PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Join me on Facebook
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
Googleplus
MYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Wuxi Fuxin Pharmaceutical Research and Development , in collaboration with Wuxi Apptec , is investigating a tablet formulation of WXFL-10203614 , a JAK1 tyrosine kinase inhibitor, for the oral treatment of rheumatoid arthritis. In January 2019, a phase I trial was planned.
Novel crystalline forms of 7h-pyrrolo[2,3-D]pyrimidine compounds (designated as forms A to E) useful as JAK1 and JAK2 inhibitors for treating arthritis, inflammation and autoimmune diseases.
JAK belongs to the family of tyrosine kinases involved in inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6. The present invention also provides a method for preparing the compound or a pharmaceutical composition comprising the compound, and a method for preventing and/or treating inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6 by administrating the compound of the present invention.
Janus kinase (JAK) is a cytoplasmic tyrosine kinase that transduces a cytokine signal from a membrane receptor to an STAT transcription factor. The prior art has described four members of the JAK family: JAK1, JAK2, JAK3 and TYK2. When cytokines bind to their receptors, JAK family members are auto-phosphorylated and/or trans-phosphorylated from each other, followed by STATs phosphorylation, and then are migrated into the cell nucleus to regulate the transcription. JAK-STAT intracellular signal transduction is suitable for interferons, most interleukins, as well as various cytokines and endocrine factors, such as EPO, TPO, GH, OSM, LIF, CNTF, GM-CSF and PRL (Vainchenker W. et al. (2008)).
A combinatorial study of a genetic model and a small molecule JAK inhibitor has revealed the therapeutic potential of several JAKs. It has been confirmed by mouse and human genetics that JAK3 is an immunosuppressive target (O’Shea J. et al. (2004)). A JAK3 inhibitor has been successfully used in clinical development. At first, it was used in organ transplant rejection, and later also used in other immunoinflammatory indications such as rheumatoid arthritis (RA), psoriasis and Crohn’s disease (http://clinicaltrials.gov/). It has been confirmed by human genetics and mouse knockout studies that TYK2 is a potential target for immunoinflammatory diseases (Levy D. and Loomis C. (2007)). JAK1 is a new target in the field of immunoinflammatory diseases. The heterodimerization of JAK1 and other JAKs arouses a transduction of cytokine-driven pro-inflammatory signaling. Thus, it is expected that inhibition of JAK1 and/or other JAKs has a therapeutic benefit for a series of inflammatory diseases and other diseases driven by JAK-mediated signal transduction.
transduction.
Example 1: Preparation of Compound 1
Step 1: 2-chloro-4-nitro-1-oxo-pyridin-1-ium (40.0 g, 229.2 mmol) and (4-methoxyphenyl)methylamine (63 g, 458.4 mmol) were dissolved in EtOH (400 mL), and the resulting solution was stirred at reflux for 5 hours. TLC (PE:EA=2:1) showed that the reaction was complete. The EtOH was concentrated to half of its volume and was cooled in an ice bath for 2-3 hours. The resulting cold mixture was filtered, and the isolated solid was washed with PE (60 mL*3) and ice water (60 mL*3), respectively. Drying in vacuum given an orange solid, N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (2) (38.6 g, 140.2 mmol, with a yield of 61.2%). MS (ESI) calcd. For r C 13H 13N 3O 4 [M+H] + 275, found 276.
Step 2: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (5.0 g, 18.16 mmol) in CHCI 3 (50 mL) was dropwise added PCI 3 (8.4 g, 60.8 mmol) at 0° C. After the addition, the reaction mixture was heated to 25° C. and stirred vigorously for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was filtered, and the resulting solid was washed with PE (30 mL*3) to give a yellow solid compound, N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (3) (4.2 g, a crude product) which was directly used in the next step without further purification. MS (ESI) calcd. For C 15H 18N 6 [M+H] +259, found 260.
Step 3: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (4.2 g, 16.2 mmol) in toluene (10 mL) was dropwise added TFA (5.0 mL) at atmospheric temperature. Then, the mixture was stirred at 80° C. for 2 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The mixture was concentrated under reduced pressure to remove the solvent. The residue was diluted with H 2O (50 mL), and its pH was adjusted to be neutral with solid NaHCO 3. The aqueous phase was extracted with EA (50 mLE*3). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica, petroleum ether/ethyl acetate=1/0-1:1) to obtain an orange solid compound, 4-nitropyridine-2-amine (4) (700 mg, 5.0 mmol, with a yield of 31.1%). MS (ESI) calcd. For C 5H 5N 3O 2 [M+H] + 139, found 140.
Step 4: to a solution of 4-nitropyridine-2-amine (200 mg, 1.4 mmol) in DME (5 mL) was added 3-bromo-2-oxo-propanoate (280 mg, 1.4 mmol) at atmospheric temperature. The resulting mixture was stirred at 25° C. for 1 hour, and then was concentrated under reduced pressure to remove the solvent. The residue was dissolved with EtOH (10 mL); and then was refluxed for 3 hours. TLC showed that the reaction was complete. The reaction solution was cooled to room temperature, and the solvent was concentrated under reduced pressure. The residue was basified with saturated NaHCO 3 aqueous solution (25 mL). The aqueous phase was extracted with DCM (15 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (EA:PE=10-60%) to obtain a light yellow solid compound, ethyl 7-nitroimidazo[1,2-]pyridin-2-carboxylate (5) (302 mg, with a yield of 88.9%). MS (ESI) calcd. For C 10H 9N 3O 4 [M+H] + 235, found 236.
Step 5: a solution of ethyl 7-nitroimidazo[1,2-a]pyridin-2-carboxylate (150 mg, 637.8 mmol) in ethanol (20 mL) was added HCl (7 mg, 0.2 mmol) and PtO 2 (15 mg, 0.6 mmol) at atmospheric temperature. The reaction system was repeatedly vacuumed and filled with N 2 for three times, then filled with H 2(50 psi), and was stirred at 50° C. for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was concentrated to half of its volume, and filtered to obtain a white solid compound, ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (6) (120 mg, a crude product). MS (ESI) calcd. For C 10H 15N 3O 2 [M+H] + 209, found 210.
Step 6: ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (100 mg, 0.4 mmol) and 4-chloro-7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidine (137 mg, 0.4 mmol) were dissolved in n-BuOH (5 mL), and DIEA (158 mg, 1.2 mmol) were added to the above solution. The resulting mixture was stirred under reflux for 16 hours. LC-MS showed that the reaction was complete. The reaction mixture was concentrated under reduced pressure, and the resulting residue was diluted with H 2O (10 mL). The aqueous phase was extracted with EA (20 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (PE:EA=0:1) to obtain a light yellow solid compound, ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl] amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (7) (55 mg, 0.11 mmol, with a yield of 28.1%). MS (ESI) calcd. For C 23H 24N 6O 4S [M+H] + 480, found 481.
Step 7: to a solution of ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (3.0 g, 6.2 mmol) in THF (150 mL) was added NaH (499 mg, 12.5 mmol) in portions under N 2 atmosphere at 0° C. The mixture was stirred at that temperature for 1 hour, and then was dropwise added MeI (7.1 g, 50.2 mmol). After the addition, the mixture was stirred at atmospheric temperature for 1 hour. TLC showed that the reaction was complete. The reaction was quenched by the addition of saturated NH 4Cl (10 mL), and then was diluted by the addition of ice water (50 mL). The aqueous phase was extracted with a mixed solvent of DCM/MeOH (3:1, 50 mL*3). The combined organic phase was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude product was purified by flash column chromatography (DCM:MeOH=10:1) to obtain a light yellow solid, ethyl 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (8) (1.5 g, with a yield of 45%). MS (ESI) calcd. For C 24H 26N 6O 4S [M+H] + 494, found 495.
Step 8: to a solution of 7-[methyl-[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (4.0 g, 8.1 mmol) in THF (40 mL) and H 2O (8 mL) was added LiOH.H 2O (509 mg, 12.1 mmol), and the mixture was stirred at 20° C. for 10 hours. TLC showed that the reactants were completely consumed. THF in the reaction mixture was removed under reduced pressure; and the pH of the residue was adjusted to 2-3 with 2M HCl (4 mL) to form a white solid. The solid was filtered out, and was concentrated under reduced pressure to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (9) as a white solid (3.6 g, with a yield of 95.4%). MS (ESI) calcd. For C 22H 22N 6O 4S [M+H] + 466, found 467.
Step 9: to a solution of 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (1.8 g, 3.9 mmol) in DMF (20 mL) was added CDI (751 mg, 4.6 mmol) at 0° C. The reaction solution was heated to 25° C. and stirred for 2 hours, and after that, solid ammonium chloride (2.1 g, 38.6 mmol) was added, and then the reaction was kept overnight at atmospheric temperature. LC-MS showed that the reactants were completely consumed. The reaction mixture was poured into ice water (50 mL), and a white solid was precipitated. The solid was filtered out, washed with water (20 mL), and was dried under reduced pressure in a rotating manner to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (10) as a white solid (2.5 g, a crude product) which product was directly used in the next step. MS (ESI) calcd. For C 22H 23N 7O 3S [M+H] + 465, found 466.
Step 10: 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxamide (2.5 g, 5.4 mmol) was dissolved in a mixture of THF (20 mL), MeOH (10 mL) and H 2O (6 mL), and NaOH (429.6 mg, 10.7 mmol) was added. The mixture was heated to 60° C. and stirred for 30 minutes. LC-MS showed that the reactants were completely consumed. The reaction mixture was concentrated under reduced pressure to obtain 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxamide (11) as a white solid (2.5 g, a crude product) which was directly used in the next step. MS (ESI) calcd. For C 15H 17N 7O [M+H] + 311, found 312.
Step 11: to a solution of 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (2.0 g, 6.4 mmol) and triethylamine (3.9 g, 38.5 mmol) in THF (20 mL) was dropwise added TFAA (4.1 g, 19.3 mmol) at 0° C. After the addition, the reaction solution was stirred at atmospheric temperature for 30 minutes. LC-MS showed the starting materials were completely consumed. The reaction mixture was poured into ice water (20 mL), and extracted with DCM/MeOH (5:1, 100 mL*2). The combined organic layer was washed with saturated saline (20 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a residue. The residue was purified by column chromatography (DCM/MeOH=40/1 to 20:1) to obtain 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (12,378 mg, with a yield of 19.8%). MS (ESI) calcd. For C 15H 15N 7 [M+H] + 293, found 294. 1H NMR (400 MHz, DMSO-d6) 11.44-11.71 (m, 1H), 7.99-8.17 (m, 2H), 7.11-7.20 (m, 1H), 6.63 (dd, J=1.76, 3.26 Hz, 1H), 5.33 (br. s., 1H), 4.21-4.21-4.31 (m, 1H), 4.13 (dt, J=4.14, 12.49 Hz, 1H), 3.27 (s, 3H), 2.91-3.11 (m, 2H), 2.31-2.44 (m, 1H), 2.07 (d, J=11.54 Hz, 1H).
Step 12: racemic 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (30 mg, 102.3 umol) was separated by a chiral column to obtain the compound 1 (10 mg, with a yield of 32.8%).
Compound 1: retention time 6.407 min; MS (ESI) calcd. For C 15H 15N 7 [293, found 294 M+H]+. Purity 98.8%, e.e. was 98.9%; [α] D20=+78.4° (c=0.6, DMSO). MS ESI calcd. For C 15H 15N 7 [M+H] + 294, found 294. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.02-2.15 (m, 1H) 2.39 (qd, J=12.42, 5.90 Hz, 1H) 2.92-3.12 (m, 2H) 3.28 (s, 3H) 4.05-4.36 (m, 2H) 5.20-5.45 (m, 1H) 6.64 (dd, J=3.39, 1.88 Hz, 1H) 7.17 (dd, J=3.26, 2.51 Hz, 1H) 8.02-8.17 (m, 2H) 11.69 (br s, 1H).
16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
Phase 1 Clinical, Acute myelogenous leukemia, Protein cereblon modulator
Useful for treating chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia or acute myeloid leukemia.
Celgene is developing CC-90009, a cereblon E3 ligase modulator, for treating AML; in January 2019, data from a phase I trial were expected later that year.
0iginator Celgene Corporation
Class Antineoplastics
Mechanism of Action CRBN protein modulators; Ubiquitin protein ligase complex modulators
Phase I Acute myeloid leukaemia
28 Mar 2019 No recent reports of development identified for clinical-Phase-Unknown development in Acute-myeloid-leukaemia in USA (IV)
01 Sep 2016 Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in Canada (IV) (NCT02848001)
04 Aug 2016 Celgene plans a phase I trial for Acute Myeloid Leukaemia in USA and Canada (NCT02848001)
In September 2016, Celgene initiated a phase I dose-finding trial of CC 90009 in patients with relapsed or refractory acute myeloid leukaemia (NCT02848001; CC-90009-AML-001). The open-label study intends to enrol 60 patients in the US and Canada
CC-90009 is a cereblon modulator. CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. .
