
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Indian Regulators promote two levels of GMP
GMP deviations and even data falsification have been identified in a number of companies in India. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? Read more in our GMP News
GMP deviations and even data falsification have been identified in a number of companies in India. The FDA has issued numerous Warning Letters, the EU has published GMP Non Compliance Reports in its EudraGMDP database and EDQM has withdrawn various CEPs because of GMP inspection findings.
In an article published by Regulatory Focus on 28 January 2014 the question has been raised whether Indian companies have a chronic data falsification problem. The article lists 7 companies in India which have received a Warning Letter in the past months – all of them because of GMP deviations and because of “actually or potentially tampering with their data”. In addition to the 7 companies the Ranbaxy case is a story of its own. Not only one facility was found to manipulate data but several sites of the company are involved. For this reason the US FDA has issued a consent decree of permanent injunction against Ranbaxy. All manufactured products in the facilities concerned are now subject to an FDA import alert. In a press release the FDA states: “Because this company continued to violate current good manufacturing practice regulations and falsify information on drug applications, the FDA took these actions in an effort to protect consumers.” Dara Corrigan, FDA associate commissioner for regulatory affairs goes on: “The FDA continues to be committed to protecting consumers from potentially unsafe products that may be offered on the market.” On January 23, 2014 the FDA added an additional facility of Ranbaxy to the existing consent decree.
So far, the Indian Authority did not initiate the same measures like US and European counterparts. This also questions the supervision system in India. If inspections have been performed by Indian Inspectors at the concerned facilities why did they fail to make the same findings? The Drug Controller General of India, Mr. G.N. Singh, gave an interesting interpretation: According to an interview published by live mint & Wall Street Journal he said: “…it must be stated that every country has different measures and we cannot judge Ranbaxy by standards set up by the American drug regulator“. When Mr Singh was asked about the problems identified at three Ranbaxy plants he stated: “Some of those were found to be true and my office had told Ranbaxy to take corrective measures. Similar procedures will be followed in this case as well. But I do not think this is a situation which will warrant withdrawal of drugs from the domestic market. Our biggest objective is to maintain good quality of medicines and we are doing that. There are no drugs in the Indian market that are not up to the standards stated under the Drugs and Cosmetics Act.” In a final statement in the interview he also mentioned that he is “not worried about issues of quality.” In another interview with the Business Standard Press Mr Singh made an alarming statement for all customers of medicinal products and APIs in Europe and the US. “If I follow US standards, I will have to shut almost all drug facilities“. If this is the truth EU and US customers are in big trouble because products not complying to EU/US GMP standard (e.g. ICH Q7 GMP for APIs) would need to be taken from the market immediately.
This all looks like it will not fit together. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? It can only mean that dual standards exist. This would result in two quality levels, an international and a domestic quality level. Such a policy possibly causes questions by Indian patients who have to accept a different and probably lower quality standard.
It does not look like the Indian Regulators will re-think the GMP inspection approach and the quality standard in their country. Instead of acting in his own country the Drug Controller General of India announced inspections in the US and the EU.
But what are the international implications of this strategy? European Regulators need to react as they require from the Indian Authority to issue Written Confirmations of GMP compliance. Without a Written Confirmation APIs can not enter EU market. Currently more than 200 Written Confirmations have been issued by Indian Authority. If the inspections which have been performed as a prerequisite for issuing a Written confirmation were not based on the international standard ICH Q7 (GMP for APIs) the Written Confirmations are no longer valid documents. This issue might be raised by an EU court if a substandard API in a medicinal product will cause a health risk to patients in Europe.

Lysosomal Storage Disorders: Orphan Drugs For Niemann-Pick Disease
This is the sixth Blog Post in a series that will examine Lysosomal Storage Disorders (LSDs) in the rare disease and orphan drug space. This Blog Post reviews orphan drugs for the treatment of Niemann-Pick Disease (NPD).
Introduction
Niemann-Pick Diseases (NPDs) are a subgroup of LSDs that affect metabolism and are caused by genetic mutations. NPD is named after the two doctors who described the symptoms in the early part of the 20th century – Dr. Albert Niemann and Dr. Luddwick Pick. NPDs are characterized by the harmful accumulation of quantities of fatty substances, or lipids, in the cells of the spleen, lungs, bone marrow, liver, and brain. The three most commonly recognized forms of NPD are:
• Niemann-Pick Types A & B (ASMD or Acid Sphingomyelinase Deficiency)
• Niemann-Pick Type C (NPC).
Niemann-Pick types A and B are caused by a deficiency of an enzyme called acid sphingomyelinase. The enzyme deficiency leads to enlargement…
View original post 278 more words
Cidofovirסידופוביר سيدوفوفير


CIDOFOVIR
(S)-1-(3-Hydroxy-2-phosphonylmethoxypropyl)cytosine
[(S)-2-(4-Amino-2-oxo-1,2-dihydropyrimidin-2-yl)-1-(hydroxymethyl)ethoxymethyl]phosphonic acid
113852-37-2 CAS
120362-37-0 (Na salt)
149394-66-1 (dihydrate)
launched 1996 Gilead
SYNTHESIS.. CHEMDRUG
Rega Instituut (Originator)
For the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS)
US5142051 PATENT
| Canada | 1340856 | 1999-12-21 | EXPIRY 2016-12-21 |
| United States | 5142051 | 1993-06-26 | 2010-06-26 |
Cidofovir is a DNA polymerase inhibitor that was launched in 1996 by Gilead for the intravenous treatment of cytomegaloviral (CMV) retinitis in AIDS patients. Early clinical trials are underway at the National Institute for Allergy & Infectious Disease (NIAID) for the treatment of BK virus nephropathy (BKVN) in patients who have undergone kidney transplants.
Cidofovir suppresses CMV replication by selective inhibition of viral DNA synthesis. Biochemical data support selective inhibition of CMV DNA polymerase by cidofovir diphosphate, the active intracellular metabolite of cidofovir. Cidofovir diphosphate inhibits herpesvirus polymerases at concentrations that are 8- to 600-fold lower than those needed to inhibit human cellular DNA polymerases alpha, beta, and gamma1, 2, 3. Incorporation of cidofovir into the growing viral DNA chain results in reductions in the rate of viral DNA synthesis.
Cidofovir was originally developed under a collaboration between the Academy of Sciences of the Czech Republic and the Rega Institute for Medical Research. In 1991 and 1992, Gilead entered into license agreements with the Rega Institute that covered a large number of nucleotide analogue compounds and structures, including cidofovir. The drug became the subject of a marketing collaboration between Gilead and Pfizer (formerly Pharmacia & Upjohn) in August 1996 that covers all countries outside the U.S.
Cidofovir (brand name Vistide) is an injectable antiviral medication primarily used as a the treatment for cytomegalovirus (CMV) retinitis (an infection of the retina of the eye) in patients with AIDS.[1][2]
Its only indication that has received regulatory approval worldwide is cytomegalovirus retinitis.[1][2] Cidofovir has also shown efficacy in the treatment ofaciclovir-resistant HSV infections.[3] Cidofovir has also been investigated as a treatment for progressive multifocal leukoencephalopathy with successful case reports of its use.[4] Despite this meta-analyses have failed to demonstrate any efficacy in AIDS patients,[5] and the limited data in non-AIDS patients fail to demonstrate any efficacy either.[6] Cidofovir might have anti-smallpox efficacy and might be used on a limited basis in the event of a bioterror incident involving smallpox cases.[7] A cidofovir derivative with much higher activity against smallpox that can be taken orally has been developed.[8] It has inhibitory effects on varicella-zoster virus replication in vitro although no clinical trials have been done to date, likely due to the abundance of safer alternatives such as aciclovir.[9] Cidofovir shows anti-BK virus activity in a subgroup of transplant patients.[10] Cidofovir is being investigated as a complementary intralesional therapy against papillomatosis caused by HPV.[11][12]
It first received FDA approval on the 26th of June 1996,[13] TGA approval on the 30th of April 1998[2] and EMA approval on the 23rd of April 1997.[14]
Other
It has been suggested as an antitumour agent, due to its suppression of FGF2.[15][16]
Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, by Antonín Holý, and developed by Gilead Sciences[20] and is marketed with the brand name Vistide by Gilead in the USA, and by Pfizerelsewhere.
The chemical name of cidofovir is 1-[(S)-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC), with the molecular formula of C8H14N3O6P•2H2O and a molecular weight of 315.22 (279.19 for anhydrous). The chemical structure is:

Cidofovir is a white crystalline powder with an aqueous solubility of ≥ 170 mg/mL at pH 6 to 8 and a log P (octanol/aqueous buffer, pH 7.1) value of -3.3.
Cidofovir Injection is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL.
The formulation is pH-adjusted to 7.4 (range 7.1 to 7.7) with sodium hydroxide and/or hydrochloric acid and contains no preservatives. The appropriate volume of Cidofovir Injection must be removed from the single-use vial and diluted prior to administration
INTRODUCTION
Cidofovir’s chemical formula is C8H14N3O6P and its IUPAC name is ({[(S)-1-(4-amino-2-oxo-1,2-dihydropyrimidin-1-yl)-3-hydroxypropan-2-yl]oxy}methyl)phosphonic acid. Cidofovir has also been described as (S)-(1-(4-amino-2-oxopyrimidin-1(2H)-yl)-3-hydroxypropan-2-yloxy)methylphosphonic acid as well as possibly by other chemical names. Its chemical structure is:

Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, and developed by Gilead Sciences. Today, cidofovir is an injectable antiviral medication for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS. It suppresses CMV replication by selective inhibition of viral DNA polymerase and therefore prevention of viral replication and transcription. It is an acyclic nucleoside phosphonate, and is therefore independent of phosphorylation by viral enzyme, in contrast to, for instance, acyclovir.
Cidofovir is marketed with the brand name Vistide® by Gilead in the United States and by Pfizer in other parts of the world. Vistide® is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL. The formulation is pH-adjusted to 7.4 with sodium hydroxide and/or hydrochloric acid and contains no preservatives. Renal impairment is the major toxicity of Vistide®.
Presently, there are no Orange Book patents listed as having claims which cover Vistide®, although previously U.S. Pat. No. 5,142,051 was listed in the Orange Book for Vistide®. The ‘051 patent is not directed specifically to cidofovir or its crystalline forms. Instead, it broadly discloses N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases.
Cytomegalovirus (Cytomegaoviyns, CMV) is one of the biggest dangers of the herpes virus, the body’s infection rates as high as 50% to 80% of the current adult prevalence rate of more than 95%, generally showed a latent infection, most infections had no clinical symptoms, but under certain conditions, the invasion of organs and systems to produce more severe disease. The virus can invade the lung, liver, kidney, salivary gland, mammary gland and other polymorphonuclear leukocytes and lymphocytes, and, since the long-term or intermittent saliva, milk sweat, blood, urine, semen, exclude uterine secretions of the virus. Spread through a variety of ways in the mouth, genital tract, placenta, blood transfusion or organ transplantation.
When the body’s immune dysfunction, such as infected with HIV, cancer patients undergoing radiotherapy, chemotherapy, organ or bone marrow transplantation immunosuppressive anti-rejection etc will stimulate active infection, can cause acute retinitis, interstitial pneumonia, gastroenteritis and encephalitis, blindness or death without treatment rate of over 70%. With the rise in HIV infection rates and organ transplants extensively for anti-CMV drugs is also increasing demand.
cidofovir (cidofovir, HPMPC) are novel ether derivatives of cytidine phosphono chemical name
[5]-NL [(3 – hydroxy-2 – methoxy-phosphonic acid) glycerol]-N4-cytosine, Molecular structure of the formula (I):

Gilead developed by the United States, in May 1996 the FDA approved injectable celecoxib Duofu Wei listed, France and Canada also continued with the approval of the use of the trade name Vistide. Its CAS number is 113852-37-2, formula C8H14N3O6P, the structure of formula (I). Cidofovir for CMV is highly inhibitory activity of certain ganciclovir or foscarnet resistant strains of the virus are also active. And herpes simplex virus (HSV), herpes zoster virus (VZV), human papillomavirus (HPV), also has a strong activity.
Its mechanism of action: cidofovir having a phosphoric acid group, a ring-opening mechanism of the antiviral nucleoside phosphonate compound (ANP) and the consistent cyclic nucleoside analogues are nucleosides or virus in vivo kinase activation into triphosphate metabolite, thereby inhibiting viral replication by DNA polymerase and reverse transcriptase. Unlike the three-step cyclic nucleoside analogues must phosphorylation reaction, ring opening nucleoside phosphonate group containing phosphorus compound itself, eliminating the first step of the phosphorylation reaction speed, and thus a higher activity. Cidofovir is absorbed when the cells in the cell pyrimidine nucleoside phosphorylase kinase (P bandit kinase and NDP kinase) to effect conversion of the active metabolite monophosphate (HPMPCp), diphosphate (HPMPCpp) and a bile acid base adducts. Cidofovir diphosphate inhibits viral DNA polymerase or reverse transcriptase activity, and its corresponding natural dNTP incorporated into the viral DNA chain competition, since no 3 – hydroxy end, continue to extend the DNA chain termination. Can slow the synthesis of DNA, viral DNA and to the loss of stability, thereby inhibiting viral replication, transcription of the ability to reduce viral DNA to exert antiviral activity. Compared with other anti-CMV drugs, cidofovir characteristics: significant and lasting effect, started the first two weeks administered once a week, then only administered once every two weeks, easy to use, and to reduce its toxicity side effects.
Several major techniques are based on the synthesis of cidofovir cytosine as starting material, mainly carried out to improve the synthesis of the side chain.
(I) J. Med Chem, 1989,32,1457 ~ 1463 discloses a synthetic process:

The route to cytosine as the raw material, with a chiral side chain by condensation, deprotection and reduction can be obtained in three steps cidofovir.However, chiral side chain subject to a six-step reaction system. The total yield is low, adverse side. And using Me3SiBr, so that the costs and the risk of surge, is not conducive to industrial production.
(2) US 5591852,1995-1-7; US 2005/023833 & WO 2006/014429 and US 2009/0270618, Tetrahedron Lett 1994,35,3243-3246 and “Chinese Journal of New Drugs”, 2007,16. , 1272-1274 for the synthesis of a lot of improvements:

Benzoyl cytosine with a chiral starting material and trityloxymethyl ethylene oxide condensation, deprotection and hydrolysis was then prepared by deprotection cidofovir group. The synthetic steps to make some shorter, but still use expensive Me3SiBr, adverse ones, the low yield of the security at the cost of industrial production is still unfavorable. (Several different patent protection only in the order of the amino cytosine different!)
(3) Patent Publication No. CN1690065A, CN1690066A, CN1690067A (2005 年 11 月 2 Publication Date) and the “Chinese Journal of Medicinal Chemistry” 2007,17,41-46, reported a new synthetic route:

The route of process steps is too long, the total yield is low, side effects side. But not conducive to industrial production.
(4) Patent No. CN 101205215A (25 June 2008 publicly) announced a halogen epoxy propane as a starting material for the synthesis route:
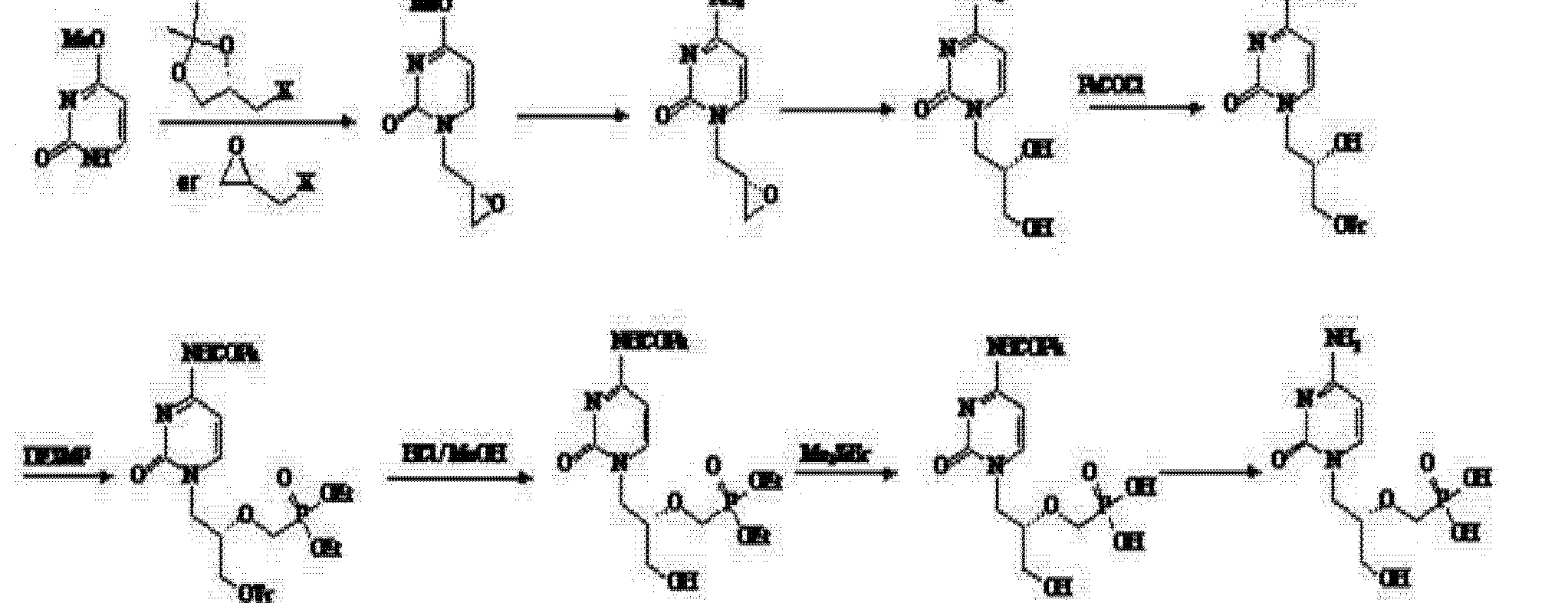
Use of the route (R) – epihalohydrin reaction with cytosine, cytosine ring because alkaline easily cause epoxy ring-opening reaction of the ring, but side reactions, the purified product is not, nor is suitable for industrial production.
Subsequently, the patent number CN 101525352A (2009 年 9 月 9 Publication Date) discloses (4) based on the modified route through epoxypropionate alkane ether in the form of a direct reaction with cytosine, after a series of similar steps obtain the final product cidofovir.
In view of the clinical application of cidofovir more favorable therapeutic effect in, looking for a high yield and because of economic and practical, easy to control, the risk of small synthetic methods and technology is now more urgent needs.
Synthesis

Brodfuehrer, P; Howell, Henry G.; Sapino, Chester; Vemishetti, Purushotham (1994). “A practical synthesis of (S)-HPMPC”. Tetrahedron Letters 35 (20): 3243. doi:10.1016/S0040-4039(00)76875-4.
………………………………………
CN 102268040

