PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
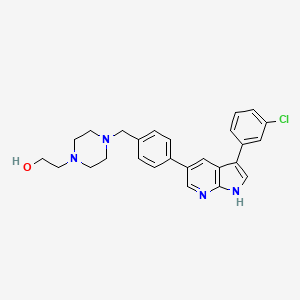
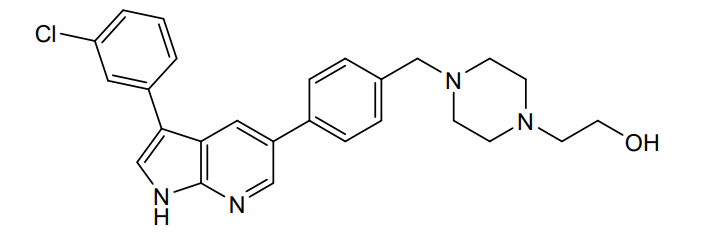
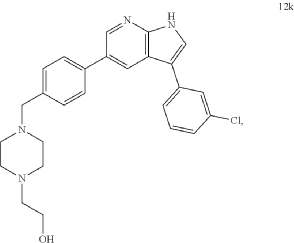
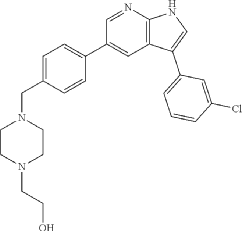
Prosetin is an orally administered blocker of MAP4K under investigation for the treatment of amyotrophic lateral sclerosis.
Famlasertib (also known as Prosetin or Prostetin/12k) is a highly potent, small-molecule inhibitor targeting the mitogen-activated protein kinase kinase kinase kinase (MAP4K) family. It is an experimental drug primarily under investigation for its neuroprotective capabilities in treating neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) and as an anti-invasive agent in certain cancers
Famlasertib functions by blocking the activation of the MAP4K protein family, specifically demonstrating powerful inhibitory values (\(\text{IC}_{50}\)) against subfamilies like HGK (MAP4K4), MLK3, and MLK1. By inhibiting these kinases, the compound: [1]
Reduces Endoplasmic Reticulum (ER) Stress: It helps mitigate the unfolded protein response that triggers programmed cell death in neurons affected by misfolded protein accumulation.
Suppresses Inflammation: It blocks inflammatory pathways associated with neurodegeneration and cell damage.
Restrains Cell Motility: In oncology contexts, it disrupts kinase signaling linked to actin cytoskeleton remodeling, preventing malignant cells from migrating.
