
Home » Posts tagged 'Antiinflammatory'
Tag Archives: Antiinflammatory
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fosrugocrixan
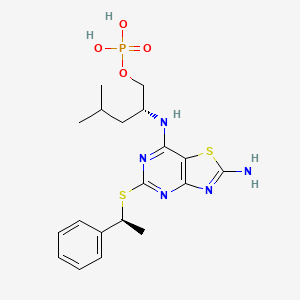
Fosrugocrixan
CAS 2408145-38-8
MF C19H26N5O4PS2, MW483.5 g/mol
[(2R)-2-[[2-amino-5-[(1S)-1-phenylethyl]sulfanyl-[1,3]thiazolo[4,5-d]pyrimidin-7-yl]amino]-4-methylpentyl] dihydrogen phosphate
- (2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
- 1-Pentanol, 2-[[2-amino-5-[[(1S)-1-phenylethyl]thio]thiazolo[4,5-d]pyrimidin-7-yl]amino]-4-methyl-, 1-(dihydrogen phosphate), (2R)-
(2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
CX3C chemokine receptor 1 (CX3CR1) antagonist, antiinflammatory, 4ZXD25SC4S, KAND-145, KAND 145
- OriginatorKancera
- DeveloperNovakand Pharma
- ClassAnti-inflammatories; Antineoplastics; Small molecules
- Mechanism of ActionChemokine CXCL13 inhibitors
- Phase IOvarian cancer
- PreclinicalChronic lymphocytic leukaemia
- No development reportedInflammation
- 22 Sep 2025Kancera is now called Novakand Pharma
- 28 Apr 2025No recent reports of development identified for preclinical development in Ovarian-cancer in Sweden (IV)
- 03 May 2024Efficacy and adverse event data from a phase I trials in healthy volunteers released by Kancera
Fosrugocrixan (also known by its developmental code KAND145) is a novel, small-molecule drug candidate acting as a selective antagonist for CX3C chemokine receptor 1 (CX3CR1), commonly known as the fractalkine receptor.
Key Characteristics and Mechanism
- Drug Class: It represents a first-in-class small molecule immune modulator.
- Phosphate Prodrug: Fosrugocrixan is designed as a soluble phosphate prodrug. Once inside the body (in vivo), it converts into its active drug form, rugocrixan (formerly KAND567).
- Mechanism of Action: By blocking the CX3CR1 fractalkine pathway, it controls and prevents the trafficking of disease-promoting immune cells. This blockage provides potent anti-inflammatory activity.
Clinical Development and Targets
The drug is being actively developed by Novakand Pharma (a company formerly known as Kancera). Its primary therapeutic targets span several conditions driven by runaway inflammation and immune responses:
- Cardiovascular Diseases: Specifically targeted to manage conditions where hyper-inflammation damages tissue (such as post-myocardial infarction or heart conditions).
- Autoimmune & Inflammatory Diseases: Evaluated for broad anti-inflammatory potential.
- Oncology: Investigated for its ability to regulate the tumor microenvironment.