, Example 1:
1 Synthesis of 4,4 ‘- dimethoxytrityl methyl – (R) – glycidol (Compound III): The 5 04 g (15 mmoDDMT-Cl grain port 0 20 g (1 52 mmol… ) 4_ dimethylaminopyridine (DMAP) was dissolved in 100 mL CH2C12 cooled to 0 ° C, was added dropwise 10 mL TEA was slowly added 2. 00 g (27mmol) hydroxymethyl chiral oxirane (Compound II ) addition was completed, the reaction warmed to room temperature naturally. fly 4 h, until TLC until the disappearance of the detection DMT-Cl, the reaction was stopped by filtration, the filtrate was washed with saturated NaHC03 solution (50mLX2), saturated NaCl solution (50 mLX2), anhydrous Na2S04 dried, filtered, and concentrated to a viscous colorless directly, i.e., 5 08 g of 4,4 ‘-dimethoxy-triphenylmethyl _ -.. (R) – glycidol (Compound III), yield 90 %, HPLC purity 99%.
2, Synthesis (S)-N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl] cytosine (Compound IV):. Under nitrogen to 3 56 g (32 mmol) of cytosine was added 150 mL of anhydrous N, N-dimethylformamide (DMF), and at room temperature, was added portionwise 1. 24 g (31 mmol, molar concentration of 60%) NaH, 0. 5 h after adding 11 92 g (31 mmol) 4,4 ‘-. dimethoxytrityl methyl – (R) – glycidol (Compound III), plus finished warming up to 10 (Tll (TC reaction . 6-8 h and then filtered, and the filtrate evaporated under reduced pressure DMF, the remaining solid phase was added 500 mL of ethyl acetate and 50 mL of water, separated and the organic layer was washed with saturated NaHC03 solution (50 mL X 2), saturated NaCl solution (50 mL X 2), dried over anhydrous Na2S04 filtered and dried, and concentrated to give 13 90 g of a white solid, S Jie (S)-Nl-[(2 -.. hydroxy-3 – (methoxy-dimethoxytrityl ) propyl] cytosine (Compound IV), yield 92%, HPLC purity 98%.
3 Synthesis ⑶-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V):
75 ~ 80 ° C under the conditions, 48 76 g (0 100 mol.) (S) _N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl]. Cytosine (Compound IV) was added to 150 mL anhydrous DMF, and then inputs 8. 5g (0. 050 mol) tert-butoxide, magnesium reaction 0.5-1 h, tosyloxy added diethyl 32 methylsulfinyl . 2 g (0. 100 mol), the reaction epileptic 8 h, p-toluenesulfonic acid was added to neutralize the excess alkali to neutral distilled DMF, ethyl acetate (300 mLX 3) washing the combined ethyl acetate phase was concentrated to give a solid, i.e., synthetic 58 18 g (S)-Nl-. {[2 – (diethoxy-phosphono-methoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V), yield 89%, HPLC purity greater than 95%.
4 Synthesis of (S)-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – hydroxy] propyl} cytosine (Compound VI): The 10 g (S)-Nl- {[2 – (phosphono-methoxy ethoxy) -3 – (methoxy-dimethoxytrityl)] propyl}-cell
Pyrimidine (compound V) was dissolved in a concentration of 70 mL of 80% acetic acid solution, 90 ° C reaction. After 5 h, cooled to room temperature, 50 mL of water and 30 mL of dichloromethane, and the organic phase washed with water (30 mL X2) and the combined aqueous phase was concentrated to give crude 9. 5 g, can be performed directly in the next reaction.
can also be separated by flash column chromatography (CH2C12 = MeOH = 10: 1), 4.6 g obtained as a pale yellow oil, i.e. (S)-Nl-{[2 – (methoxy diethoxy phosphono ) -3 – hydroxy] propyl} cytosine (Compound VI), yield 90%.
5 was synthesized ⑶-Nl-{[2_ (diphosphonic acid methoxy) -3 – hydroxy] propyl} cytosine (Compound I):
The 9.5g (S)-Nl-{[2 – (methoxy diethoxy phosphonomethyl) -3 – hydroxy] propyl} cytosine (Compound VI) into a crude product containing 5 76 g (0.. 045 mol) solution of hydrogen iodide, hydroiodic acid, and after reflux for 4-5 h. (50 mLX 2) wash solution was separated with ethyl acetate. The aqueous phase was added sodium hydroxide to adjust pH between 3 Γ3 6, filtered, recrystallized from methanol to give 3.81 g of white crystalline solid, S Jie (S)-Ni-{[2 -.. (Diphosphonic acid methoxy yl) -3 – hydroxy] propyl} cytosine (Compound I), yield 88% (containing two crystal water), HPLC purity greater than 99%.
…………………………………………
POLYMORPHS
Example 7 Amorphous Cidofovir
Intermediate 5 (FIG. 7; 0.5 g, 0.054 mol) was heated with a solution of sodium methoxide in methanol (0.5 M, 15 mL, 7.5 mmol) at 72° C. for 14.5 h then at 90° C. for 5.5 h. The reaction mixture was quenched with water (10 mL) and filtered through a bed of ion exchange resin Dowex® 50WX8 100-200 (H). The filtrate was cycled through the ion exchange bed (2 times) then washed successively with 1:1 methanol:water (40 mL), methanol (40 mL) and 4% triethylamine:methanol (50 mL). This ion-exchange bed was further washed with 48:48:4 methanol:water:triethylamine (100 mL) until no UV absorbance was detected in the filtrate. This reaction produced intermediate 7 (FIG. 7) together with cyclic cidofovir impurity. This mixture was then dissolved in 6 N HCl and heated to 65° C. After cooling the reaction mixture to room temperature, ethyl acetate was charged and stirred and the aqueous layer separated. The aqueous was stirred with ethanol (50 mL). The precipitated material was filtered and the solid was washed with ethanol. The ethanol filtrate was concentrated. The concentrated material was taken up in acetonitrile and stirred with trimethylsilyl bromide (19 mL) at room temperature for 18 h. The reaction mixture was filtered and the filtrate concentrated. The residue was taken up in toluene (30 mL) and ammonium hydroxide (28%, 50 mL) was charged and stirred at room temperature. The organic phase was separated and the aqueous phase was concentrated to dryness. Water (20 mL) and ethanol (15 mL) were added to the residue. The mixture pH was 6 and was adjusted to pH 3 with concentrated HCl (2 mL) then adjusted to pH 4 to 4.5 with 28% NH4OH. After stirring for 0.5 h, the mixture was cooled, filtered and the solids washed with 2:1 EtOH:H2O and dried under vacuum for 18 h. The isolated solid was taken up in water (10 mL) and 28% NH4OH added to give a solution. Concentrated HCl was added to the solution until pH 4 was reached. Ethanol (13 mL) was charged and the mixture stirred at −17° C. for 18 h, filtered and the solids washed with 2:1 EtOH:water (2×8 mL), dried under vacuum at 35° C. The cidofovir isolated in this manner was determined to be in the amorphous form by XRPD.

……………………………..
Journal of the American Chemical Society, 2011 , vol. 133, 7 p. 2264 – 2274
http://pubs.acs.org/doi/abs/10.1021/ja109823e

………………………………………………..
READ ALSO
Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457
http://pubs.acs.org/doi/abs/10.1021/jm00127a010
References
- “Vistide (cidofovir) dosing, indications, interactions, adverse effects, and more”.Medscape Reference. WebMD. Retrieved 4 February 2014.
- “Product Information VISTIDE®”. TGA eBusiness Services. Gilead Sciences Pty Ltd. 3 September 2013. Retrieved 5 February 2014.
- Chilukuri, S; Rosen, T (2003 Apr). “Management of acyclovir-resistant herpes simplex virus.”.Dermatologic clinics 21 (2): 311–20. doi:10.1016/S0733-8635(02)00093-1.PMID 12757254.
- Segarra-Newnham M, Vodolo KM (June 2001). “Use of cidofovir in progressive multifocal leukoencephalopathy”. Ann Pharmacother 35 (6): 741–4. doi:10.1345/aph.10338.PMID 11408993.
- De Luca, A; Ammassari, A; Pezzotti, P; Cinque, P; Gasnault, J; Berenguer, J; Di Giambenedetto, S; Cingolani, A; Taoufik, Y; Miralles, P; Marra, CM; Antinori, A; Gesida 9/99, IRINA, ACTG 363 Study, Groups (September 2008). “Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis.”. AIDS (London, England) 22 (14): 1759–67.doi:10.1097/QAD.0b013e32830a5043. PMID 18753934.
- Langer-Gould, A; Atlas, SW; Green, AJ; Bollen, AW; Pelletier, D (28 July 2005). “Progressive Multifocal Leukoencephalopathy in a Patient Treated with Natalizumab”. New England Journal of Medicine 353 (4): 375–381. doi:10.1056/NEJMoa051847. PMID 15947078.
- De Clercq E (July 2002). “Cidofovir in the treatment of poxvirus infections”. Antiviral Res. 55(1): 1–13. doi:10.1016/S0166-3542(02)00008-6. PMID 12076747.
- Bradbury, J (March 2002). “Orally available cidofovir derivative active against smallpox.”.Lancet 359 (9311): 1041. doi:10.1016/S0140-6736(02)08115-1. PMID 11937193.
- Magee, WC; Hostetler, KY; Evans, DH (August 2005). “Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate”. Antimicrobial Agents and Chemotherapy49 (8): 3153–3162. doi:10.1128/AAC.49.8.3153-3162.2005. PMC 1196213.PMID 16048917.
- Araya CE, Lew JF, Fennell RS, Neiberger RE, Dharnidharka VR (February 2006).“Intermediate-dose cidofovir without probenecid in the treatment of BK virus allograft nephropathy”. Pediatr Transplant 10 (1): 32–7. doi:10.1111/j.1399-3046.2005.00391.x.PMID 16499584.
- Broekema FI, Dikkers FG (August 2008). “Side-effects of cidofovir in the treatment of recurrent respiratory papillomatosis”. Eur Arch Otorhinolaryngol 265 (8): 871–9. doi:10.1007/s00405-008-0658-0. PMC 2441494. PMID 18458927.
- Soma MA, Albert DM (February 2008). “Cidofovir: to use or not to use?”. Curr Opin Otolaryngol Head Neck Surg 16 (1): 86–90. doi:10.1097/MOO.0b013e3282f43408.PMID 18197029.
- “Cidofovir Monograph for Professionals – Drugs.com”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 5 February 2014.
- “Vistide : EPAR -Product Information” (PDF). European Medicines Agency. Gilead Sciences International Ltd. 7 November 2013. Retrieved 5 February 2014.
- Liekens S, Gijsbers S, Vanstreels E, Daelemans D, De Clercq E, Hatse S (March 2007). “The nucleotide analog cidofovir suppresses basic fibroblast growth factor (FGF2) expression and signaling and induces apoptosis in FGF2-overexpressing endothelial cells”. Mol. Pharmacol.71 (3): 695–703. doi:10.1124/mol.106.026559. PMID 17158200.
- Liekens S (2008). “Regulation of cancer progression by inhibition of angiogenesis and induction of apoptosis”. Verh. K. Acad. Geneeskd. Belg. 70 (3): 175–91. PMID 18669159.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3. edit
- “Vistide (cidofovir)” (package insert). Gilead Sciences. September 2010. DOSAGE AND ADMINISTRATION: Dosage.
- Safrin, S; Cherrington, J; Jaffe, HS (September 1997). “Clinical uses of cidofovir”. Reviews in Medical Virology 7 (3): 145–156. doi:10.1002/(SICI)1099-1654(199709)7:3<145::AID-RMV196>3.0.CO;2-0. PMID 10398479.
- “Press Releases: Gilead”. Retrieved 2007-12-05.
- Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457 - Synthesis and antiherpesvirus activity of (S)-1-((3-hydroxy-2-phosphonylmethoxy)propyl)cytosine (HPMPC) and related nucleotide analoguesNucleosides Nucleotides 1989, 8(5-6): 923
- Journal of Pharmaceutical Sciences, 2012 , vol. 101, 9 p. 3249 – 326
- BRODFUEHRER P R ET AL: “A Practical Synthesis of (S)-HPMPC“, 19940101, vol. 35, no. 20, 1 January 1994 (1994-01-01), pages 3243-3246, XP002012084
- http://www.chemdrug.com/databases/8_0_nhnpseknoegbdisx.html CHEMDRUG SYNTHESIS
| 2 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of nucleotide analogs bearing the (S)-(3-hydroxy-2-phosphonylmethoxy)propyl moiety attached to adenine, guanine, and cytosine“, ACS SYMPOSIUM SERIES , 401(NUCLEOTIDE ANALOGUES ANTIVIRAL AGENTS), 88-102 CODEN: ACSMC8; ISSN: 0097-6156, 1989, XP002677024, |
| 3 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of the nucleotide analog (S)-1-[3-hydroxy-2-(phosphonylmethoxy)prop yl]cystosine“, JOURNAL OF MEDICINAL CHEMISTRY , 32(7), 1457-63 CODEN: JMCMAR; ISSN: 0022-2623, 1989, XP002677026, |
| 4 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; JIANG, XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir”, XP002677148, retrieved from STN Database accession no. 2009:1568897 -& JIANG XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir“, JIEFANGJUN YAOXUE XUEBAO – PHARMACEUTICAL JOURNAL OF CHINESE PEOPLE’S LIBERATION ARMY, ZHONGGUO RENMIN JIEFANGJUN, ZONGHOUQINBU, WEISHENGBU, YAOPIN YIQI JIANYANSUO, CN, vol. 25, no. 5, 1 January 2009 (2009-01-01), pages 395-397, XP008152443, ISSN: 1008-9926 |
| 5 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JIANFENG ET AL: “Improved synthesis of cidofovir”, XP002677149, retrieved from STN Database accession no. 2008:1052727 -& LIU JIANFENG ET AL: “Improved synthesis of cidofovir“, ZHONGGUO YAOWU HUAXUE ZAZHI – CHINESE JOURNAL OF MEDICINAL CHEMISTRY, GAI-KAI BIANJIBU, SHENYANG, CN, vol. 17, no. 1, 1 February 2007 (2007-02-01), pages 41-43, XP008152444, ISSN: 1005-0108 |
| 6 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LU, NING ET AL: “Synthesis of cidofovir”, XP002677151, retrieved from STN Database accession no. 2007:1124844 -& LU NING ET AL: “Synthesis of cidofovir“, ZHONGGUO XIN YAO ZAZHI – CHINESE NEW DRUGS JOURNAL, GAI-KAN BIANJIBU, BEIJING, CN, vol. 16, no. 16, 1 January 2007 (2007-01-01), pages 1272-1274, XP008152436, ISSN: 1003-3734 |
| 7 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; YI, HONG ET AL: “Synthesis of antiviral agent cidofovir”, XP002677150, retrieved from STN Database accession no. 2007:1249882 -& YI HONG ET AL: “Synthesis of antiviral agent cidofovir“, ZHONGGUO KANGSHENGSU ZAZHI/ CHINESE JOURNAL OF ANTIBIOTICS, SICHUAN, CN, vol. 31, no. 7, 1 January 2006 (2006-01-01), pages 412-413, XP008152413, ISSN: 1001-8689 |
| 8 | * | FROMTLING R A ET AL: “Cidofovir. HPMPC. GS-504. GS-0504. Viside“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 21, no. 10, 1 January 1996 (1996-01-01), pages 1003-1013, XP008152410, ISSN: 0377-8282 |
| 9 | * | HOLY A: “SYNTHESES OF ENANTIOMERIC N-(3-HYDROXY-2-PHOSPHONOMETHOXYPROPYL) DERIVATIVES OF PURINE AND PYRIMIDINE BASES“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 3, 1 January 1993 (1993-01-01), pages 649-674, XP009042514, ISSN: 0010-0765, DOI: 10.1135/CCCC19930649 |
| 10 | * | JOANNE J BRONSONA ET AL: “A New Synthesis of the Potent and Selective Anti-Herpesvirus Agent (S)-1-[3-Hydroxy-2-(Phosphonylmethoxy)Prop yl]Cytosine“, NUCLEOSIDES, NUCLEOTIDES AND NUCLEIC ACIDS, TAYLOR & FRANCIS, PHILADELPHIA, PA , vol. 9, no. 6 1 January 1990 (1990-01-01), pages 745-769, XP008152412, ISSN: 1525-7770, DOI: 10.1080/15257779008043142 Retrieved from the Internet: URL:http://www.tandfonline.com/doi/abs/10.1080/15257779008043142 [retrieved on 2006-10-04] |
| 11 | * | PETR ALEXANDER AND ANTONÍN HOL: “General Method of Preparation of N-[(S)-(3-Hydroxy-2-phosphonomethoxypropyl )] Derivatives of Heterocyclic Bases“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 5, 1 May 1993 (1993-05-01), pages 1151-1163, XP008152404, ISSN: 0010-0765, DOI: 10.1135/CCCC19931151 |
| 12 | * | SNOECK, ROBERT ET AL: “(S)-1-(3-Hydroxy-2-phosphonylmethoxypropy l)cytosine, a potent and selective inhibitor of human cytomegalovirus replication“, ANTIMICROBIAL AGENTS AND CHEMOTHERAPY , 32(12), 1839-44 CODEN: AMACCQ; ISSN: 0066-4804, 1988, XP002677023, |
| 13 | * | SYMERSK AND A HOL J: “Structure of 1-(S)-(3-hydroxy-2-phosphonylmethoxypropyl )cytosine; an antiviral agent“, ACTA CRYSTALLOGRAPHICA SECTION C. CRYSTAL STRUCTURE COMMUNICATIONS, MUNKSGAARD, COPENHAGEN, DK, vol. 47, no. 10, 15 October 1991 (1991-10-15), pages 2104-2107, XP008152403, ISSN: 0108-2701, DOI: 10.1107/S0108270190013257 |
| 14 | * | ULRIKA ERIKSSON: “Synthesis and biological evaluation of novel cidofovir prodrugs targeting HPepT1“, SYNTHESIS AND BIOLOGICAL EVALUATION OF NOVEL CIDOFOVIR PRODRUGS TARGETING HPEPT1- A DISSERTATION PRESENTED TO THE FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA IN PARTIAL FULFILLMEN , 1 January 2005 (2005-01-01), pages 1-287, XP008152406, Retrieved from the Internet: URL:http://search.proquest.com/pqdtscieng/docview/305421788/fulltextPDF/1371C222DB27F96BE2D/2?accountid=29404 |
| 15 | * | WEBB, ROBERT R., II ET AL: “Synthesis of (S)-N1-(3-hydroxy-2-phosphonylmethoxy)prop ylcytosine, (S)-HPMPC“, TETRAHEDRON LETTERS , 29(43), 5475-8 CODEN: TELEAY; ISSN: 0040-4039, 1988, XP002677025 |
| CN1559429A * | Feb 18, 2004 | Jan 5, 2005 | 肖广常 | 注射用西多福韦冻干粉针剂 |
| CN1690066B * | Apr 19, 2004 | Apr 28, 2010 | 横店集团成都分子实验室有限公 | 抗病毒剂西多福韦新衍生物 |
| CN101525352A * | Feb 16, 2009 | Sep 9, 2009 | 苏州凯达生物医药技术有限公司 | 西多福韦及其中间体的制备方法 |
| CN102268040A * | Sep 5, 2011 | Dec 7, 2011 | 扬州三友合成化工有限公司 | 抗病毒药物西多福韦的一种合成方法 |
| US5142051 | Jul 17, 1987 | Aug 25, 1992 | Ceskoslovenska Akademie Ved | N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases and a therapeutical composition therefrom with antiviral activity |
Trial to test laser acupuncture treatment for osteoarthritis
The potential for laser acupuncture to provide painless and effective treatment for osteoarthritis knee pain is being put to the test in a clinical trial beginning in Sydney. Traditional Chinese medicine practitioner Meikin Li Rees is recruiting 60 participants for the trial, being undertaken as part of her PhD research at UTS.
“Osteoarthritis (OA) is the most common form of arthritis and the major cause of musculoskeletal pain and immobility in elderly people worldwide,” Ms. Rees said. “In Australia, arthritis affects some 3.4 million people – nearly 17 per cent of the population.
“Of the proportion of Australians affected, 60 per cent are women and 60 per cent of all people living with arthritis are of working age. “If the current trend continues, one in five people, or around 4.6 million Australians, are forecast to be living with arthritis by 2020.”
Ms. Rees said the Osteoarthritis Research Society International already recommends needle acupuncture
View original post 151 more words
Uncialamycin

Uncialamycin
(1aS,11S,11aR,18R)-3,18-Dihydroxy-11a-[1(R*)-hydroxyethyl]-9,10,11,11a-tetrahydro-4H-11,1a-[3]heptene[1,5]diynonaphtho[2,3-h]oxireno[c]quinoline-4,9-dione
439.4163
C26 H17 N O6
870471-83-3 cas
WO2007038868A2, WO2013122823A1,
University of British Columbia (Originator)
uncialamycin, an enediyne natural product isolated from the Streptomyces uncialis, bacteria present on the surface of the lichen Cladonia uncialis.
Laboratory cultures of an undescribed streptomycete obtained from the surface of a British Columbia lichen produce uncialamycin (1), a new enediyne antibiotic.Uncialamycin exhibits potent in vitro antibacterial activity against Gram-positive and Gram-negative human pathogens, including Burkholderia cepacia, a major cause of morbidity and mortality in patients with cystic fibrosis.
Uncialamycin is an enediyne antibiotic with some unprecedented activity. The isolationists have filed a patent application almost right away. The total synthesis by Nicolaou [ACIE2007, 46, 4704] goes along nearly the same lines that have been predicted, and similar to Myers’ synthesis of dynemicin A [JACS 1997, 119, 6072], only it is not paper chemistry but the real one.

