
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

IOHEXOL
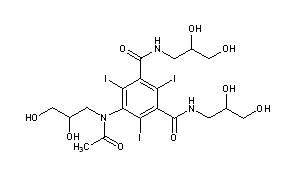
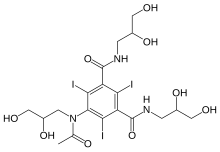
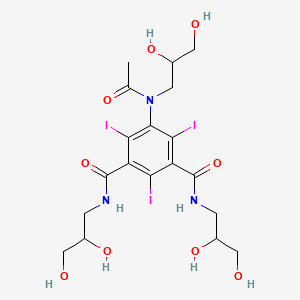
IOHEXOLCAS Registry Number: 66108-95-0N1,N3-bis(2,3-dihydroxypropyl)-5-[N-(2,3-dihydroxypropyl)acetamido]-2,4,6-triiodobenzene-1,3-dicarboxamide
CAS Name: 5-[Acetyl(2,3-dihydroxypropyl)amino]-N,N¢-bis(2,3-dihydroxypropyl)-2,4,6-triiodo-1,3-benzenedicarboxamideAdditional Names:N,N¢-bis(2,3-dihydroxypropyl)-5-[N-(2,3-dihydroxypropyl)acetamido]-2,4,6-triiodoisophthalamide
Manufacturers’ Codes: Win-39424; Compd 545Trademarks: Omnipaque (GE Healthcare)
Molecular Formula: C19H26I3N3O9Molecular Weight: 821.14Percent Composition: C 27.79%, H 3.19%, I 46.36%, N 5.12%, O 17.54%
Literature References: Nonionic radio-contrast medium. Prepn: V. Nordal, H. Holtermann, DE2726196; eidem,US4250113 (1977, 1981 both to Nyegaard). HPLC-UV determn in plasma: R. S. Soman et al., J. Chromatogr. B816, 339 (2005).
Pharmacology and toxicology: Acta Radiol.Suppl. 362, 1-134 (1980). Acute toxicity: S. Salvesen, ibid. 73. Fibrillatory potential in dogs: G. L. Wolf et al.,Invest. Radiol.16, 320 (1981).Comparative clinical studies in coronary angiography: G. B. J. Mancini et al.,Am. J. Cardiol.51, 1218 (1983); I. D. Sullivan et al.,Br. Heart J.51, 643 (1984); M. A. Bettmann et al.,Radiology153, 583 (1984). Review: T. Almén, Acta Radiol.Suppl. 366, 9-19 (1983).
Properties: Crystals from butanol, mp 174-180°. Sol in water. Stable in aqueous solutions. Viscosity (cP): 6.2 at 37°; 12.6 at 20° (c = 200 mg Iodine/ml). LD50 in male, female rats, mice (g Iodine/kg): 15.0, 12.3, 24.3, 25.1 i.v. (Salvesen).
Melting point: mp 174-180°
Toxicity data: LD50 in male, female rats, mice (g Iodine/kg): 15.0, 12.3, 24.3, 25.1 i.v. (Salvesen)Therap-Cat: Diagnostic aid (radiopaque medium).Keywords: Diagnostic Aid (Radiopaque Medium).
Synthesis ReferenceXiu C. Wang, Steve A. Chamberlin, Ashok V. Bhatia, Gregg E. Robinson, John Hufnagel, “Process for the preparation of iohexol.” U.S. Patent US5705692, issued December, 1985.
Iohexol, sold under the trade name Omnipaque among others, is a contrast agent used for X-ray imaging.[1] This includes when visualizing arteries, veins, ventricles of the brain, the urinary system, and joints, as well as during computed tomography (CT scan).[1] It is given by mouth, injection into a vein, or into a body cavity.[2]
Iohexol is a contrast agent for intrathecal administration used in myelography and contrast enhancement for computerized tomography.
Side effects include vomiting, skin flushing, headache, itchiness, kidney problems, and low blood pressure.[1] Less commonly allergic reactions or seizures may occur.[1] Allergies to povidone-iodine or shellfish do not affect the risk of side effects more than other allergies.[3] Use in the later part of pregnancy may cause hypothyroidism in the baby.[4] Iohexol is an iodinated non-ionic radiocontrast agent.[1] It is in the low osmolar family.[5]
Iohexol was approved for medical use in 1985.[6] It is on the World Health Organization’s List of Essential Medicines.[7][2]
Chemistry
The osmolality of iohexol ranges from 322 mOsm/kg—approximately 1.1 times that of blood plasma—to 844 mOsm/kg, almost three times that of blood.[8] Despite this difference, iohexol is still considered a low-osmolality contrast agent; the osmolality of older agents, such as diatrizoate, may be more than twice as high.[9]
Society and culture
Names
It is sold under the brand names Omnipaque[10] and Hexopaque. It is also sold as a density gradient medium under the names Accudenz, Histodenz and Nycodenz.[11][12]
Formulations
It is available in various concentrations, from 140[citation needed] to 350[13] milligrams of iodine per milliliter.
PATENT
https://patents.google.com/patent/WO2005003080A1/en#:~:text=Primary%20production%20of%20iohexol%20involves,and%20a%20thorough%20purification%20stage.&text=The%20solvent%20is%20then%20evaporated,and%20recrystallised%20twice%20from%20butanol.The present invention relates to a process for the manufacture of iohexol, 5-[N- (2,3- dihydroxypropyl) -acetamido]-N,N’-bis(2,3 -dihydroxypropyl)-2,4,6- triiodoisophtalamide.Iohexol is the non-proprietory name of the chemical drug substance of a non-ionic iodinated X-ray contrast agent marketed under the trade name OMNIPAQUE®. OMNIPAQUE® is one of the most used agents in diagnostic X-ray procedures.The manufacture of such non-ionic contrast agents involves the production of the chemical drug substance (referred to as primary production) followed by formulation into the drug product (referred to as secondary production). Primary production of iohexol involves a multistep chemical synthesis and a thorough purification stage. For a commercial drug product it is important for the primary production to be efficient and economical and to provide a drug substance fulfilling the specifications.The final step in the synthesis of iohexol is a N-alkylation step in which 5-(acetamido)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6 triiodoisophtalamide (hereinafter 5- Acetamide) is reacted in the liquid phase with an alkylating agent to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group. Following this reaction, iohexol is isolated from the reaction mixture and purified by crystallisation and treatment with ion exchange resins.The manufacture of iohexol is disclosed for example in US-4,250,113 which is hereby incorporated by reference. In the last step of the multistep chemical synthesis crude iohexol is obtained from the reaction between 5-Acetamide and 1-chloro-2,3- propandiol at ambient temperature in propylene glycoi and in the presence of sodium methoxide. The solvent is then evaporated and crude iohexol is obtained. The crude product is evaporated to dryness and recrystallised twice from butanol.Several suggestions to improve the N-alkylation and the purification steps have been published. WO-A-98/08804 discloses the use of 2-methoxy-ethanol and optionally isopropanol both in the alkylation step of 5-Acetamide and in the purification of crude iohexol. WO-A-02/083623 discloses the purification of crude iohexol using 1- methoxy-2-propanol as the solvent optionally in a mixture with other solvents.The N-alkylation step where 5-Acetamide in solution is reacted with an alkylation agent such as e.g. 1-chloro-2,3-propandiol to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group is illustrated in Scheme 1 :

5-Acetamide Iohexol5-acatamido-N,N’-bis(2,3-dihydroxypropyl)- 5-[N-(2,3-dihydroxypropyl)acetamido]- 2,4,6-triiodoisophtalamide N,N’-bis(2,3-dihydroxypropyl)- 2,4,6-triiodoisophtalamideScheme 1.The N-alkylation step is challenging because O-alkylated by-products can also be formed when the alkylation occurs at the oxygen atoms of the hydroxy groups. It is therefore a desire to limit the formation of these O-alkylated by-products and thereby to limit their presence in the final purified iohexol. The upper limit for values for O- alkylated by-products in the end product is fixed by the European Pharmacopea to 0.6% (HPLC by area).The O-alkylated by-products are removed to the degree desired or necessary by recrystallisation steps. Further unidentified by-products also referred to as impurities are also formed during the alkylation reaction and must be reduced to a tolerable level. In addition the solvents used should be easily available, be environmentally friendly and be of low toxicity.There is therefore a need to identify a solvent that can be used in the N-alkylation reaction and that fulfil the desiderata mentioned above. It is further desired to improve the overall process including the N-alkylation step and the purification step in the manufacture of iohexol. If the crude product obtained by the N-alkylation step is to be re-crystallised from a solvent that is different from the solvent used in the N- alkylation step, then the reaction solvent must first be removed e.g. by evaporation to dryness. It is known from crystallisation theory and experience that even small quantities of residual solvents from previous steps may cause a crystallisation process to get out of control due to changes in its supersaturation conditions, and thorough removal of the reaction solvent is an important step. Solvent removal is an energy consuming operation which also risks degradation of the product due to exposure to elevated temperature.Example 1 : Synthesis of iohexol in 1-methoxy-2-propanol/methanol1-methoxy-2-propanol (44 ml), methanol (19 ml) and sodium hydroxide (4.87 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 25°C. 1-chloro-2,3-propanediol (12.43 g) was added to the solution. After 1.5 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.1 %5-Acetamide 1.17 % O-alkylated substances 0.58 %Other impurities 0.1 %Example 2: Synthesis of iohexol in 1 -methoxy-2-propanol/water1-methoxy-2-propanol (63 ml), water (7 ml) and sodium hydroxide (4.50 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 35°C. 1-chloro-2,3-propanediol (11.39 g) was added to the solution. After 3 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.3 % 5-Acetamide 0.68 %O-alkylated substances 0.81 %Other impurities 0.3 % Example 3: Alkylation and crystallisation in solutions containing 1-methoxy-2- propanol1-methoxy-2-propanol (63 L), methanol (27 L) and sodium hydroxide (6.96 kg) was added to a 500 L reactor and stirred until all solids were dissolved and the temperature was below 30°C. 5-Acetamide (100 kg) was added to the reactor, and the mixture stirred overnight at 45°C before it was allowed to cool to 25°C. 1-chloro- 2,3-propanediol (16.76 kg) was added to the clear solution. After 1.5 hours, more 1- chloro-2,3-propanediol (1.18 kg) was added, and the reaction was allowed to proceed for 30 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 97.9 % 5-Acetamide 0.9 %O-alkylated substances 0.83 %Other impurities 0.4 %The reaction was stopped by addition of hydrochloric acid (650 ml), and the reaction mixture diluted with a mixture of 1-methoxy-2-propanol (53 L) and methanol (13 L). The mixture was filtered, and the salts on the filter washed with methanol (3×10 L). The combined filtrate and wash was diluted with water (22 L) and treated with cationic ion exchange resin (AMB 200C, 80 L) and anionic ion exchange resin (IRA 67, 80 L) to a salt content of 0.006 w/w %. The solution was filtered, and the ion exchange resins washed in several stages with a mixture of water (160 L) and methanol (85 L). The combined filtrate and wash was concentrated under reduced pressure to a volume of 155 L. One half of this was taken further to crystallisation as described below.Water was removed from the solution by azeotropic distillation. The volume was held at a constant level by replacing the distillate by 1-methoxy-2-propanol (80 L). At water content of 0.16 Ukg iohexol, further 1-methoxy-2-propanol (159 L) was added, and the solution seeded with iohexol crystals (0.26 kg). After stirring at reflux overnight, the volume of the solution was reduced by 42 L by distillation under reduced pressure (300-600 mbar). The temperature was set to 90°C, which was held for 3 hours before cooling to 60°C over 3 hours. The crystallisation mixture was stirred overnight at 60°C, filtered and washed with isopropanol (90 L, 6 portions). The yield was 48.4 kg (as dry powder), corresponding to 88-weight % corrected for seeding material and samples. HPLC analysis (water/acetonitrile) of the crystals gave the following results:Iohexol 99.3 %5-Acetamide 0.15 %O-alkylated substances 0.45 %Other impurities 0.11 %
PAPERhttps://www.quickcompany.in/patents/a-new-process-for-the-synthesis-of-high-pure-iohexol-and-its-intermediatesPATENThttps://patents.google.com/patent/WO2005003080A1/enThe present invention relates to a process for the manufacture of iohexol, 5-[N- (2,3- dihydroxypropyl) -acetamido]-N,N’-bis(2,3 -dihydroxypropyl)-2,4,6- triiodoisophtalamide.Iohexol is the non-proprietory name of the chemical drug substance of a non-ionic iodinated X-ray contrast agent marketed under the trade name OMNIPAQUE®. OMNIPAQUE® is one of the most used agents in diagnostic X-ray procedures.The manufacture of such non-ionic contrast agents involves the production of the chemical drug substance (referred to as primary production) followed by formulation into the drug product (referred to as secondary production). Primary production of iohexol involves a multistep chemical synthesis and a thorough purification stage. For a commercial drug product it is important for the primary production to be efficient and economical and to provide a drug substance fulfilling the specifications.The final step in the synthesis of iohexol is a N-alkylation step in which 5-(acetamido)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6 triiodoisophtalamide (hereinafter 5- Acetamide) is reacted in the liquid phase with an alkylating agent to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group. Following this reaction, iohexol is isolated from the reaction mixture and purified by crystallisation and treatment with ion exchange resins.The manufacture of iohexol is disclosed for example in US-4,250,113 which is hereby incorporated by reference. In the last step of the multistep chemical synthesis crude iohexol is obtained from the reaction between 5-Acetamide and 1-chloro-2,3- propandiol at ambient temperature in propylene glycoi and in the presence of sodium methoxide. The solvent is then evaporated and crude iohexol is obtained. The crude product is evaporated to dryness and recrystallised twice from butanol.Several suggestions to improve the N-alkylation and the purification steps have been published. WO-A-98/08804 discloses the use of 2-methoxy-ethanol and optionally isopropanol both in the alkylation step of 5-Acetamide and in the purification of crude iohexol. WO-A-02/083623 discloses the purification of crude iohexol using 1- methoxy-2-propanol as the solvent optionally in a mixture with other solvents.The N-alkylation step where 5-Acetamide in solution is reacted with an alkylation agent such as e.g. 1-chloro-2,3-propandiol to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group is illustrated in Scheme 1 :
5-Acetamide Iohexol5-acatamido-N,N’-bis(2,3-dihydroxypropyl)- 5-[N-(2,3-dihydroxypropyl)acetamido]- 2,4,6-triiodoisophtalamide N,N’-bis(2,3-dihydroxypropyl)- 2,4,6-triiodoisophtalamideScheme 1.The N-alkylation step is challenging because O-alkylated by-products can also be formed when the alkylation occurs at the oxygen atoms of the hydroxy groups. It is therefore a desire to limit the formation of these O-alkylated by-products and thereby to limit their presence in the final purified iohexol. The upper limit for values for O- alkylated by-products in the end product is fixed by the European Pharmacopea to 0.6% (HPLC by area).The O-alkylated by-products are removed to the degree desired or necessary by recrystallisation steps. Further unidentified by-products also referred to as impurities are also formed during the alkylation reaction and must be reduced to a tolerable level. In addition the solvents used should be easily available, be environmentally friendly and be of low toxicity.There is therefore a need to identify a solvent that can be used in the N-alkylation reaction and that fulfil the desiderata mentioned above. It is further desired to improve the overall process including the N-alkylation step and the purification step in the manufacture of iohexol. If the crude product obtained by the N-alkylation step is to be re-crystallised from a solvent that is different from the solvent used in the N- alkylation step, then the reaction solvent must first be removed e.g. by evaporation to dryness. It is known from crystallisation theory and experience that even small quantities of residual solvents from previous steps may cause a crystallisation process to get out of control due to changes in its supersaturation conditions, and thorough removal of the reaction solvent is an important step. Solvent removal is an energy consuming operation which also risks degradation of the product due to exposure to elevated temperature.Example 1 : Synthesis of iohexol in 1-methoxy-2-propanol/methanol1-methoxy-2-propanol (44 ml), methanol (19 ml) and sodium hydroxide (4.87 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 25°C. 1-chloro-2,3-propanediol (12.43 g) was added to the solution. After 1.5 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.1 %5-Acetamide 1.17 % O-alkylated substances 0.58 %Other impurities 0.1 %Example 2: Synthesis of iohexol in 1 -methoxy-2-propanol/water1-methoxy-2-propanol (63 ml), water (7 ml) and sodium hydroxide (4.50 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 35°C. 1-chloro-2,3-propanediol (11.39 g) was added to the solution. After 3 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.3 % 5-Acetamide 0.68 %O-alkylated substances 0.81 %Other impurities 0.3 % Example 3: Alkylation and crystallisation in solutions containing 1-methoxy-2- propanol1-methoxy-2-propanol (63 L), methanol (27 L) and sodium hydroxide (6.96 kg) was added to a 500 L reactor and stirred until all solids were dissolved and the temperature was below 30°C. 5-Acetamide (100 kg) was added to the reactor, and the mixture stirred overnight at 45°C before it was allowed to cool to 25°C. 1-chloro- 2,3-propanediol (16.76 kg) was added to the clear solution. After 1.5 hours, more 1- chloro-2,3-propanediol (1.18 kg) was added, and the reaction was allowed to proceed for 30 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 97.9 % 5-Acetamide 0.9 %O-alkylated substances 0.83 %Other impurities 0.4 %The reaction was stopped by addition of hydrochloric acid (650 ml), and the reaction mixture diluted with a mixture of 1-methoxy-2-propanol (53 L) and methanol (13 L). The mixture was filtered, and the salts on the filter washed with methanol (3×10 L). The combined filtrate and wash was diluted with water (22 L) and treated with cationic ion exchange resin (AMB 200C, 80 L) and anionic ion exchange resin (IRA 67, 80 L) to a salt content of 0.006 w/w %. The solution was filtered, and the ion exchange resins washed in several stages with a mixture of water (160 L) and methanol (85 L). The combined filtrate and wash was concentrated under reduced pressure to a volume of 155 L. One half of this was taken further to crystallisation as described below.Water was removed from the solution by azeotropic distillation. The volume was held at a constant level by replacing the distillate by 1-methoxy-2-propanol (80 L). At water content of 0.16 Ukg iohexol, further 1-methoxy-2-propanol (159 L) was added, and the solution seeded with iohexol crystals (0.26 kg). After stirring at reflux overnight, the volume of the solution was reduced by 42 L by distillation under reduced pressure (300-600 mbar). The temperature was set to 90°C, which was held for 3 hours before cooling to 60°C over 3 hours. The crystallisation mixture was stirred overnight at 60°C, filtered and washed with isopropanol (90 L, 6 portions). The yield was 48.4 kg (as dry powder), corresponding to 88-weight % corrected for seeding material and samples. HPLC analysis (water/acetonitrile) of the crystals gave the following results:Iohexol 99.3 %5-Acetamide 0.15 %O-alkylated substances 0.45 %Other impurities 0.11 %
PatentCN109134289https://patents.google.com/patent/CN109134289A/en

N-Acylation of 5-amino-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (1) with acetic anhydride (2) in the presence of p-TsOH gives 5-(acetylamino)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (3) , which upon condensation with glycidol using NaOMe in 2-methoxyethanol at 90 °C or epichlorohydrin by means of NaHCO3 in propylene glycol at 85 °C or 3-chloropropane-1,2-diol (5) using aqueous NaOH furnishes the iohexol .(7) synthesis of IodixanolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (207g, 2.03mol), acetic acid (103.3mL), p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), finishes reaction solution being heated to 60 DEG C Start to react, keep the temperature 30 minutes after reacting liquid temperature reaches 120-125 DEG C, cooling is concentrated into after can just stirring thereto It is added 50%v/v (600mL), is slowly added dropwise thereto into 50%w/v sodium hydrate aqueous solution, by adding in reaction process The mode of 50%w/v sodium hydrate aqueous solution keeps the pH of reaction solution between 11~12, and reaction temperature is maintained at 40-45 DEG C, Reaction is finished, and concentrated hydrochloric acid is added into reaction solution and adjusts pH3-4, and stirring filters after 3.0 hours, and filter cake is washed with water to neutrality, dries It is dry, obtain white solid 187g, yield 88.2%, HPLC98.14%.Go step obtained solid (150g, 0.2mol) be added there-necked flask in, thereto be added sodium hydroxide (14.4g, 0.36mol), purified water (300mL), epoxychloropropane (27.9g, 0.30mol) finish 30-35 DEG C of reaction 72.0 hours, instead It should finish, adjust pH3-4, Iodixanol HPLC purity 72.5%, Iohexol HPLC11.3% with concentrated hydrochloric acid.(4) synthesis of IohexolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (432g, 4.23mol) flows back 3.0 hours, be then concentrated under reduced pressure into p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), agitating and heating It can just stir, be added portionwise into reaction solution methanol (25g), methanol is added after 1.0 hours in stirring thereto again (140g) is finished and is stirred to react 1.0 hours, and being concentrated under reduced pressure into can just stir, and purified water (20g) then is added thereto, 60 DEG C are finished to be stirred overnight.Reaction solution is cooled to 30 DEG C hereinafter, extracting reaction solution 200mL, stirring is lower will with 50%w/v sodium hydrate aqueous solution Reaction solution pH is adjusted to 12, the addition 1- chloro- 2 into reaction solution, 3-propanediol (20g, 0.18mol), passes through benefit in reaction process The mode of 50%w/v sodium hydrate aqueous solution is added to keep the pH of reaction solution between 11~12, after reaction 12.0 hours thereto Add 1- chloro- 2,3-propanediol (3g, 29.29mmol) finishes that the reaction was continued 48.0 hours, and reaction solution samples HPLC detection, iodine Mykol purity is 89.9%.(5) synthesis of IoversolModus ponens (I) compound (200g, 0.28mol) is added in 1L there-necked flask, and N-Methyl pyrrolidone is added thereto Chloracetyl chloride (200mL) is added in (200mL) thereto under stirring, finish 50-53 DEG C and react 3.0 hours, and reaction is finished, and is cooled to 20 DEG C, reaction solution is slowly added in methanol (2000mL).It finishing, flows back 9.0 hours, reaction is finished, and is cooled to 25 DEG C, it filters, Filter cake is washed with methanol, and drying obtains white solid 177g, yield 79.8%, HPLC purity 98.3%.It takes previous step obtained solid (150g, 0.19mol) to be added in 1L there-necked flask, purified water 300mL is added thereto, Acetic acid sodium trihydrate (183g, 1.34mol) finishes back flow reaction, by adding 50%w/v sodium hydroxide water in reaction process The mode of solution keeps the pH of reaction solution between 5-6, and reaction is finished, and concentrated hydrochloric acid is added into reaction solution, adjusts pH3-4, stirring It being filtered after 3.0 hours, filter cake is with purifying water washing to neutrality, and drying obtains white solid 127g, yield 86.7%, HPLC98.4%.It takes step obtained solid (100g, 0.13mol), is added in 1L there-necked flask, purified water 300mL, chlorine are added thereto Change sodium (46.5g, 0.796mol), finish, be warming up to 50 DEG C, 10N sodium hydrate aqueous solution (39.3mL) and 2- are added thereto Chlorethanol (63.5g, 0.79mol) finishes 48-52 DEG C of heat preservation and reacts 5.0 hours, and reaction is finished, and concentrated hydrochloric acid is added thereto and adjusts PH6.5, reaction solution HPLC detection, Iohexol purity 89.7%.(6) synthesis of IopentolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (432g, 4.23mol) flows back 3.0 hours, be then concentrated under reduced pressure into p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), agitating and heating It can just stir, be added portionwise into reaction solution methanol (25g), methanol (140g) is added thereto again after stirring 1.0 hours, It finishes and is stirred to react 1.0 hours, being concentrated under reduced pressure into can just stir, and purified water (20g) then is added thereto, finishes 60 DEG C It is stirred overnight.Reaction solution is cooled to 30 DEG C hereinafter, extracting reaction solution 200mL, stirring is lower will with 50%w/v sodium hydrate aqueous solution Reaction solution pH is adjusted to 12, and the chloro- 3- methoxy-2-propanol (22.5g, 0.18mol) of 1-, reaction process are added into reaction solution In keep the pH of reaction solution between 11~12 by way of adding 50%w/v sodium hydrate aqueous solution, react 12.0 hours Add 1- chloro- 2 thereto afterwards, 3-propanediol (3.4g, 29.29mmol) finishes that the reaction was continued 48.0 hours, reaction solution sampling HPLC detection, Iopentol purity are 91.3%.(7) synthesis of IodixanolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (207g, 2.03mol), acetic acid (103.3mL), p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), finishes reaction solution being heated to 60 DEG C Start to react, keep the temperature 30 minutes after reacting liquid temperature reaches 120-125 DEG C, cooling is concentrated into after can just stirring thereto It is added 50%v/v (600mL), is slowly added dropwise thereto into 50%w/v sodium hydrate aqueous solution, by adding in reaction process The mode of 50%w/v sodium hydrate aqueous solution keeps the pH of reaction solution between 11~12, and reaction temperature is maintained at 40-45 DEG C, Reaction is finished, and concentrated hydrochloric acid is added into reaction solution and adjusts pH3-4, and stirring filters after 3.0 hours, and filter cake is washed with water to neutrality, dries It is dry, obtain white solid 187g, yield 88.2%, HPLC98.14%.Go step obtained solid (150g, 0.2mol) be added there-necked flask in, thereto be added sodium hydroxide (14.4g, 0.36mol), purified water (300mL), epoxychloropropane (27.9g, 0.30mol) finish 30-35 DEG C of reaction 72.0 hours, instead It should finish, adjust pH3-4, Iodixanol HPLC purity 72.5%, Iohexol HPLC11.3% with concentrated hydrochloric acid.To sum up, method of the invention is easy to operate, and (III) three obtained formula (I), formula (II) or formula intermediate can be made For the raw material for synthesizing diodone, not by-product truly;Importantly, general sieve of synthesis iodine that can be convenient Amine does not have the generation of two acylated by-products, and compared with original grinds the production technology of medicine, process route is entirely different, high income, cost It is low, a kind of very effective, completely new approach is provided for industrialized production Iopromide, is had a extensive future.
Patent
Publication numberPriority datePublication dateAssigneeTitleWO1998008804A1 *1996-08-291998-03-05Nycomed Imaging AsProcess for iohexol manufactureUS5847212A *1997-04-211998-12-08Abbott LaboratoriesProcess for the preparation of iohexolWO1999026916A1 *1997-11-261999-06-03Nycomed Imaging AsN-alkylation of 5-amino-2,4,6-triiodo-isophthalamidesFamily To Family CitationsITMI20010773A1 *2001-04-112002-10-11Chemi SpaProcess for the production of high purity iohexole
Non-Patent
TitleHAAVALDSEN J ET AL: “X-RAY CONTRAST AGENTS. I. SYNTHESIS OF SOME DERIVATIVES OF 5-AMINO-2, 4, 6-TRIIODOISOPHTHLAMIDE”, ACTA PHARMACEUTICA SUECICA, XX, XX, vol. 20, no. 3, 1983, pages 219 – 232, XP002052827, ISSN: 0001-6675 *
Publication numberPriority datePublication dateAssigneeTitleWO2007013816A1 *2005-07-292007-02-01Ge Healthcare AsContinuous crystallisation process of iodinated phenyl derivativesWO2007060380A1 *2005-11-242007-05-31Hovione Inter LtdProcess for the manufacture of iohexolJP2009502910A *2005-07-292009-01-29ジーイー・ヘルスケア・アクスイェ・セルスカプMethod for continuous crystallization of iodinated phenyl derivativesCN101195587B *2006-12-192010-07-21浙江尖峰海洲制药有限公司Production method for lodixanol hydrolysateUS8766002B22009-11-262014-07-01Imax Diagnostic Imaging Holding LimitedPreparation and purification of iodixanolNO342021B1 *2005-07-292018-03-12Ge Healthcare AsContinuous crystallization processFamily To Family CitationsWO2011041275A1 *2009-09-302011-04-07Mallinckrodt Inc.Alkylation of triiodo-substituted arylamides in an aqueous mixed solvent systemES2680019T3 *2010-12-212018-09-03Ge Healthcare AsDesalination of a composition comprising a contrast agentUS20140065076A1 *2012-08-302014-03-06Otsuka Pharmaceutical Co. Ltd.Container with concentrated substance and method of using the same* Cited by examiner, † Cited by third party, ‡ Family to family citation
Similar Documents
PublicationPublication DateTitleEP1641743B12008-11-12Process for iohexol manufactureKR101188596B12012-10-05Preparation of iodixanolEP1960349B12015-11-18Purification of iodixanolEP1966110B12013-04-24Purification process of iodixanolJP5536087B22014-07-02Method for producing iodinated contrast agentUS5948940A1999-09-07Process for iohexol manufactureUS7541494B22009-06-02Process for the manufacture of iohexolEP2277855B12011-11-09Crystallization of iodixanol using millingCA2707173C2011-08-02Crystallization of iodixanol in isopropanol and methanolRU2173315C22001-09-10Method of preparing ionexolCA2710577C2012-09-18Crystallization of iodixanol using milling

