhos), reagent grade solvent and is selective for primary alcohols. Successful methylation of amines using methanol and the direct dimethylamination of alcohols using commercial dimethylamine solution are reported. The synthesis of two pharmaceutical agents Piribedil (5) and Buspirone (25) were accomplished in good yields employing these new methods.

http://www.rsc.org/suppdata/c8/gc/c8gc03328e/c8gc03328e2.pdf
8-(4-hydroxybutyl)-8-azaspiro[4.5]decane-7,9-dione (23): A solution of 3,3-tetramethyleneglutaric anhydride (0.25 mol/L in THF) was combined in a tee piece with a solution of 4-amino-1-butanol (0.25 mol/L in THF) and reacted in a 20 mL reactor coil (stainless steel, 20 min residence time) heated at 250 °C. The output was concentrated in vacuo and the residue purified by column chromatography on silica gel to afford the product in 84% yield (Rf = 0.31, 63% DCM/AcOEt). 1H NMR (400 MHz, CDCl3) δ = 3.78 (t, J = 7.2 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.58 (s, 4H), 1.77 – 1.64 (m, 4H), 1.64 – 1.53 (m, 4H), 1.53 – 1.43 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 172.33, 62.28, 44.87, 39.47, 39.14, 37.54, 29.81, 24.35, 24.17. HRMS for [C13H22NO3] + calculated 240.1594 found 240.1605.

8-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione (Buspirone, 25): The flow system was flushed with THF, the back-pressure regulator was set to 50 bar, and the coil reactor heated to 250 °C. Then a solution (10 mL overall volume) containing 1-(2-pyrimidyl)piperazine (2 mmol), 8-(4-hydroxybutyl)- 8-azaspiro[4.5]decane-7,9-dione (23) (2 mmol), dichloro(p-cymene)ruthenium(II) dimer (0.08 mmol) and bis[(2- diphenylphosphino)phenyl] ether (DPEPhos, 0.17 mmol) was pumped at 0.8 ml/min through a heated coil (8 mL, Phoenix reactor). The output solution obtained in steady state (monitored using the FlowUV) was concentrated in vacuo and purified by column chromatography on silica gel to afford the desired product in 76% yield (Rf = 0.29, 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 4.7 Hz, 2H), 6.48 (t, J = 4.7 Hz, 1H), 3.84 (t, J = 5.1 Hz, 4H), 3.79 (t, J = 6.8 Hz, 2H), 2.60 (s, 4H), 2.50 (t, J = 5.1 Hz, 4H), 2.40 (t, J = 6.8 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.65 – 1.42 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 172.19, 161.63, 157.68, 109.77, 58.31, 53.06, 44.92, 43.60, 39.48, 39.35, 37.56, 26.04, 24.19, 24.19. HRMS for [C21H32N5O2] + calculated 386.2551 found 386.2570.


PAPER
Organic Preparations and Procedures International, 40(4), 391-394; 2008
https://www.tandfonline.com/doi/abs/10.1080/00304940809458099



PATENTS
US 3907801
ES 526304
EP 395192
EP 565274
EP 634411
EP 680961
US 5521313
Indian Pat. Appl., 2011MU01860,
PATENTS
WO 2014152737
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152737
Syn
J Med Chem 1972,15(5),477-479
DE 2057845; FR 2073406; GB 1332194; US 3717634
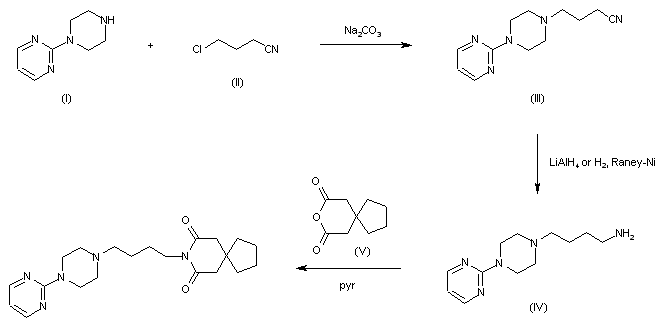
The condensation of 1-(2-pyrimidinyl)piperazine (I) with 3-chloro-1-cyanopropane (II) by means of Na2CO3 in n-butanol gives 4-(2-pyrimidinyl)-1-(3-cyanopropyl)piperazine (III). This product is reduced with LiAlH4 or with H2 and Raney-Ni yielding 4-(2-pyrimidinyl)-1-(4-aminobutyl)piperazine (IV), which is finally condensed with 8-oxaspiro[4.5]decane-7,9-dione-(3,3-tetramethylene-glutaric anhydride) (V) in pyridine.
CLIP
Anxiolytics (Tranquilizers)
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Buspirone
Buspirone, 8-[4-[4-(2-pyrimidyl)-1-piperazinyl]butyl]-8-azaspiro [4,5] decan-7,9-dione (5.2.6), is synthesized by the reaction of 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) with 8-oxaspiro[4,5]decan-7,9-dione (5.2.5). In turn, 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) is synthesized by the reaction of 1-(2-pyrimidyl)piperazine with 4-chlorobutyronitrile, giving 4-(2-pyrimidyl)-1-(3-cyanopropyl)piperazine (5.2.3), which is hydrogenated with Raney nickel into buspirone (5.2.4) [51–55].

Buspirone is an extremely specific drug that could possibly represent a new chemical class of anxiolytics—azaspirones. As an anxiolytic, its activity is equal to that of benzodiazepines; however, it is devoid of anticonvulsant and muscle relaxant properties, which are characteristic of benzodiazepines. It does not cause dependence or addiction. The mechanism of its action is not conclusively known. It does not act on the GABA receptors, which occurs in benzodiazepine use; however, it has a high affinity for seratonin (5-HT) receptors and a moderate affinity for dopamine (D2) receptors. Buspirone is effective as an anxiolytic. A few side effects of buspirone include dizziness, drowsiness, headaches, nervousness, fatigue, and weakness. This drug is intended for treatment of conditions of anxiety in which stress, muscle pain, rapid heart rate, dizziness, fear, etc. are observed; in other words, conditions of anxiety not associated with somewhat common, usual, and everyday stress. Synonyms for buspirone are anizal, axoren, buspar, buspimen, buspinol, narol, travin, and others.
CLIP
Applications of Biocatalysis for Pharmaceuticals and Chemicals
Ramesh N. Patel, in Organic Synthesis Using Biocatalysis, 2016
5.2 Enzymatic Preparation of 6-Hydroxybuspirone
Buspirone (Buspar®, 59, Figure 11.17) is a drug used for the treatment of anxiety and depression, thought to produce its effects by binding to the serotonin 5HT1A receptor [114–116]. Mainly as a result of hydroxylation reactions, it is extensively converted to various metabolites and blood concentrations return to low levels a few hours after dosing [117]. A major metabolite, 6-hydroxybuspirone, produced by the action of liver cytochrome P450 CYP3A4, was present at much higher concentrations in human blood than buspirone itself. For development of 6-hydroxybuspirone as a potential antianxiety drug, preparation and testing of the two enantiomers as well as the racemate was of interest. An enantioselective microbial reduction process was developed for the reduction of 6-oxobuspirone 60 to (R)-6-hydroxybuspirone 61a or (S)-6-hydroxybuspitone 61b. About 150 microbial cultures were screened for the enantioselective reduction of 60. Rhizopus stolonifer SC 13898, Neurospora crassa SC 13816, Mucor racemosus SC 16198, and Pseudomonas putida SC 13817 gave >50% reaction yields and >95% ee of (S)-6-hydroxybuspirone 61a. The yeast strains Hansenula polymorpha SC 13845 and Candida maltosa SC 16112 gave (R)-6-hydroxybuspirone in >60% reaction yield and >97% ee [118]. The NADPH-dependent (R)-reductase (RHBR) from H. polymorpha SC 13845 was purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADPH required for reduction, glucose-6-phosphate dehydrogenase gene from Saccharomyces cerevisiae was cloned and coexpressed in the same E. coli strain. Recombinant cultures coexpressing (R)-reductase (RHBR) and glucose 6-phosphate dehydrogenase catalyzed the reduction of 6-ketobuspirone to (R)-6-hydroxybuspirone 61a in 99% yield and 99.9% ee at 50 g/L substrate input [119].