They have easily constructed the quinoline system with required functionality and subjected it to AllocCl-assisted acetylide addition (if I interpreted correctly “92% yield based on 80% conversion”). 5-alkoxyquinoline system was later advanced to iminoquinone and the two remaining rings were again attached by Hauser annulation with 3-cyanophthalide. The final product turned out be different from the one reported, more precisely, it was a C26-epimer. It is funny that I have accidentally drawn the correct structure with R-configuration at C-26 in the previous post.
The synthetic scheme allowed to easily invert this stereocenter via oxidation/reduction sequence on the last compound shown on the scheme below. The spectral properties of the final product thus obtained matched the reported data, and the structure of uncialamycin was confirmed by X-ray, despite it was isolated as an oil. The structure on the right is the revised one. The remaining details, including the chemistry behind DNA-cleaving Bergmann cycloaromatization,

Total Synthesis and Stereochemistry of Uncialamycin
K. C. Nicolaou, Hongjun Zhang, Jason S. Chen, James Crawford, Laxman Dasunoori
1Department of Chemistry and, The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
A new tot synth of Uncialamycin by Nicolaou. This is a natural occurring enediyne. Because the stereochemistry of C26 was unknown, both diastereomers as shown were synthesized. The retrosyn led back to simpler fragments 2, 3, and 4.
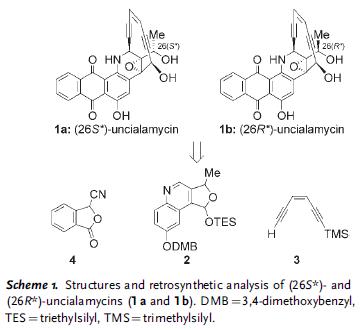
The following scheme illustrates the route to fragment 2. The key transformation was the two-step Friedlander quinoline synthesis (7 to 9).

Then fragment 2 was used in the following sequence. The key steps in the sequence involved installation of enediyne fragment 3 to give 11, the closure of the macrocycle to give 15, and the Hauser annulation in the last step to give 1a from 16.
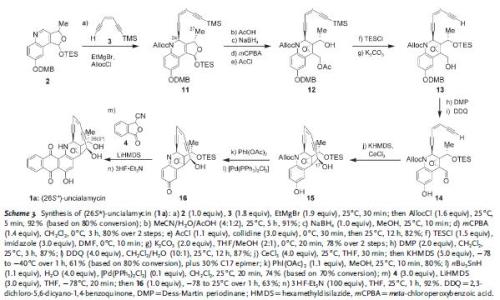
In this case, it was found that the final product’s spectrum (1a) did not match the reported value. And therefore, the other isomer was synthesized. This was easily done using fragment 12 through oxidation-reduction sequence to give 18 with the opposite stereochemistry at C26. Sequence in Scheme 3 was then repeated on this fragment.
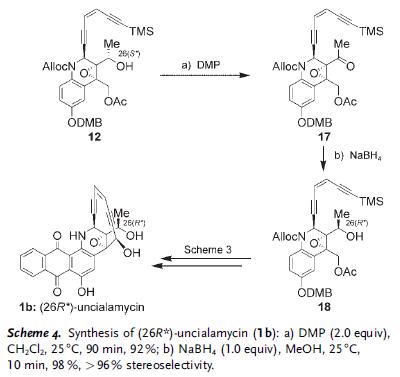
And 1b was found to match spectrum of the natural isomer. This natural compound was found to be stable as a solid and as solutions in a variety of solvents. But in presence of dray HCl in CH2Cl2 solution at rt, it rapidly converts to hexacyclic 19 through a cascade of Bergman cycloaromatization reaction. This cascade of reactions is believed to be responsible mode of action in damaging DNA and killing cells.

The enediynes are a family of antibiotics that possess a distinctive strained nine- or ten-member ring system comprising a Z-carbon-carbon double bond and two carbon- carbon triple bonds, usually arranged with the latter two flanking the former. The enediynes are potent damagers of DNA, causing single and double strand cuts. Their potency is attributed to their ability to bind to DNA and undergo a Bergmann rearrangement in which the strained ring system is converted into a highly reactive 1 ,4-benzenoid diradical, which damages the DNA by abstracting hydrogens from it.

Uncialamycin is an enediyne isolated from a Streptomyces strain found on the lichen Cladonia uncialis (Davies et al. 2005; 2007). (Full citations for references cited in this specification by first named author or inventor and year are provided in the section entitled “REFERENCES” later herein.)
Uncialamycin

The structure of uncialamycin has been confirmed by total synthesis (Nicolaou et al. 2007a; 2007b). In the course of the synthesis, it was noted that the unnatural 26(S) epimer was almost as active as the natural 26(R) epimer – that is, the stereochemistry of the C27 methyl had a minor effect on biological activity. Both epimers were active against several ovarian tumor cell lines. The IC50 values rang ed from 9 x 10“12 to 1 x 10“10, depending on the epimer and cell line or sub-line (Nicolaou et al, 2008).
Conjugates are an important method for the delivery of anti-cancer drugs, which are often highly cytotoxic and might otherwise be problematic to administer due to the risk of systemic toxicity. In a conjugate, the drug is conjugated (covalently linked) to a targeting moiety that specifically or preferentially binds to a chemical entity characteristic of the cancer cell, thus delivering the drug there with high specificity. Further, the drug is held in an inactive form until released from the conjugate, usually by cleavage of the covalent linker.
Typically, the targeting moiety is an antibody or an antigen-binding portion thereof, whose antigen is overexpressed or uniquely expressed by a cancer cell (“tumor associated antigen”). In such instances, the resulting conjugate is sometimes refered to as an “immunoconjugate” or an “antibody-drug conjugate” (ADC). Preferably the tumor associated antigen is located on the surface of the cancer cell, but also can be one that is secreted into the vicinal extracellular space. Upon binding, the antigen-conjugate complex is internalized and eventually finds its way inside a vesicular body such as a lysosome, where the covalent linker is cleaved, liberating active drug to exert its chemotherapeutic effect.
Advantageously, the covalent linker is designed such that cleavage is caused by a factor prevalent inside a cancer cell but not in plasma. One such factor is the low lysosomal pH, so that the covalent linker can be an acid-sensitive group such as a hydrazone. Another such factor is the generally higher intracellular concentration of glutathione, allowing for the cleavage of a disulfide covalent linker by a disulfide exchange mechanism. Yet another such factor is the presence of lysosomal enzymes such as cathepsin B, which can cleave peptide linkers designed to be preferred substrates (Dubowchik et al. 2002).
Conjugates have been used to deliver enediyne drugs in oncology. Gemtuzumab ozogamicin (Mylotarg®) is a conjugate of an anti-CD33 monoclonal antibody and a derivative of the enediyne calicheamicin. It was approved for treatment of acute
myelogenous leukemia but was later withdrawn from the market. Several other enediyne drugs, especially in the conjugated form, have been the subject of development efforts
 If handled carefully, enediynes make powerful cancer drugs.
If handled carefully, enediynes make powerful cancer drugs.
Inventors N. S. Chowdari, S. Gangwar, and B. Sufi synthesized enediyne compounds with general formula 1 that are based on the natural enediyne uncialamycin (2) scaffold (Figure 1). These compounds, used alone or in conjugates, are potent cytotoxins that may be useful in cancer chemotherapy.
Enediynes are a class of natural antibiotics that are characterized by 9- or 10-membered rings that contain two C≡C bonds separated by a cis (Z)-substituted C=C bond. Enediynes can undergo Bergman cyclization to form 1,4-benzenoid diradicals, which abstract hydrogen atoms from other molecules. When the diradical is generated near DNA, it abstracts hydrogen atoms from the sugar backbone of the DNA molecule and results in single- and double-strand lesions.
The high reactivity of enediynes toward DNA makes them very toxic. Their potent activity may be beneficial, however, if they are used to target the DNA of cancerous tumors. Most enediynes inhibit the proliferation of various cancer cells, including those that resist other chemotherapeutic drugs. Several naturally occurring enediynes are in clinical trials against cancer.
Both epimers at C26 of the natural enediyne uncialamycin are active against several ovarian tumor cell lines, with IC50 values ranging from 9 × 10–12 to 1 × 10–10 M, depending on the epimer and the cell line or subline. The synthetic enediynes described by the inventors are derivatives of uncialamycin.
Using these toxic molecules demands specific delivery systems. Conjugates are innovative drug-delivery systems designed to target tumor cells precisely and minimize the risk of systemic toxicity. Typically, drugs are linked covalently to conjugates that act as targeting moieties, which specifically or preferentially bind to a chemical entity characteristic of the cancer cell.
The covalent linker is designed to be cleaved only by a factor that exists inside a cancer cell and not in plasma, so that the drug remains in an inactive form until it is released from the conjugate. A typical targeting moiety may be a polymer or an antibody. Polymer-conjugated and antibody-linked enediyne drugs such as gemtuzumab ozogamicin (Mylotarg) were used to deliver enediyne drugs to cancer cells. Mylotarg, however, has been withdrawn from the market because of high patient mortality.

Compounds of structure 1 may be conjugated to a targeted moiety through a chemical bond to substituent R1. Compounds 3 and 4, shown in Figure 2, are examples of the synthetic enediynes with structure 1.
The investors tested the antiproliferative activities of several compounds against cancer cell lines. EC50 data for compounds 3 and 4 against 786-0 renal cancer cells and H226 lung cancer cells are shown in the table:
| Example | 786-0 cells, EC50 (nM) |
H226 cells, EC50 (nM) |
| 3 | 1.275 | 0.986 |
| 4 | 0.058 | 0.873 |

Several assays were also conducted on conjugates derived from other compounds of formula 1. (Bristol-Myers Squibb [Princeton, NJ]. WIPO Publication 2013122823, Aug 22, 2013;
DAVIES ET AL.: ‘UNCIALAMYCIN, A NEW ENEDIYNE ANTIBIOTIC‘ ORGANIC LETTERS vol. 7, no. 23, 13 October 2005, pages 5233 – 5236
http://pubs.acs.org/doi/abs/10.1021/ol052081f
 300 μg) as a bright purple [UV(MeOH): λmaxnm (ε) 206 (25000), 254 (33000), 280 (shoulder), 320 (shoulder), 539 (9400)] optically active ([α]D +3300 (c 0.005, MeOH)) oil.
300 μg) as a bright purple [UV(MeOH): λmaxnm (ε) 206 (25000), 254 (33000), 280 (shoulder), 320 (shoulder), 539 (9400)] optically active ([α]D +3300 (c 0.005, MeOH)) oil.

Table 1. 13C and 1H NMR Assignments for Uncialamycin (1). Data were Recorded in DMSO-d6 at 600 MHz for 1H
| position | δ 13C | δ 1H (mult., J (Hz)) |
| 1 | 10.0 (d, 4.6) | |
| 2 | 143.6 | |
| 3 | 110.4 | |
| 4 | 187.0a | |
| 5 | 134.4b | |
| 6 | 126.1c | 8.23 (dd, 1.4, 7.6)c |
| 7 | 133.6d | 7.88 (ddd, 1.4, 7.6, 7.6)d |
| 8 | 134.9d | 7.94 (ddd, 1.4, 7.6, 7.6)d |
| 9 | 126.6c | 8.24 (dd, 1.4, 7.6)c |
| 10 | 132.2b | |
| 11 | 182.2a | |
| 12 | 112.7 | |
| 13 | 154.9 | |
| 14 | 129.9 | 8.51 (s) |
| 15 | 135.6 | |
| 16 | 63.5 | |
| 17 | 63.0 | 5.14 (d, 3.3) |
| 18 | 100.4 | |
| 19 | 89.7 | |
| 20 | 123.4 | 6.05 (dd, 0.8, 10) |
| 21 | 124.0 | 5.97 (ddd, 1.4, 1.5, 10) |
| 22 | 87.4 | |
| 23 | 98.9 | |
| 24 | 43.2 | 5.04 (dd, 1.5, 4.6) |
| 25 | 76.0 | |
| 26 | 63.6 | 4.31 (qd, 6.0, 6.0) |
| 27 | 22.1 | 1.30 (d, 6.0) |
| 13-OH | 13.2 (brd.s) | |
| 17-OH | 6.66 (brd.s) | |
| 26-OH | 5.39 (d,6.0) |
a−d May be interchanged.http://pubs.acs.org/doi/suppl/10.1021/ol052081f/suppl_file/ol052081fsi20051004_065853.pdf
……………..
Isolation of Uncialamvcin
[0034] As part of a screening program aimed at discovering new antibiotics active against Bcc, it was found that crude organic extracts of cultures of a previously undescribed Streptomycete showed potent in vitro inhibition of Bcc. Bioassay guided fractionation of the crude extracts led to the identification of uncialamycin (1), a new enediyne antibiotic, as the active component. Bioactivity-guided fractionation involves thin layer chromatography of the extracts and fractions thereof and detection of the activity by overlaying a sensitive tester strain. A zone of inhibition identifies the active fraction containing the active compound.
The producing strain was extracted from the surface of the lichen Cladonia uncialis collected near Pitt River, British Columbia. Characterisation by 16S RNA sequencing showed the strain to be related, but not identical, to Streptomyces cyanogenus. Antibiotic activity of the strain was assayed by cutting plugs from solid agar cultures of the strain and placing them on lawns of tester strains of bacteria. Good inhibitory activity was detected against Gram-positive and Gram-negative bacteria (including Bcc), but not against yeasts.
Production cultures of the producing strain were grown as lawns on solid agar medium ISP4 for 14 to 21 days at room temperature. The solid agar cultures were lyophilized and extracted repeatedly with EtOAc. Concentration of the combined EtOAc extracts in vacuo gave a gummy residue that was partitioned between EtOAc and H2O. The EtOAc soluble material was fractionated by sequential application of flash C- 18 reversed-phase chromatography (eluent: step gradient from H2O to MeOH) and reversed-phase HPLC (column-Inertsil ODS-2; eluent: CH3CN/H2O 40:60) to give pure uncialamycin (1) (~ 300 μg) as a bright purple [UV(MeOH): λmaxnm (ε) 206 (25,000), 254 (33,000), 280 (shoulder), 320 (shoulder), 539 (9,400)], optically active ([α]D +3,300 (c 0.005, MeOH)) oil.
Chemical Characterization of Uncialamycin
Uncialamycin (1) gave a [M + Na]+ ion at m/z 462.0956 in the
HRESIMS appropriate for a molecular formula Of C26H17NO6 (calc’d for C26H17NO6Na 462.0954) requiring 19 sites of unsaturation. NMR data for uncialamycin was recorded in DMSO-^6 at 600 MHz using a cryoprobe. The 13C NMR spectrum (Table 1) showed well-resolved resonances for 26 carbon atoms and the 1H NMR spectrum contained resonances integrating for 17 protons in agreement with the HRMS data. Inspection of the HMQC data revealed that four of the protons (δ 5.39, 6.66, 10.0, and 13.2) were not attached to carbon atoms. Two major fragments A and B (Figure 1) of uncialamycin could be identified from analysis of the COSY, HMQC, and HMBC data obtained for the molecule.
Position δ 1W WH^mult, J(Hz)) ,
1 10.0 (d, 4.6)
2 143.6
3 110.4
4 187.0
5 134.4
6 126.1 8.23 (dd, 1.4, 7.6)
7 133.6 7.88 (ddd, 1.4, 7.6, 7.6)
8 134.9 7.94 (ddd, 1.4, 7.6, 7.6)
9 126.6 8.24 (dd, 1.4, 7.6)
10 132.2
11 182.2
12 112.7
13 154.9
14 129.9 8.51 (s)
15 135.6
16 63.5
17 63.0 5.14 (d, 3.3)
18 100.4
19 89.7
20 123.4 6.05 (dd, 0.8, 10)
21 124.0 5.97 (ddd, 1.4, 1.5, 10)
22 87.4
23 98.9
24 43.2 5.04 (dd, 1.5, 4.6)
25 76
26 63.6 4.31 (qd, 6.0, 6.0)
27 22.1 1.30 (d, 6.0)
13-OH 13.2 (brd.s)
17-OH 6.66 (brd.s)
26-OH 5.39 (d,6.0)
Table 1. C and H NMR assignments for uncialamycin (1). Data were recorded in OMSO-d6 at 600 MHz for 1H. [0038] A pair of olefinic resonances at δ 5.97 (H-21 ) and 6.05 (H-20), that were strongly correlated to each other in the COSY spectrum and had a coupling constant of 10 Hz, were assigned to a cis disubsituted olefin. The upfield olefinic resonance at δ 5.97 (H-21) showed strong HMBC correlations to non-protonated carbon resonances at δ 89.7 (C- 19) and 98.9 (C-23), and the downfield olefinic resonance at δ 6.05 (H-20) showed strong correlations to non-protonated carbon resonances at δ 87.4 (C-22) and 100.4 (C- 18). This suite of HMBC correlations identified an enediyne substructure in 1 (see Fragment A in Figure 1). The olefinic resonance at δ 5.97 (H-21) showed a long range COSY correlation to a methine resonance at δ 5.04 (H- 24), indicating that the carbon bearing the methine proton (C-24: δ 43.2) was attached to the C-23 alkyne carbon. A COSY correlation observed between the methine (δ 5.04, H-24) and a broad singlet at 10.0, that was not correlated to a carbon in the HMQC spectrum, and the chemical shift of the methine carbon (C-24, δ 43.2) suggested that C-24 had an NH substituent. HMBC correlations observed between the H-24 methine (δ 5.04) and the two alkyne carbon resonances at δ 87.4 (C-22) and 98.9 (C-23) confirmed the attachment of C-24 to the C-23 alkyne carbon.
A singlet methine resonance at δ 5.14 (H- 17) showed HMBC correlations to the alkyne carbon resonances at δ 89.7 (C- 19) and 100.4 (C- 18), which demonstrated that the methine carbon (C- 17: δ 63.0) was linked to the second alkyne at C-18. Both of the methine resonances at δ 5.04 (H- 24) and 5.14 (H- 17) showed HMBC correlations to a pair of deshielded resonances at δ 63.5 (C- 16) and 76.0 (C-25), assigned to non-protonated oxygen bearing carbons. This set of four HMBC correlations indicated that the two oxygenated carbons bridged the C- 17 and C-24 carbons to form a ten membered ring (C- 16 to C-25) containing the enediyne substructure. A COSY correlation between the methine resonance at δ 5.14 and a broad singlet at 6.66 (17-OH) revealed an alcohol funtionality attached to the methine carbon.
A methyl doublet at δ 1.30 (Me-27, J = 6 Hz) was correlated in the COSY spectrum to a methine at 4.31 (H-26, q, J = 6.0 Hz)) that was further correlated to a broad singlet at 5.39 (OH-26), assigned to an alcohol. The methyl resonance (δ 1.30, Me-27) showed an HMBC correlation to the carbon resonance at 76.0 (C-25), indicating that the hydroxyethyl fragment (C-26 and C-27) was the fourth subsituent on the non-protonated carbon C- 25. Both the NH-I proton (δ 10.0) and the H-17 methine (5.14) were correlated to a carbon at δ 135.6 (C- 15), and the H-24 methine (δ 5.04) was correlated to a carbon at 143.6 (C-2) in the HMBC spectrum indicating that the NH and C- 16 were vicinal substituents on an olefin or aromatic ring. A deshielded singlet at δ 8.51 showed strong HMBC correlations into carbon resonances at δ 63.5 (C-16), 143.6 (C-2), and 112.7 (C- 12) and a weak correlation into the carbon resonance at 154.9 (C- 13). This set of HMBC correlations confirmed that the NH and C-16 were attached to a benzene ring. Based on the assumption that the intense HMBC correlations were through three bonds, these correlations also indicated that the aromatic methine (δ 8.51, H-14) was ortho to C-16 (δ 63.5) and meta to the NH (C-2, δ 143.6). The weak HMBC correlation between δ 8.51 and 154.9 was attributed to a two bond coupling, placing the carbon at 154.9 (C-13) ortho to the methine carbon (C- 14) and its chemical shift required an oxygen substituent. [0041] The second fragment B of uncialamycin contained an isolated
1H spin system comprised of four contiguous aromatic protons (δ 8.23, dd, J = 1.4, 7.6 Hz H-6; 7.88, ddd, 1.4, 7.6, 7.6 Hz H-7; 7.94, ddd, J = 1.4, 7.6, 7.6 Hz H-8; 8.24, dd, J = 1.4, 7.6 Hz H-9). HMBC correlations observed between the proton resonance at δ 8.23 (H-6) and a carbon resonance at 187.0 (C-4) and between the proton resonance at 8.24 (H-8) and a carbon resonance at 182.2 (C-11) suggested that the other two subsituents on the benzene ring were quinone carbonyls. Fragments A and B shown in Figure 1 accounted for all of the carbon, hydrogen, and nitrogen atoms in the molecular formula of uncialamycin (1), but contained one extra oxygen atom. In order to complete the quinone and satisfy the remaining aromatic valences in Fragment A, the two carbonyl carbons of fragment B (C-4 and C-I l) had to be attached to the two substituted aromatic carbons (C-3 and C- 12) of fragment A. Finally, it was apparent that the two oxygentated carbons C- 16 and C-25 had to be bridged by an epoxide to account for the number of oxygen atoms and sites of unsaturation required by the molecular formula of 1. This implied that the C- 13 oxygen substituent had to be part of a phenol functionality that would engage in intramolecular hydrogen bonding with the C-I l carbonyl consistent with the observed OH chemical shift of δ 13.2.
A ROESY correlation between δ 5.14 (H- 17) and 4.31 (H-26) showed that C-26 and C- 17 were cis oriented about the C-16/C-25 epoxide and also defined the relative stereochemistry of H- 17 as shown. Molecular models revealed that due to steric and bond angle strain the C- 17 to C-23 enediyne containing bridge could only reasonably be cis fused to the piperidine ring. Uncialamycin (1) shares structural features with dynemicin A (2) and deoxydynemicin A (3) isolated from Micromonospora chersina. The H-24 resonance in uncialamycin (1) has a chemical shift of δ 5.04 and a 4.6 Hz coupling to the NH-I proton, which is nearly identical to the chemical shift (δ 5.05) and coupling (J = 4.3 Hz) of the corresponding methine proton (H-2) in dynemicin A (2), in agreement with the relative stereochemical assigment at C-24 in 1. Comparison of the additional NMR assigments reported for dynemicin A (2) and its triacetate derivative provided further strong support for the assigned structure of uncialamycin
…………….
Angewandte Chemie – International Edition, 2008 , vol. 47, 1 p. 185 – 189
http://onlinelibrary.wiley.com/doi/10.1002/anie.200704577/abstract