Primary Areas of Research
1. Amyotrophic Lateral Sclerosis (ALS)
In motor neuron models of ALS, cellular stress frequently triggers neurodegeneration. Because famlasertib easily passes through the blood-brain barrier, it is capable of directly shielding motor neurons from ER-stress-mediated cell death, extending cell viability in laboratory models.
2. Oncology (Medulloblastoma)
Recent findings published on bioRxiv indicate that famlasertib acts as a “migrastatic” agent in medulloblastoma (a type of pediatric brain tumor). It suppresses the highly invasive behavior and single-cell motility of tumor cells without exhibiting developmental toxicity.
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
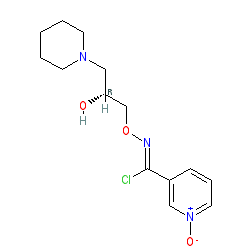
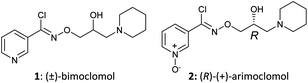
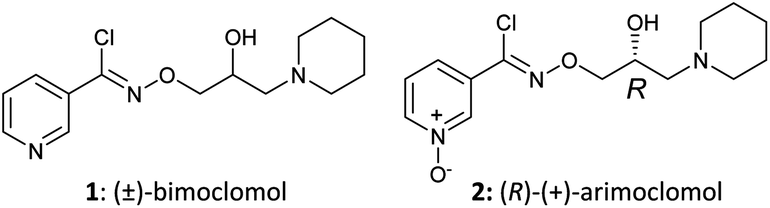
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
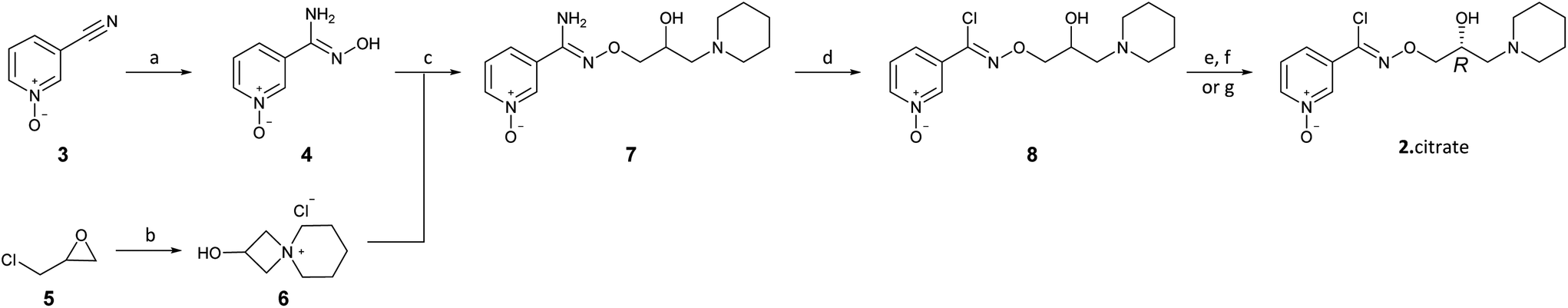
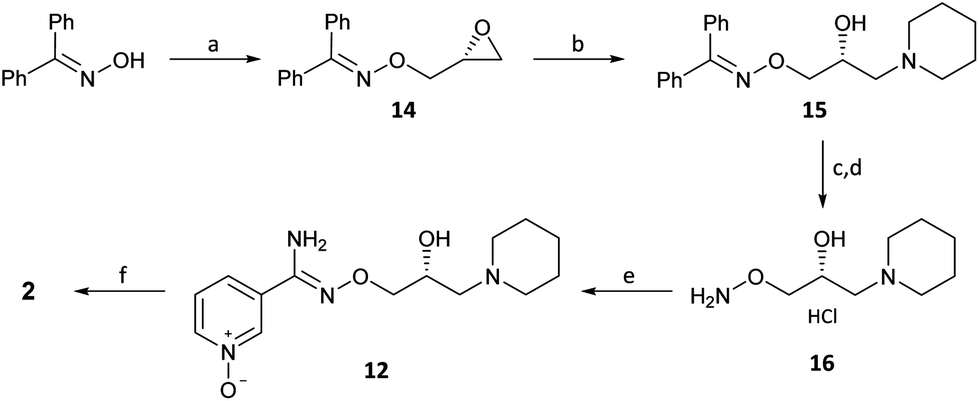
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
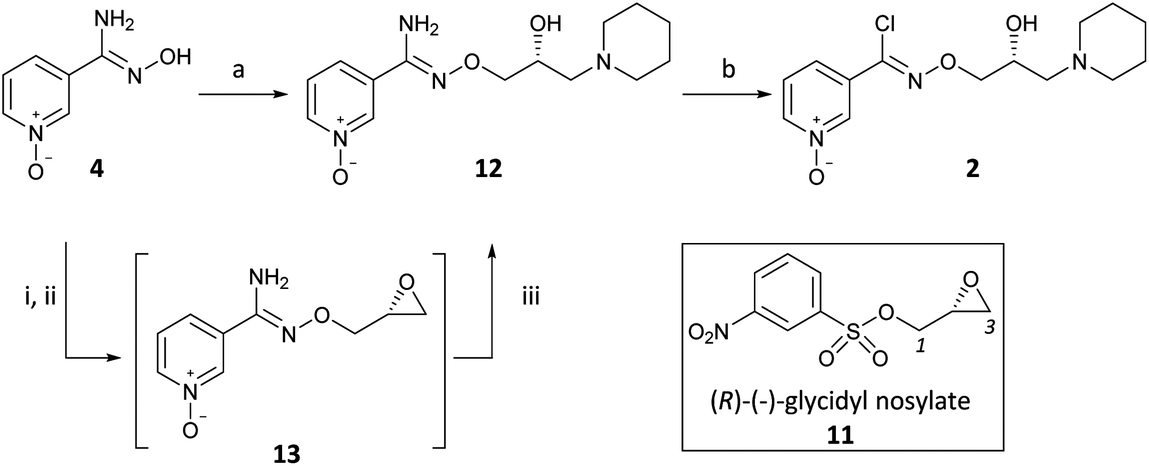
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
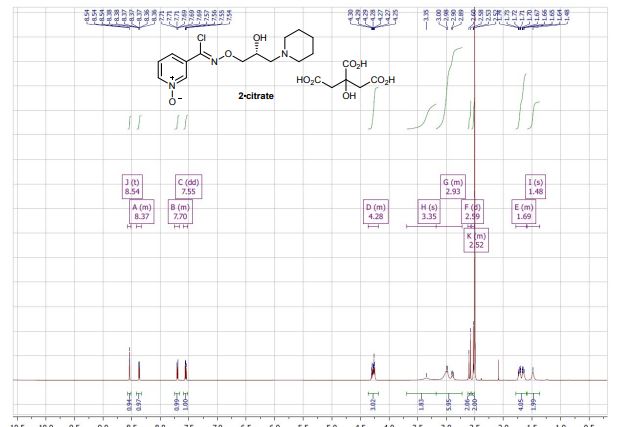
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.
Reldesemtiv, also known as CK-2127107, is a skeletal muscle troponin activator (FSTA) and is a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue such as SMA, COPD, and ALS.
