
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Selonabant
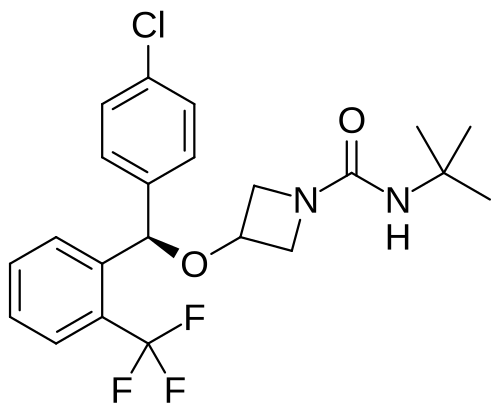
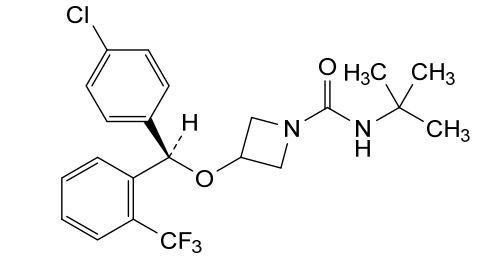
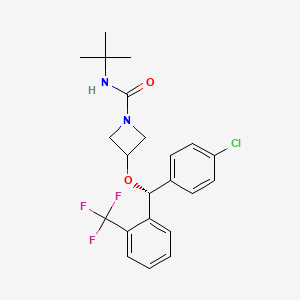
Selonabant
CAS 791848-71-0
MF C22H24ClF3N2O2 MW440.89 g/mol
N–tert-butyl-3-[(R)-(4-chlorophenyl)-[2-(trifluoromethyl)phenyl]methoxy]azetidine-1-carboxamide
N-tert-butyl-3-[(R)-(4-chlorophenyl)-[2-(trifluoromethyl)phenyl]methoxy]azetidine-1-carboxamide
(R)-N-(tert-butyl)-3-((4-chlorophenyl)(2-(trifluoromethyl)phenyl)methoxy)azetidine-1-carboxamide
N-tert-butyl-3-{(R)-(4-chlorophenyl)[2-(trifluoromethyl)phenyl]methoxy}azetidine-1-carboxamide
cannabinoid receptor 1 (CB1) antagonist, ANEB 001, V 24343, 4RNU8C6XXW, ANEB001
Drug class: Cannabinoid receptor antagonist (CB1)
The anti-obesity drug, V24343, acts by targeting the CB1 receptor in the brain and suppressing a person’s appetite.
Selonabant (INNTooltip International Nonproprietary Name, USANTooltip United States Adopted Name; developmental code names ANEB-001, V-24343) is a cannabinoid CB1 receptor antagonist which is under development for the treatment of acute cannabinoid intoxication.[1][2][3] It was also previously being developed to treat obesity, but development for this indication was discontinued.[1] The drug is administered by intravenous infusion.[1] It dramatically reduced the subjective effects of Δ9-tetrahydrocannabinol (THC) in a clinical trial.[3] Selonabant is being developed by Vernalis and Anebulo Pharmaceuticals.[1][2]
1 CLINICAL TRIALS
As of December 2024, it is in phase 2 trials.[1][2], I.V. Selonabant in Healthy Adult SubjectsCTID: NCT07211607Phase: Phase 1Status: RecruitingDate: 2026-02-12
Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of CB1 Antagonist ANEB-001 in a THC Challenge TestCTID: NCT05282797Phase: Phase 2Status: CompletedDate: 2023-08-29
Safety and Efficacy of Low Doses of V24343 in Obese SubjectsCTID: NCT00734201Phase: Phase 1Status: CompletedDate: 2011-07-25
A randomized, double-blind, placebo-controlled study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of single oral doses of CB1 antagonist ANEB-001 in healthy occasional cannabis users
EudraCT: 2021-000305-24
Phase: Phase 2, Status: Completed, Date: 2021-03-18
2. Chemical Structure Features
Key structural motifs:
• Azetidine ring (4-membered nitrogen heterocycle)
• Chiral benzhydryl ether center
• Trifluoromethyl phenyl group
• p-chlorophenyl group
• tert-butyl carboxamide
These motifs are typical of lipophilic GPCR ligands, enabling strong binding to the CB1 receptor.
Key descriptors:
| Property | Value |
|---|---|
| LogP | ~4.9 |
| H-bond donors | 1 |
| H-bond acceptors | 5 |
| TPSA | 41.6 Ų |
| Chiral centers | 1 |
3. Pharmacological Target
Primary target:
CB1 receptor (CNR1)
• GPCR located in the central nervous system
• Mediates effects of cannabinoids such as THC
Selonabant is a competitive CB1 antagonist.
Mechanism:
- THC activates CB1 receptor
- Leads to euphoria, altered cognition, tachycardia
- Selonabant blocks CB1 binding
- Reverses or prevents THC effects
4. Therapeutic Indication
Main indication
Acute cannabinoid intoxication
Potential clinical uses:
• Cannabis overdose in emergency rooms
• Pediatric accidental cannabis ingestion
• Edible THC overdose
• Severe psychiatric reactions to cannabis
A CB1 antagonist could act as a “cannabis antidote.”
5. Clinical Development
Developer:
Anebulo Pharmaceuticals (with earlier work by Vernalis)
Development timeline
| Phase | Details |
|---|---|
| Preclinical | Demonstrated CB1 antagonism |
| Phase I | Safety and PK studies |
| Phase II | THC challenge studies |
In clinical trials, the drug significantly reduced subjective effects of THC in volunteers.
Status (2024–2025): Phase II development.
6. Administration
Originally studied as:
• Intravenous infusion for rapid reversal of THC effects
Some reports indicate oral activity in research settings.
The IV route is preferred in emergency settings.
7. Comparison with Earlier CB1 Antagonists
Selonabant belongs to the same pharmacological class as:
| Drug | Status |
|---|---|
| Rimonabant | Withdrawn (psychiatric side effects) |
| Taranabant | Development stopped |
| Selonabant | Designed for short-term antidote use |
Key difference:
Earlier CB1 antagonists were chronic obesity drugs, which caused depression and suicidality.
Selonabant is designed for acute use only, reducing psychiatric risk.
8. Pharmacological Profile
Approximate characteristics (reported in literature):
• High CB1 receptor affinity
• CNS-penetrant
• Rapid onset
• Short therapeutic exposure
Effects in THC challenge studies:
• Reduced intoxication score
• Reduced cognitive impairment
• Reduced subjective “high”
9. Chemical Class
Selonabant can be classified as:
Diarylmethoxy-azetidine carbamides
Key scaffold:
Diarylmethanol → ether → azetidine carbamate.
This scaffold appears in several CB1 inverse agonists.
PATENT
WO2005080345
Title: Cannabinoid CB1 receptor antagonists
Assignee: Vernalis (R&D) Ltd
Priority: ~2004
This is the primary patent family covering the scaffold used for Selonabant.
Related Continuation Patent
WO2005080328
Follow-up Patent on CB1 Antagonists
WO2005075450
Later Use Patent (Anebulo)
US11141404
Assignee: Anebulo Pharmaceuticals
Coverage:
• use of Selonabant (ANEB-001)
• treatment of acute cannabinoid overdose
• IV formulations
The patent protection extends to ~2040
Patent Family Map
Main Selonabant IP family:
| Patent | Type | Notes |
|---|---|---|
| WO2005080345 | Composition of matter | core scaffold |
| WO2005080328 | analog compounds | synthetic examples |
| WO2005075450 | optimized CB1 antagonists | SAR |
| US11141404 | method of use | overdose treatment |
| PCT follow-ups | formulation / delivery | IV use |
SYN
https://patents.google.com/patent/US11141404B1/en
SYN
US 7,504,522
Preparation of 2-(trifluoromethyl)-4-chlorobenzhydrol (96)
Preparation of 1-benzhydryl-3-[2-(trifluoromethyl)-4′-chlorobenzhydryloxy]azetidine (97)
| This material was prepared from 1-benzhydryl-3-azetidinol (1) (40.1 mmol) and 2-(trifluoromethyl)-4-chlorobenzhydrol (96) (80.2 mmol) using the procedure described for compound (3) (13.5 g, 66%). |
Preparation of 3-[2-(trifluoromethyl)-4′-chlorobenzhydryloxy]azetidine hydrochloride (98)
| This material was prepared from 1-benzhydryl-3-[2-(trifluoromethyl)-4′-chlorobenzhydryloxy]azetidine (97) (25.6 mmol) using the procedure described for compound (9) (8.2 g, 85%). |
| NMR (400 MHz, d 6-DMSO) δ H 3.78 (1H, m), 3.97 (3H, m), 4.89 (1H, q, J 6.0 Hz), 5.85 (1H, s), 7.33 (2H, m), 7.44 (2H, m), 7.59 (1H, m), 7.76 (3H, m), 8.97 (1H, bs) |
Example 81 FINAL MOLECULE
3-[2-(trifluoromethyl)-4′-chlorobenzhydryloxy]-N-(tert-butyl)azetidine-1-carboxamide (99)
| NMR (400 MHz, d 6-DMSO) δ H 1.20 (9H, s), 3.51 (1H, m), 3.65 (1H, m), 3.84 (2H, m), 4.25 (1H, m), 5.62 (1H, s), 5.73 (1H, s), 7.31 (2H, m), 7.39 (2H, m), 7.57 (1H, m), 7.75 (3H, m) |
SYN
(R)-N-(tert-butyl)-3-((4-chlorophenyl)(2-(trifluoromethyl)phenyl)methoxy)azetidine-1-carboxamide (Compound 1):
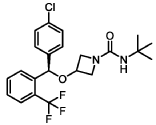
SYN
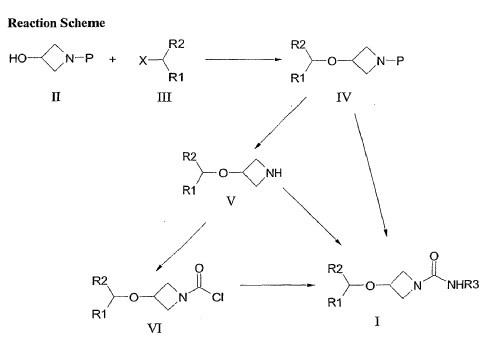
PAT
The core patent describing the chemistry for Selonabant-type molecules is:
WO2005080345
Title: Cannabinoid receptor antagonists
Assignee: Vernalis plc
Priority: ~2004
for SAR exploration
PATENTS
- Azetidinecarboxamide derivatives and their use in the treatment of cb1 receptor mediated disordersPublication Number: WO-2004096763-A1Priority Date: 2003-05-01
- Azetidinecarboxamide derivatives and their use in the treatment of CB1 receptor mediated disordersPublication Number: NZ-543016-APriority Date: 2003-05-01
- Botulinum neurotoxins for use in therapyPublication Number: US-11260114-B2Priority Date: 2017-03-22Grant Date: 2022-03-01
- Azetidinecarboxamide derivatives and their use in the treatment of cb1 receptor mediated disordersPublication Number: EP-1620395-A1Priority Date: 2003-05-01
- Azetidinecarboxamide derivatives and their use in the treatment of cb1 receptor mediated disordersPublication Number: EP-1620395-B1Priority Date: 2003-05-01Grant Date: 2009-12-30
- Azetidinecarboxamide derivatives and their use in the treatment of cb1 receptor mediated disordersPublication Number: US-7504522-B2Priority Date: 2003-05-01Grant Date: 2009-03-17
- Azetidinecarboxamide derivatives and their use in the treatment of cb1 receptor mediated disordersPublication Number: US-2007054891-A1Priority Date: 2003-05-01
- Botulinum neurotoxins production methodsPublication Number: US-2023016041-A1Priority Date: 2017-04-28
- Initiating neurotoxin treatmentsPublication Number: US-2023058666-A1Priority Date: 2017-04-21
- Initiating neurotoxin treatmentsPublication Number: EP-4137149-A1Priority Date: 2017-04-21
- Botulinum neurotoxins for treating hyperhidrosisPublication Number: US-2022370574-A1Priority Date: 2017-04-20
- Botulinum neurotoxins for use in therapyPublication Number: US-2023027850-A1Priority Date: 2017-03-22
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

| Clinical data | |
|---|---|
| Other names | ANEB-001; ANEB001; V-24343; V24343 |
| Drug class | Cannabinoid receptor antagonist; Cannabinoid CB1 receptor antagonist; Cannabinoid antidote |
| Identifiers | |
| IUPAC name | |
| CAS Number | 791848-71-0 |
| PubChem CID | 68902536 |
| DrugBank | DB18908 |
| ChemSpider | 128922145 |
| UNII | 4RNU8C6XXW |
| KEGG | D12875 |
| ChEMBL | ChEMBL5095165 |
| Chemical and physical data | |
| Formula | C22H24ClF3N2O2 |
| Molar mass | 440.89 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Anebulo Pharmaceuticals”. AdisInsight. 26 December 2024. Retrieved 25 February 2025.
- “Delving into the Latest Updates on Selonabant with Synapse”. Synapse. 23 January 2025. Retrieved 25 February 2025.
