
Home » Posts tagged 'Acute myeloid leukaemia'
Tag Archives: Acute myeloid leukaemia
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tambiciclib
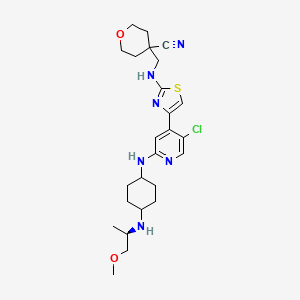
Tambiciclib
CAS 2247481-08-7
MF C25H35ClN6O2S, 519.10
4-[[[4-[5-chloro-2-[[4-[[(2R)-1-methoxypropan-2-yl]amino]cyclohexyl]amino]-4-pyridinyl]-1,3-thiazol-2-yl]amino]methyl]oxane-4-carbonitrile
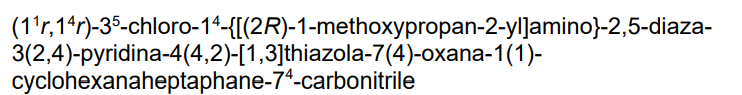
cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Tambiciclib (GFH009, JSH-009) is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib can be used for AML research.
Tambiciclib is a selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon administration, tambiciclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
- OriginatorGenFleet Therapeutics
- DeveloperGenFleet Therapeutics; Sellas Life Sciences Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 9 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia; Peripheral T-cell lymphoma
- Phase IIAcute myeloid leukaemia
- Phase I/IIDiffuse large B cell lymphoma; Haematological malignancies; Peripheral T-cell lymphoma
- Phase ISolid tumours
- PreclinicalColorectal cancer; T-cell prolymphocytic leukaemia
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Combination therapy) in USA (Parenteral)
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Monotherapy) in USA (Parenteral)
- 13 Oct 2025Pharmacodynamics data from preclinical studies in T-cell prolymphocytic leukaemia released by SELLAS Life Sciences
CLINICAL
- A Study of GFH009 in Combination With Zanubrutinib in Subjects With Relapsed or Refractory DLBCLCTID: NCT06375733Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-08-12
- A Study of GFH009 Monotherapy in Patients with Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL)CTID: NCT05934513Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-12-13
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2018-10-05
PMID: 30253346
DOI: 10.1016/j.ejmech.2018.09.025
SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018192273&_cid=P12-MJ18VV-17351-1
Example 1: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino) cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4- carboxynitrile
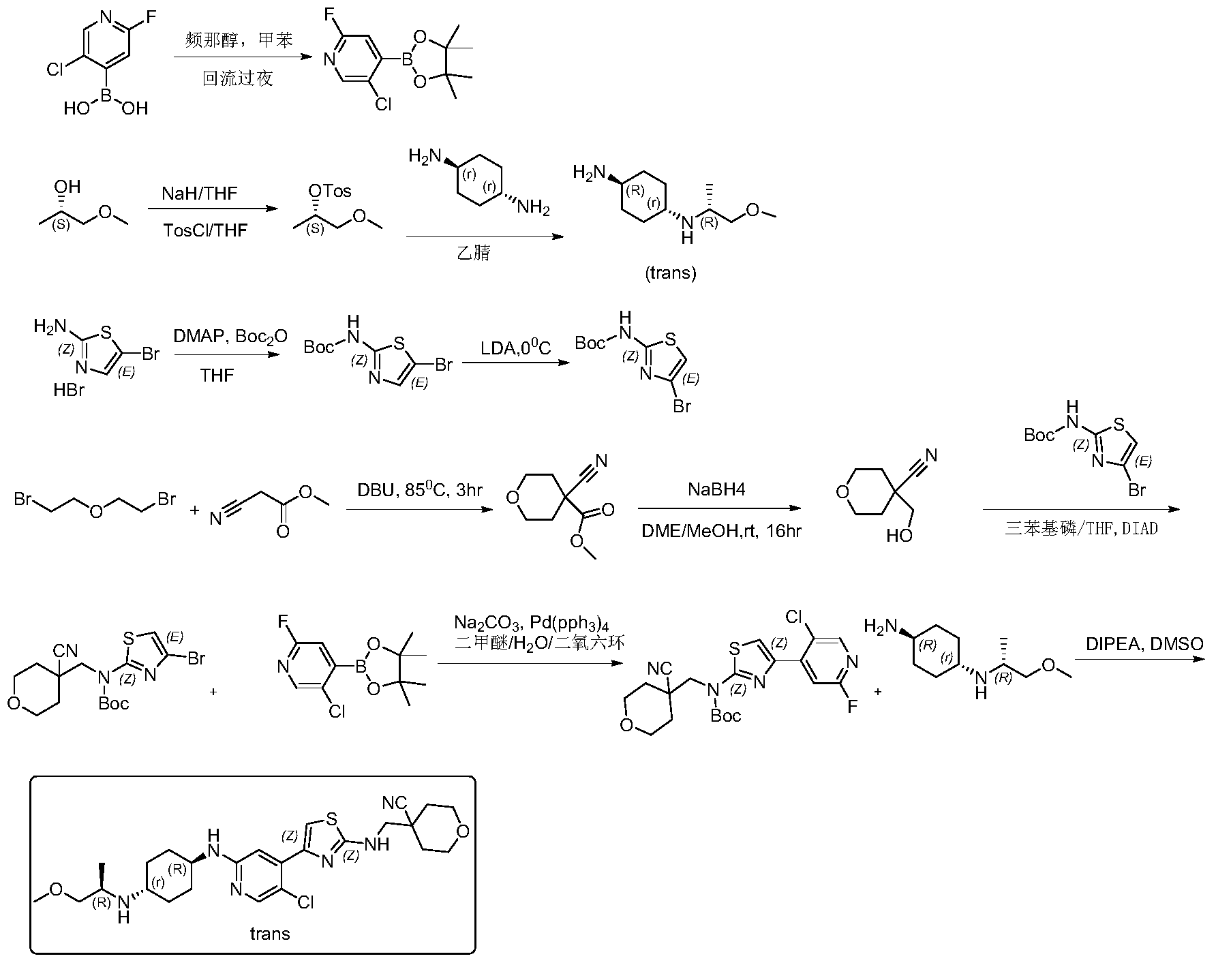
Step 1: Synthesis of 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhecyclopentan-2-yl)pyridine
[0102]5-Chloro-2-fluoropyridine-4-boronic acid (0.7 g, 4.46 mmol) and pinacol (0.63 g, 5.35 mmol) were added to 50 mL of toluene, and the mixture was refluxed at 120 °C overnight. TLC showed a small amount of starting material remaining. The reaction mixture was cooled to room temperature and concentrated, then dried by an oil pump to give 0.92 g of a white solid compound, 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine, yield 80%, MS (ESI): m/z 258.1 (M+H) + .
[0103]Step 2: Synthesis of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester
[0104]60% sodium hydride (NaH) (6.52 g, 283 mmol) was added to anhydrous tetrahydrofuran (THF) (200 mL). The mixture was cooled to 0 °C in an ice bath under nitrogen protection, and (S)-(+)-1-methoxy-2-propanol (21 g, 233 mmol) was added dropwise. After the addition was complete, the mixture was brought to room temperature and stirred for 1.5 hours. The reaction mixture was then cooled back to 0 °C, and a tetrahydrofuran (THF) solution of p-toluenesulfonyl chloride (45.3 g, 283 mmol) (200 mL) was added dropwise. After the addition was complete, the mixture was stirred overnight at room temperature. TLC showed that the starting material had reacted completely. The reaction mixture was diluted with ethyl acetate (500 mL), and the reaction was quenched by adding water (500 mL) dropwise while cooling in an ice bath. The mixture was separated, and the aqueous phase was extracted once more with ethyl acetate (200 mL). The combined organic phases were washed with water (200 mL) and then with saturated brine (200 mL). The crude product was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 43 g of a pale yellow oily substance. Column separation (petroleum ether/ethyl acetate = 5/1) yielded 37 g of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester, a pale yellow oily substance, with a yield of 65.1%. MS (ESI): m/z 245.1 (M+H) + .