Cytokinetics , in collaboration with Astellas , is developing reldesemtiv, the lead from a program of selective fast skeletal muscle troponin activators, in an oral suspension formulation, for the treatment of indications associated with neuromuscular dysfunction, including spinal muscular atrophy and amyotrophic lateral sclerosis.
Originator Cytokinetics
Developer Astellas Pharma; Cytokinetics
Class Pyridines; Pyrimidines; Pyrroles; Small molecules
05 May 2019 Safety and efficacy data from the phase II FORTITUDE-ALS trial in Amyotrophic lateral sclerosis presented at the American Academy of Neurology Annual Meeting (AAN-2019)
07 Mar 2019 Cytokinetics completes the phase III FORTITUDE-ALS trial for Amyotrophic lateral sclerosis in USA, Australia, Canada, Spain, Ireland and Netherlands (PO) (NCT03160898)
22 Jan 2019 Cytokinetics plans a phase I trial in Healthy volunteers in the first quarter of 2019
Reldesemtiv, a next-generation, orally-available, highly specific small-molecule is being developed by Cytokinetics, in collaboration with Astellas Pharma, for the improvement of skeletal muscle function associated with neuromuscular dysfunction, muscle weakness and/or muscle fatigue in spinal muscular atrophy (SMA), chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS). The drug candidate is a fast skeletal muscle troponin activator (FSTA) or troponin stimulant intended to slow the rate of calcium release from the regulatory troponin complex of fast skeletal muscle fibers. Clinical development for ALS, COPD and SMA is underway in the US, Australia, Canada, Ireland, Netherlands and Spain. No recent reports of development had been identified for phase I development for muscular atrophy in the US. Due to lack of of efficacy determined at interim analysis Cytokinetics suspended phase I trial in muscle fatigue in the elderly.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
[0003] Of the thirteen distinct classes of myosin in human cells, the myosin-II class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-II forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere’s thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-binding of the globular head from the actin filament (Ca2+ regulated) coupled with return of the catalytic domain and light chain to their starting conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin-tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the
Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
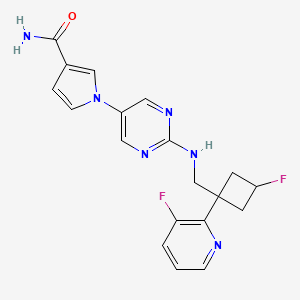
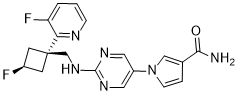
U.S. Patent No. 8962632 discloses l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide, a next-generation fast skeletal muscle troponin activator (FSTA) as a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue.
claiming the use of a similar compound for treating stress urinary incontinence.
Compound A is 1- [2-({[trans-3-fluoro-1- (3-fluoropyridin-2-yl) cyclobutyl] methyl} amino) pyrimidin-5-yl] -1H Pyrrole-3-carboxamide, which is the compound described in Example 14 of the aforementioned US Pat. The chemical structure is as shown below.
Process for preparing reldesemtiv , a myosin, actin, tropomyosin, troponin C, troponin I, troponin T modulator, useful for treating neuromuscular disorders, muscle wasting, claudication and metabolic syndrome.
Scheme 1
[0091] Scheme 1 illustrates a scheme of synthesizing the compound of Formula (1C).
Scheme 2
[0092] Scheme 2 illustrates an alternative scheme of synthesizing the compound of Formula (1C).
M
TFAA DS, toluene
Et
to
2
HCI, H20
50°C
Scheme 3
[0093] Scheme 3 illustrates a scheme of converting the compound of Formula (1C) to the compound of Formula (II).
H2
Ni Raney
NH3
Scheme 4
[0094] Scheme 4 illustrates a scheme of converting the compound of Formula (II) to the compound of Formula (1).
Examples
[0095] To a flask was added N-methylpyrrolidone (30 mL), tert-butyl cyanoacetate (8.08 g) at room temperature. To a resulting solution was added potassium tert-butoxide (7.71 g), l,3-dibromo-2,2-dimethoxy propane (5.00 g) at 0 °C. To another flask, potassium iodide (158 mg), 2,6-di-tert-butyl-p-cresol (42 mg), N-methylpyrrolidone (25 mL) were added at room temperature and then resulting solution was heated to 165 °C. To this solution, previously prepared mixture was added dropwise at 140-165 °C, then stirred for 2 hours at 165 °C. To the reaction mixture, water (65 mL) was added. A resulting solution was extracted with toluene (40 mL, three times) and then combined organic layer was washed with water (20 mL, three times) and 1N NaOH aq. (20 mL). A resulting organic layer was concentrated below 50 °C under reduced pressure to give 3, 3 -dimethoxy cyclobutane- l-carbonitrile (66% yield,
Example 2 Synthesis of methyl 3,3-dimethoxycyclobutane-l-carboxylate
[0096] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. MeOH (339.00 kg), 3-oxocyclobutanecarboxylic acid (85.19 kg, 746.6 mol, 1.0 eq.), Amberlyst-l5 ion exchange resin (8.90 kg, 10% w/w), and
trimethoxymethane (196.00 kg, 1847.3 mol, 2.5 eq.) were charged into the reactor and the resulting mixture was heated to 55±5°C and reacted for 6 hours to give methyl 3,3-dimethoxycyclobutane-l-carboxylate solution in MeOH. 1H NMR (CDCl3, 400 MHz) d 3.70 (s, 3H), 3.17 (s, 3H), 3.15 (s, 3H), 2.94-2.85 (m, 1H), 2.47-2.36 (m, 4H).
