
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

ELETRIPTAN

ELETRIPTAN
Eletriptan, UK-116044-04(HBr salt), UK-116044, Relpax
143322-58-1 CAS OF FREE BASE
143577-61-1 (hemisuccinate), 179041-30-6 (monofumarate), 177834-92-3 (monoHBr salt), 180637-87-0 (monosuccinate)
| (R)-3-[(-1-methylpyrrolidin-2-yl)methyl]-5-(2-phenylsulfonylethyl)- 1H-indole |
Eletriptan hydrobromide is a selective serotonin (5-HT1) agonist, used for the acute treatment of the headache phase of migraine attacks.
RELPAX (eletriptan hydrobromide) tablets contain eletriptan hydrobromide, which is a selective 5-hydroxytryptamine 1B/1D (5-HT1B/1D) receptor agonist. Eletriptan hydrobromide is chemically designated as (R)-3-[(1-Methyl-2-pyrrolidinyl)methyl]-5-[2-(phenylsulfonyl)ethyl]-1H-indole monohydrobromide, and it has the following chemical structure:
 |
The empirical formula is C22H26N2O2S . HBr, representing a molecular weight of 462.43. Eletriptan hydrobromide is a white to light pale colored powder that is readily soluble in water.
Each RELPAX Tablet for oral administration contains 24.2 or 48.5 mg of eletriptan hydrobromide equivalent to 20 mg or 40 mg of eletriptan, respectively. Each tablet also contains the inactive ingredients microcrystalline cellulose NF, lactose monohydrate NF, croscarmellose sodium NF, magnesium stearate NF, titanium dioxide USP, hypromellose, triacetin USP and FD&C Yellow No. 6 aluminum lake.
|
|
|||||||||||||||||||||
| Patents |
|
EP 0592438; JP 1993507288; JP 1997003063; US 5545644; WO 9206973, EP 0776323; JP 1997512283; US 6110940; WO 9606842, EP 1088817
U.S. Pat. No. 5,545,644A1 describes a synthetic process for Eletriptan. 5-Bromoindole was acylated at the 3-position by reacting the magnesium salt of 5-bromoindole. This process results in a dimer formation in the final Pd/C reduction stage which poses problems in purification which further leads to decrease in yields.
U.S. Pat. No. 7,288,662B2 discloses methods to circumvent the problems associated with dimer formation described in U.S. Pat. No. 5,545,644A1. The indole-nitrogen was acetylated prior to hydrogenation and later deacetylated to give pure Eletriptan. However, this process introduced two additional steps into the synthesis which is time consuming and subsequently costly.
WO2005/103035A1 discloses Eletriptan synthesis by a Fischer Indole process. However, enantiomeric purity of the finished product depends on the purity of an acetal intermediate which might require asymmetric synthesis or optical resolution. Eletriptan obtained in the reported procedure had about 94% enantiomeric excess.
Eletriptan (trade name Relpax, used in the form of eletriptan hydrobromide) is a second generation triptandrug intended for treatment of migraineheadaches. It is used as an abortive medication, blocking a migraine attack which is already in progress. Eletriptan is marketed and manufactured by Pfizer Inc. It is sold in the US and Canada under the brand name Relpax, and in several other countries under the brand name Relert.
Eletriptan was approved by the U.S. Food and Drug Administration (FDA) on December 26, 2002, for the acute treatment of migraine with or without aura in adults.[1] It is available only by prescription in the United States and Canada. It is not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine. It is available in 20 mg, 40 mg and 80 mg strengths.
Eletriptan is covered by U.S. Patent no. 5545644[1][2] and U.S. Patent no. 6110940;[1][3] the FDA lists the patents as scheduled for expiration on December 26, 2016, and August 29, 2017, respectively.[1]
Eletriptan is believed to reduce swelling of the blood vessels surrounding the brain. This swelling is associated with the head pain of a migraine attack. Eletriptan blocks the release of substances from nerve endings that cause more pain and other symptoms like nausea, and sensitivity to light and sound. It is thought that these actions contribute to relief of symptoms by eletriptan.
Eletriptan is a serotonin agonist. Specifically, it is a selective 5-hydroxytryptamine 1B/1D (5-HT1B) receptor agonist.
Eletriptan binds with high affinity to the 5-HT1B, 1D, 1F] receptors.
It has a modest affinity to the 5-HT[1A, 1E, 2B, 7] receptors.
And little to no affinity at the 5-HT[2A, 2C, 3, 4, 5A, 6] receptors.
Eletriptan has no significant affinity or pharmacological activity at adrenergic alpha1, alpha2, or beta; dopaminergic D1 or D2; muscarinic; or opioid receptors. Eletriptan could be efficiently co-administrated with nitric oxide synthase (NOS’s) inhibitors for the treatment of NOS-dependent diseases (US patent US 2007/0254940)
