
Home » Posts tagged 'Anti-inflammatory'
Tag Archives: Anti-inflammatory
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nelremagpran
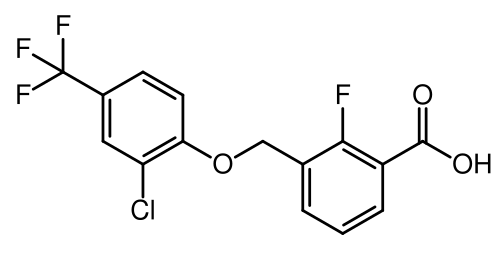
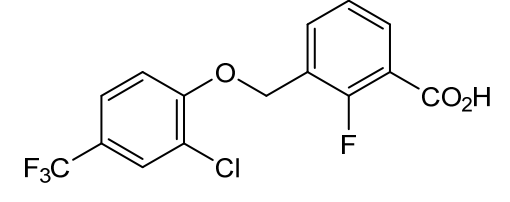

Nelremagpran
CAS 2492595-24-9
MFC15H9ClF4O3 MW 348.67 g/mol
3-[[2-Chloro-4-(trifluoromethyl)phenoxy]methyl]-2-fluorobenzoic acid
3-{[2-chloro-4-(trifluoromethyl)phenoxy]methyl}-2-fluorobenzoic acid
Mas-related G protein-coupled receptor inverse agonist, anti-inflammatory, MRGPRX4 modulator-2, BL83FAK6DZ,
Nelremagpran is an experimental drug that acts as a potent and selective antagonist (or possibly inverse agonist) of the MAS-related Gq protein-coupled receptor X4 (MRGPRX4). It has antiinflammatory effects in animal studies. This receptor is poorly characterised but is thought to be involved in immune system function, and development of selective ligands is essential for researching its role in the body.[1][2]
Nelremagpran is an experimental drug that acts as a potent and selective antagonist or inverse agonist of the Mas-related G-protein-coupled receptor X4 (MRGPRX4). It was initially developed by Escient Pharmaceuticals, Inc. for its anti-inflammatory and anti-itch properties.
🧪 Core Mechanism
The drug targets MRGPRX4, a poorly characterized receptor found in the immune and nervous systems. It functions with high potency, exhibiting an half-maximal inhibitory concentration (IC50) of less than 100 nM.
🔬 Potential Research Applications
Because the receptor plays a significant role in mediating severe itch and pain pathways, Nelremagpran is actively used as a chemical probe to study:
- Chronic Pruritus: Conditions involving persistent, debilitating itch sensations, such as cholestatic pruritus.
- Autoimmune Diseases: Underlying mechanisms in conditions like psoriasis, multiple sclerosis, and Stevens-Johnson Syndrome.
- Inflammatory Sensation: How the body signals pain and immune hypersensitivity
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020198537&_cid=P12-MREAZN-03524-1

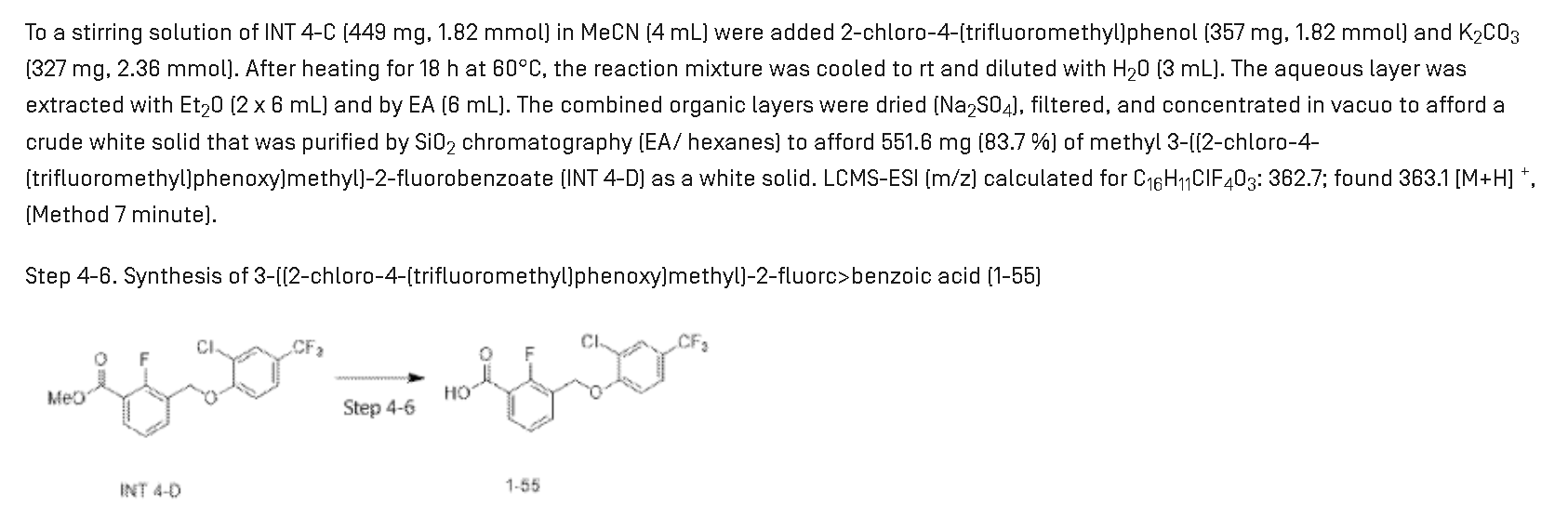
Synthesis of Compound 1-55

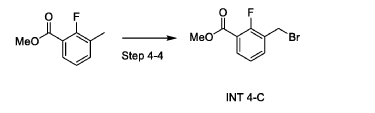
Step 4-4. Synthesis of methyl 3-(bromomethyl)-2-fluorc>benzoate (INT 4-C)



ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
 | |
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2492595-24-9 |
| PubChem CID | 155145968 |
| ChemSpider | 115008493 |
| UNII | BL83FAK6DZ |
| ChEMBL | ChEMBL4855031 |
| Chemical and physical data | |
| Formula | C15H9ClF4O3 |
| Molar mass | 348.68 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- WO 2020/198537, Yeager A, Selfridge B, Sainz M, Martinborough E, Boehm M, Huang, “Modulators of mas-related G-protein receptor x4 and related products and methods.”, published 1 October 2020, assigned to Escient Pharmaceuticals, Inc.
- “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (4). 2024.
- Modulators of Mas-related G-protein receptor X4 and related products and methodsPublication Number: US-11840521-B2Priority Date: 2019-03-28Grant Date: 2023-12-12
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: EP-3946304-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2023159474-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2023053860-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2021032213-A1Priority Date: 2019-03-28
- Modulators of mas-related G-protein receptor X4 and related products and methodsPublication Number: US-11643399-B2Priority Date: 2019-03-28Grant Date: 2023-05-09
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: US-2024300907-A1Priority Date: 2019-03-28
- Modulators of mas-related g-protein receptor x4 and related products and methodsPublication Number: WO-2020198537-A1Priority Date: 2019-03-28
- Modulators of MAS-related G protein receptor X4 and related products and methodsPublication Number: CN-113939288-BPriority Date: 2019-03-28Grant Date: 2025-04-25
//////////nelremagpran, anax labs, Mas-related G protein-coupled receptor inverse agonist, anti-inflammatory, MRGPRX4 modulator-2, BL83FAK6DZ,
Milpecitinib
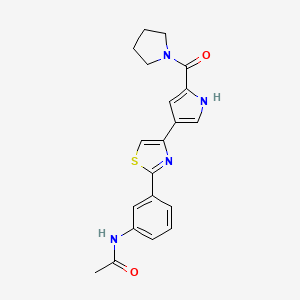
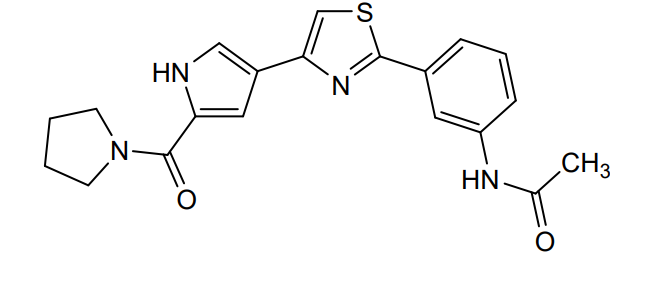
Milpecitinib
CAS 1415819-54-3
MF C20H20N4O2S MW380.5 g/mol
N-[3-[4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl]phenyl]acetamide
N-(3-{4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl}phenyl)acetamide
Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Milpecitinib is a small molecule drug. Milpecitinib has a monoisotopic molecular weight of 380.13 Da.
Milpecitinib (also known by its developmental codes PF-06263276, Ph-1012, and DNX-04013) is a potent, small-molecule Janus kinase (JAK) inhibitor used primarily in veterinary medicine and laboratory research. It functions as an ATP-competitive, broad-spectrum (pan-JAK) inhibitor that targets all four members of the JAK family: JAK1, JAK2, JAK3, and Tyrosine Kinase 2 (TYK2).
Primary Indication and Target
- Veterinary Use: Milpecitinib is designated for the control of pruritus (itching) associated with canine allergic dermatitis and the management of canine atopic dermatitis (CAD).
- Sponsorship: The United States Adopted Name (USAN) for this drug was officially adopted following sponsorship by Phibro Animal Health.
- Research Use: In laboratory settings, it is utilized to study complex inflammatory pathways, immune disorders, and certain cancers.
Mechanism of Action
Milpecitinib works by blocking the ATP-binding site of JAK enzymes. This inhibition halts the JAK-STAT signaling pathway, which plays a critical role in cellular responses to inflammatory cytokines. By blocking this cascade, the drug prevents the production and signaling of pro-inflammatory cytokines that cause severe itching and skin inflammation in dogs.