The NADH-dependent (S)-reductase (SHBR) from P. putida SC 16269 was also purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADH required for reduction, the NAD+ dependent formate dehydrogenase gene from Pichia pastoris was also cloned and co-expressed in the same E. coli strain. Recombinant E. coli coexpressing (S)-reductase and formate dehydrogenase was used to catalyze the reduction of 6-ketobuspirone to (S)-6-hydroxybuspirone 61b, in >98% yield and >99.8% ee at 50 g/L substrate input [119].
PATENT
https://patents.google.com/patent/US6686361
The present invention relates to methods of treating anxiety and depression using R-6-hydroxy-buspirone and pharmaceutical compositions containing R-6-hydroxy-buspirone.
Buspirone, chemically: 8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione, is approved for the treatment of anxiety disorders and depression by the United States Food and Drug Administration. It is available under the trade name BUSPAR® from Bristol-Myers Squibb Company.
Studies have shown that buspirone is extensively metabolized in the body. (See, for example, Mayol, et al., Clin. Pharmacol. Ther., 37, p. 210, 1985). One of the metabolites is 6-hydroxy-8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione having Formula I. This metabolite is also known as BMS 28674, BMS 442608, or

as 6-hydroxy-buspirone. This compound is believed to be the active metabolite of buspirone and its use in treating anxiety disorders and depression is disclosed in U.S. Pat. No. 6,150,365. The specific stereochemistry of 6-hydroxy-buspirone has not been described previously. Neither racemic 6-hydroxy-buspirone nor its enantiomers are commercially available at the present time.
Preclinical studies demonstrate that 6-hydroxy-buspirone, like buspirone, demonstrates a strong affinity for the human 5-HT1A receptor. In functional testing, 6-hydroxy-buspirone produced a dose-dependent anxiolytic response in the rat pup ultrasonic vocalization test, a sensitive method for assessment of anxiolytic and anxiogenic effects (Winslow and Insel, 1991, Psychopharmacology, 105:513-520).
Clinical studies in volunteers orally dosed with buspirone demonstrate that 6-hydroxy-buspirone blood plasma levels were not only 30 to 40 times higher but were sustained compared to buspirone blood plasma levels. The time course of 6-hydroxy-buspirone blood plasma levels, unlike buspirone blood plasma levels, correlate more closely with the sustained anxiolytic effect seen following once or twice a day oral dosing with buspirone.
Although buspirone is an effective treatment for anxiety disorders and depression symptomatology in a significant number of patients treated, about a third of patients get little to no relief from their anxiety and responders often require a week or more of buspirone treatment before experiencing relief from their anxiety symptomatology. Further, certain adverse effects are reported across the patient population. The most commonly observed adverse effects associated with the use of buspirone include dizziness, nausea, headache, nervousness, lightheadedness, and excitement. Also, since buspirone can bind to central dopamine receptors, concern has been raised about its potential to cause unwanted changes in dopamine-mediated neurological functions and a syndrome of restlessness, appearing shortly after initiation of oral buspirone treatment, has been reported in small numbers of patients. While buspirone lacks the prominent sedative effects seen in more typical anxiolytics such as the benzodiazepines, patients are nonetheless advised against operating potentially dangerous machinery until they experience how they are affected by buspirone.
It can be seen that it is desirable to find a medicament with buspirone’s advantages but which demonstrates more robust anxiolytic potency with a lack of the above described adverse effects.
Formation of 6-hydroxy-buspirone occurs in the liver by action of enzymes of the P450 system, specifically CYP3A4. Many substances such as grapefruit juice and certain other drugs; e.g. erythromycin, ketoconazole, cimetidine, etc., are inhibitors of the CYP3A4 isozyme and may interfere with the formation of this active metabolite from buspirone. For this reason it would be desirable to find a compound with the advantages of buspirone but without the drug—drug interactions when coadministered with agents affecting the activity level of the CYP3A4 isozyme.
EXAMPLE 3One-Step Synthesis of 6-Hydroxy-buspirone (I)
Buspirone (19.3 g, 50 mmole) was dissolved in dry THF (400 mL) and the resulting solution was cooled to −78° C. A solution of KN(SiMe3)2 in toluene (100 mL, 1 M) was added slowly. After the reaction mixture was stirred at −78° C. for 1 h, a solution of 2-(phenylsulfonyl)-3-phenyloxaziridine (Davis reagent, prepared according to literature method: F. A. Davis, et al., Org. Synth., 1988, 66, 203) (17.0 g, 65 mmole) in dry THF (150 mL, precooled to −78° C.) was added quickly via a cannular. After stirred for 30 mins at −78° C., the reaction was quenched with 1 N HCl solution (500 mL). It was extracted with EtOAc (3×500 mL). The aqueous layer was separated, neutralized with saturated sodium bicarbonate solution, and extracted with EtOAc (3×500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white solid residue which was subjected to column chromatography using CH2Cl2/MeOH/NH4OH (200:10:1) as the eluent to give pure 6-hydroxy-buspirone (I, 7.2 g) and a mixture of buspirone and 6-hydroxy-buspirone (I). The mixture was purified by above column chromatography to afford another 3.3 g of pure 6-hydroxy-buspirone (I).
1H NMR (CDCl3) δ8.30 (d, J=4.7 Hz, 2H), 6.48 (t, J=4.7 Hz, 1H), 4.20 (s, 1H), 3.83-3.72 (m, 5H), 3.55 (s, 1H), 2.80 (d, J=17.5 Hz, 1H), 2.55-2.40 (m, 7H), 2.09-2.03 (m, 1H), 1.76-1.54 (m, 10 H), 1.41-1.36 (m, 1H), 1.23-1.20 (m, 1H).
References