SYN
WO 2020008064
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020008064&_cid=P21-MPQB4Y-95682-1
SYN
Karlström et al. J. Med. Chem., 2013, 56, 3177-3190
https://pubs.acs.org/doi/10.1021/jm3012273
PAT
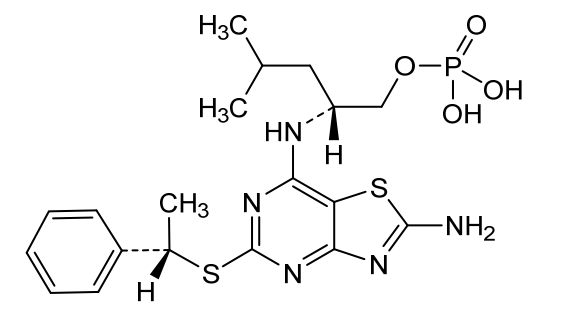
(2R)-2-[(2-Amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate (B), are known to act as antagonists of the fractalkine receptor (CX3CR1) (Karlström et al. J. Med. Chem., 2013, 56, 3177-3190; WO 2020/008064)
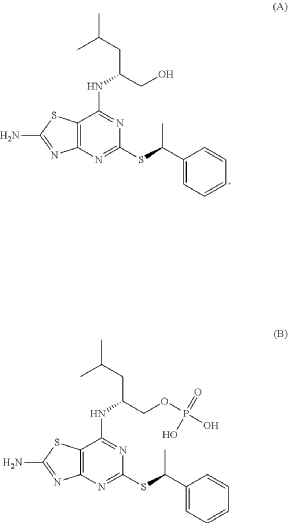
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US336567291&_cid=P21-MPQAYT-90868-1
Example 1
Preparation of (2R)-2-[(2-Amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
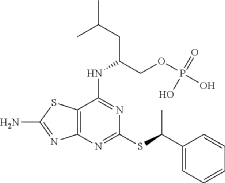
Phosphorus oxychloride (337 mg, 2.2 mmol) was dissolved in THE (0.75 mL) and water (25 mg, 1.4 mmol) was added. The mixture was cooled in an ice-bath and pyridine (111 mg, 113 μL, 1.4 mmol) was added followed by (2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}-[1,3] thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentan-1-ol hydrochloride (110 mg, 0.25 mmol) (Karlstr6m S., et al., J. Med. Chem., 2013, 56, 3177-3190; WO 2006/107258). The reaction mixture was stirred at ice-bath temperature for 1 h. To a mixture of phosphorus oxychloride (337 mg, 2.2 mmol) and water (25 mg, 1.4 mmol) in THE was added, at ice-bath temperature pyridine (111 mg, 113 μL, 1.4 mmol). Half of this mixture was added to the reaction mixture described above. The reaction mixture was stirred at ice-bath temperature for another 1 h. Water (3 mL) was added and the reaction mixture was stirred for 15 min at ice-bath temperature and 20 min at room temperature. DCM (3 mL) was added and the phases were separated. The aqueous phase was extracted with another portion of DCM (3 mL) and the organic phases were combined. At this point the product started to precipitate as a pale-yellow gum in the organic phase. MeOH was added and the now homogeneous solution was transferred to a round-bottomed flask and was evaporated to yield 120 mg of crude product, which according to HPLC was ca. 93% pure. The crude material was dissolved in a MeOH/water mixture and the pH was adjusted to about 6-7 with 1 M NaOH. The material was purified by preparative HPLC (basic method). The pure fractions were pooled, evaporated, and dried in vacuum. The product was assumed to be the diammonium salt after purification. 1H NMR (600 MHz, CD 3OD) δ H ppm 7.43-7.47 (m, 2H) 7.30-7.35 (m, 2H) 7.20-7.24 (m, 1H) 5.08 (q, J=7.03 Hz, 1H) 4.59-4.68 (m, 1H) 3.92 (ddd, J=10.12, 5.67, 4.30 Hz, 1H) 3.88 (dt, J=10.12, 4.94 Hz, 1H) 1.74 (d, J=7.03 Hz, 3H) 1.71-1.79 (m, 1H) 1.68 (ddd, J=13.87, 9.54, 5.67 Hz, 1H) 1.57 (ddd, J=13.87, 8.54, 5.33 Hz, 1H) 0.98 (d, J=6.71 Hz, 3H) 0.96 (d, J=6.56 Hz, 3H). MS (ESI +) m/z 484 [M+H] +.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: EP-3818065-B1Priority Date: 2018-07-06Grant Date: 2023-03-15
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidine and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: KR-102736870-B1Priority Date: 2018-07-06Grant Date: 2024-12-03
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: US-2021340167-A1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-D]pyrimidine and their use in the treatment of conditions associated with elevated levels of cx3cr1 and/or cx3cl1 – Patent Application 20070123333Publication Number: JP-7506653-B2Priority Date: 2018-07-06Grant Date: 2024-06-26