- Gorbenko AA, Heuberger JA, Juachon M, Klaassen E, Tagen M, Lawler JF, et al. (February 2025). “CB1 Receptor Antagonist Selonabant (ANEB-001) Blocks Acute THC Effects in Healthy Volunteers: A Phase II Randomized Controlled Trial”. Clinical Pharmacology and Therapeutics. 117 (5): 1427–1436. doi:10.1002/cpt.3581. PMC 11993283. PMID 39898464.
//////////selonabant, cannabinoid receptor 1 (CB1) antagonist, ANEB 001, V 24343, 4RNU8C6XXW, ANEB001
Rosolutamide
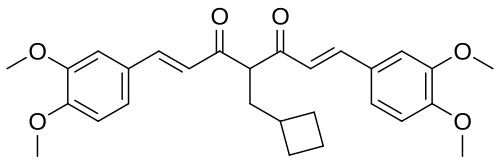
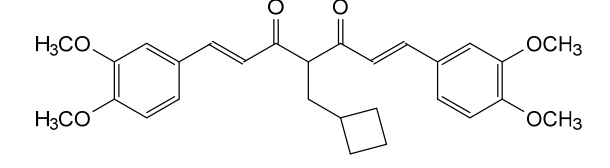
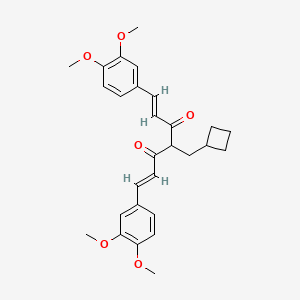
Rosolutamide
CAS 1039760-91-2
MF C28H32O6 MW464.5 g/mol
(1E,6E)-4-(cyclobutylmethyl)-1,7-bis(3,4-dimethoxyphenyl)hepta-1,6-diene-3,5-dione
(1E,6E)-4-(cyclobutylmethyl)-1,7-bis(3,4-dimethoxyphenyl)hepta-1,6-diene-3,5-dione
antiandrogen, ASC-JM-17, ASC-JM17, JM17, ALZ-003, ALZ003, 5VLL140BN9,
Rosolutamide (INNTooltip International Nonproprietary Name; developmental code name ASC-JM17, JM17, ALZ-003) is an agonist of nuclear respiratory factor 1 (NRF1), a nonsteroidal antiandrogen, and an androgen receptor degrader related to curcumin.[1][2][3][4][5] Other analogues like dimethylcurcumin (ASC-J9) are also known.[2][6]
3-hydroxy imidacloprid is an imidacloprid. It has a role as a neonicotinoid insectide and a nicotinic acetylcholine receptor agonist.
REF
- Ferroptosis-related small-molecule compounds in cancer therapy: Strategies and applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2022-12-15PMID: 36332549DOI: 10.1016/j.ejmech.2022.114861
- A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophyPublication Name: Human Molecular GeneticsPublication Date: 2016-03-08PMCID: PMC5062587PMID: 26962150DOI: 10.1093/hmg/ddw073
SYN
SYN
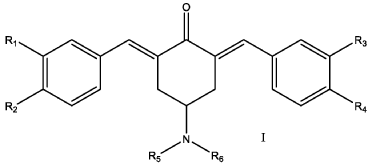
SYN
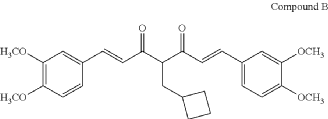
COMPD B
SYN
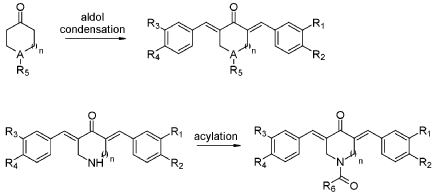
SYN
PAT
Publication Number: EP-2104659-B1
Priority Date: 2007-01-08
Grant Date: 2015-07-29
- Compounds with (1e, 6e)-1,7-bis-(3,4-dimethoxyphenyl)-4-4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-2016264539-A1Priority Date: 2007-01-08
- Compounds with (substituted phenyl)-propenal moiety, their derivatives, biological activity, and uses thereofPublication Number: DK-2104659-T3Priority Date: 2007-01-08Grant Date: 2015-11-02
- Compounds with (1E, 6E)-1,7-Bis-(3,4-dimethoxyphenyl)-4-4-distributed-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-9562025-B2Priority Date: 2007-01-08Grant Date: 2017-02-07
- Compounds with (1E, 6E)-1,7-bis-(3,4-dimethoxyphenyl)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-9000222-B2Priority Date: 2007-01-08Grant Date: 2015-04-07
- Compounds with (1 E, 6E)-1,7-bis-(3,4-dimethoxyphenyI)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-9259402-B2Priority Date: 2007-01-08Grant Date: 2016-02-16
- Compounds with (1 E, 6E)-1,7-bis-(3,4-dimethoxyphenyl)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-8710272-B2Priority Date: 2007-01-08Grant Date: 2014-04-29
- Compounds with (substituted phenyl)-propenal moiety, their derivatives, biological activity, and use thereofPublication Number: EP-2993165-A2Priority Date: 2007-01-08
- Compounds with (substituted phenyl)-propenal moiety, their derivatives, biological activity, and use thereofPublication Number: EP-2993165-B1Priority Date: 2007-01-08Grant Date: 2018-06-27
- Compounds with (1E, 6E)-1,7-bis-(3,4-dimethoxyphenyl)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-2013261121-A1Priority Date: 2007-01-08
- Compounds with (substituted phenyl)-propenal moiety, their derivatives, biological activity, and uses thereofPublication Number: CA-2674780-CPriority Date: 2007-01-08Grant Date: 2014-03-11
- Compositions including androgen receptor degradation (ard) enhancers and methods of prophylactic or therapeutic treatment of skin disorders and hair lossPublication Number: CA-2694953-CPriority Date: 2007-07-31Grant Date: 2015-12-01
- Compositions containing androgen receptor degradation enhancers and methods for preventing or treating hair loss and skin diseasesPublication Number: KR-20100051808-APriority Date: 2007-07-31
- Compositions containing androgen receptor degradation (ARD) enhancers and methods for the prophylactic or therapeutic treatment of skin diseases and hair lossPublication Number: JP-2010535213-APriority Date: 2007-07-31
- Compounds with (1E, 6E)-1,7-bis-(3,4-dimethoxyphenyl)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-2013338160-A1Priority Date: 2007-01-08
- Compounds with (1E, 6E)-1,7-bis-(3,4-dimethoxyphenyl)-4,4-disubstituted-hepta-1,6-diene-3,5-dione structural scaffold, their biological activity, and uses thereofPublication Number: US-2015190351-A1Priority Date: 2007-01-08
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
| Clinical data | |
|---|---|
| Other names | ASC-JM-17; ASC-JM17; JM17; ALZ-003; ALZ003 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1039760-91-2 |
| PubChem CID | 25183127 |
| DrugBank | DB16931 |
| ChemSpider | 64854816 |
| UNII | 5VLL140BN9 |
| ChEMBL | ChEMBL5266600 |
| Chemical and physical data | |
| Formula | C28H32O6 |
| Molar mass | 464.558 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 428. 2024.
- Chang KH, Chen CM (May 2024). “The Role of NRF2 in Trinucleotide Repeat Expansion Disorders”. Antioxidants. 13 (6): 649. doi:10.3390/antiox13060649. PMC 11200942. PMID 38929088.
- Yuan J, Zhang S, Zhang Y (December 2018). “Nrf1 is paved as a new strategic avenue to prevent and treat cancer, neurodegenerative and other diseases”. Toxicology and Applied Pharmacology. 360: 273–283. Bibcode:2018ToxAP.360..273Y. doi:10.1016/j.taap.2018.09.037. PMID 30267745.
- Bott LC, Badders NM, Chen KL, Harmison GG, Bautista E, Shih CC, et al. (May 2016). “A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy”. Human Molecular Genetics. 25 (10): 1979–1989. doi:10.1093/hmg/ddw073. PMC 5062587. PMID 26962150.
- Wu YL, Chang JC, Chao YC, Chan H, Hsieh M, Liu CS (July 2022). “In Vitro Efficacy and Molecular Mechanism of Curcumin Analog in Pathological Regulation of Spinocerebellar Ataxia Type 3”. Antioxidants. 11 (7): 1389. doi:10.3390/antiox11071389. PMC 9311745. PMID 35883884.
- Sangotra A, Lieberman AP (February 2025). “Therapeutic targeting of the polyglutamine androgen receptor in Spinal and Bulbar Muscular Atrophy”. Expert Opinion on Therapeutic Targets: 1–13. doi:10.1080/14728222.2025.2464173. PMID 39915972.
/////////rosolutamide, antiandrogen, ASC-JM-17, ASC-JM17, JM17, ALZ-003, ALZ003, 5VLL140BN9, ANAX
Rivasterat
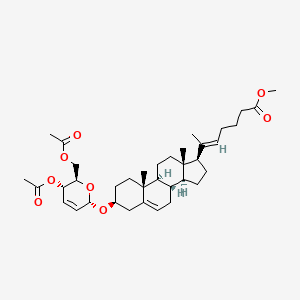
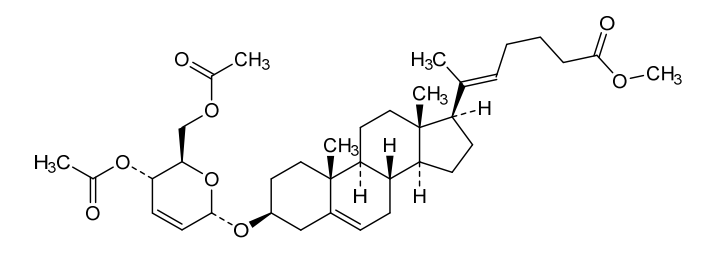
Rivasterat
CAS 2446590-96-9
MF C37H54O8 MW626.8 g/mol
methyl (E)-6-[(3S,8S,9S,10R,13S,14S,17R)-3-[[(2R,3S,6S)-3-acetyloxy-2-(acetyloxymethyl)-3,6-dihydro-2H-pyran-6-yl]oxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]hept-5-enoate
methyl (20E)-3β-[(4,6-di-O-acetyl-2,3-dideoxy-α-Derythro-hex-2-enopyranosyl)oxy]-27-norcholesta5,20(22)-dien-26-oate
cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
- OriginatorCURACLE
- ClassAnti-inflammatories; Anti-ischaemics; Antineoplastics; Eye disorder therapies; Ischaemic heart disorder therapies; Small molecules; Vascular disorder therapies
- Mechanism of ActionActin modulators; Chemokine CCL2 inhibitors; Histamine release inhibitors; Interleukin 1 beta inhibitors; Thrombin inhibitors; Vascular endothelial growth factors inhibitors
- Phase IIDiabetic macular oedema
- No development reportedAge-related macular degeneration; Cancer; Crohn’s disease; Diabetic retinopathy; Hereditary angioedema; Lung disorders; Macular degeneration; Myocardial infarction; Retinal oedema; Stroke; Ulcerative colitis; Unstable angina pectoris; Wet macular degeneration
- 28 Aug 2025No recent reports of development identified for research development in Unstable-angina-pectoris in South Korea (PO)
- 28 Jul 2025No recent reports of development identified for phase-I development in Wet macular degeneration in USA (PO)
- 28 May 2025No recent reports of development identified for phase-I development in Age-related-macular-degeneration in South Korea (PO)
Developer and Code Name
- Original code name: CU06-1004
- Developer: Curacle Co., Ltd. (South Korea)
- Drug class: Endothelial dysfunction blocker (EDB)
This class of drugs aims to restore endothelial barrier integrity rather than directly blocking VEGF like most retinal drugs.
The molecule contains:
- Steroid (cyclopenta[a]phenanthrene) core
- Unsaturated heptenoate side chain
- Acetylated sugar moiety (pyranose)
This glycosylated steroid structure is unusual for vascular-protective drugs.
