
Home » Posts tagged 'antibacterial'
Tag Archives: antibacterial
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CEFOPERAZONE
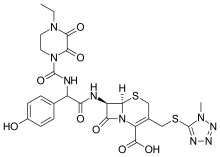
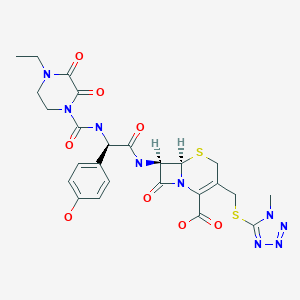
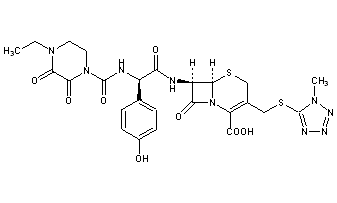
Cefoperazone
- Molecular FormulaC25H27N9O8S2
- Average mass645.667 Da
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cefoperazone sodium | 5FQG9774WD | 62893-20-3 | NCFTXMQPRQZFMZ-WERGMSTESA-M |
(6R,7R)-7-{[(2R)-2-{[(4-ethyl-2,3-dioxopiperazin-1-yl)carbonyl]amino}-2-(4-hydroxyphenyl)acetyl]amino}-3-{[(1-methyl-1H-tetrazol-5-yl)thio]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
(6R,7R)-7-[(2R)-2-[(4-ethyl-2,3-dioxopiperazine-1-carbonyl)amino]-2-(4-hydroxyphenyl)acetamido]-3-{[(1-methyl-1H-1,2,3,4-tetrazol-5-yl)sulfanyl]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
263-749-4[EINECS], 4742
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo- , (6R,7R)- [ACD/Index Name]
5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-, (6R,7R)-
62893-19-0[RN]
7-[D-(-)-a-(4-Ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic Acid
7U75I1278D
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 188-190 | Saikawa, I., Takano, S., Yoshida, C., Takashima, 0..Momonoi, K., Kuroda, S., Komatsu, M., Yasuda, T.and Kodama, Y.; British Patent 1,508,071; April 19,1978; assigned to Toyama Chemical Co., Ltd. and U.S. Patent 4,110,327; August 29,1978; also assigned to Toyama Chemical Co., Ltd. |
| logP | -0.74 | HANSCH,C ET AL. (1995) |
Cefoperazone
CAS Registry Number: 62893-19-0
CAS Name: (6R,7R)-7-[[(2R)-[[(4-Ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino](4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acidAdditional Names: 7-[D-(-)-a-(4-ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic acid
Molecular Formula: C25H27N9O8S2
Molecular Weight: 645.67
Percent Composition: C 46.50%, H 4.21%, N 19.52%, O 19.82%, S 9.93%
Literature References: Broad spectrum third generation cephalosporin antibiotic. Prepn: I. Saikawa et al.,BE837682; eidem,US4410522 (1976, 1983 both to Toyama); eidem,Yakugaku Zasshi99, 929 (1979). Stability in aq soln: eidem,ibid. 1207. In vitro activity: M. V. Borobio et al.,Antimicrob. Agents Chemother.17, 129 (1980). Kinetics in rats: J. Fabre et al.,Schweiz. Med. Wochenschr.110, 264 (1980); in humans: A. F. Allaz, ibid.109, 1999 (1979). Review of pharmacology and therapeutic efficacy: R. N. Brogden et al.,Drugs22, 423-460 (1981). Symposium on clinical studies: ibid. Suppl. 1, 1-124.
Properties: Crystals from acetonitrile/water, mp 169-171° (hydrated). Stable at pH 4.0-7.0; slightly unstable in acid; highly unstable in alkaline soln.
Melting point: mp 169-171° (hydrated)
Derivative Type: Sodium salt
CAS Registry Number: 62893-20-3
Manufacturers’ Codes: CP-52640-2; T-1551
Trademarks: Bioperazone (Biopharma); Cefazone (Firma); Cefobid (Pfizer); Cefobine (Pfizer); Cefobis (Pfizer); Cefogram (Metapharma); Cefoneg (Tosi); Cefosint (Proter); Dardum (Lisapharma); Farecef (Lafare); Kefazon (Esseti); Novobiocyl (Francia); Pathozone (Pfizer); Peracef (Pfizer); Perocef (Pulitzer); Tomabef (Aandersen)
Molecular Formula: C25H26N9NaO8S2
Molecular Weight: 667.65
Percent Composition: C 44.97%, H 3.93%, N 18.88%, Na 3.44%, O 19.17%, S 9.61%
Therap-Cat: Antibacterial., Therap-Cat-Vet: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
Cefoperazone is a third-generation cephalosporin antibiotic, marketed by Pfizer under the name Cefobid. It is one of few cephalosporin antibiotics effective in treating Pseudomonas bacterial infections which are otherwise resistant to these antibiotics.
It was patented in 1974 and approved for medical use in 1981.[1] Cefoperazone/sulbactam (Sulperazon) is a co-formulation with sulbactam.
Cefoperazone is a broad-spectrum cephalosporin antibiotic used for the treatment of bacterial infections in various locations, including the respiratory tract, abdomen, skin, and female genital tracts.
Cefoperazone is a semisynthetic broad-spectrum cephalosporin proposed to be effective against Pseudomonas infections. It is a third-generation antiobiotic agent and it is used in the treatment of various bacterial infections caused by susceptible organisms in the body, including respiratory tract infections, peritonitis, skin infections, endometritis, and bacterial septicemia. While its clinical use has been discontinued in the U.S., cefoperazone is available in several European countries most commonly under the product name, Sulperazon.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

English: I. Saikawa, S. Takano, Y. Shuntaro, C. Yoshida, 0.
Takashima, K. Momonoi, S. Kuroda, M. Komatsu, T. Yasuda, and Y. Kodama, German Offen., DE 2,600,880 (1977); Chem.
Abstr., 87_, 184533b (1977).
SYN
Following is one of the synthesis routes:
alpha-(4-Ethyl-2,3-dioxo-1-piperazinocarbonylamino)-p-hydroxyphenylacetic acid (I) is condensed with 7-amino-3-[(1-methyl-1H-tetrazol-5-yl)thiomethyl]-3-cephem-4-carboxylic acid (II) in the presence of ethyl chlorocarbonate and N,O-bis(trimethylsilyl)acetamide in acetonitrile to produce Cefoperazone sodium.

SYN
Antibiotics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cefoperazone
Cefoperazone, (6R,7R)-7-[(R)-2-(4-ethyl-2,3-dioxo-1-piperazincarboxamido)-2-(p-hydroxyphenyl)acetamido]-3-[[(1-methyl-1 H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid (32.1.2.84), is synthesized by acylating 7-amino-3-(1-methyl-1,2,3,4-tetrazol-5-yl)-thiomethyl-3-cefem-4-carboxylic acid (32.1.2.24) with a mixed anhydride synthesized from ethyl chloroformate and α-(4-ethylpiperazin-2, 3-dion-1-carbonylamino)-4-hydroxyphenylacetic acid (32.1.2.83), which in turn is synthesized from 4-ethylpiperazin-2,3-dion-1-carboxylic acid (32.1.1.29) and the sodium salt of 4-hydroxyphenylglycine [163–168].


Cefoperazone also has a broad spectrum of antimicrobial action, including most clinically significant microorganisms: Gram-positive, Gram-negative, aerobic, and anaerobic. It is stable with respect to most beta-lactamases of Gram-positive and Gram-negative bacteria.
Cefoperazone is used for bacterial infections of the lower respiratory tract, urinary and sexual tracts, bones, joints, skin, soft tissues, abdominal, and gynecological infections. Synonyms of this drug are cefazon, cefobid, cefobis, and many others.
Spectrum of bacterial susceptibility
Cefoperazone has a broad spectrum of activity and has been used to target bacteria responsible for causing infections of the respiratory and urinary tract, skin, and the female genital tract. The following represents MIC susceptibility data for a few medically significant microorganisms.
- Haemophilus influenzae: 0.12 – 0.25 µg/ml
- Staphylococcus aureus: 0.125 – 32 µg/ml
- Streptococcus pneumoniae: ≤0.007 – 1 µg/ml[2]
Adverse effects
Cefoperazone contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzyme vitamin K epoxide reductase) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.[3]
Mechanism of action
Cefoperazone exerts its bactericidal effect by inhibiting the bacterial cell wall synthesis, and sulbactam acts as a beta-lactamase inhibitor, to increase the antibacterial activity of cefoperazone against beta-lactamase-producing organisms.
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 494. ISBN 9783527607495.
- ^ “Cefoperazone (Cefobid) – The Antimicrobial Index Knowledgebase – TOKU-E”. antibiotics.toku-e.com.
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA (eds.). Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| ATC code | J01DD12 (WHO) QJ51DD12 (WHO) |
| Pharmacokinetic data | |
| Excretion | Hepatic |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62893-19-0 |
| PubChem CID | 44185 |
| DrugBank | DB01329 |
| ChemSpider | 40206 |
| UNII | 7U75I1278D |
| KEGG | D07645 |
| ChEMBL | ChEMBL507674 |
| CompTox Dashboard (EPA) | DTXSID2022759 |
| ECHA InfoCard | 100.057.936 |
| Chemical and physical data | |
| Formula | C25H27N9O8S2 |
| Molar mass | 645.67 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////cefoperazone, Antibacterial, Antibiotics, Lactams, Cephalosporins, CP-52640-2, T-1551, CP 52640-2, T 1551
[H][C@]12SCC(CSC3=NN=NN3C)=C(N1C(=O)[C@@]2([H])NC(=O)[C@H](NC(=O)N1CCN(CC)C(=O)C1=O)C1=CC=C(O)C=C1)C(O)=O

NEWDRUG APPROVALS
one time
$10.00
BIAPENEM

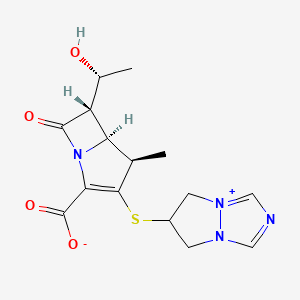
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
CLARITHROMYCIN


Clarithromycin
Synonyms:A-56268, TE-031, 6-O-methylerythromycin, ATC:J01FA09Use:macrolide antibioticChemical name:6-O-methylerythromycinFormula:C38H69NO13
- MW:747.96 g/mol
- CAS-RN:81103-11-9
- 81103-11-9
klacid XL / Klaricid XL / Macladin / Naxy / Veclam / Zeclar
(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-12,13-dihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-7-methoxy-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis Reference
Jih-Hua Liu, David A. Riley, “Preparation of crystal form II of clarithromycin.” U.S. Patent US5844105, issued May, 1997. US5844105
NEW DRUG APPROVALS
ONE TIME
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Clarithromycin citrate | 16K08R7NG0 | 848130-51-8 | MDRWXDRMSKEMRE-AZFLODHXSA-N |
ClarithromycinCAS Registry Number: 81103-11-9CAS Name: 6-O-MethylerythromycinManufacturers’ Codes: A-56268; TE-031Trademarks: Biaxin (Abbott); Clarosip (Grñenthal); Clathromycin (Taisho); Cyllind (Abbott); Klacid (Abbott); Klaricid (Abbott); Macladin (Guidotti); Naxy (Sanofi Winthrop); Veclam (Zambon); Zeclar (Abbott)Molecular Formula: C38H69NO13Molecular Weight: 747.95Percent Composition: C 61.02%, H 9.30%, N 1.87%, O 27.81%Literature References: Semisynthetic macrolide antibiotic; derivative of erythromycin, q.v. Prepn: Y. Watanabe et al.,EP41355; eidem,US4331803 (1981, 1982 both to Taisho); and in vitro antibacterial activity: S. Morimoto et al.,J. Antibiot.37, 187 (1984). In vitro and in vivo antibacterial activity: P. B. Fernandes et al.,Antimicrob. Agents Chemother.30, 865 (1986). Comparative antibacterial spectrum in vitro: C. Benson et al.,Eur. J. Clin. Microbiol.6, 173 (1987); H. M. Wexler, S. M. Finegold, ibid. 492. HPLC determn in biological fluids: D. Croteau et al.,J. Chromatogr.419, 205 (1987); in plasma: H. Amini, A. Ahmadiani, J. Chromatogr. B817, 193 (2005). Acute toxicity study: S. Abe et al.,Chemotherapy (Tokyo)36, Suppl. 3, 274 (1988). Symposium on pharmacology and comparative clinical studies: J. Antimicrob. Chemother.27, Suppl. A, 1-124 (1991). Comprehensive description: I. I. Salem, Anal. Profiles Drug Subs. Excip.24, 45-85, (1996).Properties: Colorless needles from chloroform + diisopropyl ether (1:2), mp 217-220° (dec). Also reported as crystals from ethanol, mp 222-225° (Morimoto). uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nm; (methanol): 211, 288 nm. [a]D24 -90.4° (c = 1 in CHCl3). Stable at acidic pH. LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe).Melting point: mp 217-220° (dec); mp 222-225° (Morimoto)Optical Rotation: [a]D24 -90.4° (c = 1 in CHCl3)Absorption maximum: uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nmToxicity data: LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe)Therap-Cat: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
Clarithromycin, a semisynthetic macrolide antibiotic derived from erythromycin, inhibits bacterial protein synthesis by binding to the bacterial 50S ribosomal subunit. Binding inhibits peptidyl transferase activity and interferes with amino acid translocation during the translation and protein assembly process. Clarithromycin may be bacteriostatic or bactericidal depending on the organism and drug concentration.
Clarithromycin, sold under the brand name Biaxin among others, is an antibiotic used to treat various bacterial infections.[2] This includes strep throat, pneumonia, skin infections, H. pylori infection, and Lyme disease, among others.[2] Clarithromycin can be taken by mouth as a pill or liquid.[2]
Common side effects include nausea, vomiting, headaches, and diarrhea.[2] Severe allergic reactions are rare.[2] Liver problems have been reported.[2] It may cause harm if taken during pregnancy.[2] It is in the macrolide class and works by decreasing protein production of some bacteria.[2]
Clarithromycin was developed in 1980 and approved for medical use in 1990.[3][4] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[5] Clarithromycin is available as a generic medication.[2] It is made from erythromycin and is chemically known as 6-O-methylerythromycin.[6]
Medical uses
Clarithromycin is primarily used to treat a number of bacterial infections including pneumonia, Helicobacter pylori, and as an alternative to penicillin in strep throat.[2] Other uses include cat scratch disease and other infections due to bartonella, cryptosporidiosis, as a second line agent in Lyme disease and toxoplasmosis.[2] It may also be used to prevent bacterial endocarditis in those who cannot take penicillin.[2] It is effective against upper and lower respiratory tract infections, skin and soft tissue infections and helicobacter pylori infections associated with duodenal ulcers.
Spectrum of bacterial susceptibility
Staphylococcus aureusAerobic Gram-positive bacteria
Aerobic Gram-negative bacteria
- Haemophilus parainfluenzae
- Haemophilus influenzae
- Moraxella catarrhalis
Helicobacter
Mycobacteria
Mycobacterium avium complex consisting of:
Other bacteria
Safety and effectiveness of clarithromycin in treating clinical infections due to the following bacteria have not been established in adequate and well-controlled clinical trials:[7]
Aerobic Gram-positive bacteria
- Streptococcus agalactiae
- Streptococcus (Groups C, F, G)
- Viridans group streptococci
Aerobic Gram-negative bacteria
Anaerobic Gram-positive bacteria
- Clostridium perfringens
- Peptococcus Niger
- Cutibacterium acnes
Anaerobic Gram-negative bacteria
- Prevotella melaninogenica (formerly Bacteroides melaninogenicus)
Contraindications
- Clarithromycin should not be taken by people who are allergic to other macrolides or inactive ingredients in the tablets, including microcrystalline cellulose, croscarmelose sodium, magnesium stearate, and povidone
- Clarithromycin should not be used by people with a history of cholestatic jaundice and/or liver dysfunction associated with prior clarithromycin use.[7]
- Clarithromycin should not be used in the setting of hypokalaemia (low blood potassium)
- Use of clarithromycin with the following medications: cisapride, pimozide, astemizole, terfenadine, ergotamine, ticagrelor, ranolazine or dihydroergotamine is not recommended.[7]
- It should not be used with colchicine in people with kidney or liver impairment.[7]
- Concomitant use with cholesterol medications such as lovastatin or simvastatin.[7]
- Hypersensitivity to clarithromycin or any component of the product, erythromycin, or any macrolide antibiotics.[7]
- QT prolongation or ventricular cardiac arrhythmias, including torsade de pointes.[7]
Side effects
The most common side effects are gastrointestinal: diarrhea (3%), nausea (3%), abdominal pain (3%), and vomiting (6%). It also can cause headaches, insomnia, and abnormal liver function tests. Allergic reactions include rashes and anaphylaxis. Less common side effects (<1%) include extreme irritability, hallucinations (auditory and visual), dizziness/motion sickness, and alteration in senses of smell and taste, including a metallic taste. Dry mouth, panic attacks, and nightmares have also been reported, albeit less frequently.[8]
Cardiac
In February 2018, the FDA issued a Safety Communication warning with respect to an increased risk for heart problems or death with the use of clarithromycin, and has recommended that alternative antibiotics be considered in those with heart disease.[9]
Clarithromycin can lead to a prolonged QT interval. In patients with long QT syndrome, cardiac disease, or patients taking other QT-prolonging medications, this can increase risk for life-threatening arrhythmias.[10]
In one trial, the use of short-term clarithromycin treatment was correlated with an increased incidence of deaths classified as sudden cardiac deaths in stable coronary heart disease patients not using statins.[11] Some case reports suspect it of causing liver disease.[12]
Liver and kidney
Clarithromycin has been known to cause jaundice, cirrhosis, and kidney problems, including kidney failure.[citation needed]
Central nervous system
Common adverse effects of clarithromycin in the central nervous system include dizziness, headaches. Rarely, it can cause ototoxicity, delirium and mania.
Infection
A risk of oral candidiasis and vaginal candidiasis, due to the elimination of the yeast’s natural bacterial competitors by the antibiotic, has also been noted.
Pregnancy and breastfeeding
Clarithromycin should not be used in pregnant women except in situations where no alternative therapy is appropriate.[7] Clarithromycin can cause potential hazard to the fetus hence should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.[7] For lactating mothers it is not known whether clarithromycin is excreted in human milk.[7]
Interactions
Clarithromycin inhibits a liver enzyme, CYP3A4, involved in the metabolism of many other commonly prescribed drugs. Taking clarithromycin with other medications that are metabolized by CYP3A4 may lead to unexpected increases or decreases in drug levels.
A few of the common interactions are listed below.
Colchicine
Clarithromycin has been observed to have a dangerous interaction with colchicine as the result of inhibition of CYP3A4 metabolism and P-glycoprotein transport. Combining these two drugs may lead to fatal colchicine toxicity, particularly in people with chronic kidney disease.[7]
Statins
Taking clarithromycin concurrently with certain statins (a class of drugs used to reduce blood serum cholesterol levels) increases the risk of side effects, such as muscle aches and muscle break down (rhabdomyolysis).[13]
Calcium channel blockers
Concurrent therapy with calcium channel blocker may increase risk of low blood pressure, kidney failure, and death, compared to pairing calcium channel blockers with azithromycin, a drug similar to clarithromycin but without CYP3A4 inhibition.[14] Administration of clarithromycin in combination with verapamil have been observed to cause low blood pressure, low heart rate, and lactic acidosis.[7]
Carbamazepine
Clarithromycin may double the level of carbamazepine in the body by reducing its clearance, which may lead to toxic symptoms of carbamazepine, such as double vision, loss of voluntary body movement, nausea, as well as hyponatremia.[15]
HIV medications
Depending on the combination of medications, clarithromycin therapy could be contraindicated, require changing doses of some medications, or be acceptable without dose adjustments.[16] For example, clarithromycin may lead to decreased zidovudine concentrations.[17]
Mechanism of action
Clarithromycin prevents bacteria from multiplying by acting as a protein synthesis inhibitor. It binds to 23S rRNA, a component of the 50S subunit of the bacterial ribosome, thus inhibiting the translation of peptides.[citation needed]
Pharmacokinetics
MetabolismUnlike erythromycin, clarithromycin is acid-stable, so can be taken orally without having to be protected from gastric acids. It is readily absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, clarithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations of clarithromycin are released; its concentration in the tissues can be over 10 times higher than in plasma. Highest concentrations are found in liver, lung tissue, and stool.
Clarithromycin has a fairly rapid first-pass metabolism in the liver. Its major metabolites include an inactive metabolite, N-desmethylclarithromycin, and an active metabolite, 14-(R)-hydroxyclarithromycin. Compared to clarithromycin, 14-(R)-hydroxyclarithromycin is less potent against mycobacterial tuberculosis and the Mycobacterium avium complex. Clarithromycin (20%-40%) and its active metabolite (10%-15%) are excreted in urine. Of all the drugs in its class, clarithromycin has the best bioavailability at 50%, which makes it amenable to oral administration. Its elimination half-life is about 3 to 4 hours with 250 mg administered every 12 h, but increased to 5 to 7 h with 500 mg administered every 8 to 12 h. With any of these dosing regimens, the steady-state concentration of this metabolite is generally attained within 3 to 4 days.[18]
History
Clarithromycin was invented by researchers at the Japanese drug company Taisho Pharmaceutical in 1980.[3] The product emerged through efforts to develop a version of the antibiotic erythromycin that did not experience acid instability in the digestive tract, causing side effects, such as nausea and stomachache. Taisho filed for patent protection for the drug around 1980 and subsequently introduced a branded version of its drug, called Clarith, to the Japanese market in 1991. In 1985, Taisho partnered with the American company Abbott Laboratories for the international rights, and Abbott also gained FDA approval for Biaxin in October 1991. The drug went generic in Europe in 2004 and in the US in mid-2005.
Society and culture

A pack of Clarithromycin tablets manufactured by Taisho Pharmaceutical
Available forms
Clarithromycin is available as a generic medication.[2] In the United States, clarithromycin is available as immediate release tablets, extended release tablets, and granules for oral suspension.[2]
Brand names
Clarithromycin is available under several brand names in many different countries, for example Biaxin, Crixan, Claritron, Clarihexal, Clacid, Claritt, Clacee, Clarac, Clariwin, Claripen, Clarem, Claridar, Cloff, Fromilid, Infex, Kalixocin, Karicin, Klaricid, Klaridex, Klacid, Klaram, Klabax, MegaKlar, Monoclar, Resclar, Rithmo, Truclar, Vikrol and Zeclar.
Manufacturers
In the UK the drug product is manufactured in generic form by a number of manufacturers including Somex Pharma, Ranbaxy, Aptil and Sandoz.
SYN
CN 109705180

SYN
Indian Pat. Appl., 2014DE00731, 31 Aug 2016

SYN
Heterocycles, 31(12), 2121-4; 1990

SYN
https://patents.google.com/patent/WO2006064299A1/enErythromycin A is known to be a useful macrolide antibiotic having a strong activity against Gram-positive bacteria, this compound has an undesirable property that it loses rapidly the antibacterial activity by the acid in stomach when administered orally, where- upon its blood concentration remains at a low level. 6-0-Alkyl derivatives of Erythromycin- A are well known as an useful antibacterial agents. 6-O-Methyl-Erythromycin-A (Clarithromycin) and a pharmaceutically acceptable salt is a potent macrolide antibiotic as reported in US Patent No. 4,331 ,803. Clarithromycin is stable in acidic medium and also remarkable in vivo activity and has a strong antibacterial property against Gram-positive bacteria compared to Erythromycin- A. This compound shows excellent effect for the treatment of infections by oral administration.A number of synthetic processes have been reported for preparing 6-O-alkyl erythromycin. US Patent No. 4,331 ,803 discloses a method for the preparation of Clarithromycin by methylating 6-OH group of 2′-O-3′-N-benzyloxycarbonyl erythromycinFormula (III)

21,3′-O-Protected ErythromycinMethylation of 6-OH group of the 2′,3′-benzyloxycarbonyl erythromycin was carried out using methyl iodide in the presence of a suitable base in a solvent. Clarithromycin was obtained from the compound after removing benzyloxycarbonyl group by hydrogenolysis and then subjecting to the reductive methylation in the presence of excess amount of farmaldehyde. Clarithromycin can also be synthesized by the methylation of 6-OH position of Erythromycin-A-9-OximeFormula (II)

Erythromycin-9-OximeSynthesis of Clarithromycin using 9-oxime or its derivatives are well reported in US Patent Nos. 5,274,085; 4,680,386; 4,668,776; 4,670,549 and 4,672,109. In case of Erythromycin-9-Oxime derivatives, the oxime is protected before methylation step with 2- alkenyl group (US Patent Nos. 4,670,549; 4,668,776) or benzyl group (US Patent Nos. 4,680,386 and 4,670,549). However, it has been reported (Ref. Journal of Antibiotics 46, No. 6, Page No. 647, year 1993) that when the Erythromycin-A-9-Oxime is protected by trimethylsilyl group, which is very unstable under basic condition pose potential impurities formation during methylation. There are some methods reported in US Patent Nos., e.g. , 4,680,386; 4,670,549 and US Patent No. 4,311,803 for the synthesis of Clarithromycin by using chlorobenzyloxycarbonyl group for protection at 2′ and 3′ function of of Erythromycin-A-9-Oxime derivatives.For the protection of 2′-OH group (US Patent No. 4,311 ,803) requires large amounts of benzyl chloroformate which poses problems in handling because of its severe irritating and toxic properties. This protection step also leads to the formation of 3′ -N- demethylation, which requires an additional re-methylation step. The de-protection of chlorobenzyloxy carbonyl group leads to the formation of undesired side products. In earlier reported processes, e.g. , US Patent No. 4,990,602; EP 0,272,110 Al where the methylation has been done on Erythromycin-A-9-Oxime derivatives by the protection of 2′ and 4″ hydroxyl groups using DMSO and THF as a solvent at 0° to 50C or at room temperature, smooth methylation takes place with less side product formation. However, by using the above methylation processes the formation of 6, 11-O-dimethyl erythromycin- A (Compound- A) is always more than 1.0 % in Clarithromycin. Hence, there is a need for an efficient methylation process for the production of Clarithromycin with lesser amount of 6,11-O-dimethyl erythromycin-A than reported previously.




