
Home » Posts tagged 'Antispasmodic'
Tag Archives: Antispasmodic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MEBEVERINE
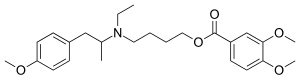
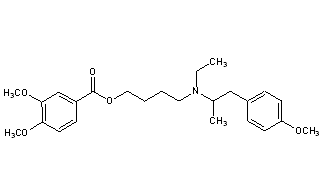
MEBEVERINE
- Molecular FormulaC25H35NO5
- Average mass429.549 Da
3,4-Dimethoxybenzoic Acid 4-[Ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl Ester
3625-06-7[RN]
222-830-4[EINECS]
3,4-Diméthoxybenzoate de 4-{éthyl[1-(4-méthoxyphényl)-2-propanyl]amino}butyle
мебеверин
ميبيفيرين
美贝维林
- EINECS:222-830-4
- LD50:24 mg/kg (M, i.v.); 995 mg/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H35NO5 • HCl
- MW:466.02 g/mol
- CAS-RN:2753-45-9
- EINECS:220-400-0
- LD50:17.7 mg/kg (R, i.v.); 1540 mg/kg (R, p.o.)
Mebeverine
CAS Registry Number: 3625-06-7
CAS Name: 3,4-Dimethoxybenzoic acid 4-[ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl ester
Additional Names: veratric acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;3,4-dimethoxybenzoic acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl 3,4-dimethoxybenzoate;4-[N-[2-(p-methoxyphenyl)-1-methylethyl]-N-ethylamino]butyl 3,4-dimethoxybenzoate
Molecular Formula: C25H35NO5
Molecular Weight: 429.55
Percent Composition: C 69.90%, H 8.21%, N 3.26%, O 18.62%
Literature References: Smooth muscle relaxant. Prepn: BE609490C.A.59, 517b (1963) and T. Kralt et al.,DE1126889; eidem,US3265577 (1962, 1962, 1966 to N. V. Philips). Pharmacology: G. Bertaccini et al.,Farmaco Ed. Sci.30, 823 (1975).
Derivative Type: Hydrochloride
CAS Registry Number: 2753-45-9
Trademarks: Colofac (Duphar); Duspatalin (Duphar); Duspatal (Duphar)
Molecular Formula: C25H35NO5.HCl
Molecular Weight: 466.01
Percent Composition: C 64.43%, H 7.79%, N 3.01%, O 17.17%, Cl 7.61%
Properties: Crystals from ethyl methyl ketone, mp 105-107° (Ger. patent); also reported as mp 129-131° (Belg. patent).
Melting point: mp 105-107° (Ger. patent); mp 129-131° (Belg. patent)
Therap-Cat: Antispasmodic.
Keywords: Antispasmodic.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PAT
Indian Pat. Appl., 201841023171

PAPER
Pharma Chemica, 2(2), 366-378; 2010
In a quest of novel antispasmodic agents with antimicrobial properties, the present study describes design and synthesis of novel analogs for veratric acid ester 4-[ethyl-{2-(4- methoxyphenyl)-1-methylethyl} amino] butan-1-ol, an antispasmodic drug which is expected to be a potent antimicrobial agent may be due to the presence of two benzene rings and a secondary or tertiary nitrogen in the basic structural framework of the molecule. The reaction between substituted 2-ethylamino-1-(4’-methoxyphenyl) propane and various haloaryl benzoates derivatives obtained from reaction between different homologs of benzoic acid and dibromoalkanes in a two step process to give corresponding structurally diverse analogs of lead compound has been achieved. The structures of these novel analogs were confirmed by different structure elucidation techniques. All the compounds have been screened for their anti-spasmodic activity and the study extended further to evaluate their sedative, antibacterial and antifungal potency. The novel analogs of lead compound exhibited pronounced antispasmodic activities and also gave encouraging results of antimicrobial and sedative activity as anticipated.
General method of preparation of veratric acid ester 4-[ethyl-{2-(4-methoxyphenyl)-1- methylethyl} amino] butan-1-ol hydrochloride (5) and its analogs (5a-5p) A mixture of compound (3) (149 g, 0.47 mol) and Compound (4) (183 g, 0.95 mol) in ethyl methyl ketone (MEK) was refluxed for a period of 30 h at 75-80oC. The progress of the reaction was monitored by TLC to ensure formation of product and complete conversion of starting. On reaction completion solvent was distilled off and water (750 ml) was added to the reaction mass followed by toluene (300 ml). The resulting solution was cooled to 30oC and stirred for 30 minutes before layer separation. The organic layer was washed further with water (2×100 ml) and dried over sodium sulphate. To the organic layer IPA-HCl (72 g, 20 %) was added till pH is acidic (2-2.5).The product precipitated as solid hydrochloride salt was isolated by filtration and recrystallized from methanol. Yield: 181 g, 82% m.p., 105-107°C.
-C-H stretching (2959-2840), -C=O stretching (1717), -C=C stretching (1605, 1514, 1459), asymmetrical -C-O-C and –C-O stretching (1265-1130), symmetrical -C-O-C stretching (1023)
Chemical Shift ð, 1.25-1.22 ppm (t, 3H, -CH3), 1.57-1.51 (m, 3H, -CH3), 1.90-1.81 (d, 2H, -CH2), 2.16-2.11(m, 2H, – CH2), 2.54-2.25 (m, 1H, -CH), 3.11-3.05 (m, 4H, -CH2), 3.58-3.54 (t, 2H, -CH2), 3.77 (s, 3H, -OCH3), 3.91 (s, 6H, – OCH3), 4.37-4.33 (d, 2H, -CH2), 6.87-6.79 (m, 3H, Ar-H), 7.14-7.12 (d, 2H, Ar-H), 7.52-7.50 (d, 1H, Ar-H), 7.68-7.65 (m, 1H, Ar-H)
Antispasmodic drugs relieve cramps or spasms of the stomach, intestines, and bladder. Antispasmodics are classes (group) of drugs that can help to control some symptoms that arise from the gut, in particular, gut spasm. There are two main types namely “Antimuscarinics” and “Smooth muscle relaxants”. Antispasmodics are commonly used in “Irritable bowel syndrome” (IBS) to help relieve some of the symptoms of IBS such as spasm (colic), bloating and abdominal (stomach) pain and to reduce the motility (movement) of the intestines (gut) [1].
After understanding further the medicinal importance of antispasmodics and their ever increasing demand worldwide, we pursue to undertake the detailed synthetic and pharmacological study of antispasmodics to identify novel candidates as potential drug substances. Our parallel interest also lies on identifying novel antimicrobials since over the years; antibiotics are known to be the major protective agents against bacterial infections. However, the usage of antibiotics and antibacterial chemotherapeutics is becoming more and more restricted in the present age, despite the fact that there exist a large number of antibiotics. This is largely attributed to the emergence of drug-resistant bacteria, which render even some of the most broad spectrum antibiotics ineffective. In addition, most antibiotics have side effects. Thus, it becomes essential to investigate newer drugs with less resistance. Different studies on search of newer antimicrobials and antibacterial have revealed that moderate to remarkable antimicrobial or antibacterial action is present in several compounds, belonging to various pharmacological categories, such as antihistamines [2-4], tranquilizers [5], antihypertensive [6], anti-psychotics [7-11] anti-spasmodic [12] and anti-inflammatory agents [13]. Such compounds, having antibacterial properties in addition to their predesignated pharmacological actions, are termed as non-antibiotics [12]. Many of these compounds possess two or three benzene rings and nitrogen in the secondary or tertiary state in their molecular structure which is expected to be one of the bases for exhibiting antimicrobial potency [14]. Based on this rationale and to pursue our interest to identify newer antispasmodic agents with sedative and antimicrobial properties
[1] M. H. Pittler, E. Ernst, Am. J. Gastroenterol., 1998, 93 (7), 1131–5. [2] S. G. Dastidar, P. K. Saha, B. Sanyamat, A. N. Chakrabarty, J. Appl. Bacteriol., 1976, 41, 209- 214. [3] D. Chattopadyay, S. G. Dastidar, A. Chakrabarty, Arzneimittelforschang, 1988, 38, 869-872. [4] A. Chakrabarty, D. P. Acharya, D. K. Neogi, S. G. Dastidar, Indian J. Med. Res., 1989, 89, 233-237. [5] S. K. Dash, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1977, 15, 324-326. [6] S. G. Dastidar, U. Mondal, S. Niyogi, A. Chakrabarty, Indian J. Med. Res., 1986, 84, 142- 147. [7] J. Molnar, Y. Mandi, J. Kiral, Acta Microbiol Acad Sci Hung., 1976, 23, 45-54. [8] J. E. Kristiansen, Acta Pathol. Microbial Immunol. Scand., 1992, 100 (Suppl. 30), 7-14 [9] S. G. Dastidar, A. Chaudhury, S. Annadurai, M. Mookerjee, A. Chakrabarty, J. Chemother., 1995, 7, 201-206. [10] V. Radhakrishnan, K. Ganguly, M. Ganguly, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1999, 37, 671-675. [11] P. Bourlioux, J. M. Moreaux, W. J. Su, H. Boureau, Acta Pathol. Microbial. Immunol Scand., 1992, 100 (Suppl. 30), 40-43. [12] S. G. Dastidar, A. Chakrabarty, J. Molnar, N. Motohashi, National Institute of Science Communication (NISCOM), New Delhi, 1998, pp. 15. [13] S. Annadurai, S. Basu, S. Ray, S. G. Dastidar, A. C
Mebeverine is a drug used to alleviate some of the symptoms of irritable bowel syndrome. It works by relaxing the muscles in and around the gut.[1]
Medical use
Mebeverine is used to alleviate some of the symptoms of irritable bowel syndrome (IBS) and related conditions; specifically stomach pain and cramps, persistent diarrhoea, and flatulence.[2]
Data from controlled clinical trials have not found a difference from placebo or statistically significant results in the global improvement of IBS.[3][4]
It has not been tested in pregnant women nor in pregnant animals so pregnant women should not take it; it is expressed at low levels in breast milk, while no adverse effects have been reported in infants, breastfeeding women should not take this drug.[1]
Adverse effects
Adverse effects include hypersensitivity reactions and allergic reactions, immune system disorders, skin disorders including hives, oedema and widespread rashes.[2]
Additionally, the following adverse effects have been reported: heartburn, indigestion, tiredness, diarrhoea, constipation, loss of appetite, general malaise, dizziness, insomnia, headache, and decreased pulse rate.[1]
It does not have systemic anticholinergic side effects.[2]
Mebeverine can, on highly rare occasions, cause drug-induced acute angle closure glaucoma.[5]
Mechanism of action
Mebeverine is an anticholinergic but its mechanism of action is not known; it appears to work directly on smooth muscle within the gastrointestinal tract and may have an anaesthetic effect, may affect calcium channels, and may affect muscarinic receptors.[2]
It is metabolized mostly by esterases, and almost completely. The metabolites are excreted in urine.[2]
Mebeverine exists in two enantiomeric forms. The commercially available product is a racemic mixture of them. A study in rats indicates that the two have different pharmacokinetic profiles.[6]
History
It is a second generation papaverine analog, and was first synthesized around the same time as verapamil.[7]
It was first registered in 1965.[8]
Availability
Mebeverine is a generic drug and is available internationally under many brand names.[9]
SYN
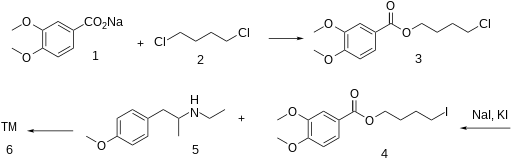
Anon., Belgian Patent 609,490 (1962); T. Kralt,
H. O. Moes, A. Lindner and W. J. Asma, German Patent 1,126,889 (1962); Chem. Abstr., 59: 517b
(1963).
SYN