Clinical Development
Phase I (Healthy Volunteers)
Key findings:
- Dose tested: 100–1200 mg
- Exposure increased more than dose proportional
- Food greatly increased absorption
- No significant drug accumulation after repeated dosing
- Minimal renal excretion detected
Phase II
Early clinical trials investigated oral therapy for diabetic macular edema with improvements in:
- Best-corrected visual acuity
- Inflammatory biomarkers
Summary
| Item | Details |
|---|---|
| Drug | Rivasterat (CU06-1004) |
| Originator | Curacle |
| Core patent | WO2013011939 |
| Chemistry | steroid glycoside |
| Key step | steroid glycosylation |
| Priority | ~2011 |
| Expiry | ~2031–2033 |
SYNTHESIS
WO2013011939
US20140148474
EP2741074
KR20130007373
SYN
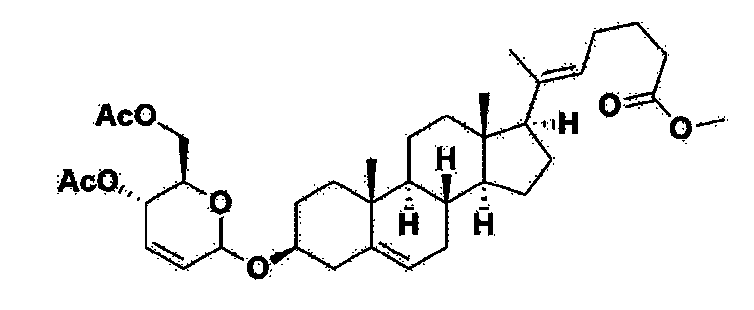
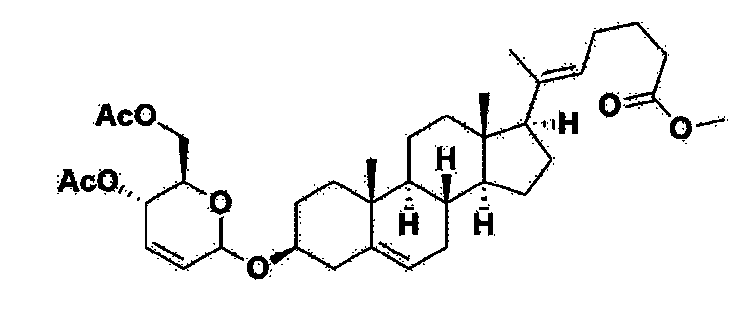
Manufacturing Example 1> Manufacturing of compound 1
[170]Compound 1 represented by the following chemical formula 1 can be manufactured using the manufacturing method described in Korean unpublished patent application number 10-2019-0166864. Specifically, it can be manufactured using the method according to the following reaction scheme 1 or reaction scheme 2.
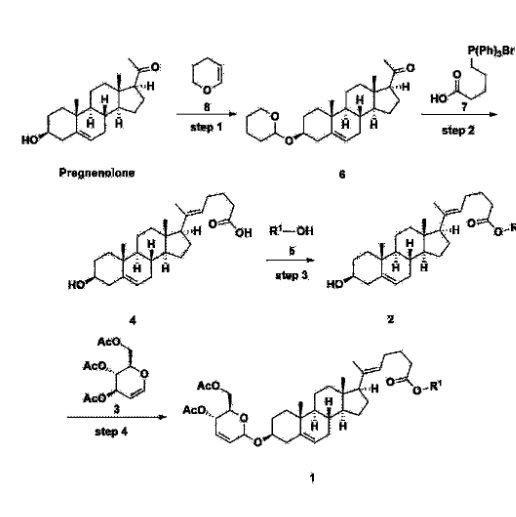
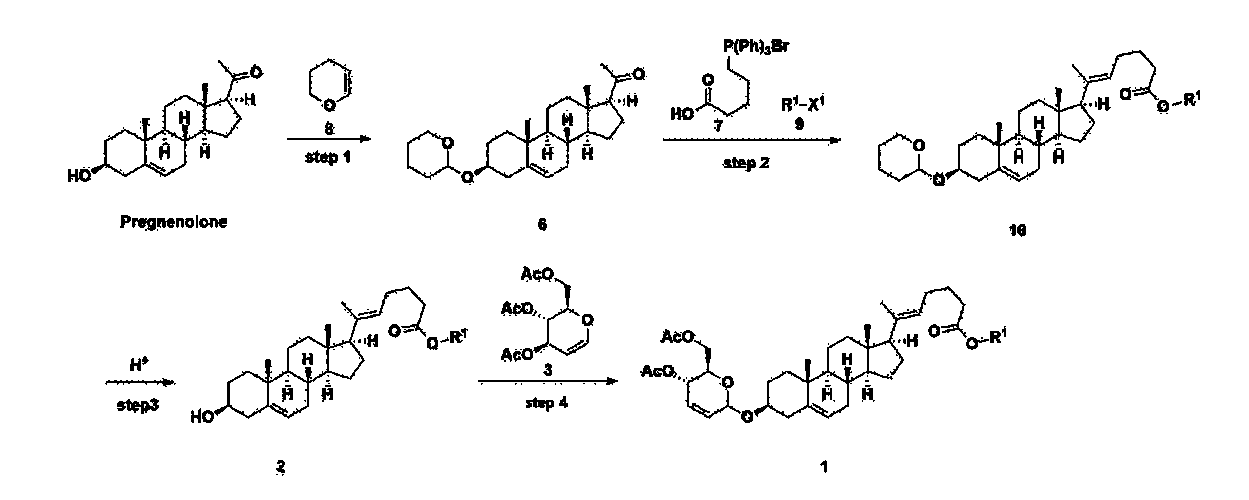
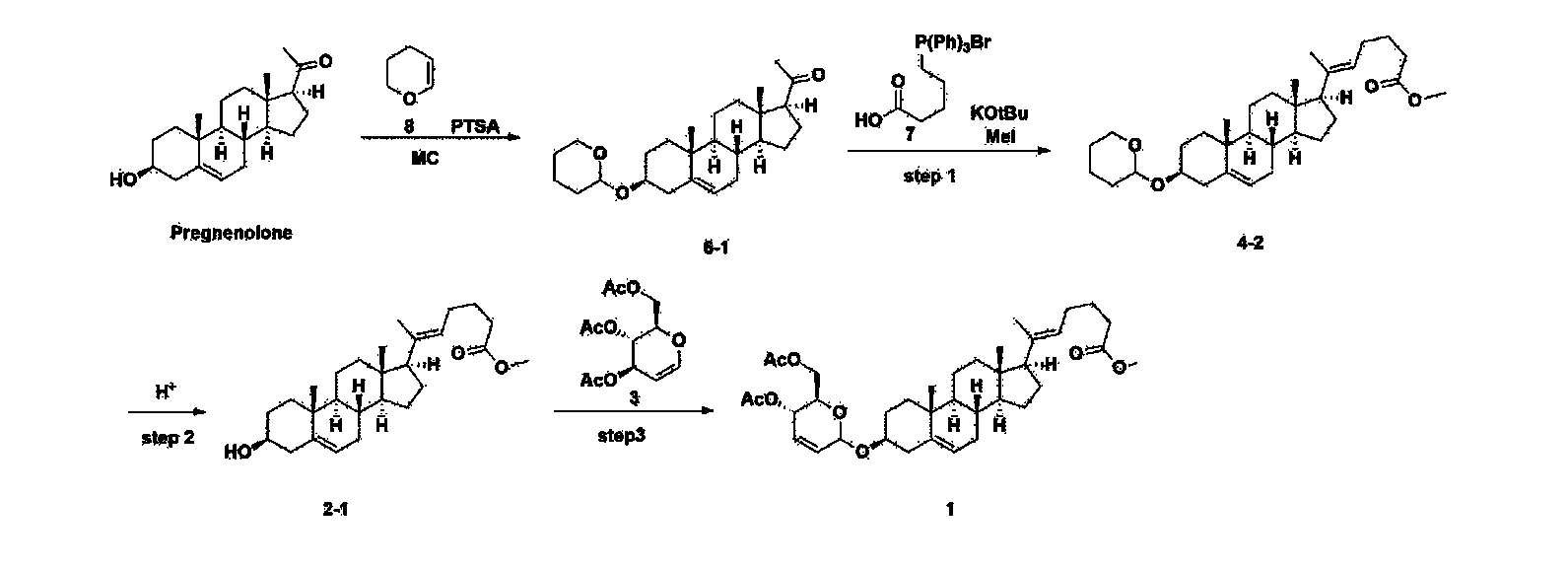
Step 1: Preparation of 6-1
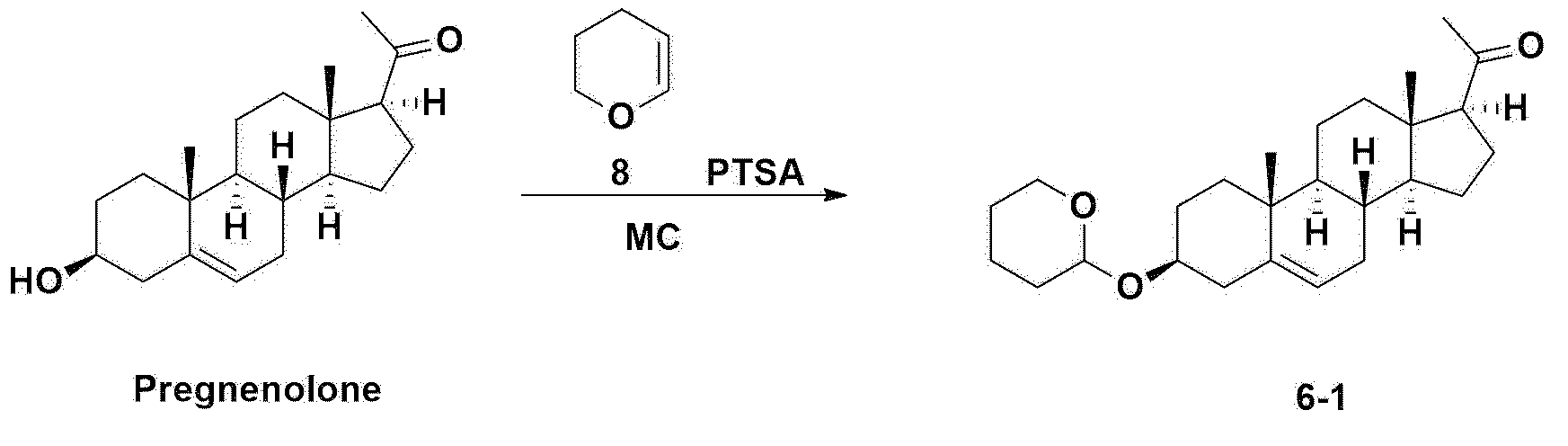
A thermometer was installed in a 5 L flask, and 200 g (0.632 mol) of pregnenolone was added to 2000 mL of dichloromethane, and 173 mL (1.896 mol) of 3,4-dihydro-2H-pyran was added. After lowering the temperature to 0-5 ℃, 3.0 g (15.8 mmol) of p-toluenesulfonic acid monohydrate dissolved in 50 mL of tetrahydrofuran (THF) was added dropwise and stirred at 0 ℃ for 1.5 hours. At 0 ℃, 800 mL of saturated sodium bicarbonate aqueous solution and 10 mL of triethylamine (TEA) were added to the reaction mixture and stirred. After separating the layers, the organic layer was washed with 800 mL of brine, and the aqueous layers were extracted again with 200 mL of dichloromethane, combined into the organic layers, dried over 200 g of anhydrous sodium sulfate, filtered, and distilled under reduced pressure. 1000 mL of MeOH and 5 mL of TEA were added to the obtained residue, heated to completely dissolve, and the temperature was lowered and stirred at -5 °C for 1 hour. The resulting solid was filtered and washed with 200 mL of MeOH to obtain 232.0 g (0.579 mol) of 6-1 (THP-Pregnenolone) as a pure white solid in a yield of 91.6%.
[204]1H-NMR (400 MHz, CDCl 3): δ 5.33-5.36 (m, 1H), 4.71-4.72 (m, 1H), 3.85-3.94 (m, 1H), 3.46-3.56 (m, 2H), 1.00-2.55 (m, 32H), 0.62 (s, 3H).
Step 2: Preparation of 4-2
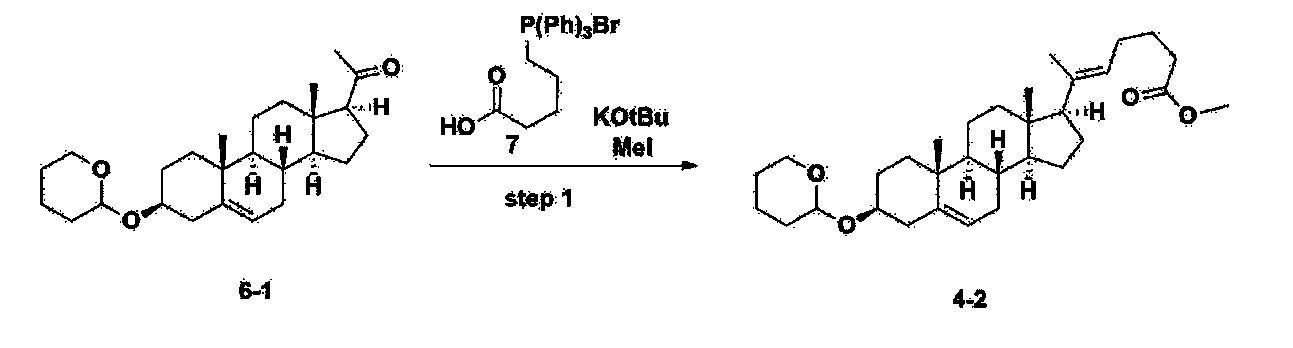
After installing a condenser, heating mantle, and mechanical stirrer in a 5L reactor, the reactor was heated to 119℃ (external temperature), cooled to room temperature while flowing nitrogen for 5 minutes, dried, and 332.5 g (0.75 mol) of 4-(carboxybutyl)triphenylphosphonium bromide and 168.1 g (1.50 mol) of potassium t-butoxide were added. Then, 2000 mL of anhydrous toluene and 750 mL of anhydrous tetrahydrofuran were added, and the reactor was heated to 119℃ (external temperature, internal mild reflux) and stirred for about 2 hours.