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: US-11339183-B2Priority Date: 2018-07-06Grant Date: 2022-05-24
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo-[4,5-D]-pyrimidines and their uses in the treatment of conditions associated with high levels of CX3CR1 and/or CX3CL1Publication Number: IL-279818-B1Priority Date: 2018-07-06
- PHOSPHATE AND PHOSPHONATE DERIVATIVES OF 7-AMINO-5-THIO-THIAZOLO[4,5-D]PYRIMIDINE AND THEIR USE IN THE TREATMENT OF CONDITIONS ASSOCIATED WITH INCREASED LEVELS OF CX3CR1 AND/OR CX3CL1Publication Number: HR-P20230532-T1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: US-12060380-B2Priority Date: 2018-07-06Grant Date: 2024-08-13
- 7-Amino-5-thio-thiazolo [4,5-D] Pyrimidine phosphate and phosphonate derivatives and their use in therapeutic conditions associated with elevated levels of CX3CR1 and / or CX3CL1Publication Number: JP-2021530474-APriority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo-[4,5-D]-pyrimidines and their uses in the treatment of conditions associated with high levels of CX3CR1 and/or CX3CL1Publication Number: IL-279818-B2Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: US-2021292349-A1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo [4,5-D ] pyrimidine modulators of CX3CR1 receptor and medical uses thereofPublication Number: CN-112867725-BPriority Date: 2018-07-06Grant Date: 2024-09-27
- New usePublication Number: US-2025195548-A1Priority Date: 2023-12-19
- Fractalkine receptor antagonists for use in the prevention of thrombus formation and/or growthPublication Number: EP-4593833-A1Priority Date: 2023-12-19
- Fractalkine receptor antagonists for use in the prevention of thrombus formation and/or growthPublication Number: WO-2025133022-A1Priority Date: 2023-12-19
- New usePublication Number: US-2025195549-A1Priority Date: 2023-12-19
- New treatments of viral infectionsPublication Number: WO-2021224494-A1Priority Date: 2020-05-08
////////fosrugocrixan, anax labs, CX3C chemokine receptor 1 (CX3CR1) antagonist, antiinflammatory, 4ZXD25SC4S, KAND-145, KAND 145
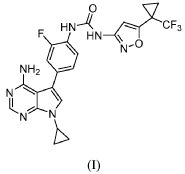
Ofirnoflast
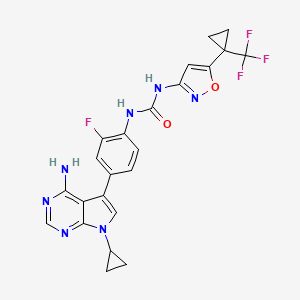
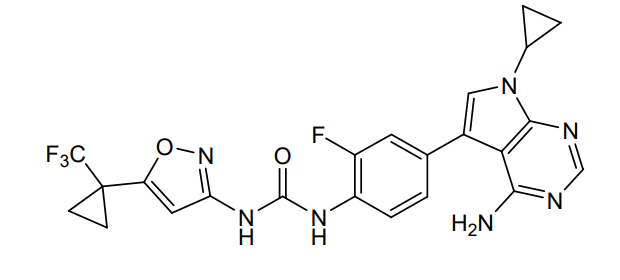
Ofirnoflast
CAS 2731294-23-6
MFC23H19F4N7O2 MW501.4 g/mol
N-[4-(4-amino-7-cyclopropyl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)-2-fluorophenyl]-N’-{5-[1-
(trifluoromethyl)cyclopropyl]-1,2-oxazol-3-yl}urea
N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-3-ISOXAZOLYL)UREA
N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-1,2-OXAZOL-3-YL)UREA
OFIRNOLAST [USAN]
OFIRNOFLAST
UREA, N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-3-ISOXAZOLYL)-
OFIRNOFLAST [INN]
serine/ threonine-protein kinase Nek7 inhibitor, antiinflammatory, HT-6184, HT 6184, 54PY2PBN7S
Ofirnoflast is an investigational drug, a NEK7 inhibitor, that targets and disrupts the formation of the NLRP3 inflammasome, a key driver of chronic inflammation. Developed by Halia Therapeutics, it is being explored for conditions like myelodysplastic syndromes (MDS), obesity, and Alzheimer’s disease. The drug’s unique mechanism aims to address inflammation at a root cause level, potentially offering a new approach to treating these diseases.
How it works
- Ofirnoflast is a “first-in-class” molecule that selectively inhibits the NEK7 protein.
- NEK7 is essential for the assembly of the NLRP3 inflammasome, a molecular complex that causes chronic inflammation.
- By inhibiting NEK7, ofirnoflast prevents the inflammasome from forming and promotes its disassembly.