- ^ Jump up to:a b Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 192–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r Loane C, Politis M (June 2012). “Buspirone: what is it all about?”. Brain Research. 1461: 111–8. doi:10.1016/j.brainres.2012.04.032. PMID 22608068. S2CID 11734819.
- ^ Jump up to:a b c “buspirone (Rx) – BuSpar, Buspirex, more.” Medscape Reference. WebMD. Retrieved 14 November 2013.
- ^ Jump up to:a b c Gammans RE, Mayol RF, LaBudde JA (March 1986). “Metabolism and disposition of buspirone”. The American Journal of Medicine. 80 (3B): 41–51. doi:10.1016/0002-9343(86)90331-1. PMID 3515929.
- ^ Jump up to:a b c d e Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 490–. ISBN 978-1-58562-309-9.
- ^ Jump up to:a b Wong H, Dockens RC, Pajor L, Yeola S, Grace JE, Stark AD, et al. (August 2007). “6-Hydroxybuspirone is a major active metabolite of buspirone: assessment of pharmacokinetics and 5-hydroxytryptamine1A receptor occupancy in rats”. Drug Metabolism and Disposition. 35 (8): 1387–92. doi:10.1124/dmd.107.015768. PMID 17494642. S2CID 25558546.
- ^ Jump up to:a b c Mahmood I, Sahajwalla C (April 1999). “Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug”. Clinical Pharmacokinetics. 36 (4): 277–87. doi:10.2165/00003088-199936040-00003. PMID 10320950. S2CID 1102318.
- ^ Jump up to:a b c d Zhu M, Zhao W, Jimenez H, Zhang D, Yeola S, Dai R, et al. (April 2005). “Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes”. Drug Metabolism and Disposition. 33 (4): 500–7. doi:10.1124/dmd.104.000836. PMID 15640381. S2CID 10142905.
- ^ Jump up to:a b c d e f “Buspirone Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d Wilson, T. K.; Tripp, J. (January 2018). “Buspirone”. StatPearls. PMID 30285372.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 338. ISBN 9780857113382.
- ^ “Buspirone Pregnancy and Breastfeeding Warnings”. Drugs.com. Retrieved 3 March 2019.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Buspirone Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b “BUSPIRONE HCL (buspirone hydrochloride) tablet [Watson Laboratories, Inc.]”. DailyMed. Watson Laboratories, Inc. July 2013. Retrieved 14 November 2013.
- ^ Jump up to:a b c “BUSPAR® (buspirone hydrochloride) Tablets 5 mg & 10 mg PRODUCT INFORMATION” (PDF). TGA eBusiness Services. Aspen Pharma Pty Ltd. January 2010. Retrieved 14 November2013.
- ^ Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Jump up to:a b “Buspirone 10mg Tablets”. electronic Medicines Compendium. Actavis UK Ltd. 10 September 2012. Retrieved 14 November 2013.
- ^ Joint Formulary Committee. British National Formulary (BNF). Pharmaceutical Press. p. 224.
- ^ Jump up to:a b Sadock BJ, Sadock VA, Ruiz P (22 September 2014). Kaplan and Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. Wolters Kluwer Health. pp. 3211–. ISBN 978-1-4698-8375-5.
- ^ Jump up to:a b c Howland RH (November 2015). “Buspirone: Back to the Future”. Journal of Psychosocial Nursing and Mental Health Services. 53 (11): 21–4. doi:10.3928/02793695-20151022-01. PMID 26535760.
- ^ Masdrakis VG, Turic D, Baldwin DS (2013). “Pharmacological treatment of social anxiety disorder”. Anxiety Disorders. Modern Trends in Pharmacopsychiatry. 29. pp. 144–53. doi:10.1159/000351960. ISBN 978-3-318-02463-0. PMID 25225024.
- ^ Goldstein I, Kim NN, Clayton AH, DeRogatis LR, Giraldi A, Parish SJ, et al. (January 2017). “Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review”. Mayo Clinic Proceedings. 92 (1): 114–128. doi:10.1016/j.mayocp.2016.09.018. PMID 27916394.
- ^ Sontheimer DL, Ables AZ (March 2001). “Is imipramine or buspirone treatment effective in patients wishing to discontinue long-term benzodiazepine use?”. The Journal of Family Practice. 50(3): 203. PMID 11252203.
- ^ Garrett AR, Hawley JS (April 2018). “SSRI-associated bruxism: A systematic review of published case reports”. Neurology. Clinical Practice. 8 (2): 135–141. doi:10.1212/CPJ.0000000000000433. PMC 5914744. PMID 29708207.
- ^ Prisco V, Iannaccone T, Di Grezia G (2017-04-01). “Use of buspirone in selective serotonin reuptake inhibitor-induced sleep bruxism”. European Psychiatry. Abstract of the 25th European Congress of Psychiatry. 41: S855. doi:10.1016/j.eurpsy.2017.01.1701.
- ^ Jump up to:a b c “Buspirone monograph”. Drugs.com. Retrieved 2011-08-27.
- ^ Geddes J, Gelder MG, Mayou R (2005). Psychiatry. Oxford [Oxfordshire]: Oxford University Press. p. 237. ISBN 978-0-19-852863-0.
- ^ Fulton B, Brogden RN (1997). “Buspirone”. CNS Drugs. 7 (1): 68–88. doi:10.2165/00023210-199707010-00007. ISSN 1172-7047.
- ^ Jump up to:a b c Dart RC (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 886–. ISBN 978-0-7817-2845-4.
- ^ Lilja JJ, Kivistö KT, Backman JT, Lamberg TS, Neuvonen PJ (December 1998). “Grapefruit juice substantially increases plasma concentrations of buspirone”. Clinical Pharmacology and Therapeutics. 64 (6): 655–60. doi:10.1016/S0009-9236(98)90056-X. PMID 9871430. S2CID 22009095.
- ^ Lamberg TS, Kivistö KT, Laitila J, Mårtensson K, Neuvonen PJ (1998). “The effect of fluvoxamine on the pharmacokinetics and pharmacodynamics of buspirone”. European Journal of Clinical Pharmacology. 54 (9–10): 761–6. doi:10.1007/s002280050548. PMID 9923581. S2CID 21939719.
- ^ Jump up to:a b c Roth BL, Driscol J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Boess FG, Martin IL (1994). “Molecular biology of 5-HT receptors”. Neuropharmacology. 33 (3–4): 275–317. doi:10.1016/0028-3908(94)90059-0. PMID 7984267. S2CID 35553281.
- ^ Jump up to:a b c d e f g h i j k l m Hamik A, Oksenberg D, Fischette C, Peroutka SJ (July 1990). “Analysis of tandospirone (SM-3997) interactions with neurotransmitter receptor binding sites”. Biological Psychiatry. 28 (2): 99–109. doi:10.1016/0006-3223(90)90627-e. PMID 1974152. S2CID 25608914.
- ^ Peroutka SJ, Switzer JA, Hamik A (1989). “Identification of 5-hydroxytryptamine1D binding sites in human brain membranes”. Synapse. 3 (1): 61–6. doi:10.1002/syn.890030109. PMID 2521959.