[209]6-1 100.0 g (0.250 mol) was dissolved in 500 mL of anhydrous toluene, added to the reaction solution, and reacted for about 20 hours. After the reaction was completed, the reaction mixture was cooled to room temperature, 320 mL (5.14 mol) of methyl iodide and 1000 mL of acetone were added, and stirred at room temperature for 15 hours. Most of the organic solvent was removed from the reaction mixture by distillation under reduced pressure, 1500 mL of ethyl acetate was added to dissolve, and the mixture was washed with 1000 mL of saturated ammonium chloride aqueous solution. The organic layer was washed twice with 1000 mL of water and 1000 mL of brine, dried with 100 g of sodium sulfate, filtered using 80 g of Celite, and concentrated.
[210]The obtained residue was dissolved in 2000 mL of methanol, stirred at 10°C for 13 hours and at 4-5°C for 1 hour, and the resulting solid was filtered, washed with 200 mL of methanol, and dried in vacuum to obtain 66.2 g of 4-2 as a white solid with a yield of 53.2%.
[211]
1H NMR(400MHz, CDCl 3): δ 5.36(t, J=5.80 Hz, 1H), 5.16(t, J=7.00 Hz, 1H), 4.71(m, 1H), 3.93(m, 1H), 3.66(s, 3H), 3.56(m, 2H), 2.37-0.88(m, 38H), 0.54(s, 3H).
Step 3: Preparation of 2-1
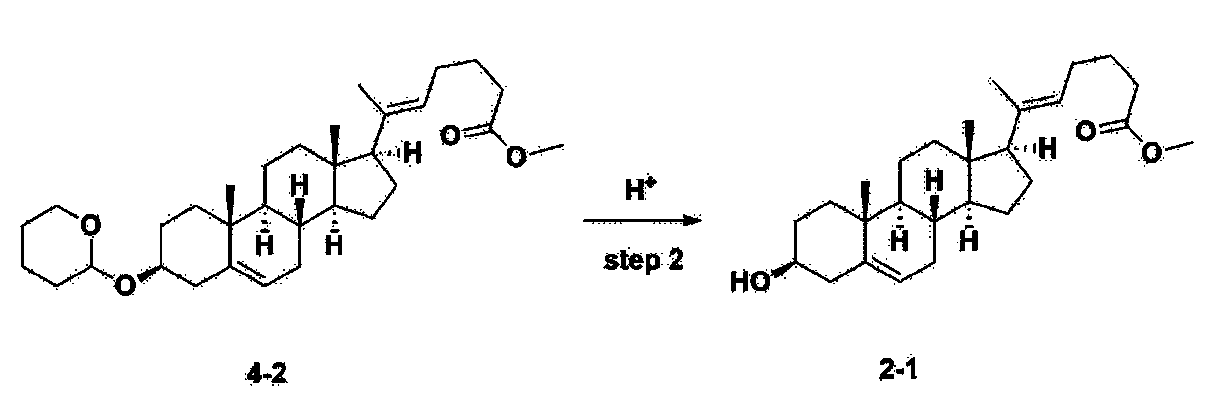
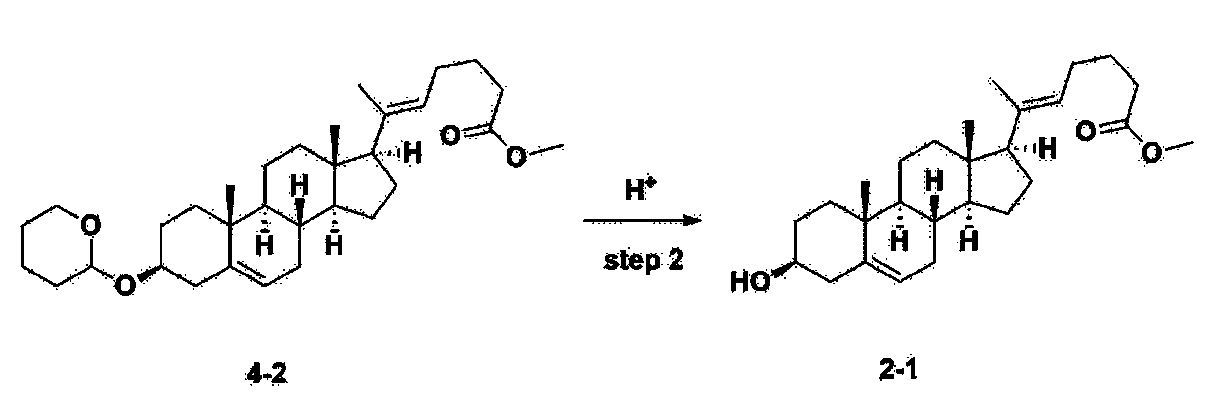
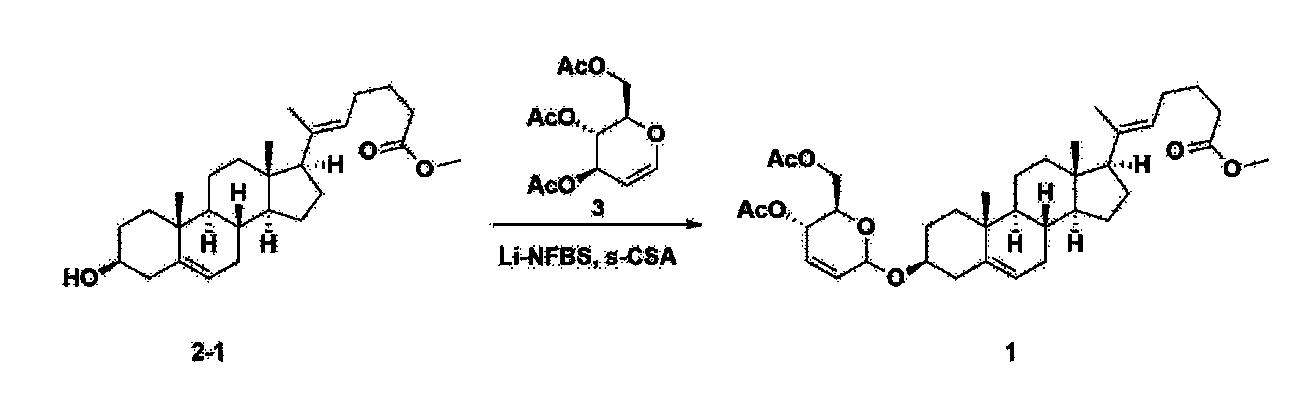
After installing a thermometer and a water bath in a 1 L flask, 42.0 g (0.101 mol) of compound 2-1 and 34.5 g (0.126 mol) of triiO-acetyl D-glucal were dissolved in 126 mL of anhydrous toluene and 252 mL of acetonitrile, and while maintaining the temperature at 30-35 ℃, 3.87 g (0.0130 mol) of lithium nonafluoro-1-butylsulfonate and 0.117 g (0.0005 mol) of (s)-camphor sulfonic acid were added and stirred for 2 hours. After completion of the reaction, the reaction was quenched with 504 mL of saturated sodium bicarbonate aqueous solution and extracted with 630 mL of heptane. The organic layer was washed twice with 504 mL of saturated sodium bicarbonate aqueous solution and then with 504 mL of brine. The organic layer was stirred with 42 g of anhydrous sodium sulfate and 34 g of charcoal, filtered with 34 g of celite, washed with 210 mL of methylene chloride, combined with the filtrate, concentrated, and dried under vacuum.
[223]
1H-NMR (400 MHz, CDCl3) : δ 5.79-5.88 (m, 2H), 5.35-5.36 (m, 1H), 5.27-5.29 (m, 1H), 5.12-5.16 (m, 2H), 4.15-4.24 (m, 3H), 3.66 (s, 3H), 3.54-3.57 (m, 1H), 0.91-2.32 (m, 38H), 0.54 (s, 3H).
PAT
- Crystalline form of vascular leakage blocker compoundPublication Number: US-12103945-B2Priority Date: 2020-05-04Grant Date: 2024-10-01
- New crystalline form of vascular leakage blocker compoundPublication Number: US-2022259256-A1Priority Date: 2020-05-04
- Preparation Method of Vascular Leakage Blockers With a High YieldPublication Number: US-2021395297-A1Priority Date: 2019-12-13
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////rivasterat, cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
Repinatrabit
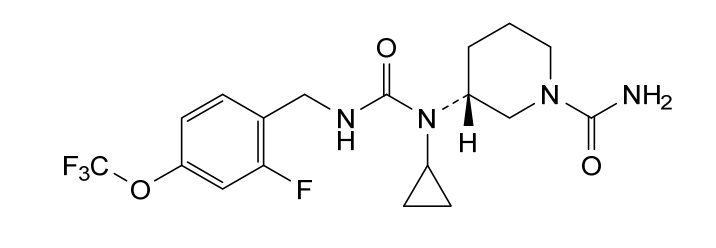
Repinatrabit
CAS 2837993-05-0
MF C18H22F4N4O3 MW 418.4 g/mol
(3R)-3-[cyclopropyl-[[2-fluoro-4-(trifluoromethoxy)phenyl]methylcarbamoyl]amino]piperidine-1-carboxamide
(3R)-3-[cyclopropyl({[2-fluoro-4-(trifluoromethoxy)phenyl]methyl}carbamoyl)amino]piperidine-1-
carboxamide
solute carrier family 6 member 19 (SLC6A19) inhibitor(phenylketonuria), JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Repinatrabit (JNT-517) is an investigational, oral, small-molecule drug developed by Jnana Therapeutics (now part of Otsuka Pharmaceutical) to treat Phenylketonuria (PKU). It acts as a selective inhibitor of the SLC6A19 transporter, reducing blood phenylalanine (Phe) levels by increasing its urinary excretion.
Key Details About Repinatrabit:
- Mechanism: It targets a novel, cryptic allosteric site to block kidney reabsorption of phenylalanine, aiming to be a first-in-class oral therapy for all PKU patients, regardless of age or genotype.
- Clinical Trials: Otsuka initiated a global Phase 3 study (NCT06971731) in December 2025 to evaluate its safety and efficacy, following positive results from earlier studies.
- Status: The FDA has granted it orphan drug and rare pediatric disease designations.
- A Study to Evaluate the Safety and Efficacy of JNT-517 in Participants With Phenylketonuria (PKU)CTID: NCT06971731Phase: Phase 3Status: RecruitingDate: 2026-02-04
- A Phase 2 Study of JNT-517 in Adolescent Participants With PhenylketonuriaCTID: NCT06637514Phase: Phase 2Status: RecruitingDate: 2025-08-19
- First-in-Human, Multiple Part Clinical Study of JNT-517 in Healthy Participants and in Participants With PhenylketonuriaCTID: NCT05781399Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-07-31
- A Study to Evaluate the Long-Term Safety and Efficacy of JNT-517 in Participants With PhenylketonuriaCTID: NCT06628128Phase: Phase 3Status: Not yet recruitingDate: 2025-06-03
EMA Drug Information,
Type, Orphan designations
(R)-3-(1-Cyclopropyl-3-(2-fluoro-4-(trifluoromethoxy)benzyl)ureido)piperidine-1-carboxamide
Intended Use, Treatment of hyperphenylalaninaemia, Status of Orphan Designation, Positive
First Published Date, 2024-08-22
PAT
SYN
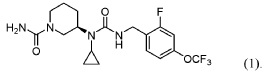
PAT
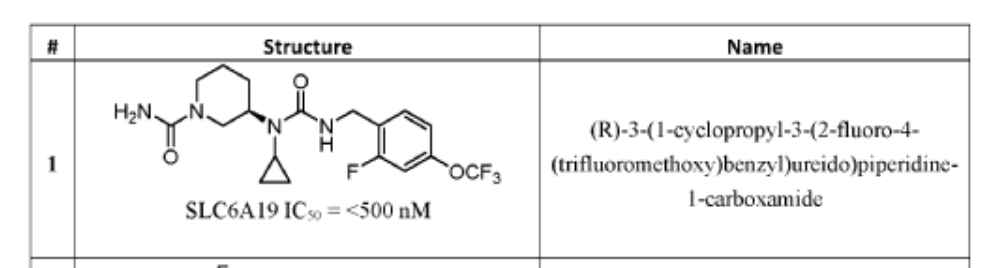
PAT
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: WO-2024118721-A1Priority Date: 2022-11-30
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: EP-4626420-A1Priority Date: 2022-11-30
- Dosing regimen for treatment of PKU with SLC6A19 functional piperidine inhibitorsPublication Number: CN-120091814-APriority Date: 2022-09-14
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2024208923-A1Priority Date: 2021-03-10
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2025289797-A1Priority Date: 2021-03-10
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////repinatrabit, ANAX, JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Remlifanserin
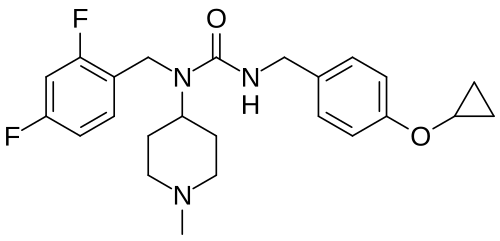
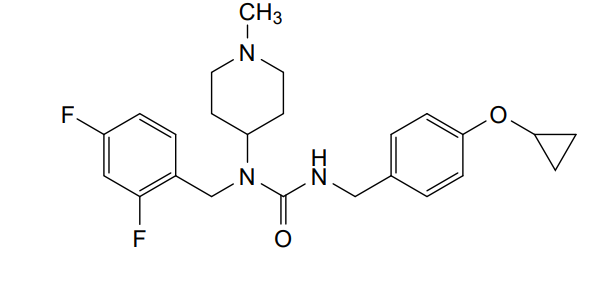
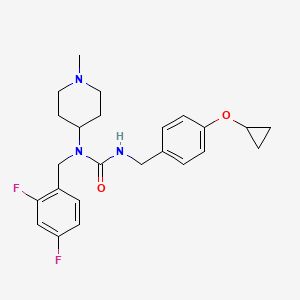
Remlifanserin
CAS 2289704-13-6
MF C24H29F2N3O2 MW 429.5 g/mol
3-[(4-cyclopropyloxyphenyl)methyl]-1-[(2,4-difluorophenyl)methyl]-1-(1-methylpiperidin-4-yl)urea
N’-{[4-(cyclopropyloxy)phenyl]methyl}-N-[(2,4-difluorophenyl)methyl]-N-(1-methylpiperidin-4-yl)urea
serotonin receptor (5-HT2A) inverse agonist, ACP-204, ACP 204, H4L2AF2XB7
Remlifanserin is a small molecule drug. The usage of the INN stem ‘-anserin’ in the name indicates that Remlifanserin is a serotonin receptor antagonist. Remlifanserin has a monoisotopic molecular weight of 429.22 Da.