Two theories have been proposed to explain the efficacy of 5-HT receptor agonists in migraine. One theory suggests that activation of 5-HT1 receptors located on intracranial blood vessels, including those on the arteriovenous anastomoses, leads to vasoconstriction, which is correlated with the relief of migraine headache. The other hypothesis suggests that activation of 5-HT1 receptors on sensory nerve endings in the trigeminal system results in the inhibition of pro-inflammatory neuropeptide release.
Common side effects include hypertension, tachycardia, headache, dizzyness, and symptoms similar to angina pectoris. Severe allergic reactions are rare.[4]
Eletriptan is contraindicated in patients with various diseases of the heart and circulatory system, such as angina pectoris, severe hypertension, and heart failure, as well as in patients that have had a stroke or heart attack. It is also contraindicated in severe renal or hepatic impairment.[4]
The drug has a relatively low potential for interactions. Notably, it is unlikely to interact to a relevant extent with beta blockers, tricyclic antidepressants and SSRI type antidepressants. Strong inhibitors of the liver enzyme CYP3A4, such as erythromycin and ketoconazole, significantly increase blood plasma concentrations and half life of eletriptan. Ergot alkaloids add to the drug’s hypertensive effect.[4]
- Merck Index: 3-[[(2R)-1-Methyl-2-pyrrolidinyl]methyl]-5-[2-(phenylsulfonyl)ethyl]-1H-indole
- 5-[2-(benzenesulfonyl)ethyl]-3-(1-methylpyrrolidin-2(R)-ylmethyl)-1H-indole
- (R)-5-[2-(phenylsulfonyl)ethyl]-3-[(1-methyl-2-pyrrolidinyl)methyl]-1H-indole
- FDA AccessData entry for Eletriptan Hydrobromide, accessed March 10, 2010.
- U.S. Patent no. 5545644, John E. Macor & Martin J. Wythes, Indole Derivatives, August 13, 1996.
- U.S. Patent no. 6110940, Valerie Denise Harding, et al., Salts of an anti-migraine indole derivative, August 29, 2000.
- Jasek, W, ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 6984–8. ISBN 978-3-85200-181-4.
- FDA label (December 2002)
- Physicians’ Desk Reference entry for Relpax
- Medline Plus Drug Information for Eletriptan
- Pfizer Relpax site
3-{[(2R)-1-methylpyrrolidin-2-yl]methyl}-5-[2-(phenylsulfonyl)ethyl]-1H-indole or Eletriptan, currently available in the market as a hydrobromide salt, is an agonist of the 5-hydroxytryptamine (5-HT1B/1D) receptor and it is used for treating migraine.
Various processes of synthesis of such molecule are known, but the one generally used is the synthesis shown in the diagram of FIG. 1, which provides for a Heck reaction (step 4 or 4b) between 5-bromo-3-{[(2R)-1-methylpyrrolidin-2-yl]methyl}-1H-indole and phenyl vinyl sulfone to obtain the 1-(3-{[(2R)-1-methylpyrrolidin-2-yl]methyl}-5-[(E)-2-(phenylsulfonyl)ethenyl]-1H-indole-1-yl)ethanone intermediate.
This reaction uses a palladium-based catalyst which is very sensitive to the impurities present in the reaction environment. It is thus essential that the 5-bromo-3{[(2R)-1-methylpyrrolidin-2-yl]methyl}-1H-indole intermediate be thoroughly purified before being reacted with phenyl vinyl sulfone.
In prior art documents (EP 0 592 438, U.S. Pat. No. 5,545,644 and U.S. Pat. No. 6,100,291) purification of the 5-bromo-3-{[(2R)-1-methylpyrrolidin-2-yl]methyl}-1H-indole intermediate is performed by means of chromatographic column, a process almost exclusively implementable in a laboratory or at a high cost in any case with long processing times alongside being ecologically unadvisable due to the large amount of solvents used.
Furthermore, it is known that the crystallisation of 5-bromo-3-{[(2R)-1-methylpyrrolidin-2-yl]methyl}-1H-indole intermediate (WO 2008/150500 and U.S. Pat. No. 5,545,644) provides a purified intermediate with assay not exceeding 98% (established through the HPLC analysis).
US5545644A1 describes a synthetic process for Eletriptan. 5-Bromoindole was acylated at the 3-position by reacting the magnesium salt of 5-bromoindole. This process results in a dimer formation in the final Pd/C reduction stage which poses problems in purification which further leads to decrease in yields.
US7288662B2 discloses methods to circumvent the problems associated with dimer formation described in US5545644A1. The indole-nitrogen was acetylated prior to hydrogenation and later deacetylated to give pure Eletriptan. However, this process introduced two additional steps into the synthesis which is time consuming and subsequently costly. WO2005/103035A1 discloses Eletriptan synthesis by a Fischer Indole process. However, enantiomeric purity of the finished product depends on the purity of an acetal intermediate which might require asymmetric synthesis or optical resolution. Eletriptan obtained in the reported procedure had about 94% enantiomeric excess.
Eletriptan and intermediates thereof, including 5-bromo-3-[(i?)-l-methyl- pyrrolidin-2-ylmethyl]-lH-indole (“BIP”) are described in US 5,545,644. Also disclosed is the synthesis of ELT, which is illustrated by the following scheme:

In the described process, intermediate I, BIP, is obtained by reacting intermediate II with lithium aluminium hydride (“LAH”). LAH spontaneously reacts with water, including atmospheric humidity, and the pure material is pyrophoric. The LAH is known as very unstable, and air-exposed samples are almost always contaminated with aluminium metal and or a mixture of lithium hydroxide and aluminium hydroxide, thus affecting the reactivity of the LAH powder. This leads to the use of a large excess of reagent in order to obtain moderate conversion. Furthermore, the described process requires heating to reflux for a long period of time (39 hours in total, according to example 29 in patent US 5,545,644) followed by a time consuming recovery process. The recovery process consists of diluting of the reaction mixture with ethyl acetate, filtering through cellulose filtration bar, as described in patent US 5,545,644 example 27, and purifying the obtained oily like residue by silica gel chromatography, wherein, dichloromethane, ethanol and concentrated aqueous ammonia are used as a mobile phase. This process provides BIP, which is then converted to ELT.
Anhydrous alpha-and beta-hydrobromide salt forms of eietriptan are disclosed in WO-A-96/06842.
………………..

5-Bromoindole under Heck reaction conditions is coupled with phenyl vinyl sulfone followed by acylation with Cbz-Proline acid chloride to obtain a compound of Formula IV which on reduction in presence of a hydride agent provide Eletriptan.
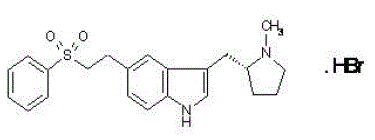
1H NMR CDCI3 δ= 8.10 (bs, NH), 7.92-7.99 5 (m, 2H), 7.62-7.69 (m, 1H), 7.53-7.61 (m, 2H), 7.30 (s, 1H), 7.22 (d, 1H), 7.03 (s, 1H), 6.93 (dd, 1 H), 3.38-3.45 (m, 2H), 3.09-3.21 (m, 4H), 2.45-2.55 (m, 2H), 2.45 (s, 3H), 2.20-2.30 (m, 1H), 1.50-1.90 (m, 4H).
ESI Mass (M+H) 383.69
………..
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
http://beilstein-journals.org/bjoc/single/printArticle.htm?publicId=1860-5397-7-57
Eletriptan (87, Relpax) is yet another indole-containing antimigraine drug. A process route for the synthesis of eletriptan published by Pfizer starts from a preformed bromo-indole 88 [28] (Scheme 20). In order to perform the acylation of the indole ring on larger scale, ethylmagnesium bromide and the corresponding acid chloride 89 are added concurrently from two different sides of the reactor to stop these reagents reacting with each other. This method of adding the reagents circumvents the necessity to isolate the magnesium salt of the indole and increases the yield from 50 to 82%. The carbonyl group of the proline side chain is then reduced simultaneously with the complete reduction of the Cbz-group to a methyl group with lithium aluminium hydride. Finally, the sulfonate side chain is introduced via a Heck-type coupling similar to that of naratriptan (Scheme 15), followed by hydrogenation of the double bond to afford eletriptan (Scheme 20).
![[1860-5397-7-57-i20]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i20.png)
A rather ingenious Mitsunobu coupling reaction has been used to create a highly functionalised substrate 96 for an intramolecular Heck reaction resulting in a very short and succinct synthesis of eletriptan and related analogues 97 [29] (Scheme 21).
![[1860-5397-7-57-i21]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i21.png)
Interestingly, it was found that the most obvious approach, the direct Fischer indole synthesis, to prepare the core of eletriptan as shown in Scheme 22 is not successful [30]. This is believed to be due to the instability of the phenyl hydrazine species 98 under the relatively harsh reaction conditions required to promote the cyclisation.
![[1860-5397-7-57-i22]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i22.png)
However, this problem could be avoided by using an acid-labile oxalate protected hydrazine 104 as depicted in Scheme 23. The yield of this step can be further improved up to 84% if the corresponding calcium oxalate is used.
![[1860-5397-7-57-i23]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i23.png)
- Macor, J. E.; Wythes, M. J. Indole Derivatives. U.S. Patent 5,545,644, Aug 13, 1996.
- Perkins, J. F. Process for the Preparation of 3-Acylindoles. Eur. Patent 1088817A2, April 4, 2001.
- Ashcroft, C. P. Modified Fischer Indole Synthesis for Eletriptan. WO Patent 2005/103035, Nov 3, 2005.
- Bischler, A. Chem. Ber. 1892, 25, 2860–2879. doi:10.1002/cber.189202502123
…………
……………..
Synthesis of compounds related to the anti-migraine drug eletriptan hydrobromide
2Engineering Chemistry Department, AU College of Engineering, Andhra University, Visakhapatnam-530003, Andhra Pradesh, India

Synthetic route of eletriptan hydrobromide. Reagents and conditions: (i) Acetic anhydride, TEA, DMF, 90–100 °C; (ii) palladium acetate, tri-(o-tolyl)phosphine, TEA, DMF, 90–100 °C; (iii) methanol, K2CO3, acetonitrile, H2O, 5–10 °C; (iv) palladium on carbon, acetone, H2O, aqueous hydrobromic acid, IPA, 25–30 °C.
………….
http://pubs.acs.org/doi/full/10.1021/op100251q

aReagents and conditions: (a) EtMgBr, Et2O. (b) 3, DCM, 50% from 1. (c) LiAlH4, THF, 72%. (d) Ac2O, TEA, DMF. (e) Phenyl vinyl sulfone (PVS), Pd(OAc)2, P(°Tol)3, TEA, DMF, 80% from 5. (f) H2, Pd/C, MeSO3H, acetone, 95%. (g) K2CO3, MeOH, 92%. (h) HBr, acetone 73%.
1H NMR (CDCl3): δ = 1.51−1.85 (m, 4H), 2.22−2.28 (m, 1H), 2.43−2.49 (m, 4H), 2.56−2.62 (m, 1H), 3.11−3.18 (m, 4H), 3.42−3.46 (m, 2H), 6.91−6.93 (s, 1H), 7.01 (s, 1H), 7.23−7.27 (d, 1H), 7.31 (s, 1H), 7.56−7.60 (m, 2H), 7.65−7.68 (m, 1H), 7.96−7.98 (d, 2H), 8.14 (s, 1H); LC/MS: Rt = 2.30 min; m/z 383 [MH]+
…………………………
ELETRIPTAN HYDROBROMIDE MONOHYDRATE
http://www.sumobrain.com/patents/wipo/Eletriptan-hydrobromide-monohydrate/WO2000032589.html
‘H-NMR (400MHz, ds-DMSO): delta = 10.90 (1H, d, J=2.2Hz), 9.35 (1 H, br s), 7.95 (2H, d, J=7.5Hz), 7.76 (1 H, t, J=7.5Hz), 7.66 (2H, t, J=7.5Hz), 7.38 (1 H, s), 7.24 (1 H, d, J=8.3Hz), 7.23 (1 H, d, J=2.2Hz), 6.92 (1 H, dd, J=8.3,1.4Hz), 3.63 (2H, m), 3.58 (2H, br m), 3.24 (1 H, m), 3.06 (1 H, m), 2.95 (2H, m), 2.86 (1 H, m), 2.83 (3H, s), 2.00 (1 H, m), 1.90 (2H, m), 1.70 (1 H, m).
Found: C, 54.85; H, 6.03; N, 5.76. C22H29N203SBr requires C, 54.87; H, 6.08; N, 5.82%.
UPDATED 29 MAR 2015
| (R)-3-[(-1-methylpyrrolidin-2-yl)methyl]-5-(2-phenylsulfonylethyl)- 1H-indole |