NEW DRUG APPROVALS
one time
$10.00
References
- ^ Jump up to:a b c d e World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. pp. 317–8. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 171. ISBN 9781284057560.
- ^ ACR Manual on Contrast Media v10.3. 2017 (PDF). American College of Radiology. 2017. p. 6. ISBN 9781559030120. Archived (PDF) from the original on 1 January 2018. Retrieved 1 January 2018.
- ^ Briggs, Gerald G.; Freeman, Roger K.; Yaffe, Sumner J. (2011). Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. Lippincott Williams & Wilkins. p. 761. ISBN 9781608317080. Archived from the original on 1 January 2017.
- ^ Sutton, David; Young, Jeremy W. R. (2012). A Short Textbook of Clinical Imaging. Springer Science & Business Media. p. 235. ISBN 9781447117551. Archived from the original on 1 January 2017.
- ^ Broe, Marc E. de; Porter, George A.; Bennett, William M.; Verpooten, G. A. (2013). Clinical Nephrotoxins: Renal Injury from Drugs and Chemicals. Springer Science & Business Media. p. 325. ISBN 9789401590884. Archived from the original on 1 January 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ GE Healthcare (May 2006). “Omnipaque (Iohexol) injection. Product label”. DailyMed. U.S. National Library of Medicine. Retrieved 28 March 2007.
- ^ Amersham Health (April 2006). “Hypaque (Diatrizoate Meglumine and Diatrizoate Sodium) injection, solution. Product label”. DailyMed. U.S. National Library of Medicine. Archived from the original on 23 May 2011. Retrieved 29 March 2007.
- ^ “Omnipaque” (PDF). Ireland: Health Products Regulatory Authority. January 2018. Retrieved 31 July 2020.
- ^ “HistoDenz (D2158)” Archived 2015-11-20 at the Wayback Machine, product information sheet, Sigma-Aldrich. Accessed on line 19 November 2015.
- ^ “Nycodenz®: A universal density gradient medium” Archived 2015-02-26 at the Wayback Machine, Axis-Shield Density Gradient Media. Accessed 19 November 2015.
- ^ Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Omnipaque 350 mg J/ml Infusionsflasche.
External links
- “Iohexol”. Drug Information Portal. U.S. National Library of Medicine.
- “Iohexol Injection, Oral, Rectal Advanced Patient Information”. Drugs.com. 13 January 2019. Retrieved 3 February 2020.
| Clinical data | |
|---|---|
| Trade names | Omnipaque, Hexopaque, Oraltag, others |
| Other names | 5-[N-(2,3-Dihydroxypropyl)acetamido]-2,4,6-triiodo-N,N’-bis(2,3-dihydroxypropyl)isophthalamide |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| License data | US DailyMed: Iohexol |
| Routes of administration | intrathecal, intravascular, by mouth, intracavital, rectal |
| ATC code | V08AB02 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | Low |
| Metabolism | Nil |
| Elimination half-life | Variable |
| Excretion | Kidney, unchanged |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 66108-95-0 |
| PubChem CID | 3730 |
| DrugBank | DB01362 |
| ChemSpider | 3599 |
| UNII | 4419T9MX03 |
| KEGG | D01817 |
| ChEBI | CHEBI:31709 |
| ChEMBL | ChEMBL1200455 |
| CompTox Dashboard (EPA) | DTXSID6023157 |
| ECHA InfoCard | 100.060.130 |
| Chemical and physical data | |
| Formula | C19H26I3N3O9 |
| Molar mass | 821.142 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 174 to 180 °C (345 to 356 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
////////////IOHEXOL, Win-39424, Compd 545, Omnipaque, Oraltag, GE Healthcare, X RAY CONTRAST AGENTS, WIN 39424
CC(=O)N(CC(O)CO)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
ONO-2910

ONO-2910
CAS 2410177-35-2
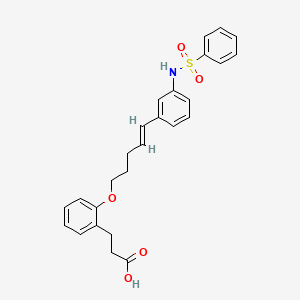
3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acidC26 H27 N O5 S465.56Benzenepropanoic acid, 2-[[(4E)-5-[3-[(phenylsulfonyl)amino]phenyl]-4-penten-1-yl]oxy]-
ONO Pharmaceuticals is developing ONO-2910 , the lead from a program of novel transient receptor potential cation channel 4/5 inhibitors, for treating peripheral neuropathy. In April 2021, a phase II trial in patients with diabetic polyneuropathy was initiated.
PATENT
CN112513011-BENZENE DERIVATIVE
| Example 84: 3-[2-[(E)-5-[3-(Benzenesulfonamido)phenyl]pent-4-enyloxy]phenyl]propionic acid |
| [Chemical formula 52] |
| |
| To a solution of the compound (146 mg) produced in Example 83 in THF (0.5 mL) and methanol (0.1 mL), 1M aqueous lithium hydroxide solution (0.5 mL) was added, and the mixture was stirred at 50°C for 8 hours. 1M hydrochloric acid was added to make it acidic, and it was extracted with ethyl acetate. After drying the organic layer over sodium sulfate, it was concentrated under reduced pressure to obtain the title compound (105 mg) having the following physical properties. |
| HPLC retention time (min): 1.10 |
| 1 H-NMR(CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61,2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75 -7.78. |

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153690
Novel crystalline forms of 3-[2-[(E)-5-[3-(benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid act as neuroprotective, useful for treating neurological disorders eg chronic inflammatory demyelinating polyneuritis, Guillain-Barre syndrome and allergic angiitis.Example 1:
Sulfuric acid (0.26 mL) is added to a solution of isopropyl 3- (2-hydroxyphenyl) propanoate 3,4-dihydrocoumarin (50.0 g) in isopropyl alcohol (500 mL), and the reaction mixture is mixed at room temperature for 2 hours. Stirred. The reaction mixture was concentrated under reduced pressure, and the obtained residue was diluted with ethyl acetate. The mixture was washed with saturated aqueous sodium hydrogen carbonate solution, water and saturated brine, dried over sodium sulfate, and concentrated under reduced pressure to give the title compound (73.2 g) having the following physical properties.
1 1 H-NMR (CDCl 3 ): δ 1.20, 2.66-2.70, 2.87-2.91, 4.95-5.08, 6.86-6.91, 7.06-7.15, 7.35.
Example 2: Isopropyl 3- (2- (pent-4-in-1-yloxy) phenyl) propanoate In a solution of the compound (3.00 g) prepared in Example 1 in N, N-dimethylacetamide (25 mL) at room temperature. Cesium carbonate (9.39 g) was added at the same temperature, and the mixture was stirred at the same temperature for 15 minutes. 5-Chloro-1-pentyne (CAS Registry Number: 14267-92-6) (1.63 g) was added to the reaction solution at room temperature, and the mixture was stirred at 60 ° C. for 3 hours. Water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 1: 0 → 5: 1) to give the title compound (2.40 g) having the following physical property values.
HPLC retention time (minutes): 1.13.Example 3: Isopropyl (E) -3- (2-((5- (4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) penta-4-en-1-yl) Il) Oxy) Phenyl) Propanoate In
a heptane (2 mL) solution of the compound (1.00 g) prepared in Example 2, 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1. 17 g) and 4-dimethylaminobenzoic acid (60.2 mg) were added, and the mixture was stirred at 100 ° C. for 4 hours. The reaction solution was cooled to room temperature and then concentrated. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 20: 1 → 4: 1) to give the title compound (503 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.38.Example 3 (1):
Pyridine (0.95 mL), N, N-dimethyl in a solution of N- (3-bromophenyl) benzenesulfonamide 3-bromoaniline (1.02 g) in dichloromethane (20 mL) at 0 ° C. Aminopyridine (hereinafter abbreviated as DMAP) (72.4 mg) and benzenesulfonyl chloride (1.10 g) were added, and the mixture was stirred at room temperature for 2 hours. After concentrating the reaction solution, the obtained residue is purified by silica gel column chromatography (hexane: ethyl acetate = 9: 1 → 2: 1) to give the title compound (1.96 g) having the following physical properties. rice field.
HPLC retention time (minutes): 0.98.
Example 4: Isopropyl (E) -3-(2-((5- (3- (phenylsulfonamide) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The
compound prepared in Example 3. In a solution of (180 mg) in THF (3 mL), the compound (168 mg) prepared in Example 3 (1), chloro (2-dicyclohexylphosphino-2′, 4′, 6′-triisopropyl-1,1′- Biphenyl) [2- (2′-amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M tripotassium phosphate aqueous solution (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. .. The reaction solution was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.24
Example 5: 3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
[Chemical 2]
A 1 M aqueous lithium hydroxide solution (0.5 mL) was added to a solution of the compound (146 mg) prepared in Example 4 in THF (0.5 mL) and methanol (0.1 mL), and the mixture was stirred at 50 ° C. for 8 hours. It was acidified by adding 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical characteristics.
Form: Amorphous
HPLC retention time (minutes): 1.101
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75-7.78.
PATENT
WO2020027150
https://patents.google.com/patent/WO2020027150A1/en
Example 83: Isopropyl (E) -3- (2-((5- (3- (phenylsulfonamido) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The compound prepared in Example 82 Compound (168 mg) prepared in Example 9 and chloro (2-dicyclohexylphosphino-2 ′, 4 ′, 6′-triisopropyl-1,1′-biphenyl) [180 mg) in THF (3 mL) solution were added. 2- (2′-Amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M aqueous solution of tripotassium phosphate (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. After cooling the reaction solution to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical data.
HPLC retention time (min): 1.24.Example 84: 3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acid
To a solution of the compound prepared in Example 83 (146 mg) in THF (0.5 mL) and methanol (0.1 mL) was added a 1 M aqueous lithium hydroxide solution (0.5 mL), and the mixture was stirred at 50 ° C. for 8 hours. The mixture was acidified with 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical data.
HPLC retention time (min): 1.10
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45 -7.49, 7.55, 7.75-7.78.

///////////ONO-2910, ONO 2910, PHASE 2,
O=S(=O)(Nc1cc(\C=C\CCCOc2ccccc2CCC(=O)O)ccc1)c1ccccc1
Bemiparin

Bemiparin
- AVE 5026
- Adomiparin
- Ardeparin
- Arteven
- Bemiparin
- CY 216
- CY 222
- Centaxarin
- Certoparin
- Clevarin
- Clivarin
- Clivarine
- Dalteparin
- Deligoparin
- F 202
- FR 860
- Fluxum
- Fragmin A
- Fragmin B
- Fraxiparin
- Gammaparin
- H 5284
- H 9399
- Hapacarin
- Heparin subcutan
- Heparin sulfate
- Heparinic acid
- Heparins
- KB 101
- Leparan
- LipoHep Forte
- Livaracine
- M 118
- M 118REH
- M 402
- M 402 (heparin)
- Mono-embolex
- Multiparin
- Nadroparin
- Nadroparine
- Necuparanib
- Novoheparin
- OP 386
- OP 622
- Octaparin
- Pabyrn
- Parnaparin
- Parvoparin
- Reviparin
- Sandoparin
- Semuloparin
- Subeparin
- Sublingula
- Tafoxiparin
- Tinzaparin
- Triofiban
- Vetren
- Vitrum AB
- α-Heparin
cas 91449-79-5
Bemiparin (trade names Ivor and Zibor, among others) is an antithrombotic and belongs to the group of low molecular weight heparins (LMWH).[1]
Bemiparin is an ultra-low molecular weight heparin (ultra-LMWH) used to prevent thromboembolism following surgery and extracorporeal clotting during dialysis.
Rovi and Archimedes (a wholly owned subsidiary of ProStrakan), have developed and launched bemiparin, a Factor Xa inhibitor for the injectable treatment and prevention of thrombosis.
low or very low molecular weight heparins (eg bemiparin sodium) with a high anti-factor Xa activity for the treatment of deep vein thrombosis.
Bemiparin is an antithrombotic and belongs to the group of drugs known as the low molecular weight heparins (LMWH). Like semuloparin, bemiparin is classified as an ultra-LMH because of its low mean molecular mass of 3600 daltons, which is a unique property of this class 1. These heparins have lower anti-thrombin activity than the traditional low molecular weight heparins and act mainly on factor-Xa, reducing the risk of bleeding due to selectivity for this specific clotting factor. Interestingly, current research is underway for the potential benefit of bemiparin in the treatment of tumors and diabetic foot ulcers 12,1.
Laboratorios Farmaceuticos Rovi has developed and launched Enoxaparina Rovi, a biosimilar version of enoxaparin sodium, an injectable low-molecular-weight fraction of heparin, for the prophylaxis of venous thromboembolism.
PATENT
WO2018015463
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018015463
claiming a method for analyzing glycosaminoglycans, heparins and their derivatives in a compound comprising a monosaccharide residues present in heparin (eg bemiparin sodium) chains by identification and relative quantification of its characteristic signals by1H NMR one-dimensional nuclear magnetic resonance and/or 1H-13C HSQC two-dimensional nuclear magnetic resonance, using dimethylmalonic acid as internal reference
PATENT
CN-110092848
https://patents.google.com/patent/CN110092848A/enEmbodiment 1Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH160712 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (5000ml) three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, freeze dryer (on Hai Dongfulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included:1. at salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 10g is poured into tank, and the purified water of 100ml is added into reactor tank, is stirred to molten Solution is complete.1.1.2 25g benzethonium chloride is added in beaker, 125ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 4.5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid Removing.1.2 washings, centrifugation, drying:1.2.1 300ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 20 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 40 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 60 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 32g, does Dry weightless 1.5%.2. degradation2.1 weighingBy above-mentioned 30g heparin-benzyl rope ammonium salt in 500ml three-necked flask, the methylene chloride of 150ml is added into reactor tank It is added in three-necked flask.2.2 dissolutions: three-necked flask is put into digital display heat-collecting magnetic stirring device, is heated to 33 DEG C and is stirred to having dissolved Entirely.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 20.4ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 34 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 180ml10% (W/V) sodium acetate methanol is added thereto Solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 300ml methanol solution, dry bemiparin crude product about 9g.3. purification3.1 will be above-mentioned dry that 9g bemiparin crude product pours into tank, and the purified water of 90ml, stirring are added into reactor tank It is complete to dissolution.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.0.54ml hydrogen peroxide is added to be stirred to react at 20 DEG C 7.5 hours, through 0.22 μm of micro porous filtration.3.3 1.8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 450ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 90ml purified water is added, and stirring adjusts PH6.5 to dissolving completely, through 0.22 μm of micro porous filtration, Sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 7g.Three, the primary quality measure statistics of gained bemiparin
| Serial number | Project | Control standard | Testing result |
| 1 | Weight average molecular weight | 3000~4200 | 3650 |
| 2 | Molecular weight is greater than 6000 constituent content | < 15% | 12.9% |
| 3 | Constituent content of the molecular weight less than 2000 | < 35% | 36.7% |
| 4 | Molecular weight is between 2000~6000 constituent contents | 50%~75% | 50.4% |
| 5 | Anti-Xa activity | 80~120IU/mg | 116IU/mg |
| 6 | Anti- IIa activity | 5~20IU/mg | 14.6IU/mg |
| 7 | The anti-anti- IIa of Xa/ | ≥7 | 7.95 |
Embodiment 2Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH180912 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (10000ml, 30000L), three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, Freeze dryer (Shanghai Dong Fulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included: 1. one-tenth salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 500g is poured into tank, the purified water of 5000ml is added into reactor tank, stirring is extremely Dissolution is complete.1.1.2 1250g benzethonium chloride is added in beaker, 6300ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid It removes.1.2 washings, centrifugation, drying:1.2.1 5000ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 30 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 45 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 70 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 1505g, Loss on drying 1.0%.2. degradation2.1 weighingBy above-mentioned 1500g heparin-benzyl rope ammonium salt in 30L glass reaction kettle, the methylene chloride of 7500ml is added thereto.2.2 dissolutions: leading to hot water for its interlayer, is heated to 33~36 DEG C and stirs complete to dissolving.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 1020ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 35 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 9000ml10% (W/V) sodium acetate first is added thereto Alcoholic solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 15000ml methanol solution, dry bemiparin crude product about 400g.3. purification3.1 will be above-mentioned dry that 400g bemiparin crude product pours into tank, and the purified water of 4000ml is added into reactor tank, Stirring is complete to dissolving.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.24ml hydrogen peroxide is added, and at 30 DEG C to be stirred to react 7 small When, through 0.22 μm of micro porous filtration.3.3 8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 20000ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 4000ml purified water is added, and stirring adjusts PH6.5, through 0.22 μm of micropore mistake to dissolving completely Filter, sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 350g.Three, the primary quality measure statistics of gained bemiparin
NEW DRUG APPROVALS
one time
$10.00
PATENT
WO-2021152192
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=9D96E01E1CE8B8107A83A95B4B344DD3.wapp2nC?docId=WO2021152192&tab=PCTDESCRIPTION
Use of a composition comprising low or very low molecular weight heparins (eg bemiparin sodium) with a high anti-factor Xa activity for the treatment of deep vein thrombosis.
Heparin belongs to the glycosaminoglycan family and is a polysaccharide of animal origin, which is extracted from the intestine or lungs of mammals (cow, lamb, pig) and is used in human therapies for the prevention and treatment of thromboembolic diseases . It is well known that the use of heparin is accompanied by very annoying bleeding effects and its daily administration, three subcutaneous or intravenous injections, constitutes a very considerable inconvenience.
During the course of the last few years, different chemical methods have been used to depolymerize heparin, such as:
– treatment with sodium nitrite in an acid medium,
– alkaline treatment of asters,
– use of free radicals generated in the presence of hydrogen peroxide,
– treatment of a quaternary ammonium salt of heparin in a non-aqueous medium with a strong base according to a beta elimination mechanism.
These methods make it possible to obtain, with variable yields, mixtures of heparin fragments in which the average molecular weight and anticoagulant activity vary according to the procedure and operating conditions. Low molecular weight heparins (LMWH) described in the state of the art or commercialized are obtained according to different depolymerization procedures. Their average molecular weights (Mw) are in the range of 3,600 and 7,500 Daltons.
It is now recognized that the antithrombotic activity of LMWH is mainly due to its ability to activate antithrombin III, a plasma protein and potent inhibitor of activated factor X and thrombin. In this way, it is possible to measure the antithrombotic activity of heparin by means of specific tests to determine the inhibition of these factors.
Research carried out by different authors shows that heparin fragments or oligosaccharides, with short chains of average molecular weight <4,800 Daltons, have a selective action on activated factor X and not on thrombin, in determinations using methods of the Pharmacopoeia. .
It has been found that if very low molecular weight fragments are required that have strong anti-factor Xa activity, it is preferable to use a selective depolymerization technique in non-aqueous medium, as described in US patent 9,981,955, which respects the antithrombin III binding site.
The document EP 1070503 A1 describes the controlled depolymerization of heparin using a process in a non-aqueous medium that makes it possible to obtain a family of LMWH that are obtained enriched in low molecular weight oligosaccharides that have a high anti-factor Xa activity and a low anti-factor lia activity, and which can be represented by the general formula:
in which:
n can vary between 1 and 12,
Ri = H or S0 3 Na,
R 2 = SOsNao COCH 3 ,
Said very low molecular weight heparin is obtained by selective depolymerization of heparin in a non-aqueous medium according to a beta elimination procedure.
Medical uses
Bemiparin is used for the prevention of thromboembolism after surgery, and to prevent blood clotting in the extracorporeal circuit in haemodialysis.[2]
Contraindications
The medication is contraindicated in patients with a history of heparin-induced thrombocytopenia with or without disseminated intravascular coagulation; acute bleeding or risk of bleeding; injury or surgery of the central nervous system, eyes or ears; severe liver or pancreas impairment; and acute or subacute bacterial endocarditis.[2]
Interactions
No interaction studies have been conducted. Drugs that are expected to increase the risk of bleeding in combination with bemiparin include other anticoagulants, aspirin and other NSAIDs, antiplatelet drugs, and corticosteroids.[2]
Chemistry
Like semuloparin, bemiparin is classified as an ultra-LMWH because of its low molecular mass of 3600 g/mol on average.[3] (Enoxaparin has 4500 g/mol.) These heparins have lower anti-thrombin activity than classical LMWHs and act mainly on factor Xa, reducing the risk of bleeding.[4]
References
- ^ Chapman TM, Goa KL (2003). “Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis”. Drugs. 63 (21): 2357–77. doi:10.2165/00003495-200363210-00009. PMID 14524738.
- ^ Jump up to:a b c Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. 2018. Ivor 2500 IE Anti-Xa/0,2 ml Injektionslösung in Fertigspritzen.
- ^ Planès A (September 2003). “Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism”. Expert Opinion on Pharmacotherapy. 4 (9): 1551–61. doi:10.1517/14656566.4.9.1551. PMID 12943485. S2CID 13566575.
- ^ Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C (October 2011). “A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin”. Thrombosis Research. 128 (4): 361–7. doi:10.1016/j.thromres.2011.03.001. PMID 21458847.
External links
- bemiparin at the US National Library of Medicine Medical Subject Headings (MeSH)
| Clinical data | |
|---|---|
| Trade names | Badyket, Ivor, Hibor, Zibor, others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Subcutaneous injection (except for haemodialysis) |
| ATC code | B01AB12 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (estimated) |
| Elimination half-life | 5–6 hours |
| Identifiers | |
| CAS Number | 91449-79-5 |
| DrugBank | DB09258 |
| ChemSpider | none |
| Chemical and physical data | |
| Molar mass | 3600 g/mol (average) |
| (what is this?) (verify) |
- Chapman TM, Goa KL: Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis. Drugs. 2003;63(21):2357-77. [Article]
- Planes A: Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism. Expert Opin Pharmacother. 2003 Sep;4(9):1551-61. [Article]
- Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C: A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin. Thromb Res. 2011 Oct;128(4):361-7. doi: 10.1016/j.thromres.2011.03.001. Epub 2011 Apr 2. [Article]
- Sanchez-Ferrer CF: Bemiparin: pharmacological profile. Drugs. 2010 Dec 14;70 Suppl 2:19-23. doi: 10.2165/1158581-S0-000000000-00000. [Article]
- Hoffman M, Monroe DM: Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007 Feb;21(1):1-11. doi: 10.1016/j.hoc.2006.11.004. [Article]
- Antonijoan RM, Rico S, Martinez-Gonzalez J, Borrell M, Valcarcel D, Fontcuberta J, Barbanoj MJ: Comparative pharmacodynamic time-course of bemiparin and enoxaparin in healthy volunteers. Int J Clin Pharmacol Ther. 2009 Dec;47(12):726-32. [Article]
- Irish Medicines Board: Bemiparin [Link]
- Hibor-Bemiparin Sodium [Link]
- Zibor 2,500 IU Solution for Injection [Link]
- Injectable drugs guide [Link]
- Thrombosis Advisors- Factor Xa inhibitor [Link]
- Anti-tumor effects of bemiparin in HepG2 and MIA PaCa-2 cells [Link]
- Bemiparin, an effective and safe low molecular weight heparin: a review [Link]
- Bemiparin sodium [Link]
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4981955A *1988-06-281991-01-01Lopez Lorenzo LDepolymerization method of heparinEP0293539B1 *1987-01-051994-06-08Laboratorios Farmaceuticos Rovi, S.A.Process for the depolymerization of heparin for obtaining heparin with a low molecular weight and having an antithrombotic activityCN1379781A *1999-10-222002-11-13阿文蒂斯药物股份有限公司Novel oligosaccharides, preparation method and pharmaceutical composition containing sameCN102399306A *2010-09-092012-04-04上海喜恩医药科技发展有限公司Preparation method of heparin-derived polysaccharide mixtureCN105693886A *2016-04-192016-06-22常州市蓝勖化工有限公司Preparation method of heparin sodiumCN106467577A *2015-08-212017-03-01苏州融析生物科技有限公司A kind of pulmonis Bovis seu Bubali Enoxaparin Sodium and preparation method and applicationCN106977627A *2017-05-162017-07-25苏州二叶制药有限公司A kind of Enoxaparin production method of sodiumCN109575156A *2018-11-052019-04-05上海宝维医药技术有限公司A kind of purification process of low molecular weight heparinFamily To Family Citations
////////////Bemiparin sodium, Bemiparin
TRK 700
![1-[4-(Dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=71738795&t=l)
TRK-700
CAS 1463432-16-7C14 H24 N4 O264.371-Propanone, 1-[4-(dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-
1-[4-(dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one
- 1-[4-(Dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-1-propanone
- OriginatorToray Industries
- ClassAnalgesics
- Mechanism of ActionUndefined mechanism
- Phase IIPostherpetic neuralgia
- PreclinicalPeripheral nervous system diseases
- 12 Sep 2018Pharmacodynamics data from a preclinical trial in Peripheral neuropathy presented at the 17th World Congress on Pain (WCP-2018)
- 01 Jul 2017Toray Industries completes a phase II trial for Postherpetic neuralgia (In adults, In the elderly) in Japan (PO) (NCT02701374)
- 21 May 2017Toray Industries completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (PO) (NCT03043248)
developed by Toray for treating neuropathic pain and investigating for fibromyalgia. In August 2021, this drug was reported to be in phase 1 clinical development.
PATENT
WO 2016136944
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016136944