EXAMPLE 1Erythromycin-A-9-OximeTo a solution of 201 Ltr water in 561 Kg isopropyl alcohol is added 282 Kg (4057 mol) of hydroxyl amine hydrochloride under stirring and the reaction mixture is brought to 10 to 200C. Caustic flakes (162 Kg, 4050 mol) is added slowly to the reaction mixture by keeping temperature between 10° to 200C. After 15 minutes of completion of addition, pH of reaction mixture is adjusted to 6.5 to 7.0 by the slow addition of glacial acetic acid (96 Ltr, 100.8 Kg, 1678.6 mole). To the stirred reaction mass is added 300 Kg (408.8 mole) of Erythromycin-A base and reaction mixture is stirred at 55° C for 28 hours. After completion of the reaction, mixture is brought to ambient temperature and to it a mixture of ammonia solution (270 Kg) and water (600 Ltr) is added within 1 hour followed by 3000 Ltr of fresh water in next two hours and stirred the reaction mass for further 1 hour. White solid product obtained is centrifuged, wet cake is washed with water and dried at 6O0C for 12 hours to give 270 Kg of erythromycin-A Oxime. Melting point = 156° to 158°C.EXAMPLE 22′,4″-O-Bis(trimethylsilyl)-erythro?nycin-A-9[O-(l-methoxy-l-methyl ethyl)oximeTo a solution of 80 Kg (106.8 mole) of Erythromycin-A-9-Oxime in 400 Ltr of dichloromethane is added 38.50 Kg (534 mole) of 2-methoxy propene at 100C temperature 19.25 Kg (166.6 mole) of pyridine hydrochloride is added under stirring and the reaction mixture is stirred at 8 to 12° C for 6 hours then to it is added 19.30 Kg (119.5 mole) of HMDS and stirring is continued for 12 to 15 hours at 15° to 18°C temperature. After completion of reaction, 400 Ltr of saturated aqueous sodium carbonate solution is added and the mixture is stirred thoroughly at room temperature. Aqueous layer is further extracted with fresh DCM (100 Ltr). Both DCM extracts are mixed together and washed with water (200 Ltr) followed by brine solution (200 Ltr). The solvent is evaporated under reduced pressure. To the obtained crude solid mass is charged isopropyl alcohol (240 Ltr) and distilled out 80 Ltr of isopropyl alcohol. To the reaction mixture 160 Ltr of water is charged and stirring continued at room temperature for 1 hour. Solid crystalline product obtained is centrifuged and dried at 60° to 650C for 8 hours under vacuum to give 85 Kg of title compound. Melting point = 125° to 126°C. HPLC Purity = More than 90 % .EXAMPLE 3Clarithromycin-9- OximeTo a solution of 80 Kg (82.98 mole) of 2′,4″-O-bis(trimethylsilyl)-erythromycin-A- 9-[O-(l-methoxy methyl ethyl)Oxime] in 1200 Ltr of a mixture of dimethyl sulfoxide and diethylether (1 : 1) are added methyl iodide (20.62 Kg, 145.2 mole) and 6.48 Kg (98.35 mole) of 85 % potassium hydroxide powder and the reaction mixture is stirred for 90 minutes at room temperature. To the reaction mass is added 53 Kg of 40 % dimethylamine solution and stirring is continued for 1 hour diethylether layer is separated and DMSO layer is further extracted with fresh diethylether (200 Ltr). Combined ether layer is washed with water and concentrated in vacuum. To the obtained semi solid mass 330 Ltr of isopropyl alcohol is charged and then distilled out 165 Ltr of isopropyl alcohol. To the obtained slurry 165 Ltr of water and 21.71 Kg formic acid (99%) are added and the mixture is stirred at room temperature for 30 minutes. 622 Ltr of water is added to the reaction mixture and pH is adjusted between 10.5 and 11.5 with 25 % aqueous sodium hydroxide solution. Solid compound obtained is centrifuged and wet cake is kept as such for further reaction on the basis of moisture content. Wet weight = 95 Kg, Moisture Content = 33 %, Dried weight = 62 KgEXAMPLE 46-O-Methyl erythromycin- A (Clarithromycin)62 Kg of 6-O-Methyl erythromycin-9-Oxime is charged into a mixture of 434 Ltr of isopropyl alcohol and water (1: 1) and to it is added 38.80 Kg of sodium metabisulphite (203 mole) and then the mixture is heated to reflux for 6 to 8 hours. To the reaction mixture is charged water (620 Ltr) at ambient temperature and then the mixture is adjusted to pH about 10.5 to 11.5 by adding 25% aqueous sodium hydroxide solution and stirred for further 1 hour. White solid crude product is centrifuged, washed with water (300 Ltr), dried at 65° to 750C for 8 hours to give 40 Kg of crude Clarithromycin which on re- crystallization with chloroform isopropyl alcohol mixture provided 20 Kg of Clarithromycin (Form II).
SYNEP 0041355; US 4331803J Antibiot 1984,37(2),187-189
| EP 0147062 |
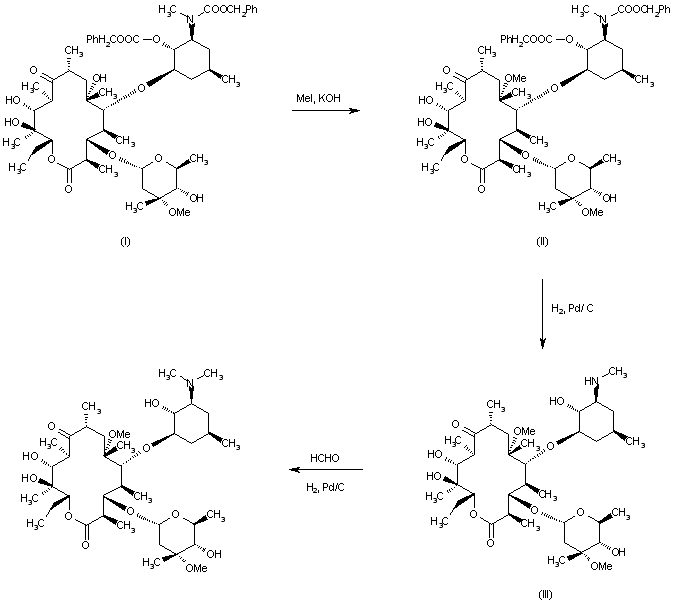
The methylation of 2′-O,N-bis(benzyloxycarbonyl)-N-demethylerythromycin A (I) with methyl iodide and KOH or NaHI in DMSO-dimethoxyethane gives the 6-O-methyl derivative (II), which is deprotected by hydrogenation with H2 over Pd/C in ethanol acetic acid affording 6-O-methyl-N-demethylerythromycin A (III). Finally, this compound is methylated with formaldehyde under reductive conditions (H2-Pd/C) in ethanol/acetic acid.
CLIP


2 Clarithromycin. Initial attempts of making clarithromycin (2) from erythromycin (1) by methylation of 8 gave approximately equal amounts of 2 and 10 by methylation at O-6 and O-11, respectively (Scheme 2, route A).[28–30] This allowed 2 to be obtained in approximately 39% yield, but it contained a small impurity of di-O-methylated 9. To improve the yields and obtain 2 in pure form, other alternatives were explored. During methylation of analogues of 8 it was observed that the conformation of the macrocyclic core plays an important role for the regioselectivity of the O-methylation.[31] As oximes are readilyhydrolysed and may have different conformations than ketone 8, oximes 11 and 13 were subjected to methylation. Interestingly, methylation of 13, but not of 11, proved to be highly selective for O-6 and provided 14 in 86% yield (Scheme2 route B); an observation which supports that 13 populates different conformations compared to 8 and 11 under the methylation conditions.[31] Compound 14 was then hydrogenated with Pd/C to deprotect the two benzyloxycarbonyl groups and the 2-chlorobenzyl group. The N-methylamine was then methylated by reductive amination and the oxime was deprotected by hydrolysis to provide clarithromycin (2). This procedure was further modified for process-scale synthesis so that clarithromycin (2) could be obtained in 70% yield starting from oxime 11 without the isolation of any intermediate.[32][28] M. Shigeo, T. Yoko, W. Yoshiaki, O. Sadafumi, J. Antibiot. 1984, 37, 187 – 189. [29] Y. Watanabe, T. Adachi, T. Asaka, M. Kashimura, S. Morimoto, Heterocycles 1990, 31, 2121 – 2124. [30] E. H. Flynn, H. W. Murphy, R. E. McMahon, J. Am. Chem. Soc. 1955, 77, 3104 – 3106. [31] Y. Watanabe, S. Morimoto, T. Adachi, M. Kashimura, T. Asaka, J. Antibiot. 1993, 46, 647 – 660.32] R. A. Dominguez, M. D. C. C. Rodriguez, L. . D. Tejo, R. N. Rib, J. S. Cebrin, J. I. B. Bilbao, 2003, US6642364B2.
References
- ^ https://www.ema.europa.eu/documents/psusa/clarithromycin-list-nationally-authorised-medicinal-products-psusa/00000788/202004_en.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “Clarithromycin”. The American Society of Health-System Pharmacists. Archivedfrom the original on September 3, 2015. Retrieved September 4, 2015.
- ^ Jump up to:a b Greenwood D (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1 ed.). Oxford: Oxford University Press. p. 239. ISBN 9780199534845. Archived from the original on 2016-03-05.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Kirst HA (2012). Macrolide Antibiotics (2 ed.). Basel: Birkhäuser Basel. p. 53. ISBN 9783034881050. Archived from the original on 2016-03-05.
- ^ Jump up to:a b c d e f g h i j k l “BIAXIN® Filmtab® (clarithromycin tablets, USP) BIAXIN® XL Filmtab® (clarithromycin extended-release tablets) BIAXIN® Granules (clarithromycin for oral suspension, USP)” (PDF). November 2, 2015. Archived (PDF) from the original on August 24, 2015. Retrieved November 2, 2015.
- ^ “Clarithromycin Side Effects in Detail – Drugs.com”. Drugs.com. Archived from the original on 2017-08-19. Retrieved 2017-08-18.
- ^ “Safety Alerts for Human Medical Products – Clarithromycin (Biaxin): Drug Safety Communication – Potential Increased Risk of Heart Problems or Death in Patients With Heart Disease”. FDA. Retrieved 24 February 2018.
- ^ Yamaguchi S, Kaneko Y, Yamagishi T, et al. [Clarithromycin-induced torsades de pointes]. Nippon Naika Gakkai Zasshi. 2003;92(1):143–5.
- ^ Winkel P, Hilden J, Fischer Hansen J, Hildebrandt P, Kastrup J, Kolmos HJ, et al. (2011). “Excess sudden cardiac deaths after short-term clarithromycin administration in the CLARICOR trial: why is this so, and why are statins protective?”. Cardiology. 118 (1): 63–7. doi:10.1159/000324533. PMID 21447948. S2CID 11873791.
- ^ Tietz A, Heim MH, Eriksson U, Marsch S, Terracciano L, Krähenbühl S (January 2003). “Fulminant liver failure associated with clarithromycin”. The Annals of Pharmacotherapy. 37 (1): 57–60. doi:10.1345/1542-6270(2003)037<0057:flfawc>2.0.co;2. PMID 12503933.
- ^ Patel AM, Shariff S, Bailey DG, Juurlink DN, Gandhi S, Mamdani M, et al. (June 2013). “Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study”. Annals of Internal Medicine. 158 (12): 869–76. doi:10.7326/0003-4819-158-12-201306180-00004. PMID 23778904. S2CID 21222679.
- ^ Gandhi S, Fleet JL, Bailey DG, McArthur E, Wald R, Rehman F, Garg AX (December 2013). “Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury”. JAMA. 310 (23): 2544–53. doi:10.1001/jama.2013.282426. PMID 24346990.
- ^ Gélisse P, Hillaire-Buys D, Halaili E, Jean-Pastor MJ, Vespignan H, Coubes P, Crespel A (November 2007). “[Carbamazepine and clarithromycin: a clinically relevant drug interaction]”. Revue Neurologique. 163 (11): 1096–9. doi:10.1016/s0035-3787(07)74183-8. PMID 18033049.
- ^ Sekar VJ, Spinosa-Guzman S, De Paepe E, De Pauw M, Vangeneugden T, Lefebvre E, Hoetelmans RM (January 2008). “Darunavir/ritonavir pharmacokinetics following coadministration with clarithromycin in healthy volunteers”. Journal of Clinical Pharmacology. 48 (1): 60–5. doi:10.1177/0091270007309706. PMID 18094220. S2CID 38368595.
- ^ Polis MA, Piscitelli SC, Vogel S, Witebsky FG, Conville PS, Petty B, et al. (August 1997). “Clarithromycin lowers plasma zidovudine levels in persons with human immunodeficiency virus infection”. Antimicrobial Agents and Chemotherapy. 41 (8): 1709–14. doi:10.1128/AAC.41.8.1709. PMC 163990. PMID 9257746.
- ^ Ferrero JL, Bopp BA, Marsh KC, Quigley SC, Johnson MJ, Anderson DJ, et al. (1990). “Metabolism and disposition of clarithromycin in man”. Drug Metabolism and Disposition. 18 (4): 441–6. PMID 1976065.
- ReferencesAllevi, P. et al.: Bioorg. Med. Chem. (BMECEP) 7, 12, 2749 (1999)Watanabe, Y. et al.: Heterocycles (HTCYAM) 31, 12, 2121 (1990).EP 158 467 (Taisho Pharmaceutical Co.; 22.3.1985; J-prior. 6.4.1984).EP 272 110 (Taisho Pharmaceutical Co.; 16.12.1987; J-prior. 17.12.1986).US 2 001 037 015 (Teva Pharm.; 15.12.2000; USA-prior. 29.2.2000).KR 2 000 043 839 (Hanmi Pharm.; ROK-prior. 29.12.1998).EP 1 150 990 (Hanmi Pharm.; 7.11.2001; ROK-prior. 29.12.1998)EP 41 355 (Taisho Pharmaceutical Co.; 27.5.1981; J-prior. 4.6.1980).Preparation of O,N-dicarbobenzoxy-N-demethylerythromycin:Flynn, E. H. et al.: J. Am. Chem. Soc. (JACSAT) 77, 3104 (1955).Process for preparation of erythromycin A oxime:US 5 808 017 (Abbott; 15.9.1998; USA-prior. 10.4.1996).Alternative synthesis of clarithromycin:Liao, G.; Zhang, G.; He, T.: Zhongguo Kangshengsu Zazhi (ZKZAEY) 27, 3, 148 (2002) (in Chinese).EP 1 134 229 (Hanmi Pharmac. Co.; 19.9.2001; ROK-prior. 15.3.2000).Crystal form 0 of clarithromycin:The Merck Index, 13th Ed., 2362, p. 408.US 5 945 405 (Abbott; 31.8.1999; USA-prior. 17.1.1997).
External links
- “Clarithromycin”. Drug Information Portal. U.S. National Library of Medicine.
- US patent 4331803, Watanabe Y, Morimoto S, Omura S, “Novel erythromycin compounds”, issued 1981-05-19, assigned to Taisho Pharmaceutical
- “FDA review finds additional data supports the potential for increased long-term risks with antibiotic clarithromycin (Biaxin) in patients with heart disease” (PDF). FDA Drug Safety Communication.
| Clinical data | |
|---|---|
| Trade names | Biaxin, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a692005 |
| License data | EU EMA: by INNUS DailyMed: Clarithromycin |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth, intravenous |
| Drug class | Macrolides |
| ATC code | J01FA09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-onlyEU: Rx-only [1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 50% |
| Protein binding | low binding |
| Metabolism | hepatic |
| Elimination half-life | 3–4 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81103-11-9 |
| PubChem CID | 84029 |
| DrugBank | DB01211 |
| ChemSpider | 10342604 |
| UNII | H1250JIK0A |
| KEGG | D00276 |
| ChEMBL | ChEMBL1741 |
| CompTox Dashboard (EPA) | DTXSID3022829 |
| ECHA InfoCard | 100.119.644 |
| Chemical and physical data | |
| Formula | C38H69NO13 |
| Molar mass | 747.964 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)OC)C)C)O)(C)O | |
| hideInChIInChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21(4)28(40)19(2)17-37(9,47-14)33(52-35-29(41)25(39(11)12)16-20(3)48-35)22(5)30(23(6)34(44)50-26)51-27-18-36(8,46-13)32(43)24(7)49-27/h19-27,29-33,35,41-43,45H,15-18H2,1-14H3/t19-,20-,21+,22+,23-,24+,25+,26-,27+,29-,30+,31-,32+,33-,35+,36-,37-,38-/m1/s1 Key:AGOYDEPGAOXOCK-KCBOHYOISA-N | |
| (what is this?) (verify) |
////////////////////CLARITHROMYCIN, Antibacterial, Antibiotics, Macrolides, A-56268, TE-031,
#CLARITHROMYCIN, #Antibacterial, #Antibiotics, #Macrolides, #A-56268, #TE-031,
ERYTHROMYCIN