- This approach aims to reduce inflammation without causing broad immunosuppression.
Therapeutic applications
- Myelodysplastic Syndromes (MDS): Ofirnoflast has completed a Phase 2 study for this condition and received Orphan Drug Designation from the FDA. It is being investigated for its potential to improve blood cell production by targeting the underlying inflammation.
- Obesity: An ongoing Phase 2 study is exploring ofirnoflast in combination with semaglutide to target inflammation and metabolic issues.
- Alzheimer’s Disease: Ofirnoflast is part of an early-stage program looking into its potential for this disease.
Ofirnoflast is a first-in-class, orally bioavailable NEK7 inhibitor currently undergoing Phase 2 clinical evaluation. It disrupts NLRP3 inflammasome assembly by targeting NEK7’s scaffolding function—blocking complex formation independently of NLRP3 activation status, upstream of caspase activation, pyroptosis, and inflammatory cytokine release. This mechanism offers a novel therapeutic approach for chronic inflammation. Unlike NSAIDs, corticosteroids, cytokine-neutralising biologics, and NLRP3-directed small molecules—which are frequently limited by off-target effects, immunosuppression, or incomplete efficacy—ofirnoflast provides a targeted approach with fewer anticipated liabilities
- A Ph2 Study to Evaluate the Safety, Efficacy and Tolerability of HT-6184 and Semaglutide in Obese Participants With T2DMCTID: NCT07172867Phase: Phase 2Status: Not yet recruitingDate: 2025-09-15
- HT-6184 in Subjects With MDSCTID: NCT07052006Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Evaluating Ability of HT-6184 to Reduce Inflammation and Pain After Third Molar ExtractionCTID: NCT06241742Phase: Phase 2Status: CompletedDate: 2025-03-30
- Study to Evaluate HT-6184 in Healthy SubjectsCTID: NCT05447546Phase: Phase 1Status: CompletedDate: 2023-08-28
SYN
https://www.tandfonline.com/doi/full/10.1080/1061186X.2025.2542856
SYN
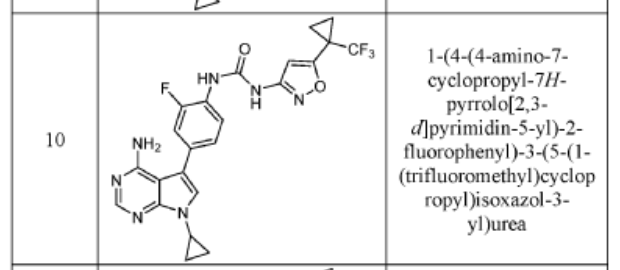
COMPD 10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021242505&_cid=P11-MHZPDU-32878-1
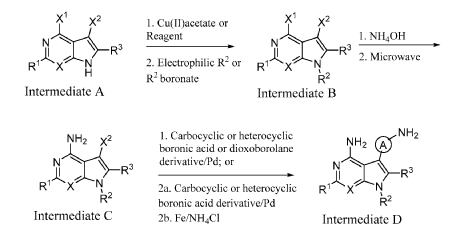
INTERMEDIATE D1
5-(4-AMINO-3-FLUOROPHENYL)-7-CYCLOPROPYL-7H-PYRROLO[2,3-D]PYRIMIDIN-4- AMINE
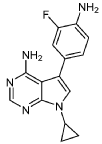
A mixture of 7-cyclopropyl-5-iodo-7H-pyrrolo[2,3-d]pyrimidin-4-amine (C1, 0.160 g, 0.533 mmol), 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (0.190 g, 0.800 mmol), and K2CO3 (0.221 g, 1.599 mmol) in 1,4-dioxane (1 mL) and water (0.3 mL) was purged with N2 for 10 min. Pd(PPh3)4 (0.062 g, 0.053 mmol) was then added and the reaction mixture was stirred at 100 °C for 12 h. Following completion of the reaction (as indicated by TLC), the mixture was filtered through a pad celite which was then rinsed with EtOAc (2 x 10 mL). The combined filtrates were concentrated under reduced pressure to yield crude material which was purified by flash chromatography (silica gel 230-400 mesh, eluting with 3% MeOH in DCM), affording
the title compound as a yellow solid (0.110 g, 73% yield).1H NMR (400 MHz, DMSO-d6) δ = 8.14 (s, 1H), 7.13 (s, 1H), 7.05-7.09 (m, 1H), 6.95-6.98 (m, 1H), 6.82-6.86 (m, 1H), 6.10 (bs, 2H), 5.22 (bs, 2H), 3.52-3.58 (m, 1H), 1.00-1.04 (m, 4H). LCMS: 284.1 [M+H].