Example 3 Synthesis of 3, 3-dimethoxycyclobutane-l -carboxamide
[0097] The methyl 3, 3 -dimethoxy cyclobutane- l-carboxylate solution in MeOH prepared as described in Example 2 was cooled to below 25°C and centrifuged. The filter cake was washed with MeOH(7.00 kg) and the filtrate was pumped to the reactor. The solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH
(139.40 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH (130.00 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. Half of the resulting solution was diluted with MeOH (435.00 kg) and cooled to below 30°C. NH3 gas (133.80 kg) was injected into the reactor below 35°C for
24 hours. The mixture was stirred at 40±5°C for 72 hours. The resulting solution was
concentrated under vacuum below 50°C until the system had no more than 2 volumes.
MTBE(l8l.OO kg) was charged into the reactor. The resulting solution was concentrated under vacuum below 50°C until the system had no more than 2 volumes. PE (318.00 kg) was charged into the reactor. The resulting mixture was cooled to 5±5°C, stirred for 4 hours at 5±5°C, and centrifuged. The filter cake was washed with PE (42.00 kg) and the wet filter cake was put into a vacuum oven. The filter cake was dried at 30±5°C for at least 8 hours to give 3,3-dimethoxycyclobutane-l-carboxamide as off-white solid (112.63 kg, 94.7% yield). 1H NMR (CDCf, 400 MHz) d 5.76 (bs, 1H), 5.64 (bs, 1H), 3.18 (s, 3H), 3.17 (s, 3H), 2.84-2.76 (m, 1H), 2.45-2.38 (m, 4H).
[0098] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Toluene (500.00 kg), 3,3-dimethoxycyclobutane-l-carboxamide (112.54kg, 706.9 mol, 1.0 eq.), and TEA (158.00 kg, 1561.3 mol, 2.20 eq) were charged into the reactor and the resulting mixture was cooled to 0+ 5°C. TFAA (164.00 kg, 781 mol, 1.10 eq.) was added dropwise at 0±5°C. The resulting mixture was stirred for 10 hours at 20±5°C and cooled below 5±5°C. H20 (110.00 kg) was charged into the reactor at below 15 °C. The resulting mixture was stirred for 30 minutes and the water phase was separated. The aqueous phase was extracted with toluene (190.00 kg) twice. The organic phases were combined and washed with H20 (111.00 kg). H20 was removed by azeotrope until the water content was no more than 0.03%. The resulting solution was cooled to below 20°C to give 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene (492.00 kg with 17.83% assay content, 87.9% yield).
Example 5 Synthesis of l-(3-fluoropyridin-2-yl)-3,3-dimethoxycyclobutane-l-carbonitrile
[0099] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene prepared as described in Example 4 (246.00 kg of a 17.8% solution of 3,3-dimethoxycyclobutane-l-carbonitrile in toluene, 1.05 eq.) and 2-chloro-3-fluoropyridine (39.17 kg, 297.9 mol, 1.00 eq.) were charged into the reactor. The reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The mixture was slowly cooled to -20±5°C. NaHDMS (2M in THF) (165.71 kg, 1.20 eq) was added
dropwise at -20±5°C. The resulting mixture was stirred at -l5±5°C for 1 hour. The mixture was stirred until the content of 2-chloro-3-fluoropyridine is no more than 2% as measured by HPLC. Soft water (16.00 kg) was added dropwise at below 0°C while maintaining the reactor temperature. The resulting solution was transferred to another reactor. Aq. NH4Cl (10% w/w, 88.60 Kg) was added dropwise at below 0°C while maintaining the reactor temperature. Soft water (112.00 kg) was charged into the reactor and the aqueous phase was separated and collected. The aqueous phase was extracted with ethyl acetate (70.00 kg) and an organic phase was collected. The organic phase was washed with sat. NaCl (106.00 kg) and collected. The above steps were repeated to obtain another batch of organic phase. The two batches of organic phase were concentrated under vacuum below 70°C until the system had no more than 2 volumes. The resulting solution was cooled to below 30°C to give a l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution. 1H NMR (CDC13, 400 MHz) d 8.42-8.38 (m, 1H), 7.50-7.45 (m, 1H), 7.38-7.33 (m, 1H), 3.28 (s, 3 H), 3.13 (s, 3H), 3.09-3.05 (m, 4H).
Example 6 Synthesis of I-(3-fluoropyridin-2-yl)-3-oxocyclohutanecarhonitrile
[0100] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Water (603.00 kg) was added to the reactor and was stirred.
