
Home » Posts tagged 'ANTICONVULSANT'
Tag Archives: ANTICONVULSANT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Divalproex sodium
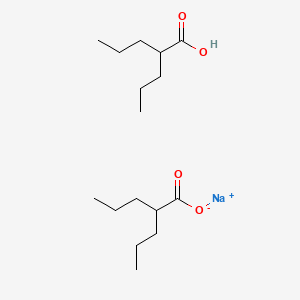
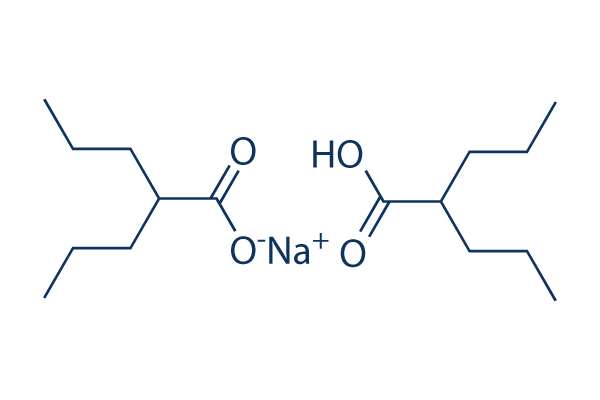
Divalproex
- 44089
WeightAverage: 144.2114
Chemical FormulaC8H16O2
UNII614OI1Z5WI, CAS number99-66-1, 76584-70-8
2-propylpentanoic acid, DIVALPROEX SODIUM, 76584-70-8, Valproate semisodium, Epival, Depakote, Sodium divalproate, Semisodium Valproate, Abbott 50711, ValdisovalValproic Acid
CAS Registry Number: 99-66-1
CAS Name: 2-Propylpentanoic acid
Additional Names: 2-propylvaleric acid; di-n-propylacetic acid
Trademarks: Convulex (Pharmacia); Depakene (Abbott)
Molecular Formula: C8H16O2
Molecular Weight: 144.21
Percent Composition: C 66.63%, H 11.18%, O 22.19%
Literature References: Antiepileptic; increases levels of g-aminobutyric acid (GABA) in the brain. Prepn: B. S. Burton, Am. Chem. J.3, 385 (1882); E. Oberreit, Ber.29, 1998 (1896); M. Tiffeneau, Y. Deux, Compte Rend.212, 105 (1941). Anticonvulsant activity: H. Meunier et al.,Therapie18, 435 (1963). Toxicity data: Jenner et al.,Food Cosmet. Toxicol.2, 327 (1964). Comprehensive description: Z. L. Chang, Anal. Profiles Drug Subs.8, 529-556 (1979). Review of teratogenicity studies: H. Nau et al.,Pharmacol. Toxicol.69, 310-321 (1991); R. Alsdorf, D. F. Wyszynski, Expert Opin. Drug Safety4, 345-353 (2005). Review of pharmacology and clinical experience in epilepsy: E. M. Rimmer, A. Richens, Pharmacotherapy5, 171-184 (1985); in psychiatric disease: D. R. P. Guay, ibid.15, 631-647 (1995); in migraine prophylaxis: C. E. Shelton, J. F. Connelly, Ann. Pharmacother.30, 865-866 (1996). Review of pharmacodynamics and mechanisms of action: W. Löscher, Prog. Neurobiol.58, 31-59 (1999).
Properties: Colorless liquid with characteristic odor. bp 219.5°. nD24.5 1.425. d40 0.9215. pKa 4.6. Very sol in organic solvents. Soly in water: 1.3 mg/ml. LD50 orally in rats: 670 mg/kg (Jenner).
Boiling point: bp 219.5°
pKa: pKa 4.6
Index of refraction:nD24.5 1.425
Density: d40 0.9215
Toxicity data: LD50 orally in rats: 670 mg/kg (Jenner)
Derivative Type: Sodium salt (1:1)
CAS Registry Number: 1069-66-5
Additional Names: Sodium valproate
Trademarks: Depacon (Abbott); Depakin (Sanofi-Synthelabo); Dépakine (Sanofi-Aventis); Epilim (Sanofi-Aventis); Ergenyl (Sanofi-Synthelabo); Leptilan (Dolorgiet); Orfiril (Desitin)
Molecular Formula: C8H15NaO2
Molecular Weight: 166.19
Percent Composition: C 57.82%, H 9.10%, Na 13.83%, O 19.25%
Properties: White, odorless, crystalline, deliquescent powder. pKa 4.8. Hygroscopic. One gram is sol in 0.4 ml water; 1.5 ml ethanol; 5 ml methanol. Practically insol in common organic solvents. LD50 orally in mice: 1700 mg/kg (Meunier).
pKa: pKa 4.8
Toxicity data: LD50 orally in mice: 1700 mg/kg (Meunier)
Derivative Type: Sodium salt (2:1)
CAS Registry Number: 76584-70-8
Additional Names: Sodium hydrogen bis(2-propylpentanoate); divalproex sodium; valproate semisodium
Manufacturers’ Codes: Abbott 50711
Trademarks: Depakote (Abbott); Valcote (Abbott)
Molecular Formula: C16H31NaO4
Molecular Weight: 310.40
Percent Composition: C 61.91%, H 10.07%, Na 7.41%, O 20.62%
Derivative Type: Magnesium salt
Trademarks: Depamag (Sigma-Tau)
Molecular Formula: C16H30MgO4
Molecular Weight: 310.71
Percent Composition: C 61.85%, H 9.73%, Mg 7.82%, O 20.60%
Therap-Cat: Anticonvulsant; antimanic; antimigraine.Keywords: Anticonvulsant; Antimigraine; Antimanic.
Synthesis Reference
Daniel Aubert, Francis Blanc, Henri Desmolin, Michel Morre, Lucette Sindely, “Valproic acid preparations.” U.S. Patent US5017613, issued January, 1965.
Patent
https://patents.google.com/patent/WO2007004238A2/enDivalproex sodium is one of the most widely used epileptic agents presently available in the market. Both the constituents, valproic acid and sodium valproate themselves have also been used for the treatment of epileptic seizures and convulsions. But their utility has remained restricted since valproic acid is a liquid and is difficult to formulate for an oral dosage form whereas sodium valproate is a hygroscopic solid with poor stability characteristics. Divalproex sodium is an oligomer having 1:1 molar ratio of valproic acid and sodium valproate containing 4 to 6 units. The relevant prior art includes US 4,988,731 (’73I) relates to a non-hygroscopic stable sodium hydrogen divalproate oligomer. Its probable structure is shown in Fig 1