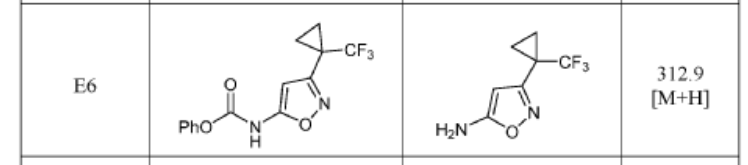
3-(1-(Trifluoromethyl)cyclopropyl)isoxazol-5-amine (precursor to E6) and 5-(1-(trifluoromethyl)cyclopropyl)isoxazol-3-amine (precursor to E7) were synthesized as reported in Synthesis 2013, 45, 171–173
EXAMPLE 5
1-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO[2,3-D]PYRIMIDIN-5-YL)-2- FLUOROPHENYL)-3-(3-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)ISOXAZOL-5-YL)UREA
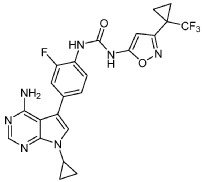
The title compound was prepared following the general procedure for urea formation (Method A), starting from 5-(4-amino-3-fluorophenyl)-7-cyclopropyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (D1, 0.080 g, 0.282 mmol) and phenyl (3-(1-(trifluoromethyl)cyclopropyl)isoxazol-5-yl)carbamate (E6, 0.088 g, 0.282 mmol), and was obtained as a white solid (0.031 g, 22% yield).1H NMR (400 MHz, DMSO-d6) δ = 10.59 (bs, 1H), 8.84 (bs, 1H), 8.11-8.17 (m, 2H), 7.26-7.37 (m, 3H), 6.20 (s, 1H), 6.16 (bs, 2H), 3.55-3.61 (m, 1H), 1.45-1.49 (m, 2H), 1.38-1.43 (m, 2H), 1.03-1.08 (m, 4H). LCMS: 502.1 [M+H].
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024249257&_cid=P11-MHZP9H-30149-1
PAT
- Targeted nek7 inhibition for modulation of the nlrp3 inflammasomePublication Number: US-2023210853-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: US-11713321-B2Priority Date: 2020-05-08Grant Date: 2023-08-01
- Inhibitors of nek7 kinasePublication Number: EP-4146348-B1Priority Date: 2020-05-08Grant Date: 2024-07-03
- Inhibitors of nek7 kinasePublication Number: US-2023416259-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: US-12091413-B2Priority Date: 2020-05-08Grant Date: 2024-09-17
- Inhibitors of nek7 kinasePublication Number: TW-202208356-APriority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: AU-2021280893-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: CN-115843272-APriority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: EP-4146348-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: KR-20230008763-APriority Date: 2020-05-08
- Polymorphs of nek 7 inhibitorsPublication Number: WO-2024249257-A1Priority Date: 2023-05-26
- Inhibitors of NEK7 kinasePublication Number: US-11161852-B1Priority Date: 2020-05-08Grant Date: 2021-11-02
- Inhibitors of nek7 kinasePublication Number: US-2021355130-A1Priority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: US-2022064173-A1Priority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: WO-2021242505-A1Priority Date: 2020-05-08

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////ofirnoflast, serine/ threonine-protein kinase Nek7 inhibitor, antiinflammatory, HT-6184, HT 6184, 54PY2PBN7S
ADX-103
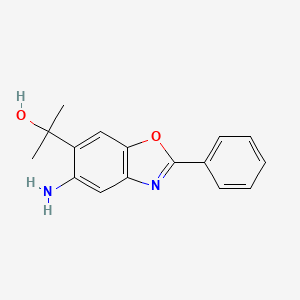
ADX-103
CAS 916056-81-0
Preclinical, Antiinflammatory Ophthalmic Agents, Diabetic Retinopathy,
Agents for Ophthalmic Drugs
MF C16 H16 N2 O2
5-Amino-α,α-dimethyl-2-phenyl-6-benzoxazolemethanol
Aldeyra Therapeutics Inc
ADX-103 , an aldehyde trap being investigated by Aldeyra for the treatment of dry eye syndrome; in May 2018, preclinical data were presented at 2018 ARVO Meeting in Honolulu, HI. Aldeyra, in collaboration with an undisclosed company, is also investigating an anti-inflammatory agent for treating ocular inflammation.