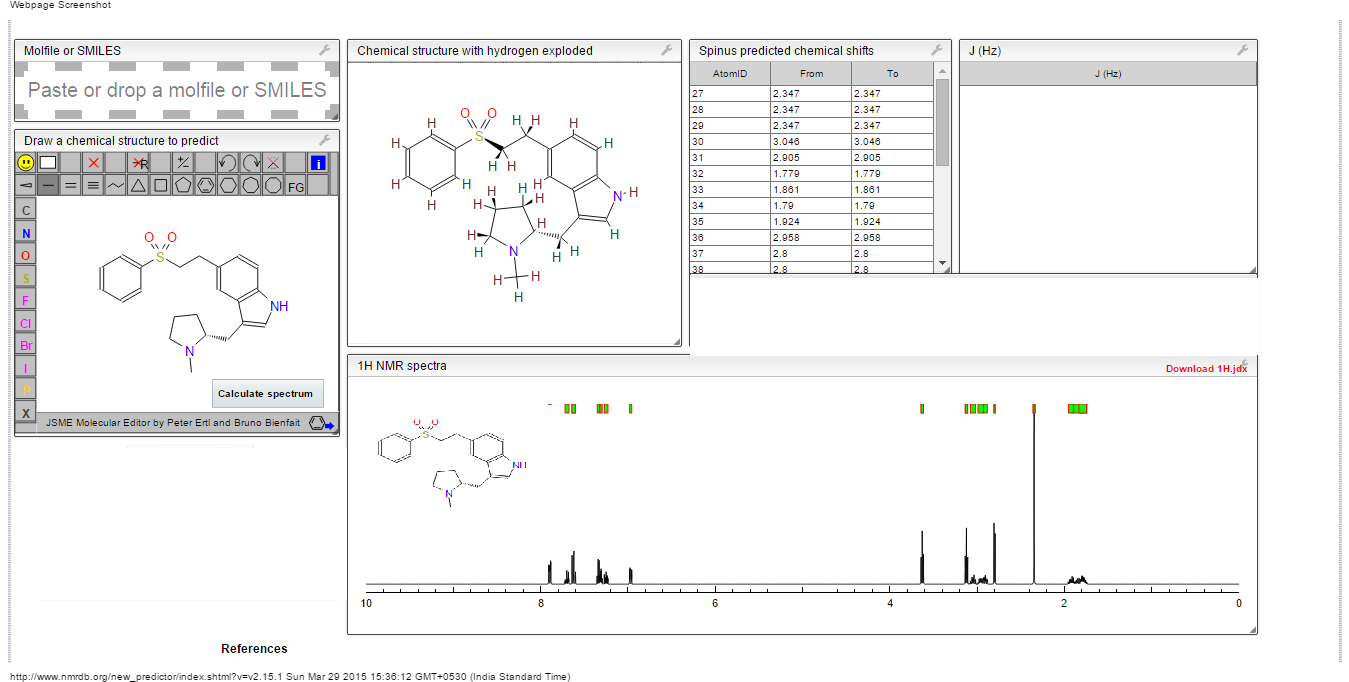
13C NMR

………….
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
- Macor, J. E.; Wythes, M. J. Indole Derivatives. U.S. Patent 5,545,644, Aug 13, 1996.
- Perkins, J. F. Process for the Preparation of 3-Acylindoles. Eur. Patent 1088817A2, April 4, 2001.
- Ashcroft, C. P. Modified Fischer Indole Synthesis for Eletriptan. WO Patent 2005/103035, Nov 3, 2005.
- Bischler, A. Chem. Ber. 1892, 25, 2860–2879. doi:10.1002/cber.189202502123
Synthesis of compounds related to the anti-migraine drug eletriptan hydrobromide
2Engineering Chemistry Department, AU College of Engineering, Andhra University, Visakhapatnam-530003, Andhra Pradesh, India
1H NMR
13C PREDICT
COSYPREDICT
SYNTHESIS
Reference:
US2012/71669 A1, ;
US2011/166364 A1, ;
WO2011/4391 A2, ;
WO2012/4811 A1, ;
US2008/287519 A1, ; Page/Page column 10 ;
WO2011/4391 A2, ; Page/Page column 19 ;
US2008/287519 A1, ; Page/Page column 8 ;
Top 10 Pharma – products in the pipeline
RALTEGRAVIR