Remlifanserin (INNTooltip International Nonproprietary Name;[4] developmental code name ACP-204) is a selective serotonin 5-HT2A receptor inverse agonist which is under development for the treatment of Alzheimer’s disease psychosis.[1][5][6][7][8][9] It is taken by mouth.[1]
The drug is an improved follow-up compound to its developer’s earlier drug pimavanserin (Nuplaizid; ACP-103).[6] It is more potent and selective than pimavanserin as a serotonin 5-HT2A receptor inverse agonist.[10] Remlifanserin shows 32- to 123-fold selectivity for antagonism and inverse agonism of the serotonin 5-HT2A receptor over the serotonin 5-HT2C receptor depending on the bioassay.[10] For comparison, pimavanserin’s selectivity was 8- to 37-fold depending on the assay.[10] Remlifanserin shows very low affinity for the serotonin 5-HT2B receptor compared to the serotonin 5-HT2A and 5-HT2C receptors.[10] It is expected to have less QT prolongation than pimavanserin.[10] The drug blocks the head-twitch response induced by the serotonergic psychedelic DOI and the hyperlocomotion induced by the NMDA receptor antagonist dizocilpine (MK-801) in rodents.[10]
Remlifanserin is under development by Acadia Pharmaceuticals.[1][5] As of January 2025, it is in phase 3 clinical trials.[1][5] Its clinicaltrials.gov identifier (nct number) is NCT06159673.[11]
SYN
Example 17: 3-[(4-cyclopropoxyphenyl)methyl]-1-[(2,4-difluorophenyl)methyl]-1-(1-methylpiperidin-4-yl)urea; hemitartrate (17)
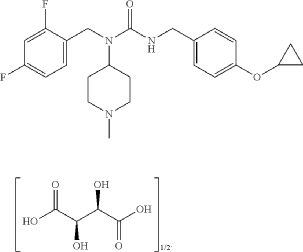
3-[(4-cyclopropoxyphenyl)methyl]-1-[(2,4-difluorophenyl)methyl]-1-(1-methylpiperidin-4-yl)urea; hemitartrate
PAT
- 3-(4-cyclo-propoxybenzyl)-1-(2,4-difluorobenzyl)-1 -(1-methylpiperidin-4-yl)urea for use in the treatment of diseases associated with the serotonin-receptor 5-htPublication Number: WO-2025029990-A1Priority Date: 2023-08-02
- Compounds, salts thereof and methods for treating diseasesPublication Number: CN-111132976-APriority Date: 2017-08-21
- COMPOUNDS, CORRESPONDING SALTS AND METHODS FOR THE TREATMENT OF DISEASESPublication Number: WO-2019040107-A1Priority Date: 2017-08-21
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: EP-3672954-A1Priority Date: 2017-08-21
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: US-2020270239-A1Priority Date: 2017-08-21
- Compounds, salts thereof and methods for treating diseasesPublication Number: CN-111132976-BPriority Date: 2017-08-21Grant Date: 2023-08-22
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: EP-4635568-A2Priority Date: 2017-08-21
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: US-12139477-B2Priority Date: 2017-08-21Grant Date: 2024-11-12
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: US-2025197385-A1Priority Date: 2017-08-21
- Compounds, salts thereof and their use for the treatment of diseasesPublication Number: EP-3672954-B1Priority Date: 2017-08-21Grant Date: 2025-08-13
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: US-11345693-B2Priority Date: 2017-08-21Grant Date: 2022-05-31
- Compounds, salts thereof and methods for treatment of diseasesPublication Number: US-2022298151-A1Priority Date: 2017-08-21
- Compounds, salts thereof and methods for treating diseasesPublication Number: CN-117466803-APriority Date: 2017-08-21
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
| Clinical data | |
|---|---|
| Other names | ACP-204; ACP204 |
| Routes of administration | Oral[1] |
| Drug class | Serotonin 5-HT2A receptor inverse agonist |
| Pharmacokinetic data | |
| Onset of action | 4–6 hours (6 hours fasted, 9 hours fed) (TmaxTooltip time to peak levels)[2][3] |
| Elimination half-life | 17.8–19.8 hours[2] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2289704-13-6 |
| PubChem CID | 137520242 |
| UNII | H4L2AF2XB7 |
| Chemical and physical data | |
| Formula | C24H29F2N3O2 |
| Molar mass | 429.512 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “ACP 204”. AdisInsight. 23 January 2025. Retrieved 22 February 2025.
- Darwish M, Feng X, Dirks B, Raether B, Pathak SS (2025). “Pharmacokinetics in Healthy Adult and Elderly Patients of ACP-204, a Novel 5-HT 2A Receptor Selective Antagonist/Inverse Agonist”. Alzheimer’s & Dementia. 21 (S5) e105732. doi:10.1002/alz70859_105732. ISSN 1552-5260. PMC 12741707.
- Darwish M, Dirks B, Feng X, Raether B, Pathak SS (2025). “Effect of Food Consumption on the Pharmacokinetics of ACP-204, a Novel 5-HT 2A Receptor Selective Antagonist/Inverse Agonist”. Alzheimer’s & Dementia. 21 (S5) e105644. doi:10.1002/alz70859_105644. ISSN 1552-5260. PMC 12741626.
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 421. 2024.
remlifanserin N’-{[4-(cyclopropyloxy)phenyl]methyl}-N-[(2,4- difluorophenyl)methyl]-N-(1-methylpiperidin-4-yl)urea serotonin receptor (5-HT2A) inverse agonist […] C24H29F2N3O2 2289704-13-6 […]
- “Delving into the Latest Updates on ACP-204 with Synapse”. Synapse. 4 February 2025. Retrieved 22 February 2025.
- “ACP-204”. ALZFORUM. 5 February 2024. Retrieved 22 February 2025.
- Imbimbo C, Cotta Ramusino M, Leone S, Mazzacane F, De Franco V, Gatti A, et al. (February 2025). “Emerging Pharmacological Approaches for Psychosis and Agitation in Alzheimer’s Disease”. CNS Drugs. 39 (2): 143–160. doi:10.1007/s40263-024-01133-9. PMC 11769872. PMID 39623197.
- IsHak WW, Meyer A, Freire L, Totlani J, Murphy N, Renteria S, et al. (2024). “Overview of Psychiatric Medications in the Pipeline in Phase III Trials as of June 1, 2024: A Systematic Review”. Innovations in Clinical Neuroscience. 21 (7–9): 27–47. PMC 11424068. PMID 39329027.
- Kwon KJ, Kim HY, Han SH, Shin CY (October 2024). “Future Therapeutic Strategies for Alzheimer’s Disease: Focus on Behavioral and Psychological Symptoms”. International Journal of Molecular Sciences. 25 (21) 11338. doi:10.3390/ijms252111338. PMC 11547068. PMID 39518892.
- Burstein E, Markus Dey P, Pathak S (December 2024). “ACNP 63rd Annual Meeting: Poster Abstracts P305-P608: P497. Nonclinical Characterization of ACP-204, a Novel Second Generation 5-HT2A Inverse Agonist” (PDF). Neuropsychopharmacology. 49 (Suppl 1): 236–417 (346–347). doi:10.1038/s41386-024-02012-z. PMID 39643634.
- ACADIA Pharmaceuticals Inc. (2025-02-21). A Master Protocol for Three Independent, Seamlessly Enrolling, Double-blind, Placebo-controlled Efficacy and Safety Studies of ACP-204 in Adults with Alzheimer’s Disease Psychosis (Report). clinicaltrials.gov.
////////////remlifanserin, ANAX, serotonin receptor (5-HT2A) inverse agonist, ACP-204, ACP 204, H4L2AF2XB7
Relicpixant
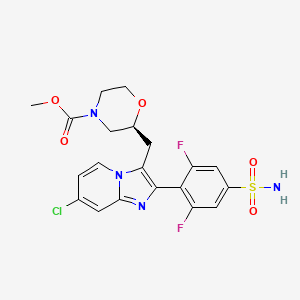
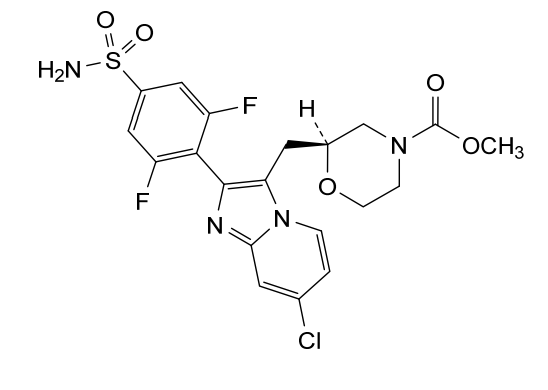
Relicpixant
CAS 2445366-94-7
MF C20H19ClF2N4O5S, Mw 500.9 g/mol
methyl (2S)-2-{[7-chloro-2-(2,6-difluoro-4-sulfamoylphenyl)imidazo[1,2-a]pyridin-3-yl]methyl}morpholine-4-
carboxylate
purinoreceptor (P2X) antagonist, 3NWJ8FHG2R
Relicpixant is a small molecule drug. The usage of the INN stem ‘-pixant’ in the name indicates that Relicpixant is a purinoreceptor (P2X) antagonist. Relicpixant has a monoisotopic molecular weight of 500.07 Da.