PATENT
WO-2020033344
Novel crystalline forms of a specific benzoxazole and it’s salts, process for their preparation, and compositions comprising them are claimed, useful for treating dry eye, inflammation and diabetes, through action as an aldehyde scavenger.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing a risk of a disease, disorder, or condition in which aldehyde toxicity is implicated in the pathogenesis. In general, salt forms or freebase forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of a variety of diseases or disorders as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A:
or a pharmaceutically acceptable salt thereof.
[0008] Compounds of the present invention, and pharmaceutically acceptable compositions thereof, are useful for treating a variety of diseases, disorders or conditions, associated with toxic aldehydes. Such diseases, disorders, or conditions include those described herein.
[0009] Compounds provided by this invention are also useful for the study of certain aldehydes in biology and pathological phenomena.
Scheme 1 – Synthesis of Compound A
Step 1: Synthesis of Compound A2
[00549] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with methanol (10L). Compound A1 (2.0kg) was added, followed by further methanol to rinse (9L). The reaction mixture was warmed to Tjacket=40°C. Once temperature had stabilized, sulfuric acid (220 mL, 0.4eq.) was slowly added. Once addition was complete, agitation was maintained for 30 mins then the vessel was heated to Tjmt=62°C. Reaction progress was
monitored by LC-MS analysis of reaction mixture. The reaction does not go to completion but is deemed complete when no change is apparent in ratio of starting material : product.
[00550] The vessel contents were cooled to Tjmt=24°C and stirred 60 minutes before filtration under vacuum. The filter cake was air dried for 2 hours and the contents then dissolved in ethyl acetate (18L) which was then washed sequentially with saturated sodium bicarbonate (8L), water (8L) and brine (8L) before drying over sodium sulfate, filtration and evaporation in vacuo. Compound A2 (1.5kg, 68.1%) was obtained as a bright orange powder.
Step 2: Synthesis of Compound A3
[00551] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with /V,/V-dimethylformamide (16L). Compound A2 (1.5kg) was added and the brown reaction mixture set to cool to Tint<20oC. Once temperature had stabilized, A-bromosucci ni mi de (l.5kg, 1.1 eq.) was added portion wise, maintaining Tint<27°C. Once addition was complete, the reaction was allowed to stir until starting material content was <1% AUC (250nm) by LCMS analysis.
[00552] A secondary jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with ethyl acetate (16L) and deionized water (22L). The reaction mixture was vacuum transferred into this vessel and held at high agitation for not less than 30 minutes. The aqueous layer was discharged and the organic layer washed with saturated sodium chloride (2 x 8L) then dried over sodium sulfate before evaporation in vacuo to Compound A3 as a deep brown oil (2.lkg, 100.8%), suitable for use in following step without purification.
Step 3: Synthesis of Compound A4
[00553] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L). Compound A3 (2.lkg) was added and the reaction mixture cooled to Tmt<l°C. A solution of Di-/er/-butyl dicarbonate (3.6kg, 2.2 eq.) in dichloromethane (0.5L) was added followed by a solution of A, A-di methyl ami nopyri di ne (92g, 0.1 eq.) in dichloromethane (0.5L). The resultant clear brown solution was stirred for 30 minutes whereupon pyridine (1.3L, 1.7 eq.) was dropwise added, maintaining Tint<5°C. Upon complete addition internal temperature was ramped from Tint=l°C to Tint=20°C over 18 hours.
[00554] The reaction mixture was sequentially washed with saturated sodium chloride (3 x 4.5L), 10 % w/v aqueous citric acid (2 x 4L), saturated sodium bicarbonate (4L), aqueous hydrochloric acid (1M, 4L), saturated sodium bicarbonate (4L) and saturated sodium chloride (4L) then dried over sodium sulfate and evaporated in vacuo with one azeotropic distillation with toluene (2L) to a very dark, heavy tar (3.4kg).