[0105]Step 3: Synthesis of (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine
[0106](S)-1-methoxypropyl-2-yl 4-toluenesulfonyl ester (5 g, 20.5 mmol) and trans-1,4-cyclohexanediamine (5.84 g, 51.2 mmol) were added to 50 mL of acetonitrile and heated to 90 °C overnight. The reaction was monitored by TLC until complete. After cooling, the reaction solution was filtered, the filtrate was concentrated, and the residue was dissolved in dichloromethane and separated by silica gel stirring column (dichloromethane/methanol = 10/1) to give 2.5 g of the pale yellow liquid compound (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine, yield 65%, MS (ESI): m/z 187.3 (M+H) + .
[0107]Step 4: Synthesis of tert-butyl 5-bromothiazol-2-ylcarbamate
[0108]105 g (403 mmol) of 5-bromothiazol-2-amine hydrobromide was suspended in 500 mL of tetrahydrofuran. Dimethylaminopyridine (2.41 g, 20 mmol) was added, resulting in a white turbidity. A tetrahydrofuran solution of di-tert-butyl dicarbonate (105.6 g, 484.6 mmol) was slowly added dropwise, and the reaction was allowed to proceed at room temperature for two days. The reaction solution was concentrated and dissolved in 300 mL of dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 10/1-6/1 gradient elution) to give 45 g of off-white solid, yield 40%. MS (ESI): m/z 278.98 (M+H) + .
[0109]Step 5: Synthesis of tert-butyl 4-bromothiazol-2-ylcarbamate
[0110]A 200 mL solution of diisopropylamine (64 mL, 446 mmol) in tetrahydrofuran was added to a dry three-necked flask. Under nitrogen protection, the mixture was cooled to 0 °C, and n-butyllithium (2.5 M, 173 mL, 431.7 mmol) was added dropwise. The reaction was allowed to proceed for 1 hour after the addition was complete. Then, a 400 mL solution of 5-bromothiazol-2-ylcarbamate in tetrahydrofuran was added dropwise at 0 °C. The reaction was allowed to proceed for 2 hours after the addition was complete. TLC showed that the reaction was complete. At 0℃, ice water (5 mL) was slowly added dropwise to quench the reaction. After stirring for 30 minutes, saturated ammonium chloride (500 mL) aqueous solution was added. The mixture was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The organic layers were combined, washed with saturated brine, dried with anhydrous sodium sulfate, filtered, concentrated, and recrystallized from petroleum ether:ethyl acetate = 30:1. 31 g of tert-butyl 4-bromothiazol-2-ylcarbamate was obtained as a white solid, yield 77.5%. MS (ESI): m/z 278.98 (M+H) + .
[0111]Step Six: Synthesis of Methyl 4-cyano-tetrahydro-2H-pyran-4-carbonate
[0112]Methyl cyanoacetate (39.1 g, 395.3 mmol) and 2,2-dibromoethyl ether (100 g, 434.8 mmol) were added to 600 mL of dimethylformamide, followed by DBU (90 g, 593 mmol). The mixture was heated to 85 °C and reacted for 3 hours. TLC showed that the starting material reacted completely. The solid was filtered off, washed with ethyl acetate (2 × 300 mL), and the mother liquor was concentrated to obtain a brown oily substance. The oil was distilled under reduced pressure at an internal temperature of 65-70 °C, and the fraction collected was a colorless liquid. Crystallization was observed to give 42 g of a white solid, 4-cyano-tetrahydro-2H-pyran-4-carbonate. Yield: 62.8%, MS (ESI): m/z 178.2 (M+H) + .
[0113]Step 7: Synthesis of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile
[0114]4-Cyano-tetrahydro-2H-pyran-4-carbonate methyl ester (42 g, 248.4 mmol) was dissolved in 400 mL of ethylene glycol dimethyl ether and 40 mL of methanol. The mixture was cooled to 0 °C in an ice bath, and sodium borohydride (11.1 g, 149 mmol) was added in portions. After the addition was complete, the mixture was allowed to rise to room temperature and stirred for 16 hours. The reaction was completed by TLC. The reaction solution was concentrated, and methanol was added to quench excess sodium borohydride. The solution was then concentrated again. Column chromatography (petroleum ether/ethyl acetate = 5/1) yielded 28 g of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow oil, yield: 79.5%, MS (ESI): m/z 142.1 (M+H) + .
[0115]Step 8: Synthesis of tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate
[0116]4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, 4-bromothiazol-2-ylcarbamate tert-butyl ester, and triphenylphosphine were added to anhydrous tetrahydrofuran (THF) and cooled to 0°C. Diisopropyl azodicarbonate (DIAD) was added dropwise. The mixture was stirred at room temperature for 10 minutes, then heated to 40°C overnight. The reaction solution was concentrated, and the residue was dissolved in dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 50/1, 30/1, 20/1) to obtain (4-bromothiazol-2-yl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester, a white solid of 365 mg, yield 50%. MS (ESI): m/z 402.1 (M+H) + .
[0117]Step Nine: Synthesis of tert-butyl (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate
[0118]5-Chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine and sodium carbonate were added to a mixture of dimethyl ether/H₂O
/ dioxane. The system was purged with nitrogen twice. Then, tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate and tetraphenylphosphine palladium Pd(pph 3 )
4 were added . The system was purged with nitrogen three times. The temperature was then raised to 70°C and the reaction was carried out for 6 hours. TLC showed that only half of the starting material remained. Heating was then stopped and the reaction was terminated. The reaction solution was cooled to room temperature, ethyl acetate and methanol were added, and the mixture was filtered. The filter cake was washed with ethyl acetate, the filtrate was concentrated, and the residue was dissolved in dichloromethane. The residue was washed with saturated brine, separated, and the organic phase was dried over anhydrous sodium sulfate. The mixture was filtered, and silica gel was added for mixing. The sample was separated by column chromatography (petroleum ether/ethyl acetate = 30/1) to give 3.2 g of (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate, a white foamy solid, with a yield of 55%. MS (ESI): m/z 453.1 (M+H) + .
[0119]Step 10: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile
[0120]The tert-butyl carbamate (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate (3.2 g, 7.1 mmol) and (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine (3.9 g, 21.2 mmol) and diisopropylethylamine (DIPEA) were added to 30 mL of dimethyl sulfoxide. Under nitrogen protection, the mixture was heated to 100-110 °C and reacted for two days. The reaction was monitored by TLC and LCMS. The starting material (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester had completely disappeared, with some BOC-free intermediate remaining. The reaction was stopped, and the reaction solution was cooled and diluted with ethyl acetate (60 mL). Water (150 mL) was added under ice bath. The mixture was separated, and the aqueous layer was extracted again with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with saturated brine (100 mL), dried with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product of yellowish-brown oil. Column separation (acetonitrile/water/trifluoroacetic acid = 80/20/0.001) yielded 700 mg of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow solid. Yield: 19.1%. ¹H NMR (400 MHz, CDCl₃
) )δ8.06(s,1H),7.38(s,1H),6.97(s,1H),5.92(brs,1H),4.45(d,J=8.0Hz,1H),4.02(dd,J 1=2.8Hz, J2=12Hz,2H),3.71-3.74(m,4H),3.54-3.56(m,1H),3.35(s,3H),3.21-3.25(m,2 H),3.00-3.05(m,1H),2.50-2.60(m,1H),2.15(d,J=9.6Hz,2H),2.04-2.07(m,1H),1.95(d ,J=12.8Hz,3H),1.74-1.82(m,3H),1.10-1.30(m,4H),1.00(d,J=.4Hz,3H),MS(ESI):m/z 519.3(M+H) + .