SYN
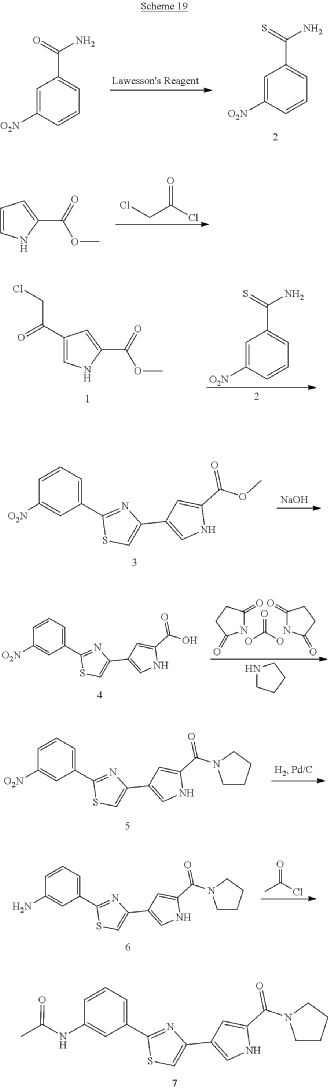
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Compositions and methods for modulating kinasesPublication Number: JP-6054379-B2Priority Date: 2011-06-07Grant Date: 2016-12-27
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-B1Priority Date: 2011-06-07Grant Date: 2016-05-04
- Compositions and methods for modulating a kinasePublication Number: CA-2837268-CPriority Date: 2011-06-07Grant Date: 2020-05-12
- Compositions and Methods for Modulating a KinasePublication Number: US-2012316148-A1Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-A2Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A9Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: US-8937065-B2Priority Date: 2011-06-07Grant Date: 2015-01-20
- Compositions and methods for modulating kinasesPublication Number: JP-2014520108-APriority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A2Priority Date: 2011-06-07
- Compositions and methods to modulate a kinasePublication Number: ES-2585244-T3Priority Date: 2011-06-07Grant Date: 2016-10-04
- Compound for modulating a kinase
- Publication Number: BR-112013031121-B1
- Priority Date: 2011-06-07
//////////milpecitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Lirucitinib
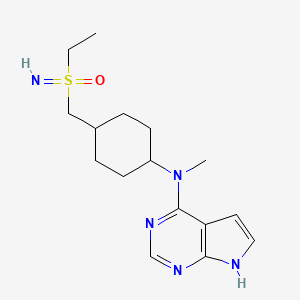
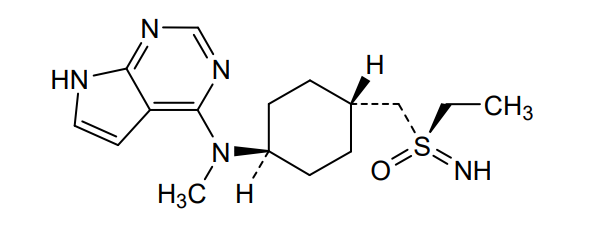
Lirucitinib
CAS 2458115-78-9
MF C16H25N5OS MW335.5 g/mol
N-[4-[(ethylsulfonimidoyl)methyl]cyclohexyl]-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
(R)-ethyl(imino)({trans-4-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclohexyl}methyl)-λ6
-sulfanone
Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Lirucitinib is a Janus kinase (JAK) inhibitor primarily known as a novel, Class I veterinary drug. It specifically targets the JAK1 enzyme to block itch-inducing (pruritic) and inflammation-causing cytokines in the body.
Core Information
- Primary Use: The drug is developed for veterinary medicine to treat acute and chronic pruritic (severe itch) skin diseases in dogs, which are commonly caused by allergies, parasites, or infections.
- Approval Status: It received a Class I New Veterinary Drug Certificate from the Ministry of Agriculture and Rural Affairs (MARA) in China.
- Human Medicine: As of 2026, there are no clinical indications or indications that Lirucitinib is being tested for use in humans.
- Chemical Profile: It is an orally active small molecule with the chemical formula C₁₆H₂₅N₅OS and acts specifically as the (R)-enantiomer of the compound.
SYN
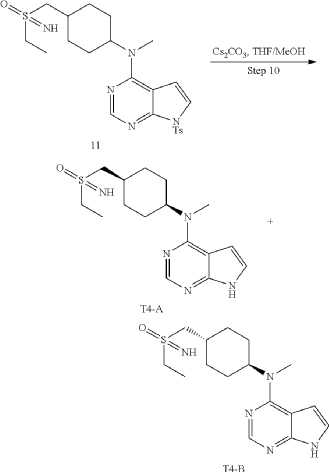
11 (1.0 g, 2.04 mmol) (prepared in step 9), tetrahydrofuran/methanol (10 mL), and cesium carbonate (1.33 g, 4.08 mmol) were added into a 25 mL single-necked flask, refluxed for 12 h, concentrated, and poured into dichloromethane and saturated salt solution, the organic phase was dried with anhydrous sodium sulfate, concentrated, and subjected to a conventional preparation method and a chiral preparation method to obtain product A as a white solid (20 mg, yield: 2.9%), LC-MS: 336 [M+H]+, H 1-NMR: 1H NMR (400 MHz, DMSO) δ 11.61 (s, 1H), 8.09 (s, 1H), 7.13 (s, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.90-3.83 (m, 1H), 3.17 (s, 3H), 3.06-2.93 (m, 4H), 2.12-2.01 (m, 3H), 1.73-1.70 (m, 4H), 1.31-1.22 (m, 5H) and product B as a white solid (25 mg, yield: 3.7%), LC-MS: 336 [M+H]+, H 1—NMR: 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 8.09 (s, 1H), 7.12 (dd, J=3.3, 2.6 Hz, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.58 (s, 1H), 3.17 (s, 3H), 3.06-2.89 (m, 4H), 2.16-1.93 (m, 3H), 1.74-1.69 (m, 4H), 1.25-1.23 (m, 5H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-B1Priority Date: 2019-01-30Grant Date: 2023-06-28
- Jak inhibitor and preparation method thereforPublication Number: US-2022106319-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: WO-2020155931-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-A1Priority Date: 2019-01-30
/////////lirucitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Tigemocoxib
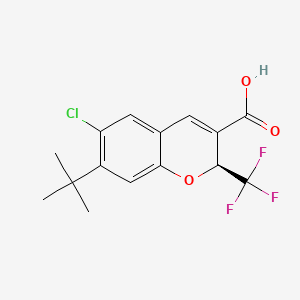
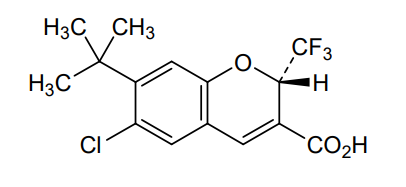
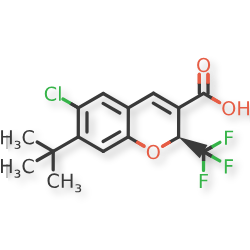
Tigemocoxib
MF C15H14ClF3O3 MW 334.72
2H-1-Benzopyran-3-carboxylic acid, 6-chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-, (2S)-
(2S)-7-tert-butyl-6-chloro-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid
(2S)-7-tert-butyl-6-chloro-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid
cyclo-oxygenase 2 (COX-2) inhibitor, anti-inflammatory, SC-75416, SC 75416, C8D42HX4WH
CAS 215122-74-0
Originator: Pfizer Inc.
Tigemocoxib is a small molecule drug. The usage of the INN stem ‘-coxib’ in the name indicates that Tigemocoxib is a selective cyclo-oxygenase inhibitor. Tigemocoxib has a monoisotopic molecular weight of 334.06 Da.
Tigemocoxib (also known as SC-75416) is a selective cyclo-oxygenase 2 (COX-2) inhibitor and nonsteroidal anti-inflammatory drug (NSAID) designed for reducing pain and inflammation. Initially developed as an orally bioavailable clinical lead for treating acute, postoperative, and chronic inflammatory pain, it acts by targeting the COX-2 enzyme
SYN
Bioorganic & Medicinal Chemistry LettersPublication Date: 2010-12-01PMID: 20709553DOI: 10.1016/j.bmcl.2010.07.054
PAT
WO2000037469
PAT
EXAMPLE 68
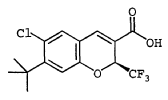
(S)-6-Chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-2H-1- benzopyran-3-carboxylic acid To a solution of 6-chloro-7-(1,1-dimethylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid (Example 8) (11.4 g, 34.1 mmol) and (S)-(-)-2-amino-3-phenyl-1-propanol (2.57 g, 17.00 mmol) was added n-heptane (200 mL) and the mixture set aside for 16 h. The resulting
suspension was filtered yielding a solid (3.8 g). This solid was recrystallized from 2-butanone (20 mL) and n-heptane (200 mL) yielding upon filtration a white solid (3.0 g).
This solid was dissolved in ethyl acetate (100 mL) and washed with 1 N hydrochloric acid (50 mL) and brine (2 × 50 mL), dried over MgSO4 and concentrated in vacuo yielding a white solid. This solid was recrystallized from n-heptane yielding the title compound of high optical purity as a crystalline solid (1.7 g, 30%): mp 175.4-176.9 °C. 1H NMR (acetone-d6/300 MHz) 7.86 (s, 1H), 7.52 (s, 1H), 7.12 (s,
1 H), 5.83 (q, 1H, J = 7.1 Hz), 1.48 (s, 9H). Anal. Calc’d for C15H14O3F3Cl : C, 53.83; H, 4.22; N, 0.0; Cl, 10.59. Found: C, 53.78; H, 4.20; N, 0.0; Cl, 10.65. This compound was determined to have an optical purity of greater than 90% ee. Chiral purity was determined as describe in Example 66.
EXAMPLE 8
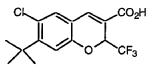
6-Chloro-7-(1,1-dimethylethyl)-2-trifluoromethyl- 2H-1-benzopyran-3-carboxylic acid
Step 1. Preparation of 4-tert-butylsalicylaldehyde.