The highly potent DNA-cleaving molecule uncialamycin (1) was prepared in an asymmetric total synthesis featuring an enantioselective Noyori reduction. Compound 1 and its C26 epimer exhibit impressive broad-spectrum antibacterial properties and highly potent antitumor activities against a variety of cell lines.


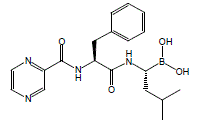
BORTEZOMIB, PS 341

BORTEZOMIB
A proteasome inhibitor.
[(1R)-3-methyl-1-({(2S)-3-phenyl-2-[(pyrazin-2-ylcarbonyl)amino]propanoyl}amino)butyl]boronic acid
Λ/-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid
179324-69-7 CAS
- Bortezomib
- HSDB 7666
- LDP 341
- LDP-341
- MG 341
- MLN-341
- NSC 681239
- PS 341
- PS 341 (pharmaceutical)
- PS-341
- UNII-69G8BD63PP
- Velcade
For treatment of multiple myeloma in patients who have not been successfully treated with at least two previous therapies.
A dipeptide boronic acid analogue with antineoplastic activity. Bortezomib reversibly inhibits the 26S proteasome, a large protease complex that degrades ubiquinated proteins. By blocking the targeted proteolysis normally performed by the proteasome, bortezomib disrupts various cell signaling pathways, leading to cell cycle arrest, apoptosis, and inhibition of angiogenesis. Specifically, the agent inhibits nuclear factor (NF)-kappaB, a protein that is constitutively activated in some cancers, thereby interfering with NF-kappaB-mediated cell survival, tumor growth, and angiogenesis. In vivo, bortezomib delays tumor growth and enhances the cytotoxic effects of radiation and chemotherapy. (NCI Thesaurus)
Bortezomib (originally PS-341 and marketed as Velcade by Millennium Pharmaceuticals) is the first therapeutic proteasome inhibitor to be tested in humans. It is approved in the U.S. for treating relapsed multiple myeloma and mantle cell lymphoma. In multiple myeloma, complete clinical responses have been obtained in patients with otherwise refractory or rapidly advancing disease.
bortezomib
Bortezomib (BAN, INN and USAN. Originally codenamed PS-341; marketed as Velcade by Millennium Pharmaceuticals and Cytomib by Venus Remedies) is the first therapeutic proteasome inhibitor to be tested in humans. It is approved in the U.S. for treating relapsed multiple myeloma[1] andmantle cell lymphoma. In multiple myeloma, complete clinical responses have been obtained in patients with otherwise refractory or rapidly advancing disease.
Bortezomib was originally synthesized in 1995 (MG-341) at a company called Myogenics, which soon changed its name to ProScript. After promising preclinical results, the drug (PS-341) was tested in a small Phase I clinical trial on patients with multiple myeloma cancer. ProScript ran out of money and was bought by Leukosite in May 1999. Leukosite in turn was bought by Millennium Pharmaceuticals in October 1999. At this point in time, the project had low priority amongst other projects at the company. This changed significantly when one of the first volunteers to receive the drug in the clinical trial achieved a complete response and were still alive four years later. At the time this was a remarkable result. Later clinical experimentation indicates the possibility of a complete response in 15% of patients in a similar condition, when treated with bortezomib.
In May 2003, seven years after the initial synthesis, bortezomib (Velcade) was approved in the United States by the Food and Drug Administration(FDA) for use in multiple myeloma, based on the results from the SUMMIT Phase II trial.[2]
Another commercially available bortezomib product – Bortenat (Natco Pharma, India), reportedly contains substantially more active entity than declared, potentially and even more resulting in increase toxicity. Moreover, Bortenat has some other chemical and formulation deviations from the registered ethic product Velcade (Millennium Pharmaceuticals and Janssen-Cilag), with unclear clinical impact.[3]

Bortezomib bound to the core particle in a yeast proteasome. The bortezomib molecule is in the center colored by atom type (boron = pink, carbon = cyan, nitrogen = blue, oxygen = red), surrounded by the local protein surface. The blue patch is catalyticthreonine residue whose activity is blocked by the presence of bortezomib.
VELCADE® (bortezomib) for Injection is an antineoplastic agent available for intravenous injection or subcutaneous use. Each single use vial contains 3.5 mg of bortezomib as a sterile lyophilized powder. Inactive ingredient: 35 mg mannitol, USP.
Bortezomib is a modified dipeptidyl boronic acid. The product is provided as a mannitol boronic ester which, in reconstituted form, consists of the mannitol ester in equilibrium with its hydrolysis product, the monomeric boronic acid. The drug substance exists in its cyclic anhydride form as a trimeric boroxine.
The chemical name for bortezomib, the monomeric boronic acid, is [(1R)-3-methyl-1-[[(2S)-1-oxo-3-phenyl-2[(pyrazinylcarbonyl) amino]propyl]amino]butyl] boronic acid.
Bortezomib has the following chemical structure:
 |
The molecular weight is 384.24. The molecular formula is C19H25BN4O4. The solubility of bortezomib, as the monomeric boronic acid, in water is 3.3 to 3.8 mg/mL in a pH range of 2 to 6.5.
Structure
The drug is an N-protected dipeptide and can be written as Pyz-Phe-boroLeu, which stands for pyrazinoic acid,phenylalanine and Leucine with a boronic acid instead of a carboxylic acid. Peptides are written N-terminus to C-terminus, and this convention is used here even though the “C-terminus” is a boronic acid instead of a carboxylic acid.
-
Boronic acid and ester compounds display a variety of pharmaceutically useful biological activities.Shenvi et al., U.S. Pat. No. 4,499,082 (1985 ), discloses that peptide boronic acids are inhibitors of certain proteolytic enzymes.Kettner and Shenvi, U.S. Pat. No. 5,187,157 (1993 ),U.S. Pat. No. 5,242,904 (1993 ), and U.S. Pat. No. 5,250,720 (1993 ), describe a class of peptide boronic acids that inhibit trypsin-like proteases. Kleeman et al., U.S. Pat. No. 5,169,841 (1992 ), disclosesN-terminally modified peptide boronic acids that inhibit the action of renin. Kinder et al., U.S. Pat. No. 5,106,948 (1992 ), discloses that certain tripeptide boronic acid compounds inhibit the growth of cancer cells.
-
More recently, boronic acid and ester compounds have displayed particular promise as inhibitors of the proteasome, a multicatalytic protease responsible for the majority of intracellular protein turnover.Ciechanover, Cell, 79: 13-21 (1994), discloses that the proteasome is the proteolytic component of the ubiquitin-proteasome pathway, in which proteins are targeted for degradation by conjugation to multiple molecules of ubiquitin. Ciechanover also discloses that the ubiquitin-proteasome pathway plays a key role in a variety of important physiological processes.
-
Adams et al., U.S. Patent No. 5,780,454 (1998 ),U.S. Patent No. 6,066,730 (2000 ), U.S. Patent No. 6,083,903 (2000 ),U.S. Patent No. 6,297,217 (2001 ), U.S. Patent No. 6,548,668 , andU.S. Patent No. 6,617,317 (2003 ), hereby incorporated by reference in their entirety, describe peptide boronic ester and acid compounds useful as proteasome inhibitors. The references also describe the use of boronic ester and acid compounds to reduce the rate of muscle protein degradation, to reduce the activity of NF-κB in a cell, to reduce the rate of degradation of p53 protein in a cell, to inhibit cyclin degradation in a cell, to inhibit the growth of a cancer cell, to inhibit antigen presentation in a cell, to inhibit NF-κB dependent cell adhesion, and to inhibit HIV replication.
-
Albanell and Adams, Drugs of the Future 27: 1079-1092 (2002), discloses that one such peptide boronic acid proteasome inhibitor, bortezomib (N-2-pyrazinecarbonyl-L-phenylalanine-L-leucineboronic acid), shows significant antitumor activity in human tumor xenograft models and is undergoing clinical evaluation. Richardson et al., New Engl. J. Med., 348:2609 (2003), reports the results of a Phase 2 study of bortezomib, showing its effectiveness in treating relapsed and refractory multiple myeloma.
-
Studies of boronic acid protease inhibitors have been greatly advanced by the development of chemistry for the preparation of functionalized boronic acid compounds, particularly alpha-halo- and alpha-aminoboronic acids. Matteson and Majumdar, J. Am. Chem. Soc., 102:7590 (1980), discloses a method for preparing alpha-chloroboronic esters by homologation of boronic esters, and Matteson and Ray, J. Am. Chem. Soc., 102:7591 (1980), reports that chiral control of the homologation reaction can be achieved by the use of pinanediol boronic esters. The preparation of alpha-aminoboronic acid and ester compounds from the corresponding alpha-chloroboronic esters has also been reported (Matteson et al., J. Am. Chem. Soc., 103:5241 (1981);Shenvi, U.S. Patent No. 4,537,773 (1985 )).
-
Matteson and Sadhu, U.S. Patent No. 4,525,309 (1985 ), describes an improved procedure for the homologation of boronic esters by rearrangement of the intermediate boron “ate” complex in the presence of a Lewis acid catalyst. The Lewis acid is reported to promote the rearrangement reaction and to minimize epimerization at the alpha-carbon atom. Rigorous exclusion of water and careful control of Lewis acid stoichiometry are required for optimum results, however. These features render the reaction difficult to perform successfully on a production scale, and limit the availability of pharmaceutically important boronic ester and acid compounds, such as bortezomib
The boron atom in bortezomib binds the catalytic site of the 26S proteasome[4] with high affinity and specificity. In normal cells, the proteasome regulates protein expression and function by degradation of ubiquitylated proteins, and also cleanses the cell of abnormal or misfolded proteins. Clinical and preclinical data support a role in maintaining the immortal phenotype of myeloma cells, and cell-culture and xenograft data support a similar function in solid tumor cancers. While multiple mechanisms are likely to be involved, proteasome inhibition may prevent degradation of pro-apoptotic factors, permitting activation of programmed cell death in neoplastic cells dependent upon suppression of pro-apoptotic pathways. Recently, it was found that bortezomib caused a rapid and dramatic change in the levels of intracellular peptides that are produced by the proteasome.[5] Some intracellular peptides have been shown to be biologically active, and so the effect of bortezomib on the levels of intracellular peptides may contribute to the biological and/or side effects of the drug.
 BORTEZOMIB
BORTEZOMIB
Bortezomib is rapidly cleared following intravenous administration.[6] Peak concentrations are reached at about 30 minutes. Drug levels can no longer be measured after an hour.Pharmacodynamics are measured by measuring proteasome inhibition in peripheral blood mononuclear cells. The much greater sensitivity of myeloma cell lines and mantle cell lines to proteasome inhibition compared with normal peripheral blood mononuclear cells and most other cancer cell lines is poorly understood.
Costs
UK
NICE recommended against Velcade in Oct 2006 due to its cost.[7]
The company proposed a cost reduction for multiple myeloma,[8] and this was taken up in the UK.[9]

Bortezomib is associated with peripheral neuropathy in 30% of patients; occasionally, it can be painful. This can be worse in patients with pre-existing neuropathy. In addition, myelosuppressioncausing neutropenia and thrombocytopenia can also occur and be dose-limiting. However, these side effects are usually mild relative to bone marrow transplantation and other treatment options for patients with advanced disease. Bortezomib is associated with a high rate of shingles,[10] although prophylactic acyclovir can reduce the risk of this.[11]
Gastro-intestinal (GI) effects and asthenia are the most common adverse events.[12]
Green tea extract epigallocatechin gallate (EGCG), which had been expected to have a synergistic effect, was found by Encouse B. Golden, et al. to reduce the effectiveness of bortezomib.[13][14][15][16]
Two open-label, phase II trials (SUMMIT and CREST) established the efficacy of bortezomib 1.3 mg/m2 (with or without dexamethasone) administered by intravenous bolus on days 1,4,8, and 11 of a 21-day cycle for a maximum of eight cycles in heavily pretreated patients with relapsed/refractory multiple myeloma.[17] The phase III APEX trial demonstrated the superiority of bortezomib 1.3 mg/m2 over a high-dose dexamethasone regimen (e.g. median TTP 6.2 vs 3.5 months, and 1-year survival 80% vs 66%).[17]
PATENTS
| Canada | 2203936 | 2005-04-12 | EXPIRY2015-10-27 |
| United States | 6713446 | 2002-01-25 | 2022-01-25 |
| United States | 6083903 | 1994-10-28 | 2014-10-28 |
Raghavendracharyulu Venkata Palle, Rajasekhar Kadaboina, Veerendeer Murki, Amarendhar Manda, Nageshwar Gunda, Ramaseshagiri Rao Pulla, Mallesha Hanmanthu, Narasimha Naidu Mopidevi, Suresh Kumar Ramdoss, “BORTEZOMIB AND PROCESS FOR PRODUCING SAME.” U.S. Patent US20100226597, issued September 09, 2010.
US20100226597 
INTRODUCTION
Bortezomib (PS-341, Velcade®; N-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid; (1R)-3-Methyl-1-[(2S)-3-phenyl-2-[(pyrazinylcarbonyl)amino]propanoyl]amino]butyl]boronic acid; CAS Registry Number: 179324-69-7) is an N-acylated dipeptide analogue of phenylalanyl-leucine in which a boronic acid functional group replaces the C-terminal carboxylic acid. It is a white to almost white crystalline powder and when appropriately formulated for injection is an anti-neoplastic agent and is a therapeutic proteosome inhibitor. In the US this active pharmaceutical ingredient (API) is approved for the treatment of multiple myeloma and mantle cell lymphoma.
Bortezomib is composed of three moieties that are fused together by two amide bonds. Two of these three units can be thought of as analogues of amino acids (viz., an α-aminoboronic acid and a pyrazinecarboxylic acid) and the third unit is a naturally occurring amino acid (viz., L-phenylalanine). Bortezomib possesses two chiral centres but is a single stereoisomer. One chiral centre exists within the α-aminoboronic acid moiety and the other exists within the naturally occurring amino acid, L-phenylalanine, moiety. In the solid state under anhydrous conditions, bortezomib can exist as a trimeric anhydride (trimeric boroxine), herein referred to as bortezomib anhydride. In the presence of water this can be hydrolysed to its monomeric boronic acid form.