CAS No…….518048-05-0 (free acid)
871038-72-1 (monopotassium salt)IUPAC Name:- N-(2-(4-(4-fluorobenzylcarbamoyl)-5-hydroxy-1-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)propan-2-yl)
Organic Process Research and Development, 2011 , vol. 15, 1 pg. 73 – 83,
143 – 144.1 °C(free acid)
MW: 444.42
………………………………………………….
K SALT
C20H20FN6O5*K, 482.513
MP..275 – 277 °C
European Journal of Medicinal Chemistry, 2012 , vol. 50, pG. 361 – 369
Drug information:- Raltegravir is an Anti-microbial drug further classified as anti-viral agent of the class integrase inhibitor. It is used either signally or in combination with other drugs for the treatment of human immunodeficiency virus (HIV) and further clinical trials are in process.
Raltegravir (RAL, Isentress, formerly MK-0518) is an antiretroviral drug produced by Merck & Co., used to treat HIV infection.[1] It received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval.[2][3]
In December 2011, it received FDA approval for pediatric use in patients ages 2–18, taken in pill form orally twice a day by prescription with two other antiretroviral medications to form the cocktail (most anti-HIV drugs regimens for adults and children use these cocktails). Raltegravir is available in chewable form but- because the two tablet formulations are not interchangeable- the chewable pills are only approved for use in children two to 11. Older adolescents will use the adult formulation.[4]
Raltegravir targets integrase, an HIV enzyme that integrates the viral genetic material into human chromosomes, a critical step in the pathogenesis of HIV. The drug is metabolized away via glucuronidation.[5]

Isentress tablets
Raltegravir is taken orally twice daily.[3] Doses of 200, 400, and 600 mg have been studied.
At the 2007 Conference on Retroviruses and Opportunistic Infections, researchers presented Phase III data showing that 77% of patients taking the 400 mg dose of raltegravir plus other antiretroviral drugs reached HIV viral loads below 400 copies, nearly twice as many compared with a control group.
Raltegravir was initially approved only for use in individuals whose infection has proven resistant to otherHAART drugs.[3] However, in July 2009, the FDA granted expanded approval for Raltegravir for use in all patients.[6] As with any HAART medication, raltegravir is unlikely to show durability if used as monotherapy.
In a study of the drug as part of combination therapy, raltegravir exhibited potent and durable antiretroviral activity similar to that of efavirenz at 24 and 48 weeks but achieved HIV-1 RNA levels below detection at a more rapid rate. After 24 and 48 weeks of treatment, raltegravir did not result in increased serum levels of total cholesterol, low-density lipoprotein cholesterol, or triglycerides.[7][8]
Raltegravir significantly alters HIV viral dynamics and decay and further research in this area is ongoing. In clinical trials patients taking raltegravir achieved viral loads less than 50 copies per millitre sooner than those taking similarly potent Non-nucleoside Reverse Transcriptase Inhibitors orProtease Inhibitors. This statistically significant difference in viral load reduction has caused some HIV researchers to begin questioning long held paradigms about HIV viral dynamics and decay.[9] Research into raltegravir’s ability to affect latent viral reservoirs and possibly aid in the eradication of HIV is currently ongoing.[10]
Research results were published in the New England Journal of Medicine on July 24, 2008. The authors concluded that “raltegravir plus optimized background therapy provided better viral suppression than optimized background therapy alone for at least 48 weeks.” [11]
Research on human cytomegalovirus (HCMV) terminase proteins demonstrated that Raltegravir may block viral replication of the herpesviruses.[12]
In January 2013, a Phase II trial was initiated to evaluate the therapeutic benefit of raltegravir in treating multiple sclerosis (MS).[13] The drug is active against Human Endogenous Retroviruses(HERVs) and possibly Epstein-Barr Virus, which have been suggested in the pathogenesis of relapsing-remitting MS.
Raltegravir was generally well tolerated when used in combination with optimized background therapy regimens in treatment-experienced patients with HIV-1 infection in trials of up to 48 weeks’ duration.[14]
Synthesis



WO 2006060730
…………………………………………………
- Savarino A (December 2006). “A historical sketch of the discovery and development of HIV-1 integrase inhibitors”. Expert Opin Investig Drugs 15 (12): 1507–22. doi:10.1517/13543784.15.12.1507.PMID 17107277.
- “FDA approval of Isentress (raltegravir)”. U.S. Food and Drug Administration (FDA). June 25, 2009. Retrieved 2009-11-15.
- “Isentress Drug Approval Package”. U.S. Food and Drug Administration (FDA). February 22, 2008. Retrieved 2009-11-15.
- http://www.everydayhealth.com/hiv-aids/1222/fda-okays-raltegravir-for-kids-teens-with-hiv.aspx?xid=aol_eh-hiv_6_20111219_&aolcat=HLT&icid=maing-grid7%7Cmain5%7Cdl10%7Csec3_lnk2%26pLid%3D122480
- HIV Antiretroviral Agents in Development
- “UPDATE 2-FDA OKs widened use of Merck’s Isentress HIV drug”. Reuters. 2009-07-10.
- Markowitz M, Nguyen BY, Gotuzzo E, et al. (2007). “Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study”. J. Acquir. Immune Defic. Syndr. 46 (2): 125–33. doi:10.1097/QAI.0b013e318157131c. PMID 17721395.
- Stephenson J (2007). “Researchers buoyed by novel HIV drugs: will expand drug arsenal against resistant virus”. JAMA 297 (14): 1535–6. doi:10.1001/jama.297.14.1535. PMID 17426263.
- Faster Viral Decay With Raltegravir
- ClinicalTrials.gov NCT00554398 Impact of MK-0518 (Raltegravir) Intensification on HIV-1 Viral Latency in Patients With Previous Complete Viral Suppression
- Steigbigel RT, Cooper DA, Kumar PN, et al. (July 2008). “Raltegravir with optimized background therapy for resistant HIV-1 infection”. N. Engl. J. Med. 359 (4): 339–54.doi:10.1056/NEJMoa0708975. PMID 18650512.
- Drug against AIDS could be effective against herpesvirus
- Raltegravir (Isentress) Pilot Study in Relapsing Multiple Sclerosis (INSPIRE)
- Croxtall JD, Keam SJ. (2009). “Raltegravir”. Drugs 69 (8): 1059–75. doi:10.2165/00003495-200969080-00007. PMID 19496631.
- Belyk, K. M.; Morrison, H. G.; Jones, P.; Summa, V.; 2007, WO 2006060730
- Manufacturer’s website
- MK-0518 at Aidsmedscom[dead link]
- Integrase Inhibitor Raltegravir (MK-0518) Doubles HIV Suppression in Treatment-Experienced Patients (aidsmap 28 February 2007)
- RMK-0518 Abstract from CROI 2007
- Interim Results From Phase II Study Of MK-0518
- World patent covering the potassium salt
- Raltegravir Pharmacokinetics
……………………………………………………………………………………………………………………………………………….
Raltegravir, also referred to as Raltegravir free-hydroxy, N-(2-(4-(4-fluorobenzyl- carbamoyl)-5-hydroxy-l-methyl-6-oxo-l ,6-dihydropyrimidin-2-yl)propan-2-yl)-5-methyl- l ,3,4-oxadiazole-2-carboxamide, having the following structure;