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376039987&_cid=P12-MJ18R0-12787-1
PAT
- A novel cyclin-dependent kinase CDK9 inhibitorPublication Number: CN-108727363-BPriority Date: 2017-04-19Grant Date: 2020-06-19
- Inhibitor of cyclin-dependent kinase CDK9Publication Number: US-10952999-B2Priority Date: 2017-04-19Grant Date: 2021-03-23
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-B1Priority Date: 2017-04-19Grant Date: 2021-12-29
- Pharmaceutical combination and use thereof in treatment of cancerPublication Number: WO-2024239512-A1Priority Date: 2023-05-22
- Polymorph of cdk9 inhibitor and preparation method for polymorph and use thereofPublication Number: WO-2020244612-A1Priority Date: 2019-06-06
- Polymorphic substance of CDK9 inhibitor and preparation method and application thereofPublication Number: CN-113966332-APriority Date: 2019-06-06
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-A1Priority Date: 2017-04-19
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: US-2020078343-A1Priority Date: 2017-04-19



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//Tambiciclib, cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
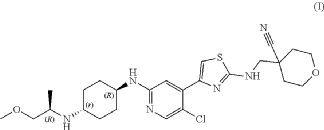
MAX 40279
MAX 40279, EX-A4057
Max 4; MAX-40279; MAX-40279-001; MAX-40279-01
UNII-DL772G3NN7
2070931-57-4
C22H23FN6OS, 438.5
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-ylpyrazol-4-yl)thieno[3,2-d]pyrimidin-2-amine
Thieno[3,2-d]pyrimidin-2-amine, 7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-

7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE SEMI-FUMARATE CAS 2388506-43-0
- 7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
- Originator Maxinovel Pharmaceuticals
- ClassAntineoplastics
- Mechanism of ActionFibroblast growth factor receptor antagonists; Fms-like tyrosine kinase 3 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia
- Phase IAcute myeloid leukaemia; Solid tumours
Most Recent Events
- 28 Nov 2019Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) in China (PO) (NCT04183764)
- 16 Apr 2019Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in China (PO) (NCT04187495)
- 23 Jan 2019Guangzhou Maxinovel Pharmaceuticals plans a phase I trial in China (ChiCTR1900020971)
- MaxiNovel Pharmaceuticals, Inc. Announces FDA Orphan Drug Designation for MAX-40279 for the Treatment of Acute Myeloid Leukemia (AML)
March 29, 2018 11:24 AM Eastern Daylight Timehttps://www.businesswire.com/news/home/20180329005826/en/MaxiNovel-Pharmaceuticals-Inc.-Announces-FDA-Orphan-Drug-Designation-for-MAX-40279-for-the-Treatment-of-Acute-Myeloid-Leukemia-AML
GUANGZHOU, China–(BUSINESS WIRE)–MaxiNovel Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (“FDA”) has granted MaxiNovel Orphan Drug Designation for MAX-40279 in the treatment of Acute Myeloid Leukemia (AML).
AML is the most common acute leukemia which accounts for approximately 25% of all adult leukemias worldwide. Approximately one-third of AML patients have a FLT3 gene mutation. Such mutation can result in faster disease progression, higher relapse rates and lower rates of survival than other forms of AML. Inhibition of FLT3 mutation is of high importance in combating AML.
In the preclinical testing, MAX-40279 demonstrated potent inhibition of both FLT3 and FGFR with excellent drug concentration in the bone marrow. It is designed to overcome the observed drug resistance of the current FLT3 inhibitors due to the bone marrow FGF/FGFR pathway activation.
“We are very pleased to receive the ODD,” commented MaxiNovel’s Vice President Dr. Elizabeth Ashraf. “Our objective is to bring the best in class medicine to the patients worldwide.”
The FDA Office of Orphan Products Development grants orphan drug designation to novel drugs and biologics that are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. The designation allows manufacturers to qualify for various incentives including federal grants, tax credits for qualified clinical trials, a waiver of PDUFA filing fees and 7 years of market exclusivity upon regulatory approval.
About MaxiNovel Pharmaceuticals, Inc:
Maxinovel Pharmaceuticals, Inc. is a biotech company focusing on the discovery and development of Immuno-oncology therapy and targeted therapy. It will use its orally active Immuno-oncology product platform to bring effective combo product of multi-components in a single oral pill to the patients worldwide. For more info: www.maxinovel.com
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) signal pathway is a signal transduction pathway stimulated by cytokines discovered in recent years, and it participates in many important biology such as cell proliferation, differentiation, apoptosis and immune regulation. Process (Aaronson, D Set al. Science 2002, 296, 1653-1655; O’Shea, J Jet al. Nat. Rev. Drug Discovery 2004, 3, 555-564). Compared with other signal pathways, the transmission process of this signal pathway is relatively simple. It mainly consists of three components, namely tyrosine kinase-related receptor, tyrosine kinase JAK and transcription factor STAT. JAK (Janus Kinase), a type of molecule in the cell, is rapidly recruited and activated on the receptor after receiving the signal from the upstream receptor molecule. The activated JAK catalyzes the receptor tyrosine phosphorylation, and the phosphorylation of tyrosine on the receptor molecule Amino acid is the recognition and binding site of a kind of signal molecule STAT SH2. Tyrosine phosphorylation occurs after STAT binds to the receptor. Tyrosine phosphorylated STAT forms a dimer and enters the nucleus. As an active transcription factor, dimeric STAT molecules directly affect the expression of related genes, thereby changing the proliferation or differentiation status of target cells.
The JAK-STAT pathway is widely present in various tissues and cells in the body, and has an important role in the differentiation, proliferation, and anti-infection of lymphocytes, and participates in the interaction of various inflammatory factors and signal transduction (Kiesseleva T. et al. . J. Gene, 2002, 285, 1-24). The abnormal activation of this pathway is closely related to a variety of diseases. Finding and screening JAK inhibitors can help in-depth study of the regulatory mechanism of JAK-STAT, thereby providing new drugs and methods for the prevention and treatment of related diseases
The occurrence, growth, invasion and metastasis of tumors are related to the JAK-STAT signal transduction pathway. In normal signal transduction, the activation of STATs is rapid and transient. The continuous activation of STATs is closely related to the process of malignant transformation of cells (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 is the focus of multiple oncogenic tyrosine kinase signal channels such as EGFR, IL-6/JAK, Src, etc. It is activated in a variety of tumor cells and tissues, such as breast cancer, ovarian cancer, and head and neck squamous cells. Like cell carcinoma, prostate cancer, malignant melanoma, multiple myeloma, lymphoma, brain tumor, non-small cell lung cancer and various leukemias, etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008 ). JAK-STAT pathway inhibitors belong to PTK inhibitors, and this enzyme is a member of the oncogene protein and proto-oncoprotein family, and plays an important role in the normal and abnormal cell proliferation. The occurrence and growth of tumors are inseparable from PTK. Therefore, JAK-STAT pathway inhibitors inhibit tumor growth by antagonizing PTK, and have obvious anti-tumor effects (Mora LBet al.J.Cancer Res.2002,62(22) , 6659-6666).
In addition, the latest research shows that: organ transplant rejection, psoriasis, tissue and organ fibrosis, bronchial asthma, ischemic cardiomyopathy, heart failure, myocardial infarction, blood system diseases, and immune system diseases are all related to JAK-STAT signaling. The pathway is closely related. This signaling pathway is not only important for maintaining the normal physiological functions of cells, but also has an important regulatory role for the occurrence and development of diseases.
The Fibroblast Growth Factor Receptor family belongs to a new type of receptor kinase family, which includes four receptor subtypes (FGFR-1,2,3) encoded by four closely related genes. And 4) and some heterogeneous molecules, which form a ternary complex with fibroblast growth factor (FGF) and heparan sulfate, and then trigger a series of signal transduction pathways to participate in the regulation of physiological processes in the organism. FGFR has a wide range of physiological and pathological effects in the body: (1) Embryo development. Studies have shown that during embryonic development, FGFR signal transduction is essential for most organ development and the formation of embryonic patterns. (2) Cell division, migration and differentiation. FGFR stimulates cell proliferation and participates in the regulation of cell transformation in the pathological process. There are many parallel pathways to achieve FGFR-mediated cell division signal transduction, which has been confirmed by many studies (JKWang et al., Oncogene 1997, 14, 1767 -1778.). (3) Bone diseases. The growth and differentiation of bones are also regulated by the FGF family, and mutations in FGFR can cause bone deformities (R. Shang et al., Cell 1994, 78, 335-342.). (4) The occurrence of tumors. FGFR can promote the migration, proliferation and differentiation of endothelial cells, and plays an important role in the regulation of angiogenesis and angiogenesis. Uncontrolled angiogenesis can lead to the occurrence of tumors and the growth of metastases (J.Folkman.Nat.Med.1995) ,1,27-31.).