Amino boronic acids – amino acids wherein terminal carboxylic groups are replaced by boronic B(OH)2 groups – are important pharmacoisosters of amino acids in various therapeutically promising molecules, mainly for treatment of cancer. For instance, talabostat contains proline boronic acid, bortezomib contains leucine boronic acid. Bortezomib, chemically Λ/-(pyrazin-2-yl)carbonyl-L-phenylalanine-L-leucine boronic acid, is an important proteasome inhibitor and has been clinically approved for use in treating mantle cell lymphoma and multiple myeloma. Recently, many novel molecules containing amino boronic acids, especially leucine boronic acid, have been prepared and biologically tested as described in WO2009/006473 A2.
The synthesis of bortezomib and other amino boronic acid and ester compounds is disclosed in
EP0788360 B1 , international patent application WO2005/097809 A2, international patent application
WO2009/004350 A1 , and international patent application WO2009/036281 A2.
EP0788360 B1 describes a general process for preparation of amino boronic acid and ester compounds using (1 S, 2S, 3R, 5S)-pinanediol leucine boronate and an amino acid or its derivative as starting materials. As coupling agents 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide hydrochloride (EDC), benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate (BOP reagent), or 0-(MH- benzotriazol-1-yl)-/V,/V,/V,/V-tetramethyluronium tetrafluoroborate (TBTU) were employed.
A synthetic process suitable for a large scale production of amino boronic acid and ester compounds is described in WO2005/097809 A2. The synthesis involves a boronate complex, which is contacted with a
Lewis acid under conditions that afford the boronic ester compounds.
WO2009/004350 A1 discloses a high yield synthesis of bortezomib and intermediates for the synthesis thereof. The procedure includes the use of a very high percentage of tetrahydrofuran in the halogenation of the starting compound (S)-pinanediol 2-m ethyl propane- 1 -boronate.
WO2009/036281 A2 describes processes for the preparation of substantially pure bortezomib and intermediates thereof. Processes for the preparation of crystalline forms of bortezomib as well as a storage system for bortezomib are also disclosed in said patent application.
In international patent application WO2005/097809 A2, in J. Biol. Chem. 1984, 259, 15106-15114 and in J. Am. Chem. Soc. 1981 , 103, 5241-5242 a route for the preparation of α-amino boronic esters, which is known to the person skilled in the art known as the Matteson’s synthetic route, is described. Homologation of boronic esters with (dichloromethyl)lithium to form α-chloro boronic esters has been shown to be efficient and result in good chiral selectivity if pinanediol was used as the chiral directing group. The use of the Lewis acid (ZnCI2) as a catalyst and chloride ion scavenger for the rearrangement of the borate intermediate improved the diastereomeric ratio in the α-chloro boronic ester product, α- Chloro boronic esters have been converted to silylated α-amino boronic esters by lithiumhexamethyldisilazane (LiHMDS), which have been desilylated and protonated in situ to the α-amino boronic esters.
An approach for the synthesis of diverse α-amino boronic esters by the highly diastereoselective copper- catalyzed addition of bis(pinacolato)diboron to N-tert-butane sulfinyl aldimines has been disclosed in the J. Am. Chem. Soc. 2008, 730, 6910-6911.
Transformation of 1 ,1-dihalogenoalkenes to corresponding alkynes and subsequent synthesis of 1- alkynylboranes have been described in Tetrahedron Letters 1972, 13, 3769-3772 and Tetrahedron Letters 1988, 29, 2631-2634.
J. Am. Chem. Soc. 1994, 116, 10302-10303 describes a process for preparing α-substituted 1- alkenyldioxaborolanes starting from 1-alkynyldioxaborolanes by hydrozirconation followed by substitution such as halogenation or carbonylation. The following α-substituted 1-alkenyldioxaborolanes are disclosed in this reference: (E)-2-(1-chloro-3,3-dimethylbut-1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2- (1-bromo-3,3-dimethylbut-1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2-(1-iodo-3,3-dimethylbut- 1-enyl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane, (E)-2,2,6,6-tetramethyl-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)hept-4-en-3-one, (E)-4,4-dimethyl-1-phenyl-2-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pent-2-en-1-one, (E)-2-(4,4-dimethylpent-2-en-2-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane zircono- cene and (E)-2-(hept-2-en-2-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane zirconocene.
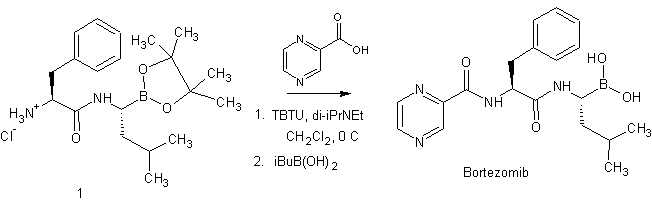

SYNTHESIS/US20120289699
The chiral centre of the α-aminoboronic acid moiety cannot, however, be derived from a chiral pool since α-aminoboronic acids are not known to be naturally occurring.
Instead, enantio-enriched α-aminoboronic acids in which the chiral centre is adjacent to the boron atom can be obtained by the use of chiral boron chemistry developed by Matteson, such as disclosed in U.S. Pat. No. 4,525,309 and a series of peer reviewed publications. Matteson’s chemistry when used for chiral applications utilises a boronic ester comprising a chiral diol auxiliary (such as 1S,2S,3R,5S)-(+)-2,3-pinanediol ((S)-(+)-pinanediol), for example) which upon reaction with the lithium salt (this salt can be prepared in situ or separately) of dichloromethane forms an α,α-dichloroboronate complex, which the boron ate functional group is chiral.
Due to induction provided by the chiral diol auxiliary, the boron ate complex undergoes a spontaneous and stereoselective internal rearrangement with displacement of one of the prochiral chloro substituents to generate an α-chloroboronic ester which possesses a newly generated chiral centre adjacent to the boron atom (See Scheme 1). This rearrangement of the boron ate complex is dramatically improved by catalysis with ZnCl2 (see J. Am. Chem. Soc., 1983, 105, 2077-2078).
α-Chloroboronic esters can be converted into the aforementioned requisite α-aminoboronic acids, preferably protected as boronic esters, possessing useful high chiral purity by reaction with LiHMDS followed by desilylation and optional salt formation of the amino group). Altogether, this reaction sequence provides a 1-carbon homologation of the original carbon backbone of the B-alkyl portion of the boronic ester in addition to a stereoselectively appended amino group. Most typically the chiral auxiliary demonstrated for this reaction sequence is homochiral pinanediol, such as the (+)-enantiomer referred to as (S)-(+)-pinanediol, or the (−)-enantiomer referred to as (R)-(−)-pinanediol.

One drawback with this stereoselective approach to α-aminoboronic acid synthesis in an industrial setting is the relatively high cost of the chiral diol auxiliary, pinanediol. Further, the use of the chiral diol imposes other synthetic restrictions, such as order of installation of the alkyl group to be homologated (i.e., the R group and dichloromethyl substituent) into the boron ate complex, and the relatively more difficult hydrolysis step required to remove stereochemically hindered diol groups afterwards. Despite this U.S. Pat. No. 7,714,159 B2, WO2009004350A1 and WO2009036281A2 disclose methods for the synthesis of bortezomib utilizing Matteson’s chemistry in conjunction with (S)-(+)-pinanediol as the chiral auxiliary.
Although a chiral auxiliary, such as (S)-(+)-pinanediol, is required for chiral induction in the homologation step in the Matteson reaction sequence, a chiral auxiliary itself is not inherently required for the Matteson homologation step to proceed, and achiral diols can also be used (Organometallics, 1983, 2, 1529-1535).
The inventors of the invention herein reasoned that the high cost of (S)-(+)-pinanediol could be circumvented in the synthesis of bortezomib by the use of a cheap, achiral diol to protect the boronic acid functional group. Since the use of an achiral diol auxiliary would not provide any stereochemical induction in the homologation step, a racemic product (that is, it would comprise equimolar amounts of each enantiomer) would be produced, which itself or a down stream synthetic derivative of it would require a classical resolution or other technique capable of separating the stereoisomers to be performed upon it.
Given that there was a need for a separation method that could separate the racemate, the inventors reasoned that one efficient approach would utilise the enantiopure API building block, L-phenylalanine as an intramolecular chiral resolving agent. L-phenylalanine or its derivatives could serve as a cost efficient in-process chiral resolving agent in this manner because i) it and its derivatives are cheap and are commercially available on large scales, and ii) it comprises part of the molecular structure of bortezomib itself. Therefore it was reasoned that its use would not be wasteful once the desired enantiomer of the racemate was separated because it would also be incorporated into the API itself.
Thus, a key characteristic of the invention herein useful for the synthesis of bortezomib is the use of a racemic diol α-aminoboronic ester salt, such as the pinacol derivative [5], as a key intermediate. This racemic key intermediate is derivatised by its reaction with L-phenylalanine to provide a mixture of diastereomers that are separated by crystallisation, or by chromatography, or by stereoselective hydrolysis.
The requisite racemic boronic esters, such as pinacol α-aminoboronic ester [5], are readily synthesized utilizing prior art chemistry disclosed by Matteson (e.g., see Pure & Appl. Chem., 1985, 57, 1741-1748), as exemplified in Scheme 2.

The racemic boronic esters, such as the pinacol α-aminoboronic ester [5], are then converted into mixtures of diastereomers [6] by reaction with a suitably protected L-phenylalanine derivative (See Scheme 3), such as N-BOC-L-phenylalanine. The protecting group of the L-phenylalanine moiety is then removed, such as by reacting the diastereomers [6] with an acid such as hydrochloric acid, to form a mixture of amine salt diastereomers [7] which is then subjected to conditions under which the desired diastereomer (R,S)-[7] is selectively isolated, such as by crystallisation, chromatography or stereoselective hydrolysis. The separated desired diastereomer (R,S)-[7] is then converted into bortezomib or bortezomib anhydride.

In this invention the need for the use of an expensive chiral auxiliary such as (S)-(+)-pinanediol to induce stereoselectivity in the Matteson homologation reaction sequence is circumvented by the use of the naturally occurring and relatively cheap amino acid L-phenylalanine in protected form. In addition to being 7-10 times cheaper than (S)-(+)-pinanediol, unlike (S)-(+)-pinanediol which is liberated from the penultimate API precursor at the end of the synthesis of bortezomib following the methods of the prior art, the amino acid, L-phenylalanine, comprises part of the final API molecular structure.
This invention differs from those disclosed in U.S. Pat. No. 7,714,159 B2, WO2009004350A1 and WO2009036281A2 which all rely on the use of the expensive chiral diol auxiliary (S)-(+)-pinanediol in conjunction with Matteson chemistry to obtain the requisite chirality.
EXAMPLES
For embodiment 1, as mentioned previously, the process has been demonstrated using pinacol as the boronate ester diol moiety and the hydrochloride salt of the diastereomeric mixture of [7].

Example 1 Synthesis of [7] Pinacol 1-chloro-3-methylbutane-1-boronate (rac-[3])
A mixture of THF (2 L) and DCM (55.3 g, 0.651 mol) was cooled to −100° C. n-BuLi (260.7 mL, 2.5 M in n-hexane, 0.651 mol) was added dropwise into the reaction mixture maintaining at −100° C. Pinacol 2-methylpropane-1-boronate ([2]; 100 g, 0.543 mol) was added into the reaction mixture. The resulting mixture was keep at −100° C. for one hour. A solution of ZnCl2 (136.3 g, 1.0 mol) in THF (500 mL) was added dropwise to the reaction over a period of 60 minutes. The resulting mixture was keep at −100° C. for one hour, the reaction mixture was warmed up to room temperature and keep at room temperature overnight. The reaction was diluted with MTBE (750 mL) and was washed twice with saturated NH4Cl (2 L each). The organic layer was dried overnight over MgSO4 before filtering and evaporating. n-Heptane (250 mL) was added into the mixture and was filtered and evaporated providing the product as an oil (119.5 g, 0.514 mol). 1H NMR (300 MHz, CDCl3) δ 3.48 (dd, J=9.8, 6.1 Hz, 1H), 1.93-1.71 (m, 2H), 1.61 (td, J=8.1, 4.0 Hz, 1H), 1.33-1.24 (m, 12H), 0.95-0.87 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 84.5, 42.8, 25.8, 24.8, 23.1, 21.5.
Pinacol 1-bis-(trimethylsilyl)-amino-3-butane-1-boronate (rac-[4])
A solution of LiHMDS (44.61 g, 0.267 mol in 217 mL THF) in THF (750 mL) was cooled to −75° C. and pinacol 1-chloro-3-methylbutane-1-boronate (rac-[3]; 77.5 g, 0.333 mol) in THF (462 mL) was added. The resulting mixture was keep at −75° C. for 1 hour. The reaction mixture was warmed up to room temperature and kept at room temperature overnight. The mixture was evaporated to provide the product as an oil (73 g, 0.204 mol). 1H NMR (300 MHz, CDCl3) δ 2.58 (t, J=7.7 Hz, 1H), 1.75 (tq, J=13.1, 6.5 Hz, 1H), 1.66-1.44 (m, 1H), 1.34-1.27 (m, 1H), 1.22 (s, 12H), 0.90-0.84 (m, 6H), 0.12-0.09 (m, 18H).
Pinacol-1-ammonium chloride-3-methylbutane-1-boronate (rac-[5])
A solution of pinacol 1-bis-(trimethylsilyl)-amino-3-butane-1-boronate (rac-[4]; 264.9 g, 0.741 mol) in n-heptane (4 L) and diethyl ether (1.6 L) was cooled to −35° C. HCl gas was bubbled through the mixture for 4 hours, and the resulting mixture was stirred at room temperature overnight and was then filtered. The filter cake was dissolved in DCM (1 L) and was stirred at room temperature for 2.5 hours, filtered and evaporated. The residue was diluted with EtOAc (713 mL) to form a slurry that was stirred for 1 hour and then filtered. The solid was dried under vacuum at 35° C. to provide the product as white crystals (123.9 g, 0.496 mol). 1H NMR (300 MHz, d6-DMSO) δ 7.75 (s, 3H), 2.70 (d, J=5.5 Hz, 1H), 1.68 (dt, J=13.5, 6.8 Hz, 1H), 1.44 (t, J=7.3 Hz, 2H), 1.24 (s, 12H), 0.86 (d, J=6.5 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 85.0, 38.6, 35.9, 25.1, 24.8, 22. 5; ESI-MS (positive) (m/z): 213, 170, 156, 128, 100, 88, 74.
Pinacol N-BOC-L-phenylalanine-D,L-leucine boronate ((R,S)-/(S,S)-[6])
To a cooled (about 0° C.) solution of BOC-L-phenylalanine (60.6 g, 0.228 mol) in DMF (670 mL) was added DIPEA (62.1 g, 0.480 mol), HATU (96.0 g, 0.252 mol) and a DMF (290 mL) solution of rac-[5] (pinacol-1-ammonium chloride-3-methylbutane-1-boronate) (60 g, 0.240 mol). The mixture was warmed to room temperature and stirred at this temperature overnight. Ethyl acetate (1 L) and a saturated aqueous solution of sodium of chloride (700 mL) were added into the reaction mixture. After mixing, the organic layer was separated and washed with a saturated aqueous solution of sodium of chloride (750 mL), then with an aqueous 0.1 N solution of KHSO4 (800 mL) and finally with an saturated aqueous solution of NaHCO3 (800 mL). The organic layer was dried over MgSO4 and concentrated at 35° C. n-Heptane (240 mL) was added to the crude product and was stirred for 45 min and was filtered. The filter cake was washed three times with n-heptane (100 mL each) and dried under vacuum at 35° C. The product was obtained as an approximately equimolar mixture of diastereomers as a white solid (92.0 g, 0.200 mol). 1H NMR (300 MHz, CDCl3) δ 7.37-7.18 (m, 5H), 6.30 (d, J=31.1 Hz, 1H), 5.07 (s, 1H), 4.45-4.27 (m, 1H), 3.06 (d, J=4.5 Hz, 2H), 2.96 (dd, J=10.8, 8.3 Hz, 1H), 1.39 (s, 9H), 1.37-1.29 (m, 3H), 1.25 (d, J=4.6 Hz, 12H), 0.85 (dt, J=11.3, 5.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 172.7, 155.6, 136.7, 129.6, 128.9, 127.1, 83.0, 80.4, 54.8, 55.4-53.8 (m), 39.9, 38.5, 37.6, 28.5, 25. 7, 25.1, 23.4, 22.0; ESI-MS (positive) (m/z): 461, 405.
Pinacol L-phenylalanine-L-leucine boronate, HCl salt ((R,S)-[7])
A MeCN (752 mL) solution of pinacol N-BOC-L-phenylalanine-D,L-leucine boronate ([6]; 94 g, 0.204 mol) was cooled to about 0° C. HCl gas was bubbled into the reaction mixture for 4 hours. The resulting mixture was stirred at room temperature overnight and was then evaporated to provide a solid. A slurry was formed by the addition of MeCN (250 mL) which was then stirred for 2 hours and was filtered and washed with MeCN (50 mL). The solid was then dried under vacuum at 35° C. furnishing the product as a white solid (56.4 g, 0.136 mol; HPLC purity 96.0% as a 1.5:1 mixture of (R,S)-[7] and (S,S)-[7])).
As mentioned previously the key upgrade step can be accomplished using:
- A) fractional crystallisation, or
- B) a reslurry/hydrolysis, or
- C) chromatography,
- D) or combinations of any of the above three techniques
These are exemplified in the following 3 examples.
Example 2 Operation A—Diastereomeric Upgrade of [7] by Fractional Crystallisation: The First Crystallisation
[7] (35.0 g, 88.2 mmol; (R,S)4(S,S)-diastereomeric ratio=1.50:1) was dissolved in a mixture of isobutyl acetate (350 mL) and ethanol (24.5 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (12.5 g, 31.5 mmol, HPLC purity 98.61% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 4.16:1).
The Second Crystallisation
[7] (12.5 g, 31.5 mmol; (R,S)-/(S,S)-diastereomeric ratio=4.16:1) was dissolved in a mixture of isobutyl acetate (125 mL) and ethanol (12.5 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (7.10 g, 17.9 mmol, HPLC purity 98.58% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 9.97:1).
The Third Crystallisation
[7] (7.1 g, 17.9 mmol; (R,S)-/(S,S)-diastereomeric ratio=9.97:1) was dissolved in a mixture of isobutyl acetate (71 mL) and ethanol (8.0 mL) at about 75° C. The solution was slowly cooled to ambient temperature and stirred overnight. The resulting mixture was cooled to about 0° C. and stirred for one hour and then filtrated and the isolated solid was dried under vacuum at 35° C. The product was obtained as white solid (5.4 g, 13.6 mmol, HPLC purity 96.69% ((S,R)-[7]+(S,S)-[7]), (S,R)-[7]/(S,S)-[7] ratio of 17.5:1). 1H NMR (300 MHz, d6-DMSO) δ 8.71 (d, J=15.6 Hz, 1H), 8.44 (d, 3H), 7.27 (m, 5H), 4.02 (s, 1H), 3.03 (m, 2H), 2.80 (d, J=4.5 Hz, 1H), 1.45 (m, 1H), 1.30-0.95 (m, 14H), 0.97-0.57 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 168.8, 134.1, 130.2, 129.0, 127.8, 83.7, 53.4, 40.3-36.7 (m), 32.0, 29.3, 25. 5, 25.0 (m), 23.4, 22.0; ESI-MS (positive) (m/z): 361, 261.
Example 3 Operation B—Diastereomeric Upgrade of [7] by Slurrying in a Wet Solvent:
A slurry of [7] (0.5 g, 1.26 mmol; (R,S)-/(S,S)-diastereomeric ratio=1.30:1) in ethyl acetate (15 mL) containing water (0.05 g, 2.78 mmol) was stirred at room temperature. After 72 hour, a sample was isolated as a white solid by filtration of the slurry and was analysed by HPLC showing a purity 97.7% and a (R,S)-/(S,S)-diastereomeric ratio of 7.0:1.
Example 4 Operation C—Diastereomeric Upgrade of [7] by Chromatography:
[7] (1.0 g, 2.52 mmol, (R,S)-/(S,S)-diastereomeric ratio=0.83:1 was dissolved in was dissolved in 1:4 i-PrOH/DCM (5.0 mL) and was purified by silica gel column chromatography eluting with 1:10 i-PrOH/DCM. Three fractions were collection providing 96.7% HPLC purity [7] (0.60 g; (R,S)-[7]/(S,S)-[7]=0.55:1), 97.2% HPLC purity [7] (0.10 g; (R,S)-[7]/(S,S)-[7]=1.98:1), and 95.9% HPLC purity [7] (0.20 g; (R,S)-[7]/(S,S)-[7]=2.25:1), after evaporation of the eluent.
For embodiment 2, examples are provided below.
Example 5 Pinacol N-(pyrazine-2-yl-carbonyl)-L-phenylalanine-L-leucine boronate ((R,S)-[8])
To a cooled (about 0° C.) solution of 2-pyrazinecarboxylic acid (1.61 g, 13 mmol) in DMF (84.6 mL) was added DIPEA (4.74 mL), HATU (5.43 g, 14.3 mmol) and recrystallised pinacol L-phenylalanine-L-leucine boronate HCl salt ((R,S)-[7]; 5.4 g, 13.6 mol, as a 17.5:1 diastereomeric mixture of (R,S)-[7]/(S,S)-[7]). The mixture was warmed to room temperature and was then stirred at this temperature overnight. Ethyl acetate (270 mL) and a saturated aqueous solution of sodium of chloride (260 mL) were added to the reaction mixture. After mixing, the organic layer was separated and washed with a saturated aqueous solution of sodium of chloride (182 mL), then an aqueous 0.1N solution of KHSO4 (273 mL) and finally a saturated aqueous solution of NaHCO3 (182 mL). The organic layer was dried over MgSO4, filtered and evaporated at 35° C. giving the product as white solid (5.77 g, 12.4 mmol; HPLC purity 87.8% ((R,S)-[8]+(S,S)-[8]) as a 22.0:1 diastereomeric mixture of (R,S)-[8])/(S,S)-[8]). 1H NMR (300 MHz, CDCl3) δ 9.34 (d, J=1.2 Hz, 1H), 8.72 (t, J=9.9 Hz, 1H), 8.53 (dd, J=2.4, 1.6 Hz, 1H), 8.36 (d, J=8.4 Hz, 1H), 7.26 (ddd, J=10.7, 6.9, 4.8 Hz, 5H), 6.06 (s, 1H), 4.83 (dd, J=14.1, 6.8 Hz, 1H), 3.24-3.15 (m, 2H), 3.06 (dd, J=12.5, 7.4 Hz, 1H), 1.51-1.31 (m, 3H), 1.30-1.22 (s, 12H), 0.83 (t, J=6.7 Hz, 6H); ESI-MS (positive) (m/z): 467.
Bortezomib (anhydride; N-(2-pyrazine)carbonyl-L-phenylalanine-L-leucine boroxine)
1 N HCl (37.1 mL) was added dropwise into a mixture of pinacol N-(pyrazine-2-yl-carbonyl)-L-phenylalanine-L-leucine boronate ([8]; 5.77 g, 12.4 mmol as a 22.0 (R,S)-[8])/(S,S)-[8] diastereomeric mixture) and 2-methylpropaneboronic acid (1.89 g, 18.5 mmol) in MeOH (57.7 mL) and n-heptane (57.7 mL). The reaction mixture was stirred at room temperature overnight. The water layer was separated and washed twice with n-heptane (30 mL each). The water layer was concentrated at 35° C. and DCM (30 mL) was added into the residue. 2 N NaOH (36.9 mL) was added dropwise into the reaction mixture. The water layer was separated and washed twice with DCM (30 mL each). After dilution with DCM (30 mL) 1 N HCl was added dropwise until the pH of the aqueous phase was about 6. The water layer was extracted twice with DCM (30 mL each). The DCM portions were collected together and concentrated at 35° C. Ethyl acetate (46 mL) was added into the residue and concentrated. Ethyl acetate (16 mL) was added into the residue and concentrated until approximately 10% of the original volume remained. n-Heptane (46 mL) was added and the resulting solid was then filtered, washed with n-heptane (20 mL) and dried under vacuum at 35° C. The crude bortezomib was obtained as yellow solid (3.7 g, 9.63 mmol).
Purification of bortezomib (anhydride; N-(2-pyrazine)carbonyl-L-phenylalanine-L-leucine boroxine), Form C Example 6
A mixture of crude bortezomib (24.2 g, 63.0 mol, HPLC purity: 97.9%), MeCN (181.5 mL) and i-PrOH (12.1 mL) was stirred at room temperature for 4 hours. The solid was filtered and dried at 30° C. under vacuum overnight providing bortezomib anhydride Form C as a white solid (18.6 g, 48.4 mmol, yield 76.9%, HPLC purity: 99.7% with no individual impurity>0.10%).
Example 7
A mixture of crude bortezomib (1.0 g, 2.36 mmol, HPLC purity: 90.7%) and MeCN (8.0 mL) was stirred at room temperature for 6 hours. The solid was filtered and dried at 35° C. under vacuum for 17 hours providing bortezomib anhydride Form C as a white solid (0.75 g, 1.94 mmol, yield 82%, HPLC purity: 99.2%).
Representative XRDP pattern, a DVS graph, 1H NMR spectrum, IR spectrum, and DSC and TGA traces of Form C are shown in FIGS. 1, 2, 3, 4, 5 and 6, respectively.
……………………………………
SYNTHESIS