Erythromycin
NSC-55929
UNII63937KV33D
CAS number114-07-8
- Molecular FormulaC37H67NO13
- Average mass733.927 Da
- эритромицин [Russian] [INN]إيريثروميسين [Arabic] [INN]红霉素 [Chinese] [INN]
IUPAC Name(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-7,12,13-trihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis ReferenceTakehiro Amano, Masami Goi, Kazuto Sekiuchi, Tomomichi Yoshida, Masahiro Hasegawa, “Process for preparing erythromycin A oxime or a salt thereof.” U.S. Patent US5274085, issued October, 1966.
US5274085ErythromycinCAS Registry Number: 114-07-8Additional Names: E-Base; E-Mycin; Erythromycin ATrademarks: Aknemycin (Hermal); Aknin (Lichtenstein); Emgel (GSK); Ery-Derm (Abbott); Erymax (Merz); Ery-Tab (Abbott); Erythromid (Abbott); ERYC (Warner-Chilcott); Erycen (APS); Erycin (Nycomed); Erycinum (Cytochemia); Ermysin (Orion); Gallimycin (Bimeda); Ilotycin (Lilly); Inderm (Dermapharm); PCE (Abbott); Retcin (DDSA); Staticin (Westwood); Stiemycin (Stiefel)Molecular Formula: C37H67NO13Molecular Weight: 733.93Percent Composition: C 60.55%, H 9.20%, N 1.91%, O 28.34%Literature References: Antibiotic substance produced by a strain of Streptomyces erythreus (Waksman) Waksman & Henrici, found in a soil sample from the Philippine Archipelago. Isoln: McGuire et al.,Antibiot. Chemother.2, 281 (1952); Bunch, McGuire, US2653899 (1953 to Lilly); Clark, Jr., US2823203 (1958 to Abbott). Properties: Flynn et al.,J. Am. Chem. Soc.76, 3121 (1954). Solubility data: Weiss et al.,Antibiot. Chemother.7, 374 (1957). Structure: Wiley et al.,J. Am. Chem. Soc.79, 6062 (1957). Configuration: Hofheinz, Grisebach, Ber.96, 2867 (1963); Harris et al.,Tetrahedron Lett.1965, 679. There are three erythromycins produced during fermentation, designated A, B, and C; A is the major and most important component. Erythromycins A and B contain the same sugar moieties, desosamine, q.v., and cladinose (3-O-methylmycarose). They differ in position 12 of the aglycone, erythronolide, A having an hydroxyl substituent. Component C contains desosamine and the same aglycone present in A but differs by the presence of mycarose, q.v., instead of cladinose. Structure of B: P. F. Wiley et al.,J. Am. Chem. Soc.79, 6070 (1957); of C: eidem,ibid. 6074. Synthesis of the aglycone, erythronolide B: E. J. Corey et al.,ibid.100, 4618, 4620 (1978); of erythronolide A: eidem,ibid.101, 7131 (1979). Asymmetric total synthesis of erythromycin A: R. B. Woodward et al.,ibid.103, 3215 (1981). NMR spectrum of A: D. J. Ager, C. K. Sood, Magn. Reson. Chem.25, 948 (1987). HPLC determn in plasma: W. Xiao et al., J. Chromatogr. B817, 153 (2005). Biosynthesis: Martin, Goldstein, Prog. Antimicrob. Anticancer Chemother., Proc. 6th Int. Congr. Chemother.II, 1112 (1970); Martin et al.,Tetrahedron31, 1985 (1975). Cloning and expression of clustered biosynthetic genes: R. Stanzak et al.,Biotechnology4, 229 (1986). Reviews: T. J. Perun in Drug Action and Drug Resistance in Bacteria1, S. Mitsuhashi, Ed. (University Park Press, Baltimore, 1977) pp 123-152; Oleinick in Antibioticsvol. 3, J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 396-419; Infection10, Suppl. 2, S61-S118 (1982). Comprehensive description: W. L. Koch, Anal. Profiles Drug Subs.8, 159-177 (1979).Properties: Hydrated crystals from water, mp 135-140°, resolidifies with second mp 190-193°. Melting point taken after drying at 56° and 8 mm. [a]D25 -78° (c = 1.99 in ethanol). uv max (pH 6.3): 280 nm (e 50). pKa1 8.8. Basic reaction. Readily forms salts with acids. Soly in water: ~2 mg/ml. Freely sol in alcohols, acetone, chloroform, acetonitrile, ethyl acetate. Moderately sol in ether, ethylene dichloride, amyl acetate.Melting point: mp 135-140°, resolidifies with second mp 190-193°pKa: pKa1 8.8Optical Rotation: [a]D25 -78° (c = 1.99 in ethanol)Absorption maximum: uv max (pH 6.3): 280 nm (e 50) Derivative Type: EthylsuccinateCAS Registry Number: 41342-53-4Trademarks: Anamycin (Chephasaar); Arpimycin (Rosemont); E.E.S. (Abbott); Eritrocina (Abbott); Eryliquid (Linden); Eryped (Abbott); Erythroped (Abbott); Esinol (Toyama); Monomycin (Grñenthal); Paediathrocin (Abbott); Pediamycin (Abbott); Refkas (Maruko)Molecular Formula: C43H75NO16Molecular Weight: 862.05Percent Composition: C 59.91%, H 8.77%, N 1.62%, O 29.70%Literature References: Prepn: GB830846; R. K. Clark, US2967129 (1960, 1961 both to Abbott).Properties: Hydrated crystals from acetone + water, mp 109-110°. [a]D -42.5°.Melting point: mp 109-110°Optical Rotation: [a]D -42.5° Therap-Cat: Antibacterial.Therap-Cat-Vet: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
NEW DRUG APPROVALS
one time
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Erythromycin estolate | XRJ2P631HP | 3521-62-8 | AWMFUEJKWXESNL-JZBHMOKNSA-N |
| Erythromycin ethylsuccinate | 1014KSJ86F | 1264-62-6 | NSYZCCDSJNWWJL-YXOIYICCSA-N |
| Erythromycin gluceptate | 2AY21R0U64 | 23067-13-2 | ZXBDZLHAHGPXIG-VTXLJDRKSA-N |
| Erythromycin lactobionate | 33H58I7GLQ | 3847-29-8 | NNRXCKZMQLFUPL-WBMZRJHASA-N |
| Erythromycin phosphate | I8T8KU14X7 | 4501-00-2 | VUEMAFLGEMYXIH-YZPBMOCRSA-N |
| Erythromycin stearate | LXW024X05M | 643-22-1 | YAVZHCFFUATPRK-YZPBMOCRSA-N |
| Erythromycin sulfate | KVW9N83AME | 7184-72-7 | XTSSJGRRFMNXGO-YZPBMOCRSA-N |
| Erythromycin thiocyanate | Y7A95YRI88 | 7704-67-8 | WVRRTEYLDPNZHR-YZPBMOCRSA-N |
Erythromycin is an antibiotic which belongs to the group of macrolide antibiotics. The pharmaceutically distributed product consists of three components: Erythromycin A, B, and C where Erythromycin A represents the main component. Naturally this antibiotic is synthesized by the grampositive bacteria Streptomyces erythreus (Saccharopolyspora erythrea).
In 1949 Erythromycin was found for the first time in a soil sample in the Philippine region Iloilo. A research team, led by J. M. McGuire, was able to isolate Erythromycin which was part of the soil sample. Under the brand name Ilosone the product was launched commercially in 1952. They named the brand after the region where the antibiotic was found. Analogically the first product name was Ilotycin. Furthermore, in 1953 the U.S. patent was granted. Since 1957 the structure of Erythromycin is known and in 1965 the X-ray structure analysis gave awareness of the absolute configuration. In 1981, almost 30 years after the detection of Erythromycin, Robert B. Woodward, the Nobel prize laureate of chemistry in 1965, and his coworkers posthumously reported the first synthesis of Erythromycin A
The structural characteristic of macrolides, to which Erythromycin affiliates, is a macrocyclic lactone ring of fourteen, fifteen or sixteen members. In case of Erythromycin the lactone ring consists of 14-members. Substituents on the mainchain are cladinose on C-3 and desosamine on C-5. Erythromycin is not a single compound but represents an alloy of structural very similar components. The main constituents are Erythromycin A, B and C. As shown in Table 1 and Figure 1 they only differ in two rests on the lactone ring or on the cladinose each case. In addition to the variants already mentioned, further variants, like Erythromycin D and E are known. They are pre- and post-stages in the biosynthesis and often do not have antibiotic effects
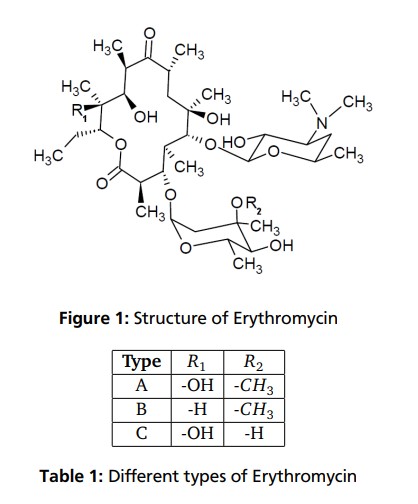
Chemical and Pharmacokinetic Properties Formula: C37H67NO13 CAS-Number: 114-07-8 Molar Mass: 733.93g/mol Half Hife 1.5 hours pkA: 8,6 – 8,8 Melting Point: 411K (hydrat) 463-466K (anhydrous)
Erythromycin is an antibiotic used for the treatment of a number of bacterial infections.[1] This includes respiratory tract infections, skin infections, chlamydia infections, pelvic inflammatory disease, and syphilis.[1] It may also be used during pregnancy to prevent Group B streptococcal infection in the newborn,[1] as well as to improve delayed stomach emptying.[3] It can be given intravenously and by mouth.[1] An eye ointment is routinely recommended after delivery to prevent eye infections in the newborn.[4]
Common side effects include abdominal cramps, vomiting, and diarrhea.[1] More serious side effects may include Clostridium difficile colitis, liver problems, prolonged QT, and allergic reactions.[1] It is generally safe in those who are allergic to penicillin.[1] Erythromycin also appears to be safe to use during pregnancy.[2] While generally regarded as safe during breastfeeding, its use by the mother during the first two weeks of life may increase the risk of pyloric stenosis in the baby.[5][6] This risk also applies if taken directly by the baby during this age.[7] It is in the macrolide family of antibiotics and works by decreasing bacterial protein production.[1]
Erythromycin was first isolated in 1952 from the bacteria Saccharopolyspora erythraea.[1][8] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[9] The World Health Organization classifies it as critically important for human medicine.[10] It is available as a generic medication.[5] In 2017, it was the 215th most commonly prescribed medication in the United States, with more than two million prescriptions.[11][12]
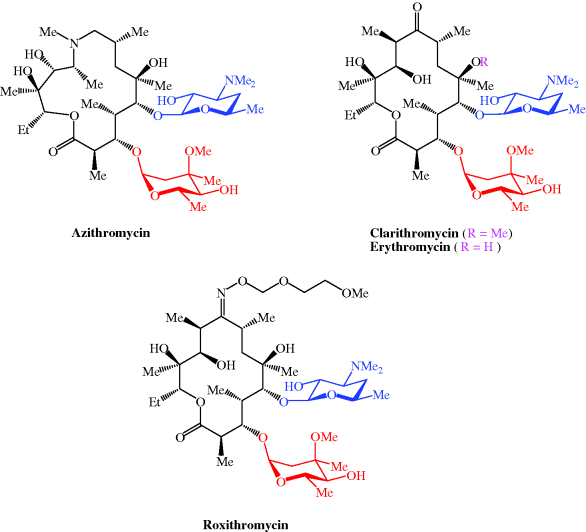
Table 4.2.1 Therapeutic indications for the macrolide antibiotics.
Medical uses
Erythromycin can be used to treat bacteria responsible for causing infections of the skin and upper respiratory tract, including Streptococcus, Staphylococcus, Haemophilus and Corynebacterium genera. The following represents MIC susceptibility data for a few medically significant bacteria:[13]
- Haemophilus influenzae: 0.015 to 256 μg/ml
- Staphylococcus aureus: 0.023 to 1024 μg/ml
- Streptococcus pyogenes: 0.004 to 256 μg/ml
- Corynebacterium minutissimum: 0.015 to 64 μg/ml
It may be useful in treating gastroparesis due to this promotility effect. It has been shown to improve feeding intolerances in those who are critically ill.[14] Intravenous erythromycin may also be used in endoscopy to help clear stomach contents.
Available forms

Enteric-coated erythromycin capsule from Abbott Labs
Erythromycin is available in enteric-coated tablets, slow-release capsules, oral suspensions, ophthalmic solutions, ointments, gels, enteric-coated capsules, non enteric-coated tablets, non enteric-coated capsules, and injections. The following erythromycin combinations are available for oral dosage:[15]
- erythromycin base (capsules, tablets)
- erythromycin estolate (capsules, oral suspension, tablets), contraindicated during pregnancy[16]
- erythromycin ethylsuccinate (oral suspension, tablets)
- erythromycin stearate (oral suspension, tablets)
For injection, the available combinations are:[15]
- erythromycin gluceptate
- erythromycin lactobionate
For ophthalmic use:
- erythromycin base (ointment)
Adverse effects
Gastrointestinal disturbances, such as diarrhea, nausea, abdominal pain, and vomiting, are very common because erythromycin is a motilin agonist.[17] Because of this, erythromycin tends not to be prescribed as a first-line drug.
More serious side effects include arrhythmia with prolonged QT intervals, including torsades de pointes, and reversible deafness. Allergic reactions range from urticaria to anaphylaxis. Cholestasis, Stevens–Johnson syndrome, and toxic epidermal necrolysis are some other rare side effects that may occur.
Studies have shown evidence both for and against the association of pyloric stenosis and exposure to erythromycin prenatally and postnatally.[18] Exposure to erythromycin (especially long courses at antimicrobial doses, and also through breastfeeding) has been linked to an increased probability of pyloric stenosis in young infants.[19][20] Erythromycin used for feeding intolerance in young infants has not been associated with hypertrophic pyloric stenosis.[19]
Erythromycin estolate has been associated with reversible hepatotoxicity in pregnant women in the form of elevated serum glutamic-oxaloacetic transaminase and is not recommended during pregnancy. Some evidence suggests similar hepatotoxicity in other populations.[21]
It can also affect the central nervous system, causing psychotic reactions, nightmares, and night sweats.[22]
Interactions
Erythromycin is metabolized by enzymes of the cytochrome P450 system, in particular, by isozymes of the CYP3A superfamily.[23] The activity of the CYP3A enzymes can be induced or inhibited by certain drugs (e.g., dexamethasone), which can cause it to affect the metabolism of many different drugs, including erythromycin. If other CYP3A substrates — drugs that are broken down by CYP3A — such as simvastatin (Zocor), lovastatin (Mevacor), or atorvastatin (Lipitor)—are taken concomitantly with erythromycin, levels of the substrates increase, often causing adverse effects. A noted drug interaction involves erythromycin and simvastatin, resulting in increased simvastatin levels and the potential for rhabdomyolysis. Another group of CYP3A4 substrates are drugs used for migraine such as ergotamine and dihydroergotamine; their adverse effects may be more pronounced if erythromycin is associated.[22] Earlier case reports on sudden death prompted a study on a large cohort that confirmed a link between erythromycin, ventricular tachycardia, and sudden cardiac death in patients also taking drugs that prolong the metabolism of erythromycin (like verapamil or diltiazem) by interfering with CYP3A4.[24] Hence, erythromycin should not be administered to people using these drugs, or drugs that also prolong the QT interval. Other examples include terfenadine (Seldane, Seldane-D), astemizole (Hismanal), cisapride (Propulsid, withdrawn in many countries for prolonging the QT time) and pimozide (Orap). Theophylline, which is used mostly in asthma, is also contraindicated.
Erythromycin and doxycycline can have a synergistic effect when combined and kill bacteria (E. coli) with a higher potency than the sum of the two drugs together. This synergistic relationship is only temporary. After approximately 72 hours, the relationship shifts to become antagonistic, whereby a 50/50 combination of the two drugs kills less bacteria than if the two drugs were administered separately.[25]
It may alter the effectiveness of combined oral contraceptive pills because of its effect on the gut flora. A review found that when erythromycin was given with certain oral contraceptives, there was an increase in the maximum serum concentrations and AUC of estradiol and dienogest.[26][27]
Erythromycin is an inhibitor of the cytochrome P450 system, which means it can have a rapid effect on levels of other drugs metabolised by this system, e.g., warfarin.
Pharmacology
Mechanism of action
Erythromycin displays bacteriostatic activity or inhibits growth of bacteria, especially at higher concentrations.[28] By binding to the 50s subunit of the bacterial rRNA complex, protein synthesis and subsequent structure and function processes critical for life or replication are inhibited.[28] Erythromycin interferes with aminoacyl translocation, preventing the transfer of the tRNA bound at the A site of the rRNA complex to the P site of the rRNA complex. Without this translocation, the A site remains occupied, thus the addition of an incoming tRNA and its attached amino acid to the nascent polypeptide chain is inhibited. This interferes with the production of functionally useful proteins, which is the basis of this antimicrobial action.
Erythromycin increases gut motility by binding to Motillin, thus it is a Motillin receptor agonist in addition to its antimicrobial properties.
Pharmacokinetics
Erythromycin is easily inactivated by gastric acid; therefore, all orally administered formulations are given as either enteric-coated or more-stable salts or esters, such as erythromycin ethylsuccinate. Erythromycin is very rapidly absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, erythromycin is actively transported to the site of infection, where, during active phagocytosis, large concentrations of erythromycin are released.
Metabolism
Most of erythromycin is metabolised by demethylation in the liver by the hepatic enzyme CYP3A4. Its main elimination route is in the bile with little renal excretion, 2%-15% unchanged drug. Erythromycin’s elimination half-life ranges between 1.5 and 2.0 hours and is between 5 and 6 hours in patients with end-stage renal disease. Erythromycin levels peak in the serum 4 hours after dosing; ethylsuccinate peaks 0.5-2.5 hours after dosing, but can be delayed if digested with food.[29]
Erythromycin crosses the placenta and enters breast milk. The American Association of Pediatrics determined erythromycin is safe to take while breastfeeding.[30] Absorption in pregnant patients has been shown to be variable, frequently resulting in levels lower than in nonpregnant patients.[29]
Chemistry
Composition
Standard-grade erythromycin is primarily composed of four related compounds known as erythromycins A, B, C, and D. Each of these compounds can be present in varying amounts and can differ by lot. Erythromycin A has been found to have the most antibacterial activity, followed by erythromycin B. Erythromycins C and D are about half as active as erythromycin A.[13][31] Some of these related compounds have been purified and can be studied and researched individually.
Synthesis
Over the three decades after the discovery of erythromycin A and its activity as an antimicrobial, many attempts were made to synthesize it in the laboratory. The presence of 10 stereogenic carbons and several points of distinct substitution has made the total synthesis of erythromycin A a formidable task.[32] Complete syntheses of erythromycins’ related structures and precursors such as 6-deoxyerythronolide B have been accomplished, giving way to possible syntheses of different erythromycins and other macrolide antimicrobials.[33] Woodward successfully completed the synthesis of erythromycin A.[34][35][36]
Erythromycin related compounds
History
In 1949 Abelardo B. Aguilar, a Filipino scientist, sent some soil samples to his employer Eli Lilly. Eli Lilly’s research team, led by J. M. McGuire, managed to isolate erythromycin from the metabolic products of a strain of Streptomyces erythreus (designation changed to Saccharopolyspora erythraea) found in the samples.[37]
Lilly filed for patent protection on the compound which was granted in 1953.[38] The product was launched commercially in 1952 under the brand name Ilosone (after the Philippine region of Iloilo where it was originally collected). Erythromycin was formerly also called Ilotycin.
The antibiotic clarithromycin was invented by scientists at the Japanese drug company Taisho Pharmaceutical in the 1970s as a result of their efforts to overcome the acid instability of erythromycin.
Scientists at Chugai Pharmaceuticals discovered an erythromycin-derived motilin agonist called mitemcinal that is believed to have strong prokinetic properties (similar to erythromycin) but lacking antibiotic properties. Erythromycin is commonly used off-label for gastric motility indications such as gastroparesis. If mitemcinal can be shown to be an effective prokinetic agent, it would represent a significant advance in the gastrointestinal field, as treatment with this drug would not carry the risk of unintentional selection for antibiotic-resistant bacteria.
Society and culture
Cost
It is available as a generic medication.[5]
In the United States in 2014 the price increased to seven dollars per tablet.[39]
The price of Erythromycin rose three times between 2010 and 2015, from 24 cents per tablet in 2010 to $8.96 in 2015.[40] In 2017, a Kaiser Health News study found that the per-unit cost of dozens of generics doubled or even tripled from 2015 to 2016, increasing spending by the Medicaid program. Due to price increases by drug manufacturers, Medicaid paid on average $2,685,330 more for Erythromycin in 2016 compared to 2015 (not including rebates).[41] By 2018, generic drug prices had climbed another 5% on average.[42]
Brand names
Brand names include Robimycin, E-Mycin, E.E.S. Granules, E.E.S.-200, E.E.S.-400, E.E.S.-400 Filmtab, Erymax, Ery-Tab, Eryc, Ranbaxy, Erypar, EryPed, Eryped 200, Eryped 400, Erythrocin Stearate Filmtab, Erythrocot, E-Base, Erythroped, Ilosone, MY-E, Pediamycin, Zineryt, Abboticin, Abboticin-ES, Erycin, PCE Dispertab, Stiemycine, Acnasol, and Tiloryth.
See also
Erythromycin/tretinoin, a combination of tretinoin and the antibiotic erythromycin
SYN
https://basicmedicalkey.com/macrolide-antibiotics/embed/#?secret=VMg8PBg4K9
Synthesis
The total synthesis of the erythromycins (Figure 4.2.2) poses a supreme challenge and has attracted the attention of some of the world’s most eminent synthetic chemists, leading to many elegant examples of the total synthesis of complex natural products. The total synthesis of the erythronolide A aglycone (lacking the sugar units) was first reported by E. J. Corey (Nobel Prize in Chemistry in 1990) in a series of articles in the late 1970s (Scheme 4.2.2) (Corey et al., 1979 and references cited therein), and the total synthesis of erythromycin (known then as erythromycin A) by R. B. Woodward (Nobel Prize in Chemistry in 1965) in a series of articles in 1981, after his death (Scheme 4.2.3) (Woodward et al., 1981 and references cited therein). The Woodward synthesis is particularly elegant, as the dithiadecalin intermediate supplies both the C3-C8 and C9-C13 fragments (Scheme 4.2.3).
Figure 4.2.2 Erythromycins A and B and their aglycones, erythronolides A and B
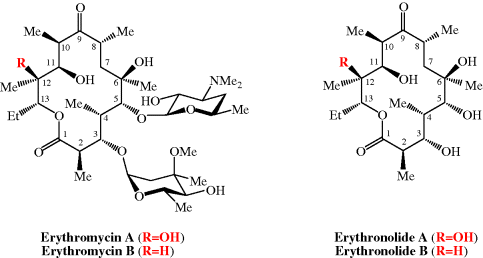
Scheme 4.2.2 Corey’s total synthesis of erythronolide A (38 steps from the cyclohexadiene fragment; 0.04% overall yield)
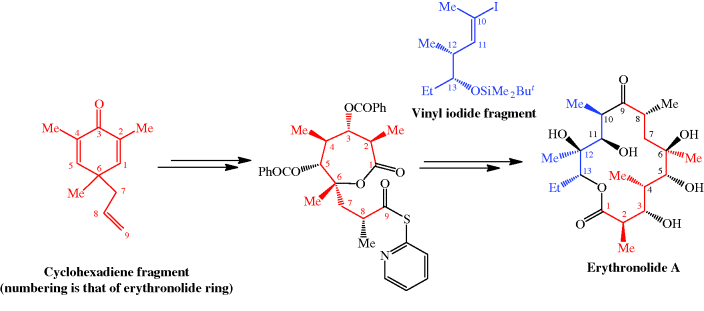
Scheme 4.2.3 Woodward’s total synthesis of erythromycin (56 steps from 4-thianone; 0.01% overall yield)
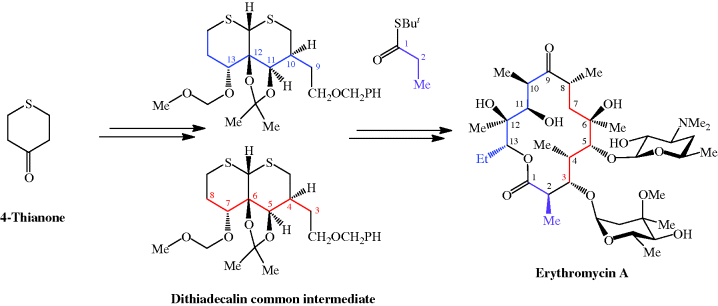
Once again, erythromycin is such a complex antibiotic that its commercial production by total synthesis will never be feasible, and it is obtained from the submerged culture of free or immobilised Saccharopolyspora erythraea (El-Enshasy et al., 2008).
We have now seen a number of examples of how very complex semi-synthetic antibiotics can be prepared through the combination of fermentation (to give the complex natural product) and chemical modification, so you will no doubt already have spotted that both clarithromycin and roxithromycin are semi-synthetic macrolide antibiotics. Clarithromycin can be obtained in a five-step synthetic procedure, from erythromycin oxime (Brunet et al., 2007), while roxithromycin can also be prepared from this oxime (Massey et al., 1970) in a single step (Scheme 4.2.4) (Gouin d’Ambrieres et al., 1982). What is not so obvious is that azithromycin is also a semi-synthetic macrolide, having originally been produced by PLIVA Pharmaceuticals from erythromycin oxime via a sequence of reactions which included the well-known Beckmann rearrangement (Djoki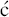 et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
Scheme 4.2.4 Preparation of the semi-synthetic macrolide antibiotic roxithromycin
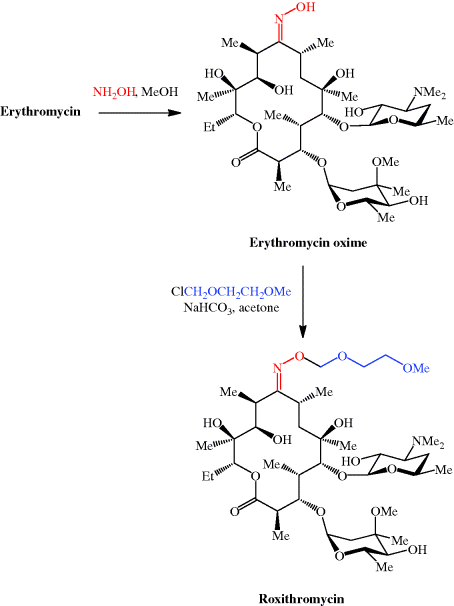
CLIP