SYN
Example 172
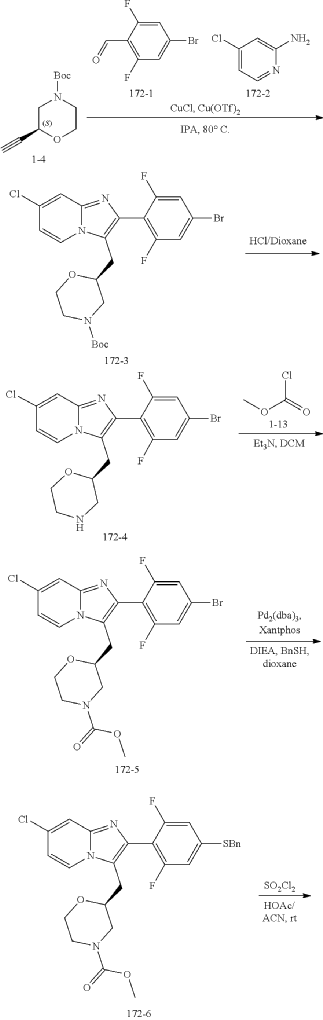
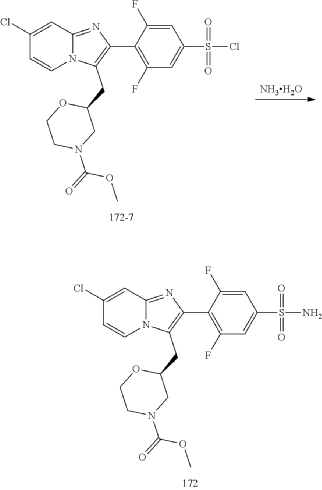
Step (8) Preparation of methyl (S)-2-((7-chloro-2-(2,6-difluoro-4-sulfamoylphenyl)imidazo[1,2-a]pyridin-3-yl)methyl)morpholine-4-carboxylate
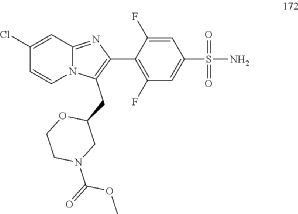
| Aqueous ammonia (2 mL) was diluted with acetonitrile (1 mL) and added dropwise to the above reaction system at 0° C. The reaction system was reacted at room temperature for 0.5 h. The starting material was consumed completely, and a target product was generated as detected by LCMS. The reaction system was extracted with water and ethyl acetate twice, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by preparative chromatography to give compound 172 (185 mg, 99.74% purity) in the form of a white solid. LC-MS: [M+H] +=501.1. |
PAT
Example 1: Preparation of Compound of Formula A
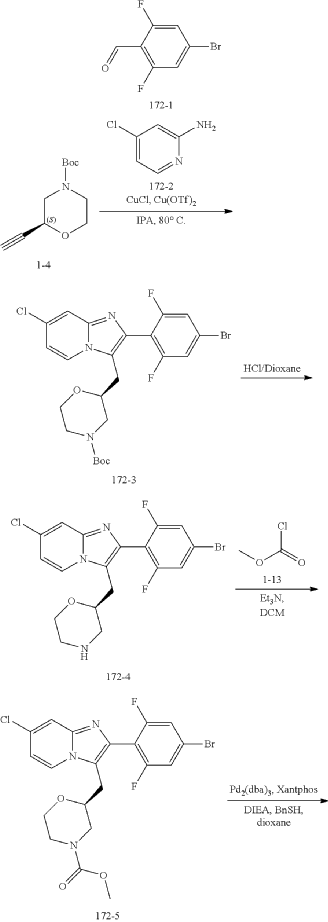
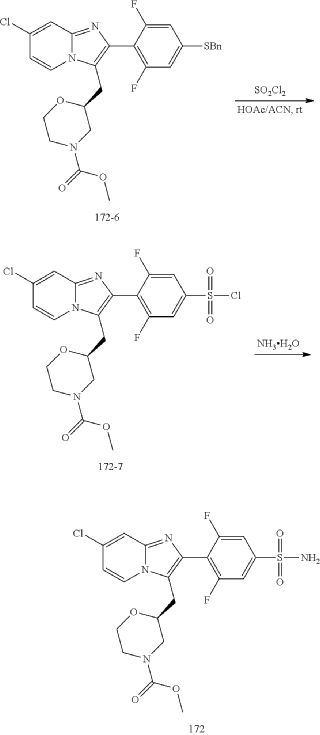
Step 6: Preparation of methyl (S)-2-((7-chloro-2-(2,6-difluoro-4-sulfamoylphenyl)imidazo[1,2-a]pyridin-3-yl)methyl)morpholine-4-carboxylate AS ABOVE
PAT
PAT
- Preparation method for heterocycloalkyl compound, and intermediate and application thereof heterocycloalkyl compoundPublication Number: WO-2022268218-A1Priority Date: 2021-06-24
- Pharmaceutical composition, preparation and preparation method and application thereofPublication Number: CN-115463133-BPriority Date: 2021-06-10Grant Date: 2024-03-01
- Pharmaceutical composition, preparation, and preparation method therefor and use thereofPublication Number: US-2024293420-A1Priority Date: 2021-06-10
- Pharmaceutical composition, preparation, and preparation method therefor and use thereofPublication Number: WO-2022258059-A1Priority Date: 2021-06-10
- Crystal form of heterocyclic compound, preparation method and application thereofPublication Number: CN-113929677-BPriority Date: 2020-06-29Grant Date: 2025-05-30
- Crystalline form of heterocyclic compound, preparation method therefor and application thereofPublication Number: EP-4169921-A1Priority Date: 2020-06-29
- Crystalline form of heterocyclic compound, preparation method therefor and application thereofPublication Number: WO-2022001820-A1Priority Date: 2020-06-29
- Crystalline form of heterocyclic compound, preparation method therefor and application thereofPublication Number: US-2023406850-A1Priority Date: 2020-06-29
- Heterocyclic compound intermediate, preparation method therefor and application thereofPublication Number: EP-3889154-A1Priority Date: 2018-12-29
- Heterocyclic compound intermediate, preparation method therefor and application thereofPublication Number: WO-2020135771-A1Priority Date: 2018-12-29
- Heterocyclic compound, intermediate, preparation method therefor and application thereofPublication Number: US-2023068538-A1Priority Date: 2018-12-29
- Heterocyclic compound, intermediate, preparation method and application thereofPublication Number: CN-113272301-BPriority Date: 2018-12-29Grant Date: 2024-04-26
- Heterocyclic compounds, intermediates, methods and applications thereof The present application applies to the Chinese patent application CN 2018116442319 with a filing date of December 29, 2018, and the Chinese patent application CN201910440214.3 with a filing date of May 24, 2019. Japan claims priority based on Chinese patent application CN200911016158.7 on October 24, 2019. In addition, the full text of the above Chinese patent application is incorporated in this application.Publication Number: JP-2022515879-APriority Date: 2018-12-29
- Heterocyclic compound, intermediate, preparation method therefor and application thereofPublication Number: US-2024034729-A9Priority Date: 2018-12-29
PAT
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////relicpixant, purinoreceptor (P2X) antagonist, 3NWJ8FHG2R
Ranosidenib
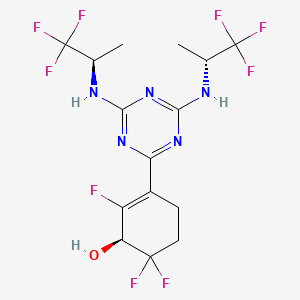
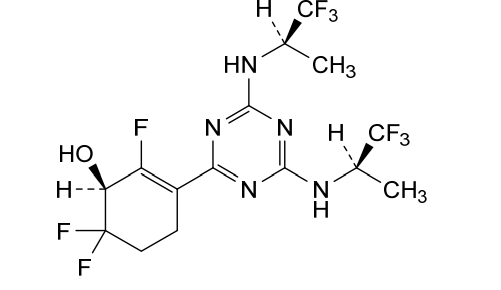
Ranosidenib
CAS 2301974-60-5
MF C15H16F9N5O MW 453.31 g/mol
(1S)-3-[4,6-bis[[(2R)-1,1,1-trifluoropropan-2-yl]amino]-1,3,5-triazin-2-yl]-2,6,6-trifluorocyclohex-2-en-1-ol
(1S)-3-(4,6-bis{[(2R)-1,1,1-trifluoropropan-2-yl]amino}-1,3,5-triazin-2-yl)-2,6,6-trifluorocyclohex-2-en-1-ol
isocitrate dehydrogenase (IDH) inhibitor, antineoplastic, [14C] HMPL-306, HMPL 306, PC64OXS2C2
- OriginatorHutchison MediPharma
- DeveloperHUTCHMED
- ClassAntineoplastics; Small molecules
- Mechanism of ActionIsocitrate dehydrogenase 1 inhibitors; Isocitrate dehydrogenase 2 inhibitors
- Phase IIIAcute myeloid leukaemia
- No development reportedHaematological malignancies; Solid tumours
- 28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in Spain (PO)
- 28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in USA (PO)
- 19 Sep 2025No development reported – Phase-I for Solid tumours (Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA (PO)
Ranosidenib is a small molecule drug. Ranosidenib is under investigation in clinical trial NCT06387069 (A Study to Evaluate HMPL-306 in Patients With IDH1- and IDH2-mutated Acute Myeloid Leukemia). Ranosidenib has a monoisotopic molecular weight of 453.12 Da.
Ranosidenib is an orally bioavailable inhibitor of mutated forms of both isocitrate dehydrogenase type 1 (IDH1, IDH1 [NADP+] soluble) in the cytoplasm and type 2 (IDH2, isocitrate dehydrogenase [NADP+], mitochondrial) in the mitochondria, with potential antineoplastic activity. Upon administration, ranosidenib specifically targets and inhibits mutant forms of IDH1 and IDH2, thereby inhibiting the formation of the oncometabolite 2-hydroxyglutarate (2HG) from alpha-ketoglutarate (a-KG). This prevents 2HG-mediated signaling and leads to both an induction of cellular differentiation and an inhibition of cellular proliferation in tumor cells expressing IDH mutations. IDH1 and 2, metabolic enzymes that catalyze the conversion of isocitrate into a-KG, play key roles in energy production and are mutated in a variety of cancer cell types. Mutant forms of IDH1 and 2 catalyze the formation of 2HG and drive cancer growth by blocking cellular differentiation and inducing cellular proliferation.
A Study of HMPL-306 in Advanced Hematological Malignancies With mIDHCTID: NCT04764474Phase: Phase 1Status: TerminatedDate: 2026-01-29
A Study of HMPL-306 in Advanced Solid Tumors With IDH MutationsCTID: NCT04762602Phase: Phase 1Status: TerminatedDate: 2025-09-16
A Study to Evaluate HMPL-306 in Patients With IDH1or IDH2-mutated Acute Myeloid LeukemiaCTID: NCT06387069Phase: Phase 3Status: RecruitingDate: 2025-08-14
Phase I Study of HMPL-306 for the Treatment of Gliomas With IDH1 and/or IDH2 MutationsCTID: NCT07025018Phase: Phase 1Status: RecruitingDate: 2025-08-01
A Study of HMPL-306 in Patients With IDH1 and/or IDH2 Mutation of Relapsed/Refractory Myeloid Leukemia/NeoplasmsCTID: NCT04272957Phase: Phase 1Status: Unknown statusDate: 2020-06-16
SYN
https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00625
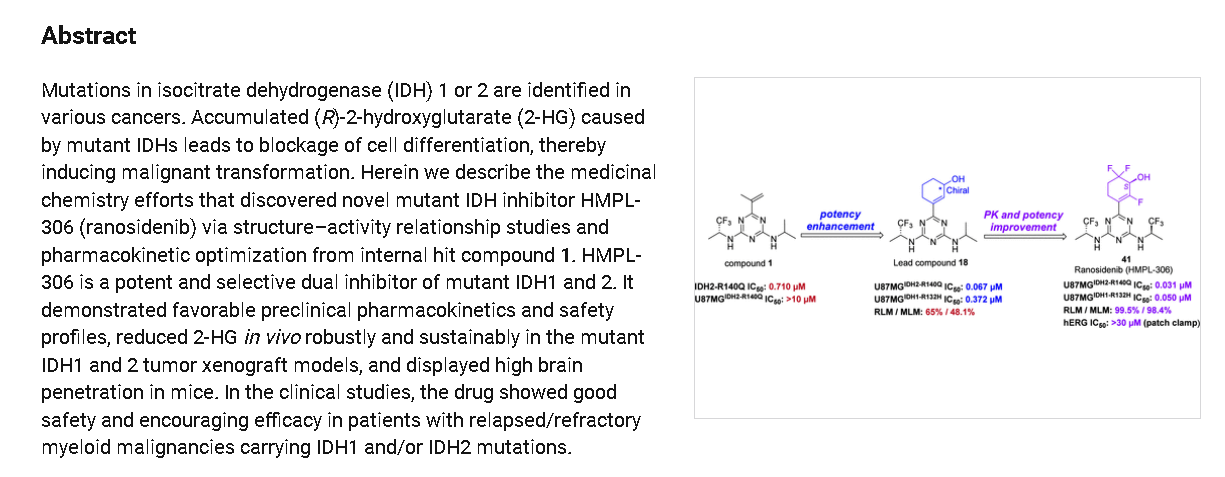
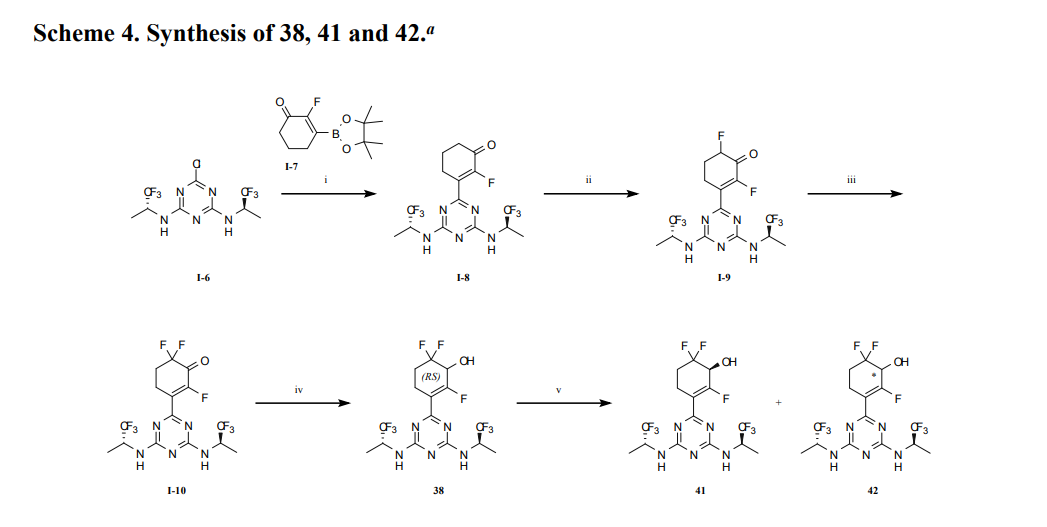
aReagents and conditions: (i) Na2PdCl4, DTBPPS, K2CO3, MeCN, H2O, 60 ℃; (ii) TBSOTf, Et3N,
DCM, 0~5 ℃; Selectfluor®, MeCN, 0~5 ℃; (iii) TBSOTf, Et3N, DCM, 0~5 ℃; Selectfluor®,
MeCN, 0~5 ℃; (iv) NaBH4, CeCl3·7H2O, EtOH, 0 ℃; (v) SFC separation.