A five liter three-neck round bottom flask equipped with overhead mechanical stirrer and condenser was charged with trifluoroacetic acid (2.4 L). A mixture of 3-tert-butylphenol (412 g, 2.8 mole) and HMTA (424 g, 3.0 mole) was added portion-wise causing an exotherm. With cooling, the temperature was maintained under 80 °C. The reaction was heated at 80 °C for one hour, then cooled, and water (2 L) added. After 0.5 hour additional water (4 L) was added and the mixture was extracted with ethyl acetate (6 L). The organic extract was washed with water and brine. The resulting organic phase was divided into 2 L volumes and each diluted with water (1 L), and solid NaHCO3 added until the mixture was neutralized. The organic phases were isolated and combined, dried over MgSO4, filtered and
concentrated in vacuo yielding an oil. This oil was
distilled at 95 °C (0.8 mm) yielding the desired
salicylaldehyde as an oil (272.9 g, 56 %) which was of sufficient purity to be used without further purification.
Step 2. Preparation of ethyl 7-(1,1-dimethylethyl)-2- (trifluoromethyl)-2H-1-benzopyran-3-carboxylate.
A one liter three-neck flask was charged with 4-tert-butylsalicylaldehyde (Step 1) (100.0 g, 0.56 mole),
dimethylformamide (110 mL), and potassium carbonate (79.9 g, 0.58 mole) causing the temperature of the mixture to rise to 40 °C. Ethyl 4,4,4-trifluorocrotonate (118.0 g, 0.70 mole) in dimethylformamide (110 mL) was added and the mixture heated to 60 °C at which time the reaction temperature rose to 70 °C. The reaction was cooled to 60 °C, maintained at 60
°C (with addedheating) for 8.5 hours and cooled to room temperature. Ethyl acetate (600 mL) and 3 N HCl (600 mL) were added, mixed, and the layers separated. The aqueous phase was extracted with ethyl acetate and the organic phases were combined. The combined organic phases were washed with brine-water (1:1), brine, dried over MgSO4, filtered and concentrated in vacuo, yielding a semi-solid. Hexane (600 mL) was added with mixing and the mixture was filtered. The filtrate was washed with brine, dried over
MgSO4, filtered and concentrated in vacuo yielding a solid. This solid was dissolved in hot ethanol (600 mL). Water (190 mL) was added which induced crystallization. Filtration of the mixture and drying of the product provided the desired ester as a crystalline solid (131.3 g, 71%): mp 91.0-94.9 °C. This material was of suitable purity to be used in subsequent steps without further purification.
Step 3. Preparation of ethyl 6-chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylate.
A one liter three-neck flask equipped with mechanical stirrer and gas inlet tube was charged with the ester (Step 2) (100 g, 0.3 mole) and acetic acid (300 mL). While cooling (water bath) the reaction mixture, chlorine gas (37.6 g, 0.53 mole) was added which caused the temperature to rise to 48 °C. After stirring for two hours, the
reaction was cooled in an ice-water bath to 15 °C. Zinc powder (19.5 g, 0.3 mole) was added in one portion which caused the temperature to rise to 72 °C. After cooling to room temperature additional zinc powder (5.0 g, 0.08 mole) was added and the mixture was stirred for 0.5 hour longer. The crude mixture was filtered through diatomaceous earth and was concentrated in vacuo yielding an oil. The oil was dissolved in ethyl acetate (700 mL) washed with brine-water (1:1, 1 L) and brine (0.5 L). The resulting aqueous phase was extracted with ethyl acetate (700 mL). This ethyl acetate phase was washed with brine-water (1:1, 1 L) and brine (0.5 L). The combined organic phases were dried over
MgSO4, filtered and concentrated in vacuo yielding the title compound as a yellow oil (116 g, 106 %). This material, which contained some entrained ethyl acetate, was of suitable purity to be used in subsequent steps without further purification.
Step 4. Preparation of 6-chloro-7-(1,1-dimethylethyl)-2- (trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid.
To a solution of the ester (Step 3) (116 g,
0.3 mole) in methanol (500 mL) and tetrahydrofuran (500 mL) in a one liter flask was added aqueous
sodium hydroxide (2.5 N, 240 mL, 0.6 mole). After
stirring overnight, the pH of the solution was
adjusted to 1 with concentrated hydrochloric acid
and the solution was extracted with ethyl acetate.
The ethyl acetate phase was dried over MgSO4,
filtered and concentrated in vacuo yielding a
solid. This solid was dissolved in hot ethanol
(500 mL). Water (500 mL) was added and upon
cooling to room temperature crystals formed which
were collected by vacuum filtration. The crystals
were washed with ethanol-water (3:7, 3 X 200 mL)
and dried providing the title acid as a
crystalline solid (91.6 g, 91 %) : mp 194.9-196.5
°C. 1H NMR (acetone-d6/300 MHz) 7.86 (s, 1H),
7.52 (s, 1H), 7.12 (s, 1H), 5.83 (q, 1H, J = 7.1
Hz), 1.48 (s, 9H). Anal. Calc’d for C15H14ClF3O3:
C, 53.83; H, 4.22; Cl, 10.59. Found: C, 53.92; H,
4.24; Cl, 10.50
REF
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Synthesis of Deuterated Benzopyran Derivatives as Selective COX-2 Inhibitors with Improved Pharmacokinetic PropertiesPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2014-08-06PMCID: PMC4190635PMID: 25313331DOI: 10.1021/ml500299q
- Leukotrienes, But Not Angiotensin II, Are Involved in the Renal Effects Elicited by the Prolonged Cyclooxygenase-2 Inhibition When Sodium Intake Is LowPublication Name: Journal of Cardiovascular PharmacologyPublication Date: 2013-04PMID: 23288201DOI: 10.1097/fjc.0b013e31828399ae
- The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-lifePublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2010-12-01PMID: 20709553DOI: 10.1016/j.bmcl.2010.07.054
- Evaluation of COX-1/COX-2 selectivity and potency of a new class of COX-2 inhibitorsPublication Name: European Journal of PharmacologyPublication Date: 2008-06-24PMID: 18457826DOI: 10.1016/j.ejphar.2008.03.057
- Modeling and Simulation to Support Dose Selection and Clinical Development of SC-75416, a Selective COX-2 Inhibitor for the Treatment of Acute and Chronic Pain
- Publication Name: Clinical pharmacology and therapeutics
- Publication Date: 2007-09-19
- PMID: 17882158
- DOI: 10.1038/sj.clpt.6100374
PAT
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: EP-0977748-A1Priority Date: 1997-04-21
- Endothermic reaction apparatusPublication Number: NO-308725-B1Priority Date: 1993-06-16
- History of Transformed Benzofiran, their preparation and pharmacological preparations for the treatment of inflammation containing themPublication Number: IL-132296-APriority Date: 1997-04-21
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: KR-100538258-B1Priority Date: 1997-04-21Grant Date: 2006-01-12
- Substituted benzopyran derivatives for the treatmentPublication Number: US-6806288-B1Priority Date: 1997-04-21Grant Date: 2004-10-19
- Benzopyran derivatives with substituents for the treatment of inflammationPublication Number: JP-4577534-B2Priority Date: 1997-04-21Grant Date: 2010-11-10
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: KR-20010020152-APriority Date: 1997-04-21
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////tigemocoxib, ANAX, cyclo-oxygenase 2 (COX-2) inhibitor, anti-inflammatory, SC-75416, SC 75416, C8D42HX4WH
Rivasterat
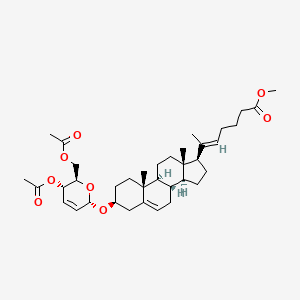
Rivasterat
CAS 2446590-96-9
MF C37H54O8 MW626.8 g/mol
methyl (E)-6-[(3S,8S,9S,10R,13S,14S,17R)-3-[[(2R,3S,6S)-3-acetyloxy-2-(acetyloxymethyl)-3,6-dihydro-2H-pyran-6-yl]oxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]hept-5-enoate
methyl (20E)-3β-[(4,6-di-O-acetyl-2,3-dideoxy-α-Derythro-hex-2-enopyranosyl)oxy]-27-norcholesta5,20(22)-dien-26-oate
cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
- OriginatorCURACLE
- ClassAnti-inflammatories; Anti-ischaemics; Antineoplastics; Eye disorder therapies; Ischaemic heart disorder therapies; Small molecules; Vascular disorder therapies
- Mechanism of ActionActin modulators; Chemokine CCL2 inhibitors; Histamine release inhibitors; Interleukin 1 beta inhibitors; Thrombin inhibitors; Vascular endothelial growth factors inhibitors
- Phase IIDiabetic macular oedema
- No development reportedAge-related macular degeneration; Cancer; Crohn’s disease; Diabetic retinopathy; Hereditary angioedema; Lung disorders; Macular degeneration; Myocardial infarction; Retinal oedema; Stroke; Ulcerative colitis; Unstable angina pectoris; Wet macular degeneration
- 28 Aug 2025No recent reports of development identified for research development in Unstable-angina-pectoris in South Korea (PO)
- 28 Jul 2025No recent reports of development identified for phase-I development in Wet macular degeneration in USA (PO)
- 28 May 2025No recent reports of development identified for phase-I development in Age-related-macular-degeneration in South Korea (PO)
Developer and Code Name
- Original code name: CU06-1004
- Developer: Curacle Co., Ltd. (South Korea)
- Drug class: Endothelial dysfunction blocker (EDB)
This class of drugs aims to restore endothelial barrier integrity rather than directly blocking VEGF like most retinal drugs.
The molecule contains:
- Steroid (cyclopenta[a]phenanthrene) core
- Unsaturated heptenoate side chain
- Acetylated sugar moiety (pyranose)
This glycosylated steroid structure is unusual for vascular-protective drugs.