Fig 1 – Divalproex sodiumWhere M is a about 2.As can be seen from the displayed structure, one mole each of the valproic acid forms coordinate bonds with the sodium of the sodium valproate molecule, and the valproate ion is ionically bonded to the sodium atom. The structure is thus consistent with the unique characteristic of the compound. However the preferred mode of representing Divalproex sodium is by reference to single compound of the formula{(CH3CH2CH2)2CHCO2} {(CH3CH2CH2)2CHCO2}Na, HThe said patent also describes two alternative processes for the preparation of the oligomer. According to one aspect, the oligomer is produced by dissolving sodium valproate and valproic acid in equimolar amount in acetone and crystallizing from chilled acetone at around O0C. Alternatively Divalproex sodium can be isolated from a two component liquid medium, which includes acetone where in half equivalent of NaOH to the valproic acid present, preferable as a solution in an acetone miscible solvent eg. water. The new compound can be recovered from the liquid phase by evaporating the solvent(s) and, if desired, the new compound can be recrystallized, for instance from acetonitrile or others or the material may be spay-dried, lyophilized or purified by chromatography.US ‘731 claims yield of 90% of theory.Drawbacks of the above mentioned reported methods for the preparation of Divalproex sodium described in US 4988731 are difficult to reproduce on a large scale and provides inconsistent yields and the material obtained is not always free flowing in nature. The process involves the crystallization of a 1:1 mixture of valproic acid and sodium valproate from a chilled solution of acetone, followed by washing with chilled acetone. Divalproex sodium is as such fairly soluble in acetone at temperatures above 1O0C and extreme care has to be. taken while performing washing with chilled acetone as any rise in temperature would lead to the loss of yield. This problem actually comes to the fore while scaling up the process during commercialization since during centrifugation of the large volume the temperature of the mixture rises and acetone has to be cooled below O0C, which require large amount of liquid nitrogen or dry ice. Moreover it was also observed that due to the cooled nature of the solvent, the isolated Divalproex sodium absorbs considerable amount of moisture and therefore requires longer time to dry eventually leading to longer time cycle for the otherwise simple single step process. Also the high moisture content in the recovered acetone makes it unsuitable for reuse. Alternatively, to avoid absorption of water, the centrifugation had to be carried out under a blanket of dry nitrogen gas. These additional infrastructural loads add to input costs eventually making the otherwise single step low cost process becoming uncompetitive and economically unviable.Similarly the other process involves the addition of half molar equivalent of sodium hydroxide dissolved in water to valproic acid and the solvent has to be evaporated to obtain crude product, which has to be recrystallized to get Divalprox of the desired specification. The process is operationally tedious and requires the reduction in the level of water in the reaction mass via evaporation of the solvent followed by re- crystallization from acetonitrile making the process lengthy and economically unviable. There is therefore a need for operationally making this single step process more efficient and high yieldingExample I:To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH ( 13g; half molar) is added & the resulting reaction mixture is stirred at 40-500C for 1 hr. Then acetonitrile(600ml) is added to obtain clear solution at 40-500C and the solution is charcoalized at 40-500C followed by filtration at 40-500C through hyflo-bed. The resultant reaction mixture was stirred at 10-200C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. (102.25g, 95%)Example II;To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH (13g; half molar) is added & the resulting reaction mixture is stirred at 30-400C for 1 hr. Then acetone (600ml) is added to obtain clear solution at 30-400C and the material is charcoalized at 30-400C followed by filtration through hyflo-bed. The resultant reaction solution was stirred at -5°C to -1O0C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. ( 55g, 51.11%) Example III:To a solution of Valproic acid (10Og) in dichloromethane (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 30- 400C for 1 hr to get clear solution. Then acetonitrile (600ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (10Og; 93%)Example IV:To a solution of Valproic acid (10Og) in diisopropyl ether(200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (104g; 96.65%)Example V:To a solution of Valproic acid (10Og) in methyl tertiary butyl ether(200ml) at 20- 300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (102g;94.79%)Example VI:To a solution of Valproic acid (10Og) in toluene (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (101g; 93.87%)Example VII: A mixture of sodium valproate (6Og) and valproic acid (52.04g) was taken in acetonitrile (800ml) and heated at reflux to obtain a clear solution, which was filtered through hyflo-bed to remove suspended particles. Then the solution was stirred at 10- 200C for 2-3 hr. The solid, thus obtained, was filtered and washed with acetonitrile (100ml). The product was dried at 40-450C for 10-12 hr. (105g ; 93.75%)Example VIII;To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C5 caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring. The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-45°C for 10-12 hr.(102g; ~ 95%) Example IX:To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -1O0C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(54g; ~ 50.11%)Example X:To a solution of valproic acid (10Og) in ethanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring.The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-450C for 10-12 hr.(101g; ~ 93.87%)Example XI: To a solution of valproic acid (10Og) in ethanol (200ml) at 20-30°C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -100C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(55g; ~ 51%)ADVANTAGES:> The process is high yielding. > The process produces Divalproex sodium with improved flowability.> The process produces Divalproex sodium that is non-hygroscopic and more stable.> The process is industrially feasible, precise, reproducible and does not require sophisticated infrastructure.
Divalproex Sodium is a stable coordination compound comprised of sodium valproate and valproic acid with anticonvulsant and antiepileptic activities. Divalproex dissociates to the valproate ion in the gastrointestinal tract. This agent binds to and inhibits gamma-aminobutyric acid (GABA) transaminase and its anticonvulsant activity may be exerted by increasing brain concentration of GABA and by inhibiting enzymes that catabolize GABA or block the reuptake of GABA into glia and nerve endings. Divalproex may also work by suppressing repetitive neuronal firing through inhibition of voltage-sensitive sodium channels.
Valproate semisodium is a mixture of valproic acid and its sodium salt in a 1:1 molar ratio. It is used for the management and treatment of seizure disorders, mania, and prophylactic treatment of migraine headache. It has a role as an antimanic drug, an anticonvulsant and a GABA agent. It contains a valproic acid and a sodium valproate.
Divalproex sodium, valproate sodium, and valproic acid, are all similar medications that are used by the body as valproic acid. Therefore, the term valproic acid will be used to represent all of these medications in this discussion.
Valproate (VPA) and its valproic acid, sodium valproate, and valproate semisodium forms are medications primarily used to treat epilepsy and bipolar disorder and prevent migraine headaches.[2] They are useful for the prevention of seizures in those with absence seizures, partial seizures, and generalized seizures.[2] They can be given intravenously or by mouth, and the tablet forms exist in both long- and short-acting formulations.[2]
Common side effects of valproate include nausea, vomiting, sleepiness, and dry mouth.[2] Serious side effects can include liver failure, and regular monitoring of liver function tests is therefore recommended.[2] Other serious risks include pancreatitis and an increased suicide risk.[2] Valproate is known to cause serious abnormalities in babies if taken during pregnancy,[2][3] and as such it is not typically recommended for women of childbearing age who have migraines.[2]
Valproate’s precise mechanism of action is unclear.[2][4] Proposed mechanisms include affecting GABA levels, blocking voltage-gated sodium channels, and inhibiting histone deacetylases.[5][6] Valproic acid is a branched short-chain fatty acid (SCFA) made from valeric acid.[5]
Valproate was first made in 1881 and came into medical use in 1962.[7] It is on the World Health Organization’s List of Essential Medicines[8] and is available as a generic medication.[2] It is marketed under the brand names Depakote, among others.[2] In 2018, it was the 131st most commonly prescribed medication in the United States, with more than 5 million prescriptions.[9][10]
Terminology
Valproic acid (VPA) is an organic weak acid. The conjugate base is valproate. The sodium salt of the acid is sodium valproate and a coordination complex of the two is known as valproate semisodium.[11]
Medical uses
It is used primarily to treat epilepsy and bipolar disorder. It is also used to prevent migraine headaches.[12]
Epilepsy
Valproate has a broad spectrum of anticonvulsant activity, although it is primarily used as a first-line treatment for tonic–clonic seizures, absence seizures and myoclonic seizures and as a second-line treatment for partial seizures and infantile spasms.[12][13] It has also been successfully given intravenously to treat status epilepticus.[14][15]
Mental illness
Bipolar disorder
Valproate products are also used to treat manic or mixed episodes of bipolar disorder.[16][17]
Schizophrenia
A 2016 systematic review compared the efficacy of valproate as an add-on for people with schizophrenia:[18]
| There is limited evidence that adding valproate to antipsychotics may be effective for overall response and also for specific symptoms, especially in terms of excitement and aggression. Valproate was associated with a number of adverse events among which sedation and dizziness appeared more frequently than in the control groups.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Dopamine dysregulation syndrome
Based upon five case reports, valproic acid may have efficacy in controlling the symptoms of the dopamine dysregulation syndrome that arise from the treatment of Parkinson’s disease with levodopa.[19][20][21]
Migraines
Valproate is also used to prevent migraine headaches. Because this medication can be potentially harmful to the fetus, valproate should be considered for those able to become pregnant only after the risks have been discussed.[22]
Other
The medication has been tested in the treatment of AIDS and cancer, owing to its histone-deacetylase-inhibiting effects.[23]
Contraindications
Contraindications include:
- Pre-existing acute or chronic liver dysfunction or family history of severe liver inflammation (hepatitis), particularly medicine related.[24]
- Known hypersensitivity to valproate or any of the ingredients used in the preparation[24]
- Urea cycle disorders[24]
- Hepatic porphyria[24]
- Hepatotoxicity[24]
- Mitochondrial disease[24]
- Pancreatitis[24]
- Porphyria[25]
Adverse effects
See also: List of adverse effects of valproic acid and List of adverse effects of valproate semisodium
Most common adverse effects include:[22]
- Nausea (22%)
- Drowsiness (19%)
- Dizziness (12%)
- Vomiting (12%)
- Weakness (10%)
Serious adverse effects include:[22]
- Bleeding
- Low blood platelets
- Encephalopathy
- Suicidal behavior and thoughts
- Low body temperature
Valproic acid has a black box warning for hepatotoxicity, pancreatitis, and fetal abnormalities.[22]
There is evidence that valproic acid may cause premature growth plate ossification in children and adolescents, resulting in decreased height.[26][27][28][29] Valproic acid can also cause mydriasis, a dilation of the pupils.[30] There is evidence that shows valproic acid may increase the chance of polycystic ovary syndrome (PCOS) in women with epilepsy or bipolar disorder. Studies have shown this risk of PCOS is higher in women with epilepsy compared to those with bipolar disorder.[31] Weight gain is also possible.[32]
Pregnancy
Valproate causes birth defects;[33] exposure during pregnancy is associated with about three times as many major abnormalities as usual, mainly spina bifida with the risks being related to the strength of medication used and use of more than one drug.[34][35] More rarely, with several other defects, including a “valproate syndrome”.[36] Characteristics of this valproate syndrome include facial features that tend to evolve with age, including a triangle-shaped forehead, tall forehead with bifrontal narrowing, epicanthic folds, medial deficiency of eyebrows, flat nasal bridge, broad nasal root, anteverted nares, shallow philtrum, long upper lip and thin vermillion borders, thick lower lip and small downturned mouth.[37] While developmental delay is usually associated with altered physical characteristics (dysmorphic features), this is not always the case.[38]
Children of mothers taking valproate during pregnancy are at risk for lower IQs.[39][40][41] Maternal valproate use during pregnancy increased the probability of autism in the offspring compared to mothers not taking valproate from 1.5% to 4.4%.[42] A 2005 study found rates of autism among children exposed to sodium valproate before birth in the cohort studied were 8.9%.[43] The normal incidence for autism in the general population is estimated at less than one percent.[44] A 2009 study found that the 3-year-old children of pregnant women taking valproate had an IQ nine points lower than that of a well-matched control group. However, further research in older children and adults is needed.[45][46][47]
Sodium valproate has been associated with paroxysmal tonic upgaze of childhood, also known as Ouvrier–Billson syndrome, from childhood or fetal exposure. This condition resolved after discontinuing valproate therapy.[48][49]
Women who intend to become pregnant should switch to a different medication if possible or decrease their dose of valproate.[50] Women who become pregnant while taking valproate should be warned that it causes birth defects and cognitive impairment in the newborn, especially at high doses (although valproate is sometimes the only drug that can control seizures, and seizures in pregnancy could have worse outcomes for the fetus than exposure to valproate). Studies have shown that taking folic acid supplements can reduce the risk of congenital neural tube defects.[22] The use of valproate for migraine or bipolar disorder during pregnancy is contraindicated in the European Union, and the medicines are not recommended for epilepsy during pregnancy unless there is no other effective treatment available.[51]
Elderly
Valproate in elderly people with dementia caused increased sleepiness. More people stopped the medication for this reason. Additional side effects of weight loss and decreased food intake were also associated with one-half of people who become sleepy.[22]
Overdose and toxicity
| Form | Lower limit | Upper limit | Unit |
| Total (including protein bound) | 50[52] | 125[52] | µg/mL or mg/l |
| 350[53] | 700[53] | μmol/L | |
| Free | 6[52] | 22[52] | µg/mL or mg/l |
| 35[53] | 70[53] | μmol/L |
Excessive amounts of valproic acid can result in sleepiness, tremor, stupor, respiratory depression, coma, metabolic acidosis, and death.[54] In general, serum or plasma valproic acid concentrations are in a range of 20–100 mg/l during controlled therapy, but may reach 150–1500 mg/l following acute poisoning. Monitoring of the serum level is often accomplished using commercial immunoassay techniques, although some laboratories employ gas or liquid chromatography.[55] In contrast to other antiepileptic drugs, at present there is little favorable evidence for salivary therapeutic drug monitoring. Salivary levels of valproic acid correlate poorly with serum levels, partly due to valproate’s weak acid property (pKa of 4.9).[56]
In severe intoxication, hemoperfusion or hemofiltration can be an effective means of hastening elimination of the drug from the body.[57][58] Supportive therapy should be given to all patients experiencing an overdose and urine output should be monitored.[22] Supplemental L-carnitine is indicated in patients having an acute overdose[59][60] and also prophylactically[59] in high risk patients. Acetyl-L-carnitine lowers hyperammonemia less markedly[61] than L-carnitine.
Interactions
Valproate inhibits CYP2C9, glucuronyl transferase, and epoxide hydrolase and is highly protein bound and hence may interact with drugs that are substrates for any of these enzymes or are highly protein bound themselves.[24] It may also potentiate the CNS depressant effects of alcohol.[24] It should not be given in conjunction with other antiepileptics due to the potential for reduced clearance of other antiepileptics (including carbamazepine, lamotrigine, phenytoin and phenobarbitone) and itself.[24] It may also interact with:[22][24][62]
- Aspirin: may increase valproate concentrations. May also interfere with valproate’s metabolism.
- Benzodiazepines: may cause CNS depression and there are possible pharmacokinetic interactions.
- Carbapenem antibiotics: reduce valproate levels, potentially leading to seizures.
- Cimetidine: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Erythromycin: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Ethosuximide: valproate may increase ethosuximide concentrations and lead to toxicity.
- Felbamate: may increase plasma concentrations of valproate.
- Mefloquine: may increase valproate metabolism combined with the direct epileptogenic effects of mefloquine.
- Oral contraceptives: may reduce plasma concentrations of valproate.
- Primidone: may accelerate metabolism of valproate, leading to a decline of serum levels and potential breakthrough seizure.
- Rifampicin: increases the clearance of valproate, leading to decreased valproate concentrations
- Warfarin: valproate may increase free warfarin concentration and prolong bleeding time.
- Zidovudine: valproate may increase zidovudine serum concentration and lead to toxicity.
Pharmacology
Pharmacodynamics
Although the mechanism of action of valproate is not fully understood,[24] traditionally, its anticonvulsant effect has been attributed to the blockade of voltage-gated sodium channels and increased brain levels of gamma-aminobutyric acid (GABA).[24] The GABAergic effect is also believed to contribute towards the anti-manic properties of valproate.[24] In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by inhibiting the re-uptake of GABA by neuronal cells.[24]
Prevention of neurotransmitter-induced hyperexcitability of nerve cells, via Kv7.2 channel and AKAP5, may also contribute to its mechanism.[63] Also, it has been shown to protect against a seizure-induced reduction in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[64]
It also has histone-deacetylase-inhibiting effects. The inhibition of histone deacetylase, by promoting more transcriptionally active chromatin structures, likely presents the epigenetic mechanism for regulation of many of the neuroprotective effects attributed to valproic acid. Intermediate molecules mediating these effects include VEGF, BDNF, and GDNF.[65][66]
Endocrine actions
Valproic acid has been found to be an antagonist of the androgen and progesterone receptors, and hence as a nonsteroidal antiandrogen and antiprogestogen, at concentrations much lower than therapeutic serum levels.[67] In addition, the drug has been identified as a potent aromatase inhibitor, and suppresses estrogen concentrations.[68] These actions are likely to be involved in the reproductive endocrine disturbances seen with valproic acid treatment.[67][68]
Valproic acid has been found to directly stimulate androgen biosynthesis in the gonads via inhibition of histone deacetylases and has been associated with hyperandrogenism in women and increased 4-androstenedione levels in men.[69][70] High rates of polycystic ovary syndrome and menstrual disorders have also been observed in women treated with valproic acid.[70]
Pharmacokinetics