(Reference Example 22) Synthesis of (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate:
[Chemical 56]
1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, Methyl (triphenylphosphoranylidene) acetate (33.4 g, 99.9 mmol) was added to a solution of 90.8 mmol) in dichloromethane (240 mL) at room temperature, and the mixture was stirred for 16 hours and then concentrated under reduced pressure. The residue was washed with a mixed solvent of hexane / dichloromethane = 19/1, and the washing liquid was concentrated. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate as a white solid (11.9 g, 71. 6 mmol, 79%).
1 H-NMR (400 MHz, CDCl 3 ) δ: 3.76 (3H, s), 3.81 (3H, s), 6.82 (1H, d, J = 15.6 Hz), 6.98 (1H, brs), 7.16 (1H, brs), 7.53 (1H, d, J = 15.6Hz).
ESI-MS: m / z = 167 (M + H) + .
(Reference Example 27) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 61]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propane. -1-one (0.179 g, 0.68 mmol, 63%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Comparative Example 1) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one hydrochloride:
[Chemical 66]
1- (4- (Dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (1.50 g, 5.67 mmol) diethyl ether (60) A dioxane solution of hydrogen chloride (4.0 M, 3.69 mL, 14.8 mmol) was added to the (0.0 mL) solution at 0 ° C. The reaction mixture was stirred at the same temperature for 1 hour and then at room temperature for 30 minutes. The precipitated white solid was collected by filtration, washed with diethyl ether (100 mL), dried at room temperature for 36 hours, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-). Imidazole-2-yl) propan-1-one hydrochloride (1.41 g, 4.18 mmol, 74%) (hereinafter, the compound of Comparative Example 1) was obtained as a white solid.
1 1 H-NMR (400 MHz, D 2 O) δ: 1.53-1.80 (2H, m), 2.12-2.23 (2H, m), 2.68-2.80 (1H, m), 2.88 (6H, s), 3.01- 3.08 (2H, m), 3.15-3.26 (3H, m), 3.47-3.58 (1H, m), 3.84 (3H, s), 4.08-4.16 (1H, m), 4.50-4.59 (1H, m), 7.29-7.33 (2H, m).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) as propan-1-one : m / z = 265 (M + H) + .
(Comparative Example 2) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one sulfate monohydrate:
[Chemical 67]
1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (6.72 g, 25.4 mmol) Concentrated sulfuric acid (2.49 g, 25.4 mmol), water (1.83 g, 102 mmol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl) in a DMSO (100 mL) solution. Seed crystals (50 mg, 0.13 mmol) of -1H-imidazol-2-yl) propan-1-one sulfate monohydrate were added at 80 ° C. The reaction was stirred at the same temperature for 2.5 hours, at 50 ° C. for 2.5 hours and at room temperature for 15 hours. The precipitated white solid was collected by filtration, washed successively with DMSO (20 mL) and methyl ethyl ketone (40 mL), dried at room temperature, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl). -1H-imidazol-2-yl) propan-1-one sulfate monohydrate (8.42 g, 22.1 mmol, 87%) (hereinafter, the compound of Comparative Example 2) was obtained as white crystals.
1 1 H-NMR (400 MHz, DMSO-d 6)) δ: 1.36 (1H, m), 1.58 (1H, m), 1.95 (2H, br), 2.44-2.57 (1H, m), 2.65 (6H, s), 2.74-2.88 (4H, m), 3.00 (1H, t, J = 12.0 Hz), 3.22 (1H, m), 3.61 (3H, s), 4.02 (1H, d, J = 14.0 Hz), 4.47 (1H, d, J = 12.8 Hz), 6.87 (1H, d, J = 1.2 Hz), 7.11 (1H, d, J = 1.2 Hz).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-) As 1H-imidazol-2-yl) propan-1-one: m / z = 265 (M + H) + .
NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153744
PATENT
WO-2021153743
Novel crystalline polymorphic form of 1-(4-(dimethylamino) piperidin-1-yl)-3-(1-methyl-1H-imidazol-2-yl)propan-1-one, useful as an analgesic in treating neuropathic pain and/or fibromyalgia.Pain is an experience with unpleasant sensations and emotions that occurs when or may cause tissue damage. Pain is mainly classified into nociceptive pain, neuropathic pain or psychogenic pain according to its cause. In addition, fibromyalgia is known as pain of unknown cause.
Neuropathic pain is pathological pain caused by dysfunction of the peripheral or central nervous system itself, and is caused by direct damage or compression of nervous tissue even though nociceptors are not stimulated. It refers to the pain that occurs. As a therapeutic agent for neuropathic pain, an anticonvulsant, an antidepressant, anxiolytic, or an antiepileptic drug such as gabapentin or pregabalin is used.
Fibromyalgia is a disease in which systemic pain is the main symptom and neuropsychiatric symptoms and autonomic nervous system symptoms are secondary symptoms. Pregabalin approved in the United States and Japan, duloxetine and milnacipran approved in the United States are mainly used as therapeutic agents for fibromyalgia, and non-approved agents for fibromyalgia are not approved. It has also been used for steroidal anti-inflammatory agents, opioid compounds, antidepressants, anticonvulsants and antiepileptic drugs. However, the therapeutic effects of non-steroidal anti-inflammatory drugs and opioid compounds are generally considered to be low (Non-Patent Document 1).
On the other hand, Patent Document 1 discloses that certain substituted piperidins have cardiotonic activity, and Patent Document 2 discloses that an imidazole derivative exhibits an FXa inhibitory effect. Patent Document 3 suggests that the substituted piperidins may have a medicinal effect on overweight or obesity, and Patent Documents 4 to 6 and Non-Patent Document 2 indicate that the imidazole derivative has an analgesic effect. It is disclosed.
In addition, the quality of pharmaceutical products needs to be maintained over a long period of time such as distribution and storage, and the compound as an active ingredient is required to have high chemical and physical stability. Therefore, as the active ingredient of a pharmaceutical product, a crystal that can be expected to have higher stability than an amorphous substance is generally adopted. Further, if crystals are obtained, a purification effect due to recrystallization during production can be expected. Further, it is preferable to have low hygroscopicity from the viewpoint of maintaining stability and handling during manufacturing, storage, formulation and analysis of the drug substance. In addition, since a drug needs to be dissolved in the digestive tract in order to exhibit its medicinal effect, it is preferable that the drug has excellent solubility, which is a physical property contrary to stability.
In order to obtain crystals of a compound that is an active ingredient of a pharmaceutical product, it is necessary to study various conditions for precipitating crystals from the solution. It is common to carry out crystallization under the condition of being dissolved in.
Patent documents
Patent Document 1: French Patent Invention No. 2567885
Patent Document 2: Japanese Patent Application Laid-Open No. 2006-0083664
Patent Document 3: International Publication No. 2003/031432
Patent Document 4: International Publication No. 2013/147160
Patent Document 5: International Publication No. 2015/046403
Patent Document 6: International Publication No. 2016/136944
Non-patent literature
Non-Patent Document 1: Okifuji et al., Pain and Therapy, 2013, Volume 2, p. 87-104
Non-Patent Document 2: Takahashi et al., Toxicological Pathology, 2019, Vol. 47. p. 494-503
Compound (I) was synthesized by the method described in the following reference example. For the compounds used in the synthesis of the reference example compounds for which the synthesis method is not described, commercially available compounds were used.
(Reference Example 4) Synthesis of amorphous compound (I):
[Chemical formula 2] 2 of
crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanol (5.00 g, 27.4 mmol) Aqueous sodium hydroxide solution (1.0N, 30.2 mL, 30.2 mmol) was added to a solution of -propanol (55 mL) at 0 ° C., and the mixture was stirred at room temperature for 12 hours. 2-Propanol (220 mL) was added to the reaction solution at room temperature, and crude 4- (dimethylamino) piperidine (3.17 g, 24.7 mmol) and DMT-MM (8.35 g, 30.2 mmol) were added at room temperature to react. The liquid was stirred at the same temperature for 3 hours. A 10% aqueous sodium chloride solution and a 1.0N aqueous sodium hydroxide solution were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give compound (I) (6.98 g) as an amorphous substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 (5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz) ), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Reference Example 5) Synthesis of crude 4- (dimethylamino) piperidine:
[Chemical
formula 3] 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (20.1 g, 77.0 mmol) in methanol (154.0 mL) Palladium-carbon (10% wet, 2.01 g) was added thereto, and the mixture was stirred at room temperature for 19 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 4- (dimethylamino) piperidine (9.86 g).
(Reference Example 6) Synthesis of crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate:
[Chemical
formula 4] Sodium hydride (55%, 4.36 g, 100 mmol) aqueous solution and tetrahydrofuran (150 mL) To the mixture was added triethylphosphonoacetate (19.1 mL, 95.0 mmol) at 0 ° C. After stirring the reaction solution for 20 minutes, a solution of 1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, 91.0 mmol) in tetrahydrofuran (150 mL) was added at 0 ° C., and then ethanol (30 mL) was added in the same manner. The mixture was added at temperature and stirred at room temperature for 2 hours. A 10% aqueous sodium chloride solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, chloroform / methanol). After adding methanol (310 mL) to the residue, palladium-carbon (10% wet, 1.40 g) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (14.2 g) of ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate.
(Reference Example 7) Synthesis of 1-benzyloxycarbonyl-4- (dimethylamino) piperidine:
[Chemical
formula 5] dichloromethane (55.7 mL) of 1-benzyloxycarbonyl-4-oxopiperidine (13.0 g, 55.7 mmol) ) Solution of dimethylamine in tetrahydrofuran (2.0 M, 34.8 mL, 69.7 mmol), acetic acid (0.32 mL, 5.6 mmol) and sodium triacetoxyborohydride (4.8 g, 22.6 mmol). Added at ° C. After stirring the reaction solution at the same temperature for 30 minutes, sodium triacetoxyborohydride (4.8 g, 22.6 mmol) was added at 0 ° C. The reaction mixture was stirred at the same temperature for 30 minutes, sodium triacetoxyborohydride (8.1 g, 38.2 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 12 hours. The reaction solution was cooled to 0 ° C. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) and then again by flash chromatography (silica gel, chloroform / methanol) to obtain 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (dimethylamino) piperidine. 13.6 g, 51.8 mmol, 93%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.34-1.46 (2H, m), 1.78-1.86 (2H, m), 2.28 (6H, s), 2.29-2.34 (1H, m), 2.75-2.85 (2H, m), 4.14-4.28 ( 2H, m), 5.12 (2H, s), 7.29-7.36 (5H, m).
ESI-MS: m / z = 263 (M + H) + .
(Reference Example 8) Synthesis of 1-benzyloxycarbonyl-4-oxopiperidine:
[Chemical
formula 6] Hydrochloride (130 mL) and water (130 mL) of 4-piperidinone hydrochloride monohydrate (10.0 g, 65.1 mmol) Sodium carbonate (13.8 g, 130.2 mmol) and benzyl chloroformate (8.79 mL, 61.8 mmol) were added to the mixed solution with and at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) to give 1-benzyloxycarbonyl-4-oxopiperidine (13.1 g, 56.2 mmol, 86%) as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 2.42-2.50 (4H, m), 3.78-3.82 (4H, m), 5.18 (2H, s), 7.32-7.38 (5H, m).
(Example 1) Production of A-type crystal of
compound (I): Amorphous compound (6.98 g) of compound (I) prepared in Reference Example 4 is purified and concentrated with chloroform / methanol by silica gel column chromatography. After that, the wall surface of the flask was rubbed with a spartel and mechanical stimulation was applied to obtain A-type crystals of compound (I) as a powder. For the obtained crystals, measurement of powder X-ray diffraction using a powder X-ray diffractometer (Rigaku Co., Ltd .; 2200 / RINT ultima + PC) and TG-DTA using a TG-DTA device (Rigaku Co., Ltd .; TG8120) Was done. The results of these measurements are shown in FIGS. 1 and 2.
Diffraction angle 2θ: 5.9, 16.5, 17.7, 20.8, 26.7 °
Endothermic peak: 55 ° C
PATENT
WO2013147160
Example 1 Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 27]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan- 1-one (0.179 g, 0.68 mmol, 63%) (hereinafter, the compound of Example 1) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016136944-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| JP-WO2013147160-A1 | Cyclic amine derivatives and their pharmaceutical use | 2012-03-29 | |
| TW-201350119-A | Cyclic amine derivatives and their medical uses | 2012-03-29 | |
| WO-2013147160-A1 | Cyclic amine derivative and use thereof for medical purposes | 2012-03-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| RU-2667062-C1 | Dynamic cyclic amine and pharmaceutical application thereof | 2015-02-27 | 2018-09-14 |
| TW-201639826-A | Cyclic amine derivatives and their medical uses | 2015-02-27 | |
| TW-I682927-B | Cyclic amine derivatives and their medical uses | 2015-02-27 | 2020-01-21 |
| US-10173999-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-01-08 |
| US-2018065950-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3263565-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| EP-3263565-B1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-06-26 |
| ES-2744785-T3 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2020-02-26 |
| JP-6569671-B2 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 | 2019-09-04 |
| JP-WO2016136944-A1 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019189781-A1 | Agent for inhibiting rise in intraneuronal calcium concentration | 2018-03-30 | |
| AU-2016224420-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| AU-2016224420-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-08-22 |
| CA-2977614-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| CN-107250128-B | Cyclic amine derivatives and its medical usage | 2015-02-27 | 2019-07-26 |
//////////TRK-700, phase 1, neuropathic pain, fibromyalgia, toray
O=C(CCc1nccn1C)N1CCC(CC1)N(C)C
Rutoside, Rutin


Rutoside
RUTIN
- Molecular FormulaC27H30O16
- Average mass610.518
- рутозид [Russian] [INN]ルチン [Japanese]روتوسيد [Arabic] [INN]芦丁 [Chinese] [INN]
CAS 153-18-4
- C.I. 75730
- NSC-9220
2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one


Rutin trihydrate | CAS 250249-75-3 RutinCAS Registry Number: 153-18-4
CAS Name: 3-[[6-O-(6-Deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-1-benzopyran-4-one
Additional Names: rutoside; quercetin-3-rutinoside; 3,3¢,4¢,5,7-pentahydroxyflavone-3-rutinoside; melin; phytomelin; eldrin; ilixathin; sophorin; globularicitrin; paliuroside; osyritrin; osyritin; myrticolorin; violaquercitrin
Trademarks: Birutan (Merck KGaA)
Molecular Formula: C27H30O16Molecular Weight: 610.52Percent Composition: C 53.12%, H 4.95%, O 41.93%
Literature References: Identity with ilixanthin: Schindler, Herb, Arch. Pharm.288, 372 (1955). Found in many plants, especially the buckwheat plant (Fagopyrum esculentum Moench., Polygonaceae) which contains about 3% (dry basis): Couch et al.,Science103, 197 (1946). From tobacco (Nicotiana tabacum L., Solanaceae) Couch, Krewson, U.S. Dept. Agr., Eastern Regional Res. Lab.AIC-52 (1944).
In forsythia [Forsythia suspensa (Thunb.) Vahl. var. fortunei (Lindl.) Rehd., Oleaceae], in hydrangea (Hydrangea paniculata Sieb., Saxifragaceae), in pansies (Viola sp., Violaceae).
General extraction procedure: BeilsteinXXXI, 376. From leaves of Eucalyptus macroryncha F. v. Muell., Myrtaceae: Attree, Perkin, J. Chem. Soc.1927, 234.
Industrial production from Eucalyptus spp.: Humphreys, Econ. Bot.18, 195 (1964).
Structure: Zemplén, Gerecs, Ber.68B, 1318 (1935).
Synthesis: Shakhova et al.,Zh. Obshch. Khim.32, 390 (1962), C.A.58, 1426e (1963). Rutin is hydrolyzed by rhamnodiastase from the seed of Rhamnus utilis Decne, Rhamnaceae (Chinese buckthorn); emulsin is not effective: Bridel, Charaux, Compt. Rend.181, 925 (1925). Toxicity data: Harrison et al.,J. Am. Pharm. Assoc.39, 557 (1950). Book: J. Q. Griffith, Jr., Rutin and Related Flavonoids (Mack, Easton, Pa., 1955).
Comprehensive description: T. I. Khalifa et al.,Anal. Profiles Drug Subs.12, 623-681 (1983).UV






Properties: Pale yellow needles from water, gradual darkening on exposure to light. The crystals contain 3 H2O and become anhydr after 12 hrs at 110° and 10 mm Hg. Anhydr rutin browns at 125°, becomes plastic at 195-197°, and dec 214-215° (with effervescence). [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine). Anhydr rutin is hygroscopic. One gram dissolves in about 8 liters water, about 200 ml boiling water, 7 ml boiling methanol. Sol in pyridine, formamide and alkaline solns; slightly sol in alcohol, acetone, ethyl acetate. Practically insol in chloroform, carbon bisulfide, ether, benzene, petr solvents. Dil solns give green color with ferric chloride. Rutin is colored brown by tobacco enzyme under experimental conditions: Neuberg, Kobel, Naturwissenschaften23, 800 (1935). LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison).
Optical Rotation: [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine)
Toxicity data: LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison)Therap-Cat: Capillary protectant.Keywords: Vasoprotectant.
C13




MASS

Rutin, also called rutoside, quercetin-3-O-rutinoside and sophorin, is the glycoside combining the flavonol quercetin and the disaccharide rutinose (α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranose). It is a citrus flavonoid found in a wide variety of plants including citrus.
Rutin, also called rutoside, is the glycoside flavonoid found in a certain fruits and vegetables. Most rutine-rich foods are capers, olives, buckwheat (whole grain flour), asparagus, raspberry.In a clinical trial, rutin was found to aid control of intraocular pressure in patients with primary open angle glaucoma. As a component of dietary supplement Phlogenzym, rutin is used for treatment of osteoarthritis. Rutin is also used for treatment of post-surgical swelling of the arm after breast cancer surgery. Traditionally, rutin is used to prevent mucositis due to cancer treatment, to treat blood vessel disease such as varicose veins, bleeding, hemorrhoids.
Occurrences
Rutin is one of the phenolic compounds found in the invasive plant species Carpobrotus edulis and contributes to the antibacterial[3] properties of the plant.
Its name comes from the name of Ruta graveolens, a plant that also contains rutin.
Various citrus fruit peels contain 32 to 49 mg/g of flavonoids expressed as rutin equivalents.[4]
Citrus leaves contain rutin at concentrations of 11 and 7 g/kg in orange and lime trees respectively.[5]
Metabolism
The enzyme quercitrinase can be found in Aspergillus flavus.[6] It is an enzyme in the rutin catabolic pathway.[7]
In food
Rutin is a citrus flavonoid glycoside found in many plants including buckwheat,[8] the leaves and petioles of Rheum species, and asparagus. Tartary buckwheat seeds have been found to contain more rutin (about 0.8–1.7% dry weight) than common buckwheat seeds (0.01% dry weight).[8] Rutin is one of the primary flavonols found in ‘clingstone’ peaches.[9] It is also found in green tea infusions.[10]
Approximate rutin content per 100g of selected foods, in milligrams per 100 milliliters:[11]
| Numeric | Alphabetic |
|---|---|
| 332 | Capers, spice |
| 45 | Olive [Black], raw |
| 36 | Buckwheat, whole grain flour |
| 23 | Asparagus, raw |
| 19 | Black raspberry, raw |
| 11 | Red raspberry, raw |
| 9 | Buckwheat, groats, thermally treated |
| 6 | Buckwheat, refined flour |
| 6 | Greencurrant |
| 6 | Plum, fresh |
| 5 | Blackcurrant, raw |
| 4 | Blackberry, raw |
| 3 | Tomato (Cherry), whole, raw |
| 2 | Prune |
| 2 | Fenugreek |
| 2 | Marjoram, dried |
| 2 | Tea (Black), infusion |
| 1 | Grape, raisin |
| 1 | Zucchini, raw |
| 1 | Apricot, raw |
| 1 | Tea (Green), infusion |
| 0 | Apple |
| 0 | Redcurrant |
| 0 | Grape (green) |
| 0 | Tomato, whole, raw |
Research
Rutin (rutoside or rutinoside)[12] and other dietary flavonols are under preliminary clinical research for their potential biological effects, such as in reducing post-thrombotic syndrome, venous insufficiency, or endothelial dysfunction, but there was no high-quality evidence for their safe and effective uses as of 2018.[12][13][14][needs update] As a flavonol among similar flavonoids, rutin has low bioavailability due to poor absorption, high metabolism, and rapid excretion that collectively make its potential for use as a therapeutic agent limited.[12]
Biosynthesis
The biosynthesis pathway of rutin in mulberry (Morus alba L.) leaves begins with phenylalanine, which produces cinnamic acid under the action of phenylalanine ammonia lyase (PAL). Cinnamic acid is catalyzed by cinnamic acid-4-hydroxylase (C4H) and 4-coumarate-CoA ligase (4CL) to form p–coumaroyl-CoA. Subsequently, chalcone synthase (CHS) catalyzes the condensation of p-coumaroyl-CoA and three molecules of malonyl-CoA to produce naringenin chalcone, which is eventually converted into naringenin flavanone with the participation of chalcone isomerase (CHI). With the action of flavanone 3-hydroxylas (F3H), dihydrokaempferol (DHK) is generated. DHK can be further hydroxylated by flavonoid 3´-hydroxylase (F3’H) to produce dihydroquercetin (DHQ), which is then catalyzed by flavonol synthase (FLS) to form quercetin. After quercetin is catalyzed by UDP-glucose flavonoid 3-O-glucosyltransferase (UFGT) to form isoquercitrin, finally, the formation of rutin from isoquercitrin is catalyzed by flavonoid 3-O-glucoside L-rhamnosyltransferase.[15]

SYN
https://www.sciencedirect.com/science/article/abs/pii/S100184171300017X
The compound 2 was synthesized for the first time by highly selective esterification reaction and fully characterized. The by-products of the reaction were complex, which brought out many considerable difficulties in separation and purification of the target product. Our work was the first in using the improved pyrogallol autoxidation method to test the antioxidant activities of these two flavonoids compounds in vitro and discovered that the compound 2 was much more effective as a free radical scavenger than the compound 1.