…………………………..
SYNTHESIS

…………………………
α-substituted boronic ester (Vl).

Scheme 1
Scheme 3.

According to the preferred embodiment of Scheme 4 (wherein R1, R2, R3, X and A are as defined as in the items above), a compound of formula VIII, for example about 3.8 mmol, in form of its (R)- or (S)- enantiomer or in form of a mixture of enantiomers, can be prepared by contacting a compound of formula Vl dissolved in an organic solvent, preferably THF, with a solution comprising a reagent for substituting X with a protected amino moiety, for example sodium bis(trimethylsilyl)amide (NaHMDS) or lithium bis(trimethylsilyl)amide (LiHMDS), at suitably low temperature such as -60 0C to -10 0C, more preferably – 40 0C to -30 0C, in an inert, preferably argon atmosphere. The solution is warmed to room temperature such as about 20 0C to 25 0C, and stirred for a suitable period of time, for example 1 to 15 hours, preferably for about 5 hours. Then, the reaction mixture is evaporated to dryness and the residue is subsequently dissolved in a suitable volume of n-heptane, for example about 10 ml_, washed with a suitable volume of H2O, for example about 8 ml_, and washed with a suitable volume of saturated aqueous solution of NaCI, for example about 4 mL. The organic phase is then dried over a suitable drying agent, most preferably MgSO4, filtrated and evaporated to dryness. Compound of formula Vila obtained in such a manner is subsequently further converted into the compound of formula VIII by dissolving the previously obtained residue in a suitable volume of n-heptane, for example 20 mL, and by adding a suitable amount of anhydrous acidic solution, for example anhydrous HCI solution in Et2O, at a suitably low temperature such as -100 0C to -10 0C, more preferably -70 0C to -50 0C, in an inert, preferably argon atmosphere. The reaction mixture is warmed to room temperature, such as about 20 0C to 25 0C, and finally the precipitating solid is isolated from reaction mixture by filtration and washed with Et2O to give α- amino boronic ester (VIII).
According to another embodiment of the present invention, the racemic mixture of the α-amino boronic ester (VIII) obtained above can be further separated to yield optically pure (R)- or (S)-enantiomer by methods known in the art, such as enantiomeric resolution by crystallization with chiral acids, e.g. malic acid, tartaric acid, mandelic acid, or by chiral chromatography. In the special case wherein the borolane part of compound of formula VIII is chiral, compound of formula VIII represents a diastereomer. Since diastereomers differ in their scalar characteristics, diastereomeric compounds of formula VIII can be separated without providing a chiral environment, e.g by crystallization or chromatographic methods on achiral supporters.
Enantiomers obtained in such manner can then be subjected to further synthesis steps to yield compounds of general formula X or free acids or esters or anhydrides or salts thereof

, wherein R1 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, or substituted or unsubstituted aralkyl;
R2 and R3 independently from each other represent substituted or unsubstituted alkyl, substituted or unsubstituted aryl, or substituted or unsubstituted aralkyl, or R2 and R3 cooperatively form a part of a 5- to 10-membered fused or unfused ring, optionally a chiral 5- to 10-membered fused or unfused ring; and peptide comprises 1-6 amino acids coupled to each other by peptide bonds with optionally acylated terminal amino group, and wherein the chiral center * is in its (R) or (S) configuration.
For example, the racemic mixture of the intermediate compound of formula VIII, for example 3-methyl-1- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)butan-1-amine hydrochloride is first separated by enantiomeric resolution method known in the art to give (R)-3-m ethyl- 1 -(4,4,5, 5-tetram ethyl- 1 , 3,2- dioxaborolan-2-yl)butan-1-amine hydrochloride, which can then be subjected to further synthesis steps to yield bortezomib by synthesis routes known to or readily devisable by a person skilled in the art. For example, the following synthesis routes may be applied:

enantiomeric resolution coupling reagent

3-methyl-1 -(4,4,5,5-tetramethyl-
1,3,2-dioxaborolan-2-yl)butan-1-amine

hydrochloride hydrochloride

Scheme 5
Another aspect of the invention is a conversion of a compound of formula V or Vl to its trifuoroborate derivative compound of formula V* and Vl*, respectively, which can be further converted to compound VIII* as depicted in Scheme 6 below.

Scheme 6
………………………
Improved process for manufacturing the proteasome inhibitor bortezomib. Thus, in one embodiment, the invention provides a large-scale process for forming a compound of formula ( XIV ):

or a boronic acid anhydride thereof. The process comprises the steps:
- (a) providing a boron “ate” complex of formula ( XV ):

wherein:
- R3
- is a nucleofugic group;
- Y
- is a nucleofugic group; and
- M+
- is an alkali metal;
- (b) contacting the boron “ate” complex of formula ( XV ) with a Lewis acid under conditions that afford a boronic ester compound of formula ( XVI ):

said contacting step being conducted in a reaction mixture comprising:
- (i) a coordinating ether solvent that has low miscibility with water; or
- (ii) an ether solvent that has low miscibility with water and a coordinating co-solvent;
- (c) treating the boronic ester compound of formula ( XVI ) with a reagent of formula M1-N(G)2, where M1 is an alkali metal and each G individually or together is an amino group protecting group, to form a compound of formula ( XVII ):

- (d) removing the G groups to form a compound of formula ( XVIII ):

or an acid addition salt thereof;
- (e) coupling the compound of formula ( XVIII ) with a compound of formula ( XIX );

wherein:
- P1 is a cleavable amino group protecting moiety; and
- X is OH or a leaving group;
to form a compound of formula ( XX ):

wherein P1 is as defined above;
- (f) removing the protecting group P1 to form a compound of formula ( XXI ):

or an acid addition salt thereof;
- (g) coupling the compound of formula ( XXI ) with a reagent of formula ( XXII )

wherein X is a OH or a leaving group, to form a compound of formula ( XXIII ):

and
- (h) deprotecting the boronic acid moiety to form the compound of formula ( XIV ) or a boronic acid anhydride thereof.









Compound 1 (1R)-(1S,2S,3R,5S)-pinanediol-1-ammoniumtrifluoroacetate-3-methylbutane-1-boronate

Compound 2(1S)-(1S,2S,3R,5S)-pinanediol-1-ammoniumtrifluoroacetate-3-methylbutane-1-boronate

SubstancesCompound 3 N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride

Compound4N-(2-Pyrazinecarbonyl)-D-phenylalanine-L-leucine boronic anhydride

Compound 5 N-(2-Pyrazinecarbonyl)-L-phenylalanine-D-leucine boronic anhydride

N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride
-
A solution of (1S,2S,3R,5S)-Pinanediol N-(2-pyrazinecarbonyl)-L-phenylalanine-L-leucine boronate (25.2 g) in 207 mL of MeOH and 190 mL of hexane was cooled to 15 °C, and 109.4 mL of 1N HCl were added in portions, keeping the temperature between 15 and 25 °C. 2-Methylpropaneboronic acid (8.67 g) was then added under vigorous stirring, and the stirring of the biphasic mixture was continued over night. After separation of the two phases, the lower layer was extracted once with 75 mL of hexane. The lower layer was then concentrated in vacuo until it became cloudy, followed by the addition of 109.4 mL of 2N NaOH and 100 mL of Et2O. The two phases were separated the lower layer was extracted with Et2O (4 · 100 mL each), and then brought to pH 6.0 by the addition of 109 mL of 1N HCl. After extraction with 100 mL of ethyl acetate, the lower layer was adjusted to pH 6.0 with 1N HCl and extracted one more time with 75 mL of ethyl acetate. The combined ethyl acetate layers were washed with semi-saturated brine (2 · 25 mL) and brine (2 · 25 mL), dried over Na2SO4, filtered, and concentrated to afford 15.3 g (81.8 %) of crude N-(2-Pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride as a foam. The crude material was dissolved in 150 mL of ethyl acetate and concentrated in vacuo to a suspension, followed by the addition of 150 mL of MTBE. The suspension was stored between 2 and 8 °C over night, filtered, washed twice with MTBE, and dried under high vacuum, yielding 10.69 g (57.2 %) of N-(2-pyrazinecarbonyl)-L-phenylalanine-L-leucine boronic anhydride as a white solid.
………………..
Synthesis
In an illustrative example of the first process of the present invention, the compound of formula (2) reacts with the compound of formula (6) ,wherein Rl is hydrogen, under presence of the coupling agent of formula (8A). The formed B-OH protected compound (4) is then deprotected to bortezomib.

The compound of formula (2) is a known compound. It may be prepared by processes known in the art, which generally start from (S)-pinanediol and 2-methylpropane boronic acid. The processes are disclosed, e.g., in WO 2005/097809, WO 2009/004350 and
WO 2009/036281. The compound (2) may be used per se or, preferably, as an acid addition salt. The most preferred acid addition salt is a trifluoroacetate salt as it is easily preparable and is crystalline.
The second reaction partner, the compound of formula (6), is advantageously prepared by a process, in which L-phenylalanine alkylester and/or its acid addition salt having the formula (5)

wherein Rl is a C1-C4 alkyl group and is preferably methyl group, is coupled in an inert solvent with pyrazine-2-carboxylic acid of formula (7) in the presence of a base.
According to one aspect of the present invention, the coupling reaction proceeds in a presence of the coupling agent of the formula (8) above, typically with the n-propylphosphonic anhydride of the formula (8A). In an advantageous embodiment, the inert solvent may be an aliphatic, cyclic or aromatic C5-C10 hydrocarbon or a halogenated C1-C4 aliphatic hydrocarbon. The base is advantageously a tertiary amine, e.g. N-methylmorpholine. The reaction temperature is typically from -20 to 0 °C, under which temperature the reaction time is about 2-4 hours. The amount of the coupling agent of formula (8) is advantageously from 1 to 2 molar equivalents in respect to the compound (5). The reaction progress may be monitored by a suitable analytical technique, e.g. by HPLC or GC. After the reaction has been terminated, the reaction mixture is elaborated by an extraction with water, by which rests of the coupling agent and the base are removed. The reaction product may be isolated from the organic phase by common means, e.g. by evaporation, or the organic phase may be used in the next step directly.
In the next step, the so formed intermediate of formula (6), in which Rl is a C1-C4 alkyl group, and is preferably methyl group, is hydrolysed by water to the compound of formula (6), in which Rl is hydrogen. Preferably, the hydrolysis is performed in a water miscible solvent in a presence of a base, e.g. an amine base. It is important to assure that essentially no epimerization occurs during the hydrolysis. Therefore, the conditions of hydrolysis must be very mild. In an advantageous mode, the basic hydrolysis under mild conditions may be performed in presence in lithium salts , for instance lithium chloride, lithium bromide, lithium nitrate, lithium trifluoroacetate , lithium tetrafluoroborate etc.
The hydrolysed compound is advantageously isolated from the reaction mixture after neutralization thereof, preferably by an extraction. The crude product may be precipitated in solid form from the extract, e.g. by using antisolvent, which typically is an aliphatic hydrocarbon such as hexane or heptanes. The crude solid may be isolated by filtration and optionally recrystallized from a suitable solvent or a solvent/antisolvent mixture.
Having the compound (2) and compound (6) available, the key step in making bortezomib according to process of the present invention comprises coupling, under presence of a base, the compound (6), in which Rl is hydrogen, with the compound (2), which preferably is charged as an acid addition salt and most preferably as trifluoroacetate salt, in an inert solvent, whereby the coupling agent necessary for the mutual reaction is the compound of formula (8), preferably of formula (8A). In an advantageous embodiment, the inert solvent may be an aliphatic, cyclic or aromatic C5-C10 hydrocarbon or a halogenated C1-C4 aliphatic hydrocarbon. The base is advantageously a tertiary amine, e.g. N-methylmorpholine. The reaction temperature is typically from -30 to 0 °C, preferably from -20 to -10°C, under which temperature the reaction time is about 1-2 hours. The amount of the coupling agent of formula (8) is advantageously from 1 to 2 molar equivalents in respect to the compound (2). The reaction progress may be monitored by a suitable analytical technique, e.g. by HPLC or GC. After the reaction has been terminated, the reaction mixture is elaborated by an extraction with water, by which rests of the coupling agent and the base are removed. The reaction product may be isolated from the organic phase by common means, e.g. by evaporation and may be optionally purified, e.g. by column chromatography.
Whenever useful, reaction products of any of the steps of the process may be used in the next step without isolation from the reaction mixture.
In the last step, the so formed protected bortezomib intermediate of the formula (4) is deprotected by yielding bortezomib of formula (1). Any of deprotecting procedures known in the art may be used. In particular, the transesterification step disclosed in WO 2005/097809, in which the protected bortezomib reacts with an organic boronic acid acceptor in acidic conditions, represents an useful deprotecting process. In an illustrative example of the second process of the present invention, the compound of formula (2) reacts in the first step with the L-phenylalanine compound of formula (5a), wherein R is a nitrogen protective group. The useful nitrogen protective group is a tert.butyloxycarbonyl group (tBOC), but is should be understood that other suitable nitrogen protective groups may be used as well.