Some metabolites of valproic acid. Glucuronidation and β-oxidation are the main metabolic pathways; ω-oxidation metabolites are considered hepatotoxic.[71][72] Details see text.
Taken by mouth, valproate is rapidly and virtually completely absorbed from the gut.[71] When in the bloodstream, 80–90% of the substance are bound to plasma proteins, mainly albumin. Protein binding is saturable: it decreases with increasing valproate concentration, low albumin concentrations, the patient’s age, additional use of other drugs such as aspirin, as well as liver and kidney impairment.[73][74] Concentrations in the cerebrospinal fluid and in breast milk are 1 to 10% of blood plasma concentrations.[71]
The vast majority of valproate metabolism occurs in the liver.[75] Valproate is known to be metabolized by the cytochrome P450 enzymes CYP2A6, CYP2B6, CYP2C9, and CYP3A5.[75] It is also known to be metabolized by the UDP-glucuronosyltransferase enzymes UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15.[75] Some of the known metabolites of valproate by these enzymes and uncharacterized enzymes include (see image):[75]
- via glucuronidation (30–50%): valproic acid β-O-glucuronide
- via beta oxidation (>40%): 2E-ene-valproic acid, 2Z-ene-valproic acid, 3-hydroxyvalproic acid, 3-oxovalproic acid
- via omega oxidation: 5-hydroxyvalproic acid, 2-propyl-glutaric acid
- some others: 3E-ene-valproic acid, 3Z-ene-valproic acid, 4-ene-valproic acid, 4-hydroxyvalproic acid
All in all, over 20 metabolites are known.[71]
In adult patients taking valproate alone, 30–50% of an administered dose is excreted in urine as the glucuronide conjugate.[75] The other major pathway in the metabolism of valproate is mitochondrial beta oxidation, which typically accounts for over 40% of an administered dose.[75] Typically, less than 20% of an administered dose is eliminated by other oxidative mechanisms.[75] Less than 3% of an administered dose of valproate is excreted unchanged (i.e., as valproate) in urine.[75] Only a small amount is excreted via the faeces.[71] Elimination half-life is 16±3 hours and can decrease to 4–9 hours when combined with enzyme inducers.[71][74]
Chemistry
Valproic acid is a branched short-chain fatty acid and the 2-n–propyl derivative of valeric acid.[5]
History
Valproic acid was first synthesized in 1882 by Beverly S. Burton as an analogue of valeric acid, found naturally in valerian.[76] Valproic acid is a carboxylic acid, a clear liquid at room temperature. For many decades, its only use was in laboratories as a “metabolically inert” solvent for organic compounds. In 1962, the French researcher Pierre Eymard serendipitously discovered the anticonvulsant properties of valproic acid while using it as a vehicle for a number of other compounds that were being screened for antiseizure activity. He found it prevented pentylenetetrazol-induced convulsions in laboratory rats.[77] It was approved as an antiepileptic drug in 1967 in France and has become the most widely prescribed antiepileptic drug worldwide.[78] Valproic acid has also been used for migraine prophylaxis and bipolar disorder.[79]
Society and culture
Valproate is available as a generic medication.[2]
Approval status
| Indications | FDA-labelled indication?[1] | TGA-labelled indication?[12] | MHRA-labelled indication?[80] | Literature support |
|---|---|---|---|---|
| Epilepsy | Yes | Yes | Yes | Limited (depends on the seizure type; it can help with certain kinds of seizures: drug-resistant epilepsy, partial and absence seizures, can be used against glioblastoma and other tumors both to improve survival and treat seizures, and against tonic–clonic seizures and status epilepticus).[81][82][83][84] |
| Bipolar mania | Yes | Yes | Yes | Limited.[85] |
| Bipolar depression | No | No | No | Moderate.[86] |
| Bipolar maintenance | No | No | No | Limited.[87] |
| Migraine prophylaxis | Yes | Yes (accepted) | No | Limited. |
| Acute migraine management | No | No | No | Only negative results.[88] |
| Schizophrenia | No | No | No | Weak evidence.[89] |
| Agitation in dementia | No | No | No | Weak evidence. Not recommended for agitation in people with dementia.[90] Increased rate of adverse effects, including a risk of serious adverse effects.[90] |
| Fragile X syndrome | Yes (orphan) | No | No | Limited.[66] |
| Familial adenomatous polyposis | Yes (orphan) | No | No | Limited. |
| Chronic pain & fibromyalgia | No | No | No | Limited.[91] |
| Alcohol hallucinosis | No | No | No | One randomised double-blind placebo-controlled trial.[92] |
| Intractable hiccups | No | No | No | Limited, five case reports support its efficacy, however.[93] |
| Non-epileptic myoclonus | No | No | No | Limited, three case reports support its efficacy, however.[94] |
| Cluster headaches | No | No | No | Limited, two case reports support its efficacy.[95] |
| West syndrome | No | No | No | A prospective clinical trial supported its efficacy in treating infantile spasms.[96] |
| HIV infection eradication | No | No | No | Double-blind placebo-controlled trials have been negative.[97][98][99] |
| Myelodysplastic syndrome | No | No | No | Several clinical trials have confirmed its efficacy as a monotherapy,[100] as an adjunct to tretinoin[100] and as an adjunct to hydralazine.[101] |
| Acute myeloid leukaemia | No | No | No | Two clinical trials have confirmed its efficacy in this indication as both a monotherapy and as an adjunct to tretinoin.[102][103][104] |
| Cervical cancer | No | No | No | One clinical trial supports its use here.[105] |
| Malignant melanoma | No | No | No | One phase II study has seemed to discount its efficacy.[106] |
| Breast cancer | No | No | No | A phase II study has supported its efficacy.[107] |
| Impulse control disorder | No | No | No | Limited.[108][109] |
Off-label uses
In 2012, pharmaceutical company Abbott paid $1.6 billion in fines to US federal and state governments for illegal promotion of off-label uses for Depakote, including the sedation of elderly nursing home residents.[110][111]
Some studies have suggested that valproate may reopen the critical period for learning absolute pitch and possibly other skills such as language.[112][113]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Formulations
| Clinical data | |
|---|---|
| Other names | valproate sodium (USAN US) |
| License data | US DailyMed: Valproate_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1069-66-5 |
| PubChem CID | 16760703 |
| DrugBank | DBSALT001257 |
| ChemSpider | 13428 |
| UNII | 5VOM6GYJ0D |
| KEGG | D00710 |
| ChEBI | CHEBI:9925 |
| ChEMBL | ChEMBL433 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C8H15NaO2 |
| Molar mass | 166.196 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
| Clinical data | |
|---|---|
| Trade names | Depakote, others |
| Other names | semisodium valproate, divalproex sodium (USAN US) |
| License data | US DailyMed: Divalproex_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76584-70-8 |
| PubChem CID | 23663956 |
| DrugBank | DBSALT000185 |
| ChemSpider | 48337 |
| UNII | 644VL95AO6 |
| KEGG | D00304 |
| ChEBI | CHEBI:4667 |
| ChEMBL | ChEMBL2105613 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C16H31NaO4 |
| Molar mass | 310.410 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Valproate exists in two main molecular variants: sodium valproate and valproic acid without sodium (often implied by simply valproate). A mixture between these two is termed semisodium valproate. It is unclear whether there is any difference in efficacy between these variants, except from the fact that about 10% more mass of sodium valproate is needed than valproic acid without sodium to compensate for the sodium itself.[114]
Brand names of valproic acid
Branded products include:
- Absenor (Orion Corporation Finland)
- Convulex (G.L. Pharma GmbH Austria)
- Depakene (Abbott Laboratories in US and Canada)[115]
- Depakine (Sanofi Aventis France)
- Depakine (Sanofi Synthelabo Romania)
- Depalept (Sanofi Aventis Israel)
- Deprakine (Sanofi Aventis Finland)
- Encorate (Sun Pharmaceuticals India)
- Epival (Abbott Laboratories US and Canada)
- Epilim (Sanofi Synthelabo Australia and South Africa)
- Stavzor (Noven Pharmaceuticals Inc.)
- Valcote (Abbott Laboratories Argentina)
- Valpakine (Sanofi Aventis Brazil)
- Orfiril (Desitin Arzneimittel GmbH Norway)
Brand names of sodium valproate
Portugal
- Tablets – Diplexil-R by Bial.
United States
- Intravenous injection – Depacon by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories. (Note Depakene capsules are valproic acid).
- Depakote tablets are a mixture of sodium valproate and valproic acid.
- Tablets – Eliaxim by Bial.
Australia
- Epilim Crushable Tablets Sanofi[116]
- Epilim Sugar Free Liquid Sanofi[116]
- Epilim Syrup Sanofi[116]
- Epilim Tablets Sanofi[116]
- Sodium Valproate Sandoz Tablets Sanofi
- Valpro Tablets Alphapharm
- Valproate Winthrop Tablets Sanofi
- Valprease tablets Sigma
New Zealand
- Epilim by Sanofi-Aventis
All the above formulations are Pharmac-subsidised.[117]
UK
- Depakote Tablets (as in USA)
- Tablets – Orlept by Wockhardt and Epilim by Sanofi
- Oral solution – Orlept Sugar Free by Wockhardt and Epilim by Sanofi
- Syrup – Epilim by Sanofi-Aventis
- Intravenous injection – Epilim Intravenous by Sanofi
- Extended release tablets – Epilim Chrono by Sanofi is a combination of sodium valproate and valproic acid in a 2.3:1 ratio.
- Enteric-coated tablets – Epilim EC200 by Sanofi is a 200-mg sodium valproate enteric-coated tablet.
UK only
- Capsules – Episenta prolonged release by Beacon
- Sachets – Episenta prolonged release by Beacon
- Intravenous solution for injection – Episenta solution for injection by Beacon
Germany, Switzerland, Norway, Finland, Sweden
- Tablets – Orfiril by Desitin Pharmaceuticals
- Intravenous injection – Orfiril IV by Desitin Pharmaceuticals
South Africa
- Syrup – Convulex by Byk Madaus[118]
- Tablets – Epilim by Sanofi-synthelabo
Malaysia
- Tablets – Epilim by Sanofi-Aventis
Romania
- Companies are SANOFI-AVENTIS FRANCE, GEROT PHARMAZEUTIKA GMBH and DESITIN ARZNEIMITTEL GMBH
- Types are Syrup, Extended release mini tablets, Gastric resistant coated tablets, Gastric resistant soft capsules, Extended release capsules, Extended release tablets and Extended release coated tablets
Canada
- Intravenous injection – Epival or Epiject by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories its generic formulations include Apo-Valproic and ratio-Valproic.
Japan
- Tablets – Depakene by Kyowa Hakko Kirin
- Extended release tablets – Depakene-R by Kyowa Hakko Kogyo and Selenica-R by Kowa
- Syrup – Depakene by Kyowa Hakko Kogyo
Europe
In much of Europe, Dépakine and Depakine Chrono (tablets) are equivalent to Epilim and Epilim Chrono above.
Taiwan
- Tablets (white round tablet) – Depakine (Chinese: 帝拔癲; pinyin: di-ba-dian) by Sanofi Winthrop Industrie (France)
Iran
- Tablets – Epival 200 (enteric coated tablet) and Epival 500 (extended release tablet) by Iran Najo
- Slow release tablets – Depakine Chrono by Sanofi Winthrop Industrie (France)
Israel
Depalept and Depalept Chrono (extended release tablets) are equivalent to Epilim and Epilim Chrono above. Manufactured and distributed by Sanofi-Aventis.
India, Russia and CIS countries
- Valparin Chrono by Torrent Pharmaceuticals India
- Valprol CR by Intas Pharmaceutical (India)
- Encorate Chrono by Sun Pharmaceutical (India)
- Serven Chrono by Leeven APL Biotech (India)
Brand names of valproate semisodium
- Brazil – Depakote by Abbott Laboratories and Torval CR by Torrent do Brasil
- Canada – Epival by Abbott Laboratories
- Mexico – Epival and Epival ER (extended release) by Abbott Laboratories
- United Kingdom – Depakote (for psychiatric conditions) and Epilim (for epilepsy) by Sanofi-Aventis and generics
- United States – Depakote and Depakote ER (extended release) by Abbott Laboratories and generics[22]
- India – Valance and Valance OD by Abbott Healthcare Pvt Ltd, Divalid ER by Linux laboratories Pvt Ltd, Valex ER by Sigmund Promedica, Dicorate by Sun Pharma
- Germany – Ergenyl Chrono by Sanofi-Aventis and generics
- Chile – Valcote and Valcote ER by Abbott Laboratories
- France and other European countries — Depakote
- Peru – Divalprax by AC Farma Laboratories
- China – Diprate OD
References