SYN
https://www.sciencedirect.com/science/article/abs/pii/S0731708502000237
References
- ^ Jump up to:a b c “Colofac data sheet” (PDF). New Zealand Medicines and Medical Devices Safety Authority. 14 June 2017. Retrieved 21 July2017.
- ^ Jump up to:a b c d e “Colofac Tablets 135mg – Summary of Product Characteristics (SPC)”. UK Electronic Medicines Compendium. 26 August 2016. Retrieved 21 July 2017.
- ^ Annaházi A, Róka R, Rosztóczy A, Wittmann T (May 2014). “Role of antispasmodics in the treatment of irritable bowel syndrome”. World Journal of Gastroenterology. 20 (20): 6031–43. doi:10.3748/wjg.v20.i20.6031. PMC 4033443. PMID 24876726.
- ^ Darvish-Damavandi M, Nikfar S, Abdollahi M (February 2010). “A systematic review of efficacy and tolerability of mebeverine in irritable bowel syndrome”. World Journal of Gastroenterology. 16(5): 547–53. doi:10.3748/wjg.v16.i5.547. PMC 2816265. PMID 20128021.
- ^ Lachkar Y, Bouassida W (March 2007). “Drug-induced acute angle closure glaucoma”. Current Opinion in Ophthalmology. 18 (2): 129–33. doi:10.1097/ICU.0b013e32808738d5. PMID 17301614. S2CID 30903966.
- ^ Hatami M, Farhadi K, Tukmechi A (August 2012). “Fiber-based liquid-phase micro-extraction of mebeverine enantiomers followed by chiral high-performance liquid chromatography analysis and its application to pharmacokinetics study in rat plasma”. Chirality. 24(8): 634–9. doi:10.1002/chir.22057. PMID 22700279.
- ^ Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 132. ISBN 9780471899792.
- ^ “Mebeverine”. druginfosys. Retrieved 1 February 2015.
- ^ “Mebeverine”. International. drugs.com. Retrieved 1 February2015.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATC code | A03AA04 (WHO) |
| Legal status | |
| Legal status | UK: P (Pharmacy medicines)US: Not approvedIn general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 3625-06-7 HCl: 2753-45-9 |
| PubChem CID | 4031 |
| ChemSpider | 3891 |
| UNII | 7F80CC3NNVHCl: 15VZ5AL4JN |
| KEGG | D04868 |
| ChEMBL | ChEMBL282121 |
| CompTox Dashboard (EPA) | DTXSID6023238 |
| ECHA InfoCard | 100.020.756 |
| Chemical and physical data | |
| Formula | C25H35NO5 |
| Molar mass | 429.557 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (verify) |
//////////mebeverine, мебеверин ,ميبيفيرين ,美贝维林 , Antispasmodic

NEW DRUG APPROVALS
ONE TIME
$10.00
Glycopyrronium bromide, гликопиррония бромид , بروميد غليكوبيرونيوم , 格隆溴铵 , グリコピロニウム臭化物
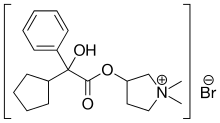
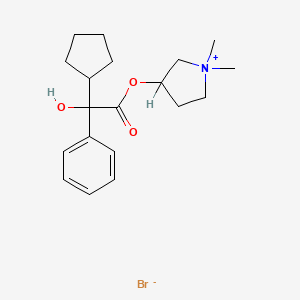

Glycopyrronium bromide
Cas 596-51-0,
- 3-Hydroxy-1,1-dimethylpyrrolidinium bromide α-cyclopentylmandelate (6CI,7CI)
- Pyrrolidinium, 3-[(cyclopentylhydroxyphenylacetyl)oxy]-1,1-dimethyl-, bromide (9CI)
- Pyrrolidinium, 3-hydroxy-1,1-dimethyl-, bromide, α-cyclopentylmandelate (8CI)
- 1,1-Dimethyl-3-hydroxypyrrolidinium bromide α-cyclopentylmandelate
- AHR-504
- Asecryl
- Copyrrolate
- Gastrodyn
- Glycopyrrolate
- Glycopyrrolate bromide
- Glycopyrrone bromide
- Glycopyrronium bromide
- NSC 250836
- NSC 251251
- NSC 251252
- NVA 237
- Nodapton
- Robanul
- Robinul
- Seebri
- Tarodyl
- Tarodyn
- β-1-Methyl-3-pyrrolidyl-α-cyclopentylmandelate methobromide
CAS FREE FORM OF ABOVE 13283-82-4
Glycopyrrolate, ATC:A03AB02
- Use:anticholinergic, antispasmodic
- Chemical name:3-[(cyclopentylhydroxyphenylacetyl)oxy]-1,1-dimethylpyrrolidinium bromide
- Formula:C19H28BrNO3, MW:398.34 g/mol
- EINECS:209-887-0
- LD50:15 mg/kg (M, i.v.); 570 mg/kg (M, p.o.);
709 mg/kg (R, p.o.)
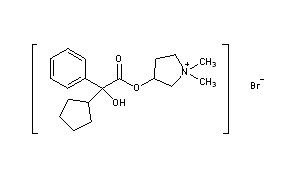
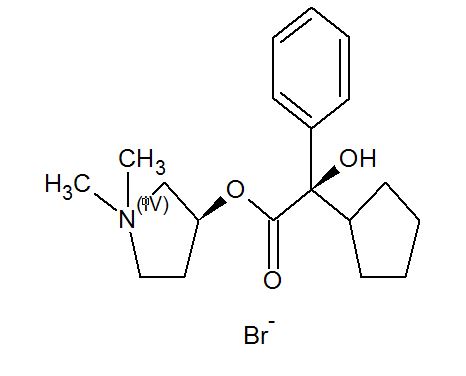
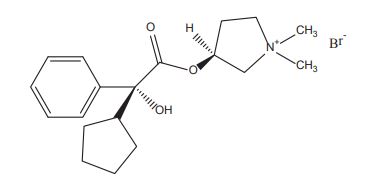
- Pyrrolidinium, 3-[(cyclopentylhydroxyphenylacetyl)oxy]-1,1-dimethyl-, bromide, (R*,S*)-(±)-
- Pyrrolidinium, 3-[[(2R)-cyclopentylhydroxyphenylacetyl]oxy]-1,1-dimethyl-, bromide, (3S)-rel- (9CI)
- erythro-Glycopyrronium bromide
FREE FORM OF ABOVE 740028-90-4
NMR analysis of the diastereomers of glycopyrronium bromide
Finnish Chemical Letters (1975), (3-4), 94-6
Michael Woehrmann, Lara Terstegen, Stefan Biel, Thomas Raschke, Svenja-Kathrin Cerv, Werner Zilz, Sven Untiedt, Thomas Nuebel, Uwe Schoenrock, Heiner Max, Helga Biergiesser, Yvonne Eckhard, Heike Miertsch, Heike Foelster, Cornelia Meier-Zimmerer, Bernd Traupe, Inge Kruse, “GLYCOPYRROLATE IN COSMETIC PREPARATIONS.” U.S. Patent US20090208437, issued August 20, 2009.US20090208437
Glycopyrrolate is a muscarinic antagonist used as an antispasmodic, in some disorders of the gastrointestinal tract, and to reduce salivation with some anesthetics.
Glycopyrronium (as the bromide salt glycopyrrolate) is a synthetic anticholinergic agent with a quaternary ammonium structure. A muscarinic competitive antagonist used as an antispasmodic, in some disorders of the gastrointestinal tract, and to reduce salivation with some anesthetics. In October 2015, glycopyrrolate was approved by the FDA for use as a standalone treatment for Chronic obstructive pulmonary disease (COPD), as Seebri Neohaler.
Medical uses
In anesthesia, glycopyrronium injection can be used as a before surgery in order to reduce salivary, tracheobronchial, and pharyngealsecretions, as well as decreasing the acidity of gastric secretion. It is also used in conjunction with neostigmine, a neuromuscular blocking reversal agent, to prevent neostigmine’s muscarinic effects such as bradycardia.
It is also used to reduce excessive saliva (sialorrhea),[3][4][5] and Ménière’s disease.[6]
It decreases acid secretion in the stomach and so may be used for treating stomach ulcers, in combination with other medications.
It has been used topically and orally to treat hyperhidrosis, in particular, gustatory hyperhidrosis.[7][8]
In inhalable form it is used to treat chronic obstructive pulmonary disease (COPD). Doses for inhalation are much lower than oral ones, so that swallowing a dose will not have an effect.[9][10]
Side effects
Since glycopyrronium reduces the body’s sweating ability, it can even cause hyperthermia and heat stroke in hot environments. Dry mouth, difficulty urinating, headaches, diarrhea and constipation are also observed side effects of the medication. The medication also induces drowsiness or blurred vision, an effect exacerbated by the consumption of alcohol.
Pharmacology
Mechanism of action
Glycopyrronium blocks muscarinic receptors,[11] thus inhibiting cholinergic transmission.
Pharmacokinetics
Glycopyrronium bromide affects the gastrointestinal tracts, liver and kidney but has a very limited effect on the brain and the central nervous system. In horse studies, after a single intravenous infusion, the observed tendencies of glycopyrronium followed a tri-exponential equation, by rapid disappearance from the blood followed by a prolonged terminal phase. Excretion was mainly in urine and in the form of an unchanged drug. Glycopyrronium has a relatively slow diffusion rate, and in a standard comparison to atropine, is more resistant to penetration through the blood-brain barrier and placenta.[12]
Research
It has been studied in asthma.[13][14]