Pat
Cycloolefin substituted heteroaromatic compounds and their use
Publication Number: US-2021363115-A2
Priority Date: 2017-09-07
PAT
Intermediate I-3
6-Chloro-N2,N4-bis((R)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine
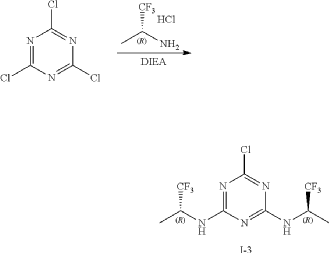
At 0° C., to a flask were added 1,4-dioxane (50 mL), 2,4,6-trichloro-1,3,5-triazine (1.84 g, 10 mmo), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (2.99 g, 20 mmol) and DIEA (5.17 g, 40 mmol). The reaction was heated to 60° C. and stirred for 4 hours. After the reaction was completed, the mixture was condensed and purified by flash column chromatography (eluting with gradient water/MeOH=100:0-0:100) to give Intermediate I-3 as yellow solid (2.50 g, yield: 74%). MS (m/z): 338.0 [M+H] +
Compounds 197 and 198
3-(4,6-Bis(((R)-1,1,1-trifluoropropan-2-yl)amino)-1,3,5-triazin-2-yl)-2,6,6-trifluorocyclohex-2-en-1-ol, optically pure diastereoisomers
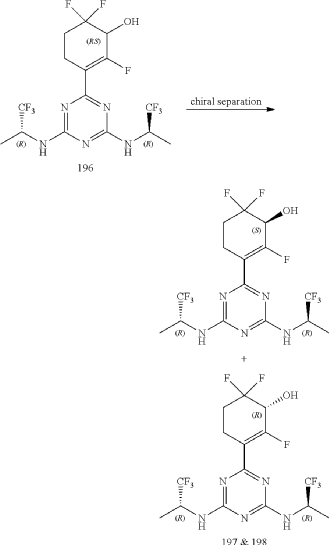
| The Compound 196 was resolved by chiral HPLC to provide a pair of optically pure diastereoisomers, Compounds 197 and 198 (Chiral HPLC conditions: Preparation instrument: Shimadzu LC-10AD vp; Column: Daicel AD-H(250 mm*30 mm, 5 um); mobile phase: n-heptane/isopropanol=90/10; flow rate: 40 mL/min; column temperature: 40° C.). The first eluent (RT=4.203 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 197, de %=99.27%, MS (m/z): 454.1 [M+1] +. The second eluent (RT=5.906 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 198, de %=97.82%, MS (m/z): 454.2 [M+1] +. |
| Compound 197: 1H NMR (400 MHz, CD 3OD): δ 5.00-4.86 (m, 2H), 4.36-4.17 (m, 1H), 2.80-2.65 (m, 1H), 2.58-2.42 (m, 1H), 2.25-2.05 (m, 2H), 1.37-1.31 (m, 6H). |
| Compound 198: 1H NMR (400 MHz, CD 3OD): δ 5.00-4.86 (m, 2H), 4.36-4.17 (m, 1H), 2.80-2.65 (m, 1H), 2.58-2.42 (m, 1H), 2.25-2.05 (m, 2H), 1.37-1.31 (m, 6H). |
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
/////////ranosidenib, isocitrate dehydrogenase (IDH) inhibitor, antineoplastic, [14C] HMPL-306, HMPL 306, PC64OXS2C2
Progerinin
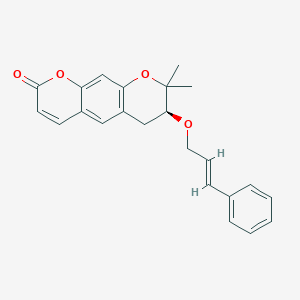
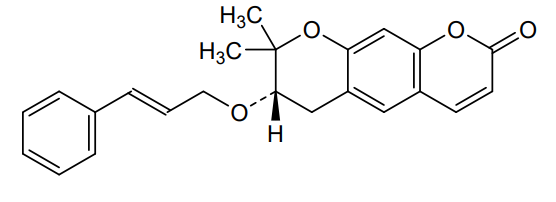
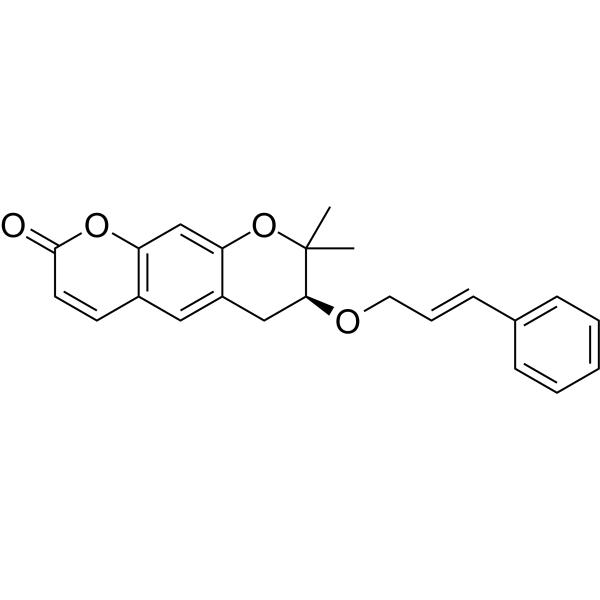
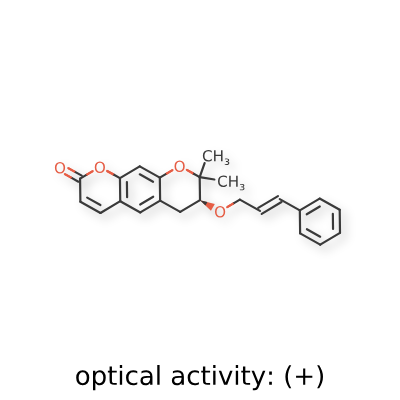
Progerinin
CAS 2249696-72-6
MF C23H22O4 MW 362.42
(3S)-2,2-dimethyl-3-[(E)-3-phenylprop-2-enoxy]-3,4-dihydropyrano[3,2-g]chromen-8-one
| (7S)-(+)-8,8-dimethyl-7-(3-phenyl-allyloxy)-7,8-dihydro-6H-pyrano[3,2-g]chromen-2-one |
| (7S)-7,8-Dihydro-8,8-dimethyl-7-[[(2E)-3-phenyl-2-propen-1-yl]oxy]-2H,6H-benzo[1,2-b:5,4-b?]dipyran-2-one |
(7S)-8,8-dimethyl-7-{[(2E)-3-phenylprop-2-en-1-yl]oxy}-7,8-dihydro-2H,6H-benzo[1,2-b:5,4-b’]dipyran-2-one
progerin-lamin A binding inhibitor, SLC-D011, SLC D011, KC 3, 426P9HSR8I
Progerinin (SLC-D011) is an orally active, targeted inhibitor designed to reduce the toxic, premature-aging protein “progerin” in Hutchinson-Gilford Progeria Syndrome (HGPS). It binds to progerin, disrupting its interaction with lamin A and promoting its degradation. Studies show it improves cardiac function, increases lifespan in mouse models, and is currently in clinical trials
Key Aspects of Progerinin:
- Mechanism of Action: Progerinin is an optimized progerin-lamin A binding inhibitor that selectively reduces progerin levels while sparing wild-type lamin A, B, and C.
- Disease Application: It targets HGPS, a rare genetic disease that causes premature, rapid aging and death due to cardiac issues.
- Preclinical Results: In Lmna mouse models, progerinin demonstrated improved physical conditions (hair morphology, body weight) and significantly extended lifespan (up to 14–21 weeks).
- Cardiac Benefits: It alleviates cardiovascular abnormalities, such as reducing cardiac muscle weakness, which is a major cause of death in HGPS patients.
- Clinical Status: A Phase I clinical trial was conducted for safety in healthy volunteers. As of 2025, trials are examining its efficacy, sometimes in combination with lonafarnib (Zokinvy).
- Administration: It is developed as a nanosuspension for oral administration. National Institutes of Health (NIH) | (.gov) +7
Progerinin was developed by Korean-based biotech company PRG Science & Technology Co., Ltd. (PRG S&T)
Progerinin (SLC-D011) is an orally active progerin-lamin A binding inhibitor. Progerinin selectively binds to the C-terminal region of progerin, disrupting its interaction with lamin A and promoting progerin degradation while sparing wild-type lamin A, B, and C. Progerinin ameliorates nuclear deformation, increases H3K9me3 levels, and reduces progerin expression in HGPS patient-derived fibroblasts. Progerinin extends lifespan in LmnaG609G/G609G mice and LmnaG609G/+ mice, improves body weight, hair morphology, cardiac function, and histological phenotypes. Progerinin can be used for the study of Hutchinson-Gilford progeria syndrome (HGPS).
- Study to Determine Optimal Dose and Evaluate Safety, Tolerability, and Pharmacokinetics of Progerinin in Patients With Hutchinson-Gilford Progeria Syndrome (HGPS)CTID: NCT06775041Phase: Phase 2Status: Active, not recruitingDate: 2026-02-09
- Phase 2, Open-Label Study to Evaluate the Safety and Tolerability of Progerinin in Werner SyndromeCTID: NCT05847179Phase: Phase 2Status: Not yet recruitingDate: 2026-01-23
- Phase I Study of Progerinin in Healthy VolunteersCTID: NCT04512963Phase: Phase 1Status: CompletedDate: 2021-09-22
PAPER
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2010-12
PMID: 20884093
DOI: 10.1016/j.ejmech.2010.09.006
SYN
<Example 1> Synthesis of Ether-Form (+)-Decursin Derivative (SLC-D011)
| (7S)-(+)-8,8-dimethyl-7-(3-phenyl-allyloxy)-7,8-dihydro-6H-pyrano[3,2-g]chromen-2-one (SLC-D011) was synthesized through the manner as in the to following Reaction Schemes 1 and 2. |
1. Synthesis Process I
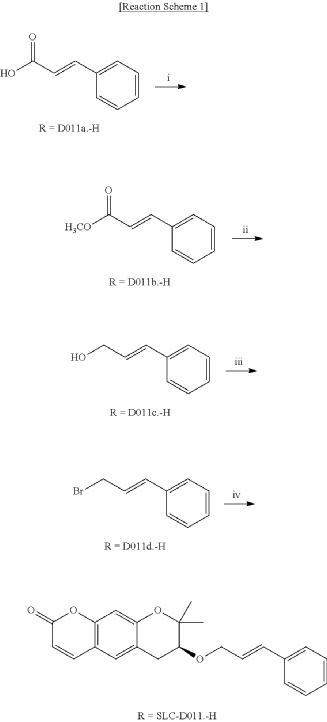
| Step (I): After dissolving trans-cinnamic acid (D0111a, 5 g, 33.7 mmol) in methanol (50 ml) in a 100 ml round bottom flask, 5 drops of concentrated H 2SO 4 was added and the mixture was refluxed by heating at 80° C. for 24 hours and was cooled to room temperature and then concentrated under reduced pressure. |
| Then, the mixture was separated with dichloromethane (300 ml) and distilled water (300 ml) to collect the organic layer and dehydrated with sodium sulfate and filtered. |
| After filtration, the filtrate was concentrated under reduced pressure to obtain 3-phenyl-acrylic acid, methyl ester (D011b, 5.39 g, yield=98.5%) as a pure product to apply to the next step. |
| The reaction solution was transferred to room temperature, stirred for 30 minutes and then a saturated aqueous solution of Rochelle’s salt (88 ml) was added thereto. |
2. Synthesis Process II
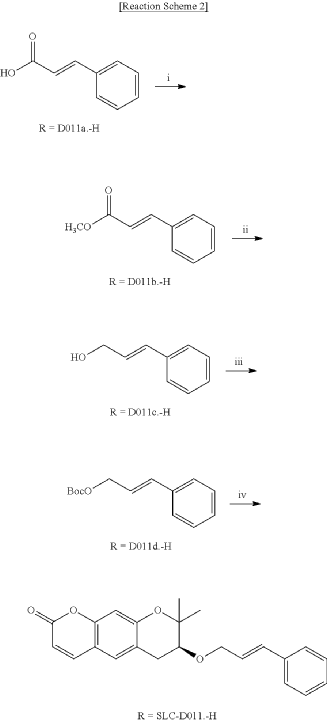
| The organic layers were collected and were dehydrated with sodium sulfate, filtered and the resulting filtrate was concentrated under reduced pressure. |
PAT
- A pharmaceutical composition for preventing or treating aging-related diseases containing a declucin derivative as an active ingredient.Publication Number: JP-6826674-B2Priority Date: 2017-04-25Grant Date: 2021-02-03
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: EP-3617211-B1Priority Date: 2017-04-25Grant Date: 2025-10-22
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: EP-3617211-A1Priority Date: 2017-04-25
- Pharmaceutical composition for preventing or treating aging-related diseases comprising decursin derivativesPublication Number: KR-20180119490-APriority Date: 2017-04-25
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: US-2020048274-A1Priority Date: 2017-04-25
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: CN-110573514-APriority Date: 2017-04-25
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: US-11008332-B2Priority Date: 2017-04-25Grant Date: 2021-05-18
- A kind of methyl isobutyl ketone low-temperature hydrogenation catalyst, preparation method and applicationPublication Number: CN-110871080-BPriority Date: 2018-08-30Grant Date: 2022-08-09