is an antiretroviral drug used to treat HIV infection. Raltegravir targets integrase, an HIV enzyme that integrates the viral genetic material into human chromosomes, a critical step in the pathogenesis of HIV. Raltegravir potassium salt is marketed under the trade name ISENTRESS™ by Merck & Co.
The processes for preparing Raltegravir that are known in the art either require a protection step for the 5-hydroxy group prior to the methylation step, or lead to an impurity resulting from the methylation of the 5-hydroxy group.
U.S. Patent No. 7, 169,780 discloses Raltegravir and preparation thereof, as described in the following reaction scheme:

Scheme 1
J. Med. Chem. 2008, 51 , 5843-5855 discloses another process for preparing Raltegravir as described in the following reaction scheme:

RLT K-salt
Scheme 2 U.S. Publication No. US 2006/0122205 describes an alternative process for preparing Raltegravir, in which the alkylation step does not include a step for protecting the 5-hydroxy group. The process is described in the following reaction scheme:

Scheme 3
Provided herein is an industrially applicable process for preparing RLT-7′, RLT-8, RLT-9 and RLT-9-OP, intermediates in the synthesis of Raltegravir, as well as processes for preparing Raltegravir and crystalline forms thereof.
US Publication No. US 2006/0122205, WO 2010/1401 56 and WO 201 1 /
024192 describe the potassium salt of Raltegravir, including amorphous and crystalline forms I, II, III and H I , as well as amorphous and crystalline forms of Raltegravir free- hydroxy. PCT publication No. WO 201 1/123754 describes certain Raltegravir salts and polymorphs, including form V of Raltegravir potassium.
|
Conditions:- i. Benzylchloroformate, N,N-diisopropylethylamine, Methyl tert-butyl ether, 20 – 25 °C, 16 h, ii. Hydroxyl amine, Water, 60 °C, 3 h, iii. Dimethyl acetylenedicarboxylate, methanol, Room temperature 2 h then Xylene 90 °C, 2 h, iv. Magnesium methoxide, dimethyl sulfoxide, Methyl iodide, 20 – 25 °C, 2 h, v. 4-fluorobenzyl amine, ethanol, 72 °C, 2 h, vi. 5% Pd/C, methanol, Molybdate sulfuric acid, Hydrogen gas, 50 °C, 3 h, vii. 5-methyl-1,3,4-oxadiazole-2-carbonylchloride, N-methylmorpholine, Tetrahydrofuran, 0 – 5 °C, 2 h |
|
preparation of Raltegravir is described in US patent 2006122205A1 and also in WO2006060730. Accordingly, 2-amino-2-methyl-propanenitrile 1 was reacted with benzylchloroformate in presence of N,N-diisopropylethylamine using methyl tert-butyl ether as solvent at ambient temperature to give benzyl N-(1-cyano-1-methyl-ethyl)carbamate 2. Treatment of 2 with hydroxyl amine using water as solvent at elevated temperature give benzyl N-[(2Z)-2-amino-2-hydroxyimino-1,1-dimethyl-ethyl]carbamate 3. The compound 3 was further cyclized with dimethyl acetylenedicarboxylate using methanol as solvent at higher temperature to give methyl 2-(1-benzyloxycarbonylamino-1-methyl-ethyl)-5-hydroxy-6-oxo-1H-pyrimidine-4-carboxylate 4. Compound 4 was then methylated with methyl iodide in presence of magnesium methoxide as base and dimethyl sulfoxide as solvent at ambient temperature to give methyl 2-(1-benzyloxycarbonylamino-1-methyl-ethyl)-5-hydroxy-1-methyl-6-oxo-pyrimidine-4-carboxylate 5. Compound 5 on condensing with 4-fluorobenzyl amine using ethanol as solvent result in to benzyl N-[1-[4-[(4-fluorophenyl)methylcarbamoyl]-5-hydroxy-1-methyl-6-oxo-pyrimidin-2-yl]-1-methyl-ethyl]carbamate 6, which underwent benzyloxy-decarboxylation on hydrogenating with hydrogen gas in presence of 5% Palladium on carbon catalyst and molybdate sulfuric acid using methanol as solvent to give 2-(1-amino-1-methyl-ethyl)-N-[(4-fluorophenyl)methyl]-5-hydroxy-1-methyl-6-oxo-pyrimidine-4-carboxamide 7. The final step involves condensation of 7 with 5-methyl-1,3,4-oxadiazole-2-carbonylchloride in presence of N-methylmorpholine as base using tetrahydrofuran as solvent at slightly lower temperature to afford N-[1-[4-[(4-fluorophenyl)methylcarbamoyl]-5-hydroxy-1-methyl-6-oxo-pyrimidin-2-yl]-1-methyl-ethyl]-5-methyl-1,3,4-oxadiazole-2-carboxamide also called Raltegravir 8. |