- ^ Waeber C, Schoeffter P, Palacios JM, Hoyer D (June 1988). “Molecular pharmacology of 5-HT1D recognition sites: radioligand binding studies in human, pig and calf brain membranes”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 337 (6): 595–601. doi:10.1007/bf00175783. PMID 2975354. S2CID 21344978.
- ^ Jump up to:a b c d e Bonhaus DW, Weinhardt KK, Taylor M, DeSouza A, McNeeley PM, Szczepanski K, et al. (1997). “RS-102221: a novel high affinity and selective, 5-HT2C receptor antagonist”. Neuropharmacology. 36 (4–5): 621–9. doi:10.1016/s0028-3908(97)00049-x. PMID 9225287. S2CID 24930608.
- ^ Nelson DR, Thomas DR (May 1989). “[3H]-BRL 43694 (Granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes”. Biochemical Pharmacology. 38 (10): 1693–5. doi:10.1016/0006-2952(89)90319-5. PMID 2543418.
- ^ Jump up to:a b Borsini F, Giraldo E, Monferini E, Antonini G, Parenti M, Bietti G, Donetti A (September 1995). “BIMT 17, a 5-HT2A receptor antagonist and 5-HT1A receptor full agonist in rat cerebral cortex”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 352 (3): 276–82. doi:10.1007/bf00168557. PMID 8584042. S2CID 19340842.
- ^ Plassat JL, Amlaiky N, Hen R (August 1993). “Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase”. Molecular Pharmacology. 44 (2): 229–36. PMID 8394987.
- ^ Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, et al. (September 1993). “A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms”. Neuron. 11 (3): 449–58. doi:10.1016/0896-6273(93)90149-l. PMID 8398139. S2CID 28729004.
- ^ Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC (September 1993). “Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation”. Proceedings of the National Academy of Sciences of the United States of America. 90 (18): 8547–51. Bibcode:1993PNAS…90.8547R. doi:10.1073/pnas.90.18.8547. PMC 47394. PMID 8397408.
- ^ Jump up to:a b Blier P, Curet O, Chaput Y, de Montigny C (July 1991). “Tandospirone and its metabolite, 1-(2-pyrimidinyl)-piperazine–II. Effects of acute administration of 1-PP and long-term administration of tandospirone on noradrenergic neurotransmission”. Neuropharmacology. 30 (7): 691–701. doi:10.1016/0028-3908(91)90176-c. PMID 1681447. S2CID 44297577.
- ^ Jump up to:a b c d Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P (March 2013). “Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors”. The International Journal of Neuropsychopharmacology. 16 (2): 445–58. doi:10.1017/S1461145712000661. PMC 5100812. PMID 22827916.
- ^ Jump up to:a b c Tunnicliff G (September 1991). “Molecular basis of buspirone’s anxiolytic action”. Pharmacology & Toxicology. 69 (3): 149–56. doi:10.1111/j.1600-0773.1991.tb01289.x. PMID 1796057.
- ^ Zuideveld KP, Rusiç-Pavletiç J, Maas HJ, Peletier LA, Van der Graaf PH, Danhof M (December 2002). “Pharmacokinetic-pharmacodynamic modeling of buspirone and its metabolite 1-(2-pyrimidinyl)-piperazine in rats”. The Journal of Pharmacology and Experimental Therapeutics. 303 (3): 1130–7. doi:10.1124/jpet.102.036798. PMID 12438536. S2CID 14139919.
- ^ Jump up to:a b c Fava M (2007). “The combination of buspirone and bupropion in the treatment of depression”. Psychotherapy and Psychosomatics. 76 (5): 311–2. doi:10.1159/000104708. PMID 17700052. S2CID 46284917.
- ^ Jump up to:a b Stern TA, Fava M, Wilens TE, Rosenbaum JF (27 April 2015). Massachusetts General Hospital Psychopharmacology and Neurotherapeutics E-Book. Elsevier Health Sciences. pp. 29–. ISBN 978-0-323-41323-7.
- ^ Nutt DJ, Ballenger JC (15 April 2008). Anxiety Disorders. John Wiley & Sons. pp. 395–. ISBN 978-0-470-98683-7.
- ^ Dockens RC, Salazar DE, Fulmor IE, Wehling M, Arnold ME, Croop R (November 2006). “Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range”. Journal of Clinical Pharmacology. 46(11): 1308–12. doi:10.1177/0091270006292250. PMID 17050795.
- ^ Jajoo HK, Mayol RF, LaBudde JA, Blair IA (1989). “Metabolism of the antianxiety drug buspirone in human subjects”. Drug Metabolism and Disposition. 17 (6): 634–40. PMID 2575499.
- ^ Taylor DP, Moon SL (July 1991). “Buspirone and related compounds as alternative anxiolytics”. Neuropeptides. 19 Suppl: 15–9. doi:10.1016/0143-4179(91)90078-w. PMID 1679210. S2CID 13730683.
- ^ Jump up to:a b Allen LE, Ferguson HC, Kissel JW (May 1972). “Psychosedative agents. 2. 8-(4-Substituted 1-piperazinylalkyl)-8-azaspiro(4.5)decane-7,9-diones”. Journal of Medicinal Chemistry. 15 (5): 477–9. doi:10.1021/jm00275a009. PMID 5035267.
- ^ US Patent 3907801 N-(8 (4-pyridyl-piperazino)-alkyl(9 -azaspiroalkanediones
- ^ United States Federal Drug Administration (September 9, 1986). Approval Type-1 New Molecular Entry.https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/018731Orig1s000rev.pdf
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 149–. ISBN 978-3-88763-075-1.
- ^ Morton IK, Hall JM (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 57–. ISBN 978-94-011-4439-1.
- ^ Jump up to:a b “Buspirone”.
- ^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2019-09-20.
- ^ “Determination That BUSPAR (Buspirone Hydrochloride) Tablets, 10 Milligrams, 15 Milligrams, and 30 Milligrams, Were Not Withdrawn From Sale for Reasons of Safety or Effectiveness”. Federal Register. 2010-10-19. Retrieved 2019-09-20.
- ^ Rabin RC (2019-02-01). “Shortage of Anxiety Drug Leaves Patients Scrambling”. The New York Times. ISSN 0362-4331. Retrieved 2019-09-20.
External links
- Media related to Buspirone at Wikimedia Commons
- “Buspirone”. Drug Information Portal. U.S. National Library of Medicine.
////////////Buspirone, буспирон , بوسبيرون , 丁螺酮 , Anxiolytic,Arylpiperazines, Serotonin Receptor Agonist, Ansial, Vita, Ansiced, Abello, Axoren, Glaxo Wellcome, Bespar, BMS, Buspar, Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
#Buspirone, #буспирон , #بوسبيرون , #丁螺酮 , #Anxiolytic, #Arylpiperazines, #Serotonin Receptor Agonist, #Ansial, #Vita, #Ansiced, #Abello, #Axoren, #Glaxo Wellcome, #Bespar, #BMS, #Buspar, #Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
MIDAZOLAM