Concentrated HC1 (157.30 kg) was charged into the reactor at below 35°C. The l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution prepared as described in Example 5 (206.00 kg) was charged into the reactor and the resulting mixture was heated to 50±5°C and reacted for 3 hours at 50±5°C. The mixture was reacted until the content of 1-(3 -fluoropyridin-2-yl)-3, 3 -dimethoxycyclobutane- l-carbonitrile was no more than 2.0% as measured by HPLC. The reaction mixture was cooled to below 30°C and extracted with ethyl acetate (771.00 kg). An aqueous phase was collected and extracted with ethyl acetate (770.00 kg). The organic phases were combined and the combined organic phase was washed with soft water (290.00 kg) and brine (385.30 kg). The organic phase was concentrated under vacuum at below 60°C until the system had no more than 2 volumes. Propan-2-ol (218.00 kg) was charged into the reactor. The organic phase was concentrated under vacuum at below
60°C until the system had no more than 1 volume. PE (191.00 kg) was charged into the reactor at 40±5 °C and the resulting mixture was heated to 60±5 °C and stirred for 1 hour at 60±5 °C. The mixture was then slowly cooled to 5±5 °C and stirred for 5 hours at 5±5 °C. The mixture was centrifuged and the filter cake was washed with PE (48.00 kg) and the wet filter cake was collected. Water (80.00 kg), concentrated HC1 (2.20 kg), propan-2-ol (65.00 kg), and the wet filter cake were charged in this order into a drum. The resulting mixture was stirred for 10 minutes at 20±5 °C. The mixture was centrifuged and the filter cake was washed with a mixture solution containing 18.00 kg of propan-2-ol, 22.50 kg of soft water, and 0.60 kg of concentrated HC1. The filter cake was put into a vacuum oven and dried at 30±5°C for at least 10 hours. The filter cake was dried until the weight did not change to give l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile as off-white solid (77.15 kg, 68.0% yield). 1H NMR (CDCl3, 400 MHz) d 8.45-8.42 (m, 1H), 7.60-7.54 (m, 1H), 7.47-7.41 (m, 1H), 4.18-4.09 (m, 2H), 4.02-3.94 (m, 2H).
Example 7 Synthesis of I-(3-fhtoropyridin-2-yl)-3-hydroxycyclobulanecarbonilrile
[0101] To a solution of l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g,
1.22 mol) in a mixture ofDCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78° C. The reaction mixture was stirred at -78°C. for 1 hour and quenched with a mixture of methanol and water (1 : 1). The organic layer was washed with water (500 mL><3), dried over Na2S04, and concentrated. The residue was purified on silica gel (50% EtO Ac/hexanes) to provide the title compound as an amber oil (185.8 g, 77.5%). Low Resolution Mass
Spectrometry (LRMS) (M+H) m/z 193.2.
Example 8 Synthesis of (ls,3s)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutane-l-carbonitrile
[0102] To a solution of 1 -(3 -fluoropyridin-2-yl)-3 -hydroxy cyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 hours. The reaction was cooled to rt and poured onto sat. NaHCCf solution. The mixture was separated and the organic layer was washed with water, dried over Na2S04, and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (116g) in a 8: 1 transxis mixture. The above brown oil (107 g) was dissolved in toluene (110 mL) and hexanes (330mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g). LRMS (M+H) m/z 195.1.
Example 9 Synthesis of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine
[0103] A mixture of ( 1.v,3.v)-3-fluoro- 1 -(3-fluoropyridin-2-yl)cyclobutane- 1 -carbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g, 97.6%). LRMS (M+H) m/z 199.2.
Example 10 Synthesis of t-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate
[0104] A mixture of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine (37.6 g, 190 mmol), 5-bromo-2-fluoropyrimidine (32.0 g, 181 mmol), DIPEA (71 mL, 407 mmol), and NMP (200 mL) was stirred at rt overnight. The reaction mixture was then diluted with EtOAc (1500 mL) and washed with saturated sodium bicarbonate (500 mL). The
organic layer was separated, dried over Na2S04, and concentrated. The resultant solid was dissolved in THF (600 mL), followed by the slow addition of DMAP (14 g, 90 mmol) and Boc20 (117.3 g, 542 mmol). The reaction was heated to 60° C. and stirred for 3 h. The reaction mixture was then concentrated and purified by silica gel chromatography
(EtO Ac/hex) to give 59.7 g oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a white solid.
Example 11 Synthesis of t-butyl 5-(3-cyano- 1 H -pyrrol- 1 -yl)pyrimidin-2-yl(((lrans)-3-fhtoro-l-(3-fluoropyridin-2-yl)cyclohutyl)methyl)carhamate
[0105] To a solution oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate (1.0 g, 2.8 mmol) in 15 mL of toluene (degassed with nitrogen) was added copper iodide (100 mg, 0.6 mmol), potassium phosphate (1.31 g, 6.2 mmol), trans-N,N’-dimethylcyclohexane-l, 2-diamine (320 mg, 2.2 mmol), and 3-cyanopyrrole (310 mg, 3.6 mmol). The reaction was heated to 100 °C and stirred for 2 h. The reaction was then concentrated and purified by silica gel chromatography (EtOAc/hexanes) to afford 1.1 g of t-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a clear oil.