Clinical Development
Phase I (Healthy Volunteers)
Key findings:
- Dose tested: 100–1200 mg
- Exposure increased more than dose proportional
- Food greatly increased absorption
- No significant drug accumulation after repeated dosing
- Minimal renal excretion detected
Phase II
Early clinical trials investigated oral therapy for diabetic macular edema with improvements in:
- Best-corrected visual acuity
- Inflammatory biomarkers
Summary
| Item | Details |
|---|---|
| Drug | Rivasterat (CU06-1004) |
| Originator | Curacle |
| Core patent | WO2013011939 |
| Chemistry | steroid glycoside |
| Key step | steroid glycosylation |
| Priority | ~2011 |
| Expiry | ~2031–2033 |
SYNTHESIS
WO2013011939
US20140148474
EP2741074
KR20130007373
SYN
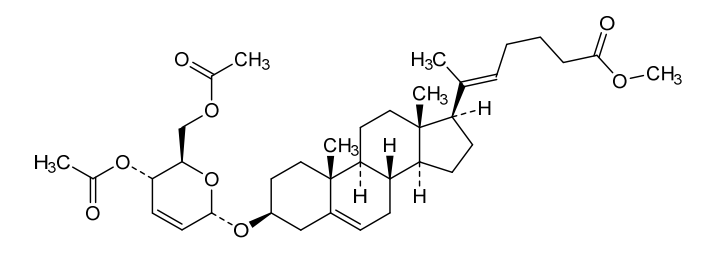
Manufacturing Example 1> Manufacturing of compound 1
[170]Compound 1 represented by the following chemical formula 1 can be manufactured using the manufacturing method described in Korean unpublished patent application number 10-2019-0166864. Specifically, it can be manufactured using the method according to the following reaction scheme 1 or reaction scheme 2.
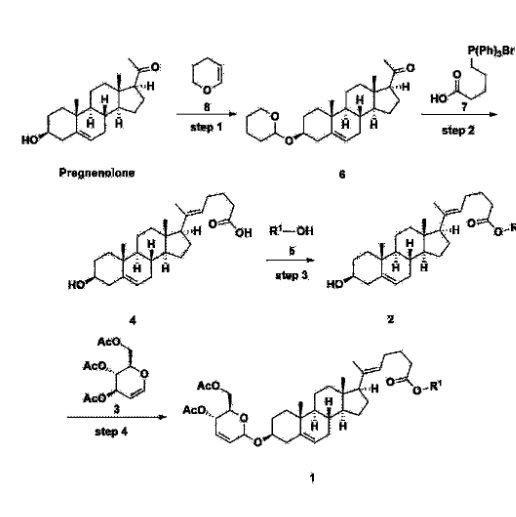
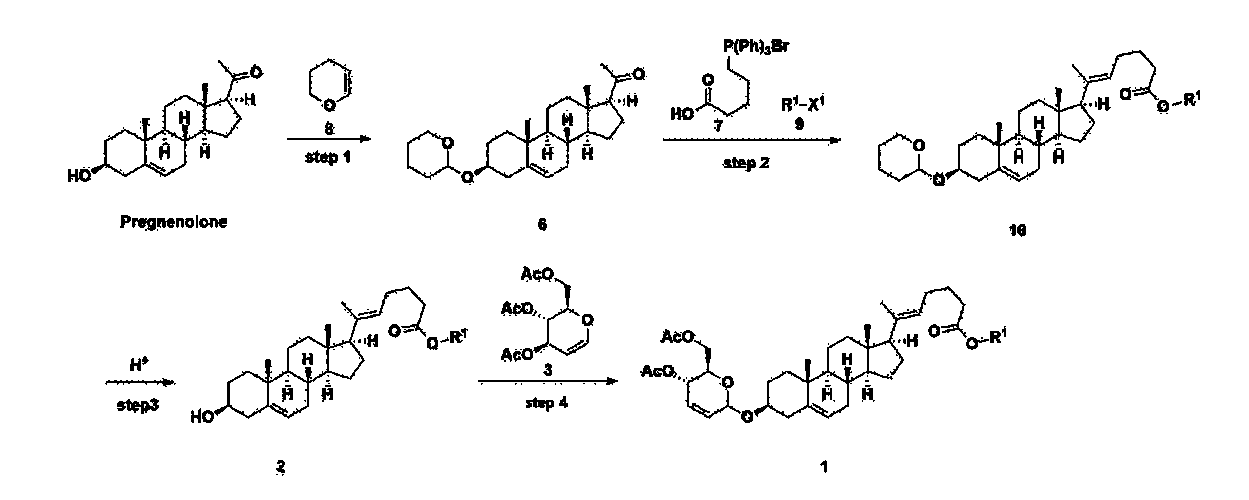
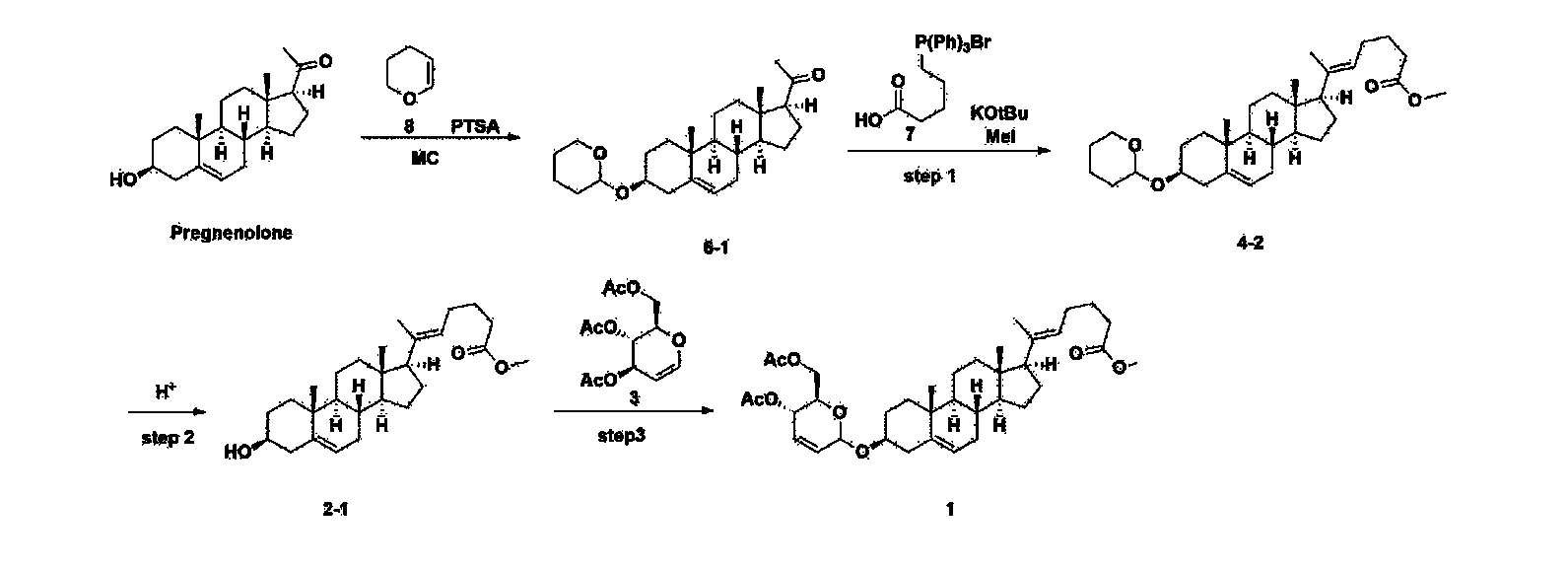
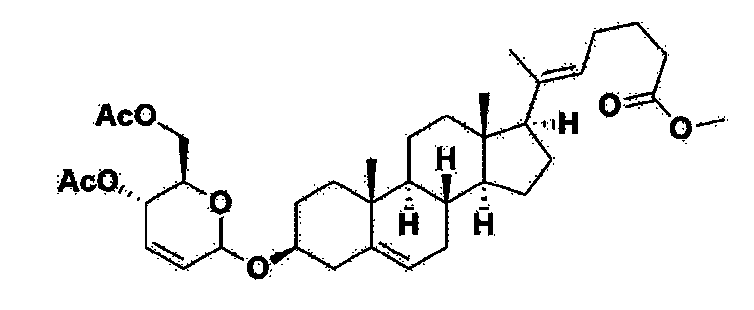
Step 1: Preparation of 6-1
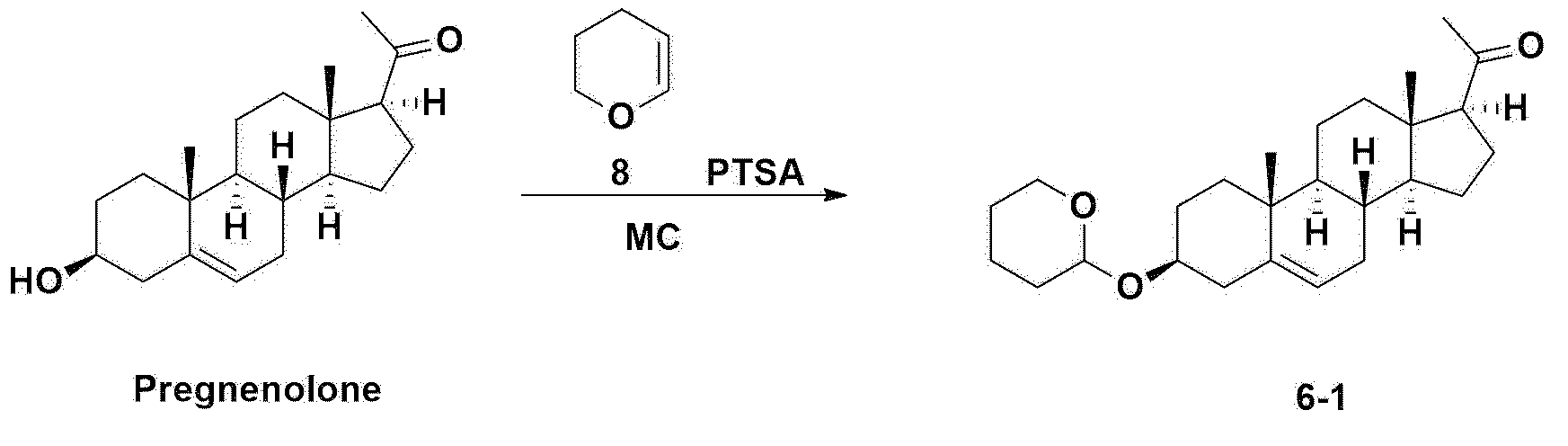
A thermometer was installed in a 5 L flask, and 200 g (0.632 mol) of pregnenolone was added to 2000 mL of dichloromethane, and 173 mL (1.896 mol) of 3,4-dihydro-2H-pyran was added. After lowering the temperature to 0-5 ℃, 3.0 g (15.8 mmol) of p-toluenesulfonic acid monohydrate dissolved in 50 mL of tetrahydrofuran (THF) was added dropwise and stirred at 0 ℃ for 1.5 hours. At 0 ℃, 800 mL of saturated sodium bicarbonate aqueous solution and 10 mL of triethylamine (TEA) were added to the reaction mixture and stirred. After separating the layers, the organic layer was washed with 800 mL of brine, and the aqueous layers were extracted again with 200 mL of dichloromethane, combined into the organic layers, dried over 200 g of anhydrous sodium sulfate, filtered, and distilled under reduced pressure. 1000 mL of MeOH and 5 mL of TEA were added to the obtained residue, heated to completely dissolve, and the temperature was lowered and stirred at -5 °C for 1 hour. The resulting solid was filtered and washed with 200 mL of MeOH to obtain 232.0 g (0.579 mol) of 6-1 (THP-Pregnenolone) as a pure white solid in a yield of 91.6%.