MIDAZOLAM
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
59467-70-8 CAS NO OF FREE BASE
59467-94-6 MALEATE, Launched – 1982, Roche (Originator)
59467-96-8 (HCl)
A short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is used in dentistry, cardiac surgery, endoscopic procedures, as preanesthetic medication, and as an adjunct to local anesthesia. The short duration and cardiorespiratory stability makes it useful in poor-risk, elderly, and cardiac patients. It is water-soluble at pH less than 4 and lipid-soluble at physiological pH.
Midazolam (/mɪˈdæzəlæm/, marketed in English-speaking countries and Mexico under the trade names Dormicum, Hypnovel, andVersed,) is a short-acting drug in the benzodiazepine class developed by Hoffmann-La Roche in the 1970s. The drug is used for treatment of acute seizures, moderate to severe insomnia, and for inducing sedation and amnesia before medical procedures. It possesses profoundly potentanxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.[6][7][8] Midazolam has a fast recovery time and is the most commonly used benzodiazepine as a premedication for sedation; less commonly it is used for induction and maintenance of anesthesia.Flumazenil, a benzodiazepine antagonist drug, can be used to treat an overdose of midazolam, as well as to reverse sedation.[7] However, flumazenil can trigger seizures in mixed overdoses and in benzodiazepine-dependent individuals, so is not used in most cases.[9][10]
midazolam
Administration of midazolam by the intranasal or the buccal route (absorption via the gums and cheek) as an alternative to rectally administereddiazepam is becoming increasingly popular for the emergency treatment of seizures in children. Midazolam is also used for endoscopyprocedural sedation and sedation in intensive care. The anterograde amnesia property of midazolam is useful for premedication before surgery to inhibit unpleasant memories. Midazolam, like many other benzodiazepines, has a rapid onset of action, high effectiveness and low toxicity level. Drawbacks of midazolam include drug interactions, tolerance, and withdrawal syndrome, as well as adverse events including cognitive impairment and sedation. Paradoxical effects occasionally occur, most commonly in children and the elderly, particularly after intravenous administration. The drug has also recently been hastily introduced for use in executions in the USA in combination with other drugs.
Midazolam is a short-acting benzodiazepine in adults with an elimination half-life of one to four hours; however, in the elderly, as well as young children and adolescents, the elimination half-life is longer. Midazolam is metabolised into an active metabolite alpha1-hydroxymidazolam. Age related deficits, renal and liver status affect the pharmacokinetic factors of midazolam as well as its active metabolite. However, the active metabolite of midazolam is minor and contributes to only 10 percent of biological activity of midazolam. Midazolam is poorly absorbed orally with only 50 percent of the drug reaching the bloodstream. Midazolam is metabolised by cytochrome P450 (CYP) enzymes and by glucuronide conjugation. The therapeutic as well as adverse effects of midazolam are due to its effects on the GABAA receptors; midazolam does not activate GABAA receptors directly but, as with other benzodiazepines, it enhances the effect of the neurotransmitter GABA on the GABAA receptors (↑ frequency of Cl− channel opening) resulting in neural inhibition. Almost all of the properties can be explained by the actions of benzodiazepines on GABAA receptors. This results in the following pharmacological properties being produced: sedation, hypnotic, anxiolytic, anterograde amnesia, muscle relaxation and anti-convulsant.Midazolam maleate is a benzodiazepine that is commercialized by Astellas Pharma and Roche as an intravenous or intramuscular injection for the long-term sedation of mechanically ventilated patients under intensive care. The drug is also available in a tablet formulation, and is currently distributed in various markets, including Germany, Japan, Switzerland and the U.K. In March 2002, two lots of a syrup formulation were recalled in the U.S. due to the potential presence of a crystalline precipitate of an insoluble complex of midazolam and saccharin. Subsequently, the injection and syrup formulations of the product were both withdrawn from the U.S. market. In 2010, a Pediatric Use Marketing Authorization (PUMA) was filed for approval in the E.U. by ViroPharma for the treatment of prolonged, acute, convulsive seizures in infants, toddlers, children and adolescents, from 3 months to less than 18 years. In 2011, a positive opinion was assigned to the PUMA and final approval was assigned in June 2011. The product was launched in the U.S. in November 2011. This product was filed for approval in Japan in 2013 by Astellas Pharma for the conscious sedation in dentistry and dental surgery. In the same year the product was approved for this indication.
In terms of clinical development, a nasal formulation of the drug is in phase III clinical trials at Upsher-Smith for rescue treatment of seizures in patients on stable anti-epileptic drug regimens who require control of intermittent bouts of increased seizure activity (seizure clusters). The Hopitaux de Paris had been developing a sublingual tablet formulation of midazolam to be used in combination with morphine for the treatment of pain in children following bone fractures; however, no recent development has been reported for this indication. NovaDel Pharma had been developing the compound preclinically for the treatment of generalized anxiety, however no recent developments have been reported.
Midazolam achieves its therapeutic effect through interaction with the gamma-aminobutyric acid benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models. GABA acts at inhibitory synapses in the brain by binding to specific transmembrane receptors in the plasma membrane of both pre- and post-synaptic neurons, opening ion channels and bringing about a hyperpolarization via either chloride or potassium ion flow.
In 2008, fast track designation was assigned to midazolam maleate in the U.S. for the treatment of seizure disorders.
In 2009, Orphan Drug Designation was received in the U.S. by for the treatment of seizure disorders in patients who require control of intermittent bouts of increased seizure activity (e.g. acute repetitive seizures, seizure clusters). This designation was assigned in the U.S. for the treatment of nerve agent-induced seizures.
In 2010, midazolam maleate was licensed to Upsher-Smith by Ikano Therapeutics for the treatment of acute repetitive seizure in patients with epilepsy. However, in 2010, Ikano closed and dissolved its business. Previously, Ikano had transferred to Upsher-Smith ownership of it nasal midazolam maleate program.
Midazolam is among about 35 benzodiazepines which are currently used medically, and was synthesised in 1975 by Walser and Fryer at Hoffmann-LaRoche, Inc in the United States.Owing to its water solubility, it was found to be less likely to cause thrombophlebitis than similar drugs.The anticonvulsant properties of midazolam were studied in the late 1970s, but not until the 1990s did it emerge as an effective treatment for convulsive status epilepticus. As of 2010, it is the most commonly used benzodiazepine in anesthetic medicine. In acute medicine, midazolam has become more popular than other benzodiazepines, such as lorazepam and diazepam, because it is shorter lasting, is more potent, and causes less pain at the injection site.Midazolam is also becoming increasingly popular in veterinary medicine due to its water solubility.
Midazolam is a water-soluble benzodiazepine available as a sterile, nonpyrogenic parenteral dosage form for intravenous or intramuscular injection. Each mL contains midazolam hydrochloride equivalent to 1 mg or 5 mg midazolam compounded with 0.8% sodium chloride and 0.01% edetate disodium with 1% benzyl alcohol as preservative, and sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 2.9-3.7.
Midazolam is a white to light yellow crystalline compound, insoluble in water. The hydrochloride salt of midazolam, which is formed in situ, is soluble in aqueous solutions. Chemically, midazolam HCl is 8-chloro-6-(2-fluorophenyl)-1-methyl-4H– imidazo[1,5-a] [1,4] benzodiazepine hydrochloride. Midazolam hydrochloride has the molecular formula C18H13ClFN3•HCl, a calculated molecular weight of 362.25 and the following structural formula:
 |
In the Netherlands, midazolam is a List II drug of the Opium Law. Midazolam is a Schedule IV drug under the Convention on Psychotropic Substances. In the United Kingdom, midazolam is a Class C controlled drug. In the United States, midazolam (DEA number 2884) is on the Schedule IV list of the Controlled Substances Act as a non-narcotic agent with low potential for abuse.
midaolam hydrochloride NDA 018654, 075154
REF
U.S. Pat. No. 4,280,957
U.S. Pat. No. 5,693,795
U.S. Pat. No. 6,512,114
Midazolam Maleate
Drugs Fut 1978, 3(11): 822
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 18 pg. 5658 – 5667
Journal of Heterocyclic Chemistry, 1983 , vol. 20, 3 pg. 551 – 558.. 32 maleate
WO 2001070744
WO 2001002402
WO 2012075286
US2011/275799 A1… no 5
Journal of Organic Chemistry, 1978 , vol. 43, p. 936,942, mp free base, nmr
| US4280957 | May 15, 1978 | Jul 28, 1981 | Hoffmann-La Roche Inc. | Imidazodiazepines and processes therefor |
| US6262260 * | Mar 23, 2000 | Jul 17, 2001 | Abbott Laboratories | Process for the preparation of midazolam |
| US6512114 | Jun 30, 1999 | Jan 28, 2003 | Abbott Laboratories | Process for the preparation of Midazolam |
……………………….
introduction
4H-imidazo[1,5-a][1,4]benzodiazepines or, more simply, imidazobenzodiazepines, are a class of benzodiazepines having the general formula (I),