SYN
Synthesis of Rutin
The synthesis of rutin can be achieved according to the following three schemes. These schemes differ in the synthesis of ouercetin (the aglycone moiety of rutin).
Scheme 1: Kostanecki — et al. 1904 (33 ). Based upon the Claisen reaction between 2-hydroxy4, 6-dimethoxyacetophenone [l] and 3, 4-dimethoxybenzaldehyde [2] to give the intermediate [3] which upon treatment with HC1, cyclization occurs to give 5, 7, 3 , which upon treatment with F2SO4 enolisation occurs to give 5, 7, 1’3 ,’4 -tetramethoxyflavonol [6]. tion with HI affords quercetin [7].
Scheme 2: Robinson et al. 1926 (34 ). , ‘4 -tetramethoxyflavonone [4]. Oximination affords [5] Demethyla- — Condensation ofw-methoxypholoroacetophenone [I] with veratric acid anhydride [2] in the presence of the potassium salt of veratric acid to give the diarylester [3]. On hydrolysis with alcoholic KOH affords 5, 7-dihydroxy-3, /3 , ‘4 -trimethoxyf lavone [ 41 , which on demethylation with HI gives quercetin [5].
Scheme 3: Shakhova et al. 1962 (35), complete synthesis of rutin. W-methoxyphloroacetophenone [2] was condensed with 0-benzylvanillinic acid, anhydride [ 13 in triethylamine to give 5 , 7-dihydroxy-4 -benzyloxy-3, /3 -dimethoxyf lavone [3]. On treatment with AcOH-HC1 mixture gave 5, 7, ‘4 -trihydroxy-3,’3 -dimethoxyflavone [4]. Demethylation of the latter with HI yielded (about 802) quercetin [5]. Ouercetin potassium salt [6] was produced upon treating [5] with AcOK in ethanol. Levoglucosan [7] was acetylated with Ac20 in the presence of AcONa to give 2, 3, 4-triacetyllevoglucosan [8] which with TIC14 gave 1-chloro-2, 3, 4-triacetyl Dglucose [9]. L-rhamnose tetraacetate [lo] treated with TiBr4 in CHC13 gave 1-bromo-2, 3, I-triacetyl-L-rhamnose [ll]. [lo] + [11] heated with Hg (OAC)~ in C6H6 gave (53x) CC – acetochloro-f3-l-L-rhamnosido-6-D-glucose [12]. [12] was treated with AgOAc and acetylated with Ac20 to prodilce (68.703 B-heptaacet yl-f3-1-L-rhamnos ido-6-D-glucose [13]. This with 33% HBr in AcOH gave (61%) d – acetobromo-~-l-L-rhamnosido-6-D-glucose [14]. [14] and quercetin potassium salt [6] were dissolved in NH40H which was evaporated and treated with methanol andpurified over a chromatographic column packed with polycaprolactum resin to give rutin [151.









References
- ^ Merck Index, 12th Edition, 8456
- ^ Krewson CF, Naghski J (Nov 1952). “Some physical properties of rutin”. Journal of the American Pharmaceutical Association. 41 (11): 582–7. doi:10.1002/jps.3030411106. PMID 12999623.
- ^ van der Watt E, Pretorius JC (2001). “Purification and identification of active antibacterial components in Carpobrotusedulis L.”. Journal of Ethnopharmacology. 76 (1): 87–91. doi:10.1016/S0378-8741(01)00197-0. PMID 11378287.
- ^ [1] p. 280 Table 1
- ^ [2] p.8 fig. 7
- ^ quercitrinase on www.brenda-enzymes.org
- ^ Tranchimand S, Brouant P, Iacazio G (Nov 2010). “The rutin catabolic pathway with special emphasis on quercetinase”. Biodegradation. 21 (6): 833–59. doi:10.1007/s10532-010-9359-7. PMID 20419500. S2CID 30101803.
- ^ Jump up to:a b Kreft S, Knapp M, Kreft I (Nov 1999). “Extraction of rutin from buckwheat (Fagopyrum esculentumMoench) seeds and determination by capillary electrophoresis”. Journal of Agricultural and Food Chemistry. 47 (11): 4649–52. doi:10.1021/jf990186p. PMID 10552865.
- ^ Chang S, Tan C, Frankel EN, Barrett DM (Feb 2000). “Low-density lipoprotein antioxidant activity of phenolic compounds and polyphenol oxidase activity in selected clingstone peach cultivars”. Journal of Agricultural and Food Chemistry. 48 (2): 147–51. doi:10.1021/jf9904564. PMID 10691607.
- ^ Malagutti AR, Zuin V, Cavalheiro ÉT, Henrique Mazo L (2006). “Determination of Rutin in Green Tea Infusions Using Square-Wave Voltammetry with a Rigid Carbon-Polyurethane Composite Electrode”. Electroanalysis. 18 (10): 1028–1034. doi:10.1002/elan.200603496.
- ^ “foods in which the polyphenol Quercetin 3-O-rutinoside is found”. Phenol-Explorer v 3.6. June 2015.
- ^ Jump up to:a b c “Flavonoids”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. November 2015. Retrieved 25 February 2018.
- ^ Morling, J. R; Yeoh, S. E; Kolbach, D. N (November 2018). “Rutosides for treatment of post-thrombotic syndrome”. Cochrane Database of Systematic Reviews. 11 (11): CD005625. doi:10.1002/14651858.CD005625.pub4. PMC 6517027. PMID 30406640.
- ^ Martinez-Zapata, M. J; Vernooij, R. W; Uriona Tuma, S. M; Stein, A. T; Moreno, R. M; Vargas, E; Capellà, D; Bonfill Cosp, X (2016). “Phlebotonics for venous insufficiency”. Cochrane Database of Systematic Reviews. 4: CD003229. doi:10.1002/14651858.CD003229.pub3. PMC 7173720. PMID 27048768.
- ^ Yu X, Liu J, Wan J, Zhao L, Liu Y, Wei Y, Ouyang Z. Cloning, prokaryotic expression, and enzyme activity of a UDP-glucose flavonoid 3-o-glycosyltransferase from mulberry (Morus alba L.) leaves. Phcog Mag 2020;16:441-7
| Names | |
|---|---|
| IUPAC name3′,4′,5,7-Tetrahydroxy-3-[α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranosyloxy]flavone | |
| Preferred IUPAC name(42S,43R,44S,45S,46R,72R,73R,74R,75R,76S)-13,14,25,27,43,44,45,73,74,75-Decahydroxy-76-methyl-24H-3,6-dioxa-2(2,3)-[1]benzopyrana-4(2,6),7(2)-bis(oxana)-1(1)-benzenaheptaphane-24-one | |
| Other namesRutoside (INN) Phytomelin Sophorin Birutan Eldrin Birutan Forte Rutin trihydrate Globularicitrin Violaquercitrin Quercetin rutinoside | |
| Identifiers | |
| CAS Number | 153-18-4 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 4444362 |
| DrugBank | DB01698 |
| ECHA InfoCard | 100.005.287 |
| KEGG | C05625 |
| PubChem CID | 5280805 |
| RTECS number | VM2975000 |
| UNII | 5G06TVY3R7 |
| CompTox Dashboard (EPA) | DTXSID3022326 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H30O16 |
| Molar mass | 610.521 g·mol−1 |
| Appearance | Solid |
| Melting point | 242 °C (468 °F; 515 K) |
| Solubility in water | 12.5 mg/100 mL[1] 13 mg/100mL[2] |
| Pharmacology | |
| ATC code | C05CA01 (WHO) |
| Hazards | |
| NFPA 704 (fire diamond) |  200 200 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////Rutoside, RUTIN, рутозид , ルチン , روتوسيد , 芦丁 , C.I. 75730, NSC 9220,
CC1C(C(C(C(O1)OCC2C(C(C(C(O2)OC3=C(OC4=CC(=CC(=C4C3=O)O)O)C5=CC(=C(C=C5)O)O)O)O)O)O)O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Anifrolumab
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYIFT NYWIAWVRQM PGKGLESMGI IYPGDSDIRY
SPSFQGQVTI SADKSITTAY LQWSSLKASD TAMYYCARHD IEGFDYWGRG TLVTVSSAST
KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF PEPVTVSWNS GALTSGVHTF PAVLQSSGLY
SLSSVVTVPS SSLGTQTYIC NVNHKPSNTK VDKRVEPKSC DKTHTCPPCP APEFEGGPSV
FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA SIEKTISKAK GQPREPQVYT LPPSREEMTK
NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG
NVFSCSVMHE ALHNHYTQKS LSLSPGK
(Lihgt chain)
EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSFFAWYQQK PGQAPRLLIY GASSRATGIP
DRLSGSGSGT DFTLTITRLE PEDFAVYYCQ QYDSSAITFG QGTRLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-96, H144-H200, H220-L215, H226-H’226, H229-H’229, H261-H321, H367-H425, H’22-H’96, H’144-H’200, H’220-L’215, H’261-H’321, H’367-H’425, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Anifrolumab
| アニフロルマブ (遺伝子組換え) |
FDA APPROVED 2021/7/30, Saphnelo
- MEDI 546
| Formula | C6444H9964N1712O2018S44 |
|---|---|
| Cas | 1326232-46-5 |
| Mol weight | 145117.1846 |
| Immunomodulator, Anti-IFN-type 1 receptor antibody | |
| Disease | Systemic lupus erythematosus |
|---|
Monoclonal antibody
Treatment of systemic lupus erythematosus (SLE)
- OriginatorMedarex
- DeveloperAstraZeneca; Medarex; MedImmune
- ClassAntirheumatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterferon alpha beta receptor antagonists
- RegisteredSystemic lupus erythematosus
- Phase IILupus nephritis
- DiscontinuedRheumatoid arthritis; Scleroderma
- 02 Jul 2021Phase-III clinical trials in Systemic lupus erythematosus in USA (SC) (NCT04877691)
- 25 Jun 2021AstraZeneca plans a phase III trial in Systemic lupus erythematosus (Adjunctive treatment) in the China, Hong Kong, South Korea, Philipines, Taiwan and Thailand (IV, Infusion), in July 2021 (NCT04931563)
- 02 Jun 2021Pharmacokinetic, efficacy and adverse events data from a phase II TULIP-LN1 trial in Lupus nephritis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
Anifrolumab, sold under the brand name Saphnelo, is a monoclonal antibody used for the treatment of systemic lupus erythematosus (SLE).[1][2] It binds to the type I interferon receptor, blocking the activity of type I interferons such as interferon-α and interferon-β.[medical citation needed]
Anifrolumab was approved for medical use in the United States in August 2021.[1][3][4][5]
Anifrolumab is a monoclonal antibody that inhibits type 1 interferon receptors, indicated in the treatment of moderate to severe systemic lupus erythematosus.
Anifrolumab, or MEDI-546, is a type 1 interferon receptor (IFNAR) inhibiting IgG1κ monoclonal antibody indicated in the treatment of adults with moderate to severe systemic lupus erythematosus.7,11 The standard therapy for systemic lupus erythematosus consists of antimalarials like hydroxychloroquine, glucocorticoids like dexamethasone, and disease modifying antirheumatic drugs like methotrexate.8,11
Three monoclonal antibodies (anifrolumab, rontalizumab, and sifalimumab) that target the type 1 interferon pathway entered clinical trials as potential treatments for systemic lupus erythematosus, but so far only anifrolumab has been approved.3
The design of early clinical trials of anti-interferon treatments such as anifrolumab, rontalizumab, and sifalimumab have come under criticism.3 The design of the clinical trials use different definitions of autoantibody positivity, making comparison between trials difficult; all trials involve large portions of patients also using corticosteroids, which may alter patient responses in the experimental and placebo groups; and patient populations were largely homogenous, which may have increased the odds of success of the trial.3
Anifrolumab has also been investigated for the treatment of Scleroderma.1
Anifrolumab was granted FDA approval on 30 July 2021.11
Adverse effects
The most common adverse effect was shingles, which occurred in 5% of patients in the low-dose group, to 10% in the high-dose group, and to 2% in the placebo group. Overall adverse effect rates were comparable in all groups.[6]
History
The drug was developed by MedImmune, a unit of AstraZeneca, which chose to move anifrolumab instead of sifalimumab into phase III trials for lupus in 2015.[7][8][9]
Clinical trial results
Anifrolumab failed to meet its endpoint of significant reduction in disease as assessed by the SLE Responder Index 4 instrument in the TULIP 1 phase III trial.[10] This multi-center, double-blind, placebo-controlled study followed adults with moderate to severe SLE over the course of one year. Preliminary results were announced on 31 August 2018.
Names
Anifrolumab is the international nonproprietary name (INN).[11]
References
- ^ Jump up to:a b chttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761123s000lbl.pdf
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Anifrolumab, American Medical Association.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761123Orig1s000ltr.pdf
- ^ https://www.astrazeneca.com/media-centre/press-releases/2021/saphnelo-approved-in-the-us-for-sle.html
- ^ “Saphnelo (anifrolumab) Approved in the US for Moderate to Severe Systemic Lupus Erythematosus” (Press release). AstraZeneca. 2 August 2021. Retrieved 2 August 2021 – via Business Wire.
- ^ Spreitzer H (29 August 2016). “Neue Wirkstoffe – Anifrolumab”. Österreichische Apothekerzeitung (in German) (18/2016).
- ^ “Press release: New Hope for Lupus Patients”. MedImmune. 11 August 2015. Archived from the original on 31 July 2017.
- ^ “Anifrolumab”. NHS Specialist Pharmacy Service. Retrieved 31 July 2017.
- ^ “Anifrolumab”. AdisInsight. Retrieved 31 July 2017.
- ^ “Update on TULIP 1 Phase III trial for anifrolumab in systemic lupus erythematosus”. http://www.astrazeneca.com. Retrieved 2019-02-05.
- ^ World Health Organization (2014). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 71”. WHO Drug Information. 28 (1). hdl:10665/331151.
Further reading
- Anderson E, Furie R (April 2020). “Anifrolumab in systemic lupus erythematosus: current knowledge and future considerations”. Immunotherapy. 12 (5): 275–86. doi:10.2217/imt-2020-0017. PMID 32237942.
External links
- “Anifrolumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01438489 for “A Study of the Efficacy and Safety of MEDI-546 in Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446912 for “Efficacy and Safety of Two Doses of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446899 for “Efficacy and Safety of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interferon α/β receptor |
| Clinical data | |
| Trade names | Saphnelo |
| Other names | MEDI-546, anifrolumab-fnia |
| License data | US DailyMed: Anifrolumab |
| Routes of administration | Intravenous |
| Drug class | type I interferon receptor antagonist (IFN) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1326232-46-5 |
| DrugBank | DB11976 |
| ChemSpider | none |
| UNII | 38RL9AE51Q |
| KEGG | D11082 |
| Chemical and physical data | |
| Formula | C6444H9964N1712O2018S44 |
| Molar mass | 145119.20 g·mol−1 |
- Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, Peng SL, Yao Y, Elgeioushi N, Chang L, Wang B, Yoo S: Dose-escalation of human anti-interferon-alpha receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther. 2014 Feb 24;16(1):R57. doi: 10.1186/ar4492. [Article]
- Peng L, Oganesyan V, Wu H, Dall’Acqua WF, Damschroder MM: Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs. 2015;7(2):428-39. doi: 10.1080/19420862.2015.1007810. [Article]
- Massarotti EM, Allore HG, Costenbader K: Editorial: Interferon-Targeted Therapy for Systemic Lupus Erythematosus: Are the Trials on Target? Arthritis Rheumatol. 2017 Feb;69(2):245-248. doi: 10.1002/art.39985. [Article]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S: Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017 Feb;69(2):376-386. doi: 10.1002/art.39962. [Article]
- Tummala R, Rouse T, Berglind A, Santiago L: Safety, tolerability and pharmacokinetics of subcutaneous and intravenous anifrolumab in healthy volunteers. Lupus Sci Med. 2018 Mar 23;5(1):e000252. doi: 10.1136/lupus-2017-000252. eCollection 2018. [Article]
- Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, de Los Reyes M, Schifferli K, Liang M, Sanjuan MA, Sims GP, Kolbeck R: Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018 Apr 5;5(1):e000261. doi: 10.1136/lupus-2018-000261. eCollection 2018. [Article]
- Bui A, Sanghavi D: Anifrolumab . [Article]
- Trindade VC, Carneiro-Sampaio M, Bonfa E, Silva CA: An Update on the Management of Childhood-Onset Systemic Lupus Erythematosus. Paediatr Drugs. 2021 Jul;23(4):331-347. doi: 10.1007/s40272-021-00457-z. Epub 2021 Jul 10. [Article]
- Ryman JT, Meibohm B: Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol. 2017 Sep;6(9):576-588. doi: 10.1002/psp4.12224. Epub 2017 Jul 29. [Article]
- Koh JWH, Ng CH, Tay SH: Biologics targeting type I interferons in SLE: A meta-analysis and systematic review of randomised controlled trials. Lupus. 2020 Dec;29(14):1845-1853. doi: 10.1177/0961203320959702. Epub 2020 Sep 22. [Article]
- FDA Approved Drug Products: Saphnelo (Anifrolumab-fnia) Intravenous Injection [Link]
 //////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
//////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
NEW DRUG APPROVALS
one time
$10.00
Ezutromid
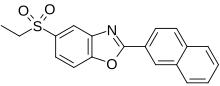
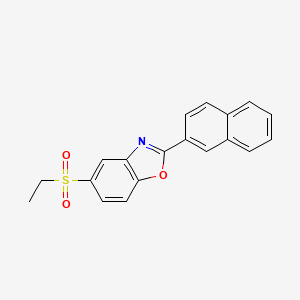
Ezutromid
945531-77-1
Chemical Formula: C19H15NO3S
Molecular Weight: 337.39
945531-77-1, SMT c1100, BMN-195, BMN 195, C 1100
5-(ethylsulfonyl)-2-(naphthalen-2-yl)benzo[d]oxazole
BMN-195; BMN 195; BMN195; SMTC-1100; SMTC1100; SMTC 1100; VOX-C1100; Ezutromid
Ezutromid, also known as BMN-195 and SMTC-1100, is a first orally bioavailable utrophin’s translation modulator. Duchenne muscular dystrophy (DMD) is a lethal, progressive muscle wasting disease caused by a loss of sarcolemmal bound dystrophin, which results in the death of the muscle fibers leading to the gradual depletion of skeletal muscle.
Ezutromid is an orally administered small molecule utrophin modulator currently involved in a Phase 2 clinical trial produced by Summit Therapeutics for the treatment of Duchenne muscular dystrophy (DMD).[1][2] DMD is a fatal x-linked recessive disease affecting approximately 1 in 5000 males and is a designated orphan disease by the FDA and European Medicines Agency.[3] Approximately 1/3 of the children obtain DMD as a result of spontaneous mutation in the dystrophin gene and have no family history of the disease.[3] Dystrophin is a vital component of mature muscle function, and therefore DMD patients have multifarious forms of defunct or deficient dystrophin proteins that all manifest symptomatically as muscle necrosis and eventually organ failure.[3][4] Ezutromid is theorized to maintain utrophin, a protein functionally and structurally similar to dystrophin that precedes and is replaced by dystrophin during development.[3][5] Utrophin and dystrophin are reciprocally expressed, and are found in different locations in a mature muscle cell.[4][6] However, in dystrophin-deficient patients, utrophin was found to be upregulated and is theorized to replace dystrophin in order to maintain muscle fibers.[7] Ezutromid is projected to have the potential to treat all patients suffering with DMD as it maintains the production of utrophin to counteract the lack of dystrophin to retard muscle degeneration.[7][8] Both the FDA and European Medicines Agency has given ezutromid an orphan drug designation.[5][9] The FDA Office of Orphan Products and Development offers an Orphan Drug Designation program (ODD) that allows drugs aimed to treat diseases that affect less than 200,000 people in the U.S. monetary incentives such as a period of market exclusivity, tax incentives, and expedited approval processes.[5][10]
The Phase 2 clinical trial was ended in 2018 and the medication discontinued after it failed to show any benefit in slowing the disease.[11]
Clinical trials
The first Phase 1b trial (NCT02056808) began on November 2013 and involved 12 patients aged 5–11 years old.[12] The patients were divided into three groups given escalating oral doses testing the safety and tolerability after each increase over the course of 10 days.[12]
Another completed Phase 1b trial (NCT02383511) began February 2015 and involved 12 patients aged 5–13 years old.[13] The goal was to determine the safety, tolerability, and pharmacokinetic parameters by measuring plasma concentration and major metabolite levels over 28 days for three sequence groups.[13] Each sequence involved placebo, 1250 mg, and 2500 mg BID (twice a day) doses given for one week each.[4][13]
A PhaseOut DMD, Phase 2, Proof of Concept (NCT02858362) clinical trial is underway that tests the clinical safety and efficacy of an oral suspension of ezutromid.[2] The 48-week open-label trial is enrolling 40 boys, ages 5–10, living in the U.K. or U.S.[2] MRI leg muscle change will be measured as well as ezutromid plasma concentration levels, with a secondary goal of obtaining quantifiable images of utrophin membrane stained biopsies at baseline and either 24 or 48 weeks.[2]
Commercial aspects
As of 2016, ataluren was the only approved drug in the EU to treat a specific subpopulation of patients with nmDMD, or DMD caused by a nonsense mutation.[14] However, nonsense mutations only account for approximately 15% of all patients with DMD.[15] Therefore, Summit Therapeutics projects to file for regulatory approval in the US and EU by 2019 and to reach market in 2020.[8] They expect to profit just over £24,046 in 2020 and £942,656 in 2025, which amounts to ~10% CGR for the first 7 years on the basis of treating all DMD patients in the US, EU, Iceland, Norway, Switzerland and Russia.[8]
Furthermore, Summit Therapeutics has entered an agreement with Sarepta Theraputics as of October 2016 regarding the commercialization of ezutromid.[16] The agreement consists of a collaboration between Sarepta and Summit to share the research and developing costs for the development of novel therapies to treat DMD patients.[16]
PAPER
https://onlinelibrary.wiley.com/doi/10.1002/anie.201906080
4-(ethylthio)Phenol S2: To a 250 mL round bottle, 4-mercaptophenol S1 (12.6 g, 100 mmol), K2CO3 (15.3 g, 110 mmol), acetone (100 mL) were added, then, iodoethane (15.6 g, 8.0 mL, 130 mmol) was added slowly at 0 oC. The system was stirred at room temperature overnight. After filtration, distillation of solvent, and flash chromatography, S2 (10.780 g) was obtained with 70% yield.
4-(ethylthio)-2-Nitrophenol S3: To a 250 mL round bottle, 4-(ethylthio)Phenol S2 (3.084 g, 20 mmol), 300-400 mesh silica gel (2 g), distilled water (2 g), and CH3CN (60 mL) was added. The system was then cooled by an ice water bath. Subsequently, citric acid (3.842 g, 20 mmol), NaNO2 (2.760 g, 40 mmol) were separately added slowly in portionwise. The system was reacted at room temperature overnight. After filtration and distillation of solvent, EA (50 mL) and water (50 mL) was added, after separation, the aqueous phase was extraction with EA (30 mL) twice. The combined organic phase was dried with MgSO4. Following by filtration and chromatography, S3 (3.590 g) was obtained with 90% yield.
4-(ethylthio)-2-Nitrophenol S4: To a 100 mL round bottle, S3 (2.46 g, 12.3 mmol), reductive iron powder (2.07 g, 36.9 mmol), and EtOH (50 mL) was added. Then, HCl (aq.) (0.15 M) (12 mL, 1.85 mmol) was added slowly. The system was refluxed overnight. After filtration, distillation of solvent, and flash chromatography, S4 (1.040 g) was obtained with 50% yield.
5-(ethylthio)-2-(naphthalen-2-yl)Benzo[d]oxazole S6 (Ezutromid-S): S4 (324 mg1.91 mmol), 2-naphthoyl chloride S5 (545.7 mg, 2.87 mmol), dry 1,4-dioxane (5 mL) was added into a sealing tube. Then, the system was vacuumed and filled with nitrogen for three times. Subsequently, the reaction was run at 160 oC for 10 hours. After distillation of solvent and flash chromatography, S6 (361.7 mg) was obtained with 62% yield. 1H NMR (500 MHz, Chloroform-d) δ 8.74 (s, 1H), 8.28 (dd, J = 8.5, 1.7 Hz, 1H), 7.96 (t, J = 7.5 Hz, 2H), 7.92 – 7.84 (m, 1H), 7.81 (d, J = 1.8 Hz, 1H), 7.57 (pd, J = 6.8, 3.4 Hz, 2H), 7.51 (d, J = 8.3 Hz, 1H), 7.39 (dd, J = 8.4, 1.8 Hz, 1H), 2.99 (q, J = 7.3 Hz, 2H), 1.33 (t, J = 7.3 Hz, 3H).13C NMR (126 MHz, Chloroform-d) δ 163.79, 149.70, 142.92, 134.76, 132.89, 132.38, 128.92, 128.78, 128.20, 127.87, 127.85, 126.91, 124.11, 123.84, 121.38, 110.72, 29.17, 14.40