Example 12 Synthesis of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide
[0106] To a solution oft-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate (1.1 g, 3.1 mmol) in DMSO (10 mL) was added potassium carbonate (1.3 g, 9.3 mmol). The mixture was cooled to 0 °C and hydrogen peroxide (3 mL) was slowly added. The reaction was warmed to rt and stirred for 90 min. The reaction was diluted with EtO Ac (75 mL) and washed three times with brine (50 mL). The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% MeOH/CH2Cl2) to afford 1.07 g of a white solid compound. This compound was dissolved in 25% TFA/CH2CI2 and stirred for 1 hour. The reaction was then concentrated, dissolved in ethyl acetate (75 mL), and washed three times with saturated potassium carbonate solution. The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was triturated with 75% ethyl acetate/hexanes. The resultant slurry was sonicated and filtered to give 500 mg of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3 -carboxamide as a white solid. LRMS (M+H=385).
REFERENCES
1: Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018 May;57(5):729-734. doi: 10.1002/mus.26017. Epub 2017 Dec 11. PubMed PMID: 29150952.
2: Gross N. The COPD Pipeline XXXII. Chronic Obstr Pulm Dis. 2016 Jul 14;3(3):688-692. doi: 10.15326/jcopdf.3.3.2016.0150. PubMed PMID: 28848893; PubMed Central PMCID: PMC5556764.
//////////////CK-2127107, CK 2127107, CK2127107, Reldesemtiv, Cytokinetics, Astellas, neuromuscular disorders, muscle wasting, claudication, metabolic syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, Orphan Drug Status, Spinal muscular atrophy, Phase II
The U.S. Food and Drug Administration today approved Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease.
May 5, 2017
Release
The U.S. Food and Drug Administration today approved Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease.
“After learning about the use of edaravone to treat ALS in Japan, we rapidly engaged with the drug developer about filing a marketing application in the United States,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This is the first new treatment approved by the FDA for ALS in many years, and we are pleased that people with ALS will now have an additional option.”
ALS is a rare disease that attacks and kills the nerve cells that control voluntary muscles. Voluntary muscles produce movements such as chewing, walking, breathing and talking. The nerves lose the ability to activate specific muscles, which causes the muscles to become weak and leads to paralysis. ALS is progressive, meaning it gets worse over time. The Centers for Disease Control and Prevention estimates that approximately 12,000-15,000 Americans have ALS. Most people with ALS die from respiratory failure, usually within three to five years from when the symptoms first appear.
Radicava is an intravenous infusion given by a health care professional. It is administered with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
The efficacy of edaravone for the treatment of ALS was demonstrated in a six-month clinical trial conducted in Japan. In the trial, 137 participants were randomized to receive edaravone or placebo. At Week 24, individuals receiving edaravone declined less on a clinical assessment of daily functioning compared to those receiving a placebo.
The most common adverse reactions reported by clinical trial participants receiving edaravone were bruising (contusion) and gait disturbance.
Radicava is also associated with serious risks that require immediate medical care, such as hives, swelling, or shortness of breath, and allergic reactions to sodium bisulfite, an ingredient in the drug. Sodium bisulfite may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The FDA granted this drug orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Radicava to Mitsubishi Tanabe Pharma America, Inc,
On June 26, 2015, Mitsubishi Tanabe Pharma Corporation announced it has received approval to market Radicut for treatment of ALS in Japan. The phase III clinical trial began in 2011 in Japan. The company was awarded Orphan Drug Designation for Radicut by the FDA and EU in 2015. Radicut is an intravenous drug and administrated 14 days followed by 14 days drug holiday.
The biotech company Treeway is developing an oral formulation of edaravone (TW001) and is currently in clinical development. Treeway was awarded orphan drug designation for edaravone by the EMA in November 2014 and FDA in January 2015.
Edaravone (CAS NO.: 89-25-8), with other name of 3-Methyl-1-phenyl-2-pyrazolin-5-one, could be produced through many synthetic methods.
Following is one of the synthesis routes: By direct cyclization of phenylhydrazine (I) with ethyl acetoacetate (II) in refluxing ethanol.
SYNTHESIS
Edaravone, chemical name: 3-methyl-1-phenyl-2-pyrazoline-5-one, of the formula: Formula: CiciHltlN2O, molecular weight: 174.20, the formula:
[0004] Edaravone is a brain-protecting agent (free radical scavenger).Clinical studies suggest that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction.In patients with acute cerebral infarction Edaravone suppressed reduce peri-infarct regional cerebral blood flow, so that the first concept of days after the onset of brain NAA glycerol content than the control group significantly increased. Preclinical studies suggest that rats after ischemia / reperfusion of ischemic intravenous edaravone, can prevent the progress of cerebral edema and cerebral infarction, and relieve the accompanying neurological symptoms, suppress delayed neuronal death.Mechanism studies suggest that edaravone can scavenge free radicals, inhibiting lipid peroxidation, thereby inhibiting brain cells, endothelial cells, oxidative damage nerve cells.