Erythromycin. Erythromycin (1) was discovered in 1952 during the investigation of soil samples from Iloilo, Philippines for antibiotic activity[18, 19] and its molecular structure was assigned in 1957.[20] The microorganism that produced erythromycin was isolated and characterised as Streptomyces erythreus, strain NRRL 2338.[18, 19] Over the years, strain improvements and genetic engineering has allowed the yield of erythromycin to be increased so that 8–10 g L1 can now be produced from a tryptic soy broth.[21–25] Erythromycin forms anhydro-erythromycin 6 and 6:9, 9:12 spiroketal 7 under the acidic conditions in the stomach (Scheme 1), which results in the loss of its antibacterial activity and induction of abdominal pain.[26, 27] Generation of by-products 6 and 7 occurs through an acid-catalysed intramolecular reaction of the C-6 hydroxyl group with the C-9 keto moiety. To avoid this by-product formation several different semi-synthetic derivatives of erythromycin have been prepared in which either of these two functionalities are modified. They led to the discovery of clarithromycin (2) by O-6 methylation of erythromycin (Figure 3). Removal of the C-9 ketone by the formation of an oxime followed by Beckmann rearrangement and reduction led to azithromycin (3), which belongs to a new class of macrolides called “azalides”. Alternatively, conversion of the C-9 ketone to an amine, followed by reaction with an aldehyde, gave dirithromycin (4). Yet another approach involved the transformation of clarithromycin to the conformationally restricted telithromycin
SYN
Chemical Synthesis
Erythromycin, (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-Cmethyl-3-O-methyl-α-L-ribo-hexopyranosyl)-oxy]-14-ethyl-7,12,13-trihydroxy- 3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy ]oxacyclotetradecan-2,10-dione (32.2.1), is more specifically called erythromycin A. It was first isolated in 1952 from the culture liquid of microorganisms of the type Streptomyces erytherus. Minor amounts of erythromycin B and C were also found in the culture fluid. Erythromycin B differs from A in that a hydrogen atom is located at position 12 in the place of a hydroxyl group, while erythromycin C differs from A in that the residue of a different carbohydrate, micarose (2-6-di-deoxy-3-C-methyl-L-ribohexose), is bound to the macrocycle in position 3 in the place of cladinose (4-methoxy-2,4-dimethyl-tetrahydropyran-3,6-diol).
Erythromycin A is produced only microbiologically using active strains of microorganisms of the type Saccharopolospora erythraea.
SYN

Erythromycin synthesis by modular polyketide synthases. The three genes EryAI-III encode three proteins of PKS: DEBS1 (the loading module, modules 1, 2) DEBS2 (modules 3, 4), DEBS3 (modules 5, 6, TE domain). Thus, PKS consists of the loading module, six extension modules, and TE domain. Each module includes from three to six domains: AT-acyl transferase, ACP-acyl carrier protein, KS-ketosynthase, KR-ketoreductase, DH-dehydratase, ERenoyl reductase.
CLIP
The chemical synthesis of Erythromycin poses a huge challenge. The molecule contains ten stereogenic centers of which five are arranged consecutively. R. B. Woodward and his research team first succeeded in synthesizing Erythromycin A. The reaction sequence, however, is so complicated that the yield was only about 0,02 % and, thus, the synthesis is not utilizable comercially. This is the reason for the preferred use of the biosynthesis of Erythromycin via fermentation of Streptomyces erythreus. Other scientists and research teams dealt with the synthesis of Erythromycin as well and developed very similar approaches. Most methods for the Erythromycin synthesis are based on the construction of the aglycon from secoic acid via glycosylation. Indeed the process is also possible inversely: first, a glycosylation, then a lactonization occurs. The yield, however, is considerably less. While earlier scientist mainly dealt with the production of the different secoic acids, the lactonization process is the major problem today because there is no fully developed method for it yet. A lot of side reactions such as dimerization and polymerization appear, because a 14 membered ring is hard to enclose. Even if the chemical synthesis of Erythromycin has no importance for the comercial fabrication of the antibiotic, it is still important for the development and fabrication of its derivatives.
References
- ^ Jump up to:a b c d e f g h i j k “Erythromycin”. The American Society of Health-System Pharmacists. Archived from the original on 2015-09-06. Retrieved Aug 1, 2015.
- ^ Jump up to:a b “Prescribing medicines in pregnancy database”. Australian Government. August 23, 2015. Archived from the original on April 8, 2014.
- ^ Camilleri M, Parkman HP, Shafi MA, Abell TL, Gerson L (January 2013). “Clinical guideline: management of gastroparesis”. The American Journal of Gastroenterology. 108 (1): 18–37, quiz 38. doi:10.1038/ajg.2012.373. PMC 3722580. PMID 23147521.
- ^ Matejcek A, Goldman RD (November 2013). “Treatment and prevention of ophthalmia neonatorum”. Canadian Family Physician. 59 (11): 1187–90. PMC 3828094. PMID 24235191.
- ^ Jump up to:a b c Hamilton RJ (2013). Tarascon pocket pharmacopoeia(2013 delux lab-coat ed., 14th ed.). [Sudbury, Mass.]: Jones & Bartlett Learning. p. 72. ISBN 9781449673611.
- ^ Kong YL, Tey HL (June 2013). “Treatment of acne vulgaris during pregnancy and lactation”. Drugs. 73 (8): 779–87. doi:10.1007/s40265-013-0060-0. PMID 23657872. S2CID 45531743.
- ^ Maheshwai N (March 2007). “Are young infants treated with erythromycin at risk for developing hypertrophic pyloric stenosis?”. Archives of Disease in Childhood. 92 (3): 271–3. doi:10.1136/adc.2006.110007. PMC 2083424. PMID 17337692. Archived from the original on 7 November 2012.
- ^ Vedas JC (2000). Biosynthesis : polyketides and vitamins. Berlin [u.a.]: Springer. p. 52. ISBN 9783540669692.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN 9789241515528. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2020”. ClinCalc. Retrieved 11 April 2020.
- ^ “Erythromycin – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ Jump up to:a b “Erythromycin Susceptibility and Minimum Inhibitory Concentration (MIC) Data” (PDF). TOKU-E.
- ^ Lewis K, Alqahtani Z, Mcintyre L, Almenawer S, Alshamsi F, Rhodes A, et al. (August 2016). “The efficacy and safety of prokinetic agents in critically ill patients receiving enteral nutrition: a systematic review and meta-analysis of randomized trials”. Critical Care. 20 (1): 259. doi:10.1186/s13054-016-1441-z. PMC 4986344. PMID 27527069.
- ^ Jump up to:a b “Erythromycin Oral, Parenteral Advanced Patient Information”. Drugs.com. Archived from the original on 2009-11-30.
- ^ Sexually Transmitted Diseases Treatment Guidelines 2006Archived 2010-02-11 at the Wayback Machine Centers for Disease Control and Prevention. MMWR 2006;55
- ^ Weber FH, Richards RD, McCallum RW (April 1993). “Erythromycin: a motilin agonist and gastrointestinal prokinetic agent”. The American Journal of Gastroenterology. 88 (4): 485–90. PMID 8470625.
- ^ “Pregnancy and lactation”. Archived from the original on 2014-04-20.
- ^ Jump up to:a b Maheshwai N (March 2007). “Are young infants treated with erythromycin at risk for developing hypertrophic pyloric stenosis?”. Archives of Disease in Childhood. 92 (3): 271–3. doi:10.1136/adc.2006.110007. PMC 2083424. PMID 17337692.
- ^ Lund M, Pasternak B, Davidsen RB, Feenstra B, Krogh C, Diaz LJ, et al. (March 2014). “Use of macrolides in mother and child and risk of infantile hypertrophic pyloric stenosis: nationwide cohort study”. BMJ. 348: g1908. doi:10.1136/bmj.g1908. PMC 3949411. PMID 24618148.
- ^ McCormack WM, George H, Donner A, Kodgis LF, Alpert S, Lowe EW, Kass EH (November 1977). “Hepatotoxicity of erythromycin estolate during pregnancy”. Antimicrobial Agents and Chemotherapy. 12 (5): 630–5. doi:10.1128/AAC.12.5.630. PMC 429989. PMID 21610.
- ^ Jump up to:a b “Erythromycine”. Belgisch Centrum voor Farmacotherapeutische Informatie. Archived from the original on 2015-10-06.
- ^ Hunt CM, Watkins PB, Saenger P, Stave GM, Barlascini N, Watlington CO, et al. (January 1992). “Heterogeneity of CYP3A isoforms metabolizing erythromycin and cortisol” (PDF). Clinical Pharmacology and Therapeutics. 51 (1): 18–23. doi:10.1038/clpt.1992.3. hdl:2027.42/109905. PMID 1732074. S2CID 28056649.
- ^ Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM (September 2004). “Oral erythromycin and the risk of sudden death from cardiac causes”. The New England Journal of Medicine. 351 (11): 1089–96. doi:10.1056/NEJMoa040582. PMID 15356306.
- ^ Pena-Miller R, Laehnemann D, Jansen G, Fuentes-Hernandez A, Rosenstiel P, Schulenburg H, Beardmore R (April 23, 2013). “When the most potent combination of antibiotics selects for the greatest bacterial load: the smile-frown transition”. PLOS Biology. 11 (4): e1001540. doi:10.1371/journal.pbio.1001540. PMC 3635860. PMID 23630452.
- ^ Blode H, Zeun S, Parke S, Zimmermann T, Rohde B, Mellinger U, Kunz M (October 2012). “Evaluation of the effects of rifampicin, ketoconazole and erythromycin on the steady-state pharmacokinetics of the components of a novel oral contraceptive containing estradiol valerate and dienogest in healthy postmenopausal women”. Contraception. 86 (4): 337–44. doi:10.1016/j.contraception.2012.01.010. PMID 22445438.
- ^ Simmons KB, Haddad LB, Nanda K, Curtis KM (January 2018). “Drug interactions between non-rifamycin antibiotics and hormonal contraception: a systematic review”. American Journal of Obstetrics and Gynecology. 218 (1): 88–97.e14. doi:10.1016/j.ajog.2017.07.003. PMID 28694152. S2CID 36567820.
- ^ Jump up to:a b Katzung PHARMACOLOGY, 9e Section VIII. Chemotherapeutic Drugs Chapter 44. Chloramphenicol, Tetracyclines, Macrolides, Clindamycin, & Streptogramins
- ^ Jump up to:a b “unknown”. Archived from the original on 2014-04-19. Cite uses generic title (help)
- ^ American Academy of Pediatrics Committee on Drugs (September 2001). “Transfer of drugs and other chemicals into human milk” (PDF). Pediatrics. 108 (3): 776–89. doi:10.1542/peds.108.3.776. PMID 11533352. Archived from the original (PDF) on 2016-03-05.
- ^ Kibwage IO, Hoogmartens J, Roets E, Vanderhaeghe H, Verbist L, Dubost M, et al. (November 1985). “Antibacterial activities of erythromycins A, B, C, and D and some of their derivatives”. Antimicrobial Agents and Chemotherapy. 28 (5): 630–3. doi:10.1128/aac.28.5.630. PMC 176346. PMID 4091529.
- ^ Pal S (2006). “A journey across the sequential development of macrolides and ketolides related to erythromycin”. Tetrahedron. 62(14): 3171–3200. doi:10.1016/j.tet.2005.11.064.
- ^ Evans DA, Kim AS (1997). “Synthesis of 6-Deoxyerythronolide B. Implementation of a General Strategy for the Synthesis of Macrolide Antibiotics”. Tetrahedron Lett. 38: 53–56. doi:10.1016/S0040-4039(96)02258-7.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 1. Synthesis of an Erythronolide A Seco Acid Derivative via Asymmetric Induction”. J. Am. Chem. Soc. 103 (11): 3210–3213. doi:10.1021/ja00401a049.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 2. Synthesis of an Erythronolide A Lactone System”. J. Am. Chem. Soc. 103 (11): 3213–3215. doi:10.1021/ja00401a050.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 3. Total Synthesis of Erythromycin”. J. Am. Chem. Soc. 103 (11): 3215–3217. doi:10.1021/ja00401a051.
- ^ Hibionada, F. Remembering the battle of Dr. Abelardo Aguilar: Cure for millions, deprived of millions. The News Today. Retrieved 22 September 2015
- ^ U.S. Patent 2,653,899
- ^ Stahl, Stephanie (September 26, 2014) Health: Generic Drugs Prices Increasing Archived 2016-04-09 at the Wayback Machine CBS Philadelphia. Retrieved March 24, 2016.
- ^ “Some Generic Drugs See Huge Price Increases”. http://www.medscape.com. Retrieved 29 June 2018.
- ^ “Climbing cost of decades-old drugs threatens to break Medicaid bank”. Philly.com. Retrieved 29 June 2018.
- ^ “Are Drugs Really Getting More Expensive? Yes”. The GoodRx Prescription Savings Blog. 27 February 2018. Retrieved 29 June2018.
External links
- “Erythromycin”. Drug Information Portal. U.S. National Library of Medicine.
- U.S. Patent 2,653,899
| Clinical data | |
|---|---|
| Trade names | Eryc, Erythrocin, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682381 |
| License data | US DailyMed: Erythromycin |
| Pregnancy category | AU: A[2] |
| Routes of administration | By mouth, intravenous (IV), intramuscular (IM), topical, eye drops |
| Drug class | Macrolide antibiotic |
| ATC code | D10AF02 (WHO) J01FA01 (WHO) S01AA17 (WHO) QJ51FA01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | Depends on the ester type between 30% – 65% |
| Protein binding | 90% |
| Metabolism | liver (under 5% excreted unchanged) |
| Elimination half-life | 1.5 hours |
| Excretion | bile |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 114-07-8 |
| PubChem CID | 12560 |
| IUPHAR/BPS | 1456 |
| DrugBank | DB00199 |
| ChemSpider | 12041 |
| UNII | 63937KV33D |
| KEGG | D00140 |
| ChEBI | CHEBI:42355 |
| ChEMBL | ChEMBL532 |
| PDB ligand | ERY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID4022991 |
| ECHA InfoCard | 100.003.673 |
| Chemical and physical data | |
| Formula | C37H67NO13 |
| Molar mass | 733.937 g·mol−1 |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)O)C)C)O)(C)O | |
| hideInChIInChI=1S/C37H67NO13/c1-14-25-37(10,45)30(41)20(4)27(39)18(2)16-35(8,44)32(51-34-28(40)24(38(11)12)15-19(3)47-34)21(5)29(22(6)33(43)49-25)50-26-17-36(9,46-13)31(42)23(7)48-26/h18-26,28-32,34,40-42,44-45H,14-17H2,1-13H3/t18-,19-,20+,21+,22-,23+,24+,25-,26+,28-,29+,30-,31+,32-,34+,35-,36-,37-/m1/s1 Key:ULGZDMOVFRHVEP-RWJQBGPGSA-N | |
| (verify) |
//////////erythromycin, NSC-55929, NSC 55929, эритромицин , إيريثروميسين , 红霉素 , ANTIBACTERIAL, MACROLIDES, ANTIBIOTICS
#erythromycin, #NSC-55929, #NSC 55929, #эритромицин , #إيريثروميسين , #红霉素 , #ANTIBACTERIAL, #MACROLIDES, #ANTIBIOTICS
Amikacin sulfate

Amikacin sulfate
アミカシン硫酸塩 , BB K 8
| Formula | C22H43N5O13. 2H2SO4 |
|---|---|
| CAS | 39831-55-5FREE 37517-28-5 |
| Mol weight | 781.7595 |
EU APPROVED, 2020/10/27, Arikayce liposomal
Antibacterial, Protein biosynthesis inhibitor
(2S)-4-amino-N-[(1R,2S,3S,4R,5S)-5-amino-2-[(2S,3R,4S,5S,6R)-4-amino-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4-[(2R,3R,4S,5S,6R)-6-(aminomethyl)-3,4,5-trihydroxyoxan-2-yl]oxy-3-hydroxycyclohexyl]-2-hydroxybutanamide;sulfuric acid AmikacinCAS Registry Number: 37517-28-5
CAS Name:O-3-Amino-3-deoxy-a-D-glucopyranosyl-(1®6)-O-[6-amino-6-deoxy-a-D-glucopyranosyl-(1®4)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine
Additional Names: 1-N-[L(-)-4-amino-2-hydroxybutyryl]kanamycin AMolecular Formula: C22H43N5O13Molecular Weight: 585.60Percent Composition: C 45.12%, H 7.40%, N 11.96%, O 35.52%
Literature References: Semisynthetic aminoglycoside antibiotic derived from kanamycin A. Prepn: Kawaguchi et al.,J. Antibiot.25, 695 (1972); H. Kawaguchi, T. Naito, DE2234315; H. Kawaguchi et al.,US3781268 (both 1973 to Bristol-Myers). Biological formation from kanamycin A: L. M. Cappelletti, R. Spagnoli, J. Antibiot.36, 328 (1983). Microbiological evaluation: Price et al.,ibid.25, 709 (1972). Pharmacokinetics: Cabana, Taggart, Antimicrob. Agents Chemother.3, 478 (1973). In vitro studies: Yu, Washington, ibid.4, 133 (1973); Bodey, Stewart, ibid. 186. Pharmacology in humans: Bodey et al.,ibid.5, 508 (1974). Toxicity studies: Fujisawa et al.,J. Antibiot.27, 677 (1974). Review: K. A. Kerridge in Pharmacological and Biochemical Properties of Drug Substancesvol. 1, M. E. Goldberg, Ed. (Am. Pharm. Assoc., Washington, DC, 1977) pp 125-153. Comprehensive description: P. M. Monteleone et al.,Anal. Profiles Drug Subs.12, 37-71 (1983).Properties: White crystalline powder from methanol-isopropanol, mp 203-204° (sesquihydrate). [a]D23 +99° (c = 1.0 in water). LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi).Melting point: mp 203-204° (sesquihydrate)Optical Rotation: [a]D23 +99° (c = 1.0 in water)Toxicity data: LD50 in mice of solns pH 6.6, pH 7.4 (mg/kg): 340, 560 i.v. (Kawaguchi)
Derivative Type: SulfateCAS Registry Number: 39831-55-5Trademarks: Amiglyde-V (Fort Dodge); Amikin (BMS); Amiklin (BMS); BB-K8 (BMS); Biklin (BMS); Lukadin (San Carlo); Mikavir (Salus); Novamin (BMS); Pierami (Fournier)Molecular Formula: C22H43N5O13.2H2SO4Molecular Weight: 781.76Percent Composition: C 33.80%, H 6.06%, N 8.96%, O 42.98%, S 8.20%Properties: Amorphous form, dec 220-230°. [a]D22 +74.75° (water).Optical Rotation: [a]D22 +74.75° (water)
Therap-Cat: Antibacterial.Therap-Cat-Vet: Antibacterial.Keywords: Antibacterial (Antibiotics); Aminoglycosides.
Amikacin Sulfate is the sulfate salt of amikacin, a broad-spectrum semi-synthetic aminoglycoside antibiotic, derived from kanamycin with antimicrobial property. Amikacin irreversibly binds to the bacterial 30S ribosomal subunit, specifically in contact with 16S rRNA and S12 protein within the 30S subunit. This leads to interference with translational initiation complex and misreading of mRNA, thereby hampering protein synthesis and resulting in bactericidal effect. This agent is usually used in short-term treatment of serious infections due to susceptible strains of Gram-negative bacteria.Amikacin disulfate is an aminoglycoside sulfate salt obtained by combining amikacin with two molar equivalents of sulfuric acid. It has a role as an antibacterial drug, an antimicrobial agent and a nephrotoxin. It contains an amikacin(4+).
Amikacin sulfate is semi-synthetic aminoglycoside antibiotic derived from kanamycin. It is C22H43N5O13•2H2SO4•O-3-amino-3-deoxy-α-D-glucopyranosyl-(1→4)-O-[6-amino-6-deoxy-α-Dglucopyranosyl-( 1→6)]-N3-(4-amino-L-2-hydroxybutyryl)-2-deoxy-L-streptamine sulfate (1:2)
|
M.W. 585.61The dosage form is supplied as a sterile, colorless to light straw colored solution for intramuscular or intravenous use. Each mL contains 250 mg amikacin (as the sulfate), 0.66% sodium metabisulfite, 2.5% sodium citrate dihydrate with pH adjusted to 4.5 with sulfuric acid.
Amikacin is an antibiotic medication used for a number of bacterial infections.[4] This includes joint infections, intra-abdominal infections, meningitis, pneumonia, sepsis, and urinary tract infections.[4] It is also used for the treatment of multidrug-resistant tuberculosis.[5] It is used by injection into a vein using an IV or into a muscle.[4]
Amikacin, like other aminoglycoside antibiotics, can cause hearing loss, balance problems, and kidney problems.[4] Other side effects include paralysis, resulting in the inability to breathe.[4] If used during pregnancy it may cause permanent deafness in the baby.[4][1] Amikacin works by blocking the function of the bacteria’s 30S ribosomal subunit, making it unable to produce proteins.[4]
Amikacin was patented in 1971, and came into commercial use in 1976.[6][7] It is on the World Health Organization’s List of Essential Medicines.[8] It is derived from kanamycin.[4]
Medical uses
Amikacin is most often used for treating severe infections with multidrug-resistant, aerobic Gram-negative bacteria, especially Pseudomonas, Acinetobacter, Enterobacter, E. coli, Proteus, Klebsiella, and Serratia.[9] The only Gram-positive bacteria that amikacin strongly affects are Staphylococcus[9] and Nocardia.[10] Amikacin can also be used to treat non-tubercular mycobacterial infections and tuberculosis (if caused by sensitive strains) when first-line drugs fail to control the infection.[4] It is rarely used alone.[11]
It is often used in the following situations:[4]
- Bronchiectasis[12]
- Bone and joint infections
- Granulocytopenia, when combined with ticarcillin, in people with cancer[13]
- Intra-abdominal infections (such as peritonitis) as an adjunct to other medicines, like clindamycin, metronidazole, piperacillin/tazobactam, or ampicillin/sulbactam
- Meningitis:
- for meningitis by E. coli, as an adjunct to imipenem
- for meningitis caused by Pseudomonas, as an adjunct to meropenem
- for meningitis caused by Acetobacter, as an adjunct to imipenem or colistin
- for neonatal meningitis caused by Streptococcus agalactiae or Listeria monocytogenes, as an adjunct to ampicillin
- for neonatal meningitis caused by Gram negative bacteria such as E. coli, as adjunct to a 3rd-generation cephalosporin
- Mycobacterial infections, including as a second-line agent for active tuberculosis.[14] It is also used for infections by Mycobacterium avium, M. abcessus, M. chelonae, and M. fortuitum.
- Rhodococcus equi, which causes an infection resembling tuberculosis
- Respiratory tract infections, including as an adjunct to beta-lactams or carbapenem for hospital-acquired pneumonia
- Sepsis, including that in neonates,[9] as an adjunct to beta-lactams or carbapenem
- Skin and suture-site infections[9]
- Urinary tract infections that are caused by bacteria resistant to less toxic drugs (often by Enterobacteriaceae or P. aeruginosa)
Amikacin may be combined with a beta-lactam antibiotic for empiric therapy for people with neutropenia and fever.[4]
Available forms[
Amikacin may be administered once or twice a day and is usually given by the intravenous or intramuscular route, though it can be given via nebulization. There is no oral form available, as amikacin is not absorbed orally. In people with kidney failure, dosage must be adjusted according to the creatinine clearance, usually by reducing the dosing frequency.[9] In people with a CNS infection such as meningitis, amikacin can be given intrathecally (by direct injection into the spine) or intraventricularly (by injection into the ventricles of brain).[4]
An liposome inhalation suspension is also available and approved to treat Mycobacterium avium complex (MAC) in the United States.[15][16] The application for Arikayce was withdrawn in the European Union because the Committee for Medicinal Products for Human Use (CHMP) was of the opinion that the benefits of Arikayce did not outweigh its risks.[17]
Special populations
Amikacin should be used in smaller doses in the elderly, who often have age-related decreases in kidney function, and children, whose kidneys are not fully developed yet. It is considered pregnancy category D in both the United States and Australia, meaning they have a probability of harming the fetus.[4] Around 16% of amikacin crosses the placenta; while the half-life of amikacin in the mother is 2 hours, it is 3.7 hours in the fetus.[9] A pregnant woman taking amikacin with another aminoglycoside has a possibility of causing congenital deafness in her child. While it is known to cross the placenta, amikacin is only partially secreted in breast milk.[4]
In general, amikacin should be avoided in infants.[18] Infants also tend to have a larger volume of distribution due to their higher concentration of extracellular fluid, where aminoglycosides reside.[3]
The elderly tend to have amikacin stay longer in their system; while the average clearance of amikacin in a 20-year-old is 6 L/hr, it is 3 L/hr in an 80-year-old.[19]
Clearance is even higher in people with cystic fibrosis.[20]
In people with muscular disorders such as myasthenia gravis or Parkinson’s disease, amikacin’s paralytic effect on neuromuscular junctions can worsen muscle weakness.[4]
Adverse effects
Side-effects of amikacin are similar to those of other aminoglycosides. Kidney damage and ototoxicity (which can lead to hearing loss) are the most important effects, occurring in 1–10% of users.[12] The nephro- and ototoxicity are thought to be due to aminoglycosides’ tendency to accumulate in the kidneys and inner ear.[3]