Dibenzoate5-(ethylsulfone)-2-(naphthalen-2- yl)benzo[d]oxazole (Ezotrumid) 5a:
5- (ethylthio)-2-(naphthalen-2-yl)Benzo[d]oxazole (30.5 mg, 0.1 mmol), UO2(OAc)2 . 2H2O (0.8 mg, 0.002 mol), H2O (10 equiv., 36 μL), o-xylene (8.3 equiv., 0.2 mL), CH3CN (1 mL) were stirred under oxygen atmosphere (1 atm, balloon) at room temperature until the total consumption of sulfide and sulfoxide under the irradiation of three 2 w blue LEDs in a paralleled reactor. 5a (27.3 mg, 81%) was obtained through column chromatography (PE/EA = 20/1-5/1) as a white solid, Rf = 0.6 (PE/EA = 2/1);
1H NMR (500 MHz, Chloroform-d) δ 8.82 (s, 1H), 8.37 (s, 1H), 8.32 (d, J = 8.5 Hz, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.99 – 7.89 (m, 2H), 7.84 – 7.76 (m, 1H), 7.61 (t, J = 7.3 Hz, 2H), 3.28 – 3.08 (m, 2H), 1.32 (dt, J = 7.3, 3.6 Hz, 3H)..
13C NMR (126 MHz, Chloroform-d) δ 165.57, 153.87, 142.86, 135.26, 135.14, 132.86, 129.09, 128.97, 128.37, 127.99, 127.19, 125.35, 123.87, 123.34, 121.00, 111.36, 51.04, 7.62.
IR (KBr) 2933, 1507, 1498, 1258, 1064, 1046, 756, 474 cm-1 .
HRMS (ESI) Calcd for C19H16NO3S 338.0851 (M+H), Found 338.0865.
PATENT
WO 2007091106
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007091107
PATENT
WO 2009021749
WO 2009019504
WO 2013167737 A
CN 110437170
CN 110483345
CN 110563619
PATENT
WO 2009021748
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009021748
It has been discovered that the compound of formula I (5-(ethylsulfonyl)-2-(naphthalen-2-yl)benzo[d]oxazole) has excellent properties for the treatment of Duchenne muscular dystrophy (see, e.g., international patent application publication no. WO 2007/091106).
The compound of formula I (R = 5-ethylsulfonyl; R9 = 2-naphthalen-2-yl) may be synthesised according to the following procedure, as disclosed in WO 2007/091106 (page 51):
Experimental
S nthesis of 5- eth lsulfon -2- na hthalen-2- l‘benzo d oxazole
Procedure:
A vessel was equipped with a retreat blade stirrer and downward pumping turbine, a five necked flange lid, seal and clamp, stirrer gland and overhead stirrer, thermometer pocket, Dean- Stark trap, dropping funnel and condenser. The water to the condenser was then switched on.
The sodium hydroxide and 0.80 L of water were then mixed (whilst cooling in an ice bath until all the sodium hydroxide has dissolved – caution exothermic). The resulting solution was then transferred to a scrubber appropriately attached to the vessel.
The 2-amino-4-(ethylsulfonyl)phenol and 2.00 L of xylenes (mixed) were then transferred to the vessel, and the reagents and solvent were stirred at 100 rpm.
Then, the 2-naphtholyl chloride was dissolved in 2.00 L of xylenes (mixed) and transferred into the vessel. The stirring rate was increased to 150 rpm.
The temperature of the solution was gradually increased to 100°C over a period of not less than 30 mins, and then maintained at that level for 10 mins. (Caution: HCl gas is evolved during this process through the gas scrubber). The stirrer speed was then increased to 315 rpm and the temperature gradually increased over a period of 30 minutes until reflux (155°C) at which level it was maintained for 90 mins. (Caution: HCl gas is evolved during this process through the gas scrubber).
The methanesulfonic acid was then added drop-wise over a period of 30 mins and relux was maintained until no further water was being collected in the Dean-Stark apparatus (approx 15 mins).
The heat was then removed and the pipe adapter from the Dean- Stark apparatus disconnected. The resulting solution was allowed to cool to 900C, and then filtered using Whatman 1 filter paper.
The resulting solution was then left at ambient temperature for 18h, after which time the product crystallised, and the product was separated by filtration using Whatman 1 filter paper. The product was then washed with Ix 1.0 L of tert-butyl methyl ether (TBME)
The product was then dried in a vacuum oven at 65°C at a pressure of 1 Ombar until constant weight was achieved (less than 0.5 g difference between consecutive measurements of mass which must be at least 1 h apart).
The product was obtained as a sandy-beige powder in a yield of 80%.
Characterisation:
5-(EthylsuIf onyl)-2-(naphthalen-2-yl)benzo [d] oxazole
LCMS RT= 6.94min, MH+ 338.1;
1H NMR (DMSO): 8.90 (IH, br), 8.34 (IH, d, J 1.4 Hz), 8.30 (IH, dd, J 8.6 1.7 Hz), 8.24-8.05 (4H, m), 7.99 (IH, dd, J 8.5 1.8 Hz), 7.73-7.64 (2H, m), 3.41 (2H, q, J 7.3 Hz), 1.15 (3H, t, J7.3 Hz);
MP = 160-1610C.
Synthesis of polymorphic forms
1. Procedure
100 mg of the compound of formula I was dissolved in the minimum amount of good solvent and then the anti-solvent was added to induce crystallisation. The supernatant liquor was then removed, and the resulting solid was dried under vacuum for 12 his.
PAPER
Journal of medicinal chemistry (2011), 54(9), 3241-50
https://pubs.acs.org/doi/10.1021/jm200135z
Abstract

A series of novel 2-arylbenzoxazoles that upregulate the production of utrophin in murine H2K cells, as assessed using a luciferase reporter linked assay, have been identified. This compound class appears to hold considerable promise as a potential treatment for Duchenne muscular dystrophy. Following the delineation of structure–activity relationships in the series, a number of potent upregulators were identified, and preliminary ADME evaluation is described. These studies have resulted in the identification of 1, a compound that has been progressed to clinical trials.
PAPER
Angewandte Chemie, International Edition (2019), 58(38), 13499-13506
Angewandte Chemie, International Edition (2020), 59(3), 1346-1353.
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b01547
Journal of medicinal chemistry (2020), 63(5), 2547-2556.
Abstract

5-(Ethylsulfonyl)-2-(naphthalen-2-yl)benzo[d]oxazole (ezutromid, 1) is a first-in-class utrophin modulator that has been evaluated in a phase 2 clinical study for the treatment of Duchenne muscular dystrophy (DMD). Ezutromid was found to undergo hepatic oxidation of its 2-naphthyl substituent to produce two regioisomeric 1,2-dihydronaphthalene-1,2-diols, DHD1 and DHD3, as the major metabolites after oral administration in humans and rodents. In many patients, plasma levels of the DHD metabolites were found to exceed those of ezutromid. Herein, we describe the structural elucidation of the main metabolites of ezutromid, the regio- and relative stereochemical assignments of DHD1 and DHD3, their de novo chemical synthesis, and their production in systems in vitro. We further elucidate the likely metabolic pathway and CYP isoforms responsible for DHD1 and DHD3 production and characterize their physicochemical, ADME, and pharmacological properties and their preliminary toxicological profiles.
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S004040201931227X
Abstract
Following on from ezutromid, the first-in-class benzoxazole utrophin modulator that progressed to Phase 2 clinical trials for the treatment of Duchenne muscular dystrophy, a new chemotype was designed to optimise its physicochemical and ADME profile. Herein we report the synthesis of SMT022357, a second generation utrophin modulator preclinical candidate, and an asymmetric synthesis of its constituent enantiomers. The pharmacological properties of both enantiomers were evaluated in vitro and in vivo. No significant difference in the activity or efficacy was observed between the two enantiomers; activity was found to be comparable to the racemic mixture.
Graphical abstract


References
- ^ “About Summit Therapeutics – Summit”. Summit. Retrieved 2016-11-14.
- ^ Jump up to:a b c d Clinical trial number NCT02858362 for “PoC Study to Assess Activity and Safety of SMT C1100 (Ezutromid) in Boys With DMD” at ClinicalTrials.gov
- ^ Jump up to:a b c d “Duchenne Muscular Dystrophy – Summit”. Summit. Archived from the original on 2016-11-15. Retrieved 2016-11-14.
- ^ Jump up to:a b c Ricotti V, Spinty S, Roper H, Hughes I, Tejura B, Robinson N, et al. (2016-01-01). “Safety, Tolerability, and Pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole Utrophin Modulator, following Single- and Multiple-Dose Administration to Pediatric Patients with Duchenne Muscular Dystrophy”. PLOS ONE. 11 (4): e0152840. Bibcode:2016PLoSO..1152840R. doi:10.1371/journal.pone.0152840. PMC 4824384. PMID 27055247.
- ^ Jump up to:a b c “Potential DMD Therapy, Ezutromid, Shows Promise in Upgraded Form”. Retrieved 2016-11-14.
- ^ Janghra N, Morgan JE, Sewry CA, Wilson FX, Davies KE, Muntoni F, Tinsley J (2016-03-14). “Correlation of Utrophin Levels with the Dystrophin Protein Complex and Muscle Fibre Regeneration in Duchenne and Becker Muscular Dystrophy Muscle Biopsies”. PLOS ONE. 11 (3): e0150818. Bibcode:2016PLoSO..1150818J. doi:10.1371/journal.pone.0150818. PMC 4790853. PMID 26974331.
- ^ Jump up to:a b “Home – Summit”. Summit. Retrieved 2016-11-14.
- ^ Jump up to:a b c Werther CA (2016). Ezutromid Has the Potential to Treat All Duchenne Patients; Initiating Coverage With a Buy. H.C. Wainwright & Co. pp. 1–29.
- ^ “Search Orphan Drug Designations and Approvals”. http://www.accessdata.fda.gov. Retrieved 2016-11-14.
- ^ Office of the Commissioner. “Developing Products for Rare Diseases & Conditions”. http://www.fda.gov. Retrieved 2016-11-14.
- ^ Inacio P (2018-06-29). “Summit Therapeutics Ends Development of Ezutromid Therapy for DMD After Trial Failure”. Muscular Dystrophy News. Retrieved 2019-11-17.
- ^ Jump up to:a b Clinical trial number NCT02056808 for “A Phase 1b Study of SMT C1100 in Subjects With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
- ^ Jump up to:a b c Clinical trial number NCT02383511 for “Modified Diet Trial: A Study of SMT C1100 in Paediatric Patients With DMD Who Follow a Balanced Diet ” at ClinicalTrials.gov
- ^ “PTC Therapeutics | ataluren”. PTC Therapeutics. Retrieved 2016-11-15.
- ^ Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Howard MT, Sampson JB, et al. (March 2011). “Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene”. Human Mutation. 32 (3): 299–308. doi:10.1002/humu.21426. PMC 3724403. PMID 21972111.
- ^ Jump up to:a b Summit Therapeutics PLC. “Sarepta Therapeutics and Summit Enter Into Exclusive License and Collaboration Agreement for European Rights to Summit’s Utrophin Modulator Pipeline for the Treatment of Duchenne Muscular Dystrophy”. GlobeNewswire News Room. Retrieved 2016-11-15.
/////////Ezutromid, BMN-195, BMN 195, BMN195, SMTC-1100, SMTC1100, SMTC 1100, VOX-C1100, Ezutromid
O=S(C1=CC=C(OC(C2=CC=C3C=CC=CC3=C2)=N4)C4=C1)(CC)=O
NEW DRUG APPROVALS
one time
$10.00
MIRDAMETINIB
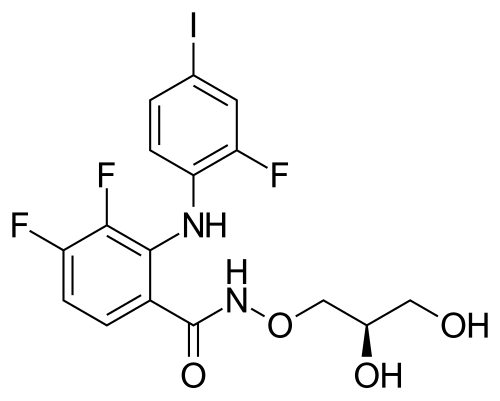

MIRDAMETINIB
391210-10-9
Chemical Formula: C16H14F3IN2O4
Molecular Weight: 482.19
PD0325901; PD 0325901; PD-325901; mirdametinib
FDA APPROVED 2/11/2025, Gomekli, To treat neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection
IUPAC/Chemical Name: (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino)benzamide
SpringWorks Therapeutics (a spin out of Pfizer ) is developing mirdametinib, a second-generation, non-ATP competitive, allosteric MEK1 and MEK2 inhibitor derived from CI-1040, for treating type 1 neurofibromatosis (NF1) and advanced solid tumors. In June 2021, a phase I/II trial was initiated in patients with low grade glioma.
- OriginatorPfizer
- DeveloperAstraZeneca; BeiGene; BIOENSIS; Pfizer; SpringWorks Therapeutics; St. Jude Childrens Research Hospital; University of Oxford
- ClassAniline compounds; Anti-inflammatories; Antineoplastics; Benzamides; Immunotherapies; Small molecules
- Mechanism of ActionMAP kinase kinase 1 inhibitors; MAP kinase kinase 2 inhibitors
- Orphan Drug StatusYes – Neurofibromatosis 1
- Phase IINeurofibromatosis 1
- Phase I/IIGlioma
- Phase ISolid tumours
- PreclinicalChronic obstructive pulmonary disease
- No development reportedCervical cancer
- DiscontinuedBreast cancer; Cancer; Colorectal cancer; Malignant melanoma; Non-small cell lung cancer
- 22 Jul 2021SpringWorks Therapeutics receives patent allowance for mirdametinib from the US Patent and Trademark Office for the treatment of Neurofibromatosis type 1-associated plexiform neurofibromas
- 16 Jun 2021SpringWorks Therapeutics and St. Jude Children’s Research Hospital agree to develop mirdametinib in USA for glioma
- 15 Jun 2021Efficacy and safety data from the phase IIb RENEU trial for Neurofibromatosis type 1-associated plexiform neurofibromas released by SpringWorks Therapeutics
Mirdametinib, sold under the brand name Gomekli, is a medication used for the treatment of people with neurofibromatosis type 1.[1] Mirdametinib is a kinase inhibitor.[1][2] It is taken by mouth.[1]
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib was approved for medical use in the United States in February 2025.[1][3]
SCHEME
SIDE CHAIN

MAIN
Medical uses
Mirdametinib is indicated for the treatment of people with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection.[1]
Adverse effects
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib can cause left ventricular dysfunction and ocular toxicity including retinal vein occlusion, retinal pigment epithelial detachment, and blurred vision.[3]
History
The efficacy of mirdametinib was evaluated in ReNeu (NCT03962543), a multicenter, single-arm trial in 114 participants aged two years of age and older (58 adults, 56 pediatric participants) with symptomatic, inoperable NF1-associated plexiform neurofibromas causing significant morbidity.[3] An inoperable plexiform neurofibromas was defined as a plexiform neurofibromas that could not be completely surgically removed without risk for substantial morbidity due to encasement or close proximity to vital structures, invasiveness, or high vascularity.[3]
The US Food and Drug Administration (FDA) granted the application for mirdametinib priority review, fast track, and orphan drug designations along with a priority review voucher.[3]
Society and culture
Legal status
Mirdametinib was approved for medical use in the United States in February 2025.[3][4][5]
PATENT
US-11066358
On July 20, 2021, SpringWorks Therapeutics announced that the United States Patent and Trademark Office (USPTO) has issued US11066358 , directed to mirdametinib , the Company’s product candidate in development for several oncology indications, including as a monotherapy for patients with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN) and was assigned to Warner-Lambert Company (a subsidiary of Pfizer ).This patent was granted on July 20, 2021, and expires on Feb 17, 2041. Novel crystalline forms of mirdametinib and compositions comprising them are claimed.
| N—((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (“mirdametinib”, or “PD-0325901”) is a small molecule drug which has been designed to inhibit mitogen-activated protein kinase kinase 1 (“MEK1”) and mitogen-activated protein kinase kinase 2 (“MEK2”). MEK1 and MEK2 are proteins that play key roles in the mitogen-activated protein kinase (“MAPK”) signaling pathway. The MAPK pathway is critical for cell survival and proliferation, and overactivation of this pathway has been shown to lead to tumor development and growth. Mirdametinib is a highly potent and specific allosteric non-ATP-competitive inhibitor of MEK1 and MEK2. By virtue of its mechanism of action, mirdametinib leads to significantly inhibited phosphorylation of the extracellular regulated MAP kinases ERK1 and ERK2, thereby leading to impaired growth of tumor cells both in vitro and in vivo. In addition, evidence indicates that inflammatory cytokine-induced increases in MEK/ERK activity contribute to the inflammation, pain, and tissue destruction associated with rheumatoid arthritis and other inflammatory diseases. |
Example 1: Production of Essentially Pure Form IV
Lab Scale Production of Essentially Pure Form IV
| All reactions were performed in toluene other than otherwise stated. Triflic anhydride gave the best yield. |
[TABLE-US-00002]TABLE 1 Coupling Agents for Step 1Entry No.Coupling AgentYieldNotes 1Mesyl Chloridedid not react 2Benzyl chloride27Had to heat 70° C. for 166 hr34-fluorobenzensulfonylchloride27Ran 93 hrs. at 70° C.44-chlorobenzensulfonylchloride35Complete after 68 hrs. 50° C.5Tosyl Chloride36Had to heat to 70° C. for 164 hrs6Benzyl chloride52study solvent effects: DMF, DMSO, NMP – all similar DMSO fastest all complete after 110 hrs., heated to 70° C. after 66 hrs.7Triflic anhydride91Cooled to −74° C. |
| [TABLE-US-00004]TABLE 3 Yields for base deprotection ReagentYield* Methyl hydrazine85-95% Anhydrous NH3 (sparged)78-90% Anhydrous NH3 (50 psi)80-92% Aqueous NH390-97% *from PD-0333760 |
Step 2: Fluoride Displacement
Pilot Plant Preparation of Essentially Pure Form IV
Step 1: Preparation of “Side Chain”, PD-0337792
Step 2: Preparation of PD-0315209
Step 3: Preparation of PD-0325901
Polymorph Transformation
| 21.4 kg PD-0315209, 9.7 kg CDI (1.05 equiv.), 91 kg solution of 9.7% PD-0337792 in Toluene (1.1 equiv.) were used and resulted in 12.74 kg of PD-0325901 (assay 99.4%, 100% Form IV, Yield 48%). |
PATENT
WO2006134469 , claiming methods of preparing MEK inhibitor, assigned to Warner-Lambert Co .
https://patents.google.com/patent/WO2006134469A1/enThe compound Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide represented by formula 1

i is a highly specific non-ATP-competitive inhibitor of MEK1 and MEK2. The compound of formula ± (Compound I) is also known as the compound PD 0325901. Compound I is disclosed in WO 02/06213; WO 04/045617; WO 2005/040098; EP 1262176; U.S. Patent Application Pub. No. 2003/0055095 A1 ; U.S. Patent Application Pub. No. 2004/0054172 A1; U.S. Patent Application Pub. No. 2004/0147478 A1 ; and U.S. Patent Application No. 10/969,681, the disclosures of which are incorporated herein by reference in their entireties.Numerous mitogen-activated protein kinase (MAPK) signaling cascades are involved in controlling cellular processes including proliferation, differentiation, apoptosis, and stress responses. Each MAPK module consists of 3 cytoplasmic kinases: a mitogen-activated protein kinase (MAPK), a mitogen-activated protein kinase kinase (MAPKK), and a mitogen-activated protein kinase kinase kinase (MAPKKK). MEK occupies a strategic downstream position in this intracellular signaling cascade catalyzing the phosphorylation of its MAP kinase substrates, ERK1 and ERK2. Anderson et al. “Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase.” Nature 1990, v.343, pp. 651-653. In the ERK pathway, MAPKK corresponds with MEK (MAP kinase ERK Kinase) and the MAPK corresponds with ERK (Extracellular Regulated Kinase). No substrates for MEK have been identified other than ERK1 and ERK2. Seger et al. “Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells.” J. Biol. Chem., 1992, v. 267, pp. 14373-14381. This tight selectivity in addition to the unique ability to act as a dual-specificity kinase is consistent with MEK’s central role in integration of signals into the MAPK pathway. The RAF-MEK-ERK pathway mediates proliferative and anti-apoptotic signaling from growth factors and oncogenic factors such as Ras and Raf mutant phenotypes that promote tumor growth, progression, and metastasis. By virtue of its central role in mediating the transmission of growth- promoting signals from multiple growth factor receptors, the Ras-MAP kinase cascade provides molecular targets with potentially broad therapeutic applications.One method of synthesizing Compound I is disclosed in the above-referenced WO 02/06213 andU.S. Patent Application Pub. No. 2004/0054172 A1. This method begins with the reaction of 2-fluoro-4- iodo-phenylamine and 2,3,4-trifluoro-benzoic acid in the presence of an organic base, such as lithium diisopropylamide, to form 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzoic acid, which is then reacted with (R)-0-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of a peptide coupling agent (e.g., diphenylphosphinic chloride) and a tertiary amine base (e.g., diisopropylethylamine). The resulting product is hydrolyzed under standard acidic hydrolysis conditions (e.g., p-TsOH in MeOH) to provide Compound 1. (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine is prepared by reaction of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol with N-hydroxyphthalimide in the presence of Ph3P and diethyl azodicarboxylate.Another method of synthesizing Compound I, which is disclosed in the above-referenced U.S.Patent Application No. 10/969,681, comprises reaction of 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzoic acid with (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of N1N1– carbonyldiimidazole. The resulting product is hydrolyzed with aqueous acid and crystallized to provide polymorphic form IV of Compound I.Although the described methods are effective synthetic routes for small-scale synthesis of Compound I, there remains a need in the art for new synthetic routes that are safe, efficient and cost effective when carried out on a commercial scale.The present invention provides a new synthetic route including Steps I through Step III to the MEK inhibitor Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I).Step I: Preparation of 0-{r(4RV2.2-dimethyl-1.3-dioxolan-4-ynmethyl}hydroxylanπine (6) The method of the present invention comprises a novel Step I of preparing of 0-{[(4R)-2,2- dimethyl-1 ,3-dioxolan-4-yl]methyl}hydroxylamine (6) from [(4S)-2,2-dimethyl-1 ,3-dioxoIan-4-yl]methanol (1) through the formation of [(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methyl trifluoromethanesulfonate (3) and its coupling with N-hydroxyphthalimide (4) to afford 2-{[(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methoxy}-1 H- isoindole-1 ,3(2H)-dione (5), which is subsequently de-protected to give 6 as shown in Scheme 1.Scheme 1



The reaction of compound (1) with trifluoromethanesulfonic anhydride (2) is carried out in the presence of a non-nucleophilic base, such as, for example, a tertiary organic amine, in an aprotic solvent at a temperature of from -5O0C to 50C, preferably, at a temperature less than -150C, to form triflate (3). A preferred tertiary organic amine is triethylamine, and a preferred solvent is toluene. Treatment of triflate (3) with N-hydroxyphthalimide (4) furnishes phthalimide (5), which can be isolated if desired. However, in order to minimize processing time and increase overall yield, 0-{[(4R)- 2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) can be prepared in a one-pot process with no phthalimide (S) isolation. Cleavage of the phthalimide function could be achieved by methods known in the art, for example, by hydrazinolysis. However, the use of less hazardous aqueous or anhydrous ammonia instead of methyl hydrazine (CH3NHNH2) is preferred.Step II: Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) As shown in Scheme 2, Step Il of the method of the present invention provides 3,4-difluoro-2-(2- fluoro-4-iodophenylamino)-benzoic acid (9).Scheme 2