Synthesis
PATENT
https://patents.google.com/patent/CN103819384A/en


PAtent
https://patents.google.com/patent/CN103159659A/en

glycopyrrolate (I)
Methyl ethyl ketone (20mL) IOOmL three-necked flask was added 8 (4.6g, 15mmol) was, at (Γ5 ° C was added dropwise dibromomethane (2.9g, 30mmol) in butanone (5 mL) was added dropwise completed, continued The reaction was stirred for 15min, and a white solid precipitated, was allowed to stand 36h at room temperature, filtered off with suction, the filter cake was sufficiently dried to give crude ketone was recrystallized twice to give a white powdery crystals I (3.9g, 66%) mp 191~193 ° C chromatographic purity 99.8% [HPLC method, mobile phase: lmol / L triethylamine acetate – acetonitrile – water (1: 150: 49); detection wavelength: 230nm, a measurement of the area normalization method] .MS m / z: 318 ( m-BrO 1HNMR (CD3OD) δ:! 1.33~1.38 (m, 2H), 1.55~1.70 (m, 6H), 2.11~2.21 (m, 1H), 2.67~2.80 (m, 1H), 3.02 (m, 1H), 3.06 (s, 3H), 3.23 (s, 3H), 3.59~3.71 (m, 3H), 3.90 (dd, /=13.8,1H), 5.47 (m, 1H), 7.27 (t, 1H) , 7.35 (t, 2H), 7.62 (dd, 2H) .13C bandit R (DMSO) δ: 27.0, 27.4, 28.0, 31.3, 47.8, 53.8, 54.3, 66.0, 71.3, 74.6, 81.1, 126.9,128.7,129.3 , 143.2 17 5.00
Patent
https://patents.google.com/patent/WO2016204998A1/en

PATENT
https://patents.google.com/patent/EP2417106B1/en
-
Glycopyrronium bromide, also known as 3-[(cyclopentylhydroxyphenylacetyl)oxy]-1,1-dimethylpyrrolidinium bromide or glycopyrrolate, is an antimuscarinic agent that is currently administered by injection to reduce secretions during anaesthesia and or taken orally to treat gastric ulcers.
- [0003]
It has the following chemical structure:

- [0004]
United States patent US 2,956,062 discloses that 1-methyl-3-pyrrolidyl alpha-cyclopentyl mandelate and can be prepared from methyl alpha cyclopentylmandelate and that the methyl bromide quaternary salt can be prepared by saturating a solution of 1-methyl-3-pyrrolidyl alpha-cyclopentyl mandelate in dry ethyl acetate with methyl bromide and filtering the crystalline solid that appears on standing.
- [0005]
The process of US 2,956,062 for preparing 1-methyl-3-pyrrolidyl alpha-cyclopentyl mandelate involves transesterifying methyl glycolate with an amino alcohol under the influence of metallic sodium to give a glycolate intermediate. Metallic sodium is highly reactive, which poses health and safety risks that make its use undesirable on an industrial scale for commercial manufacture.
- [0006]
The process of US 2,956,062 requires preparing the methylester in a previous step and alkylating the amino esters in a later step to form the desired quaternary ammonium salts.
- [0007]
The process of US 2,956,062 provides a mixture of diastereoisomers. The relative proportions of the diastereoisomers can vary widely between batches. This variation can give rise to surprising differences when preparing dry powder formulations from glycopyrronium bromide, which can cause problems when formulating such dry powders for pharmaceutical use.
- [0008]
United States patent application US 2007/0123557 discloses 1-(alkoxycarbonylmethyl)-1-methylpyrrolidyl anticholinergic esters. It describes coupling (R)-cyclopentylmandelic acid with (R,S)-1-methyl-pyrrolidin-3-ol under Mitsunobu conditions to give pure (R)-stereoisomeric compounds that are reacted with a bromoacetate to give the desired esters. It should be noted however that the chemicals used in Mitsunobu reactions, typically dialkyl azodicarboxylates and triphenylphosphine, pose health, safety and ecological risks that make their use undesirable on an industrial scale for commercial manufacture. They are also generally too expensive to source and too laborious to use in commercial manufacture.
- [0009]
United States patent application US 2006/0167275 discloses a process for the enrichment of the R, R- or S, S-configured glycopyrronium isomers and their thienyl analogues having R, S or S, R configuration.
- [0010]
WO 03/087094 A2 discloses new therapeutically useful pyrrolidinium derivatives, processes for their preparation and pharmaceutical compositions containing them.

EXAMPLE Example 1 Preparation of (3S,2’R)- and (3R,2’S)-3-[(cyclopentyl-hydroxyphenylacetyl)-oxy]-1,1-dimethylpyrrolidinium bromide
- [0071]
30 g of cyclopentyl mandelic acid, dissolved in 135 g dimethylformamide (DMF), were treated with 27 g carbonyldiimidazole at 18°C (in portions) to form the “active amide”. After the addition of 16.9 g of 1-methyl-pyrrolidin-3-ol, the mixture was heated to 60°C within 1 hour and stirred for 18 hours at this temperature. After checking for complete conversion, the mixture was cooled and 200 g water was added. The mixture was extracted with 200 g toluene and the extract was washed with water three times. The organic phase was concentrated to obtain cyclopentyl-hydroxy-phenyl-acetic acid 1-methyl-pyrrolidin-3-yl ester as an about 50% solution in toluene, ready to use for the next step.
- [0072]
This solution was diluted with 120 g of n-propanol and cooled to 0°C. 16.8 g methyl bromide was introduced and the mixture was stirred for 2 hours and then gradually heated to 60°C to evaporate the excess methyl bromide into a scrubber. The mixture was then cooled to 50°C and seed crystals were added to facilitate crystallisation. The temperature was then slowly reduced over 18 hours to 15°C. The solid was then isolated by filtration to obtain 22.7 g after drying. It was composed mainly of one pair of enantiomers, a racemic mixture of (3S,2’R)- and (3R,2’S)-3-[(cyclopentyl-hydroxyphenylacetyl)-oxy]-1,1-dimethylpyrrolidinium bromide, with a purity greater than 90% (by HPLC). The other pair of diastereoisomers ((3R,2’R)- and (3S,2’S)-3-[(cyclopentyl-hydroxyphenyl-acetyl)-oxyl-1,1-dimethylpyrrolidinium bromide) remains mainly in the filtrate as those compounds are significantly more soluble in n-propanol than the other stereoisomers.
- [0073]
The solid obtained is further recrystallised in n-propanol (1:10 wt) to give pure (3S,2’R)- and (3R,2’S)-3-[(cyclopentyl-hydroxyphenylacetyl)-oxy]-1,1-dimethylpyrrolidinium bromide i.e. purity > 99.9% as determined by high performance liquid chromatography (HPLC).
- [0074]
This process is summarised in the following reaction scheme:

Reference Example 2 Preparation of cyclopentyl-hydroxy-phenyl-acetic acid 1-methyl-pyrrolidin-3yl-ester in toluene
- [0075]
1 g of cyclopentyl mandelic acid was suspended in 4.7 g of toluene and 1.5 g of carbonyldiimidazole were added as a solid. After 30 minutes 0.69 g of 1-methyl-pyrrolidin-3-ol and 20 mg of sodium tert-butylate were added. The mixture was stirred at room temperature for 18 hours then water was added. After stirring the phases were separated and the organic phase was washed with water twice and evaporated to obtain an approximately 50% solution of cyclopentyl-hydroxy-phenyl-acetic acid 1-methyl-pyrrolidin-3yl-ester in toluene.
Example 3 Preparation of 2-cyclopentyl-2-hydroxy-1-imidazol-1-yl-2-phenyl-ethanone, the active intermediate
- [0076]
The imidazolidyl derivative of cyclopentylmandelic acid was prepared and isolated as a solid by the following method:
- [0077]
10 g of cyclopentylmandelic acid were suspended in 30 ml of acetonitrile and the mixture was cooled to 0°C. 10.3 g of carbonyldiimidazole were added as a solid and the mixture was warmed to room temperature for 2 hours. Carbon dioxide evolved as a gas as a precipitate formed. The mixture was then cooled to 5°C and the solid was filtered, washed with acetonitrile and dried in vacuum at 40°C to obtain 7.3 g of pure 2-cyclopentyl-2-hydroxy-1-imidazol-1-yl-2-phenyl-ethanone.
- [0078]
This process is summarised in the following reaction scheme:

- [0079]
High resolution MS-spectroscopy revealed the molecular formula of the compound (as M+H) to be C16H19O2N2 with an exact mass of 271.14414 (0.14575ppm deviation from the calculated value).
1H-NMR-spectroscopy (600MHz, DMSO-d6): 1.03-1.07 (m, 1H), 1.25-1.30 (m, 1H), 1.35-1.40 (m, 1H), 1.40-1.50 (m, 1H), 1.53-1.56 (m, 2H), 1-60-1.67 (m, 1H), 1.75-1.84 (m, 1H), 1.03 – 1.85 (8H, 8 secondary CH2-protons in the cyclopentylring, H-C11, H-C12, H-C13, H-C14); 2.7-2.9 (m, 1H, H-C10); 6.76 (1H, H-C5); 6.91 (1H, H-C4); 7.29 (1H, H-C18); 7.39 (2H, H-C17, H-C19); 7.49 (2H, H-C16, H-C20); 7.65 (1H, H-C2). - [0080]
The compound was characterised by IR-spectroscopy (measured as a solid film on a BRUKER TENSOR 27 FT-IR spectrometer over a wave number range of 4000-600 cm-1 with a resolution of 4 cm-1). An assignment of the most important bands is given below:
Wavenumber (cm-1) Assignments 3300 ∼ 2500 O-H stretching 3167, 3151, 3120 Imidazole CH stretching 2956, 2868 Cyclopentyl CH stretching 1727 C=O stretching 1600, 1538, 1469 Aromatic rings stretching 735 Mono-subst. benzene CH o.o.p. bending 704 Mono-subst. benzene ring o.o.p. bending
SYN
PAPER
https://link.springer.com/article/10.1007/s41981-018-0015-4
Journal of Flow Chemistry, pp 1–8| Cite as
Sequential α-lithiation and aerobic oxidation of an arylacetic acid – continuous-flow synthesis of cyclopentyl mandelic acid
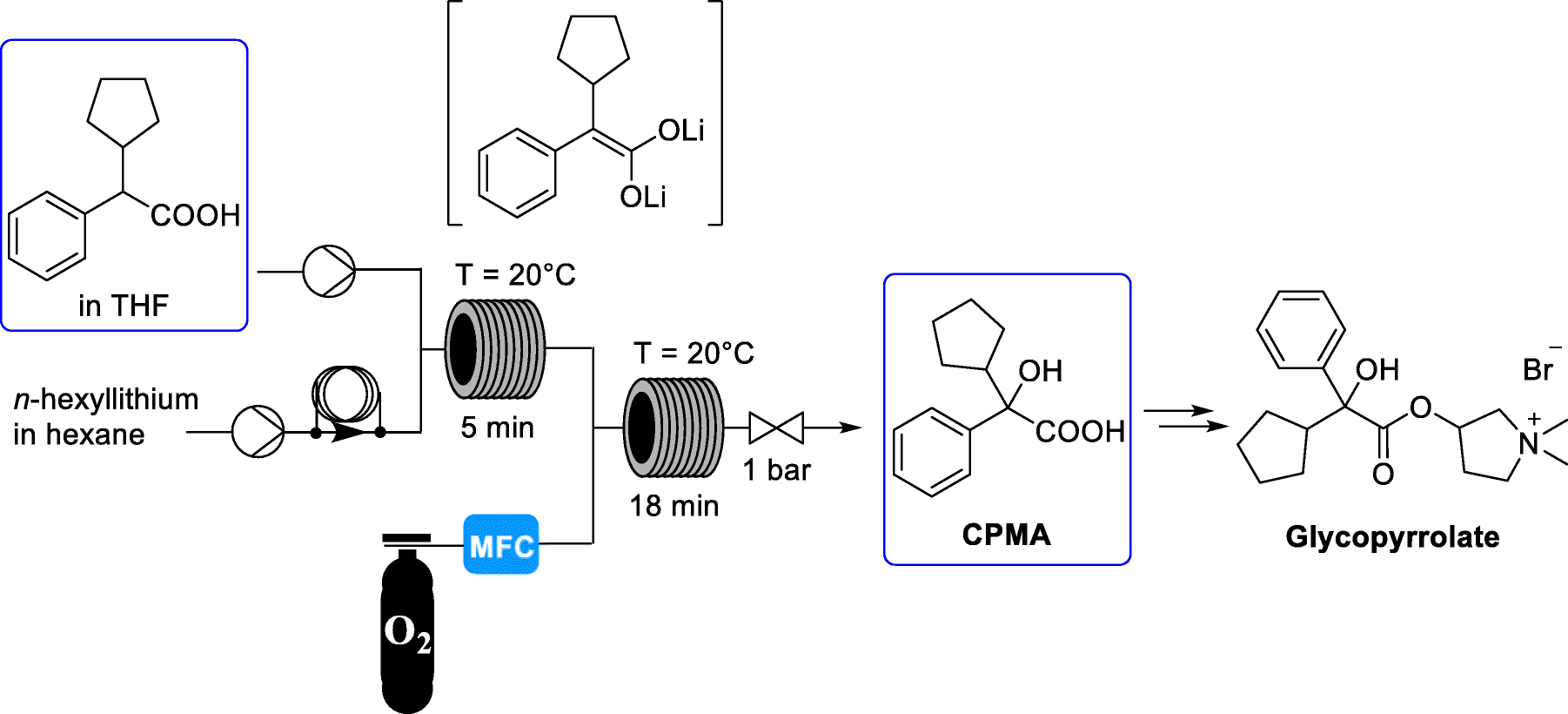
The medicinal properties of glycopyrronium bromide (glycopyrrolate, 4) were first identified in the late 1950s [1]. Glycopyrrolate is an antagonist of muscarinic cholinergic receptors and is used for the treatment of drooling or excessive salivation (sialorrhea) [2], excess sweating (hyperhidrosis) [3], and overactive bladder and for presurgery treatment. In addition, it has recently been introduced as an effective bronchodilator for the treatment of chronic obstructive pulmonary disease (COPD) for asthma patients [4]. Glycopyrrolate displays few side effects because it does not pass through the blood brain barrier. Cyclopentyl mandelic acid (CPMA, 1), or its corresponding ester derivatives, are key intermediates in the synthetic routes to 4. CPMA (1) reacts with 1-methyl-pyrrolidin-3-ol (2) to form tertiary amine 3. N-Methylation of 3 by methyl bromide gives quaternary ammonium salt glycopyrrolate 4 as a racemate (Scheme 1) [5].
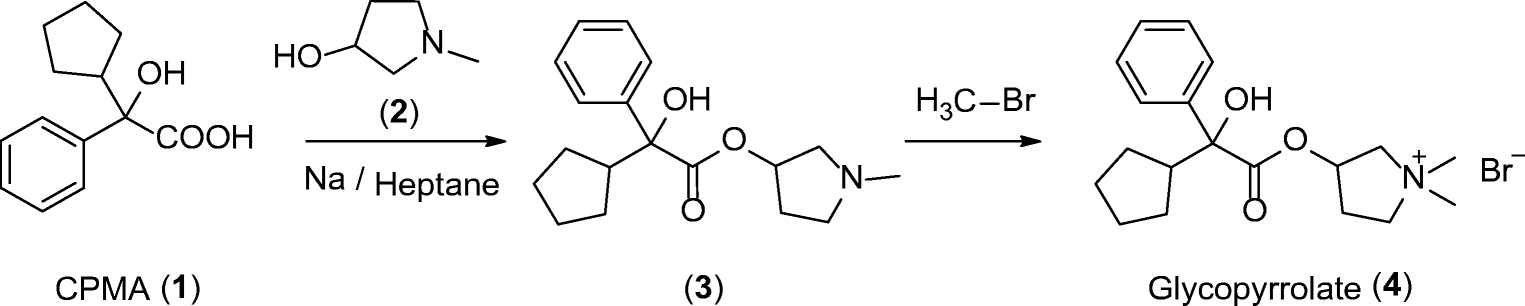
Scheme 1
Synthesis of glycopyrrolate 4 from CPMA (1)
CPMA (1) is a synthetically challenging intermediate to prepare (Scheme 2). Routes A to D are most likely to be the commercially applied methods because these procedures are described in patents [5]. The published descriptions for the yields of 1 range from 28 to 56% for routes A to D. Ethyl phenylglyoxylate is reacted with cyclopentyl magnesium bromide to form an ester which is then hydrolyzed (route A) [6]. Phenylglyoxylic acid can be reacted in a similar manner with cyclopentyl magnesium bromide to directly form 1 (route B) [7]. Alternatively, the inverse addition of phenyl-Grignard reagent to cyclopentyl glyoxylic acid ester is reported (route C) [8]. Cyclopentyl glyoxylic acid ester can also be reacted with cyclopentadienyl magnesium bromide which is followed by an additional hydrogenation step with Pd/C and H2 to afford 1 (route D) [9, 10].
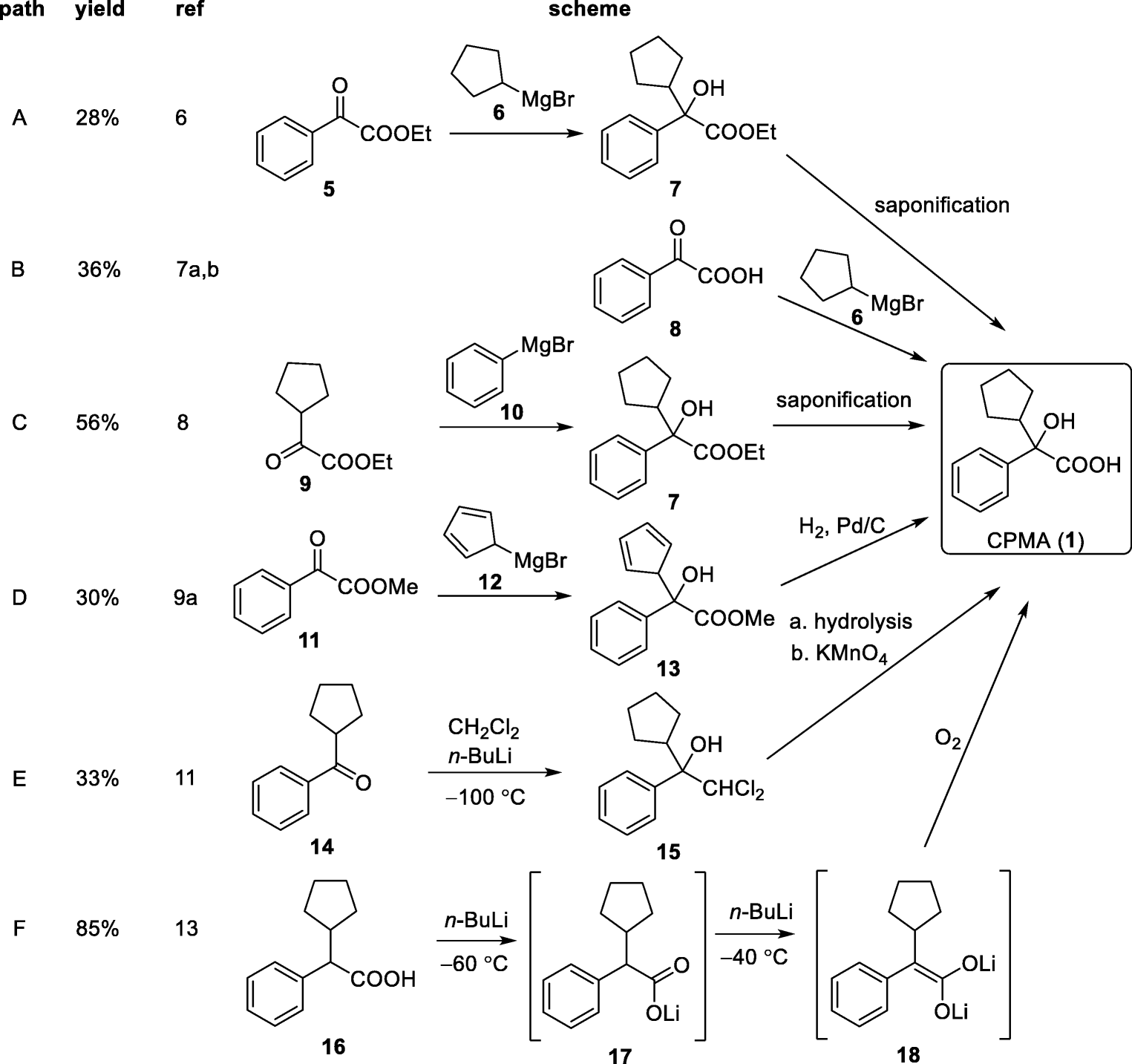
Scheme 2
Existing synthetic pathways to CPMA (1)
PATENT
EXA M PL E S
EXAM PL E 1
Scheme 1
ST E P I
To a stirred solution of N-methyl pyrrol i din- 3-ol (2, 1 equiv) and Et3N (1.2 equiv) in dichloromethane was added a solution of 2-cyclopentyl-2-oxoacetyl chloride (1, 1.1 equiv) in DCM at O °C under nitrogen atmosphere for 20 min. The resulting solution was allowed to stir at room temperature over 10h. After completion, the mixture was quenched with water and extracted with diethyl ether to afford the pure product (3A).
Similarly, the product 3A is also obtained by reaction of 2 with other reagents, phenyl oxalic acid, methyl phenyl oxalate, and phenyl hemi-oxaldehyde respectively as shown in Scheme 1.
ST E P II
3A
To a mixture of bromobenzene (2.2 equiv) and Mg metal (2.2 equiv) in TH F (15 mL) was stirred over a period of 30 min at 0 · C. To this mixture, a solution of 1 -methyl pyrrol idin-3-yl 2-cyclopentyl-2-oxoacetate (3, 1 equiv) in T HF was added in portions over a period of 30 min. Up on completion, the reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was separated and concentrated in vacuo. The resulting residue was purified by column chromatography to afford the pure product (5).
ST E P III
To a solution of compound 5 (1 equiv) in acetonitrile and chloroform mixture (10 mL, 2:3) was added methyl bromide (4 equiv). The mixture was stirred at room temperature for 72h. The solvents were evaporated, and the resulting residue was washed with diethyl ether to afford the pure product (6) as a white solid.
EXAM PL E 2
Scheme 2
ST E P I
To a stirred solution of N-methyl pyrrol i din- 3-ol (2, 1 equiv) and Et3N (1.2 equiv) in dichloromethane was added a solution of 2- oxo-2- phenyl acetyl chloride (1.1 equiv) in dichloromethane at 0 °C under nitrogen atmosphere for 15 min. The resulting solution was allowed to stir at room temperature over 12h. After completion, the mixture was quenched with water and extracted with diethyl ether to afford the pure product (3B).