FMS-like tyrosine kinase 3 (FMS-like tyrosine kinase 3, FLT3) belongs to the type III receptor tyrosine kinase (receptor tyrosine kinase III, RTK III) family member, it is composed of extracellular domain, intracellular domain and The transmembrane region is composed of 3 parts, which are first expressed in human hematopoietic stem cells. FLT3 interacts with its ligand FL to stimulate or act on stem cells, which is of great significance to the growth and differentiation of stem cells. FLT3 kinase has wild-type FLT3-WT and its main activation mutant FLT3-ITD and FLT3-D835Y. FLT3 is mainly expressed in the precursors of normal myeloid cells, but its abnormal expression is also found in a large part of acute myeloid leukemia (AML) cells.
In recent years, many large-scale studies have confirmed that activating mutations of FLT3 play a very important pathological role in the occurrence and progression of acute myeloid leukemia. FLT3 has become an important target for the treatment of acute myeloid leukemia.
rc family kinase (SFK) is a family of non-receptor tyrosine kinases, including c-Src, LYN, FYN, LCK, HCK, FGR, BLK, YES and YRK, among which LYN kinase has LYNα and LYNβ Both subtypes, LYN kinase and its two subtypes can cause similar intracellular tyrosine phosphorylation. According to the amino acid sequence, SFK can be divided into two sub-families: one family is c-Src, FYN, YES and FGR, which are widely expressed in different tissues; the other family is LCK, BLK, LYN and HCK, which are closely related to hematopoietic cells. SFK is connected to multiple signal transduction pathways in the body, and can be activated by growth factors, cytokines and immune cell receptors, G protein-coupled receptors, integrins and other cell adhesion molecules, and then activate the corresponding signal transduction pathways , Causing a variety of physiological effects of cells. The activity of SFK mainly includes the regulation of cell morphology, cell movement, cell proliferation and survival. The abnormal activation and expression of these kinases leads to the occurrence and development of a wide range of diseases, such as a large number of solid tumors, various hematological malignancies and some neuronal pathologies. Therefore, looking for SFK inhibitors is a promising research topic in the field of medicinal chemistry.


NEW DRUG APPROVALS
ONE TIME
$10.00
Patent
CN106366093A
PATENT
WO 2017012559
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559Example 31
N-[7-(4-Fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)- 1H-pyrazole-4-amine (Compound 31)
Synthesis of compound 31-e
2,4-Dichloro-6-methylthiophene [3,2-d] pyrimidine (10g, 45.6mmol) was dissolved in tetrahydrofuran (100mL) and ethanol (100mL), and the reaction solution was cooled to 0°C and divided Sodium borohydride (12.5 g, 198 mmol) was added in batches. The reaction solution was raised to room temperature and continued to stir for 16 hours, diluted with water (500 mL), and then adjusted to pH=7 with 1N aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate (150 mL×3). The organic phase was washed sequentially with water (100mL×3) and saturated brine (100mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-e (7.5g, yield: 88%). The product does not require further purification. LC-MS(ESI): m/z=187[M+H] + .[0492]Synthesis of compound 31-d[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0°C, active manganese dioxide (35 g, 400 mmol) was added, the reaction solution was raised to room temperature and stirring was continued for 16 hours. The reaction solution was filtered through Celite, and the filter cake was washed with chloroform (100 mL×3). The combined filtrates were concentrated under reduced pressure to obtain white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS(ESI): m/z=185[M+H]+.[0494]Synthesis of compound 31-c[0495]Compound 31-d (3.1g, 16.8mmol) was dissolved in trifluoroacetic acid (30mL) at 0℃, N-iodosuccinimide (5.7g, 25.3mmol) was added in batches, and the reaction solution was raised to Keep stirring at room temperature for 1 hour. Water (50 mL) was added to the reaction solution to quench the reaction, and it was extracted with dichloromethane (50 mL×3). The organic phase was washed successively with water (50mL×3) and saturated brine (50mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-c (4.9g, yield: 94%). The product does not require further purification. LC-MS(ESI): m/z=311[M+H] + .[0496]Synthesis of compound 31-b[0497]Compound 31-c (615mg, 1.98mmol), 2-methoxy-4-fluorophenylboronic acid (405mg, 2.38mmol) and sodium carbonate (630mg, 5.94mmol) were suspended in dioxane (5mL) water (5mL) ), add [1,1′-bis(diphenylphosphorus)ferrocene]dichloropalladium dichloromethane complex (163mg, 0.2mmol). Replace with nitrogen 3 times, and heat to 80°C to react for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was partitioned with dichloromethane (50mL) and water (50mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum Ether: dichloromethane=1:1) to obtain a white solid 31-b (240 mg, yield: 39%). LC-MS(ESI): m/z=309[M+H] + .[0498]Synthesis of compound 31-a[0499]Compound 31-b (240mg, 0.78mmol) and compound 32-c (208mg, 0.78mmol) were dissolved in N,N-dimethylformamide (3mL), potassium carbonate (323mg, 2.34mmol) was added, 2- Dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol) and tris(dibenzylideneacetone) dipalladium (134 mg, 0.24 mmol). Under the protection of nitrogen, heat to 110°C to react for 16 hours. After cooling to room temperature, the reaction solution was partitioned with dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin layer chromatography preparation plate (petroleum Ether: ethyl acetate = 1:1) to obtain a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z=539[M+H] + .[0500]Synthesis of compound 31[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), trifluoroacetic acid (3 mL) was added, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. The residue was layered with ethyl acetate (50mL) and 1N aqueous hydrochloric acid (50mL). The aqueous phase was adjusted to pH=10 with saturated aqueous potassium carbonate solution. 3) Washing and vacuum drying the solid to obtain a light yellow solid 31 (22 mg, yield: 14%). LC-MS(ESI): m/z=439[M+H] + .[0502]1 H-NMR (400MHz, MeOD) δ: 8.78 (d, J = 5Hz, 1H), 7.87 (s, 1H), 7.48 (s, 1H), 7.35 (m, 1H), 7.05 (dd, J = 11Hz) ,J = 2Hz, 1H), 6.91 (m, 1H), 4.10 (m, 1H), 3.79 (s, 3H), 3.22 (m, 2H), 2.77 (m, 2H), 2.47 (s, 3H), 2.03(m,2H),1.73(m,2H)ppm
PATENT
WO 2019228171
Example 1 Preparation of fumarate of fused ring pyrimidine compound as shown in formula 2
Weigh the compound N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidine-4- Base)-1H-pyrazol-4-amine (synthesized according to Example 31 of patent CN106366093A) 100mg (0.228mmol, 1eq) into the vial, add 10mL 88% acetone-water solution, add the vial at about 50°C and stir until dissolved clear. 1.1 mL of fumaric acid with a concentration of 0.25 mol/L in ethanol (0.275 mmol, 1.2 eq) was slowly added dropwise to the free base solution of fused ring pyrimidine compounds, and stirred at 50 ℃ for 1 hour, and then the solution was The rate of 5°C/h was slowly reduced to room temperature, and the solid was collected and dried under vacuum at 40°C overnight.
1 H-NMR (400MHz, DMSO-d 6 ) δ: 9.45 (s, 1H), 8.94 (s, 1H), 7.75 (s, 1H), 7.78-7.33 (m, 2H), 7.15 (d, J = 6.4Hz, 1H), 6.99 (dd, J = 7.6 Hz, J = 7.2 Hz, 1H), 6.42 (s, 1H), 4.10 (m, 1H), 3.73 (s, 3H), 3.17 (d, J = 12.4 Hz, 2H), 2.77 (dd, J = 12.4 Hz, J = 11.6 Hz, 2H), 2.40 (s, 3H), 1.94 (d, J = 11.6 Hz, 2H), 1.73 (m, 2H) ppm.