Development Overview
cereblon modulator CC-90009A modulator of cereblon (CRBN), which is part of the cullin 4-RING E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase; CUL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and pro-apoptotic activities. Upon administration, CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Check for active clinical trials using this agent. (NCI Thesaurus)
Provided herein are methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy with 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of
stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof. Further provided is a compound for use in methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy, wherein the compound is 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
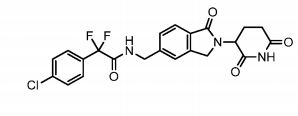
The term Compound 1 refers to”2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide” having the structure:
and its stereoisomers or mixture of stereoisomers, isotopologues, pharmaceutically acceptable salts, tautomers, solvates, hydrates, co-crystals, clathrates, or polymorphs thereof. In certain embodiments, Compound 1 refers to 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide and its tautomers. In certain embodiments, Compound 1 refers to a polymorph of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-
oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In certain embodiments, Compound 1 refers to polymorph Form C of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In one embodiment, the stereoisomer is an enantiomer.
PATENT
WO-2019136016
Novel isotopologs of the compound presumed to be CC-90009 , processes for their preparation and compositions comprising them are claimed.
SOLID FORMS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE, AND THEIR PHARMACEUTICAL COMPOSITIONS AND USES
Percent Composition: C 51.92%, H 3.87%, N 13.45%, O 23.05%, S 7.70%
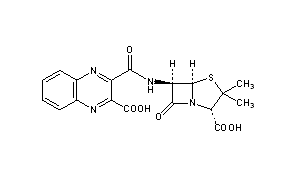
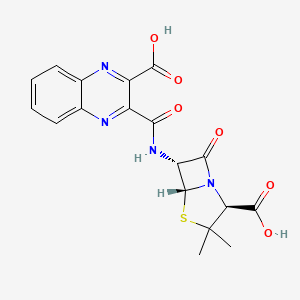
Literature References: Semi-synthetic antibiotic related to penicillin. Prepd by condensation of quinoxaline-2,3-dicarboxylic anhydride with 6-aminopenicillanic acid: Richards et al.,Nature199, 354 (1963).
Derivative Type: Disodium salt
CAS Registry Number: 985-32-0
Molecular Formula: C18H14N4Na2O6S
Molecular Weight: 460.37
Percent Composition: C 46.96%, H 3.07%, N 12.17%, Na 9.99%, O 20.85%, S 6.97%
Properties: Crystals, dec 261-262°. [a]D23 +183.5° (water). Very hygroscopic. uv max (containing 9.2% water): 242, 326 nm (e32,100; 7280). Acquires a bright yellow color on exposure to strong sunlight but is stable at 100° for at least 3 months. Freely sol in water; a 25% aq soln is stable for 2 months at 0°. Antimicrobial activity is highest against Staphylococcus aureus.
Optical Rotation: [a]D23 +183.5° (water)
Absorption maximum: uv max (containing 9.2% water): 242, 326 nm (e 32,100; 7280)
cas 13549-27-4
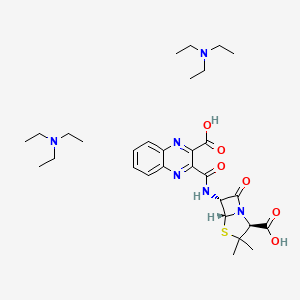
Derivative Type: Bistriethylammonium salt monohydrate
Molecular Formula: C30H46N6O6S.H2O
Molecular Weight: 636.80
Percent Composition: C 56.58%, H 7.60%, N 13.20%, O 17.59%, S 5.04%
Properties: Crystals from acetone, dec 135-137°. [a]D20 +142° (c = 0.376 in water).
Optical Rotation: [a]D20 +142° (c = 0.376 in water)
Quinacillin is a semisynthetic penicillase-resistant penicillin with antibacterial activity. Quinacillin binds to and inactivates penicillin-binding proteins (PBPs) located on the inner membrane of the bacterial cell wall. Inactivation of PBPs interferes with the cross-linkage of peptidoglycan chains necessary for bacterial cell wall strength and rigidity. This interrupts bacterial cell wall synthesis and results in the weakening of the bacterial cell wall, eventually causing cell lysis.
Selinexor is an orally available, small molecule inhibitor of CRM1 (chromosome region maintenance 1 protein, exportin 1 or XPO1), with potential antineoplastic activity. Selinexor modifies the essential CRM1-cargo binding residue cysteine-528, thereby irreversibly inactivates CRM1-mediated nuclear export of cargo proteins such as tumor suppressor proteins (TSPs), including p53, p21, BRCA1/2, pRB, FOXO, and other growth regulatory proteins. As a result, this agent, via the approach of selective inhibition of nuclear export (SINE), restores endogenous tumor suppressing processes to selectively eliminate tumor cells while sparing normal cells. CRM1, the major export factor for proteins from the nucleus to the cytoplasm, is overexpressed in a variety of cancer cell types.
Selinexor has been used in trials studying the treatment of AML, Glioma, Sarcoma, Leukemia, and Advanced, among others.
Selinexor, also known as KPT-330, is an orally bioavailable, potent and selective XPO1/CRM1 Inhibitor. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Selinexor potentiates the antitumor activity of gemcitabine in human pancreatic cancer through inhibition of tumor growth, depletion of the antiapoptotic proteins, and induction of apoptosis. Selinexor has strong activity against primary AML cells while sparing normal stem and progenitor cells.
SYN
Medical uses
Selinexor is restricted for use in combination with the steroid dexamethasone in people with relapsed or refractory multiple myelomawhich has failed to respond to at least four or five other therapies (so-called “quad-refractory” or “penta-refractory” myeloma),[5] for whom no other treatment options are available.[3][4] It is the first drug to be approved for this indication.[6]
Adverse effects
In the clinical study used to support FDA approval, selinexor was associated with high rates of pancytopenia, including leukopenia(28%), neutropenia (34%, severe in 21%), thrombocytopenia (74%, severe in 61% of patients), and anemia (59%).[4][7] The most common non-hematological side effects were gastrointestinal reactions (nausea, anorexia, vomiting, and diarrhea), hyponatremia (low blood sodium levels, occurring in up to 40% of patients), and fatigue.[7][8] More than half of all patients who received the drug developed infections, including fatal cases of sepsis.[7] However, these data are from an open-label trial, and thus cannot be compared to placebo or directly attributed to treatment.
Mechanism of action
Schematic illustration of the Ran cycle of nuclear transport. Selinexor inhibits this process at the nuclear export receptor (upper right).
Like other so-called selective inhibitors of nuclear export (SINEs), selinexor works by binding to exportin 1 (also known as CRM1). CRM1 is a karyopherin which performs nuclear transport of several proteins, including tumor suppressors, oncogenes, and proteins involved in governing cell growth, from the cell nucleus to the cytoplasm; it is often overexpressed and its function misregulated in several types of cancer.[1] By restoring nuclear transport of these proteins to normal, SINEs lead to a buildup of tumor suppressors in the nucleus of malignant cells and reduce levels of oncogene products which drive cell proliferation. This ultimately leads to cell cycle arrest and death of cancer cells by apoptosis.[1][2][7]In vitro, this effect appeared to spare normal (non-malignant) cells.[1][8]
Because CRM1 is a pleiotropicgene, inhibiting it affects many different systems in the body, which explains the high incidence of adverse reactions to selinexor.[2] Thrombocytopenia, for example, is a mechanistic and dose-dependent effect, occurring because selinexor causes a buildup of the transcription factor STAT3 in the nucleus of hematopoietic stem cells, preventing their differentiation into mature megakaryocytes (platelet-producing cells) and thus slowing production of new platelets.[2]
Selinexor was developed by Karyopharm Therapeutics of Newton, Massachusetts, a pharmaceutical company devoted entirely to the development of drugs that target nuclear transport. It was approved by the FDA on July 3, 2019, on the basis of a single uncontrolled clinical trial. The decision was controversial, and overruled the previous recommendation of an FDA Advisory Panel which had voted 8–5 against approving the drug, due to concerns about efficacy and toxicity.[3]
As of 2019, phase I/II and III trials are ongoing,[3][9] including the use of selinexor in other cancers and in combinations with other drugs used for multiple myeloma.[2]
International Publication No. WO 2013/019548 describes a series of compounds that are indicated to have inhibitory activity against chromosomal region maintenance 1 (CRM1, also referred to as exportin 1 or XPO1) and to be useful in the treatment of disorders associated with CRM1 activity, such as cancer. (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N’-(pyrazin-2-yl)acrylohydrazide (also referred to as selinexor) is one of the compounds disclosed in International Publication No. WO 2013/019548. Selinexor has the chemical structure shown in Structural Formula I:
Example 1. Preparation of Selinexor Lot No.1305365 (Form A).
[00274] Selinexor for Lot No. 1305365 was made in accordance with the following reaction scheme:
[00275] A solution of propane phosphonic acid anhydride (T3P®, 50% in ethyl acetate, 35Kg) in THF (24.6Kg) was cooled to about -40 °C. To this solution was added a solution of KG1 (13.8Kg) and diisopropylethylamine (12.4Kg) in tetrahydrofuran (THF, 24.6Kg). The resulting mixture was stirred at about -40°C for approximately 2.5 hours.
[00276] In a separate vessel, KJ8 (4.80Kg) was mixed with THF (122.7Kg), and the resulting mixture cooled to about -20°C. The cold activated ester solution was then added to the KJ8 mixture with stirring, and the reaction was maintained at about -20°C. The mixture was warmed to about 5°C, water (138.1Kg) was added and the temperature adjusted to about 20°C. After agitating for about an hour, the lower phase was allowed to separate from the mixture and discarded. The upper layer was diluted with ethyl acetate (EtOAc). The organic phase was then washed three times with potassium phosphate dibasic solution (~150Kg), then with water (138.6Kg).
[00277] The resulting organic solution was concentrated under reduced pressure to 95L, EtOAc (186.6Kg) was added and the distillation repeated to a volume of 90L. Additional EtOAc (186.8Kg) was added and the distillation repeated a third time to a volume of 90L. The batch was filtered to clarify, further distilled to 70L, then heated to about 75°C, and slowly cooled to 0 to 5°C. The resulting slurry was filtered and the filter cake washed with a mixture of EtOAc (6.3Kg) and toluene (17.9Kg) before being dried in a vacuum oven to provide selinexor designated Lot No. 1305365 (Form A).
Example 2. Preparation of Selinexor Lot No.1341-AK-109-2 (Form A).
[00278] The acetonitrile solvate of selinexor was prepared in accordance with Example 6.
[00279] The acetonitrile solvate of selinexor (2.7g) was suspended in a mixture of isopropanol (IPA, 8mL) and water (8mL), and the resulting mixture heated to 65 to 70 °C to effect dissolution. The solution was cooled to 45 °C, and water (28mL) was added over 15 minutes, maintaining the temperature between 40 and 45 °C. The slurry was cooled to 20 to 25 °C over an hour, then further cooled to 0 to 5 °C and held at that temperature for 30 minutes before being filtered. The filter cake was washed with 20% v/v IPA in water and the product dried under suction overnight, then in vacuo (40°C).
Example 3. Preparation of SelinexorSelinexorSelinexor Lot No. PC-14-005 (Form A).
[00280] The acetonitrile solvate of selinexor (Form D) was prepared in accordance with the procedure described in Example 6.
[00281] The acetonitrile solvate of selinexor (1.07Kg) was suspended in a mixture of IPA (2.52Kg) and water (3.2Kg) and the mixture heated to 70 to 75 °C to dissolve. The temperature was then adjusted to 40 to 45 °C and held at that temperature for 30 minutes. Water (10.7Kg) was added while maintaining the temperature at 40 to 45 °C, then the batch was cooled to 20 to 25 °C and agitated at that temperature for 4 hours before being further cooled to 0 to 5 °C. After a further hour of agitation, the slurry was filtered and the filter cake washed with a cold mixture of IPA (0.84Kg) and water (4.28Kg) before being dried.
Example 4. Preparation of SelinexorSelinexorSelinexor Lot No. PC-14-009 (Form A).
[00282] The acetonitrile solvate of selinexor (Form D) was prepared in accordance with the procedure described in Example 6.
[00283] The acetonitrile solvate of selinexor (1.5Kg) was suspended in IPA (3.6Kg) and water (4.5Kg) and warmed to 37 to 42 °C with gentle agitation. The suspension was agitated at that temperature for 4 hours, and was then cooled to 15 to 20 °C over 1 hour. Water (15.1Kg) was added, maintaining the temperature, then the agitation was continued for 1 hour and the batch was filtered. The filter cake was washed with a mixture of IPA (1.2Kg) and water (6Kg), then dried under a flow of nitrogen.
Example 5. Preparation of Selinexor Lot Nos.1339-BS-142-1, 1339-BS-142-2 and PC-14-008 (Form A).