- ^ Jump up to:a b c d “Depakene, Stavzor (valproic acid) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Archived from the original on 21 February 2014. Retrieved 13 February 2014.
- ^ Jump up to:a b c d e f g h i j k l “Valproic Acid”. The American Society of Health-System Pharmacists. Archived from the original on 2017-07-31. Retrieved Oct 23, 2015.
- ^ “Valproate banned without the pregnancy prevention programme”. GOV.UK. Retrieved 26 April 2018.
- ^ Owens MJ, Nemeroff CB (2003). “Pharmacology of valproate”. Psychopharmacol Bull. 37 Suppl 2: 17–24. PMID 14624230.
- ^ Jump up to:a b c Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE (April 2013). “Valproic acid pathway: pharmacokinetics and pharmacodynamics”. Pharmacogenet. Genomics. 23 (4): 236–241. doi:10.1097/FPC.0b013e32835ea0b2. PMC 3696515. PMID 23407051.
- ^ “Valproic acid”. DrugBank. University of Alberta. 29 July 2017. Archived from the original on 31 July 2017. Retrieved 30 July2017.
- ^ Scott, D.F. (1993). The history of epileptic therapy : an account of how medication was developed (1. publ. ed.). Carnforth u.a.: Parthenon Publ. Group. p. 131. ISBN 9781850703914.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Divalproex Sodium – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Brayfield, Alison (ed.). Martindale: The Complete Drug Reference. London: Pharmaceutical Press. Retrieved March 3,2018.
- ^ Jump up to:a b c Rossi, S, ed. (2013). Australian Medicines Handbook(2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Löscher W (2002). “Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy”. CNS Drugs. 16 (10): 669–694. doi:10.2165/00023210-200216100-00003. PMID 12269861. S2CID 67999301.
- ^ Olsen KB, Taubøll E, Gjerstad L (2007). “Valproate is an effective, well-tolerated drug for treatment of status epilepticus/serial attacks in adults”. Acta Neurol. Scand. Suppl. 187: 51–4. doi:10.1111/j.1600-0404.2007.00847.x. PMID 17419829. S2CID 11159645.
- ^ Kwan SY (2010). “The role of intravenous valproate in convulsive status epilepticus in the future” (PDF). Acta Neurol Taiwan. 19(2): 78–81. PMID 20830628.[permanent dead link]
- ^ “Valproate Information”. Fda.gov. Archived from the original on 2015-05-03. Retrieved 2015-04-24.
- ^ Jochim, Janina; Rifkin-Zybutz, Raphael; Geddes, John; Cipriani, Andrea (7 October 2019). “Valproate for acute mania”. Cochrane Database of Systematic Reviews. 10: CD004052. doi:10.1002/14651858.CD004052.pub2. PMC 6797024. PMID 31621892.
- ^ Jump up to:a b Wang, Y; Xia, J; Helfer, B (2016). “Valproate for schizophrenia”. Cochrane Database of Systematic Reviews. 11: CD004028.pub4. doi:10.1002/14651858.CD004028.pub4. PMC 6734130. PMID 27884042. Archived from the originalon 2017-07-29. Retrieved 2017-07-27.
- ^ Pirritano D, Plastino M, Bosco D, Gallelli L, Siniscalchi A, De Sarro G (2014). “Gambling disorder during dopamine replacement treatment in Parkinson’s disease: a comprehensive review”. Biomed Res Int. 2014: 1–9. doi:10.1155/2014/728038. PMC 4119624. PMID 25114917.
- ^ Connolly B, Fox SH (2014). “Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease”. Neurotherapeutics. 11 (1): 78–91. doi:10.1007/s13311-013-0238-x. PMC 3899484. PMID 24288035.
- ^ Averbeck BB, O’Sullivan SS, Djamshidian A (2014). “Impulsive and compulsive behaviors in Parkinson’s disease”. Annu Rev Clin Psychol. 10: 553–80. doi:10.1146/annurev-clinpsy-032813-153705. PMC 4197852. PMID 24313567.
- ^ Jump up to:a b c d e f g h i “Depakote- divalproex sodium tablet, delayed release”. Archived from the original on 5 March 2016. Retrieved 10 November 2015.
- ^ Činčárová L, Zdráhal Z, Fajkus J (2013). “New perspectives of valproic acid in clinical practice”. Expert Opin Investig Drugs. 22(12): 1535–1547. doi:10.1517/13543784.2013.853037. PMID 24160174. S2CID 11855893.
- ^ Jump up to:a b c d e f g h i j k l m n o “Valpro sodium valproate” (PDF). TGA eBusiness Services. Alphapharm Pty Limited. 16 December 2013. Retrieved 14 February 2014.
- ^ “Depakote 250mg Tablets – Summary of Product Characteristics”. electronic Medicines Compendium. Sanofi. 28 November 2013. Archived from the original on 1 February 2014. Retrieved 18 January 2014.
- ^ Wu S, Legido A, De Luca F (2004). “Effects of valproic acid on longitudinal bone growth”. J Child Neurol. 19 (1): 26–30. doi:10.1177/088307380401900105011. PMID 15032379. S2CID 19827846.
- ^ Robinson PB, Harvey W, Belal MS (1988). “Inhibition of cartilage growth by the anticonvulsant drugs diphenylhydantoin and sodium valproate”. Br J Exp Pathol. 69 (1): 17–22. PMC 2013195. PMID 3126792.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ “Could Depakote cause Mydriasis”. eHealthMe.com. 2014-11-18. Archived from the original on 2014-12-05. Retrieved 2015-04-24.
- ^ Bilo, Leonilda; Meo, Roberta (October 2008). “Polycystic ovary syndrome in women using valproate: a review”. Gynecological Endocrinology. 24 (10): 562–70. doi:10.1080/09513590802288259. PMID 19012099. S2CID 36426338.
- ^ Chukwu, J; Delanty, N; Webb, D; Cavalleri, GL (January 2014). “Weight change, genetics and antiepileptic drugs”. Expert Review of Clinical Pharmacology. 7 (1): 43–51. doi:10.1586/17512433.2014.857599. PMID 24308788. S2CID 33444886.
- ^ New evidence in France of harm from epilepsy drug valproateArchived 2017-04-21 at the Wayback Machine BBC, 2017
- ^ Koch S, Göpfert-Geyer I, Jäger-Roman E, et al. (February 1983). “[Anti-epileptic agents during pregnancy. A prospective study on the course of pregnancy, malformations and child development]”. Dtsch. Med. Wochenschr. (in German). 108 (7): 250–7. doi:10.1055/s-2008-1069536. PMID 6402356.
- ^ Moore SJ, Turnpenny P, Quinn A, et al. (July 2000). “A clinical study of 57 children with fetal anticonvulsant syndromes”. J. Med. Genet. 37 (7): 489–97. doi:10.1136/jmg.37.7.489. PMC 1734633. PMID 10882750.
- ^ Ornoy A (2009). “Valproic acid in pregnancy: how much are we endangering the embryo and fetus?”. Reprod. Toxicol. 28 (1): 1–10. doi:10.1016/j.reprotox.2009.02.014. PMID 19490988.
- ^ Kulkarni ML, Zaheeruddin M, Shenoy N, Vani HN (2006). “Fetal valproate syndrome”. Indian J Pediatr. 73 (10): 937–939. doi:10.1007/bf02859291. PMID 17090909. S2CID 38371596.
- ^ Adab N, Kini U, Vinten J, et al. (November 2004). “The longer term outcome of children born to mothers with epilepsy”. J. Neurol. Neurosurg. Psychiatry. 75 (11): 1575–83. doi:10.1136/jnnp.2003.029132. PMC 1738809. PMID 15491979.
This argues that the fetal valproate syndrome constitutes a real clinical entity that includes developmental delay and cognitive impairments, but that some children might exhibit some developmental delay without marked dysmorphism.
- ^ Umur AS, Selcuki M, Bursali A, Umur N, Kara B, Vatansever HS, Duransoy YK (2012). “Simultaneous folate intake may prevent adverse effect of valproic acid on neurulating nervous system”. Childs Nerv Syst. 28 (5): 729–737. doi:10.1007/s00381-011-1673-9. PMID 22246336. S2CID 20344828.
- ^ Cassels, Caroline (December 8, 2006). “NEAD: In Utero Exposure To Valproate Linked to Poor Cognitive Outcomes in Kids”. Medscape. Archived from the original on July 31, 2011. Retrieved 2007-05-23.
- ^ Meador KJ, Baker GA, Finnell RH, Kalayjian LA, Liporace JD, Loring DW, Mawer G, Pennell PB, Smith JC, Wolff MC (2006). “In utero antiepileptic drug exposure: fetal death and malformations”. Neurology. 67 (3): 407–412. doi:10.1212/01.wnl.0000227919.81208.b2. PMC 1986655. PMID 16894099.
- ^ Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M (2013). “Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism”. JAMA. 309 (16): 1696–1703. doi:10.1001/jama.2013.2270. PMC 4511955. PMID 23613074.
- ^ Rasalam AD, Hailey H, Williams JH, et al. (August 2005). “Characteristics of fetal anticonvulsant syndrome associated autistic disorder”. Dev Med Child Neurol. 47 (8): 551–5. doi:10.1017/S0012162205001076. PMID 16108456.
- ^ Autism Society of America: About Autism Archived 2011-01-10 at the Wayback Machine
- ^ I.Q. Harmed by Epilepsy Drug in Utero Archived 2015-12-29 at the Wayback Machine By RONI CARYN RABIN, New York Times, April 15, 2009
- ^ Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW (2009). “Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs”. N. Engl. J. Med. 360 (16): 1597–1605. doi:10.1056/NEJMoa0803531. PMC 2737185. PMID 19369666.
- ^ Valproate Products: Drug Safety Communication – Risk of Impaired Cognitive Development in Children Exposed In Utero (During Pregnancy) Archived 2011-09-02 at the Wayback Machine. FDA. June 2011
- ^ Luat, AF (20 September 2007). “Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy”. Epileptic Disorders. 9(3): 332–6. doi:10.1684/epd.2007.0119 (inactive 31 May 2021). PMID 17884759.
- ^ Ouvrier, RA (July 1988). “Benign paroxysmal tonic upgaze of childhood”. Journal of Child Neurology. 3 (3): 177–80. doi:10.1177/088307388800300305. PMID 3209843. S2CID 38127378.
- ^ Valproate Not To Be Used for Migraine During Pregnancy, FDA Warns Archived 2013-07-09 at the Wayback Machine
- ^ “New measures to avoid valproate exposure in pregnancy endorsed”. European Medicines Agency. 31 May 2018.
- ^ Jump up to:a b c d Bentley, Suzanne (Dec 11, 2013). “Valproic Acid Level”. Medscape. Archived from the original on 2015-05-04. Retrieved 2015-06-05.
- ^ Jump up to:a b c d “Free Valproic Acid Assay (Reference – 2013.03.006) Notice of Assessment” (PDF). Canadian Agency for Drugs and Technologies in Health (CADTH) with INESSS’s permission. April 2014. Archived from the original (PDF) on 2016-03-03. Retrieved 2015-06-05.
- ^ Rissardo, Jamir Pitton; Caprara, Ana Letícia Fornari; Durante, Ícaro (2021). “Valproate-associated Movement Disorder: A Literature Review”. Prague Medical Report. 122 (3): 140–180. doi:10.14712/23362936.2021.14. ISSN 1214-6994.
- ^ Sztajnkrycer MD (2002). “Valproic acid toxicity: overview and management”. J. Toxicol. Clin. Toxicol. 40 (6): 789–801. doi:10.1081/CLT-120014645. PMID 12475192. S2CID 23031095.
- ^ Patsalos PN, Berry DJ (2013). “Therapeutic drug monitoring of antiepileptic drugs by use of saliva”. Ther Drug Monit. 35 (1): 4–29. doi:10.1097/FTD.0b013e31827c11e7. PMID 23288091. S2CID 15338188.
- ^ Thanacoody RH (2009). “Extracorporeal elimination in acute valproic acid poisoning”. Clin Toxicol. 47 (7): 609–616. doi:10.1080/15563650903167772. PMID 19656009. S2CID 13592043.
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1622–1626.
- ^ Jump up to:a b Lheureux PE, Penaloza A, Zahir S, Gris M (2005). “Science review: carnitine in the treatment of valproic acid-induced toxicity – what is the evidence?”. Crit Care. 9 (5): 431–440. doi:10.1186/cc3742. PMC 1297603. PMID 16277730.
- ^ Mock CM, Schwetschenau KH (2012). “Levocarnitine for valproic-acid-induced hyperammonemic encephalopathy”. Am J Health Syst Pharm. 69 (1): 35–39. doi:10.2146/ajhp110049. PMID 22180549.
- ^ Matsuoka M, Igisu H (1993). “Comparison of the effects of L-carnitine, D-carnitine and acetyl-L-carnitine on the neurotoxicity of ammonia”. Biochem. Pharmacol. 46 (1): 159–164. doi:10.1016/0006-2952(93)90360-9. PMID 8347126.
- ^ Herzog, Andrew; Farina, Erin (June 9, 2005). “Serum Valproate Levels with Oral Contraceptive Use”. Epilepsia. 46 (6): 970–971. doi:10.1111/j.1528-1167.2005.00605.x. PMID 15946343. S2CID 7696039.
- ^ Kay, Hee Yeon; Greene, Derek L.; Kang, Seungwoo; Kosenko, Anastasia; Hoshi, Naoto (2015-10-01). “M-current preservation contributes to anticonvulsant effects of valproic acid”. The Journal of Clinical Investigation. 125 (10): 3904–3914. doi:10.1172/JCI79727. ISSN 0021-9738. PMC 4607138. PMID 26348896.
- ^ Chang P, Walker MC, Williams RS (2014). “Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid”. Neurobiol. Dis. 62: 296–306. doi:10.1016/j.nbd.2013.10.017. PMC 3898270. PMID 24148856.
- ^ Kostrouchová M, Kostrouch Z, Kostrouchová M (2007). “Valproic acid, a molecular lead to multiple regulatory pathways” (PDF). Folia Biol. (Praha). 53 (2): 37–49. PMID 17448293. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Jump up to:a b Chiu CT, Wang Z, Hunsberger JG, Chuang DM (2013). “Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder”. Pharmacol. Rev. 65 (1): 105–142. doi:10.1124/pr.111.005512. PMC 3565922. PMID 23300133.
- ^ Jump up to:a b Death AK, McGrath KC, Handelsman DJ (2005). “Valproate is an anti-androgen and anti-progestin” (PDF). Steroids. 70 (14): 946–53. doi:10.1016/j.steroids.2005.07.003. hdl:10453/16875. PMID 16165177. S2CID 25958985.