Similarly as in the first process of the present invention, the compound of formula (2) may be charged in the reaction as an acid addition salt, preferably the trifluoroacetate salt. The coupling reaction with the tBOC-protected compound (5a) typically proceeds in an inert solvent, in the presence of base and is, in accordance with the present invention, mediated by the action of the coupling agent of the formula (8), preferably (8A). The inert solvent is advantageously a chlorinated C1-C4 hydrocarbon or C5-C10 aliphatic, cyclic or aromatic hydrocarbon, the base is preferably a tertiary amine. The reaction conditions and workup of the reaction mixture are essentially the same as disclosed in the first process of the present invention.
In a next step, the protective group R in the so formed compound (3) is removed to yield a compound of formula (3) in which R is hydrogen. In case of the tBOC protective group, the deprotection is typically performed by treating the substrate with HC1 in ethyl acetate. After termination of the reaction, the product may be isolated from the reaction mixture by diluting with a hydrocarbon, e.g. with heptanes, and precipitating the product as a hydrochloride salt.
In the second coupling step, the compound (3), in which R is hydrogen, reacts with the 2-pyrazinecarboxylic acid of formula (7) in the presence of the coupling agent of the formula (8), preferably (8a), in an inert solvent and in the presence of a base.

EXAMPLES
EXAMPLE-1 : PROCESS FOR PREPARING N-[(1 S)-2-[[(1 R)-1-[(3aS,4S,6S,7aR)- hexahydro-3a,5,5-thmethyl-4,6-methano-1 ,3,2-benzodioxaborol-2-yl]-3-methyl butylamino]-2-oxo-1 -(phenylmethyl)ethyl] Pyrazinecarboxamide (FORMULA IX)

The process for preparing compound of formula IX comprises of the steps from Step a) to step h), which are individually demonstrated below:
Step-a) Preparation of 2-(2-Methylpropyl)-(3aS,4S,6S,7aR)-hexahydro-3a,5,5- trimethyl -4,6-methano-1 ,3,2-benzodioxaborole (Formula II):

To a stirred solution of isobutyl boronic acid (50.0 g) in n-heptane (250 ml) at 25.- 300C, was added (+)-Pinanediol (83.3 g) and stirred for I hour at 25-300C. To the reaction mass was added brine solution and the mixture was stirred. The layers were allowed to separate and the organic layer was concentrated under reduced pressure till no more solvent distills off to give the title compound (Formula II).
Step-b) Preparation of 2-((1 S)-1 -Chloro-3-methylbutyl)-(3aS,4S,6S,7aR)- hexahydro-3a,5,5- trimethyl-4,6-nnethano-1 ,3,2-benzodioxaborol (Formula III):

I. preparing a mixture of zinc chloride with tetrahydrofuran
II. preparing LDA mixture
III. preparing a solution of compound of formula-ll

in a solvent mixture comprising dichloromethane and water miscible ether solvent
IV. adding solution of step Il into the solution of step III followed by maintaining the solution at a temperature of about -40 to -700C
V. adding the mixture of step I into the product of step IV followed by maintaining the reaction mass at a temperature of about -40 to -700C
VI. raising the reaction temperature up to about 10°C to ambient temperature
VII. adding the aqueous acid solution VIII. separating the organic layer containing the compound of formula-Ill, and isolating the product.
I. Preparing a mixture of zinc chloride with tetrahydrofuran
Charged ZnCI2 (115 g) to tetrahydrofuran (805 ml) into a 1st Round bottom flask (R. B. flask) under nitrogen atmosphere at 25 to35°Cand the temperature of the resulting mixture was raised to 35 to 400C, maintained for 3-4 hours to give ZnCI2 solution.
II. Preparing LDA mixture
Charged diisopropyl amine (86 ml) to tetrahydrofuran (345 ml) into a 2nd R. B. flask under nitrogen atmosphere and resultant mixture was cooled to -7 to -150C, charged n-hexyl lithium to the above mixture and maintained for 30-40 minutes to give LDA mixture.
III. Preparing a solution of compound of formula-ll Compound of Formula Il (115.O g) was charged to a dichloromethane (161 ml) and tetrahydrofuran (690 ml) into a 3rd R. B. flask under nitrogen atmosphere at 25 to35°C and the mixture was cooled to -55 to -600C.
IV. Adding solution of step Il into the solution of step III Charged LDA mixture from the 2nd R. B. flask to the reaction mixture at -55 to –
600C and maintained for 30 minutes. The temperature was raised to -500C.
V. Adding the mixture of step I into the product of step IV
Charged ZnCI2 solution from the 1st R. B. flask at -45 to -500C and maintained for I hour. Vl. Raising the reaction temperature up to about 100C
The reaction mixture was warmed to 100C. VII. Adding the aqueous acid solution
Charged 10% H2SO4, stirred for 10-15 minutes and the organic layer was separated. VIII. Separating the Organic layer containing the compound of formula-Ill, and isolating the product.
The organic layer separated under step VII was subjected to next step, however, the aqueous layer was discarded.
Washed the organic layer with brine solution under stirring, till the aqueous layer reached to a pH around 6-7.
The organic layer was concentrated to isolate the compound of formula-Ill, under reduced pressure.
Step-c) Preparation of N,N-Bis(thmethylsilyl)-(1 R)-1 -[(3aS,4S,6S,7aR)- hexahydro-3a,5,5-thmethyl-4,6-methano-1 ,3,2-benzodioxaborol-2-yl]-3- methylbutylamine (Formula IV):

iv
Hexamethyldisilazane (101.3 ml) was charged to tetrahydrofuran (414 ml) under nitrogen atmosphere and the mixture was cooled to -20 to -300C. Charged n- hexyllithium slowly to the above mixture under stirring by maintaining the temperature at -20 to -30 0C. The reaction mixture was stirred for 1 -2 hours at -20 to -250C, charged compound of Formula III (138 g) to the above freshly prepared lithium HMDS in THF by maintaining the temperature at -15 to -200C. The reaction mixture was warmed to a temperature of 25-300C and maintained for 2-3 hours. Filtered the reaction mixture through silica bed and washed the bed with diisopropyl ether. The filtrate was concentrated under reduced pressure to a residue to give the title compound (Formula IV).
Step-d) Preparation of 4,6-Methano-1 ,3,2-benzodioxaborole-2-methanamine, hexahydro-3a,5,5-thmethyl-α-(2-methylpropyl)-,(αR,3aS,4S,6S,7aR)-,trifluoro acetate (Formula V):

Charged thfluoroacetic acid (129 ml) to diisopropyl ether (1980 ml) under nitrogen atmosphere at 25-300C and the reaction mass cooled to -100C. Charged compound of Formula IV (198 g) to the reaction mass slowly at -100C and maintained at the same temperature for 8 hours. The reaction mass was filtered, washed with diisopropyl ether (198 ml) and the obtained solid was slurry washed with water (1500 ml) at 25-300C. The slurry was filtered washed with water and the solid obtained was dried at 40-500C under reduced pressure for 8 hours to give 74.Og of the title compound (Formula V). Purity (by GC) = 99.54%
Step-e) Preparation of L-Phenylalanine methyl ester hydrochloride (Formula Vl):

To a stirred mixture of L-phenyl Alanine (25 g) in methanol (125 ml) at 25-300C, was charged thionyl chloride (13.2 ml) under stirring and the mixture was maintained at 55-600C for 2-3 hours. The reaction mass was cooled to 25-300C and concentrated under reduced pressure up to 2 volumes with respect to the staring material. Charged isopropyl alcohol (125 ml) to the reaction mass and concentrated up to 2 volumes with respect to the starting material. Cooled the reaction mass to 0-50C and maintained under stirring for 1-2 hours. Filtered the reaction mass, washed with isopropyl alcohol, suck dried for 30 minutes and the solid obtained was dried at 45- 500C for 3-4 hours to give 28.8 g of the title compound (Formula Vl). Chiral purity by HPLC: 100%.
Step-f) Preparation of L-Phenylalanine, N-(pyrazinylcarbonyl)-methyl ester (Formula VII):

To a stirred mixture of Pyrazine-2-carboxylic acid (3.45 g) in DMF (50 ml) at 25-300C, was charged N-hydroxy succinimide (3.2 g) under stirring and was cooled to 0-50C. Charged N,N’-dicyclohexylcarbodiimide (DCC) (5.75 g) to the reaction mass at 0-5 0C and stirred for 15-20 minutes. Charged compound of Formula Vl (5.0 g) to the reaction mass at 0-50C and stirred for 15-20 minutes. Further, charged NMM (3.8 ml) to the reaction mass at 0-50C and stirred for 15-20 minutes. The reaction mixture was warmed to 25-300C and maintained under stirring for 2-3 hours. The reaction mass was filtered and the solid was separated. The filtrate obtained was diluted with ethylacetate (100 ml) and washed with demineralized water. The organic layer was washed with 1 N HCI, followed by washing with sodium bicarbonate solution. Concentrated the organic layer up to 2 volumes with respect to Formula Vl under reduced pressure and cooled to 25- 300C. Charged n-heptane (20 ml) to precipitate the compound, cooled the reaction mass to 0-50C, maintained for 1 -2 hours and filtered under vacuum. The solid obtained was dried at 40-450C for 3-4 hours to give 5.7 g of the title compound (Formula VII). Purity by HPLC: 99.73%, chiral purity by HPLC: 99.97%.
Step-g) Preparation of N-(pyrazinylcarbonyl)-L-Phenylalanine (Formula VIII):

To a stirred mixture of Formula VII (100 g) in acetone (500 ml) at 25-300C, was charged NaOH solution (obtained by dissolving 15.4 g of NaOH in 500 ml of water) and maintained at the same temperature for 30-50 minutes. Adjusted the pH of the reaction mass to 2 by using 1 N HCI and cooled the reaction mass to 0-50C. Maintained the reaction mass at 0-5 0C under stirring for 1 -2 hours, filtered under vacuum and dried the material obtained at 45-500C for 4-5 hours to give 84.4 g of the title compound (Formula VIII). Purity by HPLC: 99.94 % by weight. Chiral purity by HPLC: 100 %
Alternately, N-(pyrazinylcarbonyl)-L-Phenylalanine (Formula VIII) may also be prepared by
(a) Using ethylchloroformate according to the process as described below: A mixture of acetone (40 ml), pyrazine carboxylic acid (5 g) and thethylamine
(6.77 ml) was cooled to about -5°C to about 00C and ethylchloroformate (4.76 ml) was charged. The reaction mass was stirred for about 30 minutes. The reaction suspension was allowed to reach the temperature of about 25°C to about 300C and maintained for about 3 hours. The reaction suspension was cooled to about 00C to about 5°C. In the second flask the aqueous sodium hydroxide (1.68 g in 70 ml water) solution was cooled to about 00C to about 5°C and to that acetone (30 ml) and L-phenyl alanine (6.6 g) were added and the mixture was stirred for about 1 hour at that temperature. The reaction mass of the second flask was added to the reaction mass of the first flask at a temperature of about 00C to about 5°C and then stirred for about 2 hours followed by raising the temperature to about 25°C to about 300C. The reaction mass was further stirred for about 16 hours at a temperature of about 25°C to about 300C. Ethyl acetate (150 ml) was charged to the reaction solution and stirred for about 30 minutes. The layers were separated and 1 N hydrochloric acid (35 ml) was added to the separated aqueous layer. The reaction solution was cooled to about 00C to about 5°C and stirred for about 2 hours. The obtained suspension was filtered and the solid was washed with water (10 ml). The solid was then dried at a temperature of about 500C for about 4 hours to afford 2.6 g of title compound. Purity by HPLC: 99.2% by weight. Chiral purity by HPLC: 100% (b) or by using combination of EDCHCI, HOBt, according to the process as described below:
A mixture of pyrazine carboxylic acid (168.7 g), dimethylformamide (1.4 lit), hydroxybenzotriazole (HOBt:220 g), and N-methyl morpholine (221 ml) was cooled to a temperature of about 00C to about 5°C. EDC. hydrochloride (1 -Ethyl-3-(3- dimethylaminopropyl) carbodiimide-HCI; 278 g) was added to the reaction solution at a temperature of about 00C and stirred for about 30 minutes. L-phenylalanine methyl ester hydrochloride (240 g) obtained from above was dissolved in DMF (1 lit) and then added to the reaction mixture. N-methyl morpholine (110 ml) was added to the reaction mixture and the reaction mixture was maintained at a temperature of about 00C to about 5°C for about 1 hour. The reaction mixture was allowed to warm to the temperature to about 25°C to about 35°C and diluted with water (3.6 lit). The reaction mass was extracted with ethyl acetate (3×2.4 lit). The separated ethyl acetate layer was washed with 1 N hydrochloric acid (1.2 lit) and two layers were then separated. The organic layer was washed with saturated sodium bicarbonate solution (4.8 lit) and brine solution (2.4 lit). The organic layer was concentrated completely at a temperature of about 45°C to afford 260 g of pyrazine-2- carbonylphenylalanine methyl ester.
Pyrazine-2-carbonylphenylalanine methyl ester (5 g) was dissolved in acetone (25 ml) and stirred for about 5 minutes. Sodium hydroxide solution (701 mg of sodium hydroxide in 25 ml of water) was added to the reaction solution and stirred for about 3 hours at a temperature of about 25°C, and the pH was then adjusted withi N hydrochloric acid (11 ml) to a pH of about 2. The reaction mixture was cooled to a temperature of about 00C to about 5°C and stirred for about 1 hour. The suspension was filtered and suck dried to afford 4.0 g of pyrazine-2- carbonylphenylalanine.
Chiral purity by chiral HPLC: 100% Chemical purity by HPLC: 99.88%.
Step-h) Preparation of Formula IX:

To a stirred mixture of compound of Formula VIII (28.6 g) in dichloromethane (400 ml) at 25-300C under nitrogen atmosphere, were charged N-hydroxysuccinimide (13.3 g) and DCC (23.9 g) and stirred for 10-20 minutes. Charged compound of Formula-V (40 g) to the reaction mass was and stirred for 15-20 minutes.
Charged diisopropylethylamine (DIPEA) (27 ml) and maintained the reaction mass at 25-300C for 2-3 hours. The reaction mass was filtered and the solid was washed with dichloromethane (80 ml). The filtrate obtained was washed with 1 N HCI, followed by washing with sodium bicarbonate solution. Concentrated the organic layer up to 2 volumes with respect to Formula-V. Charged methanol (200 ml) and concentrated up to 2 volumes with respect to Formula-V. The concentrated mass obtained is the title compound (Formula IX).
Alternately, compound of Formula IX may also be prepared by using EDCHCI, and Hydroxybenzotriazole by a process as described below: N-(2-pyrazinecarbonyl)-L-phenylalanine (500 mg) was suspended in dichloromethane (10 ml) and cooled to about -5°C to about 00C. Hydroxybenzotriazole (HOBt:310 mg) was charged in to the reaction mass followed by the addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCHCI, 385 mg) and stirred for 15 minutes. (1 R)-(S)-pinanediol 1 -ammonium trifluoroacetate-3-methylbutane-i -boronate (695 mg) was added to the reaction mixture and stirred for about 10 minutes at a temperature of about 5°C Diisopropyl ethyl amine (0.6 ml) was charged to the reaction mixture and stirred for about 30 minutes at a temperature of about 5°C The reaction mixture was allowed to warm to a temperature of about 25°C to about 300C and stirred for about 1 hour followed by the addition of 1 N hydrochloric acid (30 ml). The layers were separated and the organic layer was washed with 1 N hydrochloric acid (15 ml) and saturated sodium bicarbonate solution (2×30 ml). The organic layer was concentrated completely to afford title compound. Purity by HPLC: 84.98% Note that it is believed that 9.01 % measured by HPLC is Bortezomib that is formed prior to the final Bortezomib step. Thus, overall purity should be 84.98% + 9.0% or 93.99% as measured by HPLC.
EXAMPLE-2: PROCESS FOR PREPARING BORTEZOMIB (FORMULA I)
To a stirred mixture of compound of Formula IX (13.6 g) in methanol (272 ml) at 25- 300C, was charged n-heptane (272 ml), and isobutylboronic acid (3.2 g). Charged 2N HCI (272 ml) to the reaction mass under vigorous stirring and maintained the reaction mass at 25-300C for 1 -2 hours. After the completion of the reaction, separated the n-heptane layer and discarded. Charged n-heptane (272 ml * 2) to the aqueous layer and stirred vigorously for 10-15 minutes. Separated the n-heptane layer and the aqueous layer obtained was concentrated under vacuum at 35 to 480C. The aqueous layer was extracted with dichloromethane (272 ml) under vigorous stirring. The extraction process is repeated (272 ml *2) and the obtained dichloromethane layers were pooled and washed with saturated sodium bicarbonate solution, followed brine solution. The organic layer is separated, concentrated under vacuum to give 6 ml of the reaction mass and allowed to cool to 25-300C. Purity: 95.13% by HPLC.
Charged Toluene (102 ml) to the above reaction mass and stirred at 25-300C for 2-3 hours. Filtered the solid obtained under vacuum washed with 5% dichloromethane in toluene and dried at 45-500C under vacuum for 5 hours to give crude Bortezomib.
Yield: 7.O g (70%)
Purity by HPLC: 99.22%
Impurity-B by HPLC: 0.43%
Polymorphic Form: Form-B
XRD Pattern: As Illustrated in Fig -5 EXAMPLE-3: PROCESS FOR PURIFICATION OF BORTEZOMIB USING METHANOL AND WATER
Bortezomib (5.0 g, purity 99.22%) and methanol (15 ml) were taken into a round bottom flask and stirred at 25 to 35°c. Demineralized water (15 ml) was added to the obtained solution and stirred for 2 hours at a temperature of about 27°C. The reaction suspension was filtered and washed the solid with aqueous methanol (30 ml; water: methanol 1 :1 ). The obtained solid was dried at a temperature of about
500C for about 5 hours to afford 3.4 g of title compound.
Purity by HPLC: 99.57%
Impurity-B by HPLC: 0.30%
Further purification of the product obtained by reproducing the same process resulted in a Bortezomib having a purity of 99.6% by HPLC. Impurity-B by HPLC: 0.23%
Chiral Purity by HPLC: 99.83%
EXAMPLE-4: PROCESS FOR PREPARING BORTEZOMIB FOLLOWED BY PURIFICATION To a stirred mixture of compound of formula IX (68.3 g) in methanol (1.22 L) at 25- 300C, was charged n-heptane (1.36 L), and isobutyl boron ic acid (16.13 g). Charged 1 N HCI (13.6 L) to the reaction mass under stirring and maintained the reaction mass at 25-300C for 1 -2 hours. After the completion of the reaction, separated the n- heptane layer and discarded. Charged n-heptane (1.36 L * 2) to the aqueous layer and stirred vigorously for 10-15 minutes. Separated the n-heptane layer and the aqueous layer obtained was concentrated under vacuum. The aqueous layer was extracted with dichloromethane (13.6 L) under vigorous stirring. The extraction process is repeated (13.6 L *2) and the obtained dichloromethane layers were pooled and washed with saturated sodium bicarbonate solution, followed by brine solution. The organic layer was separated, concentrated under vacuum to give crude Bortezomib (47.0 g)
Purity by HPLC: 95.62% Impurity-B by HPLC: 0.59% Purification 1 : Bortezomib (25 g, Purity: 95.62%) and 5% ethylacetate in Toluene (250 ml) were taken into a round bottom flask and stirred at 25 to 350C for 2-3 hours. Filtered the solid obtained under vacuum washed with 5% ethylacetate in toluene and dried at 500C under vacuum for 5 hours to give Bortezomib.
Yield: 18.O g (72%) Purity by HPLC: 99.68% Impurity-B by HPLC: 0.27%
Purification 2: Bortezomib (18.0 g, purity 99.68%) and methanol (54 ml) were taken into a round bottom flask and stirred. Filtered the reaction mass through scinted funnel and washed the bed with 18 ml methanol. Demineralized water (72 ml) was added to the obtained filtrate and stirred for 2 hours at a temperature of about 27°C. The reaction suspension was filtered and washed the solid with aqueous methanol (108 ml; Water : methanol 1 :1 ). The obtained solid was dried at a temperature of about 500C for about 5 hours to afford 14 g of title compound.
Yield: 14.O g (77%) Purity by HPLC: 99.83%
Impurity B: 0.15% (by HPLC) Chiral Purity by HPLC: 99.85%