Similarly, the product 3B is also obtained by reaction of 2 with other reagents, phenyl oxalic acid, methyl phenyl oxalate, and phenyl hemi-oxaldehyde respectively as shown in Scheme 2.
ST E P II
To a mixture of cyclopentyl bromide (4, 2.2 equiv) and Mg metal (2.2 equiv) in THF (15 mL) was stirred over a period of 30 min at 0 – C. To this mixture, a solution of 1-methylpyrrolidin-3-yl-2-oxo-2-phenylacetate (3B, 1 equiv) in TH F was added in portions over a period of 30 min. Up on completion, the reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was separated and concentrated in vacuo. The resulting residue was purified by column chromatography to afford the pure product (5).
ST E P III
To a solution of compound 5 (1 equiv) in acetonitrile and chloroform mixture (10 mL, 2:3) was added methyl bromide (4 equiv). The mixture was stirred at room temperature for 75h. The solvents were evaporated, and the resulting residue was washed with diethyl ether to afford the pure product (6) as a white solid.
The invention has been described in detail with reference to preferred embodiments thereof. However, it will be appreciated by those skilled in the art that changes may be made in these embodiments without departing from the principles and nature of the invention, the scope of which is defined in the appended claims and their equivalents.
References
- Jump up^ Bajaj V, Langtry JA (July 2007). “Use of oral glycopyrronium bromide in hyperhidrosis”. Br. J. Dermatol. 157 (1): 118–21. doi:10.1111/j.1365-2133.2007.07884.x. PMID 17459043.
- Jump up^ “FDA OKs first drug made to reduce excessive sweating”. AP News. Retrieved 2018-07-02.
- Jump up^ Mier RJ, Bachrach SJ, Lakin RC, Barker T, Childs J, Moran M (December 2000). “Treatment of sialorrhea with glycopyrrolate: A double-blind, dose-ranging study”. Arch Pediatr Adolesc Med. 154 (12): 1214–8. doi:10.1001/archpedi.154.12.1214. PMID 11115305.
- Jump up^ Tscheng DZ (November 2002). “Sialorrhea – therapeutic drug options”. Ann Pharmacother. 36 (11): 1785–90. doi:10.1345/aph.1C019. PMID 12398577.[permanent dead link]
- Jump up^ Olsen AK, Sjøgren P (October 1999). “Oral glycopyrrolate alleviates drooling in a patient with tongue cancer”. J Pain Symptom Manage. 18 (4): 300–2. doi:10.1016/S0885-3924(99)00080-9. PMID 10534970.
- Jump up^ Maria, Sammartano Azia; Claudia, Cassandro; Pamela, Giordano; Andrea, Canale; Roberto, Albera (1 December 2012). “Medical therapy in Ménière’s disease”. Audiological Medicine. 10 (4): 171–177. doi:10.3109/1651386X.2012.718413 – via Taylor and Francis+NEJM.
- Jump up^ Kim WO, Kil HK, Yoon DM, Cho MJ (August 2003). “Treatment of compensatory gustatory hyperhidrosis with topical glycopyrrolate”. Yonsei Med. J. 44 (4): 579–82. doi:10.3349/ymj.2003.44.4.579. PMID 12950111.
- Jump up^ Kim WO, Kil HK, Yoon KB, Yoon DM (May 2008). “Topical glycopyrrolate for patients with facial hyperhidrosis”. Br. J. Dermatol. 158 (5): 1094–7. doi:10.1111/j.1365-2133.2008.08476.x. PMID 18294315.
- Jump up^ “EPAR – Product information for Seebri Breezhaler” (PDF). European Medicines Agency. 28 September 2012.
- Jump up^ Tzelepis G, Komanapolli S, Tyler D, Vega D, Fulambarker A (January 1996). “Comparison of nebulized glycopyrrolate and metaproterenol in chronic obstructive pulmonary disease”. Eur. Respir. J. 9 (1): 100–3. doi:10.1183/09031936.96.09010100. PMID 8834341.
- Jump up^ Haddad EB, Patel H, Keeling JE, Yacoub MH, Barnes PJ, Belvisi MG (May 1999). “Pharmacological characterization of the muscarinic receptor antagonist, glycopyrrolate, in human and guinea-pig airways”. Br. J. Pharmacol. 127 (2): 413–20. doi:10.1038/sj.bjp.0702573. PMC 1566042
 . PMID 10385241.
. PMID 10385241. - Jump up^ Rumpler, M.J.; Colahan, P.; Sams, R.A. (2014). “The pharmacokinetics of glycopyrrolate in Standardbred horses”. J. Vet Pharmacol Ther. 37 (3): 260–8. doi:10.1111/jvp.12085. PMID 24325462.
- Jump up^ Hansel TT, Neighbour H, Erin EM, et al. (October 2005). “Glycopyrrolate causes prolonged bronchoprotection and bronchodilatation in patients with asthma”. Chest. 128 (4): 1974–9. doi:10.1378/chest.128.4.1974. PMID 16236844.
- Jump up^ Gilman MJ, Meyer L, Carter J, Slovis C (November 1990). “Comparison of aerosolized glycopyrrolate and metaproterenol in acute asthma”. Chest. 98 (5): 1095–8. doi:10.1378/chest.98.5.1095. PMID 2225951.
 |
|
| Clinical data | |
|---|---|
| Trade names | Robinul, Cuvposa, Seebri, Qbrexza, others |
| License data | |
| Pregnancy category |
|
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| ECHA InfoCard | 100.008.990 |
| Chemical and physical data | |
| Formula | C19H28BrNO3 |
| Molar mass | 398.335 g/mol |
| 3D model (JSmol) | |
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602014 |
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous, inhalation |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 0.6–1.2 hours |
| Excretion | 85% renal, unknown amount in the bile |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.008.990 |
| Chemical and physical data | |
| Formula | C19H28NO3+ |
| Molar mass | 318.431 g/mol |
| 3D model (JSmol) | |
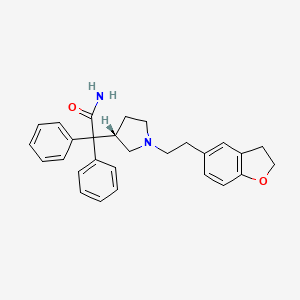
Darifenacin Hydrobromide, 臭化水素酸ダリフェナシン
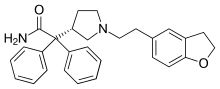
Darifenacin
2-[(3S)-1-[2-(2,3-dihydro-1-benzofuran-5-yl)ethyl]pyrrolidin-3-yl]-2,2-diphenylacetamide
Darifenacin; Emselex; Enablex; CAS 133099-04-4; UNII-APG9819VLM;
US 2004-12-22 APPROVED
EU 2004-10-22 APPROVED
| Molecular Formula: | C28H30N2O2 |
|---|---|
| Molecular Weight: | 426.56 g/mol |