[00284] A reactor, under nitrogen, was charged with KG1 (1Kg, 1.0 Eq), KJ8 (0.439 Kg, 1.4 Eq) and MeTHF (7L, 7 parts with respect to KG1). Diisopropylethylamine (0.902Kg, 2.45 Eq with respect to KG1) was added to the reaction mixture at -20 °C to -25 °C with a MeTHF rinse. To the reaction mixture, 50% T3P® in ethyl acetate (2.174Kg, 1.2 Eq with respect to KG1) was then charged, maintaining the temperature at -20 °C to -25 °C with a MeTHF rinse. After the completion of the addition, the reaction mixture was stirred briefly
and then warmed to 20 °C to 25 °C. Upon completion, the reaction mixture was washed first with water (5L, 5 parts with respect to KG1) and then with dilute brine (5L, 5 parts with respect to KG1). The organic layer was concentrated by vacuum distillation to a volume of 5 L (5 parts with respect to KG1), diluted with acetonitrile (15L, 15 parts with respect to KG1) at approximately 40 °C and concentrated again (5L, 5 parts with respect to KG1). After solvent exchange to acetonitrile, the reaction mixture was then heated to approximately 60 °C to obtain a clear solution. The reaction mixture was then cooled slowly to 0-5 °C, held briefly and filtered. The filter cake was washed with cold acetonitrile (2L, 5 parts with respect to KG1) and the filter cake was then dried under a stream of nitrogen to provide the acetonitrile solvate of selinexor (Form D) as a slightly off-white solid.
[00285] Form D of selinexor (0.9Kg) was suspended in IPA (2.1Kg, 2.7L, 3 parts with respect to Form D) and water (2.7Kg, 2.7L, 3 parts with respect to Form D) and warmed to approximately 40 °C. The resulting suspension was agitated for about 4 hours, selinexor, cooled to approximately 20 °C, and diluted with additional water (9Kg, 10 parts with respect to Form D). The mixture was stirred for a further 4-6 hours, then filtered, and the cake washed with a mixture of 20% IPA and water (4.5L, 5 parts with respect to Form D). The filter cake was then dried under vacuum to provide selinexor designated Lot No. PC-14-008 as a white crystalline powder with a >99.5% a/a UPLC purity (a/a=area to area of all peaks; UPLC-ultra performance HPLC).
Example 6. Preparation of Selinexor Lot No.1405463 (Form A).
[00286] Selinexor Lot No. 1405463 was prepared in accordance with the following reaction scheme:
.
[00287] A reactor was charged with KG1 (15.8Kg), KJ8 (6.9Kg) and MeTHF (90Kg). Diisopropylethylamine (14.2Kg) was added to the reaction mixture over approximately 35 minutes at about -20 °C. Following the addition of the diisopropylethylamine, T3P® (50%
solution in EtAOc, 34.4Kg) was added maintaining the temperature at -20 °C. The mixture stirred to complete the reaction first at -20 °C, then at ambient temperature.
[00288] Upon completion of the reaction, water (79Kg) was added over about 1 hour. The layers were separated and the organic layer was washed with a mixture of water (55Kg) and brine (18Kg), The mixture was filtered, and the methyl-THF/ethyl acetate in the mixture distillatively replaced with acetonitrile (volume of approximately 220L). The mixture was warmed to dissolve the solids, then slowly cooled to 0 to 5 °C before being filtered. The filter cake was washed with acetonitrile to provide the acetonitrile solvate of
selinexorSelinexorSelinexor (Form D).
[00289] The acetonitrile solvate of selinexorSelinexorSelinexor was dried, then mixed with isopropanol (23Kg) and water (55Kg). The slurry was warmed to about 38 °C and held at that temperature for approximately 4 hours before being cooled to 15 to 20 °C. Water (182Kg) was added. After a further 5 hours of agitation, the mixture was filtered and the filter cake washed with a mixture of isopropanol (14Kg) and water (73Kg), before being dried under vacuum (45 °C). The dried product was packaged to provide
selinexorSelinexorSelinexor Lot No. 1405463 (Form A).
Example 7. Polymorphism Studies of Selinexor.
[00290] A comprehensive polymorphism assessment of selinexor was performed in a range of different solvents, solvent mixtures and under a number of experimental conditions based on the solubility of selinexor. Three anhydrous polymorphs of
selinexorSelinexorSelinexor were observed by XRPD investigation, designated Form A, Form B and Form C. Form A is a highly crystalline, high-melting form, having a melting point of 177 °C, and was observed to be stable from a physico-chemical point of view when exposed for 4 weeks to 25 °C/97% relative humidity (RH) and to 40 °C/75% RH. A solvated form of selinexor was also observed in acetonitrile, designated Form D. A competitive slurry experiment confirmed Form A as the stable anhydrous form under the conditions investigated, except in acetonitrile, in which solvate formation was observed. It was further found that in acetonitrile, below 50 °C, only Form D is observed, at 50 °C both Form A and Form D are observed, and at 55 °C, Form A is observed .
Selinexor is an orally bioavailable selective nuclear export inhibitors, 2012 for the first time in clinical, so far carried out a total of 21 trials, indications include chronic myelogenous leukemia, acute myelogenous leukemia, acute lymphatic leukemia, prostate cancer, melanoma, non-small cell lung cancer, glioma, neuroblastoma into, gynecological cancer, diffuse large B-cell lymphoma, squamous cell carcinoma, colorectal cancer and the like.May 2014, FDA granted orphan drug designation Selinexor treatment of acute myeloid leukemia and diffuse large B-cell lymphoma, in June 2014, EMA is also granted orphan drug designation Selinexor treatment of both diseases.January 2015, received FDA orphan drug to treat multiple myeloma identified.
[0003] Currently, the synthesis process has been disclosed, the following reaction equation:
[0006] wherein the compound is 5 Selinexor drug.
[0007] In this method, however, easy to produce Intermediate 1-2 double bond is easily reversed when synthetically produced from trans impurities, in addition to more difficult to impact yield; Intermediate 3 Intermediate 4 Synthesis APIs 5 when required ultra-low temperature, and the product was purified by column required, only a yield of 20%.
SUMMARY
[0008] The object of the present invention to provide a novel compound Selinexor drug synthesis of 5, in order to solve technical problems.
[0009] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0010] A, Compound 7
[0011] Compound 6, dichloromethane and ethyl acetate mixture, stirred and dissolved, compound 4, T3P (n-propyl phosphoric anhydride) and DIPEA (N, N- diisopropylethylamine) at a low temperature; the reaction was stirred for 25-35min at a low temperature, dichloromethane and water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0012] B, Synthesis of Compound 8
[0013] the compound obtained in Step 7, and mixed sodium iodide acetic acid, warmed to 110-120 ° C, the reaction 2.5-3.5h; After completion of the reaction, the system cooled to room temperature, water and dichloromethane were added, stirred for 8 after -15min, standing layered organic phase was washed with saturated sodium bicarbonate and saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in DMF (dimethyl fumarate) to give compound in DMF 8;
Synthesis [0014] C, of Compound 5
[0015] Compound 1, DBAC0 (triethylenediamine), the DMF mixed and dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring was continued for 3-4 hours; the reaction after completion, water and ethyl acetate were added to the system, the organic phase is evaporated to dryness and petroleum ether and recrystallized from ethyl acetate to give compound 5.
[0016] Preferably, said step A, the low temperature is 0-2 ° C.
[0017] Preferably, said step B in DMF, the crude compound 8 concentration of less than 1%.
[0018] The novel synthetic methods of the present invention Selinexor drug, the chemical equation is as follows:
〇
[0020] The present invention has the following advantages: novel synthetic method Selinexor drug of the present invention to overcome the conventional synthesis process, is easy to produce trans impurities, more difficult in addition, the influence the yield and the need for ultra-low temperature, and the product requires problems purified by column, the yield is very low, reducing the synthetic steps, increased yield, there is provided a new process for the synthesis of the drug Selinexor.
[0021] In addition to the above-described objects, features and advantages of the present invention as well as other objects, features and advantages.Below the invention will be described in further detail present.
Example 1
[0024] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0025] A, Compound 7
[0026] 50ml three □ flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 0 ° C; the system at 0 ° C the reaction was stirred for 30min, 50ml of dichloromethane and 30ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0027] B, Synthesis of Compound 8
[0028] 50ml three-necked flask, added the compound obtained in Step 7,40ml of glacial acetic acid and 1.38g of sodium iodide was heated to 115.(:, The reaction 3H; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 50ml of water was added and IOOml dichloromethane, after stirring IOmin, standing separation, the organic phase was washed with saturated sodium bicarbonate and saturated washed with sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in IOmL DMF to give DMF solution of compound 8;
Synthesis [0029] C, of Compound 5
[0030] After 50ml 3-necked flask was added 0.2g compound 1,0.24gDBAC0,20mlDMF, dissolved with stirring, dropwise adding to the reaction system in DMF compound obtained in Step 8, after the addition was complete, stirring continued for 3.5 hours; after completion of the reaction, 20ml water was added to the system and 50ml ethyl acetate, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.158g of compound 5, yield 50.9%.
[0031] Example 2
[0032] – new type Se Iinexor drug synthesis, comprising the steps of:
Synthesis [0033] A, Compound 7
[0034] 50ml three □ flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 1 ° C; system at 1 ° C the reaction was stirred for 35min, 50ml of dichloromethane and 30ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0035] B, Synthesis of Compound 8
Three-neck flask [0036] 50ml of addition of the compound obtained in Step 7,40ml glacial acetic acid and 1.38g of sodium iodide was heated to 120.(:, The reaction for 2.5 h; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 60ml water and 120ml dichloromethane was added, after stirring for 15min, allowed to stand for separation, the organic phase was washed with saturated sodium bicarbonate and washed with saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, 12mLDMF was dissolved in DMF to give a solution of compound 8;
Synthesis [0037] C, of Compound 5
[0038] After 50ml 3-necked flask was added 0.2g compound 1,0.24gDBAC0,20mlDMF, dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring continued for 3 hours; after completion of the reaction, 25ml of water and 50ml of ethyl acetate was added to the system, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.152g of compound 5, yield 49.0% billion
[0039] Example 3
[0040] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0041] A, Compound 7
Three [0042] 50ml of flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 2 ° C; system from 0 ° C the reaction was stirred for 25min, 40ml of dichloromethane and 35ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0043] B, Synthesis of Compound 8
Three-neck flask [0044] 50ml of addition of the compound obtained in Step 7,35ml glacial acetic acid and 1.38g of sodium iodide was heated to 110.(:, The reaction for 3.5 h; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 50ml of water was added and dichloromethane IOOml After Smin of stirring, standing separation, the organic phase was washed with saturated sodium bicarbonate and washed with saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in IOmL DMF to give DMF solution of compound 8;
Synthesis [0045] C, of Compound 5
[0046] 50ml three-neck flask was added 0.2g compound 1,0.24gDBA⑶, 20mlDMF, and dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring was continued for 4 hours; after completion of the reaction, 20ml of water and 40ml ethyl acetate were added to the system, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.155g of compound 5, yield 49.9% billion
The drug compound having the adopted name “Selinexor” has chemical name:(Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-IH-l,2,4-triazol-1 -yl)-N’-(pyrazin-2yl) acrylohydrazide as below.
Selinexor (KPT-330) is a first-in-class, oral Selective Inhibitor of Nuclear Export / SINE™ compound. Selinexor functions by binding with and inhibiting the nuclear export protein XP01 (also called CRM1 ), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. Over 1 ,200 patients have been treated with Selinexor in company and investigator-sponsored Phase 1 and Phase 2 clinical trials in advanced hematologic malignancies and solid tumors. Karyopharm has initiated four later-phase clinical trials of Selinexor, including one in older patients with acute myeloid leukemia (SOPRA), one in patients with Richter’s transformation (SIRRT), one in patients with diffuse large B-cell lymphoma (SADAL) and a single-arm trial of Selinexor and lose-dose dexamethasone in patients with multiple myeloma (STORM). Patients may receive a twice-weekly combination of Selinexor in combination with low dose dexamethasone. Randomized 1 :1 , Selinexor will be dosed either at 60mg + dexamethasone or at 100 mg + dexamethasone.
US 8999996 B2 discloses Selinexor and a pharmaceutically acceptable salt thereof, pharmaceutical compositions and use for treating disorders associated with CRM1 activity. Further, it discloses preparative methods for the preparation of compounds disclosed therein including Selinexor by reacting (Z)-3-(3- (3,5-
bis(trifluoromethyl)phenyl)-IH-l,2,4-triazol-l-yl)acrylic acid in 1 :1 CH2CI2: AcOEt with 2-Hydrazinopyrazine at -40 °C followed by addition of T3P[Propylphosphonic anhydride] (50%) and DIPEA. After 30 minutes, the reaction mixture was concentrated and the crude oil was purified by preparative TLC using 5% MeOH in CH2CI2 as mobile phase (under ammonia atmosphere) to afford 40 mg of Selinexor with purity: 95.78%. However, it is not disclosed about the nature of the compound obtained therein.
WO 2016025904 A1 discloses various crystalline forms of Selinexor namely Form A, Form B, Form C, Form D, compositions and MoU thereof for the treatment of disorder associated with CRM1 activity and their preparative processes.
Prior art process for the preparation of Selinexor suffers from disadvantages interms of process such as the use of lengthy procedures to practice and resulting in low yields, which may not be viable at industrial scale. Synthetic product obtained therein has very low purity and contains significant amounts of unreacted starting materials and trans-isomer of Selinexor, which are further purified by time consuming and expensive chromatographic separations leading to loss of yield. Hence, there remains a need for improved process for the preparation of Selinexor which is industrially viable and reproducible. Particularly, it is desirable to have a process avoiding purification steps still meeting desired pharmaceutical quality.