PATENT
7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-yl)-1hydro-pyrazol-4-yl)thieno[3,2 -D]pyrimidine-2-amino is a strong JAK, FGFR, FLT3 kinase inhibitor, and has a good application prospect in the treatment of tumors, immune system diseases, allergic diseases and cardiovascular diseases. This compound is described in patent CN106366093A and has the following chemical structure:
CN106366093A discloses the preparation method of the compound:
In the above synthetic route, NaBH 4 is sodium borohydride, MnO 2 is manganese dioxide, NIS is N-iodosuccinimide, TFA is trifluoroacetic acid, and Pd(dppf)Cl 2 is [1,1′- Bis(diphenylphosphino)ferrocene]palladium dichloride, DIAD is diisopropyl azodicarboxylate, PPh 3 is triphenylphosphine, Pd/C is palladium on carbon, Pd 2 (dba) 3 is Tris(dibenzylideneacetone)dipalladium, RuPhos is 2-bicyclohexylphosphine-2′,6′-diisopropoxybiphenyl.
However, the above method has the problems of a large number of reaction steps, low yield, and requires column chromatography for separation and purification, and is not suitable for industrial scale-up production. Therefore, it is necessary to improve its preparation method.
The present invention provides a method for preparing a compound represented by formula B, which comprises the following steps: under a protective gas atmosphere, in a solvent, in the presence of a catalyst and a base, a compound represented by formula C is combined with a compound represented by formula K The compound can be subjected to the coupling reaction shown below; the catalyst includes a palladium compound and a phosphine ligand;
The preparation method of the compound represented by formula B may further include the following steps: in an organic solvent, in the presence of a base, the compound represented by formula E and the compound represented by formula D are subjected to the substitution reaction shown below, To obtain the compound represented by formula C;
The present invention provides a method for preparing a compound represented by formula C, which comprises the following steps: in an organic solvent, in the presence of a base, a compound represented by formula E and a compound represented by formula D are subjected to the following steps: Substitution reaction is enough;
Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 500L reactor, add 10% palladium on carbon (4.6Kg), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (24.2Kg, 109.5mol), and tetrahydrofuran (150Kg) in sequence And N,N-diisopropylethylamine (17.0Kg, 131.5mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.5 MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 120 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (58Kg) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (60Kg) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 360Kg of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was separated out, centrifuged, and the filter cake was vacuum dried to obtain the product 2-chloro-6-methylthieno[3,2-D]pyrimidine 18.94Kg, yield: 93.2%. LC-MS(ESI): m/z=185.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.30 (s, 1H), 7.34 (s, 1H), 2.73 (s, 3H).
Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
To a 100mL reaction flask, add 10% palladium on carbon (0.17g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), tetrahydrofuran (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 20 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 2.4 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 82%. The LC-MS and 1 H NMR are the same as in Example 1.
Example 3: 7-Bromo 2-chloro-6-methylthieno[3,2-D]pyrimidine (Compound E)
Add trifluoroacetic acid (150Kg) and 2-chloro-6-methylthieno[3,2-D]pyrimidine (18.90Kg, 102.4mol) into a 500L enamel reactor. Add N-bromosuccinimide (18.33Kg, 103.0mol) under temperature control at 15±5℃. After the addition, the temperature is controlled at 25±5℃ to react for 2 hours. Sampling to monitor the reaction, there is still a small amount of raw materials remaining. Additional N-bromosuccinimide (1.0 Kg, 5.6 mol) was added, stirring was continued for 1 hour, sampling and monitoring showed that the reaction was complete. Control the temperature at 10±5°C, and add 274Kg of water dropwise. After the addition, stir at 10±5°C for 2 hours. After centrifugation, the solid was vacuum-dried to obtain the product, 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine, 24.68Kg, yield: 91.4%. LC-MS(ESI): m/z=265.0[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.33 (s, 1H), 2.64 (s, 3H).
Example 4: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Add pyridine (176Kg) and N-BOC-4-hydroxypiperidine (36.00Kg, 178.9mol) to a 500L enamel reactor. Add p-toluenesulfonyl chloride (50.5Kg, 264.9mol) in batches under temperature control at 10±10°C. After the addition, the temperature is controlled at 25±5°C to react for 18 hours. The reaction solution was transferred to a 1000L reactor, the temperature was controlled at 15±5°C, and 710Kg of water was added dropwise. After the addition, stir at 15±5°C for 2 hours. After filtration, the solid was washed with water and dried in vacuum to obtain the product 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester, 59.3Kg, yield: 93.3%. LC-MS(ESI): m/z=378.0[M+Na] + .
Example 5: 4-(4-Nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound F)
Add N,N-dimethylformamide (252Kg), 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester (59.3Kg, 166.8mol), 4-nitro to the reaction kettle Pyrazole (21.5Kg, 190.1mol), and anhydrous potassium carbonate (34.3Kg, 248.2mol). The temperature was controlled at 80±5°C and the reaction was stirred for 18 hours. Cool down to 15±5°C, add 900Kg of water dropwise, control the dropping rate, and keep the temperature at 15±5°C. After the addition, stir at 5±5°C for 2 hours. After filtering, the solid was washed twice with water and dried in vacuum to obtain the product 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 39.92Kg, yield: 80.8%. LC-MS (ESI): m/z=319.1 [M+Na] + .
1 H NMR (400MHz, d 6 -DMSO): δ8.96(s,1H), 8.27(s,1H), 4.44-4.51(m,1H), 4.06-4.08(m,2H), 2.75-2.91( m, 2H), 2.04-2.07 (m, 2H), 1.80-1.89 (m, 2H), 1.41 (s, 9H).
Example 6: 4-(4-Amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound D)
Add 10% palladium-carbon (2.00Kg), 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (39.94Kg, 134.09mol) to the reaction kettle, nothing Water ethanol (314Kg) and ammonia (20.0Kg, 134.09mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.2MPa. Turn on the stirring and keep the temperature at 45±5°C to react for 4 hours. Filter, collect the filtrate, and concentrate the filtrate under reduced pressure. Add ethyl acetate (40Kg) and n-heptane (142Kg) to the concentrate, stir at 25±5°C for 1 hour, and then lower the temperature to 5±5°C and stir for 2 hours. After filtration, the solid was vacuum dried to obtain the product 4-(4-amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 31.85Kg, yield: 88.6%. LC-MS(ESI): m/z=267.2[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ7.06 (s, 1H), 6.91 (s, 1H), 4.08-4.15 (m, 1H), 3.98-4.01 (m, 2H), 3.81 (brs, 2H), 2.83-2.87 (m, 2H), 1.88-1.91 (m, 2H), 1.63-1.72 (m, 2H), 1.41 (s, 9H).
Example 7: 4-(4-(7-Bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl)piperidine-1 -Tert-butyl carbonate (compound C)
Add n-butanol (117Kg), N,N-diisopropylethylamine (15.00Kg, 116.06mol), 4-(4-amino-1hydro-pyrazol-1-yl)piperidine to the reaction kettle 1-tert-butyl carbonate (32.02Kg, 120.22mol) and 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine (24.68Kg, 93.65mol). Turn on the stirring and keep the temperature at 100±5°C to react for 42 hours. Concentrate under reduced pressure. Methanol was added to the concentrate to be beaten. The solid was filtered and dried under vacuum to obtain the product 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl ) Piperidine-1-tert-butyl carbonate 37.26Kg, yield: 80.6%. LC-MS(ESI): m/z=493.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.73 (s, 1H), 8.97 (s, 1H), 8.18 (s, 1H), 7.68 (s, 1H), 4.30-4.36 (m, 1H) ,4.01-4.04(m,2H),2.87-2.93(m,2H),2.53(s,3H),2.00-2.03(m,2H),1.70-1.80(m,2H),1.41(s,9H) .