The formation of the hydroxypyrimidone core (3.22) of raltegravir deserves further discussion as its unexpected mechanism was only recently fully elucidated in a joint effort between Merck process chemists and the Houk group at UCLA [91]. These studies combined B3LYP density functional theory with labelling studies and revealed that the most likely pathway involves the formation of a tightly bound polar radical pair 3.31 resulting from thermal homolysis of the N–O bond (Scheme 35). This species subsequently recombines under formation of a C–N bond and a C=O double bond (3.32) allowing for the final cyclocondensation to occur with liberation of methanol. Furthermore these studies were able to disprove a potential alternative [3,3]-sigmatropic rearrangement step by incorporating 15N enriched precursors leading to the formation of pyrimidone 3.22, which is only consistent with a formal [1,3]-sigmatropic rearrangement. Subsequent calculations demonstrated the high energy barrier for such a concerted [1,3]-shift, ultimately leading to the finding of the before-mentioned polar radical pair pathway which is about 8 kcal/mol lower in energy. This is consistent with the experimentally observed rate acceleration in case of the Z-isomer of 3.33 over the E-isomer which was also confirmed by calculations showing an energy gap of 3 kcal/mol.
An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles
 and Ian R. Baxendale Corresponding author email
and Ian R. Baxendale Corresponding author emailhttp://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
check beilstein journals as per link above ……………
this publication allows free usage of data if given proper ref…………………..
any objections email me amcrasto@gmail.com or cal +91 9323115463
………………..
nmr
Imp roved synthesis of raltegravir
GUO D i2liang et al
Department ofM edicinal Chem istry, China PharmaceuticalUniversity, N anjing 210009;
Journal of China Pharmaceutical University 2009, 40 (4) : 297 – 301
http://star.sgst.cn/upload/attach/attach20091230100028d4masjzgcv.pdf
1H NMR (CD3OD) δ: 7.40 (m, 2H) , 7.04 (m , 2H) ,
4.56 (s, 2H ) , 3.46 ( s, 3H ) , 2.65 (s, 3H ) , 1.83 (s,
6H);
13C NMR (CD3OD ) δ: 168.4, 164.8, 163.2,
162.0, 161.9, 160.1, 155.3, 145.8, 136.0, 134.9,
131.0, 116.7, 116.6, 60.2, 43.8, 41.3, 34.8, 27.6,
11.4;
ESI2MS m /z 443 (M )-; LR2MS (EI) m /z 444(M )+; HR2MS ( E I) m /z C20 H21 FN6O5(M )+
calcd444, 155, 7, found 444, 154, 2
second set
lH NMR (399.87 MHz5 CDCI3) δ 12.04 (s, IH), 8.45 (s, IH), 7.94 (t, J = 6.2 Hz, IH), 7.41-736 (m, 2H), 7.08-7.02 (m, 2H)5 4.61 (d, J – 6.2 Hz, 2H), 3.68 (s, 3H), 2.63 (s, 3H), 1.87 (s, 6H).
13C NMR (100.55 MHz, CDCI3) δ 168.3, 166.7, 162.6 (d, JCF=245.7 Hz), 159.6, 159.1, 152.O5 150.4, 147.2, 133.4 (d, JCP=3.2 Hz)5 129.9 (d, JcF=8.0 Hz), 124.1, 115.9 (d, JcF=21.7 Hz), 58.0, 42.7, 33.5, 26.7, 11.4.
…………………….
IR
absorption bandsKBR (cm“1) at 832, 1017, 1248, 1350, 1510, 1682, 2995, and 3374
……………….
K SALT
http://pubs.acs.org/doi/full/10.1021/op100257r
mp 274.2−275.2 °C. 1H NMR (500 MHz, DMSO-d6) δ: 11.65 (t, J = 6.0 Hz, 1 H), 9.75 (s, 1 H), 7.36 (dd, J = 8.6, 5.7 Hz, 2 H), 7.14 (app. t, J = 8.6 Hz, 2 H), 4.48 (d, J = 6.0 Hz, 2 H), 3.43 (s, 3 H), 2.58 (s, 3 H), 1.73 (s, 6 H);
13C NMR (125 MHz, DMSO-d6) δ: 168.7, 167.0, 166.6, 162.1 (d, JCF = 243 Hz), 159.7, 158.3, 153.1, 139.6, 138.0 (d, JCF = 3 Hz), 130.2 (d, JCF = 8 Hz), 123.7, 116.0 (d, JCF = 22), 58.4, 42.1, 33.3, 28.1 (2 C), 11.7.
……………………………
impurities
Org. Process Res. Dev., 2012, 16 (8), pp 1422–1429