EXAMPLES
Example-1 : Preparation of isopropyl (Z)-3-(3-(3,5-bis(trifluoromethyl) phenyl)-1 H- -triazol-1 -yl)acrylate
3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazole (250 g) was dissolved in tetrahydrofuran (2 I) under nitrogen atmosphere at 27°C and cooled to -5°C. 1 ,4- diazabicyclo[2.2.2]octane (DABCO, 1 99.5 g) was added to the reaction mixture at -5°C and stirred at the same temperature for 40 minutes. Isopropyl (Z)-3- iodoacrylate (234.8 g in 500 mL of tetrahydrofuran) was added drop wise to the reaction mixture in 1 hour 1 0 minutes at -5°C and stirred at the same temperature for 2 hours. After the completion of the reaction, the reaction mixture was added to ice cold water (2 I) and separated the organic layer. The aqueous layer was extracted with ethyl acetate (2 x 1 I). The combined organic layer was washed with brine solution (1 I) and dried over sodium sulphate. The dried solution was evaporated completely under vacuum at 40°C to obtain crude product with HPLC purity of 93.53% The crude product was triturated with hexane (700 mL) and stirred for 20 minutes at -30°C and filtered the solid. Trituration of crude product with hexane was repeated for three times and dried under vacuum to obtain the title compound with HPLC purity of 97.46% and trans-isomer content of 0.66%. Yield: 297 g Example-2: Preparation of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4- triazol-1 -yl)acr lic acid.
To a mixture of tetrahydrofuran (300 mL) and water (300 mL), Isopropyl (Z)-3-(3- (3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylate (30 g) was added and cooled to 0°C. Lithium hydroxide monohydrate (16.03 g) under cooling condition at 0°C was added to the reaction mixture and stirred the reaction mixture at same temperature for 7 hours. After completion of the reaction, 2 N HCI (180 mL) was added to adjust the pH of the reaction mixture to 2 and extracted it with ethyl acetate (300 mL). Organic layer was dried over sodium sulphate and evaporated under vacuum at 40°C. The crude compound was stirred with hexane (150 mL) and filtered the solid. Dried the compound under vacuum at 40°C for 0.5 hour to obtain the title compound with HPLC purity of 97.25% with trans-isomer content of 3 %. Yield: 24 g
Example-3: Purification of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4- tria
A mixture of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid (24 g) and acetone (240 mL) was stirred for complete dissolution at 30°C. Dicyclohexyl amine (1 5 mL) was added drop wise for 20 minutes under stirring at the same temperature. Acetone (50 mL) was added to the reaction mixture and stirred for 2 hours at 27°C. Filtered the solid and washed with hot acetone (150 mL) and dried in vacuum drier at 30°C for 1 hour to obtain the Dicyclohexyl amine salt of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid. To the above salt, dichloromethane (150 mL) and water (1 00 mL) was added and stirred for complete dissolution at 30and adjusted the pH of the solution with 2 N sulphuric acid (100 mL) to 2. Filtered the reaction mixture and washed the product with water (1 00 mL) and then with hexane (150 mL). The solid was dried under vacuum at 40°C for 0.5 hour to obtain title compound with HPLC purity 99.98% with no detectable content of trans-isomer. Yield: 17 g
Example-4: Preparation of Selinexor
(Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid (10 g) was combined with a mixture of acetonitrile (1 00 mL) and ethyl acetate (50 mL) then added the 2-hydrazinylpyrazine (3.76 g) and stirred for 5 min. Reaction mixture was cooled to 0°C and diisopropyl ethyl amine (16.63 ml) and then Propylphosphonic anhydride (T3P, 33.31 mL) was added at 0°C and stirred the reaction mixture for 2.5 hours at the same temperature. After completion of the reaction, the reaction mixture was quenched with cold water (100 mL) and extracted the product with ethyl acetate (2 x 150 mL). The combined organic layer was dried over sodium sulphate and evaporated the solvent under vacuum at 40°C to obtain the crude product as yellow syrup. The obtained crude product was combined with dichloromethane (1 00 mL) and filtered the solid and washed with dichloromethane (2 x 50 mL). The solid was dried under vacuum at 40°C to obtain the title compound with purity by HPLC of 99.86%. Yield : 7 g
^ Jump up to:abChen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N (2018). “Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia”. Blood. 131 (8): 855–863. doi:10.1182/blood-2017-08-797886. PMID29203585.
1: Wang AY, Weiner H, Green M, Chang H, Fulton N, Larson RA, Odenike O, Artz AS, Bishop MR, Godley LA, Thirman MJ, Kosuri S, Churpek JE, Curran E, Pettit K, Stock W, Liu H. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol. 2018 Jan 5;11(1):4. doi: 10.1186/s13045-017-0550-8. PubMed PMID: 29304833.
2: Crochiere ML, Hannus S, Hansen K, Becker F, Baloglu E, Lee M, Kauffman M, Shacham S, Landesman Y. XPO1 target occupancy measurements confirm the selinexor recommended phase 2 dose. Oncotarget. 2017 Nov 30;8(66):110503-110516. doi: 10.18632/oncotarget.22801. eCollection 2017 Dec 15. PubMed PMID: 29299164; PubMed Central PMCID: PMC5746399.
3: Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, Giralt S, Mateos MV, Leleu X, Anderson KC. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2017 Jan 16. doi: 10.1038/leu.2017.329. [Epub ahead of print] Review. PubMed PMID: 29257139.
4: Bobillo S, Abrisqueta P, Carpio C, Raheja P, Castellví J, Crespo M, Bosch F. Promising activity of selinexor in the treatment of a patient with refractory diffuse large B-cell lymphoma and central nervous system involvement. Haematologica. 2017 Dec 14. pii: haematol.2017.181636. doi: 10.3324/haematol.2017.181636. [Epub ahead of print] PubMed PMID: 29242296.
5: Chen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N, Baz R, Mau-Sorensen M, Berdeja JG, Savona M, Savoie L, Trudel S, Areethamsirikul N, Unger TJ, Rashal T, Hanke T, Kauffman M, Shacham S, Reece D. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom’s macroglobulinemia. Blood. 2017 Dec 4. pii: blood-2017-08-797886. doi: 10.1182/blood-2017-08-797886. [Epub ahead of print] PubMed PMID: 29203585.
6: Corno C, Stucchi S, De Cesare M, Carenini N, Stamatakos S, Ciusani E, Minoli L, Scanziani E, Argueta C, Landesman Y, Zaffaroni N, Gatti L, Perego P. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem Pharmacol. 2018 Jan;147:93-103. doi: 10.1016/j.bcp.2017.11.009. Epub 2017 Nov 16. PubMed PMID: 29155058.
7: Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman MG, Philip PA, Mohammad RM. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget. 2017 Jul 17;8(47):82144-82155. doi: 10.18632/oncotarget.19285. eCollection 2017 Oct 10. PubMed PMID: 29137251; PubMed Central PMCID: PMC5669877.
8: Chen Y, Zhang L, Huang J, Hong X, Zhao J, Wang Z, Zhang K. Dasatinib and chemotherapy in a patient with early T-cell precursor acute lymphoblastic leukemia and NUP214-ABL1 fusion: A case report. Exp Ther Med. 2017 Nov;14(5):3979-3984. doi: 10.3892/etm.2017.5046. Epub 2017 Aug 28. PubMed PMID: 29067094; PubMed Central PMCID: PMC5647690.
9: Body S, Esteve-Arenys A, Miloudi H, Recasens-Zorzo C, Tchakarska G, Moros A, Bustany S, Vidal-Crespo A, Rodriguez V, Lavigne R, Com E, Casanova I, Mangues R, Weigert O, Sanjuan-Pla A, Menéndez P, Marcq B, Picquenot JM, Pérez-Galán P, Jardin F, Roué G, Sola B. Cytoplasmic cyclin D1 controls the migration and invasiveness of mantle lymphoma cells. Sci Rep. 2017 Oct 24;7(1):13946. doi: 10.1038/s41598-017-14222-1. PubMed PMID: 29066743; PubMed Central PMCID: PMC5654982.
10: Broccoli A, Argnani L, Zinzani PL. Peripheral T-cell lymphomas: Focusing on novel agents in relapsed and refractory disease. Cancer Treat Rev. 2017 Nov;60:120-129. doi: 10.1016/j.ctrv.2017.09.002. Epub 2017 Sep 18. Review. PubMed PMID: 28946015.
11: Soung YH, Kashyap T, Nguyen T, Yadav G, Chang H, Landesman Y, Chung J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget. 2017 May 18;8(32):52935-52947. doi: 10.18632/oncotarget.17987. eCollection 2017 Aug 8. PubMed PMID: 28881784; PubMed Central PMCID: PMC5581083.
12: Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, S S, Nagare RP, Said JW, Doan NB, Ding LW, Baloglu E, Shacham S, Kauffman M, Koeffler HP. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep. 2017 Aug 29;7(1):9749. doi: 10.1038/s41598-017-10325-x. PubMed PMID: 28852098; PubMed Central PMCID: PMC5575339.
13: Conforti F, Zhang X, Rao G, De Pas T, Yonemori Y, Rodriguez JA, McCutcheon JN, Rahhal R, Alberobello AT, Wang Y, Zhang YW, Guha U, Giaccone G. Therapeutic Effects of XPO1 Inhibition in Thymic Epithelial Tumors. Cancer Res. 2017 Oct 15;77(20):5614-5627. doi: 10.1158/0008-5472.CAN-17-1323. Epub 2017 Aug 17. PubMed PMID: 28819023.
14: Arango NP, Yuca E, Zhao M, Evans KW, Scott S, Kim C, Gonzalez-Angulo AM, Janku F, Ueno NT, Tripathy D, Akcakanat A, Naing A, Meric-Bernstam F. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017 Aug 15;19(1):93. doi: 10.1186/s13058-017-0878-6. PubMed PMID: 28810913; PubMed Central PMCID: PMC5557476.
15: Schaffer M, Chaturvedi S, Davis C, Aquino R, Stepanchick E, Versele M, Liu Y, Yang J, Lu R, Balasubramanian S. Identification of potential ibrutinib combinations in hematological malignancies using a combination high-throughput screen. Leuk Lymphoma. 2017 Jul 28:1-10. doi: 10.1080/10428194.2017.1349899. [Epub ahead of print] PubMed PMID: 28750570.
16: Muz B, Azab F, de la Puente P, Landesman Y, Azab AK. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl Oncol. 2017 Aug;10(4):632-640. doi: 10.1016/j.tranon.2017.04.010. Epub 2017 Jun 29. PubMed PMID: 28668761; PubMed Central PMCID: PMC5496204.
17: Gupta A, Saltarski JM, White MA, Scaglioni PP, Gerber DE. Therapeutic Targeting of Nuclear Export Inhibition in Lung Cancer. J Thorac Oncol. 2017 Sep;12(9):1446-1450. doi: 10.1016/j.jtho.2017.06.013. Epub 2017 Jun 21. PubMed PMID: 28647672; PubMed Central PMCID: PMC5572747.
18: Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E, Unger TJ, Kashyap T, Klebanov B, Sola-Visner M, Crochiere M, Italiano JE Jr, Landesman Y. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood. 2017 Aug 31;130(9):1132-1143. doi: 10.1182/blood-2016-11-752840. Epub 2017 Jun 19. PubMed PMID: 28630120; PubMed Central PMCID: PMC5580272.
19: Podar K, Pecherstorfer M. Current and developing synthetic pharmacotherapy for treating relapsed/refractory multiple myeloma. Expert Opin Pharmacother. 2017 Aug;18(11):1061-1079. doi: 10.1080/14656566.2017.1340942. Epub 2017 Jul 5. Review. PubMed PMID: 28604120.
20: Tandon N, Kumar SK. Highlights of Multiple Myeloma at the Annual Meeting of American Society of Hematology, 2016. Indian J Hematol Blood Transfus. 2017 Jun;33(2):153-158. doi: 10.1007/s12288-017-0796-x. Epub 2017 Feb 28. Review. PubMed PMID: 28596644; PubMed Central PMCID: PMC5442069.
Karyopharm’s Selinexor Receives Fast Track Designation from FDA for the Treatment of Patients with Penta-Refractory Multiple Myeloma
NEWTON, Mass., April 10, 2018 (GLOBE NEWSWIRE) — Karyopharm Therapeutics Inc. (Nasdaq:KPTI), a clinical-stage pharmaceutical company, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to the Company’s lead, oral Selective Inhibitor of Nuclear Export (SINE) compound selinexor for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy. The FDA’s statement, consistent with the design of Karyopharm’s Phase 2b STORM study, noted that the three prior lines of therapy include regimens comprised of an alkylating agent, a glucocorticoid, Velcade® (bortezomib), Kyprolis® (carfilzomib), Revlimid® (lenalidomide), Pomalyst® (pomalidomide) and Darzalex® (daratumumab). In addition, the patient’s disease must be refractory to at least one proteasome inhibitor (Velcade or Kyprolis), one immunomodulatory agent (Revlimid or Pomalyst), glucocorticoids and to Darzalex, as well as to the most recent therapy. The Company expects to report top-line data from the STORM study at the end of April 2018.