Preparation of compound (9) can be carried out by reacting compound (7), wherein X is halogen, or O-SC^R^ or 0-P(3O)(OR^, wherein R^ is alkyl or aryl, with compound (8) optionally in a solvent, and in the presence of from about 1 mol equivalent to about 10 mol equivalents of at least one base, wherein the base is selected from: a Group I metal cation hydride or a Group 2 metal cation hydride, including lithium hydride, sodium hydride, potassium hydride, and calcium hydride, a Group I metal cation dialkylamide or a Group 2 metal cation dialkylamide, including lithium diisopropylamide, a Group I metal cation amide or a Group 2 metal cation amide, including lithium amide, sodium amide, potassium amide, a Group I metal cation alkoxide or a Group 2 metal cation alkoxide, including sodium ethoxide, potassium terf-butoxide, and magnesium ethoxide, and a Group I metal cation hexamethyldisilazide, including lithium hexamethyldisilazide; for a time, and at a temperature, sufficient to yield compound (9).Preferably, preparation of compound (9) is carried out by reacting compound (7), wherein X is halogen, more preferably, X is fluorine, in an aprotic solvent with compound (8) in the presence of from about 3 mol equivalents to about 5 mol equivalents of a Group I metal cation amide at a temperature of from 2O C to 55°C, more preferably, at a temperature from 45°C to 55°C. A catalytic amount of Group I metal cation dialkylamide can be added if necessary. A preferred Group I metal cation amide is lithium amide, a preferred Group I metal cation dialkylamide is lithium diisopropylamide, and a preferred solvent is tetrahydrofuran. Preferably, the reaction is performed by adding a small amount of compound (7) and compound (8) to lithium amide in tetrahydrofuran followed by slow continuous addition of the remaining portion. This procedure minimizes the risk of reactor over-pressurization due to gas side product (ammonia) generation.Step III: Preparation of N-((RV2.3-dihydroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I)Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using a carboxylic acid activating reagent such as, for example, COCI2, S(O)C^, S(O)2Cl2, P(O)Cl3, triphenylphosphine/diethylazodicarboxylate, diphenylphosphinic chloride, N, N’-dicyclohexylcarbodiimide, (benzotriazol-1 -yloxy)tripyrolidinophosphonium hexafluorophosphate, (benzotriazol-1 – yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, N-ethyl-N’-(3- dimethylaminopropyl)carbodiimide hydrochloride, or 1,1′-carbonyldiimidazole (CDI).A preferred carboxylic acid activating reagent is 1,1′-carbonyldimidazole (CDI) shown in Scheme 3. Preparation of the desirable polymorphic Form IV of Compound I using CDI is described in the above- referenced U.S. Patent Application No. 10/969,681.Scheme 3

10

10 11 Compound IIn according to the present invention, the method was modified to include the advantageous procedure for product purification and isolation, which procedure is performed in single-phase systems such as, for example, toluene/acetonitrile for the first isolation/crystallization and ethanol/toluene for the second recrystallization. Water addition, implemented in the previous procedure, was omitted to avoid the two-phase crystallization from the immiscible water-toluene system that caused inconsistent product purity. The one-phase procedure of the present invention provides consistent control and removal of un- reacted starting material and side products. Alternatively, Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using thionyl chloride (SOCI2) as shown in Scheme 4.Scheme 4


Compound IExamplesThe reagents and conditions of the reactions described herein are merely illustrative of the wide variety of starting materials, their amounts and conditions which may be suitably employed in the present invention as would be appreciated by those skilled in the art, and are not intended to be limiting in any way.HPLC (Conditions A): 10 μL injection volume onto Agilent Zorbax RX-C18 150 mm x 4.6 mm x 3.5 μm column at 30°C column temperature, 1.0 mL/min flow rate and detection at 246 nm. Mobile phase A (v/v): 25 mM Acetate Buffer, pH 6.0; Mobile phase B (v/v): Acetonitrile, and Linear Gradient Table:

Sample Preparation: Dilute 100 μL reaction mixture to 10 mL with acetonitrile. Mix in a vial 200 μL of this sample solution with 300 μL carbonate buffer pH 10.0 and 300 μL solution of 2-mercaptopyridine in acetonitrile (18 mM), heat the vial for 10 minutes at 500C and dilute to 1:1 ratio in mobile phase A.GC (Conditions B): 1 μL injection onto an RTX-5 column (30 m x 0.25 mm x 0.25 μm) with initial oven temperature of 120°C for 2 min. to final temperature of 250°C in 15°C/minute ramping and a final time of 2.33 min; Flow rate: 1 mL/min.HPLC (Conditions C): 5 μL injection onto Phenomenex Luna C18(2) 150 mm x 4.6 mm x 3μm column ; flow rate : 1.0 mL/min; detection at 225 nm; mobile phase A: 95/5 v/v Water/Acetonitrile with 0.1% Trifluoroacetic acid (TFA), mobile phase B: 5/95 v/v Water/Acetonitriie with 0.1% TFA; Linear Gradient Table:

Sample preparation: Dilute 1 ml_ reaction mixture to 100 mL with acetonitrile and dilute 1 mL of this solution to 10 mL with 50:50 Water/Acetonitrile.HPLC (Conditions D): 5 μL injection onto Waters SymmetryShield RP 18, 150 mm x 4.6 mm x 3.5 μm column; flow rate: 1.0 mL/min; detection at 235 nm; mobile phase A: 25 mM Acetate Buffer adjusted to pH 5.5, mobile phase B: Acetonitrile; Linear Gradient Table:

Sample preparation: Dilute 40 μL of reaction mixture in 20 mL acetonitrile.HPLC (Conditions E): 10 μL sample injection onto YMC ODS-AQ 5 μm, 250 mm x 4.6 mm column; flow rate: 1.0 ml_/min; detection at 280 nm; temperature 30°C; mobile phase : 75/25 v/v Acetonitrile/Water with 0.1% Formic acid.Sample preparation: Quench reaction mixture sample with dipropylamine and stir for about 5 minutes before further dilution with mobile phase.DSC measurement was performed using a Mettler-Toledo DSC 822, temperature range 25° to 150°C with 5°C/min heating rate in a 40 μL aluminum pan. Experimental Conditions for Powder X-Rav Diffraction (XRD):A Rigaku Miniflex+ X-ray diffractometer was used for the acquisition of the powder XRD patterns. The instrument operates using the Cu Ka1 emission with a nickel filter at 1.50451 units. The major instrumental parameters are set or fixed at:X-ray: Cu / 30 kV (fixed) / 15 mA (fixed)Divergence Slit: Variable Scattering Slit: 4.2° (fixed) Receiving Slit: 0.3 mm (fixed) Scan Mode: FT Preset Time: 2.0 s Scan Width: 0.050° Scan Axis: 2Theta/Theta Scan Range: 3.000° to 40.000°Jade Software Version: 5.0.36(SP1) 01/05/01 (Materials Data, Inc.) Rigaku Software: Rigaku Standard Measurement for Windows 3.1 Version 3.6(1994-1995) Example 1. Preparation of 0-ffl4R)-2.2-dimethyl-1.3-dioxolan-4-vπmethyl}hvdroxylamine (6)A solution containing [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol (1) (13.54 ml_, 0.109 mol) (DAISO Co., Ltd., CAS# 22323-82-6) and triethylamine (18.2 ml_, 0.131 mol) in 115 mL toluene was cooled to -15 C, then trifluoromethanesulfonic anhydride (2) (18.34 mL, 30.75 g, 0.109 mol) (Aldrich, Catalog # 17,617-6 ) was added drop wise while maintaining the temperature at less than -15°C. The mixture was then stirred for 2 hours, and transferred to a separate flask containing a mixture (slurry) of N- hydroxyphthalimide (4) (18.99 g, 0.116 mol) (Aldrich, Catalog # H5.370-4) and 18.2 mL (0.13 mol) triethylamine in 95 mL toluene. The resulting mixture was warmed to 20-25°C and stirred for at least 5 hours or until reaction completion (determined by HPLC (Conditions A)). Water (93 mL) was then added to quench the reaction mixture, the phases were separated, and the bottom aqueous layer was discarded. The water quench was repeated two more times resulting in a pale yellow organic layer. The organic layer was heated to 35 C and treated with 36.7 mL ammonium hydroxide solution (contains about 28-29% wt/wt ammonia). The mixture was stirred for at least 12 hours or until the reaction was deemed complete as determined by GC (Conditions B). The water was then removed under reduced pressure by co- distilling it with toluene to about half of the original volume at temperatures around 35-45 C. Toluene (170 mL) was added to the concentrated solution and the distillation was repeated. A sample was drawn for water content determination by Karl Fisher method (using EM Science Aquastar AQV-2000 Titrator with a sample injected to a pot containing methanol and salicylic acid). The distillation was repeated ifl water content was more than 0.1%. The concentrated solution was filtered to remove the white solid side product, and the filtrate was stored as 112mL (98 g) product solution containing 9.7% w/w compound 6 in toluene. This solution was ready for use in the final coupling step (Example 3). Overall chemical yield was 59%. A small sample was evaporated to yield a sample for NMR identification.1H NMR (400 MHz, CDCI3): δ 5.5 (bs, 2H), 4.35 (m, 1H), 4.07 (dd, 1H), 3.77 (m, 2H), 3.69 (dd, 1H), 1.44 (s, 3H), 1.37 (s, 3H).Example 2. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9)A solution of 2-fluoro-4-iodoaniline (8) (16.4 g, 0.069 mol) (Aldrich, Catalog # 30,660-6) and 2,3,4- trifluorobenzoic acid (7) (11.98 g, 0.068 mol) (Aldrich, Cat # 33,382-4) in 38 mL tetrahydrofuran (THF) was prepared and a portion (about 5%) of this solution was added to a stirring slurry of lithium amide (5 g, 0.22 mol) in 40 mL THF at 50-55 C. After about 15-30 min. an exotherm followed by gas release and color change are observed. The remaining portion of the (8) and (7) solution was added slowly over 1-2 hr while maintaining temperatures within 45-55°C. The mixture was stirred until the reaction was deemed complete (by HPLC (Conditions C). The final mixture was then cooled to 20-25°C and transferred to another reactor containing 6 N hydrochloric acid (47 mL) followed by 25 mL acetonitrile, stirred, and the bottom aqueous phase was discarded after treatment with 40 mL 50% sodium hydroxide solution. The organic phase was concentrated under reduced pressure and 57 mL acetone was added. The mixture was heated to 50°C, stirred, and added with 25 mL warm (40-50°C) water and cooled to 25-30°C to allow crystallization to occur (within 1-4 hours). Once the crystallization occurred, the mixture was further cooled to 0 to -5°C and stirred for about 2 hours. The solid product was filtered and the wet cake was dried in vacuum oven at about 55°C. Overall chemical yield was 21.4 g, 80%. 1H NMR (400 MHz, (CD3)2SO): δ 13.74 (bs, 1H), 9.15 (m, 1 H), 7.80 (dd, 1H), 7.62 (d, 1H), 7.41 (d, 1H), 7.10 (q, 1H), 6.81 (m, 1H).Example 2B. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) by the solid addition of lithium amide methodTo a stirring solution of 2,3,4-trifluorobenzoic acid (13) (5.0 g, 28.4 mmol) and 2-fluoro-4- iodoaniline (14) (6.73 g, 28.4 mmol) in MeCN (100 mL), under N2 atmosphere was added lithium amide (2.61 g, 113.6 mmol) in small portions. The reaction mixture was heated to reflux for 45 minutes, cooled to ambient temperature and quenched with 1 N HCI and then water. The yellowish white precipitate was filtered, washed with water. The solid was triturated in CH2CI2 (30 mL) for 1h, filtered and dried in a vacuum oven at 45°C for 14 hours to give 8.Og (72%) of compound (9) as an off-white solid, mp 201.5-203 °C.Example 3. Preparation of N-((R)-2.3-dihvdroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound \)3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (20 g, 0.051 mol) in 100 mL acetonitrile was treated with 1,1′-carbonyldiimidazole (CDI) (8.66 g, 0.053 mol) (Aldrich, Cat # 11,553-3) and stirred for about 2 hours at 20-25°C until the reaction was deemed complete by HPLC (Conditions D). 94 mL (84.9 g) of 9.7% w/w solution of O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) in toluene was then added and stirred for about 4 hours or until the reaction was deemed complete by HPLC (Conditions D). To this mixture was added 66 mL of 5.6 % hydrochloric acid solution, and after stirring, the bottom aqueous phase was discarded. Again 66 mL of 5.6 % hydrochloric acid solution was added to the organic phase and stirred at 20-25°C for 12-18 hours or until the reaction was deemed complete by HPLC (Conditions D). The bottom layer was then discarded and the remaining organic layer was concentrated under reduced pressure to remove about 10-20% solvent, and the volume was adjusted to about 9-11 mL/g with toluene (80 mL). Crude product was then crystallized at 10-15°C. The slurry was allowed to stir for about 2 hours and the crude solid product was filtered, and dried. The dried crude product was recharged to the reactor and dissolved into 150 mL of 5% v/v ethanol/toluene mixture at 55- 67°C. The solution was then clarified at this temperature through filter (line filter) to remove any remaining particulate matter. The solution was then cooled slowly to 5°C to crystallize and stirred for at least 2 h, filtered and dried. The dried solid product was redissolved in EtOH (60 mL) at 35°C, and product was precipitated out by adding water (300 mL) at 35°C followed by cooling to 200C. The slurry was stirred for at least 2 hours to transform the crystals to the desired polymorphic Form IV as determined by DSC and Powder X-ray Diffraction pattern (PXRD). The slurry was filtered and dried under vacuum oven at 70- 90°C to yield the final N-((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I) product. Overall chemical yield was 13 g, 53%. Melting point (DSC): 112+1° C. Appearance: White to off-white crystals.Shown in Figure 1, PXRD conforms to polymorphic crystal Form IV disclosed in the above mentioned U.S. Patent Application No. 10/969,681 1H NMR (400 MHz, (CD3)2SO): δ 11.89 (bs, 1H), 8.71 (bs, 1H), 7.57 (d, 1H), 7.37 (m, 2H), 7.20 (q, 1H), 6.67 (m, 1H), 4.84 (bs, 1H), 4.60 (m, 1H), 3.87 (m, 1 H), 3.7 (m, 2H), 3.34 (m, 2H).Example 4. Preparation of N-((R)-2.3-dihydroxypropoxyV3.4-difluoro-2-(2-fluoro-4-iodo-phenylanrιinoV benzamide (Compound \)To a stirring solution of 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (120 g, 0.30 mol) in a mixture of 1 mL N,N-dimethylformamide and 1000 mL toluene was added thionyl chloride (55 g, 0.462 mol). The mixture was heated to 50-65 C and stirred for 2 hours or until reaction completion as determined by HPLC (Conditions E). The final reaction mixture was then cooled and concentrated under reduced pressure to a slurry keeping the temperature below 35°C. Toluene (600 mL) was added to dissolve the slurry and vacuum distillation was repeated. Additional toluene (600 mL) was added to the slurry dissolving all solids and the solution was then cooled to 5° -10°C. The solution was then treated with O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) (63 g, 0.43 mol) solution in 207 mL toluene followed by potassium carbonate (65 g) and water (200 mL), stirred for at least 2 hours at 20- 25°C. The stirring was stopped to allow phase separation and the bottom phase was discarded. The remaining organic layer was treated with hydrochloric acid solution (7.4%, 240 mL) until pH was less than 1 and stirred for 2 hours. The final reaction mixture was slightly concentrated under vacuum collecting about 100 mL distillate and the resulting organic solution was cooled to 5°C to crystallize the product and filtered. The filter cake was washed with toluene (1000 mL) followed by water (100 mL) and the wet cake (crude product Compound I) was charged back to the flask. Toluene (100 mL), ethanol (100 mL) and water (100 mL) are then added, stirred at 30-35°C for about 15 min, and the bottom aqueous phase was discarded. Water (200 mL) was then added to the organic solution and the mixture was stirred at about 3O C to allow for crystallization. The stirring was continued for 2 hours after product crystallized, then it was further cooled to about 0°C and stirred for at least 2 hours. The slurry was filtered and wet cake was dried under reduced pressure at 55-85°C to yield the final product N-((R)-2,3-dihydroxypropoxy)-3,4- difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I) product. Overall chemical yield was 86 g, 58%.
PATENT
WO2002/006213 describes crystalline Forms I and II. U.S. Pat. No. 7,060,856 (“the ‘856 patent”)
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2002006213
| Clinical data | |
|---|---|
| Trade names | Gomekli |
| Other names | PD-0325901 |
| AHFS/Drugs.com | Gomekli |
| License data | US DailyMed: Mirdametinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EE05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 391210-10-9 |
| PubChem CID | 9826528 |
| IUPHAR/BPS | 7935 |
| DrugBank | DB07101 |
| ChemSpider | 10814340 |
| UNII | 86K0J5AK6M |
| KEGG | D11675 |
| ChEBI | CHEBI:9826528 |
| ChEMBL | ChEMBL507361 |
| PDB ligand | 4BM (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C16H14F3IN2O4 |
| Molar mass | 482.198 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Gomekli- mirdametinib capsule; Gomekli- mirdametinib tablet, for suspension”. DailyMed. 27 February 2025. Retrieved 2 April 2025.
- ^ Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ (June 2023). “Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas”. BMC Cancer. 23 (1): 553. doi:10.1186/s12885-023-10996-y. PMC 10273716. PMID 37328781.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection”. U.S. Food and Drug Administration (FDA). 11 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “UPDATE: SpringWorks Therapeutics Announces FDA Approval of Gomekli (mirdametinib) for the Treatment of Adult and Pediatric Patients with NF1-PN” (Press release). SpringWorks Therapeutics. 12 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025 – via GlobeNewswire News Room.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025.
External links
- “Mirdametinib (Code C52195)”. NCI Thesaurus.
- Clinical trial number NCT03962543 for “MEK Inhibitor Mirdametinib (PD-0325901) in Patients With Neurofibromatosis Type 1 Associated Plexiform Neurofibromas (ReNeu)” at ClinicalTrials.gov
- Moertel CL, Hirbe AC, Shuhaiber HH, Bielamowicz K, Sidhu A, Viskochil D, Weber MD, Lokku A, Smith LM, Foreman NK, Hajjar FM, McNall-Knapp RY, Weintraub L, Antony R, Franson AT, Meade J, Schiff D, Walbert T, Ambady P, Bota DA, Campen CJ, Kaur G, Klesse LJ, Maraka S, Moots PL, Nevel K, Bornhorst M, Aguilar-Bonilla A, Chagnon S, Dalvi N, Gupta P, Khatib Z, Metrock LK, Nghiemphu PL, Roberts RD, Robison NJ, Sadighi Z, Stapleton S, Babovic-Vuksanovic D, Gershon TR: ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025 Feb 20;43(6):716-729. doi: 10.1200/JCO.24.01034. Epub 2024 Nov 8. [Article]
- Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, Allen JC, Schorry E, Korf B, Robison NJ, Goldman S, Vinks AA, Emoto C, Fukuda T, Robinson CT, Cutter G, Edwards L, Dombi E, Ratner N, Packer R, Fisher MJ: NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol. 2021 Mar 1;39(7):797-806. doi: 10.1200/JCO.20.02220. Epub 2021 Jan 28. [Article]
- Ioannou M, Lalwani K, Ayanlaja AA, Chinnasamy V, Pratilas CA, Schreck KC: MEK Inhibition Enhances the Antitumor Effect of Radiotherapy in NF1-Deficient Glioblastoma. Mol Cancer Ther. 2024 Sep 4;23(9):1261-1272. doi: 10.1158/1535-7163.MCT-23-0510. [Article]
- FDA Approved Drug Products: GOMEKLI (mirdametinib) capsules and tablets for oral and oral suspension use (Feb 2024) [Link]
- FDA News: FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection [Link]
////////MIRDAMETINIB, Orphan Drug Status, Neurofibromatosis 1, PHASE 2, PD0325901, PD 0325901, PD-325901, FDA 2025, GOMEKLI, APPROVALS 2025
O=C(NOC[C@H](O)CO)C1=CC=C(F)C(F)=C1NC2=CC=C(I)C=C2F
OTESECONAZOLE
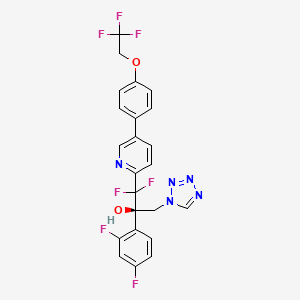

OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
| C23H16F7N5O2 527.4 | |
| Synonyms | VT 1161 Oteseconazole CAS1340593-59-0 |
|---|
Other Names
- (αR)-α-(2,4-Difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(2,2,2-trifluoroethoxy)phenyl]-2-pyridineethanol
- (2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-1,2,3,4-tetrazol-1-yl)- 1-{5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl}propan-2-ol
UPDATE MAY 2022… FDA APPROVED 2022/4/26, Vivjoa
Oteseconazole, sold under the brand name Vivjoa, is a medication used for the treatment of vaginal yeast infections.[1]
It was approved for medical use in the United States in April 2022.[2][3] It was developed by Mycovia Pharmaceuticals.[3]
Names
Oteseconazole is the international nonproprietary name (INN).[4]
Oteseconazole is an azole antifungal used to prevent recurrent vulvovaginal candidiasis in females who are not of reproductive potential.
Oteseconazole, also known as VT-1161, is a tetrazole antifungal agent potentially for the treatment of candidal vaginal infection. VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model.
Oteseconazole has been used in trials studying the treatment of Tinea Pedis, Onychomycosis, Candidiasis, Vulvovaginal, and Recurrent Vulvovaginal Candidiasis.
Mycovia Pharmaceuticals is developing oteseconazole, the lead from a program of metalloenzyme Cyp51 (lanosterol demethylase) inhibitors, developed using the company’s Metallophile technology, for treating fungal infections including onychomycosis and recurrent vulvovaginal candidiasis (RVVC). In July 2021, oteseconazole was reported to be in phase 3 clinical development. Licensee Jiangsu Hengrui Medicine is developing otesaconazole, as an oral capsule formulation, for treating fungal conditions, including RVVC, onychomycosis and invasive fungal infections, in Greater China and planned for a phase 3 trial in April 2021 for treating VVC.
- OriginatorViamet Pharmaceuticals
- DeveloperMycovia Pharmaceuticals; Viamet Pharmaceuticals
- ClassAntifungals; Foot disorder therapies; Pyridines; Small molecules; Tetrazoles
- Mechanism of Action14-alpha demethylase inhibitors
- PreregistrationVulvovaginal candidiasis
- Phase IIOnychomycosis
- No development reportedTinea pedis
- 01 Jun 2021Preregistration for Vulvovaginal candidiasis (In adolescents, In adults, In children, Recurrent) in USA (PO)
- 01 Jun 2021Mycovia intends to launch otesaconazole (Recurrent) for Vulvovaginal candidiasis in the US in early 2022
- 06 Jan 2021Interim efficacy and adverse events data from a phase III ultraVIOLET trial in Vulvovaginal candidiasis released by Mycovia Pharmaceuticals