Diagram of the inner ear. Amikacin causes damage to the cochlea and vestibules.
Amikacin can cause neurotoxicity if used at a higher dose or for longer than recommended. The resulting effects of neurotoxicity include vertigo, numbness, tingling of the skin (paresthesia), muscle twitching, and seizures.[4] Its toxic effect on the 8th cranial nerve causes ototoxicity, resulting in loss of balance and, more commonly, hearing loss.[3] Damage to the cochlea, caused by the forced apoptosis of the hair cells, leads to the loss of high-frequency hearing and happens before any clinical hearing loss can be detected.[9][21] Damage to the ear vestibules, most likely by creating excessive oxidative free radicals. It does so in a time-dependent rather than dose-dependent manner, meaning that risk can be minimized by reducing the duration of use.[22]
Amikacin causes nephrotoxicity (damage to the kidneys), by acting on the proximal renal tubules. It easily ionizes to a cation and binds to the anionic sites of the epithelial cells of the proximal tubule as part of receptor-mediated pinocytosis. The concentration of amikacin in the renal cortex becomes ten times that of amikacin in the plasma;[18] it then most likely interferes with the metabolism of phospholipids in the lysosomes, which causes lytic enzymes to leak into the cytoplasm.[3] Nephrotoxicity results in increased serum creatinine, blood urea nitrogen, red blood cells, and white blood cells, as well as albuminuria (increased output of albumin in the urine), glycosuria (excretion of glucose into the urine), decreased urine specific gravity, and oliguria (decrease in overall urine output).[9][21] It can also cause urinary casts to appear.[3] The changes in renal tubular function also change the electrolyte levels and acid-base balance in the body, which can lead to hypokalemia and acidosis or alkalosis.[22] Nephrotoxicity is more common in those with pre-existing hypokalemia, hypocalcemia, hypomagnesemia, acidosis, low glomerular filtration rate, diabetes mellitus, dehydration, fever, and sepsis, as well as those taking antiprostaglandins.[4][18][3][22] The toxicity usually reverts once the antibiotic course has been completed,[3] and can be avoided altogether by less frequent dosing (such as once every 24 hours rather than once every 8 hours).[18]
Amikacin can cause neuromuscular blockade (including acute muscular paralysis) and respiratory paralysis (including apnea).[4]
Rare side effects (occurring in fewer than 1% of users) include allergic reactions, skin rash, fever, headaches, tremor, nausea and vomiting, eosinophilia, arthralgia, anemia, hypotension, and hypomagnesemia. In intravitreous injections (where amikacin is injected into the eye), macular infarction can cause permanent vision loss.[9][12]
The amikacin liposome inhalation suspension prescribing information includes a boxed warning regarding the increased risk of respiratory conditions including hypersensitivity pneumonitis (inflamed lungs), bronchospasm (tightening of the airway), exacerbation of underlying lung disease and hemoptysis (spitting up blood) that have led to hospitalizations in some cases.[15][16] Other common side effects in patients taking amikacin liposome inhalation suspension are dysphonia (difficulty speaking), cough, ototoxicity (damaged hearing), upper airway irritation, musculoskeletal pain, fatigue, diarrhea and nausea.[15][16]
Contraindications
Amikacin should be avoided in those who are sensitive to any aminoglycoside, as they are cross-allergenic (that is, an allergy to one aminoglycoside also confers hypersensitivity to other aminoglycosides). It should also be avoided in those sensitive to sulfite (seen more among people with asthma),[9] since most amikacin usually comes with sodium metabisulfite, which can cause an allergic reaction.[4]
In general, amikacin should not be used with or just before/after another drug that can cause neurotoxicity, ototoxicity, or nephrotoxicity. Such drugs include other aminoglycosides; the antiviral acyclovir; the antifungal amphotericin B; the antibiotics bacitracin, capreomycin, colistin, polymyxin B, and vancomycin; and cisplatin, which is used in chemotherapy.[4]
Amikacin should not be used with neuromuscular blocking agents, as they can increase muscle weakness and paralysis.[4]
Interactions
Amikacin can be inactivated by other beta-lactams, though not to the extent as other aminoglycosides, and is still often used with penicillins (a type of beta-lactam) to create an additive effect against certain bacteria, and carbapenems, which can have a synergistic against some Gram-positive bacteria. Another group of beta-lactams, the cephalosporins, can increase the nephrotoxicity of aminoglycoside as well as randomly elevating creatinine levels. The antibiotics chloramphenicol, clindamycin, and tetracycline have been known to inactivate aminoglycosides in general by pharmacological antagonism.[4]
The effect of amikacin is increased when used with drugs derived from the botulinum toxin,[12] anesthetics, neuromuscular blocking agents, or large doses of blood that contains citrate as an anticoagulant.[4]
Potent diuretics not only cause ototoxicity themselves, but they can also increase the concentration of amikacin in the serum and tissue, making the ototoxicity even more likely.[4] Quinidine also increases levels of amikacin in the body.[12] The NSAID indomethacin can increase serum aminoglycoside levels in premature infants.[4] Contrast mediums such as ioversol increases the nephrotoxicity and otoxicity caused by amikacin.[12]
Amikacin can decrease the effect certain vaccines, such as the live BCG vaccine (used for tuberculosis), the cholera vaccine, and the live typhoid vaccine by acting as a pharmacological antagonist.[12]
Pharmacology
Mechanism of action

The 30S subunit of the prokaryotic ribosome. The orange represents the 16S rRNA, and the blue represents the various proteins attached.
Amikacin irreversibly binds to 16S rRNA and the RNA-binding S12 protein of the 30S subunit of prokaryotic ribosome and inhibits protein synthesis by changing the ribosome’s shape so that it cannot read the mRNA codons correctly.[9][23] It also interferes with the region that interacts with the wobble base of the tRNA anticodon.[24] It works in a concentration-dependent manner, and has better action in an alkaline environment.[3]
At normal doses, amikacin-sensitive bacteria respond within 24–48 hours.[9]
Resistance
Amikacin evades attacks by all antibiotic-inactivating enzymes that are responsible for antibiotic resistance in bacteria, except for aminoacetyltransferase and nucleotidyltransferase.[25] This is accomplished by the L-hydroxyaminobuteroyl amide (L-HABA) moiety attached to N-1 (compare to kanamycin, which simply has a hydrogen), which blocks the access and decreases the affinity of aminoglycoside-inactivating enzymes.[25][26][27] Amikacin ends up with only one site where these enzymes can attack, while gentamicin and tobramycin have six.[11]
Bacteria that are resistant to streptomycin and capreomycin are still susceptible to amikacin; bacteria that are resistant to kanamycin have varying susceptibility to amikacin. Resistance to amikacin also confers resistance to kanamycin and capreomycin.[28]
Resistance to amikacin and kanamycin in Mycobacterium, the causative agent of tuberculosis, is due to a mutation in the rrs gene, which codes for the 16S rRNA. Mutations such as these reduce the binding affinity of amikacin to the bacteria’s ribosome.[29] Variations of aminoglycoside acetyltransferase (AAC) and aminoglycoside adenylyltransferase (AAD) also confer resistance: resistance in Pseudomonas aeruginosa is caused by AAC(6′)-IV, which also confers resistance to kanamycin, gentamicin, and tobramycin, and resistance in Staphylococcus aureus and S. epidermidis is caused by AAD(4′,4), which also confers resistance to kanamycin, tobramycin, and apramycin.[26] Some strains of S. aureus can also inactivate amikacin by phosphorylating it.[13]
Pharmacokinetics
Amikacin is not absorbed orally and thus must be administered parenterally. It reaches peak serum concentrations in 0.5–2 hours when administered intramuscularly. Less than 11% of the amikacin actually binds to plasma proteins. It is distributed into the heart, gallbladder, lungs, and bones, as well as in bile, sputum, interstitial fluid, pleural fluid, and synovial fluids. It is usually found at low concentrations in the cerebrospinal fluid, except when administered intraventricularly.[4] In infants, amikacin is normally found at 10–20% of plasma levels in the spinal fluid, but the amount reaches 50% in cases of meningitis.[9] It does not easily cross the blood-brain barrier or enter ocular tissue.[3]
While the half-life of amikacin is normally two hours, it is 50 hours in those with end-stage renal disease.[11]
The vast majority (95%) of amikacin from an IM or IV dose is secreted unchanged via glomerular filtration and into the urine within 24 hours.[4][11] Factors that cause amikacin to be excreted via urine include its relatively low molecular weight, high water solubility, and unmetabolized state.[18]
Chemistry
Amikacin is derived from kanamycin A:[30][31]

Veterinary use
While amikacin is only FDA-approved for use in dogs and for intrauterine infection in horses, it is one of the most common aminoglycosides used in veterinary medicine,[32] and has been used in dogs, cats, guinea pigs, chinchillas, hamsters, rats, mice, prairie dogs, cattle, birds, snakes, turtles and tortoises, crocodilians, bullfrogs, and fish.[3][33][34] It is often used for respiratory infections in snakes, bacterial shell disease in turtles, and sinusitis in macaws. It is generally contraindicated in rabbits and hares (though it has still been used) because it harms the balance of intestinal microflora.[3]
In dogs and cats, amikacin is commonly used as a topical antibiotic for ear infections and for corneal ulcers, especially those that are caused by Pseudomonas aeruginosa. The ears are often cleaned before administering the medication, since pus and cellular debris lessen the activity of amikacin.[32] Amikacin is administered to the eye when prepared as an ophthalmic ointment or solution, or when injected subconjunctivally.[35] Amikacin in the eye can be accompanied by cephazolin. Despite its use there amikacin (and all aminoglycosides) are toxic to intraocular structures.[36]
In horses, amikacin is FDA-approved for uterine infections (such as endometriosis and pyometra) when caused by susceptible bacteria.[37] It is also used in topical medication for the eyes and arthroscopic lavage; when combined with a cephalosporin, is used to treat subcutaneous infections that are caused by Staphylococcus. For infections in the limbs or joints, it is often administered with a cephalosporin via limb perfusion directly into the limb or injected into the joint.[32][38] Amikacin is also injected into the joints with the anti-arthritic medication Adequan in order to prevent infection.[39]
Side effects in animals include nephrotoxicity, ototoxicity, and allergic reactions at IM injection sites. Cats tend to be more sensitive to the vestibular damage caused by ototoxicity. Less frequent side effects include neuromuscular blockade, facial edema, and peripheral neuropathy.[3][32]
The half-life in most animals is one to two hours.[40]
Treating overdoses of amikacin requires kidney dialysis or peritoneal dialysis, which reduce serum concentrations of amikacin, and/or penicillins, some of which can form complexes with amikacin that deactivate it.[3]
Liposome inhalation suspension
Amikacin liposome inhalation suspension was the first drug approved under the US limited population pathway for antibacterial and antifungal drugs (LPAD pathway).[15] It also was approved under the accelerated approval pathway.[15] The U.S. Food and Drug Administration (FDA) granted the application for amikacin liposome inhalation suspension fast track, breakthrough therapy, priority review, and qualified infectious disease product (QIDP) designations.[15] The FDA granted approval of Arikayce to Insmed, Inc.[15]
The safety and efficacy of amikacin liposome inhalation suspension, an inhaled treatment taken through a nebulizer, was demonstrated in a randomized, controlled clinical trial where patients were assigned to one of two treatment groups.[15] One group of patients received amikacin liposome inhalation suspension plus a background multi-drug antibacterial regimen, while the other treatment group received a background multi-drug antibacterial regimen alone.[15] By the sixth month of treatment, 29 percent of patients treated with amikacin liposome inhalation suspension had no growth of mycobacteria in their sputum cultures for three consecutive months compared to 9 percent of patients who were not treated with amikacin liposome inhalation suspension.[15]

SYN

SYN
https://www.mdpi.com/1420-3049/22/12/2267/htm
Scheme 1. Original chemical reactions sequence to obtain amikacin by modification of kanamycin A.PATENThttps://patents.google.com/patent/CN105440090A/zh
Amikacin is a semi-synthetic aminoglycoside antibiotic with a broad antibacterial spectrum and strong antibacterial activity against a variety of bacteria; its sulfate has become a clinically commonly used first-line anti-infective drug in the world and continues to Develop new dosage forms and uses.
[0003] Amikacin sulfate is suitable for Pseudomonas aeruginosa and other Pseudomonas, Escherichia coli, Proteus, Klebsiella, Enterobacter, Serratia, Acinetobacter Severe infections caused by other sensitive gram-negative bacilli and Staphylococcus (methicillin-sensitive strains), such as bacteremia or sepsis, bacterial endocarditis, lower respiratory tract infections, bone and joint infections, biliary tract infections, abdominal infections, Complex urinary tract infections, skin and soft tissue infections, etc. Because it is stable to most aminoglycoside inactivating enzymes, it is especially suitable for the treatment of serious infections caused by gram-negative bacilli against kanamycin, gentamicin or tobramycin-resistant strains.
[0004] Amikacin, also known as amikacin, has a molecular weight of 585. The most commonly used synthetic route is a silyl protecting routes, such as the document “amikacin by New Method” (Author: Jiangzhong Liang, Wang Yu; Fine & Specialty Chemicals, 2004, 12 (10), 26- 28) The main process recorded is: (1) Using kanamycin A (KMA) as a raw material to protect the 11 amino groups and hydroxyl groups of kanamycin to obtain methylsilyl kanamycin; (2) ) Using YN-phthalimido-α-hydroxybutyric acid (PHBA) and N-hydroxy-phthalimide (NOP) as raw materials in dicyclohexylcarbodiimide (DCC) The active ester compound is prepared in the presence; (3) acylation (transesterification reaction) with methylsilyl kanamycin and active ester, and then acidolysis and hydrazinolysis reactions to obtain amikacin. As shown in the following route:
[0005] 1. Silanization protection reaction:
[0006]
[0007] 2. Preparation of Living King®:

[0008]

[0009] 3. Acylation reaction:
U
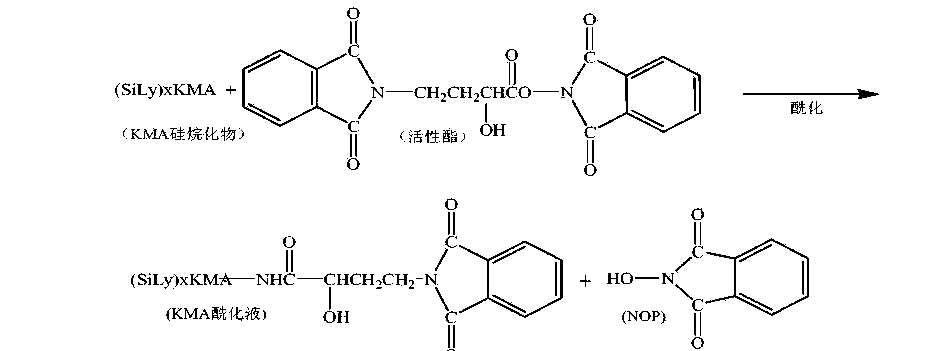
[0011] 4. Acidolysis reaction:
[0012]

[0013] 5. Hydrazine reaction:
[0014]