The FDA’s Fast Track program facilitates the development of drugs intended to treat serious conditions and that have the potential to address unmet medical needs. A drug program with Fast Track status is afforded greater access to the FDA for the purpose of expediting the drug’s development, review and potential approval. In addition, the Fast Track program allows for eligibility for Accelerated Approval and Priority Review, if relevant criteria are met, as well as for Rolling Review, which means that a drug company can submit completed sections of its New Drug Application (NDA) for review by FDA, rather than waiting until every section of the NDA is completed before the entire application can be submitted for review.
“The designation of Fast Track for selinexor represents important recognition by the FDA of the potential of this anti-cancer agent to address the significant unmet need in the treatment of patients with penta-refractory myeloma that has continued to progress despite available therapies,” said Sharon Shacham, PhD, MBA, Founder, President and Chief Scientific Officer of Karyopharm. “We are fully committed to working closely with the FDA as we continue development of this potential new, orally-administered treatment for patients who currently have no other treatment options of proven benefit.”
About the Phase 2b STORM Study
In the multi-center, single-arm Phase 2b STORM (Selinexor Treatment of Refractory Myeloma) study, approximately 122 patients with heavily pretreated, penta-refractory myeloma receive 80mg oral selinexor twice weekly in combination with 20mg low-dose dexamethasone, also dosed orally twice weekly. Patients with penta-refractory disease are those who have previously received an alkylating agent, a glucocorticoid, two immunomodulatory drugs (IMiDs) (Revlimid® (lenalidomide) and Pomalyst® (pomalidomide)), two proteasome inhibitors (PIs) (Velcade® (bortezomib) and Kyprolis® (carfilzomib)), and the anti-CD38 monoclonal antibody Darzalex® (daratumumab), and their disease is refractory to at least one PI, at least one IMiD, Darzalex, glucocorticoids and their most recent anti-myeloma therapy. Overall response rate is the primary endpoint of the study, with duration of response and clinical benefit rate being secondary endpoints. All responses will be adjudicated by an Independent Review Committee (IRC).
About Selinexor
Selinexor (KPT-330) is a first-in-class, oral Selective Inhibitor of Nuclear Export (SINE) compound. Selinexor functions by binding with and inhibiting the nuclear export protein XPO1 (also called CRM1), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. To date, over 2,300 patients have been treated with selinexor, and it is currently being evaluated in several mid- and later-phase clinical trials across multiple cancer indications, including in multiple myeloma in a pivotal, randomized Phase 3 study in combination with Velcade® (bortezomib) and low-dose dexamethasone (BOSTON), in combination with low-dose dexamethasone (STORM) and as a potential backbone therapy in combination with approved therapies (STOMP), and in diffuse large B-cell lymphoma (SADAL), and liposarcoma (SEAL), among others. Additional Phase 1, Phase 2 and Phase 3 studies are ongoing or currently planned, including multiple studies in combination with one or more approved therapies in a variety of tumor types to further inform Karyopharm’s clinical development priorities for selinexor. Additional clinical trial information for selinexor is available at www.clinicaltrials.gov.
About Karyopharm Therapeutics
Karyopharm Therapeutics Inc. (Nasdaq:KPTI) is a clinical-stage pharmaceutical company focused on the discovery, development and subsequent commercialization of novel first-in-class drugs directed against nuclear transport and related targets for the treatment of cancer and other major diseases. Karyopharm’s SINE compounds function by binding with and inhibiting the nuclear export protein XPO1 (or CRM1). In addition to single-agent and combination activity against a variety of human cancers, SINE compounds have also shown biological activity in models of neurodegeneration, inflammation, autoimmune disease, certain viruses and wound-healing. Karyopharm, which was founded by Dr. Sharon Shacham, currently has several investigational programs in clinical or preclinical development.
/////////Selinexor, FDA 2019, セリネクソル ,KPT-330, KPT 330 , KPT330, AML, Glioma, Sarcoma, Leukemia, Fast Track, CANCER
Reldesemtiv, also known as CK-2127107, is a skeletal muscle troponin activator (FSTA) and is a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue such as SMA, COPD, and ALS.
Cytokinetics , in collaboration with Astellas , is developing reldesemtiv, the lead from a program of selective fast skeletal muscle troponin activators, in an oral suspension formulation, for the treatment of indications associated with neuromuscular dysfunction, including spinal muscular atrophy and amyotrophic lateral sclerosis.
Originator Cytokinetics
Developer Astellas Pharma; Cytokinetics
Class Pyridines; Pyrimidines; Pyrroles; Small molecules
05 May 2019 Safety and efficacy data from the phase II FORTITUDE-ALS trial in Amyotrophic lateral sclerosis presented at the American Academy of Neurology Annual Meeting (AAN-2019)
07 Mar 2019 Cytokinetics completes the phase III FORTITUDE-ALS trial for Amyotrophic lateral sclerosis in USA, Australia, Canada, Spain, Ireland and Netherlands (PO) (NCT03160898)
22 Jan 2019 Cytokinetics plans a phase I trial in Healthy volunteers in the first quarter of 2019
Reldesemtiv, a next-generation, orally-available, highly specific small-molecule is being developed by Cytokinetics, in collaboration with Astellas Pharma, for the improvement of skeletal muscle function associated with neuromuscular dysfunction, muscle weakness and/or muscle fatigue in spinal muscular atrophy (SMA), chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS). The drug candidate is a fast skeletal muscle troponin activator (FSTA) or troponin stimulant intended to slow the rate of calcium release from the regulatory troponin complex of fast skeletal muscle fibers. Clinical development for ALS, COPD and SMA is underway in the US, Australia, Canada, Ireland, Netherlands and Spain. No recent reports of development had been identified for phase I development for muscular atrophy in the US. Due to lack of of efficacy determined at interim analysis Cytokinetics suspended phase I trial in muscle fatigue in the elderly.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
[0003] Of the thirteen distinct classes of myosin in human cells, the myosin-II class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-II forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere’s thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-binding of the globular head from the actin filament (Ca2+ regulated) coupled with return of the catalytic domain and light chain to their starting conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin-tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the
Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
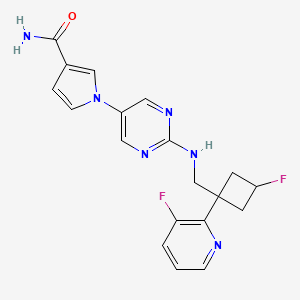
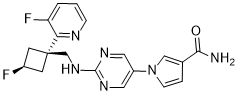
U.S. Patent No. 8962632 discloses l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide, a next-generation fast skeletal muscle troponin activator (FSTA) as a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue.
claiming the use of a similar compound for treating stress urinary incontinence.
Compound A is 1- [2-({[trans-3-fluoro-1- (3-fluoropyridin-2-yl) cyclobutyl] methyl} amino) pyrimidin-5-yl] -1H Pyrrole-3-carboxamide, which is the compound described in Example 14 of the aforementioned US Pat. The chemical structure is as shown below.
Process for preparing reldesemtiv , a myosin, actin, tropomyosin, troponin C, troponin I, troponin T modulator, useful for treating neuromuscular disorders, muscle wasting, claudication and metabolic syndrome.
Scheme 1
[0091] Scheme 1 illustrates a scheme of synthesizing the compound of Formula (1C).
Scheme 2
[0092] Scheme 2 illustrates an alternative scheme of synthesizing the compound of Formula (1C).
M
TFAA DS, toluene
Et
to
2
HCI, H20
50°C
Scheme 3
[0093] Scheme 3 illustrates a scheme of converting the compound of Formula (1C) to the compound of Formula (II).
H2
Ni Raney
NH3
Scheme 4
[0094] Scheme 4 illustrates a scheme of converting the compound of Formula (II) to the compound of Formula (1).
Examples
[0095] To a flask was added N-methylpyrrolidone (30 mL), tert-butyl cyanoacetate (8.08 g) at room temperature. To a resulting solution was added potassium tert-butoxide (7.71 g), l,3-dibromo-2,2-dimethoxy propane (5.00 g) at 0 °C. To another flask, potassium iodide (158 mg), 2,6-di-tert-butyl-p-cresol (42 mg), N-methylpyrrolidone (25 mL) were added at room temperature and then resulting solution was heated to 165 °C. To this solution, previously prepared mixture was added dropwise at 140-165 °C, then stirred for 2 hours at 165 °C. To the reaction mixture, water (65 mL) was added. A resulting solution was extracted with toluene (40 mL, three times) and then combined organic layer was washed with water (20 mL, three times) and 1N NaOH aq. (20 mL). A resulting organic layer was concentrated below 50 °C under reduced pressure to give 3, 3 -dimethoxy cyclobutane- l-carbonitrile (66% yield,
Example 2 Synthesis of methyl 3,3-dimethoxycyclobutane-l-carboxylate
[0096] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. MeOH (339.00 kg), 3-oxocyclobutanecarboxylic acid (85.19 kg, 746.6 mol, 1.0 eq.), Amberlyst-l5 ion exchange resin (8.90 kg, 10% w/w), and
trimethoxymethane (196.00 kg, 1847.3 mol, 2.5 eq.) were charged into the reactor and the resulting mixture was heated to 55±5°C and reacted for 6 hours to give methyl 3,3-dimethoxycyclobutane-l-carboxylate solution in MeOH. 1H NMR (CDCl3, 400 MHz) d 3.70 (s, 3H), 3.17 (s, 3H), 3.15 (s, 3H), 2.94-2.85 (m, 1H), 2.47-2.36 (m, 4H).
Example 3 Synthesis of 3, 3-dimethoxycyclobutane-l -carboxamide
[0097] The methyl 3, 3 -dimethoxy cyclobutane- l-carboxylate solution in MeOH prepared as described in Example 2 was cooled to below 25°C and centrifuged. The filter cake was washed with MeOH(7.00 kg) and the filtrate was pumped to the reactor. The solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH
(139.40 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH (130.00 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. Half of the resulting solution was diluted with MeOH (435.00 kg) and cooled to below 30°C. NH3 gas (133.80 kg) was injected into the reactor below 35°C for
24 hours. The mixture was stirred at 40±5°C for 72 hours. The resulting solution was
concentrated under vacuum below 50°C until the system had no more than 2 volumes.
MTBE(l8l.OO kg) was charged into the reactor. The resulting solution was concentrated under vacuum below 50°C until the system had no more than 2 volumes. PE (318.00 kg) was charged into the reactor. The resulting mixture was cooled to 5±5°C, stirred for 4 hours at 5±5°C, and centrifuged. The filter cake was washed with PE (42.00 kg) and the wet filter cake was put into a vacuum oven. The filter cake was dried at 30±5°C for at least 8 hours to give 3,3-dimethoxycyclobutane-l-carboxamide as off-white solid (112.63 kg, 94.7% yield). 1H NMR (CDCf, 400 MHz) d 5.76 (bs, 1H), 5.64 (bs, 1H), 3.18 (s, 3H), 3.17 (s, 3H), 2.84-2.76 (m, 1H), 2.45-2.38 (m, 4H).
[0098] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Toluene (500.00 kg), 3,3-dimethoxycyclobutane-l-carboxamide (112.54kg, 706.9 mol, 1.0 eq.), and TEA (158.00 kg, 1561.3 mol, 2.20 eq) were charged into the reactor and the resulting mixture was cooled to 0+ 5°C. TFAA (164.00 kg, 781 mol, 1.10 eq.) was added dropwise at 0±5°C. The resulting mixture was stirred for 10 hours at 20±5°C and cooled below 5±5°C. H20 (110.00 kg) was charged into the reactor at below 15 °C. The resulting mixture was stirred for 30 minutes and the water phase was separated. The aqueous phase was extracted with toluene (190.00 kg) twice. The organic phases were combined and washed with H20 (111.00 kg). H20 was removed by azeotrope until the water content was no more than 0.03%. The resulting solution was cooled to below 20°C to give 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene (492.00 kg with 17.83% assay content, 87.9% yield).
Example 5 Synthesis of l-(3-fluoropyridin-2-yl)-3,3-dimethoxycyclobutane-l-carbonitrile
[0099] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene prepared as described in Example 4 (246.00 kg of a 17.8% solution of 3,3-dimethoxycyclobutane-l-carbonitrile in toluene, 1.05 eq.) and 2-chloro-3-fluoropyridine (39.17 kg, 297.9 mol, 1.00 eq.) were charged into the reactor. The reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The mixture was slowly cooled to -20±5°C. NaHDMS (2M in THF) (165.71 kg, 1.20 eq) was added
dropwise at -20±5°C. The resulting mixture was stirred at -l5±5°C for 1 hour. The mixture was stirred until the content of 2-chloro-3-fluoropyridine is no more than 2% as measured by HPLC. Soft water (16.00 kg) was added dropwise at below 0°C while maintaining the reactor temperature. The resulting solution was transferred to another reactor. Aq. NH4Cl (10% w/w, 88.60 Kg) was added dropwise at below 0°C while maintaining the reactor temperature. Soft water (112.00 kg) was charged into the reactor and the aqueous phase was separated and collected. The aqueous phase was extracted with ethyl acetate (70.00 kg) and an organic phase was collected. The organic phase was washed with sat. NaCl (106.00 kg) and collected. The above steps were repeated to obtain another batch of organic phase. The two batches of organic phase were concentrated under vacuum below 70°C until the system had no more than 2 volumes. The resulting solution was cooled to below 30°C to give a l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution. 1H NMR (CDC13, 400 MHz) d 8.42-8.38 (m, 1H), 7.50-7.45 (m, 1H), 7.38-7.33 (m, 1H), 3.28 (s, 3 H), 3.13 (s, 3H), 3.09-3.05 (m, 4H).