…………………
intermediates
http://www.google.com/patents/WO2013098854A2

N-[(1Z)-1 -amino-1 -(hydroxyimino)-2-memylpropan-2-yl]-5-methyl-l ,3 ,4- oxadiazole-2-carboxamide (IVa) (198 gms) was suspended in methanol (1188 ml) and cooled to 15 to 25°C. Dimethyl acetyl enedicarboxylate (DMAD; 152.8 gms) was added and the reaction mass was stirred for 2 to 3 hours at 25°C. The reaction mass was concentrated under reduced pressure and xylene was added and stirred between 135°C and 125°C for 6 hour. After completion of reaction, the mixture was cooled to 60°C and methanol (170 ml) & methyl tert-butyl ether (MTBE) were added to the reaction mass and stirred for 1 hour. The resultant slurry was filtered and washed with a 9:1 mixture of methanol & methyl tert-butyl ether (MTBE) and dried to give methyl 2-(2-(5-methyl-l ,3,4-oxadiazole-2-carboxamido)propan-2-yl)-l ,6-dihydro-5- hydroxy-6-oxopyrimidine-4-carboxylate (V a).
Yield: 198 gms (66 %).
1H NMR (400 MHz, DMSO d6): δ 12.74 (s, 1H), 10.35 (s, 1H), 9.12 (s, 1 H), 3.81 (s, 3H), 2.58 (s, 3H), 1.59 (s, 6 H);
13C NMR (100 MHz, DMSO d6): δ 166.60, 166.15, 160.19, 159.23, 153.26, 152.87, 145.65, 128.30, 56.60, 52.91 , 26.26, 11.34;
retroviral drugs

elvitegravir
Å for chemical synthesis from carboxylic acids elvitegravir 1 starts, the NIS transformed into acid chloride iodide, 2 , and with 3 condensation 4 . 4 and amino alcohols 5 addition-elimination reaction occurs 6 , 6 in alkaline conditions Shimonoseki ring hydroxyl group protected with TBS after seven , seven and zinc reagent 8 occurred Negishi coupling get nine , the last ninehydrolysis and methoxylated get angstrom for elvitegravir.
The US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat)
13 dec 2013
Genzyme’s Cerdelga NDA receives US FDA priority review designationpharmabiz.comThe US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat), an investigational oral therapy for adult patients with Gaucher disease
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
old article cut paste
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
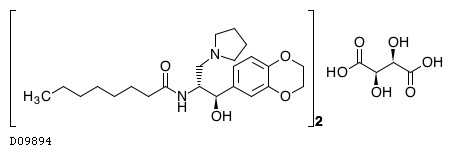
Eliglustat tartrate (USAN)
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.




Merck Statement on FDA Advisory Committee for GRASTEK® (Timothy Grass Pollen Allergen Extract), Merck’s Investigational Sublingual Allergy Immunotherapy Tablet

pollen
An allergen is a type of antigen that produces an abnormally vigorous immune response in which the immune system fights off a perceived threat that would otherwise be harmless to the body. Such reactions are called allergies. In technical terms, an allergen is an antigen capable of stimulating a type-I hypersensitivity reaction in atopic individuals through Immunoglobulin E (IgE) responses.[1] Most humans mount significant Immunoglobulin E responses only as a defense against parasitic infections. However, some individuals may respond to many common environmental antigens. This hereditary predisposition is called atopy. In atopic individuals, non-parasitic antigens stimulate inappropriate IgE production, leading to type I hypersensitivity. Sensitivities vary widely from one person (or other animal) to another. A very broad range of substances can be allergens to sensitive individuals.

WHITEHOUSE STATION, N.J., Dec. 12, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today issued the following statement after the conclusion of the Allergenic Products Advisory Committee of the U.S. Food and Drug Administration (FDA) meeting to discuss GRASTEK® (Timothy grass pollen allergen extract). GRASTEK is the proposed trade name for the company’s investigational sublingual tablet for the treatment of Timothy grass induced allergic rhinitis, with or without conjunctivitis, in appropriate adult and pediatric patients who are candidates for immunotherapy.
An FDA committee has recommended a grass pollen allergen extract for the treatment of Timothy grass pollen-induced allergic rhinitis, with or without conjunctivitis, in individuals aged 5 to 65 years.
CHYAVAN PRASH ; AN EXCELLENT AYURVEDA PREPARATION FOR ALL AGES TO BE TAKEN IN COLD SEASON

AYURVEDA, the Indian System of Medicine have full of hundreds of thousands of formulea and have almost a great hidden treasures of remedies for all ailing conditions. Among these formulea, CHARAK have mentioned an excellent combination of Herbs and food articles, which is used since centuries in India and in Indian continents. CHARAK SAMAHITA, the great Olden Book of CLASSICAL AYURVEDA PHILOSOPHY, THE INDIAN SYSTEM OF MEDICINE ; ORIGINALLY WRITTEN IN “SANSAKRIT LANGUAGE”,
CHARAK SAMAHITA, the great Olden Book of CLASSICAL AYURVEDA PHILOSOPHY, THE INDIAN SYSTEM OF MEDICINE ; ORIGINALLY WRITTEN IN “SANSAKRIT LANGUAGE”,
CHYAVAN PRASH is one of them, widely used in all seasons , specially in Cold for maintaining Health and to protect from cold and cold exposure related anomalies.
Truely to say, CHYAVAN PRAS is a RASAYANA. Rasayan literally have wide means in AYURVEDA. To understand RASAYAN, one have to understand the SAPTA DHATU, which is a fundamental principles subject. SAPATA DHATU is considered equivalent to AYURVEDA PATHOLOGY.
 PAGES FROM CHARAK SAMAHITA MENTIONING THE FORMULA OF HERBS AND METHODS…
PAGES FROM CHARAK SAMAHITA MENTIONING THE FORMULA OF HERBS AND METHODS…
View original post 555 more words
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