Example 8: 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1 Hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound B)
Add purified water (113Kg), dioxane (390Kg), 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) into the reactor -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (37.26Kg, 93.65mol), 2-methoxy-4-fluorophenylboronic acid pinacol ester (23.05Kg, 120.22mol) , Anhydrous potassium carbonate (20.95Kg, 151.8mol), palladium acetate (0.18Kg, 0.80mol) and 2-dicyclohexylphosphine-2,4,6-triisopropylbiphenyl (0.90Kg, 1.89mol). Under the protection of nitrogen, the temperature is controlled at 70±5℃ to react for 4 hours. Cool down to 40±5°C, add ammonia water (68Kg), and stir for 8 hours. Cool down to 20±5°C and dilute with water (1110Kg). Dichloromethane extraction twice (244Kg, 170Kg). Combine the organic phases, wash sequentially with water and then with saturated brine. Add 3-mercaptopropyl ethyl sulfide-based silica (4.0Kg, used to remove heavy metal palladium) into the organic phase, and stir at 40±5°C for 20 hours. After filtration, the filtrate was concentrated under reduced pressure. The remainder was slurried sequentially with methyl tert-butyl ether and ethanol. Filter and dry in vacuo to obtain 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 34.6Kg, yield: 68.6%. LC-MS(ESI): m/z=539.3[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.46 (s, 1H), 8.94 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.33 to 7.35 (m, 1H) ,7.08-7.11(m,1H),6.91-6.95(m,1H),4.03-4.12(m,3H),3.73(s,3H),2.85-2.89(m,2H),2.39(s,3H) ,1.90-1.93(m,2H),1.55-1.60(m,2H),1.41(s,9H).
Comparative Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 100mL reaction flask, add 10% palladium on carbon (0.1g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), methanol (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 21 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 1.6 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 54%. Methoxy substituted impurities in 20% yield.
Comparative Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
After replacing the solvent tetrahydrofuran in Example 2 with ethyl acetate, the solubility of 2-chloro-6-methylthieno[3,2-D]pyrimidine in ethyl acetate was poor, and only a small amount of product was formed, which was not calculated Specific yield.
Comparative example 3: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Triethylamine (25mL), N-BOC-4-hydroxypiperidine (5g) were added to a 100mL reaction flask. P-toluenesulfonyl chloride (7.1g) was added in batches while controlling the temperature at 10±10°C. After the addition, the temperature is controlled at 25±5℃ to react for 25 hours. Monitoring by LC-MS showed a large amount of unreacted raw materials and the reaction liquid was black and red.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019228171-A1 | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof | 2018-05-31 | |
| AU-2016295594-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| AU-2016295594-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2020-04-16 |
| EP-3354653-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| EP-3354653-B1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-09-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2018520202-A | Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereof | 2015-07-21 | |
| KR-20180028521-A | Condensed ring pyrimidine-based compounds, intermediates, methods for their preparation, compositions and applications | 2015-07-21 | |
| US-10494378-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-12-03 |
| US-2018208604-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| WO-2017012559-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03412292 | MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML) | Phase 1 | Recruiting | 2021-05-21 |
///////////////Orphan Drug, Acute myeloid leukaemia, MAX 40279, EX-A4057, Max 4, MAX-40279, MAX-40279-001, MAX-40279-01, PHASE 1, Maxinovel Pharmaceuticals
CC1=C(C2=NC(=NC=C2S1)NC3=CN(N=C3)C4CCNCC4)C5=C(C=C(C=C5)F)OC
AKN 028

AKN-028
CAS 1175017-90-9
Chemical Formula: C17H14N6
Molecular Weight: 302.33
N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine
N2-(1H-indol-5-yl)-6-(pyridin-4-yl)pyrazine-2,3-diamine
- Originator Swedish Orphan Biovitrum
- Developer Akinion Pharmaceuticals
- Class Antineoplastics; Small molecules
- Mechanism of Action Fms-like tyrosine kinase 3 inhibitors; Proto oncogene protein c-kit inhibitors
- Phase I/II Acute myeloid leukaemia
- 01 Mar 2016 Akinion Pharmaceuticals terminates phase I/II trial in Acute myeloid leukaemia in Czech Republic, Poland, Sweden and United Kingdom (NCT01573247)
- 17 Sep 2015 AKN 028 is still in phase I/II trials for Acute myeloid leukaemia in Czech Republic, Poland and Sweden
- 09 Apr 2014 AKN 028 is still in phase I/II trials for Acute myeloid leukaemia in Czech Republic, Poland and Sweden
AKN-028, a novel tyrosine kinase inhibitor (TKI), is a potent FMS-like receptor tyrosine kinase 3 (FLT3) inhibitor (IC(50)=6 nM), causing dose-dependent inhibition of FLT3 autophosphorylation. Inhibition of KIT autophosphorylation was shown in a human megakaryoblastic leukemia cell line overexpressing KIT. In a panel of 17 cell lines, AKN-028 showed cytotoxic activity in all five AML cell lines included. AKN-028 triggered apoptosis in MV4-11 by activation of caspase 3. In primary AML samples (n=15), AKN-028 induced a clear dose-dependent cytotoxic response (mean IC(50) 1 μM). However, no correlation between antileukemic activity and FLT3 mutation status, or to the quantitative expression of FLT3, was observed. Combination studies showed synergistic activity when cytarabine or daunorubicin was added simultaneously or 24 h before AKN-028. In mice, AKN-028 demonstrated high oral bioavailability and antileukemic effect in primary AML and MV4-11 cells, with no major toxicity observed in the experiment. (source: Blood Cancer J. 2012 Aug 3;2:e81. doi: 10.1038/bcj.2012.28.)
SYN
WO 2013/089636

Clip
Development of a Synthesis of Kinase Inhibitor AKN028


The novel tyrosine kinase inhibitor AKN028 has demonstrated promising results in preclinical trials. An expedient protocol for the synthesis of the compound at kilogram scale is described, including an SNAr reaction with high regioselectivity and a Suzuki coupling. Furthermore, an efficient method for purification and removal of residual palladium is described.
yellow or faint-orange powder. Mp 300 °C (dec.);
IR 3133 broad, 1689, 1597, 1554, 1480 cm–1; 1H NMR (DMSO-d6) δ 11.01 (s, 1H), 8.62–8.50 (m, 2H), 8.22 (s, 1H), 8.15 (s, 1H), 8.06 (s, 1H), 7.89–7.82 (m, 2H), 7.39 (d, J = 2.0 Hz, 2H), 7.32 (t, J = 2.7 Hz, 1H), 6.77 (s, 2H), 6.42 (dd, J1 = 8.7 Hz, J2 = 2.0 Hz, 1H);
13C NMR (DMSO-d6) δ 149.9, 145.2, 145.0, 139.6, 132.8, 132.4, 132.2, 128.4, 127.6, 125.6, 118.7, 116.1, 111.2, 111.0, 101.0.
PATENT
WO 2009095399
PATENT
WO 2013089636
https://patents.google.com/patent/WO2013089636A1/ko
Protein kinases are involved in the regulation of cellular metabolism, proliferation, differentiation and survival. The FLT-3 (fms-like tyrosine kinase) receptor is a member of the class III subfamily of receptor tyrosine kinases and has been shown to be involved in various disorders such as haematological disorders, proliferative disorders, autoimmune disorders and skin disorders.
In order to function effectively as an inhibitor, a kinase inhibitor needs to have a certain profile regarding its target specificity and mode of action. Depending on factors such as the disorder to be treated, mode of administration etc. the kinase inhibitor will have to be designed to exhibit suitable properties. For instance, compounds exhibiting a good plasma stability are desirable since this will provide a pharmacological effect of the compounds extending over time. Another example is oral administration of the inhibitor which may require that the inhibitor is transformed into a prodrug in order to improve the bioavailability.