Synthesis Reference
Hoekstra, WJ., et al. (2020). Antifungal compound process (U.S. Patent No. US 10,745,378 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/f4/62/19/5ba525b1caad0e/US10745378.pdf
PATENT
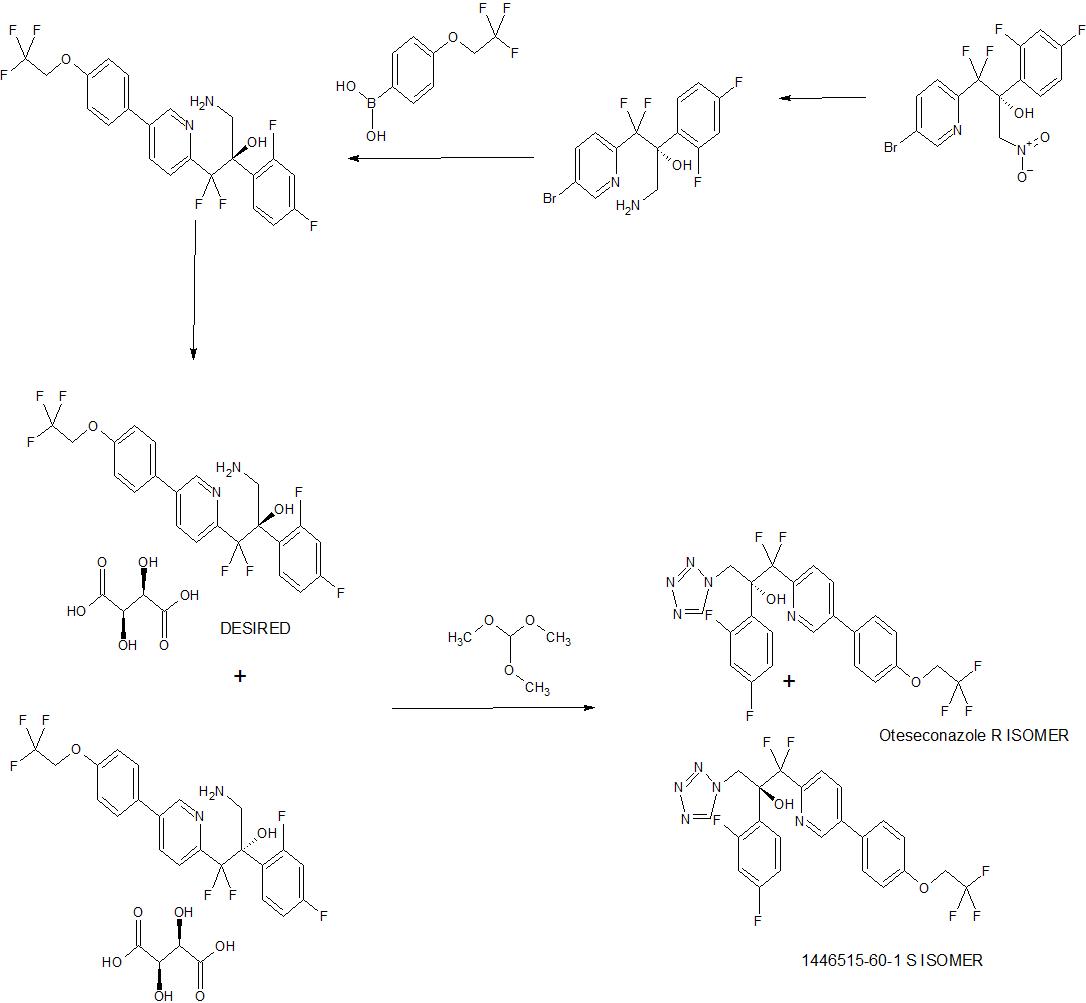
WO 2017049080
WO 2016149486
US 20150024938
WO 2015143172
WO 2015143184
WO 2015143180
WO 2015143142
WO 2013110002
WO 2013109998
WO 2011133875
PATENT
WO 2017049080,
Syn
J. Med. Chem. 2024, 67, 4376−4418
Oteseconazole was approved by the USFDA in April 2022 for the treatment of recurrent vulvovaginal candidiasis in women with a history of vulvovaginal candidiasis and who are not of reproductive
potential. Additional studies for other invasive and opportunistic infections and for onychomycosis are underway.40, The design and discovery of oteseconazole is published by a group from Viamet Pharmaceuticals, now part of Mycovia Pharmaceuticals. It details the racemic synthesis of the drug on
<1 g scale in which the metal-binding tetrazole is installed by treatment of ester 5.2 (Scheme 10) with diazomethane and tetrazole.42
A more scale-friendly asymmetric route that avoided the use of diazomethane was subsequently disclosed in patents and is detailed in Scheme 10 and Scheme 11.43
First, a mixture of ethyl bromodifluoroacetate, stoichiometric copper
powder, and 2,5-dibromopyridine (5.1) in DMSO provided ester 5.2 as an oil that was purified via distillation (Scheme10). Conversion to the aryl ketone 5.5 was achieved via direct addition of lithiated 5.3 or via a two-step process by first conversion to morpholine amide 5.4 followed by addition of
the Grignard generated from aryl bromide 5.3. The resulting ketone 5.5 was a liquid that was carried into the next step without purification.
The key step in the synthesis of 5 is an asymmetric Henry reaction using cinchona alkaloid catalyst 5.6. Addition of nitromethane to ketone 5.5 furnished alcohol 5.7 in 75% yield and ∼90:10 ratio of enantiomers. Next, reduction of the nitro group to the primary amine was accomplished using Pt
catalyzed hydrogenation. The chiral purity of the resulting amine was upgraded by classical resolution using di-p-toluoyl L-tartaric acid to provide 5.8·L-DTTA in 33% yield and >99% chiral purity.Conversion of amino alcohol 5.8 to oteseconazole (5) required two steps: cross coupling to introduce the aryltrifluoroethyl ether fragment and tetrazole formation. These steps were performed in either sequence in the patent. The route shown in Scheme 11 represents the largest scale demonstrated (>100 g input of 5.8). While the use of azide containing reagents presents significant safety risks, no information was provided on safe operation of the tetrazole forming step in the laboratory or on plant scale. Some of the
procedures for tetrazole formation described in the patent would likely require modification for safe scale-up.
To complete the synthesis of oteseconazole, resolved amino alcohol 5.8 first underwent a salt break followed by Suzuki coupling using boronic acid 5.9 to provide biaryl product 5.10 as the L-tartrate salt (Scheme 11). Conversion of 5.10 to 5 was accomplished using TMSN3 in acetic acid with sodium acetate and trimethoxy orthoformate. Treatment of the resulting solution with a Pd scavenger preceded crystallization of the product from EtOH and water after pH adjustment with potassium carbonate. The product was isolated in 85% yield as a hydrated form. Another patent described conversion of the oteseconazolehydrate totheanhydrous form byrecrystallizationfrom EtOHandn-heptanetofurnish5 in90%yield.45
(40) Hoy, S. M. Oteseconazole: First approval. Drugs 2022, 82,1017−1023.
(41) Sobel, J. D.; Nyirjesy, P. Oteseconazole: an advance in
treatment of recurrent vulvovaginal candidiasis. Future Microbiol 2021,
16, 1453−1461.
(42) Hoekstra, W. J.; Garvey, E. P.; Moore, W. R.; Rafferty, S. W.;
Yates, C. M.; Schotzinger, R. J. Design and optimization of highly
selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24,
3455−3458.
(43) Wirth, D. D.; Yates, C. M.; Hoekstra, W. J.; Bindl, M. F.;
Hartmann, E. Process for enantioselective preparation of tetrazolyl
pyridinyl diaryl propanols as antifungal drugs and their precursors.
WO 2017049080, 2017.
(44) González-Bobes, F.; Kopp, N.; Li, L.; Deerberg, J.; Sharma, P.;
Leung, S.; Davies, M.; Bush, J.; Hamm, J.; Hrytsak, M. Scale-up of
Azide Chemistry: A Case Study. Org. Process Res. Dev. 2012, 16,
2051−2057.
(45) Hoekstra, W. J.; Wirth, D. D.; Ehiwe, T.; Bonnaud, T.
Antifungal compounds and processes for making. WO 2016149486,
2016.


.
PATENT
WO-2021143811
Novel crystalline polymorphic form of VT-1161 (also known as oteseconazole) phosphate disodium salt, useful as a prodrug of oteseconazole, for treating systemic fungal infection (eg Candida albicans infection) or onychomycosis.The function of metalloenzymes is highly dependent on the presence of metal ions in the active site of the enzyme. It is recognized that reagents that bind to and inactivate metal ions at the active site greatly reduce the activity of the enzyme. Nature uses this same strategy to reduce the activity of certain metalloenzymes during periods when enzyme activity is not needed. For example, the protein TIMP (tissue inhibitor of metalloproteinases) binds to zinc ions in the active sites of various matrix metalloproteinases, thereby inhibiting enzyme activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain 1-(1,2,4-triazole) group, which exists in the active site of the target enzyme lanosterol demethylase The heme iron binds, thereby inactivating the enzyme. Another example includes zinc-bound hydroxamic acid groups, which have been introduced into most of the published inhibitors of matrix metalloproteinases and histone deacetylases. Another example is the zinc-binding carboxylic acid group, which has been introduced into most of the published angiotensin converting enzyme inhibitors.
VT-1161, the compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2, 2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol, is an antifungal drug developed by VIAMET, currently in the clinical research stage, its structure is as follows Shown:
This compound mainly acts on the CYP51 target of fungal cells. Compared with the previous triazole antifungal drugs, it has the advantages of wider antibacterial spectrum, low toxicity, high safety and good selectivity. However, this compound is not suitable for Liquid preparations (including or excluding the parenteral delivery carrier) are used to treat patients in need thereof.
2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoro Ethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate is a prodrug of VT-1161.
On the other hand, nearly half of the drug molecules are in the form of salts, and salt formation can improve certain undesirable physicochemical or biological properties of the drug. Relative to 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2- Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate, it is of great significance to develop salts with more excellent properties in terms of physical and chemical properties or pharmaceutical properties.To this end, the present disclosure provides a new pharmaceutically acceptable salt form of a metalloenzyme inhibitor.Example 1:[0161](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2, 2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate disodium salt (Compound 1)[0162]
[0163](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2 ,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate (compound 1a, prepared according to the method of patent WO2013110002, 0.28g, 0.46mmol, 1.0eq) and ethanol (5mL ) Add to the reaction flask and stir evenly. A solution of NaOH (36.90 mg, 2.0 eq) dissolved in water (1 mL) was added dropwise into the above reaction flask, stirring was continued for 2 h, and concentrated to obtain compound 1, 300 mg of white solid.[0164]After X-ray powder diffraction detection, the XRPD spectrum has no sharp diffraction peaks, as shown in FIG. 10.[0165]Ms:608.10[M-2Na+3H] + .[0166]Ion chromatography detected that the sodium ion content was 6.23%.[0167]Example 2: (R)-((2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4 -(2,2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)prop-2-yl)oxy)methyl phosphate disodium salt (compound 2)
[0169]Under ice-cooling, NaH (58mg, 0.87mmol) was added to the reaction flask, 1.5mL of N,N-dimethylformamide and 0.6mL of tetrahydrofuran were added, followed by iodine (38mg, 0.15mmol), and then Compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-tri Fluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (2b, prepared according to the method of patent WO2013110002, 158mg, 0.3mmol) tetrahydrofuran (1ml) solution was added to the reaction solution, stirred and reacted for 1-4h , And then add compound 2a (519mg, 2.01mmol) in tetrahydrofuran (1ml) solvent to the reaction, stir until the reaction is complete, 10% aqueous ammonium chloride solution to quench the reaction, extract, concentrate and drain, the crude product 2c is directly used for the next One-step reaction, Ms: 750.0[M+H] + .[0170]
[0171]Under ice-bath cooling, add trifluoroacetic acid (0.5mL) to the crude product 2c (300mg) in dichloromethane (2mL) solution, stir until the reaction is complete, and after concentration, the target compound 2d, 82mg, Ms was separated by high performance liquid phase separation. :638.0[M+H] + .[0172]
Add compound 2d (0.29g, 0.46mmol, 1.0eq) and ethanol (5mL) obtained in the previous step into the reaction flask, stir, and add NaOH (36.90mg, 2.0eq) water (1ml) solution dropwise to the aforementioned reaction solution , Stirred for 2-5 h, and concentrated to obtain 2,313 mg of the target compound.
Ms:638.10[M-2Na+3H] + .
PATENT
WO2011133875
https://patents.google.com/patent/WO2011133875A2/en
Product pat, WO2011133875 , protection in the EU states and the US April 2031.
PATENT
WO2015143184 ,
https://patents.google.com/patent/WO2015143184A1/en
Mycovia, claiming a process for preparing antifungal compounds, particularly oteseconazole.EXAMPLE 11

2-(2,4-Difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (11)Compound 11 was prepared using the conditions employed for 1: 0.33 g as a solid. The precursor l-bromo-4-(2,2,2-trifluoroethoxy)benzene was prepared as described below in one step.1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.95 (d, / = 8.0 Hz, 1 H), 7.70 (s, 1 H), 7.64 (d, / = 8.5 Hz, 1 H), 7.54 (d, / = 8.5 Hz, 2 H), 7.42- 7.37 (m, 1 H), 7.08 (d, / = 8.5 Hz, 2 H), 6.79- 6.75 (m, 1 H), 6.69- 6.66 (m, 1 H), 5.58 (d, / = 14.0 Hz, 1 H), 5.14 (d, / = 14.0 Hz, 1 H), 4.44 – 4.39 (m, 2 H). HPLC: 99.1%. MS (ESI): m/z 528 [M++l].Chiral preparative HPLC Specifications for (+)-ll:Column: Chiralpak IA, 250 x 4.6mm, 5uMobile Phase: A) w-Hexane, B) IPAIsocratic: A: B (65:35)Flow Rte: l.OO mL/minOptical rotation [a]D: + 24° (C = 0.1 % in MeOH). 1 -Bromo-4-( 2,2,2-trifluoroethoxy )benzeneTo a stirred solution of trifluoroethyl tosylate (1.5 g, 5.8 mmol) in DMF (20 mL) was added K2CO3 (4 g, 29.4 mmol) followed by addition of p-bromo phenol (1.1 g, 6.46 mmol) at RT under inert atmosphere. The reaction mixture was stirred at 120 °C for 6 h. The volatiles were evaporated under reduced pressure; the residue was diluted with water (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04, filtered and concentrated in vacuo. The crude compound was purified by silica gel column chromatography eluting with 5% EtOAc/hexane to afford the desired product (0.8 g, 3.13 mmol, 53.3%) as semi solid. 1H NMR (200 MHz, CDC13): δ 7.44 – 7.38 (m, 2 H), 6.86-6.80 (m, 2 H), 4.38- 4.25 (m, 2 H).ExamplesThe present invention will now be demonstrated using specific examples that are not to be construed as limiting.General Experimental ProceduresDefinitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo- pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester can be reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br. Alternatively, ketone 3-Br can be afforded directly from ester 2-Br as shown in Scheme 1. Scheme 1. Synthesis of ketone 3-Br r

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).Scheme 2. Synthesis of ketone 3

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.Alternatively, compound 1 can be prepared according to Scheme 3 utilizing diols 2-6b (or 2- 6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof). Olefins 2-5a and 2-5 can be prepared by reacting ketones 3 and 1-4 under Wittig olefination conditions (e.g., Ph3PCH3Br and BuLi). Also, as indicated in Scheme 5, any of pyridine compounds, 3, 2-5a, 2-6b, 2-7b, 4*, 4b, or 6 can be converted to the corresponding 4-CF3CH2O-PI1 analogs (e.g., 1-4, 2-5, 2-6a, 2-7a, 5*, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with 4,4,5, 5-tetramethyl-2- (4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (or the corresponding alkyl boronates or boronic acid or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2), and in the presence of a base (e.g., KHCO3, K2C03, Cs2C03, or Na2C03, or the like). Olefins 2-5a and 2-5 can be transformed to the corresponding chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), through exposure to Sharpless asymmetric dihydroxylation conditions: 1) commercially available AD- mix alpha or AD-mix beta with or without additional osmium oxidant and methanesulfonamide, 2) combination of a catalytic osmium oxidant (e.g., Os04 or K20sC>2(OH)4), a stoichiometric iron oxidant (e.g., K3Fe(CN)6), a base (e.g., KHCO3, K2CO3, Cs2C03, or Na2C03, or the like), and a chiral ligand (e.g., (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, (DHQ)2AQN, (DHQD)2PYR, or (DHQ)2PYR; preferably (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, and (DHQD)2PYR), or 3) option 2) with methanesulfonamide. The primary alcohol of the resultant chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), can then be activated to afford compounds 2-7b (or 2-7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof). For example, the mesylates can be prepared by exposing chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), to methanesulfonyl chloride and a base. Epoxide formation can be affected by the base-mediated (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like) ring closure of compounds 2-7b (or 2- 7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof) to provide epoxides 4* (or 4c*, the enantiomer of 4*, or mixtures thereof) and 5* (or 5-b*, the enantiomer of 5*, or mixtures thereof). The epoxides can then be converted into amino-alcohols 4b (or 4c, the enantiomer of 4b, or mixtures thereof) and 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof) through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 6 (or 6a, the enantiomer of 6, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).Scheme 3. Synthesis of 1 via Asymmetric Dihydroxylation Method


Y is -OS02-alkyl, -OS02-substituted alkyl, -OS02-aryl, -OS02- substituted aryl, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, – 0(C=0)-aryl, -0(C=0)-substituted aryl, or halogen

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or oZ-S-OHcombinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof). cheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

The following describes the HPLC method used in assessing HPLC purity of the examples and intermediates presented below:Column: Waters XBridge Shield RP18, 4.6 x 150 mm, 3.5 μιηMobile Phase: A = 0.05% TFA/H20, B = 0.05% TFA/ACNAutosampler flush: 1 : 1 ACN/H20Diluent: 1:1 ACN/H20Flow Rate: 1.0 ml/minTemperature: 45 °CDetector: UV 275 nmPump Parameters:

EXAMPLE 1Preparation of ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2-Br)

2-Br Dialkylated impurity In a clean multi-neck round bottom flask, copper powder (274.7 g, 2.05 eq) was suspended in dimethyl sulfoxide (3.5 L, 7 vol) at 20 – 35 °C. Ethyl bromodifluoroacetate (449 g, 1.05 eq) was slowly added to the reaction mixture at 20 – 25 °C and stirred for 1 – 2 h. 2, 5- dibromopyridine (500 g, 1 eq) was added to the reaction mixture and the temperature was increased to 35 – 40 °C. The reaction mixture was maintained at this temperature for 18 – 24 h and the reaction progress was monitored by GC.After the completion of the reaction, ethyl acetate (7 L, 14 vol) was added to the reaction mixture and stirring was continued for 60 – 90 min at 20 – 35 °C. The reaction mixture was filtered through a Celite bed (100 g; 0.2 times w/w Celite and 1L; 2 vol ethyl acetate). The reactor was washed with ethyl acetate (6 L, 12 vol) and the washings were filtered through a Celite bed. The Celite bed was finally washed with ethyl acetate (1 L, 2 vol) and all the filtered mother liquors were combined. The pooled ethyl acetate solution was cooled to 8 – 10 °C, washed with the buffer solution (5 L, 10 vol) below 15 °C (Note: The addition of buffer solution was exothermic in nature. Controlled addition of buffer was required to maintain the reaction mixture temperature below 15 °C). The ethyl acetate layer was washed again with the buffer solution until (7.5 L; 3 x 5 vol) the aqueous layer remained colorless. The organic layer was washed with a 1: 1 solution of 10 % w/w aqueous sodium chloride and the buffer solution (2.5 L; 5 vol). The organic layer was then transferred into a dry reactor and the ethyl acetate was distilled under reduced pressure to get crude 2-Br.The crude 2-Br was purified by high vacuum fractional distillation and the distilled fractions having 2-Br purity greater than 93 % (with the dialkylated not more than 2 % and starting material less than 0.5 %) were pooled together to afford 2-Br.Yield after distillation: 47.7 % with > 93 % purity by GC (pale yellow liquid). Another 10 % yield was obtained by re-distillation of impure fractions resulting in overall yield of ~ 55 – 60 %.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz): 8.85 (1H, d, 1.6 Hz), 8.34 (1H, dd, J = 2.0 Hz, 6.8 Hz), 7.83 (1H, d, J = 6.8 Hz), 4.33 (2H, q, J = 6.0 Hz), 1.22 (3H, t, J = 6.0 Hz). 13C NMR: 162.22 (i, -C=0), 150.40 (Ar-C-), 149.35 (t, Ar-C), 140.52 (Ar-C), 123.01 (Ar-C), 122.07 (Ar-C), 111.80 (t, -CF2), 63.23 (-OCH2-), 13.45 (-CH2CH3).EXAMPLE 2
Preparation of2-( 5-bromopyridin-2-yl )-l -(2,4-difluorophenyl )-2, 2-difluoroethanone ( 3-Br ) A. One-step Method

l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.The reaction was quenched by slow addition of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) below -65 °C. The reaction mixture was gradually warmed to 20 – 35 °C and an additional amount of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) was added. The aqueous layer was separated, the organic layer was washed with a 10 % w/w sodium bicarbonate solution (600 mL, 2 vol) followed by a 5 % sodium chloride wash (600 mL, 2 vol). The organic layer was dried over sodium sulfate (60 g; 0.2 times w/w), filtered and the sodium sulfate was washed with MTBE (300 mL, 1 vol). The organic layer along with washings was distilled below 45 °C under reduced pressure until no more solvent was collected in the receiver. The distillation temperature was increased to 55 – 60 °C, maintained under vacuum for 3 – 4 h and cooled to 20 – 35 °C to afford 275 g (73.6 % yield, 72.71 % purity by HPLC) of 3-Br as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.63 (1H, d, 1.6 Hz, Ar-H), 8.07 – 8.01 (2H, m, 2 x Ar-H), 7.72 (1H, d, J = 6.8 Hz, Ar-H), 7.07 – 6.82 (1H, m, Ar-H), 6.81 – 6.80 (1H, m, Ar-H). 13C NMR: 185.60 (t, -C=0), 166.42 (dd, Ar-C-), 162.24 (dd, Ar-C),150.80 (Ar-C), 150.35 (Ar-C), 140.02 (Ar-C), 133.82 (Ar-C), 123.06 (Ar-C), 1122.33 (Ar-C), 118.44 (Ar-C), 114.07 (-CF2-), 122.07 (Ar-C), 105.09 (Ar-C).
B. Two-step Method via 2b-Br

2-Br (147.0 g) was dissolved in n-heptane (1.21 L) and transferred to a 5-L reactor equipped with overhead stirrer, thermocouple, condenser and addition funnel. Morpholine (202 ml) was added. The solution was heated to 60 °C and stirred overnight. The reaction was complete by HPLC analysis (0.2% 2-Br; 94.7% 2b-Br). The reaction was cooled to room temperature and 1.21 L of MTBE was added. The solution was cooled to ~4 °C and quenched by slow addition of 30% citric acid (563 ml) to maintain the internal temperature <15 °C. After stirring for one hour the layers were allowed to settle and were separated (Aq. pH=5). The organic layer was washed with 30% citric acid (322 ml) and 9% NaHC03 (322 ml, aq. pH 7+ after separation). The organic layer was concentrated on the rotary evaporator (Note 1) to 454 g (some precipitation started immediately and increased during concentration). After stirring at room temperature the suspension was filtered and the product cake was washed with n-heptane (200 ml). The solid was dried in a vacuum oven at room temperature to provide 129.2 g (77%) dense powder. The purity was 96.5% by HPLC analysis.To a 1-L flask equipped with overhead stirring, thermocouple, condenser and addition funnel was added magnesium turnings (14.65 g), THF (580 ml) and l-bromo-2,4-difluorobenzene (30.2 g, 0.39 equiv). The mixture was stirred until the reaction initiated and self-heating brought the reaction temperature to 44 °C. The temperature was controlled with a cooling bath as the remaining l-bromo-2,4-difluorobenzene (86.1 g, 1.11 equiv) was added over about 30 min. at an internal temperature of 35-40 °C. The reaction was stirred for 2 hours while gradually cooling to room temperature. The dark yellow solution was further cooled to 12 °C.During the Grignard formation, a jacketed 2-L flask equipped with overhead stirring, thermocouple, and addition funnel was charged with morpholine amide 2b-Br (129.0 g) and THF (645 ml). The mixture was stirred at room temperature until the solid dissolved, and then the solution was cooled to -8.7 °C. The Grignard solution was added via addition funnel over about 30 min. at a temperature of -5 to 0 °C. The reaction was stirred at 0 °C for 1 hour and endpointed by HPLC analysis. The reaction mixture was cooled to -5 °C and quenched by slow addition of 2N HC1 over 1 hour at <10 °C. The mixture was stirred for 0.5 h then the layers were allowed to settle and were separated. The aqueous layer was extracted with MTBE (280 ml). The combined organic layers were washed with 9% NaHCC>3 (263 g) and 20% NaCl (258 ml). The organic layer was concentrated on the rotary evaporator with THF rinses to transfer all the solution to the distillation flask. Additional THF (100 ml) and toluene (3 x 100 ml) were added and distilled to remove residual water from the product. After drying under vacuum, the residue was 159.8 g of a dark brown waxy solid (>theory). The purity was approximately 93% by HPLC analysis.EXAMPLE 3Preparation of 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (±ib-Br)

4-Br (200g, 1 eq) was added into methanolic ammonia (8.0 L; 40 vol; ammonia content: 15 – 20 % w/v) in an autoclave at 10 – 20 °C. The reaction mixture was gradually heated to 60 – 65 °C and at 3 – 4 kg/cm2 under sealed conditions for 10 – 12 h. The reaction progress was monitored by GC. After completion of the reaction, the reaction mixture was cooled to 20 – 30 °C and released the pressure gradually. The solvent was distilled under reduced pressure below 50 °C and the crude obtained was azeotroped with methanol (2 x 600 mL, 6 vol) followed by with isopropanol (600 mL, 2 vol) to afford 203 g (96.98 % yield, purity by HPLC: 94.04 %) of +4b-Br. EXAMPLE 4Preparation of3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (4b-Br or 2c-Br)