Derivative Type: Hydrobromidd
CAS Registry Number: 133099-07-5
Research Code:UK-88525-04
Trade Name:Emselex® / Enablex® / Xelena®
MOA:M3 muscarinic acetylcholine receptor antagonistIndication:Overactive bladder (OAB)
Status:Approved
Company:Novartis (Originator) , Merus Labs,Warner chilcottSales:
ATC Code:G04BD10
臭化水素酸ダリフェナシン
Darifenacin Hydrobromide

C28H30N2O2 HBr : 507.46
HBr : 507.46
[133099-07-7]
Darifenacin (originally developed by Pfizer, trade name En`blex in USA and Canada, Emselex in Europe) is an effective medibatinn used for treatment of overactive bladder (OAB) symptoms.
OAB is a common condition symptomized by urinary urgency, with or without urge in continence, usually with frequency and nocturia that notably affects the lives of millions of people. Human bladder tissue contains M2 (80%) and M3 (20%) muscarinic receptors, and the latter act as the primary mediator of detrusor contraction in response to cholinergic activation.
So muscarinic receptor antagonists are the current treatment of choice for OAB. As different subtypes of muscarinic receptors are widely distributed in the human body to play key physiological roles, a very selective M3 receptor antagonist is in high demand in the market for OAB medication. Darifenacin is a potent and competitive M3 selective receptor antagonist (M3SRA) that has been shown to have high affinity and selectivity (59-fold higher) for the M3 receptor, with low selectivity for the other muscarinic receptor subtypes. Its hydrobromide salt is the active ingredient of pharmaceutical formulations. The efficacy, tolerability and safety of darifenacin in the treatment of OAB are well established.
Darifenacin (trade name Enablex in US and Canada, Emselex in Europe) is a medication used to treat urinary incontinence. It was discovered by scientists at the Pfizer research site in Sandwich, UK under the identifier UK-88,525 and used to be marketed by Novartis. In 2010 the US rights were sold to Warner Chilcott for 400 million US$.
Mechanism of action
Darifenacin works by blocking the M3 muscarinic acetylcholine receptor, which is primarily responsible for bladder muscle contractions. It thereby decreases the urgency to urinate. It is not known whether this selectivity for the M3 receptor translates into any clinical advantage when treating symptoms of overactive bladder syndrome.
It should not be used in people with urinary retention. Anticholinergic agents, such as darifenacin, may also produce constipation and blurred vision. Heat prostration (due to decreased sweating) can occur when anticholinergics such as darifenacin are used in a hot environment.[1]
Clinical uses
Darifenacin is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency and frequency in adults.
clip
http://nopr.niscair.res.in/bitstream/123456789/18844/1/IJCb%2052B(6)%20824-828.pdf





The substance was first described in EP 388 054. The method of its preparation in accordance with this document is shown in the following scheme.
Scheme 1

DARIFENACIN

wherein the substituents R and X can be

A particular preferable embodiment is shown in Scheme 2, wherein substance VII is alkylated with 5-(2-bromoethyl)-2,3-dihydrobenzofuran (VIII) in the presence of potash by reflux in acetonitrile. Crude darifenacin (IX) is purified using column chromatography and crystallized from diisopropylether
Scheme 2

Scheme 2: Synthesis of darifenacin by N-alkylation of pyrrolidine VII with 5-(2-bromoethyi)-
2,3-dihydrobenzofuran (VIII)
Darifenacin hydrobromide is prepared by precipitation of purified darifenacin base dissolved in acetone by addition of concentrated aqueous HBr.
However, in repeated reproduction these procedures did not provide a product of an adequate quality in a reasonable industrially applicable yield. It has been found out that a portion of the resulting darifenacin undergoes subsequent alkylation to the second stage, producing the twice substituted substance X. In the course of the reaction undesired reactions of 5-(2-biOmoethyl)-2,3-dihydrobenzofuran VIII also occur, namely hydrolysis producing a hydroxy derivative (XI) and elimination producing a vinyl derivative (XII). All these reactions reduce the yield of the desired substance and complicate the preparation of high-quality API.
By reproduction of the above mentioned procedure a substance was obtained with the following contents of constituents in accordance with HPLC [%] : VII 2.8 VIII 14.2 1X 57.2 X 7.8 XI 1.2 XII 8.0.

A purification procedure for darifenacin was published in WO03080599A1.
Darifenacin in t-amyl alcohol is heated with Amberlite (22 h), the solid fraction is filtered off, the solvent is evaporated from the filtrate and the residue is dissolved in toluene; a solvate of darifenacin with toluene is separated by cooling. This solvate can be directly used for the preparation of darifenacin hydrobromide (the solvate is dissolved in 2-butanol, concentrated HBr is added and the darifenacin salt is separated by cooling).
Another method of purification of darifenacin, described in the same document, is conversion of the darifenacin/toluene solvate to darifenacin hydrate (the solvate is dissolved in acetonitrile and water is added under gradual separation of darifenacin hydrate (Scheme 3)), which can be used for the preparation of salts or can be directly incorporated into pharmaceutical forms. The hydrate can be optionally converted to the hydrogen bromide in a similar way as the solvate.

(IX.W)
Scheme 3: Methods of purification of crude darifenacin and its conversion to hydrobromide
During reproduction of the purification procedure it was possible to separate a portion of substance X in the solid phase form after dissolution of crude darifenacin in toluene. However, the attempt to obtain the desired toluene solvate of darifenacin from the toluene solution was not successful during the reproduction. This means that this method does not lead to the pure substance.
WO2007076159 (TEVA) describes preparation of darifenacin from dihydrobenzofuran ethylchloride and carbamoyl(diphenylmethyl)pyrrolidine tartrate in the aqueous phase using K2CO3 as the base. After cooling of the reaction mixture n-butanol is added, the aqueous and organic phases are separated, acetanhydride is added and a reaction with concentrated hydrobromic acid (48%) is performed.
This method enables preparation of the substance with a satisfactory yield, ca. 77%; however, the reaction in the aqueous phase takes place in the melt, which is very thick, which causes techno logical problems, e.g. difficult stirring, sticking of the mixture on the walls of the reaction vessel, etc. During a reproduction of this procedure it was found that acetanhydride caused partial decomposition of the product and formation of further impurities. The crude product prepared this way cannot be converted to hydrobromide without further purification. N-butanol mentioned in the procedure is partly miscible with water, which also has a negative impact on the process yield. Contents of constituents (HPLC [%]) in the crude product within the reproduction of the procedure in accordance with WO2007076159 (TEVA):
Reaction with dihydrobenzofuran ethylchloride: VII 1.9 VIII 6.1 1X 82.0 X 6.3 XI not found XII not found
Reaction with dihydrobenzofuran ethylbromide: VII 2.8 VIII 0.5 1X 77.5 X 9.5 XI 2.0 XII 2.4
The above mentioned analysis of the described procedures and attempts to reproduce them have revealed that compound X is the major problem. During the application it was never possible to obtain the product that would contain less than 5% of this impurity. The substance is similar to the desired product in its character, it has similar solubility in most solvents and moreover it also changes to hydrogen bromide or other salts. For this reason it is very difficult to separate this substance by normal crystallization of the base or one of the salts of darifenacin.
While toluene has proved suitable for this function in the above-described procedures (WO03080599A1), after the separation of a portion of substance X it was not possible to obtain the desired toluene solvate of darifenacin. The procedure appears to be hardly usable without further modification and it does not lead to the desired pure product.
Darifenacin (la) is chemically known as (S)-2-[l-[2-(2,3-Dihydrobenzofuran-5- yl)ethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide and is approved as hydrobromide salt. Darifenacin is a potent muscarinic M3 receptor antagonist. Muscarinic receptors play an important role in several major cholinergically mediated functions, including contractions of the urinary bladder, gastrointestinal smooth muscle, saliva production, and iris sphincter function. Darifenacin has greater affinity for the M receptor than for the other known muscarinic receptors. Darifenacin hydrobromide is commercially available under the brand name Enablex® in the US. It has been approved for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency and frequency.
US 5,096,890 disclosed Darifenacin and its pharmaceutically acceptable salts. US ‘890 discloses several processes for preparing Darifenacin. According to the process disclosed in US ‘890, Darifenacin (la) may be prepared by condensing 5-(2-bromoethyl)-2,3-dihydrobenzofuran (II) with 3-(S)-(-)-(l – carbamoyl-l , l -diphenylmethyl)pyrrolidine (III) in the presence of K2C03 in acetonitrile.
The process is as shown in Scheme-I below:
1. Anhydrous K2C03

US ‘890 also discloses a variant process for the preparation of Darifenacin (la) by condensing 5-(2-bromoethyl)-2,3-benzofuran (IV) with 3-(S)-(-)-(l -carbamoyl- 1 , 1- diphenylmethyl)pyrrolidine (III) in the presence of K2C03 in acetonitrile to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)ethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide (V), which is further hydrogenated in the presence of Pd/C in acetic acid to produce Darifenacin crude, followed by purification using column chromatography.
The rocess is as shown in Scheme-II below:

Darifenacin
(la)
US ‘890 also discloses an another variant process for the preparation of Darifenacin hydrobromide (I) by condensing 5-chloroacetyl-2,3-dihydrobenzofuran (VI) with 3- (S)-(-)-(l-carbamoyl-l ,l-diphenylmethyl)pyrrolidine (III) in the presence of K2CO3 in an industrial methylated spirit to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)-2- oxoethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide hydrochloride (VII), which is further hydrogenated in the presence of Pd/C in acetic acid to produce Darifenacin crude, followed by purification using column chromatography to produce pure Darifenacin (la), which is converted to Darifenacin hydrobromide (I) using aqueous hydrobromic acid in acetone.
The rocess is as shown in Scheme-Ill below:

The disadvantage with the above processes is the use of column chromatography in the purification of Darifenacin (la). Employing column chromatography technique is tedious and laborious and also involves use of large quantities of solvents, and hence is not suitable for industrial scale operations.
US 6,930,188 discloses a process for the preparation of Darifenacin hydrobromide (I), by condensing 2-(2,3-dihydrobenzofuran-5-yl)acetic acid (VIII) with (S)-2,2- diphenyl-2-(3-pyrrolidinyl)acetonitrile hydrobromide (IX) in the presence of carbonyldiimidazole in ethyl acetate to produce (S)-3-(cyanodiphenylmethyl)-l-[2- (2,3-dihydrobenzofuran-5-yl)acetyl]pyri lidine (X), which is further reduced in the presence of sodium borohydride and boron trifluoride tetrahydrofuran complex to produce (S)-2-{l-[2-(2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl}-2,2- diphenyl acetonitrile (XI), followed by treating with HBr to produce (S)-2-{ l-[2- (2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl}-2,2-diphenyl acetonitrile hydrobromide (XIa). Compound (XIa) is treated with potassium hydroxide at 50 to 60°C to produce Darifenacin (la), followed by treating with ion-exchange resin to produce Darifenacin toluene solvate (lb), which is further converted to Darifenacin hydrobromide (I) using 48% hydrobromic acid in 2-butanone.
The rocess is as shown in Scheme-IV below:

Darifenacin HBr
(I) It has now been found that, during the condensation of 5-(2-bromoethyl)-2,3- benzofuran (IV) with 3-(S)-(-)-(l -carbamoyl- l ,l-diphenylmethyl)pyrrolidine (III) to produce (S)-2-[l-[2-(2,3-benzofuran-5-yl)ethyl]-3-pyrroIidinyl]-2,2- diphenylacetamide (V), 3-(S)-(-)-(l -carbamoyl- l , l-diphenylmethyl)pyrrolidine (III) remained unreacted to about 8 to 10% in the reaction mass. It is difficult to separate the compound (III) through crystallization from Darifenacin hydrobromide (I), which typically require two to three crystallizations to achieve desired Darifenacin hydrobromide (I) purity. The second and third crystallization adds time to the manufacturing process and thus negatively impacts product throughput. Additionally, a second and third crystallization reduces yield as some Darifenacin hydrobromide (I) remains uncrystallized and is not recovered from the liquid phase.
Hence, there is a need to develop a purification process, which removes the unreacted intermediate compound 3-(S)-(-)-(l-carbamoyl-l ,l – diphenylmethyl)pyrrolidine (III) from the reaction mass, which in turn provides Darifenacin hydrobromide of high purity with improved yield.
Further, it has been found that Darifenacin produced by the condensation of 5-(2- bromoethyl)-2,3-dihydrobenzofuran (II) with 3-(S)-(-)-( 1 -carbamoyl- 1 ,1 – diphenylmethyOp rrolidine (III) contains dimmer impurity (XII).
Formula (XII)

Hence, there is a need to develop process, which reduces the unwanted Darifenacin dimer (XII), which is influenced by controlling the quantity of compound (XIII).

Formula (XIII)
PROCESS
(a) Dunn, P. J.; Matthews, J. G.; Newbury, T. J.; O’Connor, G.US 6,930,188 B2, 2005.
(b) Narayan, K; Reddy, J. M.; Rao, G.; Chary, S.; Islam, A.; SivakumaranWO 2011/D70419 A1, 2011.
(c) Evansa, P.; Thomas, J.; Davies, R. H.US 2003/0199494 A1, 2003.
(d) Bhanu, M. N.; Naik, S.; Bodkhe, A.; Soni, A.US 2011/0144354 A1, 2011.
(e) Merli, V.; Canavesi, A.; Baverio, P.US 7,442,806 B2, 2008.
(f) Merli, V.; Canavesi, A.; Baverio, P.US 2009/0156831 A1, 2009.
(G) WO2009125426A2.

PATENT
https://www.google.com/patents/WO2011070419A1?cl=en

EXAMPLE – 1
Stage-1:
PREPARATION OF 5-(2-TOSYLOXYETHYL)-2,3-
DIHYDROBENZOFURAN
2-(2,3-Dihydrobenzofuran-5yl)ethanol (65 g, 0.39 mol) was dissolved in dichloromethane (650 ml) at 20-25°C under nitrogen atmosphere. The solution was cooled to 0-5°C and p-toluenesulfonyl chloride (79.27 g, 0.41 mol) was added in one lot. Triethylamine (60.04 g, 0.59 mol) was added slowly at 0-10°C, stirred for ~ 15 h at 20-25°C and the reaction was monitored by HPLC. Water was added and stirred for 10 min at 20-25°C. Layers were separated and the aqueous layer was extracted with dichloromethane (130 ml). The organic layer was combined and washed with water (2 x 130 ml) at 20-25°C at pH 12 – 12.5. Finally the organic layer was washed with saturated brine solution (130ml) and concentrated to complete dryness under reduced pressure at 35-45°C. The product was crystallized from ethyl acetate and n- hexanes mixture.
Yield: 96.5 g
Chromatographic purity (By HPLC): 97.85%
Stage-2:
PREPARATION OF DARIFENACIN HYDROBROMIDE
3-(S)-(-)-(l -Carbamoyl- l , l -diphenylmethyl)pyrrolidine L-(+)-tartrate (10 g, 0.02 mol), anhydrous potassium carbonate (22.50 g, 0.16 mol) and 5-(2-tosyloxyethyl)- 2,3-dihydrobenzofuran (7 g, 0.02 mol) were suspended in anhydrous acetonitrile ( 100 ml) under nitrogen atmosphere at 25 ± 2°C. The reaction suspension was heated to 70 ± 2 °C and stirred for 4 h. Reaction progress was monitored by HPLC. The reaction mass was cooled to 30 + 2°C, the salts were filtered and washed with acetonitrile (10 ml). The filtrate was concentrated under reduced pressure at 50 ± 2 °C. The residue was dissolved in dichloromethane (50 ml), water (50 ml) was added and the pH was adjusted to 2 ± 0.1 with 24% w/w aqueous hydrobromic acid at 25- 30°C. The layers were separated and the aqueous layer was extracted with aqueous dichloromethane (20 ml). Water (50 ml) was added to the combined dichloromethane layer and pH was adjusted to 9 ± 0.1 with 25% w/w aqueous potassium carbonate solution at 25 ± 2°C. The layers were separated and concentrated under reduced pressure at 35-40°C. The residue was dissolved in acetone (50 ml), cooled to 5-10°C and the pH was adjusted to acidic with 48% w/w aqueous hydrobromic acid at 5-10°C. The residue was stirred for 2 h at 20-25°C, cooled to 0-5°C and stirred for 1 h at 0-5°C. The product was filtered, washed with chilled acetone (10 ml) and dried at 50-55°C.
Yield: 9.4 g
Chromatographic purity (By HPLC): 98.2%.
5 -(2-Tosy loxyethy l)-2, 3 -dihydrobenzofuran : Nil
Darifenacin dimer impurity: 0.96%.
EXAMPLE – 2
Stage-1 :
PREPARATION OF 5-(2-BROMOETHYL)-2,3-DIHYDROBENZOFURAN
2- (2,3-Dihydrobenzofuran-5-yl)ethanol (10 g, 0.06 mol) was dissolved in acetonitrile (60 ml) at 25 ± 2°C under nitrogen atmosphere and triphenylphosphine dibromide (27.02 g, 0.06 mol) was added in one lot at 25 ± 2°C. The reaction mass was heated to 76-78°C and stirred for 2 h. Reaction progress was monitored by TLC [Ethyl acetate: n-Hexanes; 2:8 v/v], Acetonitrile was completely distilled off under reduced pressure at 76-78°C. The residue was cooled and the product was extracted with n-hexanes (4 x 30 ml) at 25 ± 2°C. The solution was filtered and diluted with ethyl acetate (50 ml) and washed with 5% w/w aqueous sodium bicarbonate solution (2 x 50 ml) at 25 ± 2°C. The organic layer was concentrated under reduced pressure at 40-50°C.
Yield: 7 g
Stage-2
PREPARATION OF DARIFENACIN HYDROBROMIDE
3- (S)-(-)-(l -Carbamoyl- l ,l-diphenylmethyl)pyrrolidine L-(+)-tartrate (5 g, 0.01 mol), anhydrous potassium carbonate (1 1.25 g, 0.08 mol) and 5-(2-bromoethyl)-2,3- dihydrobenzofuran (2.5 g, 0.01 mol) were suspended in anhydrous acetonitrile (50 ml) under nitrogen atmosphere at 25 ± 2°C. The reaction suspension was heated to 70 ± 2 °C and stirred for 4 h. Reaction progress was monitored by HPLC. The reaction mass was cooled to 30 ± 2°C, salts were filtered and washed with acetonitrile (5 ml). The filtrate was concentrated under reduced pressure at 50 ± 2 0 C. The residue was dissolved in dichloromethane (25 ml), water (25 ml) was added and the pH was adjusted to 2 ± 0.1 with 24% w/w aqueous hydrobromic acid at 25- 30°C. The layers were separated and the aqueous layer was extracted with dichloromethane (10 ml). Water (25 ml) was added to the combined dichloromethane layer and pH was adjusted to 9 ± 0.1 with 25% w/w aqueous potassium carbonate solution at 25 ± 2°C. The layers were separated and the organic layer was concentrated under reduced pressure at 35-40°C. The residue was dissolved in acetone (25 ml), cooled to 5-10°C and the pH was adjusted to acidic with 48% w/w aqueous hydrobromic acid at 5-10°C. The residue was stirred for 2 h at 20-25°C, cooled to Q-5°C and stirred for 1 h at 0-5°C. The product was filtered, washed with chilled acetone (5 ml) and dried at 50-55°C.
Yield: 4.5 g
Chromatographic purity (By HPLC): 99.24%
5-(2-bromoethyl)-2,3-dihydiObenzofuran: Nil
Darifenacin dimer impurity: 0.39%.
EXAMPLE – 3
PURIFICATION OF DARIFENACIN HYDROBROMIDE Darifenacin hydrobromide (10 g) was suspended in acetic acid (15 g) at 25 ± 2°C and heated to 65-70°C. Activated carbon (0.25 g) was added and stin-ed for 15 min at 65-70°C. Carbon was filtered off through hyflo and washed with hot acetic acid (5 g). Water (200 ml) was added to the filtrate slowly at 50-55°C, cooled to 45°C and Darifenacin hydrobromide seed (0.05 g) was added. The resulting solution was cooled to 20-25 °C and stin-ed for 1 h and further cooled to 0-5 °C and stirred for 1 h. The solid was filtered and washed with cold water (10 ml). The product was dried at 50-55°C.
Yield: 7.6 g
Chromatographic purity (By HPLC): 99.52%
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil 5-(2-Tosyloxyethyl)-253-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.20%.
EXAMPLE – 4
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (15 g) was suspended in a mixture of acetic acid (25 g) and water (25 ml) at 25 ± 2°C and heated to 65-70°C. Activated carbon (0.75 g) was added and stirred for 15 min at 65-70°C. Carbon was filtered off through hyflo and washed with a mixture of acetic acid and DM water (10 g). Water (120 ml) was added to the filtrate slowly at 50-55°C, cooled to 45°C and Darifenacin hydrobromide seed (0.05 g) was added. The resulting solution was cooled to 20- 25°C and stirred for 1 h and further cooled to 0-5°C and stirred for 1 h. The solid was filtered and washed with cold water (30 ml). The product was dried at 50-55°C. Yield: 1 1.9 g
Chromatographic purity (By HPLC): 99.71 %
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil
5-(2-Tosyloxyethyl)-2,3-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.20%. EXAMPLE – 5
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (9 g) was suspended in acetone (45 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min at 55-60°C. The resulting solution was cooled to 20-25°C and stin-ed for 30 + 5 min, which is further cooled to 0-5°C and stirred for 1 h. The solid was filtered and washed with chilled acetone (9 ml). The product was dried at 50-55°C.
Yield: 8.8 g
Chromatographic purity (By HPLC): 99.87%
5-(2-bromoethyl)-2,3-dihydrobenzofuran: Nil
5-(2-Tosyloxyethyl)-2,3-dihydrobenzofuran: Nil
Darifenacin dimer impurity: 0.08%. EXAMPLE – 6
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (9 g) was suspended in a mixture of acetone (45 ml) and DM water (1.77 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min at 55- 60°C. The resulting solution was cooled to 20-25°C and stirred for 30 + 5 min, which was further cooled to 0-5°C and stirred for 1 h. The product was filtered and washed with chilled acetone (9 ml). The product was dried at 50-55°C.
Yield: 8.4 g
Chromatographic purity (By HPLC): 99.88%
EXAMPLE – 7
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (10 g) was suspended in a mixture of acetone (50 ml) and DM water (3.95 ml) at 25 ± 2°C, heated to 55-58°C and stirred for 30 ± 5 min. The resulting solution was cooled to 20-25°C and stirred for 30 ± 5 min, which was further cooled to 0-5 °C and stirred for 1 hour. The product was filtered and washed with chilled acetone (10ml, 0-5°C). The product was dried at 50-55°C.
Yield: 8.30g
Chromatographic Purity (By HPLC): 99.83 %
Darifenacin dimmer: 0.10%
EXAMPLE – 8
PURIFICATION OF DARIFENACIN HYDROBROMIDE
Darifenacin hydrobromide (10 g) was suspended in a mixture of acetone (50 ml) and DM water (7.9 ml) at 25 ± 2°C, heated to 55-60°C and stirred for 30 + 5 min. The resulting solution was cooled to 20-25°C and stirred for 30 ± 5 min, which was further cooled to 0-5°C and stirred for 1 hour. The product was filtered and washed with chilled acetone (10 ml, 0-5°C). The product was dried at 50-55°C.
Yield: 6.70g
Chromatographic Purity (By HPLC): 99.94 %
Darifenacin dimmer: Nil.
Paper
A New Solvent System (Cyclopentyl Methyl Ether–Water) in Process Development of Darifenacin HBr