- ^ Jump up to:a b Wyllie E, Cascino GD, Gidal BE, Goodkin HP (17 February 2012). Wyllie’s Treatment of Epilepsy: Principles and Practice. Lippincott Williams & Wilkins. pp. 288–. ISBN 978-1-4511-5348-4. Archived from the original on 6 June 2014.
- ^ Uchida, Hiroshi; Maruyama, Tetsuo; Arase, Toru; Ono, Masanori; Nagashima, Takashi; Masuda, Hirotaka; Asada, Hironori; Yoshimura, Yasunori (2005). “Histone acetylation in reproductive organs: Significance of histone deacetylase inhibitors in gene transcription”. Reproductive Medicine and Biology. 4 (2): 115–122. doi:10.1111/j.1447-0578.2005.00101.x. ISSN 1445-5781. PMC 5891791. PMID 29662388.
- ^ Jump up to:a b Isojärvi, Jouko I T; Taubøll, Erik; Herzog, Andrew G (2005). “Effect of Antiepileptic Drugs on Reproductive Endocrine Function in Individuals with Epilepsy”. CNS Drugs. 19 (3): 207–223. doi:10.2165/00023210-200519030-00003. ISSN 1172-7047. PMID 15740176. S2CID 9893959.
- ^ Jump up to:a b c d e f Haberfeld H, ed. (2021). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Depakine chrono retard 300 mg Filmtabletten.
- ^ Kumar, S.; Wong, H.; Yeung, S. A.; Riggs, K. W.; Abbott, F. S.; Rurak, D. W. (2000). “Disposition of valproic acid in maternal, fetal, and newborn sheep. II: Metabolism and renal elimination”. Drug Metabolism and Disposition: The Biological Fate of Chemicals. 28(7): 857–864. PMID 10859160.
- ^ Schneemann; Young, Lloyd; Koda-Kimble, Mary Anne, eds. (2001). Angewandte Arzneimitteltherapie (in German). Springer. pp. 28–29. ISBN 3540413561.
- ^ Jump up to:a b Valproate FDA Professional Drug Information. Accessed 2021-08-06.
- ^ Jump up to:a b c d e f g h “Pharmacology”. Valproic Acid. DrugBank. University of Alberta. 31 August 2017.
- ^ Burton BS (1882). “On the propyl derivatives and decomposition products of ethylacetoacetate”. Am. Chem. J. 3: 385–395.
- ^ Meunier H, Carraz G, Neunier Y, Eymard P, Aimard M (1963). “[Pharmacodynamic properties of N-dipropylacetic acid]” [Pharmacodynamic properties of N-dipropylacetic acid]. Thérapie (in French). 18: 435–438. PMID 13935231.
- ^ Perucca E (2002). “Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience”. CNS Drugs. 16 (10): 695–714. doi:10.2165/00023210-200216100-00004. PMID 12269862. S2CID 803106.
- ^ Henry TR (2003). “The history of valproate in clinical neuroscience”. Psychopharmacol Bull. 37 Suppl 2: 5–16. PMID 14624229.
- ^ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ Rimmer EM, Richens A (May–June 1985). “An update on sodium valproate”. Pharmacotherapy. 5 (3): 171–84. doi:10.1002/j.1875-9114.1985.tb03413.x. PMID 3927267. S2CID 7700266.
- ^ Glauser, Tracy A; Cnaan, Avital; Shinnar, Shlomo; Hirtz, Deborah G; Dlugos, Dennis; Masur, David; Clark, Peggy O; Capparelli, Edmund V; Adamson, Peter C (2010). “Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy”. New England Journal of Medicine. 362 (9): 790–9. doi:10.1056/NEJMoa0902014. PMC 2924476. PMID 20200383.
- ^ Jiang, Mei (6 April 2015). “Co-Administration of Valproic Acid and Lamotrigine in the Treatment of Refractory Epilepsy (P1.238)”. Neurology. 84 (14 Supplement): P1.238 – via http://www.neurology.org.
- ^ Berendsen, S.; Kroonen, J.; Seute, T.; Snijders, T.; Broekman, M. L. D.; Spliet, W. G. M.; Willems, M.; Artesi, M.; Bours, V.; Robe, P. A. (1 September 2014). “O9.06 * Prognostic Relevance and Oncogenic Correlates of Epilepsy in Glioblastoma Patients”. Neuro-Oncology. 16 (suppl_2): ii21. doi:10.1093/neuonc/nou174.77. PMC 4185847.
- ^ Vasudev K, Mead A, Macritchie K, Young AH (2012). “Valproate in acute mania: is our practice evidence based?”. Int J Health Care Qual Assur. 25 (1): 41–52. doi:10.1108/09526861211192395. PMID 22455007.
- ^ Bond DJ, Lam RW, Yatham LN (2010). “Divalproex sodium versus placebo in the treatment of acute bipolar depression: a systematic review and meta-analysis”. J Affect Disord. 124 (3): 228–334. doi:10.1016/j.jad.2009.11.008. PMID 20044142.
- ^ Haddad PM, Das A, Ashfaq M, Wieck A (2009). “A review of valproate in psychiatric practice”. Expert Opin Drug Metab Toxicol. 5(5): 539–51. doi:10.1517/17425250902911455. PMID 19409030. S2CID 74028228.
- ^ Frazee LA, Foraker KC (2008). “Use of intravenous valproic acid for acute migraine”. Ann Pharmacother. 42 (3): 403–7. doi:10.1345/aph.1K531. PMID 18303140. S2CID 207263036.
- ^ Wang, Yijun; Xia, Jun; Helfer, Bartosz; Li, Chunbo; Leucht, Stefan (2016). “Valproate for schizophrenia”. The Cochrane Database of Systematic Reviews. 11: CD004028. doi:10.1002/14651858.CD004028.pub4. ISSN 1469-493X. PMC 6734130. PMID 27884042.
- ^ Jump up to:a b Baillon, Sarah F.; Narayana, Usha; Luxenberg, Jay S.; Clifton, Andrew V. (2018-10-05). “Valproate preparations for agitation in dementia”. The Cochrane Database of Systematic Reviews. 2018(10): CD003945. doi:10.1002/14651858.CD003945.pub4. ISSN 1469-493X. PMC 6516950. PMID 30293233.
- ^ Gill D, Derry S, Wiffen PJ, Moore RA (2011). “Valproic acid and sodium valproate for neuropathic pain and fibromyalgia in adults”. Cochrane Database Syst Rev (10): CD009183. doi:10.1002/14651858.CD009183.pub2. PMC 6540387. PMID 21975791.
- ^ Aliyev ZN, Aliyev NA (July–August 2008). “Valproate treatment of acute alcohol hallucinosis: a double-blind, placebo-controlled study”. Alcohol Alcohol. 43 (4): 456–459. doi:10.1093/alcalc/agn043. PMID 18495806.
- ^ Jacobson PL, Messenheimer JA, Farmer TW (1981). “Treatment of intractable hiccups with valproic acid”. Neurology. 31 (11): 1458–60. doi:10.1212/WNL.31.11.1458. PMID 6796902. S2CID 1578958.
- ^ Sotaniemi K (1982). “Valproic acid in the treatment of nonepileptic myoclonus”. Arch. Neurol. 39 (7): 448–9. doi:10.1001/archneur.1982.00510190066025. PMID 6808975.
- ^ Wheeler SD (July–August 1998). “Significance of migrainous features in cluster headache: divalproex responsiveness”. Headache. 38 (7): 547–51. doi:10.1046/j.1526-4610.1998.3807547.x. PMID 15613172. S2CID 27948702.
- ^ Siemes H, Spohr HL, Michael T, Nau H (September–October 1988). “Therapy of infantile spasms with valproate: results of a prospective study”. Epilepsia. 29 (5): 553–60. doi:10.1111/j.1528-1157.1988.tb03760.x. PMID 2842127. S2CID 23789333.
- ^ Smith SM (2005). “Valproic acid and HIV-1 latency: beyond the sound bite” (PDF). Retrovirology. 2 (1): 56. doi:10.1186/1742-4690-2-56. PMC 1242254. PMID 16168066. Archived from the original (PDF) on 2015-09-24. Retrieved 2014-02-13.
- ^ Routy JP, Tremblay CL, Angel JB, Trottier B, Rouleau D, Baril JG, Harris M, Trottier S, Singer J, Chomont N, Sékaly RP, Boulassel MR (2012). “Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study”. HIV Med. 13 (5): 291–6. doi:10.1111/j.1468-1293.2011.00975.x. PMID 22276680. S2CID 27571864.
- ^ Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, Eron J, Cohen M, Margolis DM (2010). “Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection”. PLOS ONE. 5 (2): e9390. Bibcode:2010PLoSO…5.9390A. doi:10.1371/journal.pone.0009390. PMC 2826423. PMID 20186346.
- ^ Jump up to:a b Hardy JR, Rees EA, Gwilliam B, Ling J, Broadley K, A’Hern R (2001). “A phase II study to establish the efficacy and toxicity of sodium valproate in patients with cancer-related neuropathic pain” (PDF). J Pain Symptom Manage. 21 (3): 204–9. doi:10.1016/S0885-3924(00)00266-9. PMID 11239739.[permanent dead link]
- ^ Candelaria M, Herrera A, Labardini J, González-Fierro A, Trejo-Becerril C, Taja-Chayeb L, Pérez-Cárdenas E, de la Cruz-Hernández E, Arias-Bofill D, Vidal S, Cervera E, Dueñas-Gonzalez A (2011). “Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial”. Ann. Hematol. 90 (4): 379–387. doi:10.1007/s00277-010-1090-2. PMID 20922525. S2CID 13437134.
- ^ Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG (2005). “Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia”. Cancer. 104 (12): 2717–2725. doi:10.1002/cncr.21589. PMID 16294345. S2CID 1802132.
- ^ Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N (2006). “The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia”. Cancer. 106 (1): 112–119. doi:10.1002/cncr.21552. PMID 16323176. S2CID 43747497.
- ^ Fredly H, Gjertsen BT, Bruserud O (2013). “Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents” (PDF). Clin Epigenetics. 5 (1): 12. doi:10.1186/1868-7083-5-12. PMC 3733883. PMID 23898968. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, Perez-Cardenas E, Taja-Chayeb L, Arias-Bofill D, Candelaria M, Vidal S, Dueñas-González A (2011). “A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results”. Med. Oncol. 28 Suppl 1: S540–6. doi:10.1007/s12032-010-9700-3. PMID 20931299. S2CID 207372333.
- ^ Rocca A, Minucci S, Tosti G, Croci D, Contegno F, Ballarini M, Nolè F, Munzone E, Salmaggi A, Goldhirsch A, Pelicci PG, Testori A (2009). “A phase I-II study of the histone deacetylase inhibitor valproic acid plus chemoimmunotherapy in patients with advanced melanoma”. Br. J. Cancer. 100 (1): 28–36. doi:10.1038/sj.bjc.6604817. PMC 2634690. PMID 19127265.
- ^ Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, Daud A, Neuger A, Minton S, Sullivan D (2009). “Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC” (PDF). Clin. Cancer Res. 15 (7): 2488–96. doi:10.1158/1078-0432.CCR-08-1930. PMID 19318486. S2CID 3230087.
- ^ Hicks CW, Pandya MM, Itin I, Fernandez HH (2011). “Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease”. Parkinsonism Relat. Disord. 17 (5): 379–81. doi:10.1016/j.parkreldis.2011.03.003. PMID 21459656.
- ^ Sriram A, Ward HE, Hassan A, Iyer S, Foote KD, Rodriguez RL, McFarland NR, Okun MS (2013). “Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson’s disease”. J. Neurol. 260 (2): 521–7. doi:10.1007/s00415-012-6669-1. PMID 23007193. S2CID 21544457.
- ^ Aizenman, N. C. (7 May 2012). “Abbott Laboratories to pay $1.6 billion over illegal marketing of Depakote”. Washington Post. Retrieved 27 June 2018.
- ^ Schmidt, Michael; Thomas, Katie (8 May 2012). “Abbott settles marketing lawsuit”. New York Times. Retrieved 27 June 2018.
- ^ Gervain J, Vines BW, Chen LM, Seo RJ, Hensch TK, Werker JF, Young AH (2013). “Valproate reopens critical-period learning of absolute pitch”. Frontiers in Systems Neuroscience. 7: 102. doi:10.3389/fnsys.2013.00102. PMC 3848041. PMID 24348349.
- ^ Thomson, Helen. “Learning drugs reawaken grown-up brain’s inner child”. New Scientist. New Scientist Ltd. Retrieved 8 May2021.
- ^ David Taylor; Carol Paton; Shitij Kapur (2009). The Maudsley Prescribing Guidelines, Tenth Edition (10, revised ed.). CRC Press. p. 124. ISBN 9780203092835.
- ^ “Depakene- valproic acid capsule, liquid filled”. DailyMed. 19 September 2019. Retrieved 14 April 2020.
- ^ Jump up to:a b c d “Australian product information epilim (sodium valproate) crushable tablets, enteric-coated tablets, syrup, liquid” (PDF). TGA eBS. 15 April 2020. Retrieved 15 April 2020.
- ^ “Sodium valproate — Pharmaceutical Schedule”. Pharmaceutical Management Agency. Archived from the originalon 4 March 2016. Retrieved 22 June 2014.
- ^ South African Electronic Package Inserts: Convulex
External links
- “Valproic acid”. Drug Information Portal. U.S. National Library of Medicine.
- “Valproate sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Divalproex sodium”. Drug Information Portal. U.S. National Library of Medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCA1144558A *1979-10-221983-04-12Francis E. FischerProcess for making sodium hydrogen divalproateUS4988731A *1979-08-201991-01-29Abbott LaboratoriesSodium hydrogen divalproate oligomerUS5212326A *1979-08-201993-05-18Abbott LaboratoriesSodium hydrogen divalproate oligomerWO2001032595A1 *1999-11-022001-05-10Cilag AgMethod for producing compounds of the valproinic acidUS20030018215A1 *2001-06-292003-01-23Procos S.P.A.Process for the preparation of sodium divalproatePublication numberPriority datePublication dateAssigneeTitleUS20110040122A1 *2009-08-112011-02-17Sci Pharmtech, Inc.Method for preparing metal salt of valproic acidCN102942467A *2012-10-172013-02-27山东方明药业集团股份有限公司Preparation method of divalproex sodiumCN103183600A *2011-12-302013-07-03北大方正集团有限公司Method for preparing divalproex sodium
////// divalproex, Anticonvulsant, Antimigraine, Antimanic, valproic acid, sodium valproate
CCCC(CCC)C(O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
MIDAZOLAM