wherein the 1,4-diazepine ring is fused with a 1,3-imidazole ring. The main compounds part of the 4H-imidazo[1,5-a][1,4]benzodiazepines are Midazolam of formula (IV):

an active ingredient currently commercially available as a hydrochloride salt under the name of Versed or Hypnovel for anaesthetic and sedative use and the maleate salt currently commercially available under the name Dormicum or Flormidal.
Other important compounds are Climazolam of formula (VII):

Imidazenil of formula (VIII):

1-Hydroxymidazolam of formula (IX):

and Desmethyl midazolam of formula (X):

all these being biologically active substances and having psychotropic and sedative action.
The synthesis of the Midazolam as described in U.S. Pat. No. 4,280,957 of Hoffmann-La Roche provides for the decarboxylation reaction of the 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid of formula (VI) according to the following scheme:

The process for preparing the intermediate (VI) via basic hydrolysis of the corresponding ester is described in such patent publication and it is well known in the art.
The thermal decarboxylation reaction in high boiling solvent such as mineral oil at 230° C. for 5 min results in a mixture of products of Midazolam of formula (IV) and of Isomidazolam of formula (IV-bis), a non-pharmacologically active isomer, at a 80:20 ratio. The two products are separated by chromatography.
At industrial level, the formation of the Isomidazolam isomer impurity requires a further isomerisation reaction performed on the mixture of the two compounds to convert the isomer into the active product. The reaction mixture obtained from the thermal decarboxylation is thus subjected to basic treatment under the action of KOH in EtOH followed by an acid treatment which thus provides a mixture of Midazolam-Isomidazolam at a 95:5 ratio. The final removal of the Isomidazolam impurity from the product occurs through crystallisation of the product from AcOEt and EtOH. In order to limit this isomerisation treatment, in the subsequent U.S. Pat. No. 5,693,795 of Hoffmann-La Roche dated 1999, there is described a process for performing the decarboxylation of the compound of formula (VI) in n-butanol in a continuous tubular reactor with a 4 minutes permanence period with a yield between 47-77%. However, the reaction, performed at high temperature and pressure (280° C., 100 bars) results in the formation of a considerable percentage of Isomidazolam (85:15 Midazolam/Isomidazolam ratio) which still requires the basic isomerisation step.
Lastly, in U.S. Pat. No. 6,512,114 of Abbott Laboratories there is described the decarboxylation of the compound of formula (VI) in mineral oil or in N,N-Dimethylacetamide (DMA) at 160-230° C. for at least 3 hours obtaining a 3/1 to 6/1 Midazolam/Isomidazolam ratio with a yield of isolated product equal to just 54%.
Though performed using dedicated apparatus and in extreme conditions, the prior art processes do not allow selectively performing the decarboxylation reaction of the intermediate (VI) to Midazolam thus requiring a further synthetic passage followed by crystallisation with ensuing reduction of the overall yield.
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is represented by the following structural formula (I):

Midazolam is a central nervous system (CNS) depressant, used for short term treatment of insomnia. Like other benzodiazepines, midazolam binds to benzodiazepine receptors in the brain and spinal cord and is thus used as a short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is currently used in dentistry, cardiac surgery, endoscopic procedures, as a preanesthetic medication, as an adjunct to local anesthesia and as a skeletal muscle relaxant. Depending on the pH value, midazolam can exist in solution as a closed ring form (I) as well as an open ring form (IA), which are in equilibrium, as shown in Scheme 1:

The amount of the open ring form (IA) is dependent upon the pH value of the solution. At a pH value of about 3, the content of the open ring form (IA) can be 40%, while at pH value of 7.5, the closed ring form (I) can be more than 90%.
Clinical studies have demonstrated that there are no significant differences in the clinical activity between midazolam hydrochloride and midazolam maleate, however the use of intravenous midazolam hydrochloride has been associated, in some cases, with respiratory depression and arrest.
U.S Pat. No. 4,280,957 (hereinafter the ‘957 patent) describes a synthetic process for preparing midazolam, which is depicted in Scheme 2 below. This process includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) with acetic anhydride in dichloromethane to produce 2-acetamido-methyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (III). The latter is heated with polyphosphoric acid at 150° C. to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine of formula (IV), which is purified by column chromatography. Compound IV is then mixed with toluene and manganese dioxide and heated to reflux to afford midazolam base, which is crystallized from ether to yield a product with mp of 152-154° C.