Darifenacin is a potent and competitive M3 selective receptor antagonist (M3SRA), and its hydrobromide salt (1) is the active ingredient of pharmaceutical formulations for oral treatment of urinary incontinence. The present work demonstrates an efficient, commercial manufacturing process for darifenacin hydrobromide (1).
1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.8 (bs, 0.7H), 9.3 (bs, 0.3 H), 7.4–7.3 (m, 10 H), 7.1–7.0 (m, 1H), 7.0–6.7 (m, 2H), 6.7 (m, 1H), 4.5 (m, 2H), 4.0–3.9 (m, 1.3 H), 3.8–3.7 (m, 0.7 H), 3.4–3.3 (m, 2H), 3.1 (m, 2H), 2.9 (m, 1.3 H), 2.8–2.7 (m, 2H), 2.6 (m, 0.7H), 2.4–2.3 (m, 0.7H), 2.2 (m, 1.3H), 1.6 (m, 0.7 H), 1.5 (m, 0.3 H).
13C NMR (DMSO-d6, 100 MHz, δ ppm): 174.4, 174.2, 158.5, 141.2, 140.7, 140.6, 129.7, 129.4, 129.5, 128.3, 128.0, 127.9, 127.5, 127.2, 127.1, 125.4, 125.2, 108.7, 70.8, 62.4, 62.1, 56.1, 55.2, 55.1, 54.7, 53.0, 52.2, 40.0, 40.8, 30.3, 30.1, 29.0, 26.9, 25.6.
Calcd for C28H30N2O2·HBr, (M+)/z: 425.56; found (M + H)/z 427.2, (M + Na)/z 449.3.
Anal. Calcd for C28H31BrN2O2: C, 66.27; H, 6.16; N, 5.52. Found: C, 66.36; H, 6.07; N, 5.68.


PATENT
https://www.google.com/patents/WO2009094957A1?cl=en
Scheme 4:

Example 1

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium phosphate (9.43 g; 0.041 mol in 20 ml of water) at the laboratory temperature. A toluene solution (20 ml) of intermediate VIII (2.41 g; 0.011 mol) is added to the mixture and the mixture is heated up in an oil bath T=90 0C while being stirred for 3.5 h. After cooling the toluene layer is separated and the aqueous layer is extracted with toluene. The combined toluene extracts are shaken with water and the solvent is distilled off at a reduced pressure. The evaporation residue is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacin hydrobromide is filtered off and dried.
Yield: 85% of theory.
Example 2

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium carbonate (6.1 g; 0.044 mol in 20 ml of water) at the laboratory temperature. A toluene solution (20 ml) of intermediate VIII (2.41 g; 0.011 mol) is added to the mixture and the mixture is heated in an oil bath T=90 °C while being stirred for 3.5 h. After cooling the toluene layer is separated and the aqueous layer is extracted with toluene. The combined toluene extracts are shaken with water and the solvent is distilled off at a reduced pressure. The evaporation residue is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacine hydrobromide is filtered off and dried.
Yield: 86% of theory.
Example 3

Advanced intermediate VII (4.3 g; 0.01 mol) is stirred up in an aqueous solution of potassium phosphate (9.43 g; 0.041 mol in 20 ml of water) at the laboratory temperature. A solution of intermediate VIII (2.41 g; 0.011 mol) in cyclohexane (20 ml) is added to the mixture and the mixture is heated in an oil bath T=90 0C while being stirred for 3.5 h. The layers are separated while hot. The cyclohexane solution is cooled to the laboratory temperature under intensive stirring. This way the darifenacin base is separated. The product is filtered off and dried. The base is dissolved in ethylmethylketone, and an equimolar amount of 48% hydrobromic acid is added. The separated darifenacin hydrobromide is filtered off and dried.
Yield: 85% of theory.
clip


References
External links
- Enablex product website, run by Warner Chilcott
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2011070419A1 * | Dec 3, 2010 | Jun 16, 2011 | Aurobindo Pharma Limited | An improved process for the preparation of darifenacin hydrobromide |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003080599A1 | Mar 17, 2003 | Oct 2, 2003 | Novartis International Pharmaceutical Ltd. | Stable hydrate of a muscarinic receptor antagonist |
| WO2007076157A2 * | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceuticals Industries Ltd. | Processes for preparing darifenacin hydrobromide |
| WO2007076158A2 * | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceutical Industries Ltd. | Processes for preparing darifenacin hydrobromide |
| WO2007076159A2 | Dec 27, 2006 | Jul 5, 2007 | Teva Pharmaceutical Industries Ltd. | Pure darifenacin hydrobromide substantially free of oxidized darifenacin and salts thereof and processes for the preparation thereof |
| EP0388054A1 | Mar 2, 1990 | Sep 19, 1990 | Pfizer Limited | Pyrrolidine derivatives |
| WO2009094957A1 * | Jan 14, 2009 | Aug 6, 2009 | Zentiva, K.S. | A method for the preparation of darifenacin hydrogen bromide |
| US5096890 | Mar 13, 1990 | Mar 17, 1992 | Pfizer Inc. | Pyrrolidine derivatives |
| US6930188 | Mar 25, 2003 | Aug 16, 2005 | Novartis International Pharmaceutical, Ltd. | Stable hydrate of a muscarinic receptor antagonist |
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Enablex |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605039 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | G04BD10 (WHO) |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 15 to 19% (dose-dependent) |
| Protein binding | 98% |
| Metabolism | Hepatic (CYP2D6– and CYP3A4-mediated) |
| Biological half-life | 13 to 19 hours |
| Excretion | Renal (60%) and biliary (40%) |
| Identifiers | |
| CAS Number | 133099-04-4 |
| PubChem (CID) | 444031 |
| IUPHAR/BPS | 321 |
| DrugBank | DB00496 |
| ChemSpider | 392054 |
| UNII | APG9819VLM |
| KEGG | D01699 |
| ChEBI | CHEBI:391960 |
| ChEMBL | CHEMBL1346 |
| ECHA InfoCard | 100.118.382 |
| Chemical and physical data | |
| Formula | C28H30N2O2 |
| Molar mass | 426.55 g/mol |
| 3D model (Jmol) | Interactive image |
/////////Darifenacin, 臭化水素酸ダリフェナシン , Antispasmodic, Antimuscarinic, UK-88525-04, Emselex® , Enablex® , Xelena®,
C1CN(CC1C(C2=CC=CC=C2)(C3=CC=CC=C3)C(=O)N)CCC4=CC5=C(C=C4)OCC5

{kind=link}
{kind=link}