MIDAZOLAM
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
59467-70-8 CAS NO OF FREE BASE
59467-94-6 MALEATE, Launched – 1982, Roche (Originator)
59467-96-8 (HCl)
A short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is used in dentistry, cardiac surgery, endoscopic procedures, as preanesthetic medication, and as an adjunct to local anesthesia. The short duration and cardiorespiratory stability makes it useful in poor-risk, elderly, and cardiac patients. It is water-soluble at pH less than 4 and lipid-soluble at physiological pH.
Midazolam (/mɪˈdæzəlæm/, marketed in English-speaking countries and Mexico under the trade names Dormicum, Hypnovel, andVersed,) is a short-acting drug in the benzodiazepine class developed by Hoffmann-La Roche in the 1970s. The drug is used for treatment of acute seizures, moderate to severe insomnia, and for inducing sedation and amnesia before medical procedures. It possesses profoundly potentanxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.[6][7][8] Midazolam has a fast recovery time and is the most commonly used benzodiazepine as a premedication for sedation; less commonly it is used for induction and maintenance of anesthesia.Flumazenil, a benzodiazepine antagonist drug, can be used to treat an overdose of midazolam, as well as to reverse sedation.[7] However, flumazenil can trigger seizures in mixed overdoses and in benzodiazepine-dependent individuals, so is not used in most cases.[9][10]
midazolam
Administration of midazolam by the intranasal or the buccal route (absorption via the gums and cheek) as an alternative to rectally administereddiazepam is becoming increasingly popular for the emergency treatment of seizures in children. Midazolam is also used for endoscopyprocedural sedation and sedation in intensive care. The anterograde amnesia property of midazolam is useful for premedication before surgery to inhibit unpleasant memories. Midazolam, like many other benzodiazepines, has a rapid onset of action, high effectiveness and low toxicity level. Drawbacks of midazolam include drug interactions, tolerance, and withdrawal syndrome, as well as adverse events including cognitive impairment and sedation. Paradoxical effects occasionally occur, most commonly in children and the elderly, particularly after intravenous administration. The drug has also recently been hastily introduced for use in executions in the USA in combination with other drugs.
Midazolam is a short-acting benzodiazepine in adults with an elimination half-life of one to four hours; however, in the elderly, as well as young children and adolescents, the elimination half-life is longer. Midazolam is metabolised into an active metabolite alpha1-hydroxymidazolam. Age related deficits, renal and liver status affect the pharmacokinetic factors of midazolam as well as its active metabolite. However, the active metabolite of midazolam is minor and contributes to only 10 percent of biological activity of midazolam. Midazolam is poorly absorbed orally with only 50 percent of the drug reaching the bloodstream. Midazolam is metabolised by cytochrome P450 (CYP) enzymes and by glucuronide conjugation. The therapeutic as well as adverse effects of midazolam are due to its effects on the GABAA receptors; midazolam does not activate GABAA receptors directly but, as with other benzodiazepines, it enhances the effect of the neurotransmitter GABA on the GABAA receptors (↑ frequency of Cl− channel opening) resulting in neural inhibition. Almost all of the properties can be explained by the actions of benzodiazepines on GABAA receptors. This results in the following pharmacological properties being produced: sedation, hypnotic, anxiolytic, anterograde amnesia, muscle relaxation and anti-convulsant.Midazolam maleate is a benzodiazepine that is commercialized by Astellas Pharma and Roche as an intravenous or intramuscular injection for the long-term sedation of mechanically ventilated patients under intensive care. The drug is also available in a tablet formulation, and is currently distributed in various markets, including Germany, Japan, Switzerland and the U.K. In March 2002, two lots of a syrup formulation were recalled in the U.S. due to the potential presence of a crystalline precipitate of an insoluble complex of midazolam and saccharin. Subsequently, the injection and syrup formulations of the product were both withdrawn from the U.S. market. In 2010, a Pediatric Use Marketing Authorization (PUMA) was filed for approval in the E.U. by ViroPharma for the treatment of prolonged, acute, convulsive seizures in infants, toddlers, children and adolescents, from 3 months to less than 18 years. In 2011, a positive opinion was assigned to the PUMA and final approval was assigned in June 2011. The product was launched in the U.S. in November 2011. This product was filed for approval in Japan in 2013 by Astellas Pharma for the conscious sedation in dentistry and dental surgery. In the same year the product was approved for this indication.
In terms of clinical development, a nasal formulation of the drug is in phase III clinical trials at Upsher-Smith for rescue treatment of seizures in patients on stable anti-epileptic drug regimens who require control of intermittent bouts of increased seizure activity (seizure clusters). The Hopitaux de Paris had been developing a sublingual tablet formulation of midazolam to be used in combination with morphine for the treatment of pain in children following bone fractures; however, no recent development has been reported for this indication. NovaDel Pharma had been developing the compound preclinically for the treatment of generalized anxiety, however no recent developments have been reported.
Midazolam achieves its therapeutic effect through interaction with the gamma-aminobutyric acid benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models. GABA acts at inhibitory synapses in the brain by binding to specific transmembrane receptors in the plasma membrane of both pre- and post-synaptic neurons, opening ion channels and bringing about a hyperpolarization via either chloride or potassium ion flow.
In 2008, fast track designation was assigned to midazolam maleate in the U.S. for the treatment of seizure disorders.
In 2009, Orphan Drug Designation was received in the U.S. by for the treatment of seizure disorders in patients who require control of intermittent bouts of increased seizure activity (e.g. acute repetitive seizures, seizure clusters). This designation was assigned in the U.S. for the treatment of nerve agent-induced seizures.
In 2010, midazolam maleate was licensed to Upsher-Smith by Ikano Therapeutics for the treatment of acute repetitive seizure in patients with epilepsy. However, in 2010, Ikano closed and dissolved its business. Previously, Ikano had transferred to Upsher-Smith ownership of it nasal midazolam maleate program.
Midazolam is among about 35 benzodiazepines which are currently used medically, and was synthesised in 1975 by Walser and Fryer at Hoffmann-LaRoche, Inc in the United States.Owing to its water solubility, it was found to be less likely to cause thrombophlebitis than similar drugs.The anticonvulsant properties of midazolam were studied in the late 1970s, but not until the 1990s did it emerge as an effective treatment for convulsive status epilepticus. As of 2010, it is the most commonly used benzodiazepine in anesthetic medicine. In acute medicine, midazolam has become more popular than other benzodiazepines, such as lorazepam and diazepam, because it is shorter lasting, is more potent, and causes less pain at the injection site.Midazolam is also becoming increasingly popular in veterinary medicine due to its water solubility.
Midazolam is a water-soluble benzodiazepine available as a sterile, nonpyrogenic parenteral dosage form for intravenous or intramuscular injection. Each mL contains midazolam hydrochloride equivalent to 1 mg or 5 mg midazolam compounded with 0.8% sodium chloride and 0.01% edetate disodium with 1% benzyl alcohol as preservative, and sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 2.9-3.7.
Midazolam is a white to light yellow crystalline compound, insoluble in water. The hydrochloride salt of midazolam, which is formed in situ, is soluble in aqueous solutions. Chemically, midazolam HCl is 8-chloro-6-(2-fluorophenyl)-1-methyl-4H– imidazo[1,5-a] [1,4] benzodiazepine hydrochloride. Midazolam hydrochloride has the molecular formula C18H13ClFN3•HCl, a calculated molecular weight of 362.25 and the following structural formula:
 |
In the Netherlands, midazolam is a List II drug of the Opium Law. Midazolam is a Schedule IV drug under the Convention on Psychotropic Substances. In the United Kingdom, midazolam is a Class C controlled drug. In the United States, midazolam (DEA number 2884) is on the Schedule IV list of the Controlled Substances Act as a non-narcotic agent with low potential for abuse.
midaolam hydrochloride NDA 018654, 075154
REF
U.S. Pat. No. 4,280,957
U.S. Pat. No. 5,693,795
U.S. Pat. No. 6,512,114
Midazolam Maleate
Drugs Fut 1978, 3(11): 822
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 18 pg. 5658 – 5667
Journal of Heterocyclic Chemistry, 1983 , vol. 20, 3 pg. 551 – 558.. 32 maleate
WO 2001070744
WO 2001002402
WO 2012075286
US2011/275799 A1… no 5
Journal of Organic Chemistry, 1978 , vol. 43, p. 936,942, mp free base, nmr
| US4280957 | May 15, 1978 | Jul 28, 1981 | Hoffmann-La Roche Inc. | Imidazodiazepines and processes therefor |
| US6262260 * | Mar 23, 2000 | Jul 17, 2001 | Abbott Laboratories | Process for the preparation of midazolam |
| US6512114 | Jun 30, 1999 | Jan 28, 2003 | Abbott Laboratories | Process for the preparation of Midazolam |
……………………….
introduction
4H-imidazo[1,5-a][1,4]benzodiazepines or, more simply, imidazobenzodiazepines, are a class of benzodiazepines having the general formula (I),