[204]1H-NMR (400 MHz, CDCl 3): δ 5.33-5.36 (m, 1H), 4.71-4.72 (m, 1H), 3.85-3.94 (m, 1H), 3.46-3.56 (m, 2H), 1.00-2.55 (m, 32H), 0.62 (s, 3H).
Step 2: Preparation of 4-2
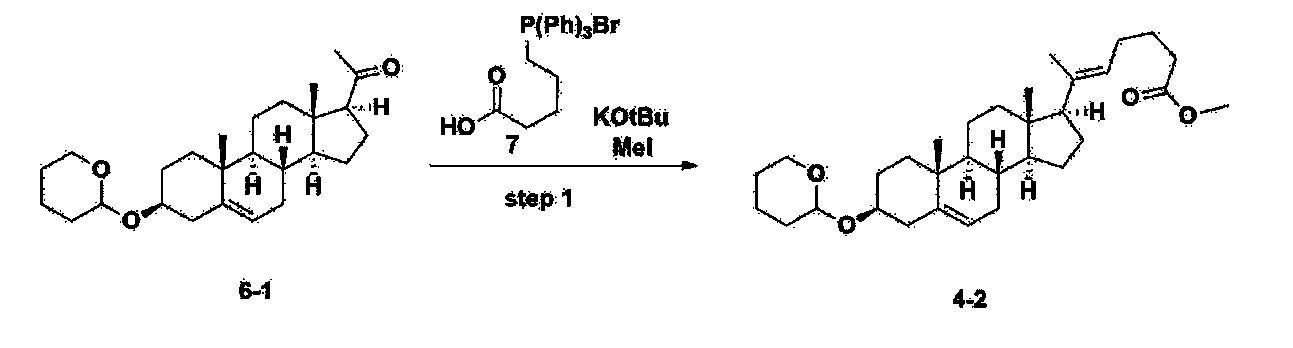
After installing a condenser, heating mantle, and mechanical stirrer in a 5L reactor, the reactor was heated to 119℃ (external temperature), cooled to room temperature while flowing nitrogen for 5 minutes, dried, and 332.5 g (0.75 mol) of 4-(carboxybutyl)triphenylphosphonium bromide and 168.1 g (1.50 mol) of potassium t-butoxide were added. Then, 2000 mL of anhydrous toluene and 750 mL of anhydrous tetrahydrofuran were added, and the reactor was heated to 119℃ (external temperature, internal mild reflux) and stirred for about 2 hours.
[209]6-1 100.0 g (0.250 mol) was dissolved in 500 mL of anhydrous toluene, added to the reaction solution, and reacted for about 20 hours. After the reaction was completed, the reaction mixture was cooled to room temperature, 320 mL (5.14 mol) of methyl iodide and 1000 mL of acetone were added, and stirred at room temperature for 15 hours. Most of the organic solvent was removed from the reaction mixture by distillation under reduced pressure, 1500 mL of ethyl acetate was added to dissolve, and the mixture was washed with 1000 mL of saturated ammonium chloride aqueous solution. The organic layer was washed twice with 1000 mL of water and 1000 mL of brine, dried with 100 g of sodium sulfate, filtered using 80 g of Celite, and concentrated.
[210]The obtained residue was dissolved in 2000 mL of methanol, stirred at 10°C for 13 hours and at 4-5°C for 1 hour, and the resulting solid was filtered, washed with 200 mL of methanol, and dried in vacuum to obtain 66.2 g of 4-2 as a white solid with a yield of 53.2%.
[211]
1H NMR(400MHz, CDCl 3): δ 5.36(t, J=5.80 Hz, 1H), 5.16(t, J=7.00 Hz, 1H), 4.71(m, 1H), 3.93(m, 1H), 3.66(s, 3H), 3.56(m, 2H), 2.37-0.88(m, 38H), 0.54(s, 3H).
Step 3: Preparation of 2-1
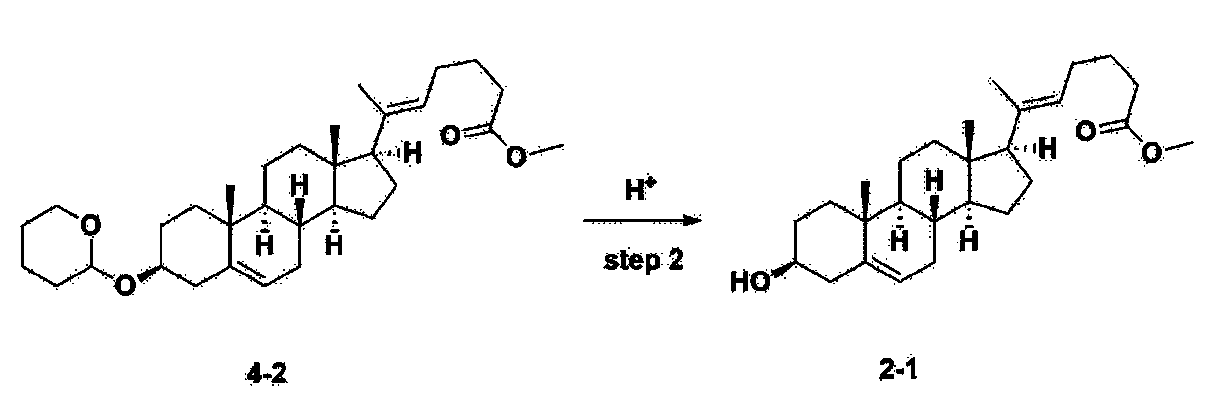
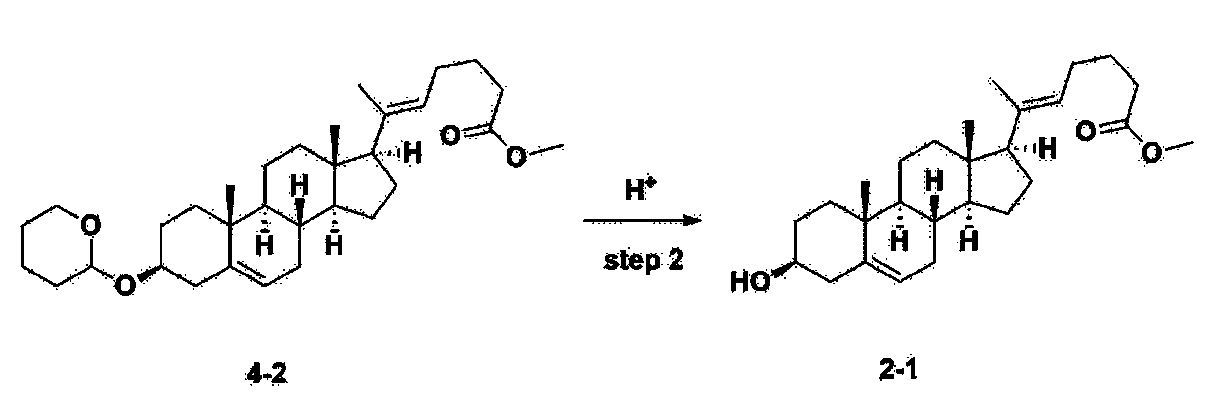
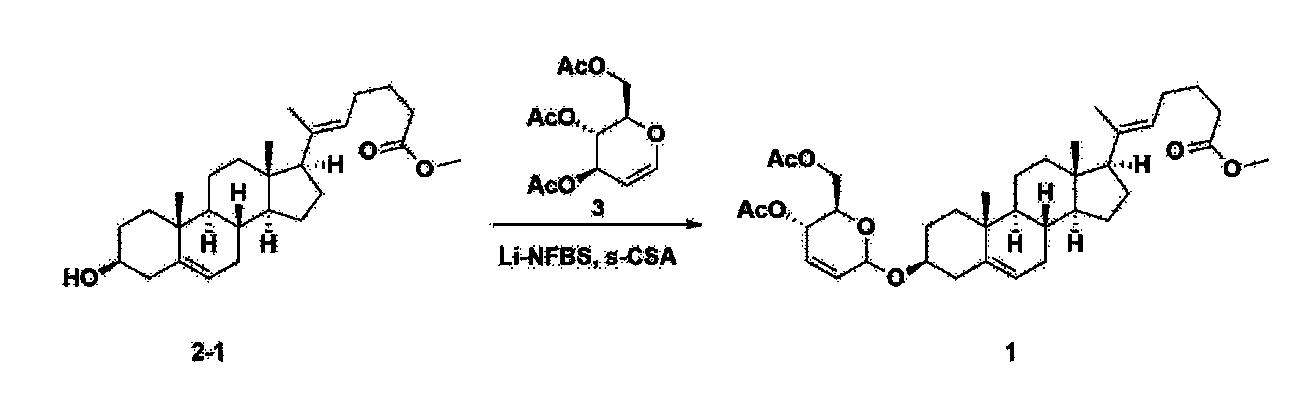
After installing a thermometer and a water bath in a 1 L flask, 42.0 g (0.101 mol) of compound 2-1 and 34.5 g (0.126 mol) of triiO-acetyl D-glucal were dissolved in 126 mL of anhydrous toluene and 252 mL of acetonitrile, and while maintaining the temperature at 30-35 ℃, 3.87 g (0.0130 mol) of lithium nonafluoro-1-butylsulfonate and 0.117 g (0.0005 mol) of (s)-camphor sulfonic acid were added and stirred for 2 hours. After completion of the reaction, the reaction was quenched with 504 mL of saturated sodium bicarbonate aqueous solution and extracted with 630 mL of heptane. The organic layer was washed twice with 504 mL of saturated sodium bicarbonate aqueous solution and then with 504 mL of brine. The organic layer was stirred with 42 g of anhydrous sodium sulfate and 34 g of charcoal, filtered with 34 g of celite, washed with 210 mL of methylene chloride, combined with the filtrate, concentrated, and dried under vacuum.