Example 6 Synthesis of I-(3-fluoropyridin-2-yl)-3-oxocyclohutanecarhonitrile
[0100] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Water (603.00 kg) was added to the reactor and was stirred.
Concentrated HC1 (157.30 kg) was charged into the reactor at below 35°C. The l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution prepared as described in Example 5 (206.00 kg) was charged into the reactor and the resulting mixture was heated to 50±5°C and reacted for 3 hours at 50±5°C. The mixture was reacted until the content of 1-(3 -fluoropyridin-2-yl)-3, 3 -dimethoxycyclobutane- l-carbonitrile was no more than 2.0% as measured by HPLC. The reaction mixture was cooled to below 30°C and extracted with ethyl acetate (771.00 kg). An aqueous phase was collected and extracted with ethyl acetate (770.00 kg). The organic phases were combined and the combined organic phase was washed with soft water (290.00 kg) and brine (385.30 kg). The organic phase was concentrated under vacuum at below 60°C until the system had no more than 2 volumes. Propan-2-ol (218.00 kg) was charged into the reactor. The organic phase was concentrated under vacuum at below
60°C until the system had no more than 1 volume. PE (191.00 kg) was charged into the reactor at 40±5 °C and the resulting mixture was heated to 60±5 °C and stirred for 1 hour at 60±5 °C. The mixture was then slowly cooled to 5±5 °C and stirred for 5 hours at 5±5 °C. The mixture was centrifuged and the filter cake was washed with PE (48.00 kg) and the wet filter cake was collected. Water (80.00 kg), concentrated HC1 (2.20 kg), propan-2-ol (65.00 kg), and the wet filter cake were charged in this order into a drum. The resulting mixture was stirred for 10 minutes at 20±5 °C. The mixture was centrifuged and the filter cake was washed with a mixture solution containing 18.00 kg of propan-2-ol, 22.50 kg of soft water, and 0.60 kg of concentrated HC1. The filter cake was put into a vacuum oven and dried at 30±5°C for at least 10 hours. The filter cake was dried until the weight did not change to give l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile as off-white solid (77.15 kg, 68.0% yield). 1H NMR (CDCl3, 400 MHz) d 8.45-8.42 (m, 1H), 7.60-7.54 (m, 1H), 7.47-7.41 (m, 1H), 4.18-4.09 (m, 2H), 4.02-3.94 (m, 2H).
Example 7 Synthesis of I-(3-fhtoropyridin-2-yl)-3-hydroxycyclobulanecarbonilrile
[0101] To a solution of l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g,
1.22 mol) in a mixture ofDCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78° C. The reaction mixture was stirred at -78°C. for 1 hour and quenched with a mixture of methanol and water (1 : 1). The organic layer was washed with water (500 mL><3), dried over Na2S04, and concentrated. The residue was purified on silica gel (50% EtO Ac/hexanes) to provide the title compound as an amber oil (185.8 g, 77.5%). Low Resolution Mass
Spectrometry (LRMS) (M+H) m/z 193.2.
Example 8 Synthesis of (ls,3s)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutane-l-carbonitrile
[0102] To a solution of 1 -(3 -fluoropyridin-2-yl)-3 -hydroxy cyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 hours. The reaction was cooled to rt and poured onto sat. NaHCCf solution. The mixture was separated and the organic layer was washed with water, dried over Na2S04, and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (116g) in a 8: 1 transxis mixture. The above brown oil (107 g) was dissolved in toluene (110 mL) and hexanes (330mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g). LRMS (M+H) m/z 195.1.
Example 9 Synthesis of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine
[0103] A mixture of ( 1.v,3.v)-3-fluoro- 1 -(3-fluoropyridin-2-yl)cyclobutane- 1 -carbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g, 97.6%). LRMS (M+H) m/z 199.2.
Example 10 Synthesis of t-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate
[0104] A mixture of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine (37.6 g, 190 mmol), 5-bromo-2-fluoropyrimidine (32.0 g, 181 mmol), DIPEA (71 mL, 407 mmol), and NMP (200 mL) was stirred at rt overnight. The reaction mixture was then diluted with EtOAc (1500 mL) and washed with saturated sodium bicarbonate (500 mL). The
organic layer was separated, dried over Na2S04, and concentrated. The resultant solid was dissolved in THF (600 mL), followed by the slow addition of DMAP (14 g, 90 mmol) and Boc20 (117.3 g, 542 mmol). The reaction was heated to 60° C. and stirred for 3 h. The reaction mixture was then concentrated and purified by silica gel chromatography
(EtO Ac/hex) to give 59.7 g oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a white solid.
Example 11 Synthesis of t-butyl 5-(3-cyano- 1 H -pyrrol- 1 -yl)pyrimidin-2-yl(((lrans)-3-fhtoro-l-(3-fluoropyridin-2-yl)cyclohutyl)methyl)carhamate
[0105] To a solution oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate (1.0 g, 2.8 mmol) in 15 mL of toluene (degassed with nitrogen) was added copper iodide (100 mg, 0.6 mmol), potassium phosphate (1.31 g, 6.2 mmol), trans-N,N’-dimethylcyclohexane-l, 2-diamine (320 mg, 2.2 mmol), and 3-cyanopyrrole (310 mg, 3.6 mmol). The reaction was heated to 100 °C and stirred for 2 h. The reaction was then concentrated and purified by silica gel chromatography (EtOAc/hexanes) to afford 1.1 g of t-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a clear oil.
Example 12 Synthesis of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide
[0106] To a solution oft-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate (1.1 g, 3.1 mmol) in DMSO (10 mL) was added potassium carbonate (1.3 g, 9.3 mmol). The mixture was cooled to 0 °C and hydrogen peroxide (3 mL) was slowly added. The reaction was warmed to rt and stirred for 90 min. The reaction was diluted with EtO Ac (75 mL) and washed three times with brine (50 mL). The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% MeOH/CH2Cl2) to afford 1.07 g of a white solid compound. This compound was dissolved in 25% TFA/CH2CI2 and stirred for 1 hour. The reaction was then concentrated, dissolved in ethyl acetate (75 mL), and washed three times with saturated potassium carbonate solution. The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was triturated with 75% ethyl acetate/hexanes. The resultant slurry was sonicated and filtered to give 500 mg of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3 -carboxamide as a white solid. LRMS (M+H=385).
REFERENCES
1: Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018 May;57(5):729-734. doi: 10.1002/mus.26017. Epub 2017 Dec 11. PubMed PMID: 29150952.
2: Gross N. The COPD Pipeline XXXII. Chronic Obstr Pulm Dis. 2016 Jul 14;3(3):688-692. doi: 10.15326/jcopdf.3.3.2016.0150. PubMed PMID: 28848893; PubMed Central PMCID: PMC5556764.
//////////////CK-2127107, CK 2127107, CK2127107, Reldesemtiv, Cytokinetics, Astellas, neuromuscular disorders, muscle wasting, claudication, metabolic syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, Orphan Drug Status, Spinal muscular atrophy, Phase II
Axelar is developing picropodophyllin, a small-molecule IGF-1 receptor antagonist for the treatment of cancer including NSCLC and malignant astrocytoma. In February 2019, a phase Ia study was planned to initiate for solid tumor in March 2019.
Picropodophyllin is a cyclolignan alkaloid found in the mayapple plant family (Podophyllum peltatum), and a small molecule inhibitor of the insulin-like growth factor 1 receptor (IGF1R) with potential antineoplastic activity. Picropodophyllin specifically inhibits the activity and downregulates the cellular expression of IGF1R without interfering with activities of other growth factor receptors, such as receptors for insulin, epidermal growth factor, platelet-derived growth factor, fibroblast growth factor and mast/stem cell growth factor (KIT). This agent shows potent activity in the suppression o f tumor cell proliferation and the induction of tumor cell apoptosis. IGF1R, a receptor tyrosine kinase overexpressed in a variety of human cancers, plays a critical role in the growth and survival of many types of cancer cells.
Picropodophyllotoxin is an organic heterotetracyclic compound that has a furonaphthodioxole skeleton bearing 3,4,5-trimethoxyphenyl and hydroxy substituents. It has a role as an antineoplastic agent, a tyrosine kinase inhibitor, an insulin-like growth factor receptor 1 antagonist and a plant metabolite. It is a lignan, a furonaphthodioxole and an organic heterotetracyclic compound.
Picropodophyllin has been investigated for the treatment of Non Small Cell Lung Cancer.
One of the largest challenges in pharmaceutical drug development is that drug compounds often are poorly soluble, or even insoluble, in aqeous media. Insufficient drug solubility means insufficient bioavailability, as well as poor plasma exposure of the drug when administered to humans and animals. Variability of plasma exposure in humans is yet a problem when developing drugs which are poorly soluble, or even insoluble, in aqeous media.
It is estimated that between 40% and 70 % of all new chemical entities identified in drug discovery programs, are insufficiently soluble in aqeous media (M. Lindenberg, S et al: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004). Scientists have investigated various ways of solving the problem with poor drug solubility in order to enhance bioavailability of poorly absorbed drugs, aiming at increasing their clinical efficacy when administered orally.
Technologies such as increase of the surface area and hence dissolution may sometimes solve solubility problems. Other techniques that may also solve bioavailability problems are addition of surfactants and polymers. However, each chemical compound has its own unique chemical and physical properties, and hence has its own unique challenges when being formulated into a pharmaceutical product that can exert its clinical efficacy.
Picropodophyllin is an insulin-like growth factor-1 receptor inhibitor fiGF-lR inhibitor) small-molecule compound belonging to the class of compounds denominated cyclolignans, having the chemical structure:
The patent applicant is presently entering clinical phase II development with its development compound picropodophyllin (AXL1717). However, picropodophyllin is poorly soluble in aqueous media. In a phase I clinical study performed by the applicant in 2012 (Ekman S et al; Acta Oncologica, 2016; 55: pp. 140-148), it was discovered that picropodophyllin, when administered as an oral suspension to lung cancer patients, resulted in unacceptable variability in drug exposure. A large variability in plasma exposure of the active drug picropodophyllin occurred not only within certain patients, but also between several patients.
Yet a problem with administering picropodophyllin as an aqeous solution, is that due to the poor solubility in aqueous media, it is difficult or even impossible to reach the required therapeutic doses.
The compound picropodophyllin is furthermore physically unstable, and transforms from amorphous picropodophyllin into crystalline picropodophyllin. Yet a stability problem with picropodophyllin is that it is chemically unstable in solution.
Novel amorphous forms of picropodophyllin , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating cancers, such as neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, and eye cancers. , claiming picropodophyllin derivatives as modulators of insulin-like growth factor-1 receptor (IGF-1), useful for treating cancers, assigned to Axelar AB ,
A nickel-catalyzed reductive cascade approach to the efficient construction of diastereodivergent cores embedded in podophyllum lignans is developed for the first time. Their gram-scale access paved the way for unified syntheses of naturally occurring podophyllotoxin and other members.
he first catalytic enantioselective total synthesis of (−)-podophyllotoxin is accomplished by a challenging organocatalytic cross-aldol Heck cyclization and distal stereocontrolled transfer hydrogenation in five steps from three aldehydes. Reversal of selectivity in hydrogenation led to the syntheses of other stereoisomers from the common precursor.
(-)-Picropodophyllin 4. The lactone 5 (0.2 g, 0.38 mmol) was taken in 1-pentanol (5 mL) in a double neck RB flask at rt. Water (0.14 mL, 7.6 mmol) was added to above mixture and it was then degassed with argon followed by addition of Pd/C (0.04 g, 20% by wt.) and HCO2Na (0.78g, 11.4 mmol). The reaction mixture was heated at 40 °C for 12 h. On completion, the reaction mixture was diluted with EtOAc (200 mL), filtered through a celite pad and solvent was removed under vacuum. This crude mixture was dissolved in THF (3.8 mL), TBAF (1.9 mL, 1.9 mmol, 1M in THF) was added and stirred for 6 h at 27 °C. On completion, EtOAc (250 mL) was added, washed with water (100 mL), brine and dried over Na2SO4. After removal of solvent, the crude product was purified by column chromatography (hexanes-EtOAc, 3:2) to get the title compound as a white solid (0.082 g, 52%): Rf 0.32 (hexanes/EtOAc, 1:1); [α]25 D = -10.6 (c = 0.4, CHCl3) [lit. -10 (c = 0.3, CHCl3), -11 (c = 0.41, CHCl3)]3a,b;
The formylation of 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) with BuLi and DMF gives the 6-formyl derivative (II), which is reduced with NaBH4 in ethanol to yield the corresponding carbinol (III). The cyclization of (III) with dimethyl acetylenedicarboxylate (V) in hot acetic acid (through the nonisolated intermediate (IV)) affords dimethyl 1,4-epoxy-6,7-(methylenedioxy)naphthalene-2,3-dicarboxylate (VI), which is hydrogenated with H2 over Pd/C in ethyl acetate to give the (1R*,2S*,3R*,4S*)-tetrahydro derivative (VII). The reduction of (VII) with LiAlH4 in refluxing ethyl ether affords the corresponding bis carbinol (VIII), which is treated with acetic anhydride to afford the diacetate (IX). The enzymatic monodeacetylation of (VIII) with PPL enzyme in DMSO/buffer gives (1R,2R,3S,4S)-2-(acetoxymethyl)-1,4-epoxy-3-(hydroxymethyl)-6,7-(methylenedioxy)-1,2,3,4-tetrahydronaphthalene (X), which is silylated with TBDMS-Cl and imidazole in DMF yielding the silyl ether (XI). The hydrolysis of the acetoxy group of (XI) with K2CO3 in methanol affords the carbinol (XII), which is oxidized with oxalyl chloride in dichloromethane affording the carbaldehyde (XIII). The exchange of the silyl protecting group of (XIII) (for stability problems) provided the triisopropylsilyl ether (XIV), which is treated with sodium methoxide in methanol to open the epoxide ring yielding the hydroxy aldehyde (XV). The protection of the hydroxy group of (XV) with 2-(trimethylsilyl)ethoxymethyl chloride and DIEA in dichloromethane provides the corresponding ether (XVI). The carbinol (III) can also be obtained directly from 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) by reaction with formaldehyde and BuLi in THF.