[0015] The acylation reaction in the above route adopts a transesterification reaction between a silyl group protection reactant and an independently prepared active ester. Due to the active transesterification reaction, a large excess of reactant active ester is needed to improve the reaction yield, and there is an independent unit reaction for preparing active ester, and the raw material N-hydroxy-phthalimide is used. (NOP), increasing the usage amount of reaction solvent, the solvent in the process is volatile, the loss is large, the environment is affected, and the production cost is increased.
[0016] How to find a direct one-step acylation reaction between the silyl group protection and YN-phthalimido-α-hydroxybutyric acid (PHBA), which can not only ensure the synthesis yield, but also reduce the synthesis The steps are easy to operate, and the N-hydroxy-phthalimide (NOP), the raw material for preparing active esters, is no longer used, and the acylation reaction conditions that reduce solvent consumption are a very beneficial synthetic process line.
Example 1
[0046] 600mL of acetonitrile was put into the silanization reaction flask, and 0.1 billion kanamycin A (KMA) was added. After the feeding port was closed and stirred for 10 minutes, hexamethyldisilazane (HMDS) was added. 400mL, heated to reflux, refluxed at 75~80°C for 7hr. Use drinking water to cool the outside of the reaction flask to lower the temperature to below 35°C, and let it stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0047] Add 1000mL acetone to the silyl group protection product, start stirring, add 60g γ-N-phthalimido-α-hydroxybutyric acid (PHBA), and then add 2.5g catalyst 4-N, N -Lutidine (DMAP), cooled to -15~-1 (TC〇
[0048] Dissolve 60gN, N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactant, control the flow rate of 5mL/min, and control the temperature of the reactant to -15~-10°C; the flow is completed Continue the reaction for 1 hour.
[0049] After the completion of the acylation reaction, the material was transferred to the acidolysis bottle, the stirring was turned on, and 400mL of 4.0mol/L hydrochloric acid was added for acidolysis, and the feed solution was pH 3.0 and allowed to stand for 60 minutes. The lower acid hydrolysis solution was collected by suction filtration, and the filter cake (DCU) was washed three times with 150 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0050] The acid hydrolysate was transferred to a distillation flask. Turn on the vacuum, the degree of vacuum: <0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time: 2.5 hours after the distillation is complete; transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, and add 7.Omol/ L ammonia water 200mL, so that the pH of the material solution reaches 8.0; add 180mL hydrazine hydrate, increase the temperature, the temperature is 85~95°C, hydrazinolysis 3.5 hours, use drinking water to cool outside the hydrazinolysis bottle, and cool to 40 °C.
[0051] Add 4.0111〇1/1 hydrochloric acid 12001^ to the hydrazinolysis bottle, adjust? !1 is 4.0. Turn on the vacuum filtration. With 5001 ^ deionized water top washing filter, 1510mL of amikacin synthetic solution, amikacin content 5.8% (g/mL), relative to the synthetic yield of kanamycin A is 72.5 %.
[0052] Example 2
[0053] 600mL of acetonitrile was put into the silanization reaction flask, 0.1 billion kanamycin A (KMA) was added, the feeding port was closed and stirred for 10 minutes, and hexamethyldisilazane (HMDS) was added 500mL, heated to reflux, refluxed at 75~80°C for 8hr. After the reaction is completed, cool down to 40°C with drinking water and let stand for natural layering. Separate and collect the lower layer to obtain a silyl group protected product.
[0054] Add 1000mL acetone to the silyl group protection product, start stirring, add 70g Y-N-phthalimido-α-hydroxybutyric acid (PHBA), and add 3.0g catalyst 1-hydroxybenzo Triazole (HOBT), after the material is dissolved, the temperature is reduced to -15~-10°C.
[0055] Dissolve 70g of N,N-bicyclohexylcarbodiimide with 300mL of acetone, add its flow to the above-mentioned reactants, control the flow rate of 6mL/min, and control the temperature of the reactants from -15 to -10°C; the flow is completed Continue the reaction for 1.5 hours.
[0056] After the acylation reaction is completed, the material is transferred to the acidolysis bottle, the stirring is turned on, and 6.0m〇l/L hydrochloric acid 300mL is added for acidolysis, the feed solution is pH 2.0, and the acidolysis is completed, and it is allowed to stand for 50 minutes. The lower acid hydrolysis solution was collected by suction filtration, the filter cake (DCU) was washed three times with 200 mL of deionized water, and the washing water was incorporated into the acid hydrolysis solution.
[0057] Transfer the acid hydrolysate into a distillation flask. Turn on the vacuum, vacuum degree: <-0.07Mpa, the distillation temperature is controlled at 40~68°C, the distillation time is 3.0 hours, except for acetone. After the distillation is completed, transfer the PKS concentrate in the distillation flask into the hydrazinolysis flask, add 150 mL of 10.0 mol/L ammonia water, the pH of the feed solution is 8.5; add 200 mL of hydrazine hydrate, increase the temperature at 85~95 °C, hydrazinolysis 4 After hours, use drinking water to cool down outside the hydrazinolysis bottle to 45°C.
[0058] Add 6.0111〇1/1 hydrochloric acid 10001^ to the hydrazinolysis bottle, adjust? !1 is 3.0. Turn on the vacuum filtration, use 8001^ deionized water to wash and filter the fish, to obtain 1620 mL of amikacin synthetic solution, and the amikacin content is 5.5% (g/mL). The synthetic yield relative to kanamycin A was 73.7%.
References
- ^ Jump up to:a b c “Amikacin Use During Pregnancy”. Drugs.com. 2 December 2019. Retrieved 13 March 2020.
- ^ “Amikacin 250 mg/ml Injection – Summary of Product Characteristics (SmPC)”. (emc). 16 September 2015. Retrieved 13 March 2020.
- ^ Jump up to:a b c d e f g h i j k l m n Plumb, Donald C. (2011). “Amikacin Sulfate”. Plumb’s Veterinary Drug Handbook (7th ed.). Stockholm, Wisconsin; Ames, Iowa: Wiley. pp. 39–43. ISBN 978-0-470-95964-0.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa “Amikacin Sulfate”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 8 December 2016.
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 137. hdl:10665/44053. ISBN 9789241547659.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 507. ISBN 9783527607495. Archived from the original on 20 December 2016.
- ^ Oxford Handbook of Infectious Diseases and Microbiology. OUP Oxford. 2009. p. 56. ISBN 9780191039621. Archived from the original on 24 November 2015.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g h i j k l m “Amikacin sulfate injection, solution”. DailyMed. 10 April 2019. Retrieved 13 March 2020.
- ^ Scholar, Eric M.; Pratt, William B. (22 May 2000). The Antimicrobial Drugs (2nd ed.). Oxford University Press, USA. pp. 15–19. ISBN 978-0-19-975971-2. Archived from the original on 10 September 2017.
- ^ Jump up to:a b c d Cunha, Burke A. (1 November 2006). “New Uses for Older Antibiotics: Nitrofurantoin, Amikacin, Colistin, Polymyxin B, Doxycycline, and Minocycline Revisited”. Medical Clinics of North America. Antimicrobial Therapy. 90 (6): 1089–1107. doi:10.1016/j.mcna.2006.07.006. ISSN 0025-7125. PMID 17116438.
- ^ Jump up to:a b c d e f g “amikacin (Rx)”. Medscape. WebMD. Archived from the original on 9 August 2017. Retrieved 9 August 2017.
- ^ Jump up to:a b Aronson J. K., ed. (2016). “Amikacin”. Meyler’s Side Effects of Drugs (16th ed.). Oxford: Elsevier. pp. 207–209. ISBN 978-0-444-53716-4.
- ^ Vardanyan, Ruben; Hruby, Victor (2016). “Chapter 32: Antimicobacterial Drugs”. Synthesis of Best-Seller Drugs. Boston: Academic Press. pp. 669–675. ISBN 978-0-12-411492-0.
- ^ Jump up to:a b c d e f g h i j “FDA approves a new antibacterial drug to treat a serious lung disease using a novel pathway to spur innovation”. U.S. Food and Drug Administration (FDA)(Press release). Retrieved 12 November 2018.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Arikayce- amikacin suspension”. DailyMed. 30 September 2018. Retrieved 13 March 2020.
- ^ “Arikayce: Withdrawn application”. European Medicines Agency (EMA). 14 October 2016. Retrieved 13 March 2020.
- ^ Jump up to:a b c d e Ettinger, Stephen J.; Feldman, Edward C. (24 December 2009). Textbook of Veterinary Internal Medicine. Elsevier Health Sciences. pp. 1976, 523. ISBN 978-1-4377-0282-8. Archived from the original on 10 September 2017.
- ^ Maire, P.; Bourguignon, L.; Goutelle, S.; Ducher, M.; Jelliffe, R. (2017). “Chapter 20 – Individualizing Drug Therapy in the Elderly”. Individualized Drug Therapy for Patients. Boston: Academic Press. pp. 373–382. ISBN 978-0-12-803348-7.
- ^ Eghianruwa, Kingsley (2014). Essential Drug Data for Rational Therapy in Veterinary Practice. Author House. p. 16. ISBN 978-1-4918-0000-3. Archived from the original on 10 September 2017.
- ^ Jump up to:a b Morris, Daniel O.; Kennis, Robert A. (11 October 2012). Clinical Dermatology, An Issue of Veterinary Clinics: Small Animal Practice, E-Book. Elsevier Health Sciences. p. 29. ISBN 978-1-4557-7377-0. Archived from the original on 10 September 2017.
- ^ Jump up to:a b c Corti, Natascia; Taegtmeyer, Anne; Imhof, Alexander (1 January 2011). “Miscellaneous antibacterial drugs”. Side Effects of Drugs Annual. A worldwide yearly survey of new data in adverse drug reactions. 33: 509–540. doi:10.1016/B978-0-444-53741-6.00026-X. ISBN 9780444537416. ISSN 0378-6080.
- ^ Bauman, Robert W. (2015). Microbiology: with diseases by body system (4th ed.). Boston: Pearson. ISBN 978-0-321-91855-0.
- ^ “Amikacin”. DrugBank. 2 August 2017. Archived from the original on 16 August 2017. Retrieved 10 August 2017.
- ^ Jump up to:a b Mudd, Efrain (7 August 2017). “O Aminoglycosides”. Pharmacological Sciences. Archived from the original on 16 August 2017. Retrieved 14 August 2017.
- ^ Jump up to:a b Kondo, Shinichi; Hotta, Kunimoto (1 January 1999). “Semisynthetic aminoglycoside antibiotics: Development and enzymatic modifications”. Journal of Infection and Chemotherapy. 5 (1): 1–9. doi:10.1007/s101560050001. ISSN 1341-321X. PMID 11810483. S2CID 38981498.
- ^ Park, Je Won; Ban, Yeon Hee; Nam, Sang-Jip; Cha, Sun-Shin; Yoon, Yeo Joon (1 December 2017). “Biosynthetic pathways of aminoglycosides and their engineering”. Current Opinion in Biotechnology. Chemical biotechnology: Pharmaceutical biotechnology. 48: 33–41. doi:10.1016/j.copbio.2017.03.019. ISSN 0958-1669. PMID 28365471.
- ^ Caminero, José A; Sotgiu, Giovanni; Zumla, Alimuddin; Migliori, Giovanni Battista (1 September 2010). “Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis”. The Lancet Infectious Diseases. 10 (9): 621–629. doi:10.1016/S1473-3099(10)70139-0. ISSN 1473-3099. PMID 20797644.
- ^ Ahmad, Suhail; Mokaddas, Eiman (1 March 2014). “Current status and future trends in the diagnosis and treatment of drug-susceptible and multidrug-resistant tuberculosis”. Journal of Infection and Public Health. 7 (2): 75–91. doi:10.1016/j.jiph.2013.09.001. ISSN 1876-0341. PMID 24216518.
- ^ Kawaguchi, H.; Naito, T.; Nakagawa, S.; Fujisawa, K. I. (December 1972). “BB-K 8, a new semisynthetic aminoglycoside antibiotic”. The Journal of Antibiotics. 25 (12): 695–708. doi:10.7164/antibiotics.25.695. ISSN 0021-8820. PMID 4568692. Archived from the original on 16 August 2017.
- ^ Monteleone, Peter M.; Muhammad, Naseem; Brown, Robert D.; McGrory, John P.; Hanna, Samir A. (1 January 1983). Amikacin Sulfate. Analytical Profiles of Drug Substances. 12. pp. 37–71. doi:10.1016/S0099-5428(08)60163-X. ISBN 9780122608124. ISSN 0099-5428.
- ^ Jump up to:a b c d Forney, Barbara. “Amikacin for Veterinary Use”. Wedgewood Pharmacy. Archived from the original on 16 August 2017. Retrieved 9 August 2017.
- ^ Riviere, Jim E.; Papich, Mark G. (13 May 2013). Veterinary Pharmacology and Therapeutics. John Wiley & Sons. p. 931. ISBN 978-1-118-68590-7. Archived from the original on 10 September 2017.
- ^ Mader, Douglas R.; Divers, Stephen J. (12 December 2013). Current Therapy in Reptile Medicine and Surgery – E-Book. Elsevier Health Sciences. p. 382. ISBN 978-0-323-24293-6. Archived from the original on 10 September 2017.
- ^ Maggs, David; Miller, Paul; Ofri, Ron (7 August 2013). Slatter’s Fundamentals of Veterinary Ophthalmology – E-Book. Elsevier Health Sciences. p. 37. ISBN 978-0-323-24196-0. Archived from the original on 10 September 2017.
- ^ Hsu, Walter H. (25 April 2013). Handbook of Veterinary Pharmacology. John Wiley & Sons. p. 486. ISBN 978-1-118-71416-4.
- ^ US National Library of Medicine (9 March 2017). “Amiglyde-V- amikacin sulfate injection”. DailyMed. Archived from the original on 16 August 2017. Retrieved 8 August2017.
- ^ Orsini, James A. (1 August 2017). “Update on Managing Serious Wound Infections in Horses: Wounds Involving Joints and Other Synovial Structures”. Journal of Equine Veterinary Science. 55: 115–122. doi:10.1016/j.jevs.2017.01.016. ISSN 0737-0806.
- ^ Wanamaker, Boyce P.; Massey, Kathy (25 March 2014). Applied Pharmacology for Veterinary Technicians – E-Book. Elsevier Health Sciences. p. 392. ISBN 978-0-323-29170-5.
- ^ Papich, Mark G. (October 2015). “Amikacin”. Saunders Handbook of Veterinary Drugs: Small and Large Animal (4th ed.). Elsevier Health Sciences. pp. 25–27. ISBN 978-0-323-24485-5. Archived from the original on 10 September 2017.
External links
- “Amikacin”. Drug Information Portal. U.S. National Library of Medicine.
- “Amikacin sulfate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Amikin, Amiglyde-V, Arikayce, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682661 |
| License data | US DailyMed: Amikacin |
| Pregnancy category | AU: D[1]US: D (Evidence of risk)[1] |
| Routes of administration | intramuscular, intravenous |
| Drug class | Aminoglycoside |
| ATC code | D06AX12 (WHO) J01GB06 (WHO), S01AA21 (WHO), J01RA06 (WHO), QD06AX12 (WHO), QJ01GB06 (WHO), QS01AA21 (WHO), QJ01RA06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) [2]US: ℞-onlyEU: Rx-only |
| Pharmacokinetic data | |
| Bioavailability | >90%[3] |
| Protein binding | 0–11% |
| Metabolism | Mostly unmetabolized |
| Elimination half-life | 2–3 hours |
| Excretion | Kidney |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 37517-28-5 |
| PubChem CID | 37768 |
| DrugBank | DB00479 |
| ChemSpider | 34635 |
| UNII | 84319SGC3C |
| KEGG | D02543 as salt: D00865 |
| ChEBI | CHEBI:2637 |
| ChEMBL | ChEMBL177 |
| CompTox Dashboard (EPA) | DTXSID3022586 |
| ECHA InfoCard | 100.048.653 |
| Chemical and physical data | |
| Formula | C22H43N5O13 |
| Molar mass | 585.608 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=C(N[C@H]3[C@H](O[C@H]1O[C@@H]([C@@H](O)[C@H](N)[C@H]1O)CO)[C@@H](O)[C@H](O[C@H]2O[C@H](CN)[C@@H](O)[C@H](O)[C@H]2O)[C@@H](N)C3)[C@@H](O)CCN | |
| InChI[hide]InChI=1S/C22H43N5O13/c23-2-1-8(29)20(36)27-7-3-6(25)18(39-22-16(34)15(33)13(31)9(4-24)37-22)17(35)19(7)40-21-14(32)11(26)12(30)10(5-28)38-21/h6-19,21-22,28-35H,1-5,23-26H2,(H,27,36)/t6-,7+,8-,9+,10+,11-,12+,13+,14+,15-,16+,17-,18+,19-,21+,22+/m0/s1 Key:LKCWBDHBTVXHDL-RMDFUYIESA-N |
/////////Amikacin sulfate, Arikayce liposomal, EU 2020, 2020 APPROVALS, Antibacterial, Protein biosynthesis inhibitor, アミカシン硫酸塩 , BB K 8, AMIKACIN
C1C(C(C(C(C1NC(=O)C(CCN)O)OC2C(C(C(C(O2)CO)O)N)O)O)OC3C(C(C(C(O3)CN)O)O)O)N.OS(=O)(=O)O.OS(=O)(=O)O
AZITHROMYCIN, アジスロマイシン;
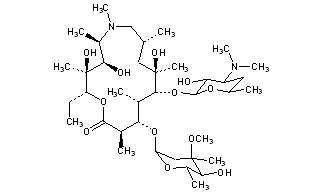
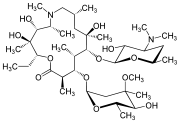
AZITHROMYCIN
C38H72N2O12,
748.9845
アジスロマイシン;
| CAS: | 83905-01-5 |
| PubChem: | 51091811 |
| ChEBI: | 2955 |
| ChEMBL: | CHEMBL529 |
| DrugBank: | DB00207 |
| PDB-CCD: | ZIT[PDBj] |
| LigandBox: | D07486 |
| NIKKAJI: | J134.080H |
Azithromycin is an antibiotic used for the treatment of a number of bacterial infections.[3] This includes middle ear infections, strep throat, pneumonia, traveler’s diarrhea, and certain other intestinal infections.[3] It can also be used for a number of sexually transmitted infections, including chlamydia and gonorrhea infections.[3] Along with other medications, it may also be used for malaria.[3] It can be taken by mouth or intravenously with doses once per day.[3]
Common side effects include nausea, vomiting, diarrhea and upset stomach.[3] An allergic reaction, such as anaphylaxis, QT prolongation, or a type of diarrhea caused by Clostridium difficile is possible.[3] No harm has been found with its use during pregnancy.[3] Its safety during breastfeeding is not confirmed, but it is likely safe.[4] Azithromycin is an azalide, a type of macrolide antibiotic.[3] It works by decreasing the production of protein, thereby stopping bacterial growth.[3]
Azithromycin was discovered 1980 by Pliva, and approved for medical use in 1988.[5][6] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] The World Health Organization classifies it as critically important for human medicine.[8] It is available as a generic medication[9] and is sold under many trade names worldwide.[2] The wholesale cost in the developing world is about US$0.18 to US$2.98 per dose.[10] In the United States, it is about US$4 for a course of treatment as of 2018.[11] In 2016, it was the 49th most prescribed medication in the United States with more than 15 million prescriptions.[12]
Medical uses
Azithromycin is used to treat many different infections, including:
- Prevention and treatment of acute bacterial exacerbations of chronic obstructive pulmonary disease due to H. influenzae, M. catarrhalis, or S. pneumoniae. The benefits of long-term prophylaxis must be weighed on a patient-by-patient basis against the risk of cardiovascular and other adverse effects.[13]
- Community-acquired pneumonia due to C. pneumoniae, H. influenzae, M. pneumoniae, or S. pneumoniae[14]
- Uncomplicated skin infections due to S. aureus, S. pyogenes, or S. agalactiae
- Urethritis and cervicitis due to C. trachomatis or N. gonorrhoeae. In combination with ceftriaxone, azithromycin is part of the United States Centers for Disease Control-recommended regimen for the treatment of gonorrhea. Azithromycin is active as monotherapy in most cases, but the combination with ceftriaxone is recommended based on the relatively low barrier to resistance development in gonococci and due to frequent co-infection with C. trachomatis and N. gonorrhoeae.[15]
- Trachoma due to C. trachomatis[16]
- Genital ulcer disease (chancroid) in men due to H. ducrey
- Acute bacterial sinusitis due to H. influenzae, M. catarrhalis, or S. pneumoniae. Other agents, such as amoxicillin/clavulanate are generally preferred, however.[17][18]
- Acute otitis media caused by H. influenzae, M. catarrhalis or S. pneumoniae. Azithromycin is not, however, a first-line agent for this condition. Amoxicillin or another beta lactam antibiotic is generally preferred.[19]
- Pharyngitis or tonsillitis caused by S. pyogenes as an alternative to first-line therapy in individuals who cannot use first-line therapy[20]
Bacterial susceptibility
Azithromycin has relatively broad but shallow antibacterial activity. It inhibits some Gram-positive bacteria, some Gram-negative bacteria, and many atypical bacteria.
A strain of gonorrhea reported to be highly resistant to azithromycin was found in the population in 2015. Neisseria gonorrhoeae is normally susceptible to azithromycin,[21] but the drug is not widely used as monotherapy due to a low barrier to resistance development.[15] Extensive use of azithromycin has resulted in growing Streptococcus pneumoniae resistance.[22]
Aerobic and facultative Gram-positive microorganisms
- Staphylococcus aureus (Methicillin-sensitive only)
- Streptococcus agalactiae
- Streptococcus pneumoniae
- Streptococcus pyogenes
Aerobic and facultative Gram-negative microorganisms
- Haemophilus ducreyi
- Haemophilus influenzae
- Moraxella catarrhalis
- Neisseria gonorrhoeae
- Bordetella pertussis
- Legionella pneumophila
Anaerobic microorganisms
- Peptostreptococcus species
- Prevotella bivia
Other microorganisms
- Chlamydophila pneumoniae
- Chlamydia trachomatis
- Mycoplasma genitalium
- Mycoplasma pneumoniae
- Ureaplasma urealyticum
Pregnancy and breastfeeding
No harm has been found with use during pregnancy.[3] However, there are no adequate well-controlled studies in pregnant women.[23]
Safety of the medication during breastfeeding is unclear. It was reported that because only low levels are found in breast milk and the medication has also been used in young children, it is unlikely that breastfed infants would suffer adverse effects.[4] Nevertheless, it is recommended that the drug be used with caution during breastfeeding.[3]
Airway diseases
Azithromycin appears to be effective in the treatment of COPD through its suppression of inflammatory processes.[24] And potentially useful in asthma and sinusitis via this mechanism.[25] Azithromycin is believed to produce its effects through suppressing certain immune responses that may contribute to inflammation of the airways.[26][27]
Adverse effects
Most common adverse effects are diarrhea (5%), nausea (3%), abdominal pain (3%), and vomiting. Fewer than 1% of people stop taking the drug due to side effects. Nervousness, skin reactions, and anaphylaxis have been reported.[28] Clostridium difficile infection has been reported with use of azithromycin.[3] Azithromycin does not affect the efficacy of birth control unlike some other antibiotics such as rifampin. Hearing loss has been reported.[29]
Occasionally, people have developed cholestatic hepatitis or delirium. Accidental intravenous overdose in an infant caused severe heart block, resulting in residual encephalopathy.[30][31]
In 2013 the FDA issued a warning that azithromycin “can cause abnormal changes in the electrical activity of the heart that may lead to a potentially fatal irregular heart rhythm.” The FDA noted in the warning a 2012 study that found the drug may increase the risk of death, especially in those with heart problems, compared with those on other antibiotics such as amoxicillin or no antibiotic. The warning indicated people with preexisting conditions are at particular risk, such as those with QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or those who use certain drugs to treat abnormal heart rhythms.[32][33][34]
Pharmacology
Mechanism of action
Azithromycin prevents bacteria from growing by interfering with their protein synthesis. It binds to the 50S subunit of the bacterial ribosome, thus inhibiting translation of mRNA. Nucleic acid synthesis is not affected.[23]
Pharmacokinetics
Azithromycin is an acid-stable antibiotic, so it can be taken orally with no need of protection from gastric acids. It is readily absorbed, but absorption is greater on an empty stomach. Time to peak concentration (Tmax) in adults is 2.1 to 3.2 hours for oral dosage forms. Due to its high concentration in phagocytes, azithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations are released. The concentration of azithromycin in the tissues can be over 50 times higher than in plasma due to ion trapping and its high lipid solubility.[citation needed] Azithromycin’s half-life allows a large single dose to be administered and yet maintain bacteriostatic levels in the infected tissue for several days.[35]
Following a single dose of 500 mg, the apparent terminal elimination half-life of azithromycin is 68 hours.[35] Biliary excretion of azithromycin, predominantly unchanged, is a major route of elimination. Over the course of a week, about 6% of the administered dose appears as unchanged drug in urine.
History
A team of researchers at the pharmaceutical company Pliva in Zagreb, SR Croatia, Yugoslavia, — Gabrijela Kobrehel, Gorjana Radobolja-Lazarevski, and Zrinka Tamburašev, led by Dr. Slobodan Đokić — discovered azithromycin in 1980.[6] It was patented in 1981. In 1986, Pliva and Pfizer signed a licensing agreement, which gave Pfizer exclusive rights for the sale of azithromycin in Western Europe and the United States. Pliva put its azithromycin on the market in Central and Eastern Europe under the brand name Sumamed in 1988. Pfizer launched azithromycin under Pliva’s license in other markets under the brand name Zithromax in 1991.[36] Patent protection ended in 2005.[37]
Society and culture
_tablets.jpg)
Zithromax (azithromycin) 250 mg tablets (CA)
Cost
It is available as a generic medication.[9] The wholesale cost is about US$0.18 to US$2.98 per dose.[10] In the United States it is about US$4 for a course of treatment as of 2018.[11] In India, it is about US$1.70 for a course of treatment.[citation needed]
Available forms
Azithromycin is commonly administered in film-coated tablet, capsule, oral suspension, intravenous injection, granules for suspension in sachet, and ophthalmic solution.[2]
Usage
In 2010, azithromycin was the most prescribed antibiotic for outpatients in the US,[38] whereas in Sweden, where outpatient antibiotic use is a third as prevalent, macrolides are only on 3% of prescriptions.[39]
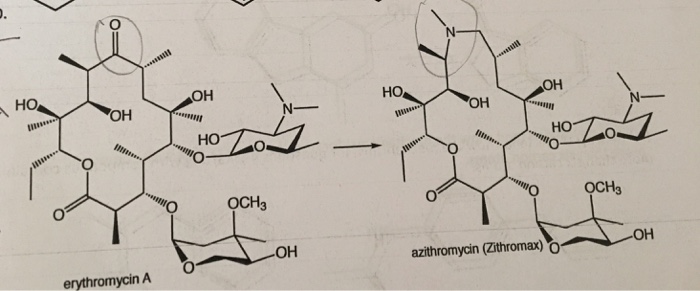
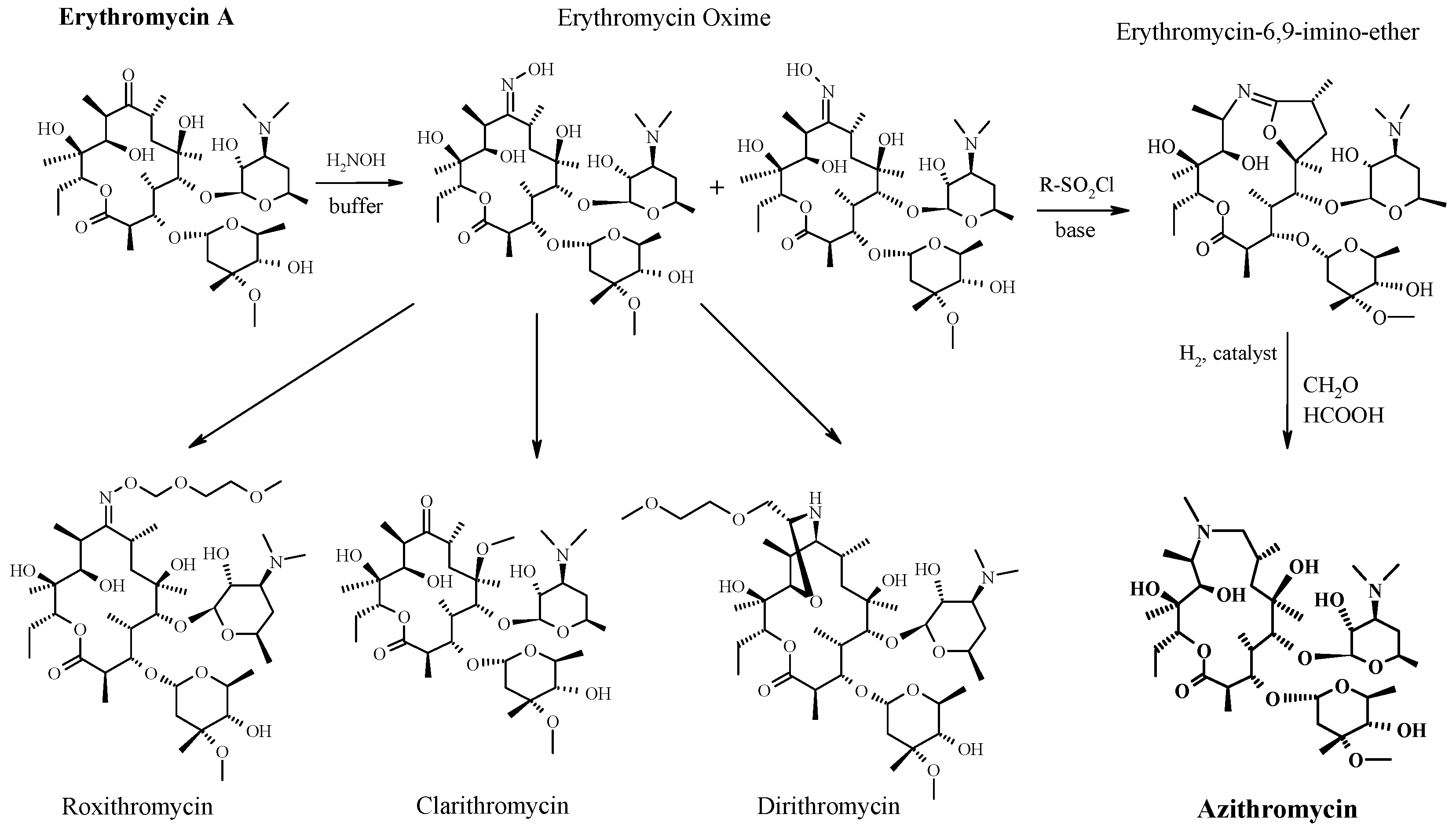
READ
References
- ^ Jump up to:ab “Azithromycin Use During Pregnancy”. Drugs.com. 2 May 2019. Retrieved 24 December 2019.
- ^ Jump up to:abcdef “Azithromycin International Brands”. Drugs.com. Archived from the original on 28 February 2017. Retrieved 27 February 2017.
- ^ Jump up to:abcdefghijklm “Azithromycin”. The American Society of Health-System Pharmacists. Archived from the original on 5 September 2015. Retrieved 1 August 2015.
- ^ Jump up to:ab “Azithromycin use while Breastfeeding”. Archived from the original on 5 September 2015. Retrieved 4 September 2015.
- ^ Greenwood, David (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1. publ. ed.). Oxford: Oxford University Press. p. 239. ISBN9780199534845. Archived from the original on 5 March 2016.
- ^ Jump up to:ab Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN9789241515528. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:ab Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. ISBN9781284057560.
- ^ Jump up to:ab “Azithromycin”. International Drug Price Indicator Guide. Retrieved 4 September 2015.
- ^ Jump up to:ab “NADAC as of 2018-05-23”. Centers for Medicare and Medicaid Services. Retrieved 24 May 2018.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December2018.
- ^ Taylor SP, Sellers E, Taylor BT (2015). “Azithromycin for the Prevention of COPD Exacerbations: The Good, Bad, and Ugly”. Am. J. Med. 128 (12): 1362.e1–6. doi:10.1016/j.amjmed.2015.07.032. PMID26291905.
- ^ Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, Dowell SF, File TM, Musher DM, Niederman MS, Torres A, Whitney CG (2007). “Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults”. Clin. Infect. Dis. 44 Suppl 2: S27–72. doi:10.1086/511159. PMID17278083.
- ^ Jump up to:ab “Gonococcal Infections – 2015 STD Treatment Guidelines”. Archived from the original on 1 March 2016.
- ^ Burton M, Habtamu E, Ho D, Gower EW (2015). “Interventions for trachoma trichiasis”. Cochrane Database Syst Rev. 11 (11): CD004008. doi:10.1002/14651858.CD004008.pub3. PMC4661324. PMID26568232.
- ^ Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, Orlandi RR, Palmer JN, Patel ZM, Peters A, Walsh SA, Corrigan MD (2015). “Clinical practice guideline (update): adult sinusitis”. Otolaryngol Head Neck Surg. 152 (2 Suppl): S1–S39. doi:10.1177/0194599815572097. PMID25832968.
- ^ Hauk L (2014). “AAP releases guideline on diagnosis and management of acute bacterial sinusitis in children one to 18 years of age”. Am Fam Physician. 89 (8): 676–81. PMID24784128.
- ^ Neff MJ (2004). “AAP, AAFP release guideline on diagnosis and management of acute otitis media”. Am Fam Physician. 69 (11): 2713–5. PMID15202704.
- ^ Randel A (2013). “IDSA Updates Guideline for Managing Group A Streptococcal Pharyngitis”. Am Fam Physician. 88 (5): 338–40. PMID24010402.
- ^ The Guardian newspaper: ‘Super-gonorrhoea’ outbreak in Leeds, 18 September 2015Archived 18 September 2015 at the Wayback Machine
- ^ Lippincott Illustrated Reviews : Pharmacology Sixth Edition. p. 506.
- ^ Jump up to:ab “US azithromycin label”(PDF). FDA. February 2016. Archived(PDF) from the original on 23 November 2016.
- ^ Simoens, Steven; Laekeman, Gert; Decramer, Marc (May 2013). “Preventing COPD exacerbations with macrolides: A review and budget impact analysis”. Respiratory Medicine. 107 (5): 637–648. doi:10.1016/j.rmed.2012.12.019. PMID23352223.
- ^ Gotfried, Mark H. (February 2004). “Macrolides for the Treatment of Chronic Sinusitis, Asthma, and COPD”. CHEST. 125 (2): 52S–61S. doi:10.1378/chest.125.2_suppl.52S. ISSN0012-3692. PMID14872001.
- ^ Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. (May 2012). “Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases”. European Journal of Clinical Pharmacology. 68 (5): 479–503. doi:10.1007/s00228-011-1161-x. ISSN1432-1041. PMID22105373.
- ^ Steel, Helen C.; Theron, Annette J.; Cockeran, Riana; Anderson, Ronald; Feldman, Charles (2012). “Pathogen- and Host-Directed Anti-Inflammatory Activities of Macrolide Antibiotics”. Mediators of Inflammation. 2012: 584262. doi:10.1155/2012/584262. PMC3388425. PMID22778497.
- ^ Mori F, Pecorari L, Pantano S, Rossi M, Pucci N, De Martino M, Novembre E (2014). “Azithromycin anaphylaxis in children”. Int J Immunopathol Pharmacol. 27 (1): 121–6. doi:10.1177/039463201402700116. PMID24674687.
- ^ Dart, Richard C. (2004). Medical Toxology. Lippincott Williams & Wilkins. p. 23.
- ^ Tilelli, John A.; Smith, Kathleen M.; Pettignano, Robert (2006). “Life-Threatening Bradyarrhythmia After Massive Azithromycin Overdose”. Pharmacotherapy. 26 (1): 147–50. doi:10.1592/phco.2006.26.1.147. PMID16506357.
- ^ Baselt, R. (2008). Disposition of Toxic Drugs and Chemicals in Man (8th ed.). Foster City, CA: Biomedical Publications. pp. 132–133.
- ^ Denise Grady (16 May 2012). “Popular Antibiotic May Raise Risk of Sudden Death”. The New York Times. Archived from the original on 17 May 2012. Retrieved 18 May 2012.
- ^ Ray, Wayne A.; Murray, Katherine T.; Hall, Kathi; Arbogast, Patrick G.; Stein, C. Michael (2012). “Azithromycin and the Risk of Cardiovascular Death”. New England Journal of Medicine. 366(20): 1881–90. doi:10.1056/NEJMoa1003833. PMC3374857. PMID22591294.
- ^ “FDA Drug Safety Communication: Azithromycin (Zithromax or Zmax) and the risk of potentially fatal heart rhythms”. FDA. 12 March 2013. Archived from the original on 27 October 2016.
- ^ Jump up to:ab “Archived copy”. Archived from the original on 14 October 2014. Retrieved 10 October 2014.
- ^ Banić Tomišić, Z. (2011). “The Story of Azithromycin”. Kemija U Industriji. 60 (12): 603–617. ISSN0022-9830. Archived from the original on 8 September 2017.
- ^ “Azithromycin: A world best-selling Antibiotic”. http://www.wipo.int. World Intellectual Property Organization. Retrieved 18 June 2019.
- ^ Hicks, LA; Taylor TH, Jr; Hunkler, RJ (April 2013). “U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 368 (15): 1461–1462. doi:10.1056/NEJMc1212055. PMID23574140.
- ^ Hicks, LA; Taylor TH, Jr; Hunkler, RJ (September 2013). “More on U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 369 (12): 1175–1176. doi:10.1056/NEJMc1306863. PMID24047077.
External links
Keywords: Antibacterial (Antibiotics); Macrolides.
- “Azithromycin”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Zithromax, Azithrocin, others[2] |
| Other names | 9-deoxy-9α-aza-9α-methyl-9α-homoerythromycin A |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a697037 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth (capsule, tablet or suspension), intravenous, eye drop |
| Drug class | Macrolide antibiotic |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 38% for 250 mg capsules |
| Metabolism | Liver |
| Elimination half-life | 11–14 h (single dose) 68 h (multiple dosing) |
| Excretion | Biliary, kidney (4.5%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.126.551 |
| Chemical and physical data | |
| Formula | C38H72N2O12 |
| Molar mass | 748.984 g·mol−1 g·mol−1 |
| 3D model (JSmol) | |
/////////AZITHROMYCIN, Antibacterial, Antibiotics, Macrolides, CORONA VIRUS, COVID 19, アジスロマイシン ,
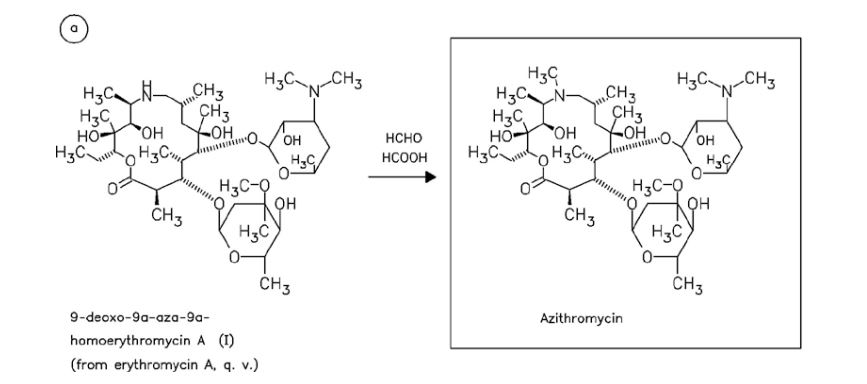
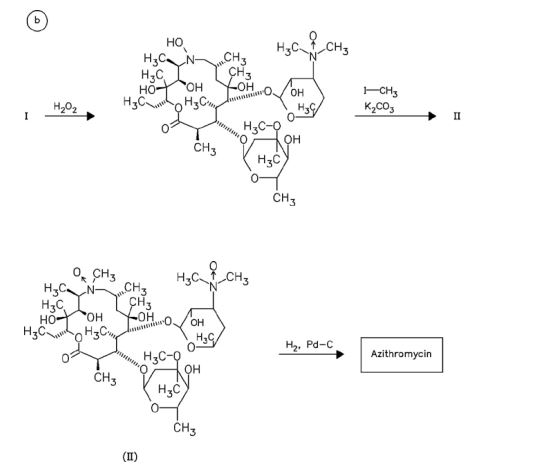
Substances Referenced in Synthesis Path
CAS-RN Formula Chemical Name CAS Index Name
76801-85-9 C37H70N2O12 2-deoxo-9a-aza-9a-homoerythromycin A 1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-2-eth- yl-3,4,10-trihydroxy-3,5,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-, [2R-(2
R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,13S*,1
4R*)]-
90503-04-1 C37H70N2O14 [2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,
13S*,14R*)]-13-[(2,6-dideoxy-3-C-methyl3-O-methyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,6,10-tetrahydroxy3,5,8,10,12,14-hexamethyl-13-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-
β-D-xylo-hexopyranosyl] oxy]-1-oxa-6-azacyclopentadecan-15-one
1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-Omethyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,6,10-tetrahydroxy3,5,8,10,12,14-hexamethyl-13-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-β-Dxylo-hexopyranosyl]oxy]-, [2R-(2R*,3S*,4R
*,5R*,8R*,10R*,11R*,12S*,13S*,14R*)]-
90503-05-2 C38H72N2O14 [2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,
13S*,14R*)]-13-[(2,6-dideoxy-3-C-methyl3-O-methyl-α-L-ribo-hexopyranosyl) oxy]-2-ethyl-3,4,10-trihydroxy3,5,6,8,10,12,14-heptamethyl-11-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-
β-D-xylo-hexopyranosyl]
oxy]-1-oxa-6-azacyclopentadecan-15-one
6-oxide
1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-Omethyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,10-trihydroxy3,5,6,8,10,12,14-heptamethyl-11-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-βD-xylo-hexopyranosyl]oxy]-, 6-oxide,
[2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,1
3S*,14R*)]-
50-00-0 CH2O formaldehyde Formaldehyde
74-88-4 CH3I methyl iodide Methane, iodoTrade Names
Country Trade Name Vendor Annotation
D Ultreon Pfizer
Zithromax Pfizer Pharma/Gödecke/Parke-Davis
numerous generic preparations
F Azadose Pfizer
Monodose Pfizer
Zithromax Pfizer
GB Zithromax Pfizer
I Azitrocin Bioindustria
Ribotrex Pierre Fabre
Trocozina Sigma-Tau
Zithromax Pfizer
J Zithromac Pfizer
USA Azasite InSite Vision
Zithromax Pfizer as dihydrate
Formulations
cps. 100 mg, 250 mg; Gran. 10%; susp. 200 mg (as dihydrate); tabl. 250 mg
References
Djokic, S. et al.: J. Antibiot. (JANTAJ) 40, 1006 (1987).
a DOS 3 140 449 (Pliva; appl. 12.10.1981; YU-prior. 6.3.1981).
US 4 517 359 (Pliva; 14.5.1985; appl. 22.9.1981; YU-prior. 6.3.1981).
b EP 101 186 (Pliva; appl. 14.7.1983; USA-prior. 19.7.1982, 15.11.1982).
US 4 474 768 (Pfizer; 2.10.1984; prior. 19.7.1982, 15.11.1982).
educt by ring expansion of erythromycin A oxime by Beckmann rearrangement:
Djokic, S. et al.: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1986, 1881-1890.
Bright, G.M. et al.: J. Antibiot. (JANTAJ) 41, 1029 (1988). US 4 328 334 (Pliva; 4.5.1982; YU-prior. 2.4.1979).
stable, non-hygroscopic dihydrate: EP 298 650 (Pfizer; appl. 28.6.1988).
medical use for treatment of protozoal infections:
US 4 963 531 (Pfizer; 16.10.1990; prior. 16.8.1988, 10.9.1987).
Pretomanid, プレトマニド;
Pretomanid
プレトマニド;
| Formula |
C14H12F3N3O5
|
|---|---|
| CAS |
187235-37-6
|
| Mol weight |
359.2574
|
- (S)-PA 824
2019/8/14 FDA 2109 APPROVED
Antibacterial (tuberculostatic),
MP 149-150 °C, Li, Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262 and Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
150-151 °C Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482
Pretomanid is an antibiotic used for the treatment of multi-drug-resistant tuberculosis affecting the lungs.[1] It is generally used together with bedaquiline and linezolid.[1] It is taken by mouth.[1]
The most common side effects include nerve damage, acne, vomiting, headache, low blood sugar, diarrhea, and liver inflammation.[1] It is in the nitroimidazole class of medications.[2]
Pretomanid was approved for medical use in the United States in 2019.[3][1] Pretomanid was developed by TB Alliance,[4] a not-for-profitproduct development partnership dedicated to the discovery and development of new, faster-acting and affordable medicines for tuberculosis (TB).[5]
Global Alliance for the treatment of tuberculosis (TB).
The compound was originally developed by PathoGenesis (acquired by Chiron in 2000). In 2002, a co-development agreement took place between Chiron (acquired by Novartis in 2005) and the TB Alliance for the development of the compound. The compound was licensed to Fosunpharma by TB Alliance in China.
History
Pretomanid is the generic, nonproprietary name for the novel anti-bacterial drug compound formerly called PA-824.[6] Pretomanid is referred to as “Pa” in regimen abbreviations, such as BPaL. The “preto” prefix of the compound’s name honors Pretoria, South Africa, the home of a TB Alliance clinical development office where much of the drug’s development took place. The “manid” suffix is used to group compounds with similar chemical structures. This class of drug is variously referred to as nitroimidazoles, nitroimidazooxazines or nitroimidazopyrans. Development of this compound was initiated because of the urgent need for new antibacterial drugs effective against resistant strains of tuberculosis. Also, current anti-TB drugs are mainly effective against replicating and metabolically active bacteria, creating a need for drugs effective against persisting or latent bacterial infections as often occur in patients with tuberculosis.[7]
Discovery and pre-clinical development
Pretomanid was first identified in a series of 100 nitroimidazopyran derivatives synthesized and tested for antitubercular activity. Importantly, pretomanid has activity against static M. tuberculosis isolates that survive under anaerobic conditions, with bactericidal activity comparable to that of the existing drug metronidazole. Pretomanid requires metabolic activation by Mycobacterium for antibacterial activity. Pretomanid was not the most potent compound in the series against cultures of M. tuberculosis, but it was the most active in infected mice after oral administration. Oral pretomanid was active against tuberculosis in mice and guinea pigs at safely tolerated dosages for up to 28 days.[7]
Limited FDA approval
FDA approved pretomanid only in combination with bedaquiline and linezolid for treatment of a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug resistant pulmonary tuberculosis. Pretomanid was approved under the Limited Population Pathway (LPAD pathway) for antibacterial and antifungal drugs. The LPAD Pathway was established by Congress under the 21st Century Cures Act to expedite development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need. Pretomanid is only the third tuberculosis drug to receive FDA approval in more than 40 years.[3][8]
PATENT
IN 201641030408
HETERO RESEARCH FOUNDATION
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
- By Reddy, Bandi Parthasaradhi; Reddy, Kura Rathnakar; Reddy, Adulla Venkat Narsimha; Krishna, Bandi Vamsi
- From Indian Pat. Appl. (2018), IN 201641030408
The nitroimidazooxazine Formula I (PA-824) is a new class of bioreductive drug for tuberculosis. The recent introduction of the nitroimidazooxazine Formula I (PA-824) to clinical trial by the Global Alliance for TB Drug Development is thus of potential significance, since this compound shows good in vitro and in vivo activity against Mycobacterium tuberculosis in both its active and persistent forms. Tuberculosis (TBa) remains a leading infectious cause of death worldwide, but very few new drugs have been approved for TB treatment ifi the past 35 years, the current drug therapy for TB is long and complex, involving multidrug combinations.
The mechanism of actiém of Pretomanid is thoughrto involve reduction of the nitro group, in a‘ process dependent on the Bacterial ‘ m E Nfilw‘fieéFPEOEPEa‘e fillyeifiaasnfi (F8189); $943“; 20mm; “q Mcyarecent Swiss on mutant strains showed that a 151-amino acid (17.37 kDa) protein of unknown function, Rv3547, also, appears to be critical for this activation. Equivalent genes are present in M. boVis and MaVium.
Pretomanid and its pharmace’utically acceptable salts Were generically disclosed in US 5,668,127 A and Specifically disclosed in US 6,087,358 A. US ‘358 patent discloses a process for the preparation of Pretomanid, which is as shown below in scheme 1:

CN 104177372 A discloses a process for the preparation of Pretomanid, which is as shown below in scheme II: 
Bioorganic & Medicinal Chemistry Letters 2008, Volume: 18, Issue: 7, Pages: 2256-2262 discloses a process for the preparation of Pretomanid, which is as shown below in scheme Ill: 
US 7,!15,736 B2-discloses_a process fdr the preparation of 3S-Hydroxy-6-nitrQ-2H-3, 4— dihydro-[2-1b]-imidazopyran which is a key intermediate of Pretomanid, which is as shown below in scheme IV:

Journal Medicinal Chemistry 2009, Volume: 52, Pages: 637 — 645 discloses a process for the preparation of ‘Pretomanid, which is as shown below in scheme V:

Joumal Organic Chemistry 2010; Volume: 75 (2]), Pages: 7479—82 discloses a process for. the preparation of Pretomanid, which is as shown below in scheme VI:


Example 3: Preparation of Pretomanid (S) 1- -(3 (tert- -Butyldomethylsilyloxy)- -2- -(-4 -(trifluoromethoxy)-71benzyloxy2‘- propyl)- 2- -methylP AT E N4Tnitro- fi-Eimigazole (Efgm Awlas (3315;501:1691 gin! %etra%1y7drofuraen (18(150 ml) at room temperature and stirred for 5 to 10 minutes then TBAF (9516 ml) was added to the reaction mixture and stirred for 2 hours, at room temperature, afler completion of the reaction removed solvent through vacuum to obtained residue, dissolved the residue in MDC (1800 ml) and water (1800 ml) stirred, separated the layers and the organic layer washed with 10% ‘ sodium bicarbonate the obtained organic solution was concentrated under atmospheric pressure to obtained residue added MeOH (1730 ml) at room temperature and the reaction mixture was cooled to 0°C to 5°C, added KOH (24.5 gm) at the same temperaturé then cooled to room temperature and stirred for 24 hours. After completion of reaction DM Water added drop wise over 30 minutes at 10°C to 15° C and stirred for 1 hour to 1 hour 30 minutes at room’lemperature, filtrated the compound and washed with DM wa‘er (133 ml) and dried under vacuum for 10 hours at 50° C. Yield: 53 gm , Chromatographic purity: 97.69% (by HPLC):
Example 4: Purification of Pretomanid Pretomanid (53 gm) was dissolved in MDC (795 ml) at room temperatur’e and stirred for 10 to 15 minutes, added charcoal (10 gm) and stirred for 30-35 minutes, remove the charcoal and concentrated to obtained residue: Dissolved the obtained residue in IPA (795 ml) and the reaction mixture was heated to 80°C maintained for 10-15 minutes, added cyclohexane (1600ml) for 30 minutes at 80° C, then cooled to room temperature and stirred the reaction mass for overnight, filtered the solid and washed with cyclohexane (265 ml), and dried under vacuum for 10 hours at 50° C. Yield: 48 gm (Percentage of Yield: 90%) Chromatographic purity: 99.97% by HPLC).
CLIP

ReferencE
CN104177372A.
WO9701562A1.
IN 201641030408
IN 201621026053
CN 107915747
CN 106632393
CN 106565744
CN 104177372
WO 9701562
US 6087358
PAPER
Science (Washington, DC, United States) (2008), 322(5906), 1392-1395.
Paper
PAPER
Huagong Shikan (2010), 24(4), 32-34, 51.
Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262
PAPER
Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
https://onlinelibrary.wiley.com/doi/abs/10.1002/adsc.200700119
https://application.wiley-vch.de/contents/jc_2258/2007/f700119_s.pdf



Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482

Scheme 1

aDHP = 3,4-dihydropyran; p-TsOH = p-toluenesulfonic acid; MsOH = methanesulfonic acid.
Scheme 3

aCl3CCN = trichloroacetonitrile; TBME = tert-butylmethyl ether; TfOH = trifluoromethanesulfonic acid.