[223]
1H-NMR (400 MHz, CDCl3) : δ 5.79-5.88 (m, 2H), 5.35-5.36 (m, 1H), 5.27-5.29 (m, 1H), 5.12-5.16 (m, 2H), 4.15-4.24 (m, 3H), 3.66 (s, 3H), 3.54-3.57 (m, 1H), 0.91-2.32 (m, 38H), 0.54 (s, 3H).
PAT
- Crystalline form of vascular leakage blocker compoundPublication Number: US-12103945-B2Priority Date: 2020-05-04Grant Date: 2024-10-01
- New crystalline form of vascular leakage blocker compoundPublication Number: US-2022259256-A1Priority Date: 2020-05-04
- Preparation Method of Vascular Leakage Blockers With a High YieldPublication Number: US-2021395297-A1Priority Date: 2019-12-13
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////rivasterat, cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
Navepdekinra
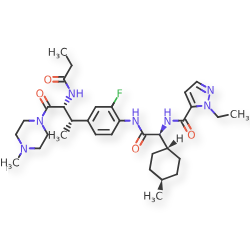
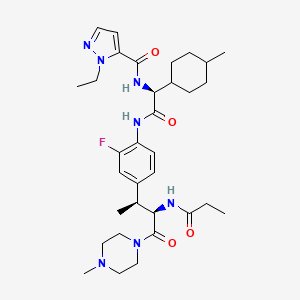
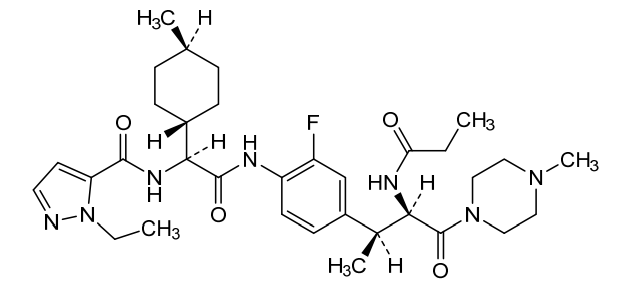
Navepdekinra
CAS 2467732-66-5
MF C33H48FN7O4 MW625.78
1H-Pyrazole-5-carboxamide, 1-ethyl-N-[(1S)-2-[[2-fluoro-4-[(1S,2R)-1-methyl-3-(4-methyl-1-piperazinyl)-3-oxo-2-[(1-oxopropyl)amino]propyl]phenyl]amino]-1-(trans-4-methylcyclohexyl)-2-oxoethyl]-
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-
yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Navepdekinra (also known as DC-806 or LY4100504) is an experimental, orally active small-molecule inhibitor of interleukin-17A (IL-17A). It was primarily developed to treat autoimmune and inflammatory conditions, such as psoriasis, by disrupting the interaction between IL-17A and its receptor.
Key Properties and Development
- Mechanism: It is a potent inhibitor with an IC50 of 10.81 nM, designed to provide an oral alternative to existing injectable IL-17 biologic therapies.
- Acquisition: The drug was originally developed by DICE Therapeutics, which was acquired by Eli Lilly and Company in 2023 for approximately $2.4 billion to bolster their immunology pipeline.
Navepdekinra (DC-806) is an orally active, potent interleukin-17A (IL-17A) inhibitor (IC50 = 10.81 nM). Navepdekinra disrupts the IL-17A protein-receptor interaction, suppressing the downstream pro-inflammatory signaling pathway. Navepdekinra inhibits arthritis in a collage-induced arthritis (CIA) rat model. Navepdekinra can be used for psoriasis, psoriatic arthritis, and ankylosing spondylitis
SYN
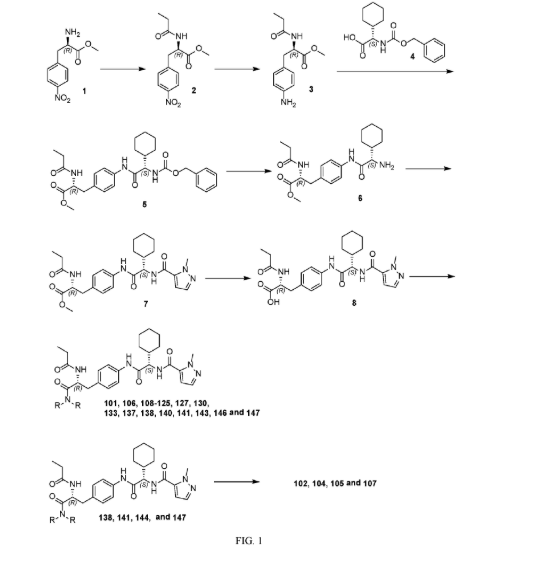
Example 210: N-[(2R,3S)-3-{4-[(2S)-2-[(1-ethyl-1H-pyrazol-5-yl)formamido]-2-[(1r,4S)-4-methylcyclo hexyl]acetamido]-3-fluorophenyl}-1-(4-methylpiperazin-1-yl)-1-oxobutan-2-yl]propanamide) (234)
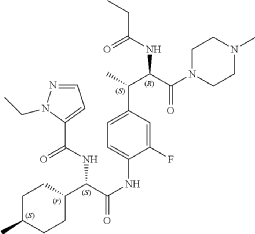
Following General Procedure R, 0.227 g, 0.310 mmol, 1.0 eq) of 82d in DMF (1 mL) were added 1-ethyl-1H-pyrazole-5-carboxylic acid (0.052 g, 0.372 mmol, 1.2 eq), DIPEA (0.43 mL, 2.482 mmol, 8.0 eq) and then HATU (0.177 g, 0.465 mmol, 1.5 eq.) and the resulting mixture was stirred at RT for 1 h. The mixture was concentrated to dryness and the residue was purified via reverse phase column chromatography on a 120 g C18 cartridge eluting with a 5-95% H 2O:MeCN eluent (0.1% ammonia) to afford 234 (0.025 g) as a white solid. 1H NMR (400 MHz, DMSO-d 6) δ 9.86 (s, 1H), 8.46 (d, J=8.3 Hz, 1H), 8.26 (d, J=8.7 Hz, 1H), 7.75 (t, J=8.3 Hz, 1H), 7.47 (d, J=2.1 Hz, 1H), 7.15-7.07 (m, 1H), 7.05-6.97 (m, 2H), 4.86 (t, J=9.4 Hz, 1H), 4.53 (t, J=8.4 Hz, 1H), 4.47 (q, J=7.2 Hz, 2H), 3.46-3.38 (m, 2H), 3.29-3.14 (m, 2H), 3.12-2.99 (m, 2H), 2.25-2.03 (m, 5H), 1.98 (s, 3H), 1.81 (ddt, J=15.0, 9.9, 5.6 Hz, 2H), 1.74-1.60 (m, 4H), 1.58-1.47 (m, 1H), 1.28 (t, J=7.1 Hz, 4H), 1.20 (d, J=7.0 Hz, 3H), 1.14-1.02 (m, 1H), 0.99 (t, J=7.6 Hz, 3H), 0.93-0.87 (m, 1H), 0.86 (d, J=6.5 Hz, 3H). UPLC-MS (basic 4 min): rt=1.76 min; m/z=626.4 for [M+H] +.