The oxidation of the aldehyde group of (XVI) with NaClO2 in tert-butanol affords the corresponding carboxylic acid (XVII), which is condensed with 2-oxazolidinone (XVIII) by means of carbonyldiimidazole (CDI) in THF to give the acyl imidazolide (XIX). The arylation of (XIX) with 3,4,5-trimethoxyphenylmagnesium bromide (XX) in THF yields the expected addition product (XXI), which is cyclized by means of TBAF in hot THF to afford the tetracyclic intermediate (XXII). Isomerization of the cis-lactone ring of (XXII) with LDA in THF affords intermediate (XXIII) with its lactone ring with the correct trans-conformation. Finally, this compound is deprotected with ethyl mercaptane and MgBr2 in ethyl ether to provide the target compound.
Synthesis 1992,719
The intermediate trans-8-oxo-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetra-hydronaphtho[2,3-d][1,3]benzodioxole-6-carboxylic acid ethyl ester (XI) has been obtained by several different ways: (a) The condensation of benzophenone (XXXVIII) with diethyl malonate (XXXIX) by means of t-BuOK gives the alkylidenemalonate (XL), which is hydrogenated with H2 over Pd/C to the alkylmalonate hemiester (XLI). The reaction of (XLI) with acetyl chloride affords the mixed anhydride (XLII), which is finally cyclized to the target (XI) by means of SnCl4. (b) The cyclization of the malonic ester derivative (XLIII) by means of Ti(CF3–CO2)3 gives the 5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho [2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLIV), which is finally oxidized and decarboxylated with NBS and NaOH in methanol to afford the target intermediate (XI). (c) The cyclization of the benzylidenemalonate (XLV) with the aryllithium derivative (XLVI) gives the 8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLVII), which is demethylated with TFA and oxidized with CrO3 and pyridine to the target compound (XI). (d) The cyclopropanation of the chalcone (XLVIII) with (ethoxycarbonyl) (dimethylsulfonium)methylide (XLIX) gives the cyclopropanecarboxylate (L), which is finally rearranged with BF3/Et2O to the target intermediate (IX).
The cyclization of 3,4,5-trimethoxycinnamic acid ethyl ester (LI) with malonic acid ethyl ester potassium salt (LII) by means of Mn(OAc)3 gives the tetrahydrofuranone (LIII), which is acylated with 1,3-benzodioxol-5-ylcarbonyl chloride (LIV) yielding the tetrahydrofuranone (LV). Finally, this compound is rearranged and decarboxylated with SnCl4 to the target intermediate (XI).
The cyclization of 6-[1-hydroxy-1-(3,4,5-trimethoxyphenyl)methyl]-1,3-benzodioxol-5-carbaldehyde dimethylacetal (LVI) by means of AcOH gives 5-(3,4,5-trimethoxyphenyl)-1,3-dioxolo[4,5-f]isobenzofuran (LVII), which is submitted to a Diels-Alder cyclization with acetylenedicarboxylic acid dimethyl ester (LVIII) yielding the epoxy derivative (LIX). The selective reduction of (LIX) with LiBEt3H and H2 affords the carbinol (LX), which is treated with H2 over RaNi in order to open the epoxide ring to give the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to (LXII) with the suitable configuration. Finally, this compound is cyclized with DCC to the desired target compound.
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl maleate (LXIII) gives the expected adduct (LXIV), which by thermal extrusion of CO2 yields the dihydronaphthodioxole (LXV). This compound is then converted to dihydroxycompound (X), which is finally cyclized by means of ZnCl2 to provide the target compound. The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl fumarate (LXVI) gives the expected adduct (LXVII), which by hydrogenation with H2 over Pd/C yields the tricarboxylic acid derivative (LXVIII). The reaction of (LXVIII) with Pb(OAc)4 affords the acetoxy derivative (LXIX), which is selectively reduced with LiBEt3H providing the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to give the previously described (X) with the suitable configuration.
The reaction of benzocyclobutane derivative (LXX) with isocyanate (LXXI) by means of Ph3SnOAc gives the carbamate (LXXII), which is cyclized by a thermal treatment with LiOH yielding the tetracyclic carboxylic acid (LXXIII). The opening of the oxazinone ring of (LXXIII) in basic medium affords the tricyclic amino acid (LXXIV), which is finally cyclized to the target compound by reaction with sodium nitrite in acidic medium (pH = 4).
J Chem Soc Chem Commun 1993,1200
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with the chiral dihydrofuranone (II) in hot acetonitrile gives the pentacyclic anhydride (III), which is opened with warm acetic acid yielding the carboxylic acid (IV). Hydrogenation of the benzylic double bond of (IV) with H2 over Pd/C affords (V), which is treated with lead tetraacetate and acetic acid in THF to give the acetoxy compound (VI). The hydrolysis of the acetoxy group and the menthol hemiacetal group with HCl in hot dioxane yields the diol (VII), which is treated with diazomethane in ether/methanol affording the aldehyde (VIII). The reduction of the aldehyde group of (VIII) with LiEt3BH in THF gives the diol (IX) as a diastereomeric mixture, which is treated with HCl in THF to afford the diol (X) with the right conformation. Finally, this compound is lactonized to the target compound with ZnCl2 in THF.
Selpercatinib is a tyrosine kinase inhibitor with antineoplastic properties.
A phase I/II trial is also under way in pediatric patients and young adults with activating RET alterations and advanced solid or primary CNS tumors.
Loxo Oncology (a wholly-owned subsidiary of Eli Lilly ), under license from Array , is developing selpercatinib, a lead from a program of RET kinase inhibitors, for treating cancer, including non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma and soft tissue sarcoma
In 2018, the compound was granted orphan drug designation in the U.S. for the treatment of pancreatic cancer and in the E.U. for the treatment of medullary thyroid carcinoma.
Trk is a high affinity receptor tyrosine kinase activated by a group of soluble growth factors called neurotrophic factor (NT). The Trk receptor family has three members, namely TrkA, TrkB and TrkC. Among the neurotrophic factors are (1) nerve growth factor (NGF) which activates TrkA, (2) brain-derived neurotrophic factor (BDNF) and NT4/5 which activate TrkB, and (3) NT3 which activates TrkC. Trk is widely expressed in neuronal tissues and is involved in the maintenance, signaling and survival of neuronal cells.
The literature also shows that Trk overexpression, activation, amplification and/or mutations are associated with many cancers including neuroblastoma, ovarian cancer, breast cancer, prostate cancer, pancreatic cancer, multiple myeloma, astrocytoma. And medulloblastoma, glioma, melanoma, thyroid cancer, pancreatic cancer, large cell neuroendocrine tumor and colorectal cancer. In addition, inhibitors of the Trk/neurotrophin pathway have been shown to be effective in a variety of preclinical animal models for the treatment of pain and inflammatory diseases.
The neurotrophin/Trk pathway, particularly the BDNF/TrkB pathway, has also been implicated in the pathogenesis of neurodegenerative diseases, including multiple sclerosis, Parkinson’s disease, and Alzheimer’s disease. The modulating neurotrophic factor/Trk pathway can be used to treat these and related diseases.
It is believed that the TrkA receptor is critical for the disease process in the parasitic infection of Trypanosoma cruzi (Chagas disease) in human hosts. Therefore, TrkA inhibitors can be used to treat Chagas disease and related protozoal infections.
Trk inhibitors can also be used to treat diseases associated with imbalances in bone remodeling, such as osteoporosis, rheumatoid arthritis, and bone metastasis. Bone metastases are a common complication of cancer, up to 70% in patients with advanced breast or prostate cancer and about 15 in patients with lung, colon, stomach, bladder, uterine, rectal, thyroid or kidney cancer Up to 30%. Osteolytic metastases can cause severe pain, pathological fractures, life-threatening hypercalcemia, spinal cord compression, and other neurostress syndromes. For these reasons, bone metastases are a serious cancer complication that is costly. Therefore, an agent that can induce apoptosis of proliferating bone cells is very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, almost all osteoblast apoptosis agents are very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, localization of NGF was observed in almost all osteoblasts. Recently, it was demonstrated that pan-Trk inhibitors in human hFOB osteoblasts inhibit tyrosine signaling activated by neurotrophic factors that bind to all three Trk receptors. This data supports the theory of using Trk inhibitors to treat bone remodeling diseases, such as bone metastases in cancer patients.
Developed by Loxo Oncology, Larotrectinib (LOXO-101) is a broad-spectrum antineoplastic agent for all tumor patients expressing Trk, rather than tumors at an anatomical location. LOXO-101 chemical name is (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)pyrazolo[1,5-a] Pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide, the structural formula is as follows. LOXO-101 began treatment of the first patient in March 2015; on July 13, 2016, the FDA granted a breakthrough drug qualification for the inoperable removal or metastatic solid tumor of adults and children with positive Trk fusion gene mutations; Key entry was completed in February 2017; in November 2018, the FDA approved the listing under the trade name Vitrakvi.
Poor absorption, distribution, metabolism, and/or excretion (ADME) properties are known to be the primary cause of clinical trial failure in many drug candidates. Many of the drugs currently on the market also limit their range of applications due to poor ADME properties. The rapid metabolism of drugs can lead to the inability of many drugs that could be effectively treated to treat diseases because they are too quickly removed from the body. Frequent or high-dose medications may solve the problem of rapid drug clearance, but this approach can lead to problems such as poor patient compliance, side effects caused by high-dose medications, and increased treatment costs. In addition, rapidly metabolizing drugs may also expose patients to undesirable toxic or reactive metabolites.
Although LOXO-101 is effective as a Trk inhibitor in the treatment of a variety of cancers and the like, it has been found that a novel compound having a good oral bioavailability and a drug-forming property for treating a cancer or the like is a challenging task. Thus, there remains a need in the art to develop compounds having selective inhibitory activity or better pharmacodynamics/pharmacokinetics for Trk kinase mediated diseases useful as therapeutic agents, and the present invention provides such compounds.
Compounds of Formula I-IV, 4-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula I); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula II); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-(6-methoxynicotinoyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula III); and 6-(2-hydroxy-2-methylpropoxy)-4-(6-(4-hydroxy-4-(pyridin-2-ylmethyl)piperidin-1-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula IV) are inhibitors of RET kinase, and are useful for treating diseases such as proliferative diseases, including cancers.
[0007] Accordingly, provided herein is a compound of Formula I-IV:
and pharmaceutically acceptable salts, amorphous, and polymorph forms thereof.
PATENT
WO 2019075114
PATENT
WO-2019120194
Novel deuterated analogs of pyrazolo[1,5-a]pyrimidine compounds, particularly selpercatinib , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, inflammation, cancer and certain infectious diseases.
Example 2(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl-2,3,3-d 3)-pyrazolo[ 1,5-a] pyrimidin-3-yl) -3-hydroxypyrazole prepared pyrrolidine-1-carboxamide (compound L-2) a.
No development reported B-cell lymphoma; Lymphoid leukaemia
26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
The kinase ataxia telangiectasia mutated and rad3 related (ATR) is a key regulator of the DNA-damage response and the apical kinase which orchestrates the cellular processes that repair stalled replication forks (replication stress) and associated DNA double-strand breaks. Inhibition of repair pathways mediated by ATR in a context where alternative pathways are less active is expected to aid clinical response by increasing replication stress. Here we describe the development of the clinical candidate 2(AZD6738), a potent and selective sulfoximine morpholinopyrimidine ATR inhibitor with excellent preclinical physicochemical and pharmacokinetic (PK) characteristics. Compound 2 was developed improving aqueous solubility and eliminating CYP3A4 time-dependent inhibition starting from the earlier described inhibitor 1 (AZ20). The clinical candidate 2 has favorable human PK suitable for once or twice daily dosing and achieves biologically effective exposure at moderate doses. Compound 2 is currently being tested in multiple phase I/II trials as an anticancer agent.
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).