[00555] The isolated tar was mixed with absolute ethanol (3.1L) for 2 days whereupon it was filtered providing light cream colored, granular solids and a black mother liquor. The solids were washed with ice-cold ethanol (3 x 1L) and dried to constant mass. Compound A4 was obtained as off- white granules (1.7 kg, 50.2%).
Step 4: Synthesis of Compound AS
[00556] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with reagent alcohol (6.1 L) and Compound A4 (0.8kg), Tmt<20°C. Iron powder (0.5kg, 5.0 eq.) was added and the suspension stirred vigorously for 30 minutes. Acetic acid (glacial, 1.6L, 15.7 eq.) was added, maintaining Tint<30C.
[00557] Once LCMS confirmed complete consumption of starting material, ethyl acetate (10.2L) and water (10.2L) were added. Sodium bicarbonate (2.3kg, 15.9 eq.) was added portion wise and the layers separated once gas evolution had ceased. The aqueous layer was washed with ethyl acetate until LCMS indicated no further product was being extracted (8 x 2L) and the combined organic layers were sequentially washed with deionized water (6L) then saturated sodium chloride (6L) before drying over magnesium sulfate and evaporation in vacuo. Compound A5 was obtained as a light orange solid (0.7kg, 91.5%).
Step 5: Synthesis of Compound A6
[00558] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L), Compound A5 (0.7kg), and the reaction mixture cooled to Tint 20°C. Benzoyl chloride (0.3L, 1.5 eq.) was added and the reaction stirred 15 minutes. N,N-dimethylaminopyridine (7g, 0.04 eq.) in dichloromethane (0.1L) was added and the reaction stirred 15 minutes. Pyridine (0.5L, 2.5 eq.) was dropwise added, maintaining Tint<20°C. Upon complete addition the reaction was stirred until LCMS indicated consumption of starting material.
[00559] The reaction mixture was washed with deionized water (11L) and the organic layer extracted sequentially with aqueous hydrochloric acid (1M, 3 x 5L), saturated aqueous sodium bicarbonate (11 L), saturated sodium chloride (11 L), dried over magnesium sulfate and evaporated in vacuo. Compound A6 was obtained as a cream colored solid, suitable for use without further purification (0.9kg, 100.7%).
Step 6: Synthesis of Compound A 7
[00560] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with l,2-dimethoxy ethane (16L) and temperature set to Tint=2l°C. Compound A6 (0.9kg) was added and stirred to dissolution. Copper iodide (0.3kg, 1.0 eq.) was added and the mixture stirred 15 minutes. l, lO-phenanthroline (0.3kg, 1.2 eq.) was added and the mixture stirred 15 minutes. Cesium carbonate (l .5kg, 3.0 eq.) was added and the reaction was stirred for 15 minutes. The reaction temperature was ramped to Tint=80-85oC and maintained for 23 hours whereupon it was cooled to Tmt=20°C.
[00561] The reaction mixture was filtered through a celite pad, washing sequentially with deionized water (8L) and ethyl acetate (8L). The organic layer was extracted sequentially with deionized water (2 x 5L), saturated sodium chloride (4L), dried over sodium sulfate and evaporated in vacuo. Compound A7 was obtained as a brown solid, suitable for use without further purification (0.8kg, 104.1%).
Step 7: Synthesis of Compound A8
[00562] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A7 (0.8kg) in dichloromethane (3.6L) and cooled to Tmt<5°C in an ice bath. Hydrochloric acid in dioxane (4M, 1 2L, 3.1 eq.) was added dropwise with vigorous stirring, maintaining Tmt<25°C. Once addition was complete, the reaction mixture was allowed to stir for 18 hours at Tint=20-25oC.
[00563] The reaction mixture was filtered and the filter cake washed with dichloromethane (2 x 1L) and dried to constant mass. The hydrochloride salt of Compound A8 was isolated as an off-white solid (0.5kg, 88.7%).