Amino alcohol ±4b-Br (150 g, 1 eq) was dissolved in an isopropanol /acetonitrile mixture (1.5L, 8:2 ratio, 10 vol) and Di-p-toluoyl-L-tartaric acid (L-DPTTA) (84.05 g, 0.55 eq) was added into the reactor at 20 – 30 °C. The reaction mixture was heated to 45 – 50 °C for 1 – 1.5 h (Note: The reaction mixture becomes clear and then became heterogeneous). The reaction mixture was gradually cooled to 20 – 30 °C and stirred for 16 – 18 h. The progress of the resolution was monitored by chiral HPLC analysis.After the completion of the resolution, the reaction mixture was gradually cooled to 20 – 35 °C. The reaction mixture was filtered and the filtered solid was washed with a mixture of acetonitrile and isopropanol (8:2 mixture, 300 mL, 2 vol) and dried to afford 75 g of the L- DPTTA salt (95.37 % ee). The L-DPTTA salt obtained was chirally enriched by suspending the salt in isopropanol /acetonitrile (8:2 mixture; 750 mL, 5 vol) at 45 – 50 °C for 24 – 48 h. The chiral enhancement was monitored by chiral HPLC; the solution was gradually cooled to 20 – 25 °C, filtered and washed with an isoporpanol /acetonitrile mixture (8:2 mixture; 1 vol). The purification process was repeated and after filtration, the salt resulted in chiral purity greater than 96 % ee. The filtered compound was dried under reduced pressure at 35 – 40 °C to afford 62 g of the enantio-enriched L-DPPTA salt with 97.12% ee as an off-white solid. The enantio-enriched L-DPTTA salt (50 g, 1 eq) was dissolved in methanol (150 mL, 3 vol) at 20 – 30 °C and a potassium carbonate solution (18.05 g K2CO3 in 150 mL water) was slowly added at 20 – 30 °C under stirring. The reaction mixture was maintained at this temperature for 2 – 3 h (pH of the solution at was maintained at 9). Water (600 mL, 12 vol) was added into the reaction mixture through an additional funnel and the reaction mixture was stirred for 2 – 3 h at 20 – 30 °C. The solids were filtered; washed with water (150 mL, 3 vol) and dried under vacuum at 40 – 45 °C to afford 26.5 g of amino alcohol 4b-Br or 4c-Br with 99.54 % chemical purity, 99.28 % ee as an off-white solid. (Water content of the chiral amino alcohol is below 0.10 % w/w).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.68 (1H, d, J = 2.0 Hz, Ar- H), 8.16 (1H, dd, J = 8.0 Hz, 2.0 Hz, Ar-H), 7.49 – 7.43 (1H, m, Ar-H), 7.40 (1H, d, J = 8 Hz, Ar-H), 7.16 – 7.11 (1H, m, Ar-H), 7.11 – 6.99 (1H, m, Ar-H), 3.39 – 3.36 (1H, m, -OCHAHB– ), 3.25 – 3.22 (1H, m, -OCHAHB-).13C NMR: 163.87 -158.52 (dd, 2 x Ar-C-), 150.88 (Ar-C), 149.16 (Ar-C), 139.21 (Ar-C), 132.39 (Ar-C), 124.49 (Ar-C), 122.17 (Ar-C), 121.87 (d, Ar- C), 119.91 (t, -CF2-), 110.68 (Ar-C), 103.97 (i, Ar-C), 77.41 (i,-C-OH), 44.17 (-CH2-NH2).EXAMPLE 5
Preparation of l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l- yl)propan-2-ol (l-6*-Br or l-7*-Br)

4b-Br or 4c-Br (20.0 g, 1 eq.) was added to acetic acid (50 mL, 2.5 vol) at 25 – 35 °C followed by the addition of anhydrous sodium acetate (4.32 g, 1 eq), trimethyl orthoformate (15.08 g, 2.7 eq). The reaction mixture was stirred for 15 – 20 min at this temperature and trimethylsilyl azide (12.74 g, 2.1 eq) was added to the reaction mixture (Chilled water was circulated through the condenser to minimize the loss of trimethylsilyl azide from the reaction mixture by evaporation). The reaction mixture was then heated to 70 – 75 °C and maintained at this temperature for 2 -3 h. The reaction progress was monitored by HPLC. Once the reaction was complete, the reaction mixture was cooled to 25 – 35 °C and water (200 mL, 10 vol) was added. The reaction mixture was extracted with ethyl acetate (400 mL, 20 vol) and the aqueous layer was back extracted with ethyl acetate (100 mL, 5 vol). The combined organic layers were washed with 10 % potassium carbonate solution (3 x 200 mL; 3 x 10 vol) followed by a 10 % NaCl wash (1 x 200 mL, 10 vol). The organic layer was distilled under reduced pressure below 45 °C. The crude obtained was azeotroped with heptanes (3 x 200 mL) to get 21.5g (94 % yield, 99.26 5 purity) of tetrazole 1-6* or 1-7* compound as pale brown solid (low melting solid).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz NMR instrument): 9.13 (1H, Ar-H), 8.74 (1H, Ar-H), 8.22 – 8.20 (1H, m, Ar-H), 7.44 (1H, d, J = 7.2 Hz, Ar-H), 7.29 (1H„Ar-H), 7.23 – 7.17 (1H, m, Ar-H), 6.92 – 6.88 (1H, Ar-H), 5.61 (1H, d, J = 1 1.2 Hz, – OCHAHB-), 5.08 (1H, d, J = 5.6 Hz, -OCHAHB-).13C NMR: 163.67 -161.59 (dd, Ar-C-), 160.60 – 158.50 (dd, Ar-C-), 149.65 (Ar-C), 144.99 (Ar-C), 139.75 (Ar-C), 131.65 (Ar-C), 124.26 (Ar-C), 122.32 (d, Ar-C), 119.16 (t, -CF2-), 118.70 (d, Ar-C), 1 11.05 (d, Ar-C) 104.29 (t, Ar-C), 76.79 (i,-C-OH), 59.72 (Ar-C), 50.23 (-OCH2N-). EXAMPLE 6Preparation of 2-(2,4-difluorophenyl)-l , 1 -difluoro-3-( 1 H-tetrazol-1 -yl)-l -(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)A. Preparation of 1 or la via l-6*-Br or l-7*-Br

Synthesis of 4,4,5, 5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane Potassium carbonate (59.7 g, 2.2 eq.) was added to a slurry of DMF (190 mL, 3.8 Vol.), 4- Bromo phenol (37.4g, 1.1 eq.) and 2,2,2-trifluroethyl tosylate (50.0 g, 1.0 eq.) at 20 – 35 °C under an inert atmosphere. The reaction mixture was heated to 115 – 120 °C and maintained at this temperature for 15 – 18 h. The reaction progress was monitored by GC. The reaction mixture was then cooled to 20 – 35 °C, toluene (200 mL, 4.0 vol.) and water (365 mL, 7. 3 vol.) were added at the same temperature, stirred for 10 – 15 minutes and separated the layers. The aqueous layer was extracted with toluene (200 mL, 4.0 vol.). The organic layers were combined and washed with a 2M sodium hydroxide solution (175 mL, 3.5 vol.) followed by a 20 % sodium chloride solution (175 mL, 3.5 vol.). The organic layer was then dried over anhydrous sodium sulfate and filtered. The toluene layer was transferred into clean reactor, spurged with argon gas for not less than 1 h. Bis(Pinacolato) diborane (47 g, 1.1 eq.), potassium acetate (49.6 g, 3.0 eq.) and 1,4-dioxane (430 mL, 10 vol.) were added at 20 -35 °C, and spurged the reaction mixture with argon gas for at least 1 h. Pd(dppf)Cl2 (6.88 g, 0.05eq) was added to the reaction mixture and continued the argon spurging for 10 – 15 minutes. The reaction mixture temperature was increased to 70 – 75 °C, maintained the temperature under argon atmosphere for 15 – 35 h and monitored the reaction progress by GC. The reaction mixture was cooled to 20 – 35 °C, filtered the reaction mixture through a Celite pad, and washed with ethyl acetate (86 mL, 2 vol.). The filtrate was washed with water (430 mL, 10 vol.). The aqueous layer was extracted with ethyl acetate (258 mL, 6 vol.) and washed the combined organic layers with a 10 % sodium chloride solution (215 mL, 5 vol.). The organic layer was dried over anhydrous sodium sulfate (43g, 1 time w/w), filtered and concentrated under reduced pressure below 45 °C to afford crude 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g; 71 % yield with the purity of 85.18 % by GC). The crude 4,4,5,5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g, 1 eq.) was dissolved in 10 % ethyl acetate – n-Heptane (455 mL, 7 vol.) and stirred for 30 – 50 minutes at 20 – 35 °C. The solution was filtered through a Celite bed and washed with 10 % ethyl acetate in n-Heptane (195 mL, 3 vol.). The filtrate and washings were pooled together, concentrated under vacuum below 45 °C to afford 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a thick syrup (45.5 g; 70 % recovery). This was then dissolved in 3 % ethyl acetate-n-heptane (4 vol.) and adsorbed on 100 – 200 M silica gel (2 times), eluted through silica (4 times) using 3 % ethyl acetate – n- heptane. The product rich fractions were pooled together and concentrated under vacuum. The column purified fractions (> 85 % pure) were transferred into a round bottom flask equipped with a distillation set-up. The compound was distilled under high vacuum below 180 °C and collected into multiple fractions. The purity of fractions was analyzed by GC (should be > 98 % with single max impurity < 1.0 %). The less pure fractions (> 85 % and < 98 % pure fraction) were pooled together and the distillation was repeated to get 19g (32% yield) of 4,4,5, 5-tetramethyl-2-(4- (2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):7.64 (2H, d, 6.8 Hz), 7.06 (2H, d, J = 6.4 Hz), 4.79 (2H, q, J = 6.8 Hz), 1.28 (12H, s).13C NMR: 159.46 (Ar-C-O-), 136.24 (2 x Ar-C-), 127.77 – 120.9 (q, -CF3), 122.0 (Ar-C-B), 114.22 (2 x Ar-C-), 64.75 (q, J = 27.5 Hz).Synthesis of 2-(2.4-difluorophenyl)-l.l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)l-6*-Br or l-7*-Br (14 g, 0.03 mol, 1 eq) was added to tetrahydrofuran (168 mL, 12 vol) at 25 – 35 °C and the resulting solution was heated to 40 – 45 °C. The reaction mixture was maintained at this temperature for 20 – 30 min under argon bubbling. Sodium carbonate (8.59 g, 0.08 mol, 2.5 eq) and water (21 mL, 1.5 vol) were added into the reaction mixture and the bubbling of argon was continued for another 20 – 30 min. 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (10.76 g, 1.1 eq) dissolved in tetrahydrofuran (42 mL, 3 vol) was added into the reaction mixture and argon bubbling was continued for 20 – 30 min. Pd(dppf)Cl2 (2.65 g, 0.1 eq) was added to the reaction mixture under argon bubbling and stirred for 20 – 30 min (Reaction mixture turned into dark red color). The reaction mixture was heated to 65 – 70 °C and maintained at this temperature for 3 – 4 h. The reaction progress was monitored by HPLC. The reaction mixture was cooled to 40 – 45 °C and the solvent was distilled under reduced pressure. Toluene (350 mL, 25 vol.) was added to the reaction mixture and stirred for 10 – 15 min followed by the addition of water (140 mL, 10 vol). The reaction mixture was filtered through Hyflo (42 g, 3 times), the layers were separated and the organic layer was washed with water (70 mL, 5 vol) and a 20 % w/w sodium chloride solution (140 mL, 10 vol). The organic layer was treated with charcoal (5.6 g, 0.4 times, neutral chalrcoal), filtered through Hyflo. (lS)-lO-Camphor sulfonic acid (7.2 g, 1 eq.) was added to the toluene layer and the resulting mixture was heated to 70 – 75 °C for 2 – 3 h. The reaction mixture was gradually cooled to 25 – 35 °C and stirred for 1 – 2 h. The solids were filtered, washed with toluene (2 x 5 vol.) and then dried under vacuum below 45 °C to afford 18.0 g of an off white solid. The solids (13.5 g, 1 eq.) were suspended in toluene (135 mL, 10 vol) and neutralized by adding 1M NaOH solution (1.48 vol, 1.1 eq) at 25 – 35 °C and stirred for 20 – 30 min. Water (67.5 mL, 5 vol) was added to the reaction mixture and stirred for 10 – 15 min, and then the layers were separated. The organic layer was washed with water (67.5 mL, 5 vol) to remove the traces of CSA. The toluene was removed under reduced pressure below 45 °C to afford crude 1 or la. Traces of toluene were removed by azeotroping with ethanol (3 x 10 vol), after which light brown solid of crude 1 or la (7.5 g, 80% yield) was obtained.The crude 1 or la (5 g) was dissolved in ethanol (90 mL, 18 vol.) at 20 – 35 °C, and heated to 40 – 45 °C. Water (14 vol) was added to the solution at 40 – 45 °C, the solution was maintained at this temperature for 30 – 45 min and then gradually cooled to 20 – 35 °C. The resulting suspension was continued to stir for 16 – 18 h at 20 – 35 °C, an additional amount of water (4 vol.) was added and the stirring continued for 3 – 4 h. The solids were filtered to afford 4.0 g (80% recovery) of 1 or la (HPLC purity >98%) as an off-white solid.1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):9.15 (1H, s, Ar-H), 8.93 (1H, d, J = 0.8 Hz, Ar-H), .8.22 – 8.20 (1H, m, Ar-H), 7.80 (2H, d, J = 6.8 Hz, Ar-H), 7.52 (1H, d, J = 6.8 Hz, Ar-H), 7.29 (1H, d,J = 3.2Hz, Ar-H), 7.27 – 7.21 (1H, m, Ar-H), 7.23 – 7.21 (2H, d, J = 6.8 Hz, Ar-H), 7.19 (1H, d, J = 6.8 Hz, Ar-H), 6.93 – 6.89 (1H, m, Ar-H), 5.68 (1H, / = 12 Hz, -CHAHB), 5.12 (2H, d, J = 11.6 Hz, -CHAHB), 4.85 (2H, q, J = 1.6 Hz).13C NMR: 163.93 – 158.33 (m, 2 x Ar-C), 157.56 (Ar-C), 149.32 (i, Ar-C), 146.40 (Ar-C), 145.02 (Ar-C), 136.20 (Ar-C), 134.26 (2 x Ar-C), 131.88 – 131.74 (m, AR-C), 129.72 (Ar-C), 128.47 (2 x Ar-C), 123.97 (q, -CF2-), 122.41 (Ar-C), 119.30 (-CF3), 118.99 (Ar-C), 115.65 (2 x Ar-C), 110.99 (d, Ar-C), 104.22 (i, Ar-C), 77.41 – 76.80 (m, Ar-C), 64.72 (q, -OCH2-CF3), 50.54 (-CH2-N-).B. Preparation of 1 or la via 4b-Br or 4c-Br


Synthesis of 3-amino-2-(2.4-difluorophenyl)-l.l-difluoro-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (8a or 8b)Potassium carbonate (30.4 g) and water (53.3 g) were charged to a 1-L flask equipped with overhead stirring, thermocouple, and nitrogen/vacuum inlet valve, and stirred until dissolved. The boronic acid (19.37 g), a solution of 4b-Br or 4c-Br in 2-butanol (103.5 g, 27.8 g theoretical 4b-Br or 4c-Br)) and 2-BuOH (147.1 g) were added and stirred to form a clear mixture. The flask was evacuated and refilled with nitrogen 3 times. Pd(d f)2Cl2 (0.30 g) was added and stirred to form a light orange solution. The flask was evacuated and refilled with nitrogen 4 times. The mixture was heated to 85 °C and stirred overnight and endpointed by HPLC analysis. The reaction mixture was cooled to 60 °C and the layers were allowed to settle. The aqueous layer was separated. The organic layer was washed with 5% NaCl solution (5 x 100 ml) at 30-40 °C. The organic layer was filtered and transferred to a clean flask with rinses of 2-BuOH. The combined solution was 309.7 g, water content 13.6 wt% by KF analysis. The solution was diluted with 2-BuOH (189 g) and water (10 g). Theoretically the solution contained 34.8 g product, 522 ml (15 volumes) of 2-BuOH, and 52.2 ml (1.5 volumes) of water. L-Tartaric acid (13.25 g) was added and the mixture was heated to a target temperature of 70-75 °C. During the heat-up, a thick suspension formed. After about 15 minutes at 70-72 °C the suspension became fluid and easily stirred. The suspension was cooled at a rate of 10 °C/hour to 25 °C then stirred at 25 °C for about 10 hours. The product was collected on a vacuum filter and washed with 10:1 (v/v) 2-BuOH/water (50 ml) and 2- butanol (40 ml). The salt was dried in a vacuum oven at 60 °C with a nitrogen purge for 2 days. The yield was 40.08 g of 8a or 8b as a fluffy, grayish-white solid. The water content was 0.13 wt% by KF analysis. The yield was 87.3% with an HPLC purity of 99.48%. Synthesis of 2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)To a 350 ml pressure bottle were charged acetic acid (73 ml), 8a or 8b (34.8 g), sodium acetate (4.58 g) and trimethylorthoformate (16.0 g). The mixture was stirred for 18 min. at room temperature until a uniform suspension was obtained. Azidotrimethylsilane (8.88 g) was added and the bottle was sealed. The bottle was immersed in an oil bath and magnetically stirred. The oil bath was at 52 °C initially, and was warmed to 62-64 °C over about ½ hour. The suspension was stirred at 62-64 °C overnight. After 20.5 hours the suspension was cooled to room temperature and sampled. The reaction was complete by HPLC analysis. The reaction was combined with three other reactions that used the same raw material lots and general procedure (total of 3.0 g additional starting material). The combined reactions were diluted with ethyl acetate (370 ml) and water (368 ml) and stirred for about ½ hour at room temperature. The layers were settled and separated. The organic layer was washed with 10% K2C03 solution (370 ml/ 397 g) and 20% NaCl solution (370 ml/ 424 g). The organic layer (319 g) was concentrated, diluted with ethanol (202 g) and filtered, rinsed with ethanol (83 g). The combined filtrate was concentrated to 74 g of amber solution.The crude 1 or la solution in ethanol (74 g solution, containing theoretically 31.9 g 1 or la) was transferred to a 2-L flask equipped with overhead stirring, thermocouple, and addition funnel. Ethanol (335 g) was added including that used to complete the transfer of the 1 or la solution. The solution was heated to nominally 50 °C and water (392 g) was added over 12 minutes. The resulting hazy solution was seeded with 1 or la crystals and stirred at 50 °C. After about ½ hour the mixture was allowed to cool to 40 °C over about ½ hour during which time crystallization started. Some darker colored chunky solid separated out from the main suspension. The pH of the crystallizing mixture was adjusted from 4.5 to 6 using 41% KOH (1.7 g). After about 1 hour a good suspension had formed. Additional water (191 g) was added slowly over ½ hour. The suspension was heated to 50 °C and cooled at 5 °C/min to room temperature. After stirring overnight the suspension was cooled in a water bath to 16 °C and filtered after 1 hour. The wet cake was washed with 55:45 (v/v) water/ethanol (2 x 50 ml) and air-dried on the vacuum filter funnel overnight. Further drying at 40 °C in a vacuum oven with a nitrogen bleed resulted in no additional weight loss. The yield was 30.2 g of off-white fine powder plus some darker granular material. By in-process HPLC analysis there was no difference in the chemical purity of the darker and lighter materials. The purity was 99.4%. The water content was 2.16 wt% by KF analysis. The residual ethanol was 1.7 wt% estimated by ‘Ft NMR analysis. The corrected yield was 29.0 g, 91.0% overall yield for tetrazole formation and crystallization. The melting point was 65 °C by DSC analysis.
FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)
– Approval of VIVJOA™ marks a significant therapeutic advancement for reducing the incidence of RVVC, a condition with substantial unmet need, in permanently infertile and postmenopausal women
– VIVJOA™ is the first FDA approval in Mycovia’s pipeline of novel treatments for fungal infections
– U.S. commercial launch of VIVJOA™ expected in Q2
April 28, 2022 07:55 AM Eastern Daylight Time
DURHAM, N.C.–(BUSINESS WIRE)–The U.S. Food and Drug Administration (FDA) approved VIVJOA™ (oteseconazole capsules), an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication for this condition and provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. VIVJOA is the first FDA-approved product for Mycovia Pharmaceuticals, Inc. (Mycovia), an emerging biopharmaceutical company dedicated to recognizing and empowering those living with unmet medical needs by developing novel therapies.
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans”Tweet this
RVVC, also known as chronic yeast infection, is defined by the Centers for Disease Control and Prevention (CDC) as three or more symptomatic acute episodes of yeast infection in 12 months. RVVC is a distinct condition from vulvovaginal candidiasis (VVC), and until now, there have been no FDA-approved medications specifically indicated for it. Nearly 75% of all adult women will have at least one yeast infection in their lifetime, with approximately half experiencing a recurrence. Of those women, up to 9% develop RVVC.
“After nearly two decades of living with chronic yeast infection and feeling like there was no hope from the itchiness, irritation and constant dread of when the next yeast infection would return, I was overjoyed to even be a part of this clinical trial,” said Leslie Ivey, RVVC patient and clinical trial participant. “It is gratifying to see RVVC finally get the attention it deserves.”
Symptoms of RVVC include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
VIVJOA’s FDA approval is based upon the positive results from three Phase 3 trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries. In the two global VIOLET studies, 93.3% and 96.1% of women with RVVC who received VIVJOA did not have a recurrence for the 48-week maintenance period compared to 57.2% and 60.6% of patients who received placebo (p <0.001). In the ultraVIOLET study, 89.7% of women with RVVC who received VIVJOA cleared their initial yeast infection and did not have a recurrence for the 50-week maintenance period compared to 57.1% of those who received fluconazole followed by placebo (p <0.001). The most common side effects reported in Phase 3 clinical studies were headache (7.4%) and nausea (3.6%). VIVJOA is contraindicated in those with a hypersensitivity to oteseconazole, and based on data from rat studies, also in females who are of reproductive potential, pregnant, or lactating. Please see additional Important Safety Information below.
Patrick Jordan, CEO of Mycovia Pharmaceuticals and Partner at NovaQuest Capital Management, stated, “We celebrate this important milestone for Mycovia, as VIVJOA is the first antifungal in our pipeline to obtain FDA approval and achieves our goal to fulfill a previously unmet medical need among women suffering from RVVC. We are honored to lead this advancement in women’s health.”
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans,” Jordan continued. “As we enter a new chapter of our history as a commercial biopharmaceutical company, we will continue driving our mission forward to develop novel therapies for overlooked conditions.”
Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA granted oteseconazole Qualified Infectious Disease Product and Fast Track designations.
“A medicine with VIVJOA’s sustained efficacy combined with the clinical safety profile has been long needed, as until now, physicians and their patients have had no FDA-approved medications for RVVC,” stated Stephen Brand, Ph.D., Chief Development Officer of Mycovia. “We are excited to be the first to offer a medication designed specifically for RVVC, a challenging and chronic condition that is expected to increase in prevalence over the next decade.”
Mycovia is planning its commercial launch of VIVJOA™ in the second quarter of 2022.
About Recurrent Vulvovaginal Candidiasis
RVVC is a debilitating, chronic infectious condition that affects 138 million women worldwide each year. RVVC, also known as chronic yeast infection, is a distinct condition from vulvovaginal candidiasis (VVC) and defined as three or more symptomatic acute episodes of yeast infection in 12 months. Primary symptoms include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
About VIVJOA™
VIVJOA™ (oteseconazole) is an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication that provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA approved VIVJOA based upon the positive results from three Phase 3 clinical trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries.
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215888s000lbl.pdf
- ^ “Vivjoa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 27 April 2022.
- ^ Jump up to:a b “FDA Approves Mycovia Pharmaceuticals’ VIVJOA (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)” (Press release). Mycovia Pharmaceuticals. 28 April 2022. Retrieved 28 April 2022 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Sobel JD, Nyirjesy P (December 2021). “Oteseconazole: an advance in treatment of recurrent vulvovaginal candidiasis”. Future Microbiology. 16: 1453–1461. doi:10.2217/fmb-2021-0173. PMID 34783586.
External links
- “Oteseconazole”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03562156 for “A Study of Oral Oteseconazole for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03561701 for “A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03840616 for “Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients With Recurrent Yeast Infections (ultraVIOLET)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Vivjoa |
| Other names | VT-1161 |
| License data | US DailyMed: Oteseconazole |
| Routes of administration | By mouth |
| Drug class | Antifungal |
| ATC code | J02AC06 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1340593-59-0 |
| PubChem CID | 77050711 |
| DrugBank | DB13055 |
| ChemSpider | 52083215 |
| UNII | VHH774W97N |
| KEGG | D11785 |
| ChEBI | CHEBI:188153 |
| ChEMBL | ChEMBL3311228 |
| ECHA InfoCard | 100.277.989 |
| Chemical and physical data | |
| Formula | C23H16F7N5O2 |
| Molar mass | 527.403 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////OTESECONAZOLE, vt 1161, fungal infection, Candida albicans infection, onychomycosis, PHASE 3,



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
C1=CC(=CC=C1C2=CN=C(C=C2)C(C(CN3C=NN=N3)(C4=C(C=C(C=C4)F)F)O)(F)F)OCC(F)(F)F
NEW DRUG APPROVALS
ONE TIME
$10.00
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}