WO 2009/095399 discloses pyrazine compounds acting as inhibitors of protein kinases, especially FTL3, useful in the treatment of haematological disorders, proliferative disorders, autoimmune disorders and skin disorders. This document discloses methods for manufacturing such compounds. However these methods are not suitable for large scale processes and the chemical yields are moderate. Furthermore, the compounds obtained by these methods are in amorphous form.
n one aspect of the invention, there is provided a process for preparing a compound of formula (I)
said process comprises the steps of:
a) reacting a compound of formula (1) with a compound of formula (2) in an inert solvent and in the presence of an (C1-6alkyl)3amine, providing a compound of formula (3):
, b) Suzuki coupling of a compound of formula (3) and a compound of formula (4) in an inert solvent and in the presence of a palladium catalyst and a base, providing a crude product comprising a compound of formula (I) and palladium
and
c) removing the palladium from the crude product in step b).
The compound of formula (I) may be obtained in amorphous or crystalline form using the processes outlined below.
Step 1:
Reaction of 2-amino-3,5-dibromopyrazine (1) and 5-aminoindole (2) in a
nucleophilic substitution reaction in the presence of a C1-6alkylamine and an inert polar solvent yields 3-bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine (3). Examples of inert polar solvents are DMSO, water and NEP. Examples of (C1-6alkyl)3amine are triethylamine, trimethylamine and tributylamine. The reaction may be performed at reflux temperature or at about 100-130°C.
Step 2:
A Suzuki coupling of 3-bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine) (3) and 4- pyridyl-boronic acid (4) in an inert polar solvent in the presence of a palladium catalyst and a base yields N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine (I) in amorphous form. Examples of inert solvents are DMF, water and DMA. Examples of palladium catalysts are Pd(dppf) and Pd(OAc)2-DTB-PPS. Example of a base is
K2CO3 The reaction may be performed under inert and oxygen-free atmosphere such as nitrogen or argon.
Heating may take place during step 1 and/or step 2. Steps 1 and 2 may be performed at reflux or in a temperature range of from 100 to 140°C, such as from 105 to 135°C, such as from 110 to 130°C, such as from 130-135°C, such as from 110-115ºC.
Step 3:
A compound of formula (I), also denominated N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine, in amorphous form may be dissolved in acetic acid (HOAc) after which potassium hydroxide (KOH) is added. The compound of formula (I) in amorphous form may be obtained from the process outlined in steps 1 and 2.
Alternatively, the compound of formula (I) may be obtained according to the process described in WO 2009/095399. The obtained crystalline form is removed from the slurry by, for instance, filtration. Step 3 may be repeated. Step 3 may be performed at a temperature of about 40°C followed by cooling to room temperature.
The process for preparing a compound according to formula (I) may comprise an additional step (step i) between step 2 and step 3 in order to remove palladium from the crude product of the compound of formula (I). The step comprises; forming a slurry comprising an acid and the compound according to formula (I) in a solvent, adding a siloxane compound to said slurry, removing the solvent from the slurry and adding an organic solvent, such as DMF and/or toluene, to the solid formed whereby a mixture is formed and then potassium hydroxide is added to the formed mixture, Alternatively, palladium may be removed from the crude product comprising (I) using a palladium scavenger such as TMT and/or 3-mercaptopropyl ethyl sulfide silica.
The crystalline form of the compound according to formula (I) may also be prepared from an amorphous form of the compound according to formula (I) by dissolving said amorphous form of the compound in a solvent mixture of
dichloromethane/methanol followed by evaporation of the solvent in a rotary evaporator. The amorphous form of the compound of formula (I) may obtained using the process disclosed in WO 2009/095399.
Example 1. Preparation of 5-Bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine (compound 3)
DMSO (10 L, 11 kg), 2-amino-3,5-dibromopyrazine (1) (4.5 kg, 17.8 mol, 1 eq.), 5- amino indole (2) (3.06 kg, 23.15 mol, 1.3 eq.) and triethylamine (7.4 L, 5.4 kg, 53.36 mol, 3 eq.) were charged to a reactor. The reaction mixture was heated to 95°C while agitated. After 12 hours, the heating was discontinued and the conversion was 88% of 2-amino-3,5-dibromopyrazine. The reaction was heated again to 95°C and
agitated for an additional 2.5 hours. There was no improvement in conversion. The reaction mixture was agitated at ambient temperature overnight. Triethylamine (3.5 kg) was removed under vacuum and the remaining reaction mixture was transferred to a stainless steel container from which it was charged into another reactor.
Subsequently, 18.4 kg of 50% acetic acid (aq.) was introduced over a period of 20 minutes under agitation, followed by purified water (61 L) charged over a period time of 60 minutes. The slurry was then filtered and the isolated material was washed with 2 x 20 L of 1% acetic acid (aq.).
The isolated 3-bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine) (3) was transferred to a drying cabinet and dried to invariable weight at 40 ±3°C, (19 hours), to afford 4.36 kg, 14.34 mol, 81 % yield, with a purity of 96% by HPLC.
The reaction temperature in the batch record was set to be 130-135°C. However, at 95°C the reaction mixture was at reflux.
Example 2. Preparation of N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3- diamine (compound I)
To a reactor was charged N,N-dimethylformamide (46.7 L, 45 kg), 4-pyridylboronic acid (4) (2.64 kg, 21.5 mol, 1.5 eq.) and 5-bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3- diamine (3) (4.36 kg, 14.3 mol). The reactor was then flushed with nitrogen prior to the charging of Pd(dppf)Cl2-catalyst (0.47 kg, 0.55 mol, 0.04 eq.). To reactor was then charged, over a period of 20 minutes, 24.9 kg of a 2 M solution of potassium carbonate (aq.). The reactor was flushed with nitrogen and heated under agitation to 110-115°C for 1.5 hours, after which 98.3% conversion of (3) was showed. The reaction mixture was quenched by addition of purified water (180 L) under vigorous agitation. The precipitated material was isolated on a hastalloy filter and washed with purified water (50 L), The isolated material was transferred to a drying cabinet and dried to invariable weight at 40 ±3°C (18 hours), to afford a compound of formula (5), i.e. a compound of formula (!) also denominated N-3-(1H-lndol-5-yl)-5-pyhdin-4-yl-pyrazine-2,3-diamine, (3.64 kg, 12.1 mol, 85 % yield).
During the process precipitated material was observed in the solutions, after the reactions, in both steps not previously seen in lab-scale. These impurities were not removed.
Example 3. Purification and crystallisation
In order to remove residual solvents from the material, two consecutive re-precipitations of the material from acetic acid were performed. This also gave crystallinity of the isolated substance. The purification is performed in order to remove palladium.
Purification
To a 1 L round bottomed flask was added 37.8 g of a compound according to formula (I) followed by 600 mL 2 M HOAc (aq.). The material was stirred at RT until a clear, dark red solution was obtained. To the solution was added 30 g Hyflo Super Celite and the slurry was filtered. The filter cake was washed with 25 mL 2 M HOAc
(aq) and 2×35 mL purified water. The obtained filtrate was transferred to a 2 L round bottomed flask containing 950 mL of Me-THF. The mixture was then stirred and heated to 40°C for 30 minutes. To the solution was then added 290 mL 8 M KOH (aq.) at 40°C and pH in the solution was 14.
The aqueous phase was removed and the organic phase washed with 2×100 mL of purified water. The remaining organic phase was then transferred to a 2 L round bottomed flask, followed by 95 mL of DMF, 20 g scavenger 3-Mercaptopropyl ethyl sulphide silica, Phosphonics LTD and 20 g scavenger 2-Mercaptoethyl ethyl sulfide silica purchased from Phosphonics LTD. The solution was vigorously stirred and heated at 60°C. A sample was withdrawn from the slurry after 12 hours, and showed 6 ppm of palladium remaining in the solution. The mixture was allowed to cool and was then filtered to remove the scavenger. The round bottomed flask and filter were rinsed with a mixture of 90 mL Me-THF and 10 mL DMF. Me-THF was then removed on a rotary evaporator and the remaining slurry was azeotropically dried with two portions of 100 mL toluene. To the remaining slurry was then added 85 mL of DMF to a total of 185 mL DMF (5ml DMF/g substance). To the clear solution was then added, slowly, while agitated, 1500 mL of toluene which produced a heavy precipitate. The slurry was filtered off and washed with 2×50 mL of toluene where after the material was dried overnight at 35°C under vacuum to afford 30.9 g of a compound according to formula (I) in a yield of 82%.