Step 8: Synthesis of Compound A
[00564] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A8 (0.5kg) in tetrahydrofuran (4.8L) and cooled to Tint<-30°C in a dry-ice / acetone bath. Methylmagnesium bromide (3.4M in 2-methyltetrahydrofuran, 2.4L, 5.0eq.) was added slowly, maintaining Tmt<-lO°C. Once addition was complete, the reaction was allowed to warm to room temperature overnight.
[00565] Saturated aqueous ammonium chloride (2L) and ethyl acetate (2L) were added and the reaction mixture stirred for 30 minutes. The aqueous layer was extracted with further ethyl acetate (2 x 2L) and the combined organic layers washed with saturated sodium chloride (2L), dried over sodium sulfate and evaporated in vacuo to a dark heavy oil. The heavy oil was purified by column chromatography on silica gel, eluting with ethyl acetate : heptane 1 : 19 to 1 : 1. Pure Compound A was obtained after evaporation and drying as a brown powder (99.8 g, 23.0%).
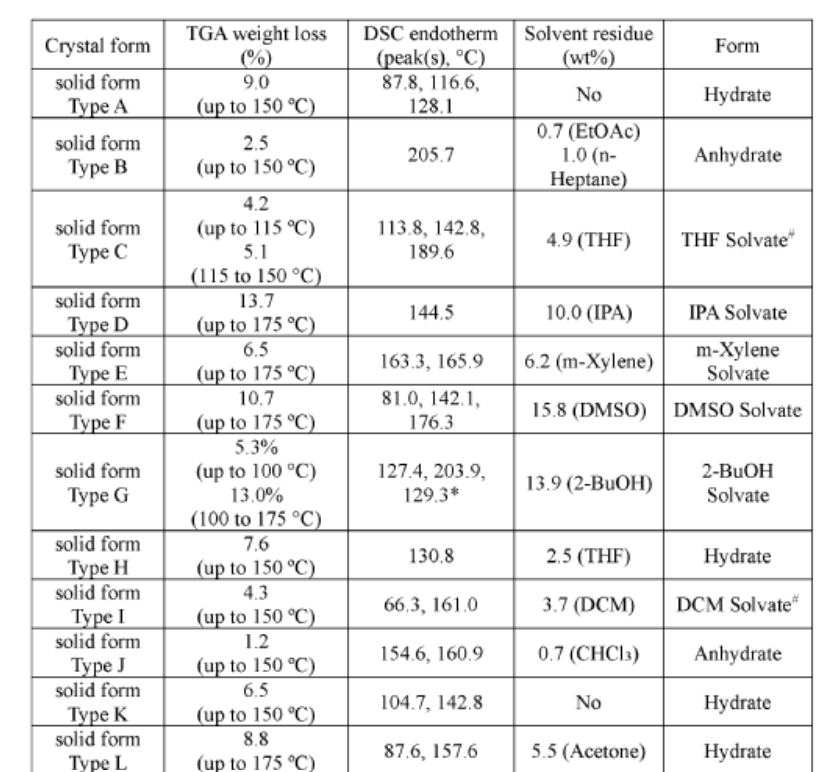
Example 1 – Preparation of Free Base Forms A, B and C of Compound A
Compound A
Primary Polymorph Screen
[00566] Based on solubility screen results, a primary polymorph screen using an initial set of 24 solvents, as shown in Table 18, was performed as follows: A) To 24 x 20 mL vials, approximately 50 mg of the received ADX-103 was added; B) The solids were then slurried in 2 mL of the solvents and left placed in an incubator/shaker to temperature cycle between ambient and 40 °C in 4 hour cycles; C) After 72 hours temperature cycling, the mother liquors were removed from the vials and split evenly between 4 x 2 mL vials. The vials were then split between evaporation, crash cooling to 2 °C and -18 °C and anti-solvent addition; and D) Any solids
recovered were analysed by XRPD, any new patterns identified were also analysed by TG/DTA and PLM.
Table 18. Solvents Selected for Initial Primary Polymorph Screen
PATENT
WO2018039197 , as compound I-8.
PATENT
WO 2006127945
WO 2011072141
WO 2014116593
US 20150344447
WO 2020028820
////////////ADX-103, Preclinical, Antiinflammatory, Ophthalmic Agents, Diabetic Retinopathy, Aldeyra Therapeutics Inc,
CC(C)(O)c1cc2oc(nc2cc1N)c3ccccc3

{kind=link}