wherein the 1,4-diazepine ring is fused with a 1,3-imidazole ring. The main compounds part of the 4H-imidazo[1,5-a][1,4]benzodiazepines are Midazolam of formula (IV):

an active ingredient currently commercially available as a hydrochloride salt under the name of Versed or Hypnovel for anaesthetic and sedative use and the maleate salt currently commercially available under the name Dormicum or Flormidal.
Other important compounds are Climazolam of formula (VII):

Imidazenil of formula (VIII):

1-Hydroxymidazolam of formula (IX):

and Desmethyl midazolam of formula (X):

all these being biologically active substances and having psychotropic and sedative action.
The synthesis of the Midazolam as described in U.S. Pat. No. 4,280,957 of Hoffmann-La Roche provides for the decarboxylation reaction of the 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid of formula (VI) according to the following scheme:

The process for preparing the intermediate (VI) via basic hydrolysis of the corresponding ester is described in such patent publication and it is well known in the art.
The thermal decarboxylation reaction in high boiling solvent such as mineral oil at 230° C. for 5 min results in a mixture of products of Midazolam of formula (IV) and of Isomidazolam of formula (IV-bis), a non-pharmacologically active isomer, at a 80:20 ratio. The two products are separated by chromatography.
At industrial level, the formation of the Isomidazolam isomer impurity requires a further isomerisation reaction performed on the mixture of the two compounds to convert the isomer into the active product. The reaction mixture obtained from the thermal decarboxylation is thus subjected to basic treatment under the action of KOH in EtOH followed by an acid treatment which thus provides a mixture of Midazolam-Isomidazolam at a 95:5 ratio. The final removal of the Isomidazolam impurity from the product occurs through crystallisation of the product from AcOEt and EtOH. In order to limit this isomerisation treatment, in the subsequent U.S. Pat. No. 5,693,795 of Hoffmann-La Roche dated 1999, there is described a process for performing the decarboxylation of the compound of formula (VI) in n-butanol in a continuous tubular reactor with a 4 minutes permanence period with a yield between 47-77%. However, the reaction, performed at high temperature and pressure (280° C., 100 bars) results in the formation of a considerable percentage of Isomidazolam (85:15 Midazolam/Isomidazolam ratio) which still requires the basic isomerisation step.
Lastly, in U.S. Pat. No. 6,512,114 of Abbott Laboratories there is described the decarboxylation of the compound of formula (VI) in mineral oil or in N,N-Dimethylacetamide (DMA) at 160-230° C. for at least 3 hours obtaining a 3/1 to 6/1 Midazolam/Isomidazolam ratio with a yield of isolated product equal to just 54%.
Though performed using dedicated apparatus and in extreme conditions, the prior art processes do not allow selectively performing the decarboxylation reaction of the intermediate (VI) to Midazolam thus requiring a further synthetic passage followed by crystallisation with ensuing reduction of the overall yield.
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is represented by the following structural formula (I):

Midazolam is a central nervous system (CNS) depressant, used for short term treatment of insomnia. Like other benzodiazepines, midazolam binds to benzodiazepine receptors in the brain and spinal cord and is thus used as a short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is currently used in dentistry, cardiac surgery, endoscopic procedures, as a preanesthetic medication, as an adjunct to local anesthesia and as a skeletal muscle relaxant. Depending on the pH value, midazolam can exist in solution as a closed ring form (I) as well as an open ring form (IA), which are in equilibrium, as shown in Scheme 1:

The amount of the open ring form (IA) is dependent upon the pH value of the solution. At a pH value of about 3, the content of the open ring form (IA) can be 40%, while at pH value of 7.5, the closed ring form (I) can be more than 90%.
Clinical studies have demonstrated that there are no significant differences in the clinical activity between midazolam hydrochloride and midazolam maleate, however the use of intravenous midazolam hydrochloride has been associated, in some cases, with respiratory depression and arrest.
U.S Pat. No. 4,280,957 (hereinafter the ‘957 patent) describes a synthetic process for preparing midazolam, which is depicted in Scheme 2 below. This process includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) with acetic anhydride in dichloromethane to produce 2-acetamido-methyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (III). The latter is heated with polyphosphoric acid at 150° C. to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine of formula (IV), which is purified by column chromatography. Compound IV is then mixed with toluene and manganese dioxide and heated to reflux to afford midazolam base, which is crystallized from ether to yield a product with mp of 152-154° C.

The ‘957 patent further describes an alternative process which includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) (optionally as a dimaleate salt) with triethylorthoacetate in ethanol and in the presence of p-toluenesulfonic acid to afford 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine (IV). This product is dissolved in xylene and treated with activated manganese dioxide to afford the crude base, which is reacted in situ with maleic acid in ethanol and crystallized by addition of ether to produce the midazolam maleate having melting point of 148-151° C. The process is depicted in Scheme 3 below.

The preparation of midazolam maleate from the isolated midazolam base is also described in a further example of the ‘957 Patent, wherein a warm solution of midazolam base in ethanol is combined with a warm solution of maleic acid in ethanol. The mixture is diluted with ether and at least part of the solvents is evaporated using a steam bath to obtain crystalline midazolam maleate having melting point of 148-151° C. The yield and the purity of the obtained midazolam maleate are not disclosed.
U.S. Pat. No. 6,512,114 (hereinafter the ‘114 patent) describes another synthetic process for preparing midazolam, which is depicted in Scheme 4 below. According to this Process, the starting material 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid (V) is heated in mineral oil for 3 hours at 230° C. until it is decarboxylated, followed by treatment with potassium tert-butoxide, to afford midazolm (I), isomidazolam (VI) and a midazolam dimmer (VII). Midazolam base is obtained in 54.5% yield after two re-crystallizations from ethyl acetate and heptane; however, the purity of the product is not disclosed.