PAT
Example 1: Exemplary Scheme—Synthesis of Intermediate Compounds 62a-62d
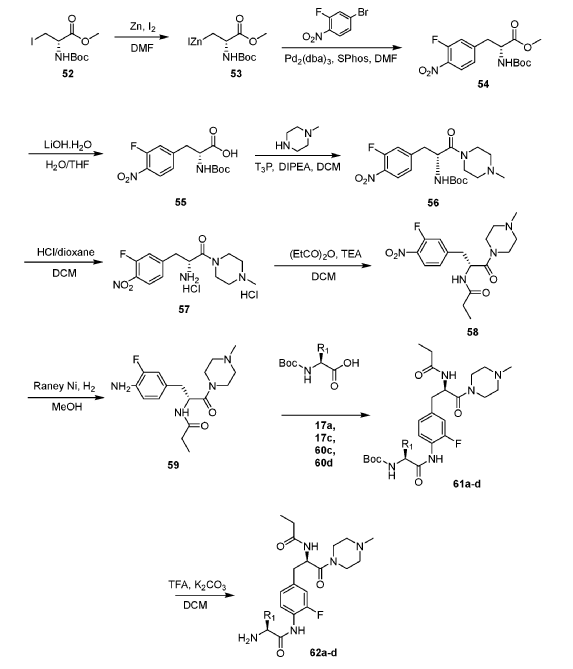
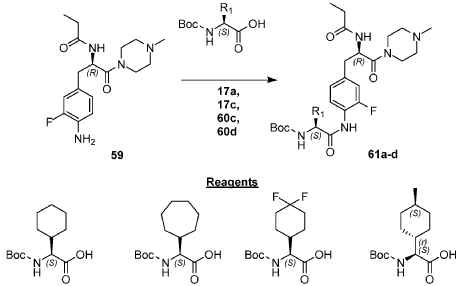
PAT
IL-17 Ligands And Uses Thereof
Publication Number: US-2020247785-A1
Priority Date: 2019-02-06
- Substituted benzenecarboxamides as IL-17A modulatorsPublication Number: US-11274094-B2Priority Date: 2019-09-16Grant Date: 2022-03-15
- Il-17a modulators and uses thereofPublication Number: US-2021101886-A1Priority Date: 2019-09-16
- IL-17 ligands and uses thereofPublication Number: US-11447468-B2Priority Date: 2019-02-06Grant Date: 2022-09-20
- Il-17 ligands and uses thereofPublication Number: US-2023053746-A1Priority Date: 2019-02-06
- IL-17 ligands and uses thereofPublication Number: US-12234225-B2Priority Date: 2019-02-06Grant Date: 2025-02-25
- Mannose 6-phosphate or asgpr receptor binding compounds for the degradation of extracellular proteinsPublication Number: WO-2023028338-A2Priority Date: 2021-08-27
- Potent asgpr-binding compounds for the degradation of immunoglobulins and other proteinsPublication Number: WO-2022235699-A2Priority Date: 2021-05-03
- Il-17a modulators and uses thereofPublication Number: WO-2021055376-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023141212-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023145481-A1Priority Date: 2019-09-16

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
- [1]. Paul R. Fatheree, et al. IL-17 Ligands And Uses Thereof. US20200247785A1.[2]. Kim D, et al. Next-Generation Anti-IL-17 Agents for Psoriatic Disease: A Pipeline Review. Am J Clin Dermatol. 2025 May;26(3):307-320. [Content Brief][3]. Xiaobing Deng, et al. The Critical and Unexpected Role of a Methyl Group in Interleukin-17A Inhibitors. bioRxiv 2025.10.02.680113
//////////navepdekinra, interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Glasmacinal



Glasmacinal
CAS 2097822-02-9
MF C37H62N2O10 MW694.90
[(2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyloxan-3-yl] benzoate
- (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[[2-O-Benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one
- 1-Oxa-6-azacyclopentadecan-15-one, 11-[[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-, (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-
(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-{[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino) -β-D-xylo-hexopyranosyl]oxy}-2-ethyl3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-
azacyclopentadecan-15-one
non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US234729681&_cid=P12-MKVZ26-57135-1
Example 2: (2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyl-tetrahydropyran-3-yl] benzoate)

To a mixture of (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one (Example 1) (0.5 g, 0.8500 mmol) and Triethylamine (428.2 mg, 4.23 mmol) in DCM (5 ml), cooled on ice, was added Benzoyl chloride (356.9 mg, 2.54 mmol). The reaction mixture was allowed to reach room temperature. After 3 days good conversion to the desired benzoylated product was obtained and the mixture was portioned between DCM and saturated sodium hydrogen carbonate solution. The organic phase was dried over magnesium sulphate and concentrated to a white foam. The product was purified using reversed phase chromatography (see general information)
PAT
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2018354981-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-10723752-B2Priority Date: 2015-11-19Grant Date: 2020-07-28
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2020317710-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-12049477-B2Priority Date: 2015-11-19Grant Date: 2024-07-30
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-11236120-B2Priority Date: 2015-11-19Grant Date: 2022-02-01
- Compounds
- Publication Number: US-2022106349-A1
- Priority Date: 2015-11-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com

ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
///////glasmacinal, ANAX, ADVECT, non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
Cenacitinib



Cenacitinib
CAS 2641636-52-2
MF C19H19F2N7O3 MW431.4
Urea, N-[(1R,2S)-2-fluorocyclopropyl]-N′-[5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl]-
N-{5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl}-N′-[(1R,2S)-2-fluorocyclopropyl]urea
Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
VTX958 for the Treatment of Moderately to Severely Active Crohn’s Disease
CTID: NCT05688852
Phase: Phase 2
Status: Terminated
Date: 2025-07-03
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750705&_cid=P22-MKEUDK-45432-1
Example 4: Synthesis of 1-(5-((7-fluoro-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl)-3-((1R,2S)-2-fluorocyclopropyl)urea (5)


| Step 1: To a solution of 1E (100 g, 288 mmol) and 2E (57 g, 345 mmol) in dry 1,4-dioxane (3000 mL) under N 2 atmosphere was added Cs 2CO 3 (141 g, 432 mmol), Pd(OAc) 2 (5.2 g, 23.3 mmol) and BINAP (28.6 g, 46.6 mmol). After stirring at 115° C. overnight, the reaction mixture was cooled to rt. and diluted with hexane (3000 mL). The solid was collected by filtration and washed with 2×1500 mL (50% hexane in DCM). The solid was suspended into 5000 mL water and stirred for 1 h. The solid was collected by filtration and dried under vacuum to afford compound 2 (90 g, 65%) as a brown solid. |
PAT
Publication Number: US-2021139486-A1
Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: EP-4054581-A1Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: US-2023348478-A1Priority Date: 2019-11-08
- Substituted pyrazolo[1,5-a]pyrimidines as TYK2 pseudokinase ligandsPublication Number: US-11753411-B2Priority Date: 2019-11-08Grant Date: 2023-09-12
- TYK2 pseudokinase ligandsPublication Number: CN-114929226-BPriority Date: 2019-11-08Grant Date: 2024-09-27
- TYK2 pseudokinase ligandPublication Number: CN-114929226-APriority Date: 2019-11-08
- Preparation of a tyk2 inhibitorPublication Number: WO-2024151992-A1Priority Date: 2023-01-13
- Crystalline forms of a tyk2 inhibitorPublication Number: US-2024010654-A1Priority Date: 2022-07-06
- Crystalline forms of TYK2 inhibitorsPublication Number: CN-119816502-APriority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: EP-4551576-A1Priority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: WO-2024011136-A1Priority Date: 2022-07-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////cenacitinib, cenacitinib, Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Zemprocitinib


Zemprocitinib
CAS 2417414-44-7
MF C16H19N5O2S MW 345.4 g/mol
N-[3-(3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-3-yl)-1-bicyclo[1.1.1]pentanyl]propane-1-sulfonamide
N-[3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl]propane-1-sulfonamide
Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Zemprocitinib (also known as LNK01001) is a selective Janus kinase (JAK) 1 inhibitor, a type of small molecule drug being developed for inflammatory and autoimmune conditions like rheumatoid arthritis, atopic dermatitis, and ankylosing spondylitis. It works by blocking the JAK1 enzyme, reducing the inflammatory signals that cause these diseases, and has shown promising results in clinical trials, with development reaching Phase 3.
Key Aspects:
- Drug Class: JAK1 Inhibitor.
- Mechanism: Blocks Janus Kinase 1, a key enzyme in inflammatory pathways.
- Developer: Initially Lynk Pharmaceuticals.
- Potential Uses: Rheumatoid Arthritis, Atopic Dermatitis, Ankylosing Spondylitis, Psoriasis, Alopecia Areata.
- Development Stage: Reached Phase 3 clinical trials for several indications.
- Chemical Info: CAS: 2417414-44-7; Formula: C16H19N5O2S.
In Summary:
Zemprocitinib is an investigational drug targeting inflammation by inhibiting JAK1, with potential to treat various autoimmune disorders, showing strong efficacy in early clinical trials for conditions like rheumatoid arthritis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US347660217&_cid=P21-MJDP3D-82397-1
Example 1



Step 1. 4-Chloro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1b)
Step 2. 4-Chloro-5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1c)
Step 3. Tert-butyl 3-((5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (Id)
Step 4. Tert-butyl 3-((5-amino-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (le)
Step 5. Tert-butyl 3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentane-1-carboxylate (1f)
Step 6. 3-(6-Tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentane-1-carboxylic acid (1g)
Step 7. Tert-butyl (3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1h)
Step 8. Tert-butyl (3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1i)
Step 9. 3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-amine 2,2,2-trifluoroacetate (1j)
Step 10. N-(3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)propane-1-sulfonamide (1)
PAT
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: ES-2993867-T3Priority Date: 2018-11-01Grant Date: 2025-01-10
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: JP-2024147699-APriority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: EP-3856742-B1Priority Date: 2018-11-01Grant Date: 2024-10-02
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2022009927-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023357247-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023339950-A1Priority Date: 2018-11-01
- Tricyclic Janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: AU-2019372677-B2Priority Date: 2018-11-01Grant Date: 2024-05-30
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: TW-202432555-APriority Date: 2018-11-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Zemprocitinib, Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Nibrozetone
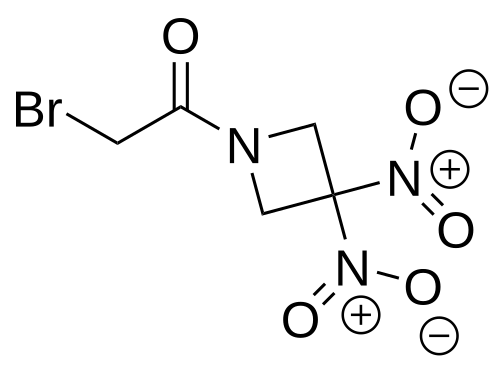
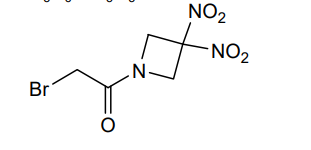
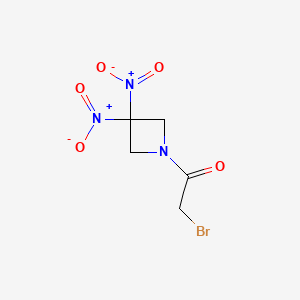
Nibrozetone
CAS 925206-65-1
MF C5H6BrN3O5 MW268.02 g/mol
2-bromo-1-(3,3-dinitroazetidin-1-yl)ethan-1-one
2-Bromo-1-(3,3-dinitroazetidin-1-yl)ethanone
2-BROMO-1-(3,3-DINITROAZETIDIN-1-YL)ETHAN-1-ONE
anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Nibrozetone is an investigational new drug that is being evaluated by EpicentRx for the treatment of oral mucositis in head and neck cancer patients. It is a small molecule that combines direct inhibition of the NLRP3 inflammasome, induction of NRF2, and release of nitric oxide under hypoxic conditions.[1][2] It has received Fast Track designation from the FDA for severe oral mucositis in head and neck cancer patients.[3]
Nibrozetone (RRx-001) is an investigational, multi-action small molecule drug that is being developed by EpicentRx for a range of conditions, including head and neck cancers, small cell lung cancer, and neurodegenerative diseases like Parkinson’s and ALS. Its mechanism involves inhibiting the NLRP3 inflammasome, activating the Nrf2 pathway, and releasing nitric oxide in hypoxic tumor environments, while also protecting healthy tissues. It is being evaluated for its potential to reduce the side effects of cancer treatments and as a disease-modifying therapy itself.