For the synthesis of edaravone reported some use of benzene and methyl ethyl ketone amide corpus obtained, but methyl ethyl ketone amide difficult to obtain and slow reaction, which now has basically been abandoned; some use benzene corpus and ethyl acetoacetate in ethanol (see US4857542A, Synthesis Example 1) or water (Dykhanov NN Ethyl and butyl acetoacetates, Med Prom SSSR, 1961,15 (1):. 42-45) refluxing the reaction of the reaction The resulting purity edaravone poor, and the yield is not high, only about 70%.
Edaravone, chemical name: 2,4_-dihydro-5-methyl-2-phenyl pyrazole -3H- – one, of the formula: CiciHltlN2O, molecular weight: 174.20, the formula:
edaravone is a clear cerebral infarction harmful factors (free radicals), protection of new therapeutic agents for cerebral infarction nerve cells.Clinical studies have shown that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction.When patients with acute cerebral infarction Edaravone, peri-infarct rCBF decrease has improved, and the first 28 days after the onset of brain NAA content was significantly higher than that in the control group glycerol.Mechanism studies suggest that edaravone can clear the brain is highly cytotoxic hydroxyl radicals, inhibiting the synthesis of lipids free radicals, which can suppress brain infarction after reperfusion edema, protecting brain from damage and improve nerve impairment symptoms, and the delayed neuronal death inhibition, to protect the brain.
The first is by phenylhydrazine and methyl ethyl ketone amide (National API process compilation, 1980.737-739) condensation reaction in water at 50 ° C, a yield of up to 97%, but the raw material ketone amide ( CH3C0CH2C0NH2) are not readily available.Formula I
Edaravone synthetic route for the reaction:
[0008] The second is to phenylhydrazine and ethyl acetoacetate in ethanol or water at reflux the reaction, sodium bisulfite as the preparation of the catalyst.From the perspective of the chemical reaction, acetyl ethyl ketone amide more than hydrazine reacted with benzene and ethyl acetoacetate more readily available, the price is cheaper, but lower reaction yield of about 70%.Formula 2 for the synthesis route Edaravone reaction formula:
[0024] (1) Weigh benzene hydrochloride corpus 13. 5g (94mmol), was added to IOOml water, stirred for 0.5 hours, sodium hydroxide was added an equimolar 3. 76g, stirred for 0.5 hours; [0025] ( 2) To the reaction solution was added dropwise ethyl acetoacetate 11. 7g (90mmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 15. 5g;
[0026] (3) The crude product was added 30ml volume ratio of 2: 1 isopropanol – water, 2g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 14 a white crystalline powder. 8g, yield 90%, mpU9 ° C, with a purity of 99.9% 0
2 Edaravone Synthesis Example [0027] Example
[0028] (1) Weigh 15g of benzene hydrochloride corpus (I (Mmmol), was added to 120ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 4. 16g, stirred for 0.5 hours;
[0029] (2) To the reaction solution was added dropwise 13g of ethyl acetoacetate (lOOmmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 16. 7g;
(3) The crude product was added 40ml volume ratio of 2: 1 isopropanol – water, 2. 5g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature to precipitate a white solid, as a white crystalline powder 16. lg, a yield of 88.9%, mpU8 ° C, with a purity of 99.9% 0
3 Edaravone Synthesis Example [0031] Example
[0032] (1) Weigh 22g of benzene hydrochloride corpus (152mm0l), was added to 200ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 6. 08g, stirred for 0.5 hours;
[0033] (2) To the reaction solution was added dropwise 19g of ethyl acetoacetate (146mm0l), the reaction exotherm, the reaction was heated to reflux for 3 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 24. Sg;
[0034] (3) The crude product was added 50ml volume ratio of 2: 1 isopropanol – water, 3g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 23 a white crystalline powder. 2g, a yield of 87. 8%, mpU8 ° C, with a purity of 99.9% 0
[0035] Comparative Example
[0036] The ethyl acetoacetate 65g (0. 5mol) and 180ml of anhydrous ethanol mixed, with stirring at 50 ° C was added dropwise benzyl corpus 54g (0. 5mol) and a solution consisting of 30ml absolute ethanol, dropwise at reflux for 2 Bi hours, ethanol was distilled off 60ml, cooled, suction filtered, washed crystals with cold absolute ethanol twice, and dried in vacuo to give pale yellow crystals 70g.Recrystallized twice from absolute ethanol to give pale yellowish white crystals 56g (yield 65%).
Example 1: Preparation of phenylhydrazine edaravone.