- Herbicidal composition containing bicyclopyrone and application thereofPublication Number: CN-113057169-BPriority Date: 2017-11-09Grant Date: 2022-07-01
- Preparation method for propylene epoxidation catalyst, and application thereofPublication Number: US-11291985-B2Priority Date: 2017-10-27Grant Date: 2022-04-05
- Pharmaceutical composition for preventing or treating aging-related diseases comprising decursin derivativesPublication Number: KR-102070328-B1Priority Date: 2017-04-25Grant Date: 2020-01-28
- Pharmaceutical composition for preventing or treating aging-related diseases containing decursin derivative as active ingredientPublication Number: WO-2018199633-A1Priority Date: 2017-04-25
- Tires comprising rubber compounds that comprise propylene-α-olefin-diene polymersPublication Number: US-12281221-B2Priority Date: 2019-07-17Grant Date: 2025-04-22
- Memory of sequences, method for creation and functioning of sequence memory, hierarchical sequence memoryPublication Number: US-2022027408-A1Priority Date: 2019-04-04
- A kind of preparation method of isovaleraldehydePublication Number: CN-111718247-BPriority Date: 2019-03-21Grant Date: 2022-08-05
- Deuterated analogs of acetyl-leucinePublication Number: CN-120157593-APriority Date: 2018-12-06
- Deuterated analogs of acetyl-leucinePublication Number: CN-113348018-BPriority Date: 2018-12-06Grant Date: 2025-04-01
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////progerinin, progerin-lamin A binding inhibitor, SLC-D011, SLC D011, KC 3, 426P9HSR8I
Lead (212Pb) bamzireotide navoxetan
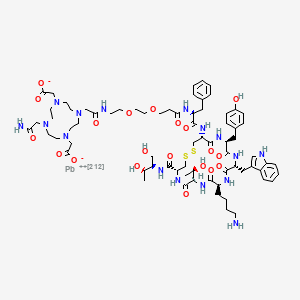
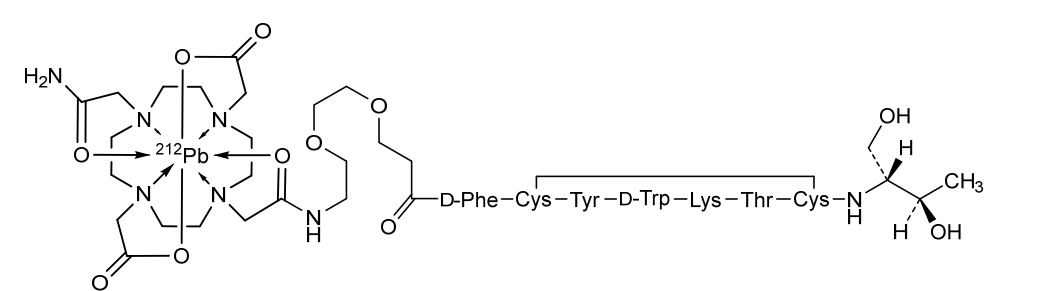
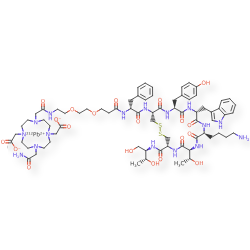
Lead (212Pb) bamzireotide navoxetan
CAS 2941523-47-1
MF C72H104N16O20212PbS2 MW1789.8 g/mol
2-[4-[2-[2-[2-[3-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(2R,3R)-1,3-dihydroxybutan-2-yl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-oxopropoxy]ethoxy]ethylamino]-2-oxoethyl]-10-(2-amino-2-oxoethyl)-7-(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lead-212(2+)

ANTINEOPLASTIC, D2A42X3LCS
SYN
US 11037690 B2
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////lead (212Pb) bamzireotide navoxetan, ANTINEOPLASTIC, D2A42X3LCS
Pixavir marboxil
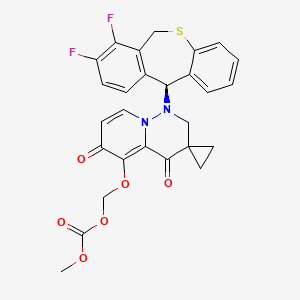
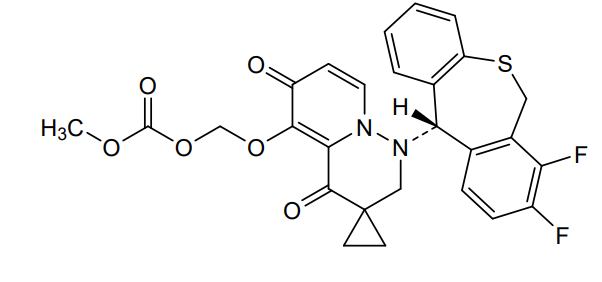
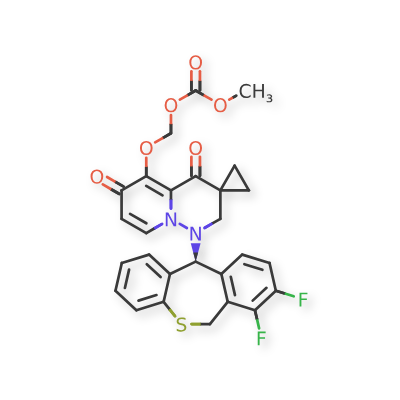
Pixavir marboxil
CAS 2365473-17-0
MF C27H22F2N2O6S MW540.535
(1-((11S)-7,8-DIFLUORO-6,11-DIHYDROBENZO(C)(1)BENZOTHIEPIN-11-YL)-4,6-DIOXOSPIRO(2H-PYRIDO(1,2-B)PYRIDAZINE-3,1′-CYCLOPROPANE)-5-YL)OXYMETHYL METHYL CARBONATE
({1′-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-4′,6′-dioxo1′,2′,4′,6′-tetrahydrospiro[cyclopropane-1,3′-pyrido[1,2-b]pyridazin]-5′-yl}oxy)methyl methyl carbonate
antiviral, TG 1000, Yi Li Kang, SV42843XSX, Cap-dependent endonuclease-IN-1, Influenza virus infections, TaiGen Biotechnology
- OriginatorTaiGen Biotechnology
- Class3-ring heterocyclic compounds; Antivirals; Benzene derivatives; Carbonates; Cyclopropanes; Dibenzothiepins; Esters; Ethers; Fluorobenzenes; Organic sulfur compounds; Pyridazines; Pyridones; Small molecules; Spiro compounds
- Mechanism of ActionEndonuclease inhibitors; Virus replication inhibitors
- MarketedInfluenza virus infections
- 27 Feb 2026Launched for Influenza virus infections (In adults, In adolescents) in China (PO), prior to February 2026 (TaiGen Biotechnology pipeline, February 2026)
- 26 Jan 2026Pixavir marboxil licensed to Boryung Biopharma for commercialization in South Korea
- 16 Dec 2025Chemical structure information added.
Pixavir marboxil (also known as TG-1000) is an investigational antiviral drug designed to treat and inhibit influenza virus infections. It belongs to a class of compounds known as cap-dependent endonuclease (CEN) inhibitors, which target a key viral enzyme necessary for influenza virus replication.
Mechanism of Action
- Blocks viral replication: Pixavir marboxil works by inhibiting the influenza virus’s cap-dependent endonuclease, a part of the viral RNA polymerase complex the virus needs to “snatch” capped RNA fragments from host cell mRNA. Without this process, the virus cannot efficiently produce its own viral proteins or replicate.
What Viruses It Targets
Pixavir marboxil has shown activity against:
- Influenza A viruses
- Influenza B viruses
- Certain drug-resistant influenza strains
This broad spectrum makes it useful for seasonal flu and potentially strains less responsive to older antiviral drugs
Clinical Development & Approval Status
Phase Trials & Results
- Completed Phase III: Clinical trials in adults and adolescents (age ≥12) showed that a single dose shortened time to symptom relief compared to placebo (e.g., median ~60.9 h vs ~87.9 h).
- Symptom relief benefits: The data indicated statistically significant improvement in flu symptoms and faster viral inactivation in treated patients versus placebo.
- Pediatric Formulation: China’s health authority approved pediatric Phase III studies for Pixavir (children <12), indicating further development for younger patients.
Regulatory Filings
- NDA (New Drug Application): Pixavir marboxil has been submitted for approval to the National Medical Products Administration (NMPA) in mainland China based on Phase III results.
- Generic Name Approved: The drug has been officially recognized with the generic name “Pixavir marboxil,” moving it closer to commercialization.
Pixavir marboxil is a small molecule drug. The usage of the INN stem ‘-xavir’ in the name indicates that Pixavir marboxil is a influenza CAP-dependent endonuclease inhibitor. Pixavir marboxil has a monoisotopic molecular weight of 540.12 Da.
SYN
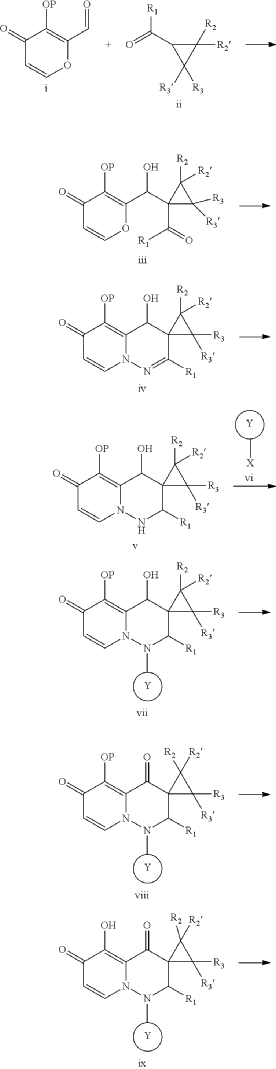
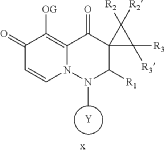
PAT
- Cap-dependent endonuclease inhibitorsPublication Number: US-10596171-B2Patent Family: AU-2019209426-A1; AU-2019209426-B2; BR-112020014810-A2; CA-3078391-A1; CA-3078391-C; CL-2020001919-A1; CN-110300753-A; CN-110300753-B; CO-2020006411-A2; EA-202090658-A1; EP-3743424-A1; EP-3743424-A4; IL-274199-A; IL-274199-B; JO-P20200159-A1; JP-2021512146-A; JP-6994121-B2; KR-102432975-B1; KR-20200086385-A; MX-2020007722-A; MY-197875-A; NZ-763248-A; PE-20211240-A1; PH-12020550921-A1; SG-11202003014V-A; TW-201938166-A; TW-I714951-B; US-10596171-B2; US-2019224198-A1; WO-2019144089-A1; ZA-202002037-BPriority Date: 2018-01-22Grant Date: 2020-03-24Inventor(s): LIN CHU-CHUNG; CHEN HUNG-CHUAN; CHIANG CHIAYN; YEN CHI-FENG; HSU MING-CHUAssignee(s): TAIGEN BIOTECHNOLOGY CO LTD; HSU MING CHUClassification: A61K31/5025; A61P31/16Abstract: Provided is a compound of Formula (I) below, or a pharmaceutically acceptable salt, metabolite, or prodrug thereof: n nwherein: A 1 is CR 4 or N; A 2 is CR 5 R 6 or NR 7 ; A 3 is CR 5 ′R 6 ′ or NR 7 ′; each of R 1 , R 2 , R 2 ′, R 3 , R 3 ′, R 4 , R 5 , R 5 ′, R 6 , R 6 ′, R 7 , and R 7 ′, independently, is hydrogen, deuterium, halogen, cyano, hydroxyl, carboxyl, amino, formyl, nitro, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 2-6 alkenyloxy, C 1-6 alkylcarbonyl, C 1-6 alkyloxycarbonyl, C 1-6 alkylamine, C 3-20 carbocyclyl, or C 3-20 heterocyclyl; or R 5 and R 6 , R 5 ′ and R 6 ′, or R 5 and R 5 ′, together with the adjacent atom to which they are each attached, form C 3-10 carbocyclyl or C 3-10 heterocyclyl. Further provided are a method of using the above-described compound, or the pharmaceutically acceptable salt, metabolite, or prodrug thereof for treating influenza and a pharmaceutical composition containing same.Linked Compounds: 274Linked Substances: 411
- Cap-dependent endonuclease inhibitorsPublication Number: US-2019224198-A1Priority Date: 2018-01-22Linked Compounds: 275Linked Substances: 411
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////pixavir marboxil, antiviral, TG 1000, Yi Li Kang, SV42843XSX, Cap-dependent endonuclease-IN-1, Influenza virus infections, TaiGen Biotechnology
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}