How it works
- Anti-inflammatory: Nibrozetone inhibits the NLRP3 inflammasome, which is a key driver of inflammation in several diseases.
- Antioxidant: It activates the Nrf2 pathway, a cellular defense mechanism that protects against oxidative stress.
- Tumor-specific delivery: It acts as a “hypoxia-activated” drug, releasing a nitric oxide-releasing radical only in the low-oxygen environment of tumors, which can be toxic to cancer cells.
- Protective to normal tissue: The drug’s protective mechanisms are thought to keep it from causing harm to healthy tissues outside of the tumor environment.
Current and potential uses
- Oral mucositis: It is being studied to prevent and treat severe mouth sores that can be a side effect of head and neck cancer radiation therapy.
- Small cell lung cancer (SCLC): It is being investigated in a Phase 3 trial for the treatment of SCLC.
- Neurodegenerative diseases: Animal studies have shown promising neuroprotective effects in models of Parkinson’s and ALS.
- Other potential applications: Research is ongoing for its use as a treatment for other conditions, including endometriosis, toxic exposures, and various types of cancers.
- RRx-001 in Lung Cancer, Ovarian Cancer and Neuroendocrine Tumors Prior to Re-administration of Platinum Based Doublet Regimens (QUADRUPLE THREAT)CTID: NCT02489903Phase: Phase 2Status: CompletedDate: 2025-03-17
- RRx-001 for Reducing Oral Mucositis in Patients Receiving Chemotherapy and Radiation for Head and Neck CancerCTID: NCT05966194Phase: Phase 2Status: RecruitingDate: 2024-11-15
- Safety and Efficacy of RRx-001 in the Attenuation of Oral Mucositis in Patients Receiving Chemoradiation for the Treatment of Oral CancersCTID: NCT03515538Phase: Phase 2Status: CompletedDate: 2024-11-04
- Safety and Pharmacokinetic Study of RRx-001 in Cancer SubjectsCTID: NCT01359982Phase: Phase 1Status: CompletedDate: 2024-11-01
- RRx-001 Given With Irinotecan and Temozolomide for Pediatric Patients With Recurrent or Progressive Malignant Solid and Central Nervous System TumorsCTID: NCT04525014Phase: Phase 1Status: TerminatedDate: 2024-10-31
REF
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
PAT
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-8927527-B2Priority Date: 2005-08-12Grant Date: 2015-01-06
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-9226915-B2Priority Date: 2005-08-12Grant Date: 2016-01-05
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: WO-2007022225-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-2022016077-A1Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-11925617-B2Priority Date: 2005-08-12Grant Date: 2024-03-12
- Methods of synthesizing and isolating N-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: US-8471041-B2Priority Date: 2010-02-09Grant Date: 2013-06-25
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: WO-2011100090-A1Priority Date: 2010-02-09
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: IL-221141-A0Priority Date: 2010-02-09
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-B1Priority Date: 2005-08-12Grant Date: 2014-12-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011100090&_cid=P11-MHTYGA-61308-1
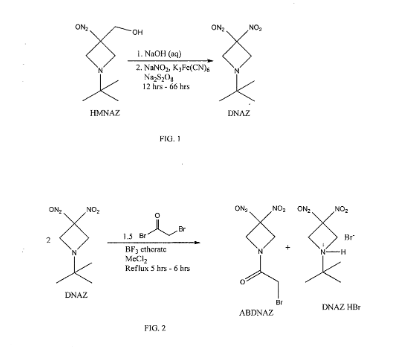
Cyclic nitro compounds, such as ABDNAZ, are being investigated for their potential use in treating cancer. Methods of synthesizing ABDNAZ have been described, such as in United States Patent No. 7,507,842 to Bednarski et al.
(“Bednarski”). In Bednarski, ABDNAZ is synthesized by reacting
l-½rt-butyl-3,3-dinitroazetidine (DNAZ) with bromoacetyl bromide and boron trifluoride etherate. For every mole of ABDNAZ produced, a mole of a hydrogen bromide salt of DNAZ (DNAZ HBr) is also produced as a coproduct. The ABDNAZ is isolated from the DNAZ HBr by cooling the reaction mixture, adding
dichloromethane, and filtering the DNAZ HBr. Solid DNAZ HBr is sensitive to impact, friction, and other external stimuli and, therefore, must be handled carefully. The dichloromethane filtrate is washed with water, dried, and then the dichloromethane is evaporated, producing a crude ABDNAZ mixture. The product is washed sequentially with diethyl ether and dried under vacuum, yielding ABDNAZ that is approximately 98% pure and at a yield of approximately 75% (based on bromoacetyl bromide). The 2% of impurities remaining in the ABDNAZ are believed to include
bromoacetic acid, unreacted DNAZ, and DNAZ HBr. This method of producing ABDNAZ is referred to herein as the Bednarski process. While the Bednarski process provides ABDNAZ at a reasonable purity and yield, the purity is not sufficient for pharmaceutical uses. In addition, solid DNAZ HBr produced during the Bednarski process is an explosive compound, which adds to the complexity of producing
Example 2
Synthesis of ABDNAZ from DNAZ
A three neck round bottom flask (3 L) equipped with a magnetic stir bar and a water jacketed reflux condenser was charged with the dichloromethane solution of DNAZ (produced as described in Example 1). A nitrogen gas purge of the apparatus was initiated and, after ten minutes, boron trifluoride diethyletherate (6.37 mL, 52 mmol) was added, followed by bromoacetyl bromide (33.77 mL, 388 mmol). The flask was sealed, except for a small vent at the top of the condenser, and the solution was heated to a mild reflux. After six hours (± 0.5 hour), heating was stopped and dichloromethane (1000 mL) and distilled water (800 mL) were added, in that order, to the heterogeneous mixture. The two-phase system was stirred vigorously for sixteen hours, until all solids (DNAZ HBr) were dissolved. The two-phase system was then transferred to a separatory funnel. The aqueous phase was removed and the organic phase was washed with additional distilled water (4 x 500 mL). The organic phase was dried with sodium sulfate (100 g – 150 g) and then transferred to a single neck, round bottom flask. The solution was concentrated on a rotary evaporator to approximately half of its initial volume and then ethanol (250 mL) was added. The remaining dichloromethane was removed by a rotary evaporator, causing precipitation of clear, colorless crystals. The flask was chilled in an ice bath for thirty minutes. The precipitate was isolated by vacuum filtration, rinsed with additional cold ethanol (5 x 150 mL), and dried to afford pure ABDNAZ (56.04 g, 81% yield): Ή NMR
(d6-acetone) δ 4.02 (s, 2H, -CH2Br ), 4.96 (br s, 2H, ring -CH2), 5.36 (br s, 2H, ring -CH2); 13C NMR (d6-acetone) δ 25.58, 58.58, 60.53, 107.69, 167.48.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007022225&_cid=P11-MHTYDP-59218-1
Example 5: Synthesis of ABDNAZ
[00139] A 25 ml, three-neck, round bottom flask was charged with 7 ml of methylene chloride and 2.50 g (12.3 mmol) of t-BuDNAZ prepared as described in Archibald et at, Journal of Organic Chemistry, 1990, 2920. Under nitrogen, 0.16 ml (1.23 mmol) of boron trifluoride etherate was added. After stirring 5 min. at ambient temperature, 0.54 ml (6.15 mol) of bromoacetyl bromide was added. The solution was heated between 50-600C for 2 h. The darkened reaction mixture was cooled to ambient temperature, diluted with 50 ml methylene chloride, and filtered. The solid was identified as the HBr salt of t-BuDNAZ. The methylene chloride filtrate was washed with two 20 ml portions of water, dried over sodium sulfate, filtered, and evaporated under reduced pressure. The resultant solid was washed with three 20 ml portions of ethyl ether and dried under vacuum to yield 1.24 g (75.2% based on bromoacetyl bromide) of BrADNAZ as a white solid (mp = 124-1250C). 1H NMR (CDCl3): δ 3.76 (s, 2H), 4.88 (br s, 2H), 5.14 (br s, 2H); 13C NMR (CDCl3): δ 165.2, 105.0, 59.72, 57.79, 23.90. CaIc. for C5H6BrN3O5: %C 22.41, %H 2.26, %N 15.68; Found: %C 22.61, %H 2.36, %N 15.58.
HPLC/MS C-8 reverse phase column with acetonitrile/water mobile phase – m/e 266.95 (100%), 268.95 (98.3%). FT-IR 3014.24 (weak), 1677.66, 1586.30, 1567.65, 1445.55 (NO2), 1367.80, 1338.00, 1251.27 cm‘1.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Oronsky B, Takahashi L, Gordon R, Cabrales P, Caroen S, Reid T (2023). “RRx-001: a chimeric triple action NLRP3 inhibitor, Nrf2 inducer, and nitric oxide superagonist”. Frontiers in Oncology. 13 1204143. doi:10.3389/fonc.2023.1204143. PMC 10258348. PMID 37313460.
- Jayabalan N, Oronsky B, Cabrales P, Reid T, Caroen S, Johnson AM, et al. (April 2023). “A Review of RRx-001: A Late-Stage Multi-Indication Inhibitor of NLRP3 Activation and Chronic Inflammation”. Drugs. 83 (5): 389–402. doi:10.1007/s40265-023-01838-z. PMC 10015535. PMID 36920652.
- Ryan C (30 March 2023). “FDA Grants Fast Track Designation to RRx-001 for Severe Oral Mucositis in Head and Neck Cancer”. OncLive.
| Clinical data | |
|---|---|
| Other names | Rrx-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 925206-65-1 |
| PubChem CID | 15950826 |
| DrugBank | DB12060 |
| ChemSpider | 13092644 |
| UNII | 7RPW6SU9SC |
| KEGG | D12720 |
| ChEMBL | ChEMBL3526802 |
| Chemical and physical data | |
| Formula | C5H6BrN3O5 |
| Molar mass | 268.023 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Nibrozetone, anti-inflammatory, RRx-001, RRx 001, ABDNAZ

{kind=link}