[0024] a. Weigh 5.1g phenylhydrazine (47mmol), was added under stirring to water containing 45mL round-bottom flask, take appropriate concentrated hydrochloric acid solution was adjusted to pH 6.0 with PH meter.
[0025] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, was added 1.5g sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0026] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 85% yield, 99.2% purity 0
[0027] Example 2: Preparation of phenylhydrazine hydrochloride edaravone.
[0028] a. Weigh 6.8g phenylhydrazine hydrochloride (47mmol), was added under stirring to water containing 45mL round-bottomed flask, the pH of the solution adjusted to 6.0 with aqueous ammonia.
[0029] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, 1.25g was added sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0030] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 84% yield, with a purity of 99.2%.[0031] Comparative Example:
Under the [0032] state of agitation will phenylhydrazine 10.2g (94mmol) added to a round bottom flask equipped with IOOmL water in an appropriate amount of concentrated hydrochloric acid was dubbed the volume ratio of 1: 1 aqueous hydrochloric acid, with a PH adjusting pH of the solution was measured 6.0.After weighing Ethylacetoacetate 11.7g (90mmol) added to the reaction mixture, the reaction was exothermic and cooling to room temperature, sodium bisulfite (NaHSO3), heated to 105 ° C under reflux for 3h, the hot solution Water was added into the beaker and mechanical stirring, cooling, filtration, and dried to give the yellow edaravone crude, 73% yield, with a purity of 99.1%.
These values are in accordance with the previous published in literature1 .
In the carbon spectrum in DMSO presented in Figure SM 4.2.3.1.8 is evident the presence of the two major tautomeric structures of edaravone, signals are identified by different colours in both structures in the figure. Also in the IR analysis of the solid material (Figure SM 4.2.3.1.9) is possible to see either the NH form (max/cm-1, 3129), the OH form (max/cm- 1 , 3431) and the C=O (max/cm-1, 1599) of the enol and keto tautomeric forms of edaravone.
1. S. Pal, J. Mareddy and N. S. Devi, J. Braz. Chem. Soc., 2008, 19, 1207.
We have shown that the short reaction time, in combination with good yields can make microwave assisted reaction of hydrazines with β-ketoesters ideal for a rapid entry to pyrazolones. All the compounds synthesized are characterized by spectroscopic (1H NMR, IR and MS) data. While determination of tautomeric composition of compound 3 is quite challenging as eight possible tautomeric forms need to be considered, interestingly, two major tautomeric forms of compound 3a was observed in two different solvents. For example, it exists as 1,2-dihydro pyrazolone (T-1, Figure 2) in DMSO and 2,4-dihydro form (T-2, Figure 2) in chloroform as indicated by 1H NMR spectra (Figure 3). The olefinic proton of T-1 appeared at 5.36 δ whereas the methylene hydrogens appeared at 3.43 δ in case of T-2. Additionally, the NH proton of T-1 at 11.40 δ was not observed incase of T-2 confirmed the absence of NH in the 2,4-dihydro form. Existence of two major tautomeric forms was also observed in case compound 3b (see 1H NMR data in the experimental section). However, X-ray study on single crystal of 2-(4-chlorophenyl)-5-methyl-1,2-dihydro pyrazol-3-one (3i) indicates that 2-aryl pyrazol-3-ones e.g. 3a-b, 3e-f and 3i exist as 1,2-dihydro form in crystal state. 27 It is mention worthy that the aryl ring of all these 2-aryl pyrazol-3-ones remain twisted with respect to the pyrazole plane as indicated by the crystallographic data of 3i [the dihedral angle between the pyrazole and benzene ring planes was found to be 15.81 (11)º].27
Jump up^Higashi Y, Jitsuiki D, Chayama K, Yoshizumi M (January 2006). “Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases”. Recent Patents on Cardiovascular Drug Discovery. 1 (1): 85–93. doi:10.2174/157489006775244191. PMID18221078.
Jump up^Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N (2006). “Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury”. CNS Drug Reviews. 12 (1): 9–20. doi:10.1111/j.1527-3458.2006.00009.x. PMID16834755.
Jump up^Yokoyama H, Takagi S, Watanabe Y, Kato H, Araki T (June 2008). “Role of reactive nitrogen and reactive oxygen species against MPTP neurotoxicity in mice”. Journal of Neural Transmission (Vienna, Austria : 1996). 115 (6): 831–42. doi:10.1007/s00702-008-0019-6. PMID18235988.
Jump up^Yokoyama H, Yano R, Aoki E, Kato H, Araki T (September 2008). “Comparative pharmacological study of free radical scavenger, nitric oxide synthase inhibitor, nitric oxide synthase activator and cyclooxygenase inhibitor against MPTP neurotoxicity in mice”. Metabolic Brain Disease. 23 (3): 335–49. doi:10.1007/s11011-008-9096-3. PMID18648914.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader





















![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)

























 . PMID 18671880.
. PMID 18671880.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}