PAPER
Journal of Medicinal Chemistry (2010), 53(1), 282-294.
Journal of Medicinal Chemistry (2009), 52(3), 637-645.
PATENT

References
- ^ Jump up to:a b c d e “FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs”. FDA. 14 August 2019. Retrieved 28 August 2019.
- ^ “Compounds | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ Jump up to:a b Abutaleb Y (14 August 2019). “New antibiotic approved for drug-resistant tuberculosis”. Washington Post.
- ^ “TB Medicine Pretomanid Enters Regulatory Review Process in the United States | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ “About TB Alliance”. TB Alliance. Retrieved 2019-04-18.
- ^ “PA-824 has a New Generic Name: Pretomanid”. TB Alliance. Retrieved 2019-04-18.
- ^ Jump up to:a b Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM (June 2005). “Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models”. Antimicrobial Agents and Chemotherapy. 49 (6): 2294–301. doi:10.1128/AAC.49.6.2294-2301.2005. PMC 1140539. PMID 15917524.
- ^ FDA News Release. FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs.
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C14H12F3N3O5 |
| Molar mass | 359.261 g·mol−1 |
| 3D model (JSmol) | |
//////////////Pretomanid, FDA 2109, プレトマニド , Antibacterial, tuberculostatic, PA-824, ANTI tuberculostatic
Novobiocin, ノボビオシン;

Novobiocin
ノボビオシン;
- Molecular FormulaC31H36N2O11
- Average mass612.624 Da
Monoisotopic: 612.231910004
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Novobiocin sodium | Q9S9NQ5YIY | 1476-53-5 | WWPRGAYLRGSOSU-RNROJPEYSA-M |
Reata Pharmaceuticals Inc
Abgentis is investigating a novobiocin analog, GYR-12 (discovery), as a re-engineered, previously-marketed-but-uncompetitive (undisclosed) antibacterial compound inhibiting ATPase activity of DNA supercoiling GyrB/ParE, for the potential broad-spectrum treatment of bacterial infections, including multi-drug resistant Gram-negative infections. In April 2017, development was underway [1924695].
Novobiocin, also known as albamycin or cathomycin, is an aminocoumarin antibiotic that is produced by the actinomycete Streptomyces niveus, which has recently been identified as a subjective synonym for S. spheroides[1] a member of the order Actinobacteria. Other aminocoumarin antibiotics include clorobiocin and coumermycin A1.[2] Novobiocin was first reported in the mid-1950s (then called streptonivicin).[3][4]
It is active against Staphylococcus epidermidis and may be used to differentiate it from the other coagulase-negative Staphylococcus saprophyticus, which is resistant to novobiocin, in culture.
Novobiocin was licensed for clinical use under the tradename Albamycin (Pharmacia And Upjohn) in the 1960s. Its efficacy has been demonstrated in preclinical and clinical trials.[5][6] The oral form of the drug has since been withdrawn from the market due to lack of efficacy.[7] Novobiocin is an effective antistaphylococcal agent used in the treatment of MRSA.[8]
Mechanism of action
The molecular basis of action of novobiocin, and other related drugs clorobiocin and coumermycin A1 has been examined.[2][9][10][11][12] Aminocoumarins are very potent inhibitors of bacterial DNA gyrase and work by targeting the GyrB subunit of the enzyme involved in energy transduction. Novobiocin as well as the other aminocoumarin antibiotics act as competitive inhibitors of the ATPase reaction catalysed by GyrB. The potency of novobiocin is considerably higher than that of the fluoroquinolones that also target DNA gyrase, but at a different site on the enzyme. The GyrA subunit is involved in the DNA nicking and ligation activity.
Novobiocin has been shown to weakly inhibit the C-terminus of the eukaryotic Hsp90 protein (high micromolar IC50). Modification of the novobiocin scaffold has led to more selective Hsp90 inhibitors.[13] Novobiocin has also been shown to bind and activate the Gram-negative lipopolysaccharide transporter LptBFGC.[14][15]
Structure
Novobiocin is an aminocoumarin. Novobiocin may be divided up into three entities; a benzoic acid derivative, a coumarin residue, and the sugar novobiose.[9] X-ray crystallographic studies have found that the drug-receptor complex of Novobiocin and DNA Gyrase shows that ATP and Novobiocin have overlapping binding sites on the gyrase molecule.[16] The overlap of the coumarin and ATP-binding sites is consistent with aminocoumarins being competitive inhibitors of the ATPase activity.[17]
Structure–activity relationship
In structure activity relationship experiments it was found that removal of the carbamoyl group located on the novobiose sugar lead to a dramatic decrease in inhibitory activity of novobiocin.[17]
Biosynthesis
This aminocoumarin antibiotic consists of three major substituents. The 3-dimethylallyl-4-hydroxybenzoic acid moiety, known as ring A, is derived from prephenate and dimethylallyl pyrophosphate. The aminocoumarin moiety, known as ring B, is derived from L-tyrosine. The final component of novobiocin is the sugar derivative L-noviose, known as ring C, which is derived from glucose-1-phosphate. The biosynthetic gene cluster for novobiocin was identified by Heide and coworkers in 1999 (published 2000) from Streptomyces spheroidesNCIB 11891.[18] They identified 23 putative open reading frames (ORFs) and more than 11 other ORFs that may play a role in novobiocin biosynthesis.
The biosynthesis of ring A (see Fig. 1) begins with prephenate which is a derived from the shikimic acid biosynthetic pathway. The enzyme NovF catalyzes the decarboxylation of prephenate while simultaneously reducing nicotinamide adenine dinucleotide phosphate (NADP+) to produce NADPH. Following this NovQ catalyzes the electrophilic substitution of the phenyl ring with dimethylallyl pyrophosphate (DMAPP) otherwise known as prenylation.[19] DMAPP can come from either the mevalonic acid pathway or the deoxyxylulose biosynthetic pathway. Next the 3-dimethylallyl-4-hydroxybenzoate molecule is subjected to two oxidative decarboxylations by NovR and molecular oxygen.[20] NovR is a non-heme iron oxygenase with a unique bifunctional catalysis. In the first stage both oxygens are incorporated from the molecular oxygen while in the second step only one is incorporated as determined by isotope labeling studies. This completes the formation of ring A.

Figure 1. Biosynthetic scheme of benzamide portion of novobiocin (4-hydroxy-3-(3-methylbut-2-en-1-yl)benzoic acid)
The biosynthesis of ring B (see Fig. 2) begins with the natural amino acid L-tyrosine. This is then adenylated and thioesterified onto the peptidyl carrier protein (PCP) of NovH by ATPand NovH itself.[21] NovI then further modifies this PCP bound molecule by oxidizing the β-position using NADPH and molecular oxygen. NovJ and NovK form a heterodimer of J2K2 which is the active form of this benzylic oxygenase.[22] This process uses NADP+ as a hydride acceptor in the oxidation of the β-alcohol. This ketone will prefer to exist in its enol tautomer in solution. Next a still unidentified protein catalyzes the selective oxidation of the benzene (as shown in Fig. 2). Upon oxidation this intermediate will spontaneously lactonize to form the aromatic ring B and lose NovH in the process.

Figure 2. Biosynthesis of 3-amino-4,7-dihydroxy-2H-chromen-2-one component of novobiocin (ring B)
The biosynthesis of L-noviose (ring C) is shown in Fig. 3. This process starts from glucose-1-phosphate where NovV takes dTTP and replaces the phosphate group with a dTDP group. NovT then oxidizes the 4-hydroxy group using NAD+. NovT also accomplishes a dehydroxylation of the 6 position of the sugar. NovW then epimerizes the 3 position of the sugar.[23] The methylation of the 5 position is accomplished by NovU and S-adenosyl methionine (SAM). Finally NovS reduces the 4 position again to achieve epimerization of that position from the starting glucose-1-phosphate using NADH.

Figure 3. Biosynthesis of L-noviose component of novobiocin (ring C)
Rings A, B, and C are coupled together and modified to give the finished novobiocin molecule. Rings A and B are coupled together by the enzyme NovL using ATP to diphosphorylate the carboxylate group of ring A so that the carbonyl can be attacked by the amine group on ring B. The resulting compound is methylated by NovO and SAM prior to glycosylation.[24] NovM adds ring C (L-noviose) to the hydroxyl group derived from tyrosine with the loss of dTDP. Another methylation is accomplished by NovP and SAM at the 4 position of the L-noviose sugar.[25] This methylation allows NovN to carbamylate the 3 position of the sugar as shown in Fig. 4 completing the biosynthesis of novobiocin.

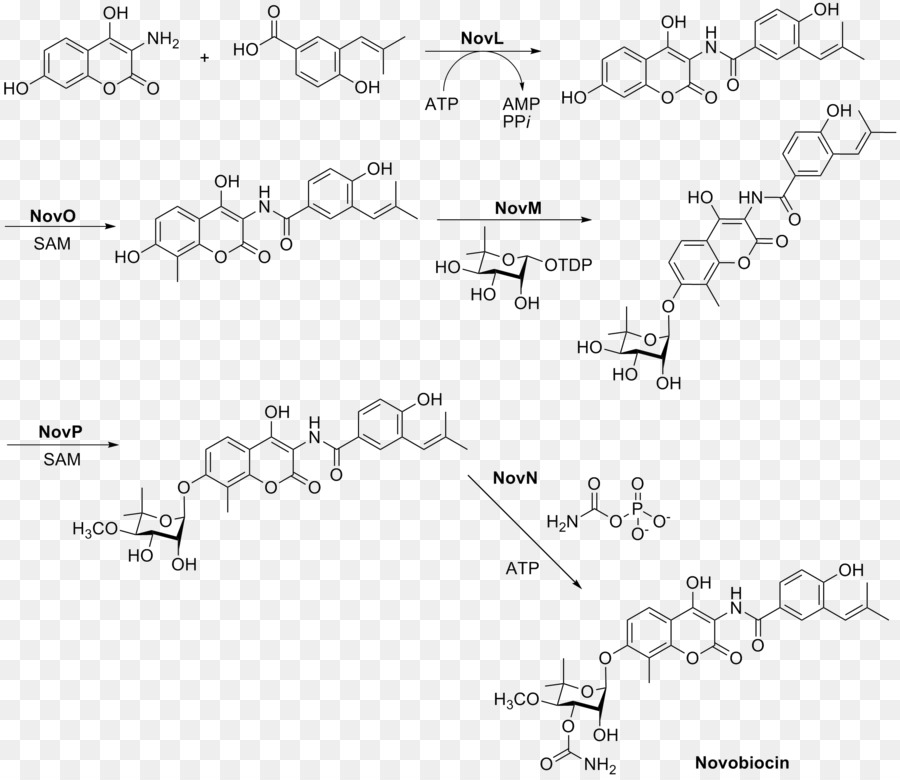
CLIP

CLIP

CLIP
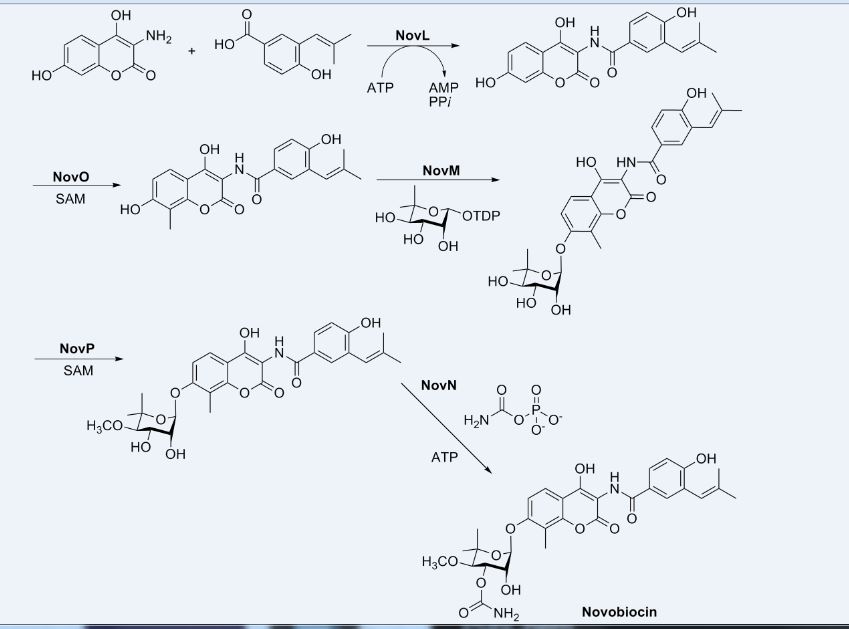

PATENT
US-20190241599
Novel co-crystal forms of novobiocin and its analogs and proline, processes for their preparation and compositions comprising them are claimed. Also claims are methods for inhibiting heat shock protein 90 and treating or preventing neurodegenerative disorders, such as diabetic peripheral neuropathy.
References
- ^ Lanoot B, Vancanneyt M, Cleenwerck I, Wang L, Li W, Liu Z, Swings J (May 2002). “The search for synonyms among streptomycetes by using SDS-PAGE of whole-cell proteins. Emendation of the species Streptomyces aurantiacus, Streptomyces cacaoi subsp. cacaoi, Streptomyces caeruleus and Streptomyces violaceus”. International Journal of Systematic and Evolutionary Microbiology. 52 (Pt 3): 823–9. doi:10.1099/ijs.0.02008-0. PMID 12054245.
- ^ Jump up to:a b Alessandra da Silva Eustáquio (2004) Biosynthesis of aminocoumarin antibiotics in Streptomyces: Generation of structural analogues by genetic engineering and insights into the regulation of antibiotic production. DISSERTATION
- ^ Hoeksema H.; Johnson J. L.; Hinman J. W. (1955). “Structural studies on streptonivicin, a new antibiotic”. J Am Chem Soc. 77 (24): 6710–6711. doi:10.1021/ja01629a129.
- ^ Smith C. G.; Dietz A.; Sokolski W. T.; Savage G. M. (1956). “Streptonivicin, a new antibiotic. I. Discovery and biologic studies”. Antibiotics & Chemotherapy. 6: 135–142.
- ^ Raad I, Darouiche R, Hachem R, Sacilowski M, Bodey GP (November 1995). “Antibiotics and prevention of microbial colonization of catheters”. Antimicrobial Agents and Chemotherapy. 39 (11): 2397–400. doi:10.1128/aac.39.11.2397. PMC 162954. PMID 8585715.
- ^ Raad II, Hachem RY, Abi-Said D, Rolston KV, Whimbey E, Buzaid AC, Legha S (January 1998). “A prospective crossover randomized trial of novobiocin and rifampin prophylaxis for the prevention of intravascular catheter infections in cancer patients treated with interleukin-2”. Cancer. 82 (2): 403–11. doi:10.1002/(SICI)1097-0142(19980115)82:2<412::AID-CNCR22>3.0.CO;2-0. PMID 9445199.
- ^ “Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn From Sale for Reasons of Safety or Effectiveness”. The Federal Register. 19 January 2011.
- ^ Walsh TJ, Standiford HC, Reboli AC, John JF, Mulligan ME, Ribner BS, Montgomerie JZ, Goetz MB, Mayhall CG, Rimland D (June 1993). “Randomized double-blinded trial of rifampin with either novobiocin or trimethoprim-sulfamethoxazole against methicillin-resistant Staphylococcus aureus colonization: prevention of antimicrobial resistance and effect of host factors on outcome”. Antimicrobial Agents and Chemotherapy. 37 (6): 1334–42. doi:10.1128/aac.37.6.1334. PMC 187962. PMID 8328783.
- ^ Jump up to:a b Maxwell A (August 1993). “The interaction between coumarin drugs and DNA gyrase”. Molecular Microbiology. 9 (4): 681–6. doi:10.1111/j.1365-2958.1993.tb01728.x. PMID 8231802.
- ^ Maxwell A (February 1999). “DNA gyrase as a drug target”. Biochemical Society Transactions. 27 (2): 48–53. doi:10.1042/bst0270048. PMID 10093705.
- ^ Lewis RJ, Tsai FT, Wigley DB (August 1996). “Molecular mechanisms of drug inhibition of DNA gyrase”. BioEssays. 18 (8): 661–71. doi:10.1002/bies.950180810. PMID 8760340.
- ^ Maxwell A, Lawson DM (2003). “The ATP-binding site of type II topoisomerases as a target for antibacterial drugs”. Current Topics in Medicinal Chemistry. 3 (3): 283–303. doi:10.2174/1568026033452500. PMID 12570764.
- ^ Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS (September 2005). “Hsp90 inhibitors identified from a library of novobiocin analogues”. Journal of the American Chemical Society. 127 (37): 12778–9. doi:10.1021/ja0535864. PMID 16159253.
- ^ Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D (June 2018). “Novobiocin Enhances Polymyxin Activity by Stimulating Lipopolysaccharide Transport”. Journal of the American Chemical Society. 140 (22): 6749–6753. doi:10.1021/jacs.8b02283. PMC 5990483. PMID 29746111.
- ^ May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, Kahne D (December 2017). “The Antibiotic Novobiocin Binds and Activates the ATPase That Powers Lipopolysaccharide Transport”. Journal of the American Chemical Society. 139 (48): 17221–17224. doi:10.1021/jacs.7b07736. PMC 5735422. PMID 29135241.
- ^ Tsai FT, Singh OM, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB (May 1997). “The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin”. Proteins. 28 (1): 41–52. doi:10.1002/(sici)1097-0134(199705)28:1<41::aid-prot4>3.3.co;2-b. PMID 9144789.
- ^ Jump up to:a b Flatman RH, Eustaquio A, Li SM, Heide L, Maxwell A (April 2006). “Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis”. Antimicrobial Agents and Chemotherapy. 50 (4): 1136–42. doi:10.1128/AAC.50.4.1136-1142.2006. PMC 1426943. PMID 16569821.
- ^ Steffensky M, Mühlenweg A, Wang ZX, Li SM, Heide L (May 2000). “Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891”. Antimicrobial Agents and Chemotherapy. 44 (5): 1214–22. doi:10.1128/AAC.44.5.1214-1222.2000. PMC 89847. PMID 10770754.
- ^ Pojer F, Wemakor E, Kammerer B, Chen H, Walsh CT, Li SM, Heide L (March 2003). “CloQ, a prenyltransferase involved in clorobiocin biosynthesis”. Proceedings of the National Academy of Sciences of the United States of America. 100 (5): 2316–21. Bibcode:2003PNAS..100.2316P. doi:10.1073/pnas.0337708100. PMC 151338. PMID 12618544.
- ^ Pojer F, Kahlich R, Kammerer B, Li SM, Heide L (August 2003). “CloR, a bifunctional non-heme iron oxygenase involved in clorobiocin biosynthesis”. The Journal of Biological Chemistry. 278 (33): 30661–8. doi:10.1074/jbc.M303190200. PMID 12777382.
- ^ Chen H, Walsh CT (April 2001). “Coumarin formation in novobiocin biosynthesis: beta-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI”. Chemistry & Biology. 8 (4): 301–12. doi:10.1016/S1074-5521(01)00009-6. PMID 11325587.
- ^ Pacholec M, Hillson NJ, Walsh CT (September 2005). “NovJ/NovK catalyze benzylic oxidation of a beta-hydroxyl tyrosyl-S-pantetheinyl enzyme during aminocoumarin ring formation in novobiocin biosynthesis”. Biochemistry. 44 (38): 12819–26. CiteSeerX 10.1.1.569.1481. doi:10.1021/bi051297m. PMID 16171397.
- ^ Thuy TT, Lee HC, Kim CG, Heide L, Sohng JK (April 2005). “Functional characterizations of novWUS involved in novobiocin biosynthesis from Streptomyces spheroides”. Archives of Biochemistry and Biophysics. 436 (1): 161–7. doi:10.1016/j.abb.2005.01.012. PMID 15752721.
- ^ Pacholec M, Tao J, Walsh CT (November 2005). “CouO and NovO: C-methyltransferases for tailoring the aminocoumarin scaffold in coumermycin and novobiocin antibiotic biosynthesis”. Biochemistry. 44 (45): 14969–76. doi:10.1021/bi051599o. PMID 16274243.
- ^ Freel Meyers CL, Oberthür M, Xu H, Heide L, Kahne D, Walsh CT (January 2004). “Characterization of NovP and NovN: completion of novobiocin biosynthesis by sequential tailoring of the noviosyl ring”. Angewandte Chemie. 43 (1): 67–70. doi:10.1002/anie.200352626. PMID 14694473.
External links
- Novobiocin bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
intravenous |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | negligible oral bioavailability |
| Metabolism | excreted unchanged |
| Elimination half-life | 6 hours |
| Excretion | renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.005.589 |
| Chemical and physical data | |
| Formula | C31H36N2O11 |
| Molar mass | 612.624 g·mol−1 |
| 3D model (JSmol) | |
4309-70-0 CAS
calcium;7-[(2R,3R,4S,5R)-4-carbamoyloxy-3-hydroxy-5-methoxy-6,6-dimethyloxan-2-yl]oxy-3-[[4-hydroxy-3-(3-methylbut-2-enyl)benzoyl]amino]-8-methyl-2-oxochromen-4-olate
///////// Novobiocin, ノボビオシン , Antibacterial, Antimicrobial, crystallinic acid, streptonivicin,
History
Novobiocin is a coumarin antibiotic obtained from Streptomyces niveus and other Streptomyces species. Novobiocin is useful primarily in infections involving staphylococci, and other gram-positive organisms. It acts by inhibiting the initiation of DNA replication in bacterial and mammanlian cells. Evidences indicated that Novobiocin blocks prokaryotic DNA gyrase and eukaryotic II topoisomerase, enzymes that relax super-coiled DNA and are crucial for DNA replication.1
Novobiocin

| UIPAC Name | 4-Hydroxy-3-4-hydroxy-3-(3-methylbut-2-enyl)benzamido-8-methylcoumarin-7-yl 3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside |
| CAS Number | 303-81-1 |
| Molecular Mass | 612.624 g / mol |
| Chemical Formular | C31H36N2O11 |
Biosynthesis
The substituted coumarin (ring B, red) and the 4-OH benzoyl moiety (ring A, aqua) in novobiocin were derived from -Tyr based on earlier labeling studies. β-OH-Tyr is proposed to be a common intermediate in these two biosynthetic pathways.2

NovH is a -Tyr specific didomain NRPS that generates the
-tyrosyl-S-NovH intermediate. NovH, isolated from E. coli is primed by a PPTase with CoA. The A domain activates
-Tyr as
-tyrosyl-AMP and then transfers the
-tyrosyl group to the HS-pant-PCP domain of NovH through thioester formation.3

-tyrosyl-S-NovH is then function as a cytochrome P450 monooxygenase that hydroxylates the β-carbon of the tethered
-tyrosyl group on NovH. While the substrate
-tyrosyl-S-NovH provides two electrons for a single round of the hydroxylation reaction, the other two electrons needed to reduce the oxygen atom are provided by NADPH via two-electron transfer effected by electron transfer proteins ferrodoxin (Fd) and ferrodoxin reductase (Fd Red).3 The electron transfer route is from NADPH→FAD in Fd Red→Fe–S center in Fd→Heme in NovI→oxygen.

Both NovJ and NovK are similar to 3-keto-ACP reductase and they may form a heterodimer and operate in the reverse direction to oxidize 3-OH to 3-keto. NovO is similar to some quinone C-methyltransferases 3 but the timing of methylation is not clear. NovC resembles flavin-dependent monooxygenases (35 and 32% similarity to dimethylaniline and cyclohexanone monooxygenases, respectively) 3 and is proposed to hydroxylate the ortho position of the phenyl ring. The nucleophilic attack of the ortho hydroxyl group on the thioester carbonyl center would release the coumarin ring and regenerate NovH. Ring B is then synthesized.

Synthesis





Mechanism of action
E.Coli DNA gyrase utilizes ATP to catalyze the negative supercoiling, or under-twisting, of duplex DNA. The energy coupling components of the supercoiling reaction includes 1) the DNA-dependent hydrolysis that converts ATP to ADP and Pi, and 2) the gyrase cleavage reaction that targets the specified DNA site. The two activities are induced by treating the stable gyrase-DNA complex trapped by the inihibitor oxolinic acid with sodium dodecyl sulfate (SDS or Sulphate). 4 Novobiocin competes with ATP in the ATPase and supercoiling assays, hence Novobiocin prevents the ATP from shifting the primary cleavage site on ColE1 DNA by places the site of action of the antibiotics at a reaction step prior to ATP hydrolysis and blocks the binding of ATP. 4 Such a simple mechanism of action represents for all effects of the drugs on DNA gyrase.
Clinical Use
Due to factors as low solubility, poor pharmacokinetics, and limited activity agasinst Gram-negative bacteria, the clinical usage of Novobiocin is not achieved. 5 Therefore, it is of interest to study the novobiocin biosynthetic pathway in order to generate analogs with enhanced solubility and pharmacokinetic properties while maintaining the gyrase inhibitory properties.
References
1 J.C. D’Halluin, M. Milleville, and P. Boulanger. “Effect of Novobiocin on adenovirus DNA synthesis and encapsidation”. Nucleic Acids Research 1980; 8: 1625-1641
2 M. Steffensky, S.M. Li and L. Heide, “Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides ” NCIB 11891. J. Biol. Chem. 275 (2000), pp. 21754–21760.
3 Huawei Chen and Christopher T. Walsh, “Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI” Chemistry and Biology; 2001; 8: 301-312
4 K. Scheirer and N. P. Higgins. “The DAN Cleavage Reaction of DNA Gyrase ” The Journal of Biological Chemistry; 1997; 272 (43): 27202-27209
5 N Pi, C. L. F. Meyers, M. Pacholec, C. T. Walsh, and J. A. Leary. “Mass spectrometric characterization of a three-enzyme tandem reacton for assembly and modification of the novobiocin skeleton” PNAS 2004;101;10036-10041
New Antibacterial oxazolidinones in pipeline by Wockhardt

WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
THIS MAY BE WCK 4086?????….WATCHOUT THIS POST FOR UPDATION
PATENTS
WO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
PATENT
WO2015173664

In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1

(I I) (I N)
Scheme 1

Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula: ![]()
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula: ![]()
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).
PATENT
http://www.google.st/patents/WO2007132314A2?cl=en



(3) (4)
Scheme -1

(6) Formula π Scheme-2

Formula II Formula in

Formula I(a) Scheme-4
Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Patent
INDIAN 3049/MUM/2010
Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
PATENT
http://www.google.co.in/patents/WO2012059823A1?cl=en


c) Converting intermediate of Formula (5) into intermediate of structure (6)



f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof



ormu a-
Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
PATENT
http://www.google.co.in/patents/WO2008038092A2?cl=en

IV

V
‘ Scheme-1 ‘
/////////
SEE FULL ZOLID SERIES…………http://drugsynthesisint.blogspot.in/p/zolid.html

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}