The ‘957 patent further describes an alternative process which includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) (optionally as a dimaleate salt) with triethylorthoacetate in ethanol and in the presence of p-toluenesulfonic acid to afford 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine (IV). This product is dissolved in xylene and treated with activated manganese dioxide to afford the crude base, which is reacted in situ with maleic acid in ethanol and crystallized by addition of ether to produce the midazolam maleate having melting point of 148-151° C. The process is depicted in Scheme 3 below.

The preparation of midazolam maleate from the isolated midazolam base is also described in a further example of the ‘957 Patent, wherein a warm solution of midazolam base in ethanol is combined with a warm solution of maleic acid in ethanol. The mixture is diluted with ether and at least part of the solvents is evaporated using a steam bath to obtain crystalline midazolam maleate having melting point of 148-151° C. The yield and the purity of the obtained midazolam maleate are not disclosed.
U.S. Pat. No. 6,512,114 (hereinafter the ‘114 patent) describes another synthetic process for preparing midazolam, which is depicted in Scheme 4 below. According to this Process, the starting material 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid (V) is heated in mineral oil for 3 hours at 230° C. until it is decarboxylated, followed by treatment with potassium tert-butoxide, to afford midazolm (I), isomidazolam (VI) and a midazolam dimmer (VII). Midazolam base is obtained in 54.5% yield after two re-crystallizations from ethyl acetate and heptane; however, the purity of the product is not disclosed.

The preparation of midazolam by conventional routes is liable to produce impurities such as isomidazolam (VI) and a midazolam dimmer (VII), and possibly other impurities. There is, therefore, a need in the art for a midazolam purification process that will provide highly pure midazolam containing minimal amounts of impurities produced. The present invention provides such a process.
This example describes the preparation of midazolam base as taught in the ‘957 patent.
16 g (0.03 mol) of 2-aminomethyl-7-chloro-5-(2-fluorophenyl)-2,3-dihydro-1H-1,4-bezodiazepine dimaleate was dissolved in 200 ml of toluene and 10 ml of 25% ammonium hydroxide solution was added and mixing was maintained for an hour. Then, the phases were separated and the toluene phase was dried by azeotropic distillation using a Dean Stark apparatus. 7 ml (0.038 mol) of triethylorthoacetate was added and the solution was heated to reflux for 4 hours, after which time the solution was left to cool to ambient temperature. 25 ml of methyl tert-butyl ether was added and the mixture was cooled overnight to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine, which was isolated by filtration. The product was mixed with 200 ml of toluene and dried by azeotropic distillation using a Dean Stark apparatus. Then, 30 g of manganese dioxide was added and the mixture was heated to reflux for two hours. The excess manganese dioxide was filtered off to afford a solution of midazolam base in toluene, which was evaporated to obtain a product having 97.9% purity and containing 0.44% of impurity VIII and 1.14% of impurity IX (according to HPLC).
…………………………
EXAMPLE 28
2-Aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine dimaleate
A suspension of 17 g (0.05 m) of 7-chloro-1,3-dihydro-5-(2-fluorophenyl)-2-nitromethylene-2H-1,4-benzodiazepine-4-oxide in 200 ml of tetrahydrofuran and 100 ml of methanol was hydrogenated in presence of 17 g of Raney nickel at an initial pressure of 155 psi for 24 hrs. The catalyst was removed by filtration and the filtrate was evaporated. The residue was dissolved in 50 ml of 2-propanol and warmed on the steambath. A warm solution of 17 g of maleic acid in 60 ml of ethanol was added and the salt was allowed to crystallize by cooling in the ice bath. The final product consisted of yellow crystals with mp 196
EXAMPLE 14
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Acetic anhydride, 7 ml., was added to a solution of 6.16 g. of crude 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine in 200 ml. of methylene chloride. The solution was layered with 200 ml. of saturated aqueous sodium bicarbonate and the mixture was stirred for 20 minutes. The organic layer was separated, washed with sodium bicarbonate, dried over sodium sulfate and evaporated to leave 6.2 g. resinous 2-acetaminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine. This material was heated with 40 g. of polyphosphoric acid at 150 water, made alkaline with ammonia and ice and extracted with methylene chloride. The extracts were dried and evaporated and the residue (5.7 g.) was chromatographed over 120 g. of silica gel using 20% methanol in methylene chloride. The clean fractions were combined and evaporated to yield resinous 8-chloro-3a,4-dihydro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[ 1,5-a][1,4]benzodiazepine. A mixture of this material with 500 ml. of toluene and 30 g. of manganese dioxide was heated to reflux for 11/2 hours. The manganese dioxide was separated by filtration over celite. The filtrate was evaporated and the residue was crystallized from ether to yield a product with m.p. 152 was recrystallized from methylene chloride/hexane
EXAMPLE 49
8-Chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.625 g. (5.5 mmol), was added to a solution of 1.625 g. (5 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 nitrogen for 10 min. at -30 ml. of glacial acetic acid and was then partitioned between aqueous bicarbonate and toluene/methylene chloride (3:1 v/v). The organic layer was separated, dried and evaporated. The residue was chromatographed over 60 g. of silica gel using 25% (v/v) methylene chloride in ethyl acetate. The less polar product was eluted first and was crystallized from ethylacetate/hexane to yield product with m.p. 180
EXAMPLE 50
8-Chloro-6-(2-fluorphenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.125 g. (1.1 mmol) was added to a solution of 0.325 g. (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 -30 by addition of 0.2 ml. of glacial acetic acid and was partitioned between aqueous sodium bicarbonate and methylene chloridetoluene (1:3). The organic phase was washed with water, dried and evaporated. The residue was chromatographed over 20 g. of silica gel using ethyl acetate for elution. After elution of starting material, product was collected and crystallized from ether/hexane, m.p. 156
hyd and dihydrochloride
EXAMPLE 24
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine dihydrochloride
A solution of 0.32 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 5 ml of ethanol was treated with excess ethanolic hydrogen chloride. The salt was crystallized by addition of 2-propanol and ether. The colorless crystals were collected, washed with ether and dried to leave a final product with mp 290
EXAMPLE 258-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride
A solution of 0.325 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 3 ml of ethanol was combined with a suspension of 0.4 g (1 mmol) of the dihydrochloride of this compound in 5 ml of ethanol. After filtration, the solution was treated with ether and heated on the steambath for 5 min to crystallize. The crystals were collected, washed with ether and dried to leave the monohydrochloride with mp 295
maleate
EXAMPLE 22
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine maleate
A warm solution of 6.5 g (0.02 m) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 30 ml of ethanol was combined with a warm solution of 2.6 g (0.022 m) of maleic acid in 20 ml of ethanol. The mixture was diluted with 150 ml of ether and heated on the steam bath for 3 min. After cooling, the crystals were collected, washed with ether and dried in vacuo to yield a final product with mp 148
…
Synthesis
Midazolam, can be described according to scheme 4 indicated below:

 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.For the reactions performed in the microreactor, the solutions containing the substrates to be decarboxylated were loaded into 5 and 10 mL gastight glass syringes (Hamilton, item n. 81527, 81627) mounted on syringe pumps (KD Scientifics, model KDS100). A pipe made of PTFE® (OD=1.58 mm, ID=0.8 mm, Supelco, item n. 58696-U) was used for making the reaction channel.A counterpressure valve sold by Swagelok (item n. SS-SS1-VH) was used for regulating the flow within the channel.Example 1Synthesis of the Compound of Formula (V)—Example of the Invention

50 g (0.135 mol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-3-carboxylic acid of formula (VI) and 250 mL of ethanol were loaded into a two-neck 500 mL flask, equipped with a magnetic stirrer. 40 mL of an aqueous solution of 1 M HCl are dripped in about 10 minutes. The open di-hydrochloride intermediate of formula (V) starts precipitating into the reaction environment already after 3 minutes from the beginning of the addition of the acid solution. The mixture is maintained stirred at RT for 3 hrs and then it is filtered on buckner washing the solid with ethanol. The moist product is dried in an oven under vacuum at 60° C. up to reaching a constant weight. A light yellow crystalline product is obtained (51.5 g, 83% yield). The crude product was used for the decarboxylation without further purifications.
ESI-MS [MeCN+0.1% HCOOH]: m/z 388 (V); 370 (VI).
1H-NMR (250 MHz, CD3OD): 2.52 (s, 3H); 4.27-4.41 (m, 2H); 7.22-8.1 (m, 7H). M.p.: 217° C.
Example 2
Synthesis of Midazolam of Formula (IV)—Performed in Batch—Example of the Invention

30 g (0.065 mol) of 5-(aminomethyl)-1-{(4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 90 mL of NMP are loaded into a three-neck 250 mL flask, equipped with a magnetic stirrer and coolant. The mass is heated using an oil bath at T=195-203° C. for one hour. Thus, 1 mL of solution is collected for performing HPLC analysis. The reaction product is Midazolam having 82% titre (w/w) (determined via HPLC titre correcting it using the solvent) and it contains 1% of Isomidazolam. The product is extracted using Isopropyl acetate after raising the pH to 10 by adding aqueous Na2CO3.
Example 3
Synthesis of Midazolam of Formula (IV)—Performed in a Micro-Reactor—Example of the Invention

3.22 g (7 mmol) of 5-(aminomethyl)-1-{4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 10 mL of NMP are loaded into a 10 mL flask equipped with a magnetic stirrer. In order to facilitate the complete solubilisation of the substrate, it is necessary to slightly heat the reaction mixture (about 40° C.) for a few minutes. The solution thus obtained is transferred into a 10 mL gastight glass syringe mounted on a KDS100 syringe pump (FIG. 1) and the flow is regulated at 1.0 mL/h so as to set a residence period of 30 minutes at 200° C. The reaction product is Midazolam having an 89% titre (w/w) (determined via HPLC titre correcting it using the solvent) and containing 3% (w/w) of Isomidazolam.
Example 4Synthesis of Midazolam of formula (IV)—Comparison of the InventionA table is reported which summarises the results of the decarboxylation of the compound of formula (V) and (V-bis) (for the latter see Examples 6 and 7) obtained according to some embodiments of the invention and those obtained by way of experiment through the decarboxylation of the intermediate of formula (VI) (process of the prior art) both performed in 3 volumes of NMP at 200° C., both in batch method (Example 4) and in continuous method with the microreactor (MR) made of PTFE of FIG. 1. (Examples 4-1, 4-2, 4-3).
| Example | substrate | Mode | Solv. | T° C. | t min. | Midazolam (p/p) | Isomidaz. (P/P) |
| 2 | (V) | Batch | NMP | 200 | 60 | 82 | 1 |
| 3 | (V) | MR | NMP | 200 | 30 | 89 | 3 |
| 7 | (V-bis) | Batch | NMP | 200 | 60 | 68 | 3 |
| 4 | (VI) | Batch | NMP | 200 | 60 | 78 | 18 |
| 4-1 | (VI) | MR | NMP | 200 | 38 | 81 | 17 |
| 4-2 | (VI) | MR | NMP | 200 | 20 | 77 | 18 |
| 4-3 | (VI) | MR | NMP | 200 | 15 | 58 | 22 |
| U.S. Pat. No. | (VI) | Tubular | n-BuOH | 290 | 4 | 85 * | 15 * |
| 5,693,795 | reactor | ||||||
| U.S. Pat. No. | (VI) | Batch | Olio | 230 | 180 | 75 * | 25 * |
| 6,512,114 | min. | 87.5 * | 12.5 * | ||||
| or DMA | |||||||
| * = Midazolam/Isomidazolam ratio only (other impurities not considered). | |||||||
The product of the comparative experiments 4, 4-1, 4-2, 4-3 and of the two USA patents should be subjected to a further isomerisation process to reduce the high amount of Isomidazolam so as to be able to obtain Midazolam free of Isomidazolam after further crystallization, which would not be required for the product obtained according to the invention (examples 2 and 3).

A 4-neck RBF was charged under nitrogen flow with: 10 g of Midazolam (IV) (prepared according to example 2) and 40 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following solution: 3.72 g of maleic acid are dissolved in 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. The maleic acid solution is dropped in 30/40 minutes and keeping T=25/30° C. into the solution containing Midazolam. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 40 mL of cool Ethanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. 12.8 g of Midazolam Maleate as white solid were collected (Molar yield=94.5%). m.p.=149-152° C. (by DSC).

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. 5 mL of a ethanolic solution of Hydrochloric acid 2N were slowly added. 20 mL of Isopropanol were added over 30 minutes at RT. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 10 mL of cool isopropanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. Midazolam dihydrochloride as white solid was collected.
MIDAZOLAM HYDROCHLORIDE
Example 10
Preparation of 8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride (Midazolam hydrochloride)

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 10 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following suspension: 1.22 g of Midazolam dihydrochloride (prepared according to example 9) and 15 mL of Ethanol. The Midazolam ethanolic solution was added to the Midazolam dihydrochloride suspension. After filtration, the solution was treated with MTBE and heated at 60° C. until crystallization. After cooling to RT, the slurry was filtered, the cake washed with MTBE and the product was dried to provide Midazolam (mono)hydrochloride as a white solid.
…..


NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}