Scheme 1. Medicinal Chemistry Route to AZD6738
Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
AZD6738 is currently being tested in multiple phase I/II trials for the treatment of cancer. Its structure, comprising a pyrimidine core decorated with a chiral morpholine, a cyclopropyl sulfoximine, and an azaindole, make it a challenging molecule to synthesize on a large scale. We describe the evolution of the chemical processes, following the manufacture of AZD6738 from the initial scale-up through to multikilos on plant scale. During this evolution, we developed a biocatalytic process to install the sulfoxide with high enantioselectivity, followed by introduction of the cyclopropyl group first in batch, then in a continuous flow plate reactor, and finally through a series of continuous stirred tank reactors. The final plant scale process to form AZD6738 was operated on 46 kg scale with an overall yield of 18%. We discuss the impurities formed throughout the process and highlight the limitations of this route for further scale-up.
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) (30.0 g) were added at 75 °C, and the reaction mixture was held for 2 h. The mixture was cooled to 20 °C, and n-heptane (141.9 kg) was added at the rate of 40 kg/h. The solid was collected by filtration, washed with a mixture of 1-butanol and n-heptane (9.3 and 22.4 kg respectively), and then given a further wash with n-heptane (32.2 kg). The solid was dried at 40 °C to give imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) as a whit solid (41.4 kg, 92% yield): Assay (HPLC) 99.9%; Assay (NMR) 99% wt/wt.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.
//////AZD6738, AZD-6738, AZD 6738, AstraZeneca, University of Pennsylvania, Phase II, Breast cancer, Gastric cancer, Non-small cell lung cancer, Ovarian cancer, Ceralasertib
Ceralasertib
CAS 1352226-88-0
(R)-imino(methyl)(1-{6-[(3R)-3-methylmorpholin-4-yl]-2-{1H-pyrrolo[2,3-c]pyridin-4-yl}pyrimidin-4-yl}cyclopropyl)-lambda6-sulfanone
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane
UNII 85RE35306Z
Molecular Weight
412.51
Formula
C20H24N6O2S
AZD 6738, ATR KINASE INHIBITOR AZD6738, AZD-6738, AZD6738, WHO 10842
Ceralasertib (AZD6738) is an orally active and bioavailable inhibitor of ATR kinase with an IC50 of 1 nM.
Ceralasertib is an investigational new drug that is being evaluated for the treatment of cancer.[1] It is an ATR kinase inhibitor.[2]
Originator AstraZeneca; University of Pennsylvania
Developer Acerta Pharma; AstraZeneca; Dana-Farber Cancer Institute; Gustave Roussy; National Cancer Institute (France); Samsung Medical Center; University of California at San Francisco; University of Pennsylvania
Class Antineoplastics; Cyclopropanes; Imines; Ketones; Morpholines; Organic sulfur compounds; Pyridines; Pyrimidines; Pyrroles; Small molecules
Mechanism of Action ATR protein inhibitors
Phase III Non-small cell lung cancer
Phase IICholangiocarcinoma; Gynaecological cancer; Malignant melanoma; Osteosarcoma; Ovarian cancer; Pancreatic cancer; Prostate cancer; Small cell lung cancer; Solid tumours; Triple negative breast cancer
PreclinicalDiffuse large B cell lymphoma; Type 1 diabetes mellitus
DiscontinuedHaematological malignancies; Non-Hodgkin’s lymphoma; Squamous cell cancer
22 Apr 2025AstraZeneca plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) (PO) in April 2025(NCT06929260)
23 Dec 2024AstraZeneca plans a phase I trial for Non-small Cell Lung Cancer, Ovarian Cancer, or Endometrial Cancer in United Kingdom(PO) In January 2025 (NCT06754761)
07 Dec 2024Updated efficacy and adverse event data from a phase I trial in Myelodysplastic syndrome presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
SCHEME
PATENT
WO2011154737
CN112939966
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00482
4-{4-[(3R)-3-methyl-4-morpholinyl]-6-[1-(S-methylsulfonimidoyl)cyclopropyl]-2-
pyrimidinyl}-1H-pyrrolo[2,3-b]pyridine (1).
Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry
Literature References: Prepn: R. Ueno et al.,EP289349; eidem,US5221763 (1988, 1993 both to Ueno). Pharmacological characterization: Y. Goh, J. Kishino, Jpn. J. Ophthalmol.38, 236 (1994). Mechanism of action study: M. Sakurai et al.,ibid.37, 252 (1993). Comparative clinical trial in glaucoma and ocular hypertension: J.-P. Nordmann et al., Am. J. Ophthalmol.133, 1 (2002).
13,14-Dihydro-15-keto-20-ethyl-PGF2
Derivative Type: Isopropyl ester
CAS Registry Number: 120373-24-2
Manufacturers’ Codes: UF-021
Trademarks: Rescula (Novartis)
Molecular Formula: C25H44O5
Molecular Weight: 424.61
Percent Composition: C 70.72%, H 10.44%, O 18.84%
Therap-Cat: Antiglaucoma; in treatment of ocular hypertension.
Unoprostone isopropyl is a prostaglandin analogue. Ophthalmic Solution 0.15% is a synthetic docosanoid. Unoprostone isopropyl has the chemical name isopropyl (+)-(Z)-7-[(1R,2R,3R,5S)-3,5 dihydroxy-2-(3-oxodecyl)cyclopentyl]-5-heptenoate. The main indication of Unoprostane is treatment of glucoma.
This compound can be prepared by two different ways: 1) The reaction of 1-benzyl-4-(hydroxymethyl)pyrrolidin-2-one (I) with SOCl2 in refluxing dichloromethane gives 1-benzyl-4-(chloromethyl)pyrrolidin-2-one (II), which is condensed with potassium phthalimide (III) in DMF yielding 1-benzyl-4-(phthalimidomethyl)pyrrolidin-2-one (IV). Finally, this compound is treated with hydrazine in ethanol and neutralized with fumaric acid. 2) The dehydration of 1-benzyl-2-oxo-pyrrolidine-4-carboxamide (V) with POCl3 in hot DMF gives 1-benzyl-4-cyanopyrrolidine-2-one (VI), which is reduced with H2 and RaNi in methanol – NH3 and neutralized with fumaric acid. EP 0289349; JP 1989151552; US 5001153; US 5106869
syn 2
The condensation of dimethyl methylphosphonate (I) with ethyl octanoate (II) by means of butyllithium in THF gives dimethyl 2-oxononylphosphonate (III), which is condensed with the protected aldehyde (IV) by means of NaH in THF, yielding the unsaturated ketone (V). The hydrogenation of (V) with H2 over Pd/C in ethyl acetate affords the corresponding saturated ketone (VI), which is treated with ethylene glycol and p-toluenesulfonic acid to give the cyclic ketal (VII). The mild hydrolysis of (VII) with K2CO3 and acetic acid gives the alcohol derivative (VIII); the reduction of the lactone group of (VIII) with dibutylaluminum hydride in toluene affords the lactol (IX), which is condensed with (4-carboxybutyl)triphenylphosphonium bromide (X) by means of NaH in DMSO yielding the protected prostaglandin (XI). Esterification of (XI) with isopropyl iodide and DBU in acetonitrile gives the precursor (XII), which is finally deprotected with acetic acid in THF – water.
Alnodesertib CAS 2267316-76-5 MF C18H24N6O2S MW388.49 4-[4-[(cyclopropyl-methyl-oxo-λ6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine 4-[4-[(cyclopropyl-methyl-oxo-lambda6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine (S)-({2-(2-aminopyridin-4-yl)-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}imino)(cyclopropyl)(methyl)-λ6-sulfanoneserine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085 Alnodesertib (formerly known as ART0380) is an investigational, orally administered drug designed to…
Zelebrudomide CAS 2416131-46-7 MF C39H45N9O5 MW 719.8 g/mol 3-[4-[1-[[(3S)-1-[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-5-yl]pyrrolidin-3-yl]methyl]piperidin-4-yl]anilino]-5-piperidin-1-ylpyrazine-2-carboxamide 3-[[4-[1-[[(3S)-1-[2-(2,6-Dioxo-3-piperidyl)-1,3-dioxo-5-isoindolinyl]-3-pyrrolidinyl]methyl]-4-piperidyl]phenyl]amino]-5-(1-piperidyl)pyrazine-2-carboxamide protein degrader, antineoplastic, NX 2127, LSC67HA8DE, NX-2127, BTK Degrader NX-2127 Zelebrudomide (NX-2127) is an investigational new drug that is…
Vormatrigine CAS 2392951-18-5 MF C16H12F6N4O2 MW406.28 g/mol 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine 3-[ethoxydi(fluoro)methyl]-6-[5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl][1,2,4]triazolo[4,3-a]pyridinesodium channel blocker, PRAX-628, PRAX 628, QU3C48T4NV, Vormatrigine is a small molecule drug. The usage of the INN…
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO



![(7R)-7-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carbonitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=134405105&t=l)

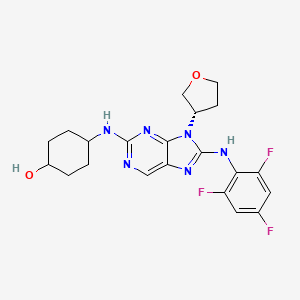

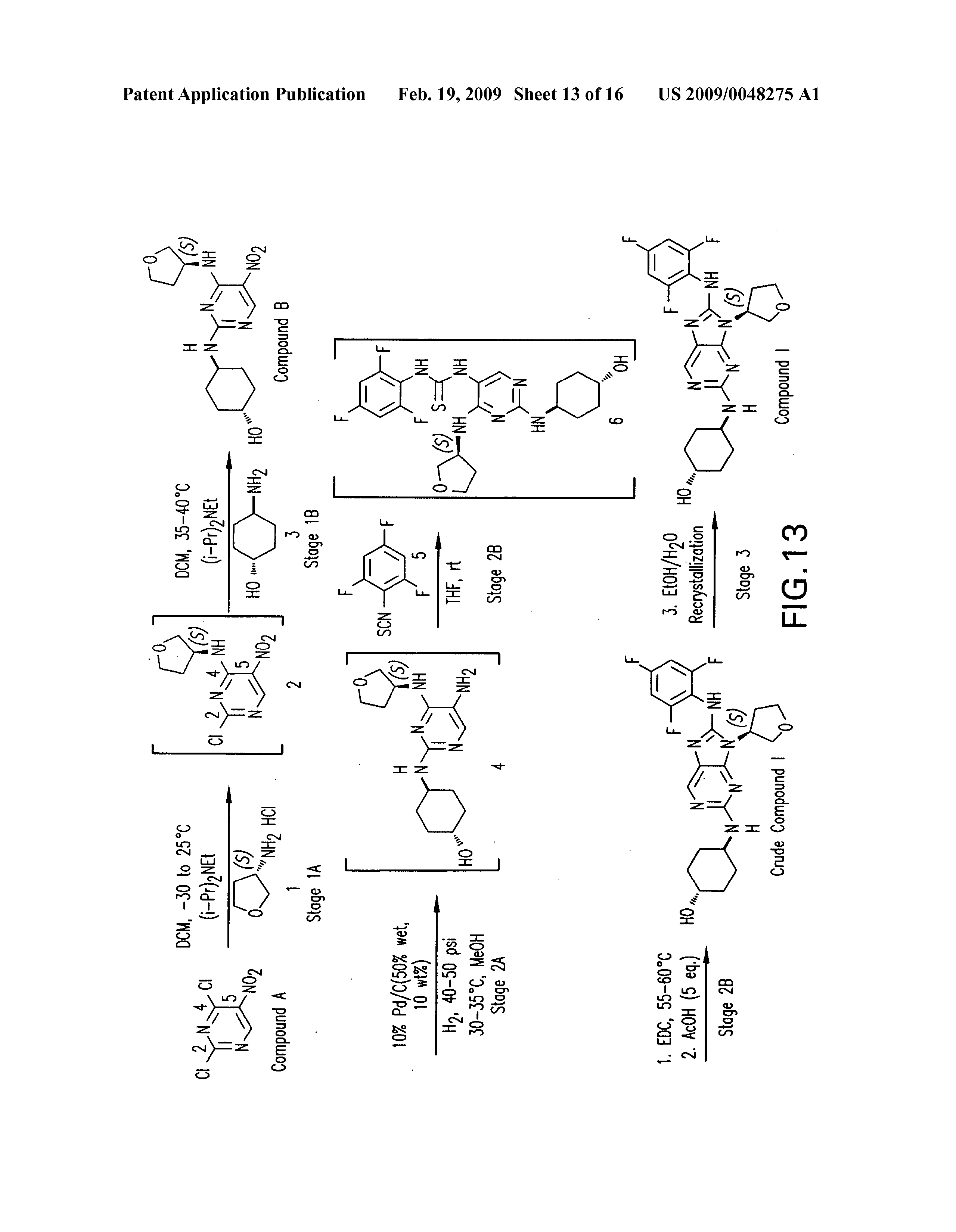


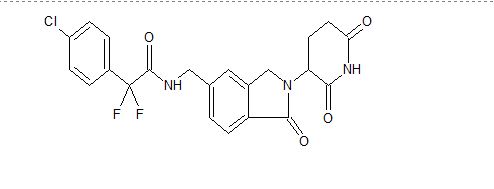
![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)