The preparation of midazolam by conventional routes is liable to produce impurities such as isomidazolam (VI) and a midazolam dimmer (VII), and possibly other impurities. There is, therefore, a need in the art for a midazolam purification process that will provide highly pure midazolam containing minimal amounts of impurities produced. The present invention provides such a process.
This example describes the preparation of midazolam base as taught in the ‘957 patent.
16 g (0.03 mol) of 2-aminomethyl-7-chloro-5-(2-fluorophenyl)-2,3-dihydro-1H-1,4-bezodiazepine dimaleate was dissolved in 200 ml of toluene and 10 ml of 25% ammonium hydroxide solution was added and mixing was maintained for an hour. Then, the phases were separated and the toluene phase was dried by azeotropic distillation using a Dean Stark apparatus. 7 ml (0.038 mol) of triethylorthoacetate was added and the solution was heated to reflux for 4 hours, after which time the solution was left to cool to ambient temperature. 25 ml of methyl tert-butyl ether was added and the mixture was cooled overnight to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine, which was isolated by filtration. The product was mixed with 200 ml of toluene and dried by azeotropic distillation using a Dean Stark apparatus. Then, 30 g of manganese dioxide was added and the mixture was heated to reflux for two hours. The excess manganese dioxide was filtered off to afford a solution of midazolam base in toluene, which was evaporated to obtain a product having 97.9% purity and containing 0.44% of impurity VIII and 1.14% of impurity IX (according to HPLC).
…………………………
EXAMPLE 28
2-Aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine dimaleate
A suspension of 17 g (0.05 m) of 7-chloro-1,3-dihydro-5-(2-fluorophenyl)-2-nitromethylene-2H-1,4-benzodiazepine-4-oxide in 200 ml of tetrahydrofuran and 100 ml of methanol was hydrogenated in presence of 17 g of Raney nickel at an initial pressure of 155 psi for 24 hrs. The catalyst was removed by filtration and the filtrate was evaporated. The residue was dissolved in 50 ml of 2-propanol and warmed on the steambath. A warm solution of 17 g of maleic acid in 60 ml of ethanol was added and the salt was allowed to crystallize by cooling in the ice bath. The final product consisted of yellow crystals with mp 196
EXAMPLE 14
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Acetic anhydride, 7 ml., was added to a solution of 6.16 g. of crude 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine in 200 ml. of methylene chloride. The solution was layered with 200 ml. of saturated aqueous sodium bicarbonate and the mixture was stirred for 20 minutes. The organic layer was separated, washed with sodium bicarbonate, dried over sodium sulfate and evaporated to leave 6.2 g. resinous 2-acetaminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine. This material was heated with 40 g. of polyphosphoric acid at 150 water, made alkaline with ammonia and ice and extracted with methylene chloride. The extracts were dried and evaporated and the residue (5.7 g.) was chromatographed over 120 g. of silica gel using 20% methanol in methylene chloride. The clean fractions were combined and evaporated to yield resinous 8-chloro-3a,4-dihydro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[ 1,5-a][1,4]benzodiazepine. A mixture of this material with 500 ml. of toluene and 30 g. of manganese dioxide was heated to reflux for 11/2 hours. The manganese dioxide was separated by filtration over celite. The filtrate was evaporated and the residue was crystallized from ether to yield a product with m.p. 152 was recrystallized from methylene chloride/hexane
EXAMPLE 49
8-Chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.625 g. (5.5 mmol), was added to a solution of 1.625 g. (5 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 nitrogen for 10 min. at -30 ml. of glacial acetic acid and was then partitioned between aqueous bicarbonate and toluene/methylene chloride (3:1 v/v). The organic layer was separated, dried and evaporated. The residue was chromatographed over 60 g. of silica gel using 25% (v/v) methylene chloride in ethyl acetate. The less polar product was eluted first and was crystallized from ethylacetate/hexane to yield product with m.p. 180
EXAMPLE 50
8-Chloro-6-(2-fluorphenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.125 g. (1.1 mmol) was added to a solution of 0.325 g. (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 -30 by addition of 0.2 ml. of glacial acetic acid and was partitioned between aqueous sodium bicarbonate and methylene chloridetoluene (1:3). The organic phase was washed with water, dried and evaporated. The residue was chromatographed over 20 g. of silica gel using ethyl acetate for elution. After elution of starting material, product was collected and crystallized from ether/hexane, m.p. 156
hyd and dihydrochloride
EXAMPLE 24
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine dihydrochloride
A solution of 0.32 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 5 ml of ethanol was treated with excess ethanolic hydrogen chloride. The salt was crystallized by addition of 2-propanol and ether. The colorless crystals were collected, washed with ether and dried to leave a final product with mp 290
EXAMPLE 258-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride
A solution of 0.325 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 3 ml of ethanol was combined with a suspension of 0.4 g (1 mmol) of the dihydrochloride of this compound in 5 ml of ethanol. After filtration, the solution was treated with ether and heated on the steambath for 5 min to crystallize. The crystals were collected, washed with ether and dried to leave the monohydrochloride with mp 295
maleate
EXAMPLE 22
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine maleate
A warm solution of 6.5 g (0.02 m) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 30 ml of ethanol was combined with a warm solution of 2.6 g (0.022 m) of maleic acid in 20 ml of ethanol. The mixture was diluted with 150 ml of ether and heated on the steam bath for 3 min. After cooling, the crystals were collected, washed with ether and dried in vacuo to yield a final product with mp 148
…
Synthesis
Midazolam, can be described according to scheme 4 indicated below:

 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.For the reactions performed in the microreactor, the solutions containing the substrates to be decarboxylated were loaded into 5 and 10 mL gastight glass syringes (Hamilton, item n. 81527, 81627) mounted on syringe pumps (KD Scientifics, model KDS100). A pipe made of PTFE® (OD=1.58 mm, ID=0.8 mm, Supelco, item n. 58696-U) was used for making the reaction channel.A counterpressure valve sold by Swagelok (item n. SS-SS1-VH) was used for regulating the flow within the channel.Example 1Synthesis of the Compound of Formula (V)—Example of the Invention

50 g (0.135 mol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-3-carboxylic acid of formula (VI) and 250 mL of ethanol were loaded into a two-neck 500 mL flask, equipped with a magnetic stirrer. 40 mL of an aqueous solution of 1 M HCl are dripped in about 10 minutes. The open di-hydrochloride intermediate of formula (V) starts precipitating into the reaction environment already after 3 minutes from the beginning of the addition of the acid solution. The mixture is maintained stirred at RT for 3 hrs and then it is filtered on buckner washing the solid with ethanol. The moist product is dried in an oven under vacuum at 60° C. up to reaching a constant weight. A light yellow crystalline product is obtained (51.5 g, 83% yield). The crude product was used for the decarboxylation without further purifications.
ESI-MS [MeCN+0.1% HCOOH]: m/z 388 (V); 370 (VI).
1H-NMR (250 MHz, CD3OD): 2.52 (s, 3H); 4.27-4.41 (m, 2H); 7.22-8.1 (m, 7H). M.p.: 217° C.
Example 2
Synthesis of Midazolam of Formula (IV)—Performed in Batch—Example of the Invention

30 g (0.065 mol) of 5-(aminomethyl)-1-{(4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 90 mL of NMP are loaded into a three-neck 250 mL flask, equipped with a magnetic stirrer and coolant. The mass is heated using an oil bath at T=195-203° C. for one hour. Thus, 1 mL of solution is collected for performing HPLC analysis. The reaction product is Midazolam having 82% titre (w/w) (determined via HPLC titre correcting it using the solvent) and it contains 1% of Isomidazolam. The product is extracted using Isopropyl acetate after raising the pH to 10 by adding aqueous Na2CO3.
Example 3
Synthesis of Midazolam of Formula (IV)—Performed in a Micro-Reactor—Example of the Invention

3.22 g (7 mmol) of 5-(aminomethyl)-1-{4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 10 mL of NMP are loaded into a 10 mL flask equipped with a magnetic stirrer. In order to facilitate the complete solubilisation of the substrate, it is necessary to slightly heat the reaction mixture (about 40° C.) for a few minutes. The solution thus obtained is transferred into a 10 mL gastight glass syringe mounted on a KDS100 syringe pump (FIG. 1) and the flow is regulated at 1.0 mL/h so as to set a residence period of 30 minutes at 200° C. The reaction product is Midazolam having an 89% titre (w/w) (determined via HPLC titre correcting it using the solvent) and containing 3% (w/w) of Isomidazolam.
Example 4Synthesis of Midazolam of formula (IV)—Comparison of the InventionA table is reported which summarises the results of the decarboxylation of the compound of formula (V) and (V-bis) (for the latter see Examples 6 and 7) obtained according to some embodiments of the invention and those obtained by way of experiment through the decarboxylation of the intermediate of formula (VI) (process of the prior art) both performed in 3 volumes of NMP at 200° C., both in batch method (Example 4) and in continuous method with the microreactor (MR) made of PTFE of FIG. 1. (Examples 4-1, 4-2, 4-3).
| Example | substrate | Mode | Solv. | T° C. | t min. | Midazolam (p/p) | Isomidaz. (P/P) |
| 2 | (V) | Batch | NMP | 200 | 60 | 82 | 1 |
| 3 | (V) | MR | NMP | 200 | 30 | 89 | 3 |
| 7 | (V-bis) | Batch | NMP | 200 | 60 | 68 | 3 |
| 4 | (VI) | Batch | NMP | 200 | 60 | 78 | 18 |
| 4-1 | (VI) | MR | NMP | 200 | 38 | 81 | 17 |
| 4-2 | (VI) | MR | NMP | 200 | 20 | 77 | 18 |
| 4-3 | (VI) | MR | NMP | 200 | 15 | 58 | 22 |
| U.S. Pat. No. | (VI) | Tubular | n-BuOH | 290 | 4 | 85 * | 15 * |
| 5,693,795 | reactor | ||||||
| U.S. Pat. No. | (VI) | Batch | Olio | 230 | 180 | 75 * | 25 * |
| 6,512,114 | min. | 87.5 * | 12.5 * | ||||
| or DMA | |||||||
| * = Midazolam/Isomidazolam ratio only (other impurities not considered). | |||||||
The product of the comparative experiments 4, 4-1, 4-2, 4-3 and of the two USA patents should be subjected to a further isomerisation process to reduce the high amount of Isomidazolam so as to be able to obtain Midazolam free of Isomidazolam after further crystallization, which would not be required for the product obtained according to the invention (examples 2 and 3).

A 4-neck RBF was charged under nitrogen flow with: 10 g of Midazolam (IV) (prepared according to example 2) and 40 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following solution: 3.72 g of maleic acid are dissolved in 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. The maleic acid solution is dropped in 30/40 minutes and keeping T=25/30° C. into the solution containing Midazolam. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 40 mL of cool Ethanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. 12.8 g of Midazolam Maleate as white solid were collected (Molar yield=94.5%). m.p.=149-152° C. (by DSC).

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. 5 mL of a ethanolic solution of Hydrochloric acid 2N were slowly added. 20 mL of Isopropanol were added over 30 minutes at RT. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 10 mL of cool isopropanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. Midazolam dihydrochloride as white solid was collected.
MIDAZOLAM HYDROCHLORIDE
Example 10
Preparation of 8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride (Midazolam hydrochloride)

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 10 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following suspension: 1.22 g of Midazolam dihydrochloride (prepared according to example 9) and 15 mL of Ethanol. The Midazolam ethanolic solution was added to the Midazolam dihydrochloride suspension. After filtration, the solution was treated with MTBE and heated at 60° C. until crystallization. After cooling to RT, the slurry was filtered, the cake washed with MTBE and the product was dried to provide Midazolam (mono)hydrochloride as a white solid.
…..


NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}