Crystallisation:
Example i
1. First re-precipitation
The N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine material (30.9 g) was added to a 1 L round bottomed flask and 450 mL 2 M HOAc (aq.) was added. The slurry was agitated and heated to 40°C for 1 hour, until the material had dissolved. To the solution was then added 158 mL 8 M KOH (aq.) at 40°C. The pH in the solution was 11.4. The slurry was then allowed to cool to 25°C and filtered. The filter cake was washed with 3x 80 mL of purified water and the material was dried overnight at 95°C under vacuum to afford 28.7g N-3-(1H-indol-5-yl)-5-pyridin-4-yl- pyrazine-2,3-diamine in a yield of 93%.
2. Second re-precipitation
N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine material (28.7 g) was added to a 1L round bottomed flask and 430 mL 2 M HOAc (aq) was added. The slurry was agitated and heated to 40°C for 1 hour, until the material had dissolved. To the solution was then added 15 mL 8M KOH (aq) at 40°C. The pH in the solution was 12.3. The slurry was then allowed to cool to 25°C and filtered. The filter cake was washed with 5×50 mL of purified water, and the solid was then dried overnight at 95°C under vacuum to afford 28.3 g N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3- diamine in a yield of 99%.
Example ii
The N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine material (2.1 kg, 7 mol) was added to a reactor, followed by 2M HOAc (aq.) (59.6 L, 60.2 kg) . The solution in the reactor was then heated to 40°C and stirred for 20 minutes. To the clear solution was then charged, slowly, 30% KOH (aq.) (25 kg) under vigorous agitation. The slurry was agitated for 15 minutes. pH in the solution was 6.2, and a total of 1.5 kg 30% KOH (aq.) was then added to the solution to give pH 12.1. The precipitated material was isolated on a Hastelloy filter and washed with purified water (5×30 L). The solid was then transferred to a drying cabinet and dried to invariable weight at 85 ±3°C under vacuum (16 hours; a sample was withdrawn after 16 hours, showing 1400 ppm HOAc and 75 ppm DMF), to afford N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine (2.0 kg, 7 mol, 95 % yield).
Hence, N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine is obtained in an uniform crystalline form, which was achieved by precipitating the product from aqueous acetic acid by introduction of aqueous potassium hydroxide.
Example 5. Synthesis of 5-Bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine (compound 3)
2-Amino-3,5-dibromopyrazine (45 g, 1.0 eq.), 5-aminoindole (30,6 g, 1.3 eq.), 67.5 mL NEP, i.e. 1-ethyl-2-pyrrolidone, and 74.5 mL triethylamine were added to a 250 mL reactor. The jacket temperature was set to 130°C and the reaction mixture was stirred for 22 h. HPLC after 22 h showed 87% conversion of the 2-amino-3,5-dibromopyrazine. After 24 h HPLC showed 92% conversion and the reaction slurry was cooled to 80°C and quenched by addition of addition of 50% HOAc(aq) and water. The obtained slurry was then allowed to cool to room temperature over night while agitated. The material was isolated on a glass filter funnel and was washed with water. The material was dried at 80 °C under vacuum until dry to afford 71% of the compound 5-bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine as a dark brown powder. The purity was 99.8% as measured by HPLC.
Example 6. Synthesis of N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine (Compound I)
5-Bromo-N-3-(1H-indol-5-yl)-pyrazine-2,3-diamine (15.0 g, 49 mmol, 1.0 eq.), 4-pyridyl boronic acid (6.6 g, 59 mmol, 1.2 eq.), Pd(OAc)2 (166 mg, 0.74 mmol, 0.015 eq.), DTB-PPS, i.e. 3-(di-tert-butylphosphino)propane-1-sulfonic acid, (199 mg, 0.74 mmol, 0.015 eq.), and DMA, i.e. N,N-dimethylacetamide, (75 mL) were added to a three-necked round-bottomed flask equipped with a mechanical stirrer,
thermometer, and a nitrogen atmosphere. Through a septa was added 2M K2CO3 (aq) (27 ml, 54 mmol, 1.1 eq.) with a syringe. The temperature was increased to 100 °C. Samples for HPLC-analysis of the conversion were drawn and when the conversion had reached 100% the temperature was cooled to 25 °C. At that temperature a water solution of 0.5 M L-cysteine (150 ml) was added by a syringe pump over 1 hour with a rate of 2.5 mL/minute. After 3 hours maturing time at room temperature the material was isolated on a glass filter funnel and was washed with water. The material was dried at 40 °C under vacuum over the weekend, and 15 grams of N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine (101%) were obtained as a brown powder.
Example 7. Purification of N-3-(1H-indol-5-yl)-5-pyridin-4-yl-pyrazine-2,3-diamine (Compound I)
The crude (7.0 g, 23 mmol) and 2M HOAc (98 mL) was added to a 250 mL round-bottomed flask. To this was added TMT, i.e. trithiocyanuric acid, (1.4 g) and SPM32, i.e. 3-mercaptopropyl ethyl sulfide silica, (1.4 g). The mixture was stirred in room temperature for 24 hours. After 24 hour a polish filtration through hyflo super cel was performed. To the clear filtrate was added 50 mL 5 M KOH(aq) under 15 minutes to precipitate the product. After 18 hours maturing time at room temperature the material was isolated on a glass filter funnel and was washed with 2×20 mL water. The first was being a slurry wash and the second a displacement wash. The material was dried at 40 °C under vacuum over the weekend, and 3.9 grams (56%) was obtained as a light yellow powder. The Pd content was 3.7 ppm.
PATENT
US 8436171
PATENT
WO 2016008433
PATENT
WO 2016015604
PATENT
WO 2016015597
PATENT
WO 2016015605
PATENT
WO 2016015598
PATENT
WO 2017146794
PATENT
WO 2017146795
https://patents.google.com/patent/WO2017146795A1/en
PATENT
US 20180071302
REFERENCES
1: Eriksson A, Hermanson M, Wickström M, Lindhagen E, Ekholm C, Jenmalm Jensen A, Löthgren A, Lehmann F, Larsson R, Parrow V, Höglund M. The novel tyrosine kinase inhibitor AKN-028 has significant antileukemic activity in cell lines and primary cultures of acute myeloid leukemia. Blood Cancer J. 2012 Aug 3;2:e81. doi: 10.1038/bcj.2012.28. PubMed PMID: 22864397; PubMed Central PMCID: PMC3432483.
////////////AKN028 , AKN-028 , AKN 028, phase 2, Swedish Orphan Biovitrum, Akinion Pharmaceuticals, Acute myeloid leukaemia
NC1=NC=C(C2=CC=NC=C2)N=C1NC3=CC4=C(NC=C4)C=C3
Phase 3-Volasertib for acute myeloid leukaemia in patients ineligible for intensive induction therapy

Volasertib (BI 6727) for Acute myeloid leukaemia. is a cell cycle kinase inhibitor of polo-like kinases 1, 2 and 3. Volasertib inhibits cancer growth by disrupting cell division and inducing cell death. Volasertib is administered as a 350mg one hour intravenous (IV) infusion on days 1 and 15 of a 28 day cycle in combination with low-dose cytarabine (LDAC), administered via subcutaneous injection (SC) at 20mg twice daily on days 1-10 of a 28 day cycle.
Acute myeloid leukaemia (AML): previously untreated; patients considered ineligible for intensive remission induction therapy – first line; in combination with low-dose cytarabine
volasertib (also known as BI 6727) is a small molecule inhibitor of the PLK1 (polo-like kinase 1) protein being developed by Boehringer Ingelheim for use as an anti-cancer agent. Volasertib is the second in a novel class of drugs called dihydropteridinone derivatives.