References
- Takimoto CH, Calvo E. “Principles of Oncologic Pharmacotherapy” in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- Adams J, Kauffman M (2004). “Development of the Proteasome Inhibitor Velcade (Bortezomib)”. Cancer Invest 22 (2): 304–11. doi:10.1081/CNV-120030218.PMID 15199612.
- Stephen R.Byrn et al (2011). “Analysis of two commercially available bortezomib products: differences in assay of active agent and impurity profile”. AAPS PharmSciTech (April 1).
- Bonvini P, Zorzi E, Basso G, Rosolen A (2007). “Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma”. Leukemia 21 (4): 838–42. doi:10.1038/sj.leu.2404528. PMID 17268529.
- Gelman JS, Sironi J, Berezniuk I, Dasgupta S, Castro LM, Gozzo FC, Ferro ES, Fricker LD (2013). “Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib”. In Gartel, Andrei L. PLoS One 8 (8): e53263.doi:10.1371/journal.pone.0053263. PMC 3538785. PMID 23308178.
- Voorhees PM, Dees EC, O’Neil B, Orlowski RZ (2003). “The proteasome as a target for cancer therapy”. Clin Cancer Res 9 (17): 6316–25. PMID 14695130.
- “NHS watchdog rejects cancer drug”. BBC News UK. 20 October 2006. Retrieved 2009-08-14.
- “Summary of VELCADE Response Scheme”. Retrieved 2009-08-14.
- “More Velcade-Style Risk-Sharing In The UK?”. Euro Pharma Today. 21 January 2009. Retrieved 2009-08-14.
- Oakervee HE, Popat R, Curry N, et al. (2005). “PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma”. Br J Haematol 129 (6): 755–62. doi:10.1111/j.1365-2141.2005.05519.x. PMID 15953001.
- Pour L., Adam Z., Buresova L., et al. (2009). “Varicella-zoster virus prophylaxis with low-dose acyclovir in patients with multiple myeloma treated with bortezomib”. Clinical Lymphoma & Myeloma 9 (2): 151–3. doi:10.3816/CLM.2009.n.036. PMID 19406726.
- Highlights Of Prescribing Information
- “Cancer drug benefits could be negated by healthy tea treatment”. Belfast Telegraph. 3 February 2009. Retrieved 2009-08-14.
- “Green tea may counteract anticancer effects of cancer therapy, bortezomib (Velcade)”. Retrieved 15 July 2013.
- “Green tea clash with bortezomib suggested”. Retrieved 26 July 2013.
- Golden EB, et al.; Lam, P. Y.; Kardosh, A.; Gaffney, K. J.; Cadenas, E.; Louie, S. G.; Petasis, N. A.; Chen, T. C.; Schonthal, A. H. (2009). “Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors”. Blood 113 (23): 5927–37.doi:10.1182/blood-2008-07-171389. PMID 19190249.
- Curran M, McKeage K. (2009). “Bortezomib: A Review of its Use in Patients with Multiple Myeloma”. Drugs 69 (7): 859–888. doi:10.2165/00003495-200969070-00006.PMID 19441872. doi:10.2165/00003495-200969070-00006.
- Journal of Pharmaceutical Sciences, 2000 , vol. 89, 6 p. 758 – 765
-
WO2009/36281 A2, ANDUS2013/85277 A1,
- [1]
- Myeloma patients campaigning for access to a life prolonging cancer drug
- Millennium Pharmaceuticals website on Velcade
- Multiple Myeloma Research Foundation article on Velcade
- International Myeloma Foundation article on Velcade
- U.S. Food and Drugs Administration on Velcade
- Dedicated website for European audience
- Presentation at 2006 ASCO of the PINNACLE Study on MCL by Dr. Goy, with video/slides
| EP2377868A1 * | Mar 24, 2005 | Oct 19, 2011 | Millennium Pharmaceuticals, Inc. | Synthesis of Bortezomib |
| WO2005097809A2 | Mar 24, 2005 | Oct 20, 2005 | Millennium Pharm Inc | Synthesis of boronic ester and acid compounds |
| WO2009004350A1 | Jul 2, 2008 | Jan 8, 2009 | Pliva Hrvatska D O O | Methods for preparing bortezomib and intermediates used in its manufacture |
| WO2009006473A2 | Jul 1, 2008 | Jan 8, 2009 | William W Bachovchin | Pro-soft polypeptide proteasome inhibitors, and methods of use thereof |
| WO2009036281A2 | Sep 12, 2008 | Mar 19, 2009 | Nageshwar Gunda | Bortezomib and process for producing same |
| EP0315574A2 | Oct 29, 1988 | May 10, 1989 | Hoechst Aktiengesellschaft | Renin inhibitors |
| EP0788360B1 | Oct 27, 1995 | May 28, 2003 | Millennium Pharmaceuticals, Inc. | Boronic ester and acid compounds, synthesis and uses |
| US4924026 | Aug 11, 1989 | May 8, 1990 | International Flavors & Fragrances Inc. | Triisobutylene alcohols and esters, uses thereof in perfumery and halogenated intermediates useful for preparing same |
| US20010012907 * | Feb 1, 2001 | Aug 9, 2001 | Nissan Chemical Industries, Ltd. | Substituted cyclopentene derivatives and method for preparing the same |
| US20030008828 * | Dec 6, 2001 | Jan 9, 2003 | Priestley E. Scott | Novel lactam inhibitors of hepatitis C virus NS3 protease |
| US20070244319 | Apr 18, 2006 | Oct 18, 2007 | Boaz Neil W | Metallocenyl P-N ligands, preparation thereof, and use for asymmetric catalysis |
| WO2010146172A2 | Jun 18, 2010 | Dec 23, 2010 | Lek Pharmaceuticals D.D. | NEW SYNTHETIC ROUTE FOR THE PREPARATION OF α-AMINO BORONIC ACID DERIVATIVES VIA SUBSTITUTED ALK-1-YNES |
| WO2011098963A1 | Feb 9, 2011 | Aug 18, 2011 | Ranbaxy Laboratories Limited | Process for the preparation of bortezomib |
| WO2011099018A1 * | Feb 15, 2010 | Aug 18, 2011 | Hetero Research Foundation | Polymorphs of bortezomib |
| WO2011107912A1 | Feb 24, 2011 | Sep 9, 2011 | Ranbaxy Laboratories Limited | Polymorphic forms of bortezomib |
| WO2012048745A1 | Oct 14, 2010 | Apr 19, 2012 | Synthon Bv | Process for making bortezomib and intermediates for the process |
| CN102206188B | Apr 8, 2011 | Feb 27, 2013 | 苏州二叶制药有限公司 | Preparation method for N-(pyrazine-2-yl carbonyl)-L-phenylalanine |
| CN102212036B | Apr 8, 2011 | Oct 17, 2012 | 上海医药工业研究院 | Preparation method of N-(pyrazine-2-radical carbonyl)-L-phenyl alanine |
| EP2270019A1 | Jun 19, 2009 | Jan 5, 2011 | LEK Pharmaceuticals d.d. | New synthetic route for the preparation of alpha-amino boronic esters |
| EP2280016A1 | Jul 27, 2009 | Feb 2, 2011 | LEK Pharmaceuticals d.d. | New synthetic route for the preparation of alpha-amino boronic esters via substituted alk-1-ynes |
| US8497374 | May 12, 2011 | Jul 30, 2013 | Scinopharm Taiwan, Ltd. | Process for preparing and purifying bortezomib |
BARDOXOLONE METHYL

BARDOXOLONE METHYL
- Molecular FormulaC32H43NO4
- Average mass505.688 Da
Methyl 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate
methyl 2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oate
2-Cyano-3,12-dioxoolean-1,9(11)-dien-28-oic acid methyl ester
(6aR,6bS,8aR,12aS,14aR,14bS)-11-Cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-hexadecahydropicene-4a(2H)-carboxylic acid methyl ester
BARD
CDDO-Me
Methyl-CDDO
NSC-713200
RTA-402
TP-155C
218600-53-4 CAS
218600-44-3 (free acid)
Treatment of pulmonary arterial hypertension (PAH), diabetic nephropathies and hereditary nephritis, Phase 3

Compounds were synthesized as below:

Scheme 1

Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
Bardoxolone methyl (also known as “RTA 402” and “CDDO-methyl ester”) is an orally-available first-in-class synthetic triterpenoid. It is an inducer of the Nrf2 pathway, which can suppress oxidative stress and inflammation, and is undergoing clinical development for the treatment of advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients.
Bardoxolone methyl was previously being investigated by Reata Pharmaceuticals, Inc. in partnership with Abbott Laboratories and Kyowa Hakko Kirin, as an experimental therapy for advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients. Reata, in consultation with the BEACON Steering Committee, has decided to terminate the Phase 3 BEACON trial of bardoxolone methyl in patients with stage 4 chronic kidney disease and type 2 diabetes. This decision was made based upon a recommendation of the Independent Data Monitoring Committee (IDMC) to stop the trial “for safety concerns due to excess serious adverse events and mortality in the bardoxolone methyl arm.” [1][2][3][4]
RTA-402 is a triterpenoid anti-inflammatory agent in phase II trials at Reata Pharmaceuticals for the treatment of pulmonary arterial hypertension.
This company and M.D. Anderson Cancer Center had been evaluating clinically the product for the treatment of lymphoma. Reata had been evaluating the compound in combination with gemcitabine in patients with unresectable pancreatic cancer and melanoma. Preclinical studies were also being conducted by Reata for the treatment of inflammatory bowel disease (IBD) and autoimmune disease. Reata Pharmaceuticals and Kyowa Hakko Kirin had been conducting phase II clinical studies for the treatment of diabetic nephropathy. Reata and Abbott also had been conducting phase III clinical trials for delaying progression to end-stage renal disease in patients with chronic kidney disease and type 2 diabetes; however, in 2012 these trials were discontinued due to serious adverse events and mortality. Phase II clinical trials for this indication were discontinued by Kyowa Hakko Kirin in Japan. The compound had been in early clinical studies for the treatment of multiple myeloma; however, no recent development has been reported for this indication. Phase I clinical trials for the treatment of solid tumors have been completed.
RTA-402 has demonstrated a wide variety of potentially therapeutic mechanisms, including inhibition of inducible nitric oxide synthase and cyclooxygenase expression, stimulation of expression of cytoprotective enzymes such as NAD(P)H quinine oxidoreductase and hemeoxygenase-1, and reduction in pSTAT3 levels. In cancer patients, the drug candidate exploits fundamental physiological differences between cancerous and non-cancerous cells by modulating oxidative stress response pathways. Due to this mechanism, RTA-402 is toxic to cancer cells, but induces protective antioxidant and anti-inflammatory responses in normal cells. In previous studies, the compound was shown to inhibit growth and cause regression of cancerous tumors as a single agent and, in combination with radiation and chemotherapy, to suppress radiation and chemotherapy-induced toxicities in normal tissues and cause minimal toxicity in non-human primates when dosed orally at very high doses for 28 consecutive days.
An analog of RTA-401, RTA-402 is a compound found in medicinal plants with a greater potency than the natural product.
RTA-401 was originally developed at Dartmouth College and M.D. Anderson Cancer Center. In November 2004, Reata completed a license agreement with these organizations, and was granted exclusive worldwide rights to this new class of anticancer compounds. In 2008, orphan drug designation was assigned by the FDA for the treatment of pancreatic cancer. In 2010, the compound was licensed to Kyowa Hakko Kirin by Reata Pharmaceuticals in China, Japan, Korea, Thailand and Southeast Asian countries for the treatment of chronic kidney disease. Abbott acquired rights to develop and commercialize the drug outside US, excluding certain Asian markets.
Phase 1
Bardoxolone methyl was first advanced into the clinic to assess its anticancer properties. In two Phase 1 trials that included 81 oncology patients, bardoxolone methyl reduced serum creatinine levels, with a corresponding improvement in estimated glomerular filtration rate (eGFR). Improvements were more pronounced in a subset of patients with established CKD and were maintained over time in patients who continued on bardoxolone methyl therapy for 5 months. Based on these observed effects and the well-described role of oxidative stress and inflammation in CKD, especially in type 2 diabetes, it was hypothesized that bardoxolone methyl could improve renal function in CKD patients with type 2 diabetes.[5]
Phase 2
A multi-center, double-blind, placebo-controlled Phase 2b clinical trial (BEAM) conducted in the US studied 227 patients with moderate to severe CKD (eGFR 20 – 45 ml/min/1.73m²) and type 2 diabetes. The primary endpoint was change in estimated GFR following 24 weeks of treatment. Following 24 weeks, patients treated with bardoxolone methyl experienced a mean increase in estimated GFR of over 10 ml/min/1.73m², compared with no change in the placebo group. Approximately three-quarters of bardoxolone methyl treated patients experienced an improvement in eGFR of 10 percent or more, including one-quarter who saw a significant improvement of 50% or more compared to less than 2% of patients on placebo. Adverse events were generally manageable and mild to moderate in severity. The most frequently reported adverse event in the bardoxolone methyl group was muscle spasm. Final data was published in The New England Journal of Medicine.
Concerns have been raised whether there is a true improvement in kidney function because of the significant weight loss of the patients in the active-treatment-group that ranged from 7.7-10.1 kg (7-10% of the initial body weight) and whether this weight loss in patients receiving bardoxolone included muscle wasting with a commensurate decrease in the serum creatinine level. In that case the decrease in creatinine would not necessarily be a true improvement in kidney function.[6][7][8][9][10]
Phase 3
A multinational, double-blind, placebo-controlled Phase 3 outcomes study (BEACON) was started in June 2011, testing bardoxolone methyl’s impact on progression to ESRD or cardiovascular death in 1600 patients with Stage 4 CKD (eGFR 15 – 30 ml/min/1.73m²) and type 2 diabetes. This phase 3 trail was halted in October 2012 because of adverse effects (namely a higher cardiovascular mortality in the treatment arm).[11]
Mechanism of action
Bardoxolone methyl is an inducer of the KEAP1–Nrf2 pathway.
PAPER
http://modernsteroid.blogspot.com/2012/04/synthetic-oleane-triterpenoids-as.html


Click to access ol400399x_si_001.pdf

1. To a stirred solution of oleanolic acid (22.8 grams, 0.05 mol, 1.0 equiv) in dimethyl formamide (200 mL) was
added powdered K2CO3 (20.7 grams, 0.15 mol, 3.0 equiv) slowly upon stirring, and the reaction mixture was allowed to
cool to 0 o
C. To the stirred suspension was added iodomethane (3.4 mL, 0.055 mol, 1.1 equiv) slowly, and after the
completion of addition, the reaction was allowed to warm to room temperature overnight. After the completion of the
reaction, dimethyl formamide was removed by distillation. The resulting solid mixture was dissolved in methylene
chloride (1 L) and washed with water (4 x 100 mL) and brine (1 x 100 mL). The organics was dried over Na2SO4 and the
solvent was removed to give the crude product 8 as a white solid, which was used directly for the next step without
further purifications.
2. To a stirred suspension of ester 8 (11.8 grams, 0.025 mol, 1.0 equiv) obtained above in anhydrous dimethyl
sulfoxide (250 mL) was added iodoxybenzoic acid (21.0 grams, 0.075 mol, 3.0 equiv) and fluorobenzene (5 mL). The
resulting suspension was heated to 85 o
C under nitrogen for 24 hours. After the completion of the reaction, it was
quenched with 20% aqueous sodium thiosulfate (200 mL). The resulting mixture was extracted with methylene chloride
(4 x 150 mL), the combined organic extracts were washed with saturated NaHCO3 (100 mL) and brine (100 mL), and
dried over Na2SO4. The solvent was removed to give the crude product 14 as yellowish solid, which was used directly for
the next step without further purifications.
3. To a stirred solution of 14 (9.32 grams, 0.02 mol, 1.0 equiv) in methylene chloride (100 mL) was slowly added mchloroperbenzoic
acid (6.4 grams, ~70% purity, 0.026 mol, 1.3 equiv) at 0 o
C. After the completion of addition, the
reaction was allowed to warm to room temperature and kept stirring for 24 hours. After the completion of the reaction,
the reaction mixture was diluted with methylene chloride (300 mL), and the resulting mixture was washed with 20%
aqueous sodium thiosulfate (3 x 100 mL), 10% potassium carbonate (2 x 100 mL), and brine (100 mL). The organics were
dried over Na2SO4 and the solvent was removed to give crude mixture of 15 and 16 as yellowish solid, which was used
directly for the next step without purifications.
4. To the resulting solution of 15 and 16 obtained above in acetic acid (50 mL) was added dropwise hydrobromic
acid (1.0 mL, 0.009 mol, 0.44 equiv) at room temperature. The reaction mixture was then heated to 35 o
C, and bromine
(5.8 mL, 0.05 mol, 2.4 equiv) was thus added dropwise. The resulting reaction mixture was kept stirring for another 24 h.
After completion of the reaction, the acid was removed under vacuum. And the residue was then quenched with 20%
aqueous sodium thiosulfate (100 mL), and extracted with methylene chloride (4 x 100 mL). The combined organic
extracts were washed with saturated sodium bicarbonate (2 x 50 mL), brine (1 x 50 mL), and dried over Na2SO4. The
solvent was removed to give crude bromo enone 17 as yellowish to yellow solid, which can be used directly for the next
step without further purification or subjected to flash column chromatography to give pure bromo enone 17 as a
yellowish solid.
5. To a stirred solution of bromo enone 17 (5.8 grams, 10.0 mmol, 1.0 equiv) in anhydrous dimethyl formamide (80
mL) was added copper (I) cyanide (1.0 grams, 11.0 mmol, 1.1 equiv) and potassium iodide (328 mg, 2.0 mmol, 0.20
equiv), and the resulting reaction mixture was heated to 120 o
C for 24 h. After the completion of reaction, it was cooled
to room temperature, quenched with water (200 mL), and diluted with ethyl acetate (500 mL). The organic phase was
washed with saturated NaHCO3 (2 x 80 mL), brine (80 mL), and dried over Na2SO4. Removal of solvent and flash column
chromatography over silica gel using hexanes:EtOAc (2:1) to give bardoxolone methyl (1) as a yellowish solid.
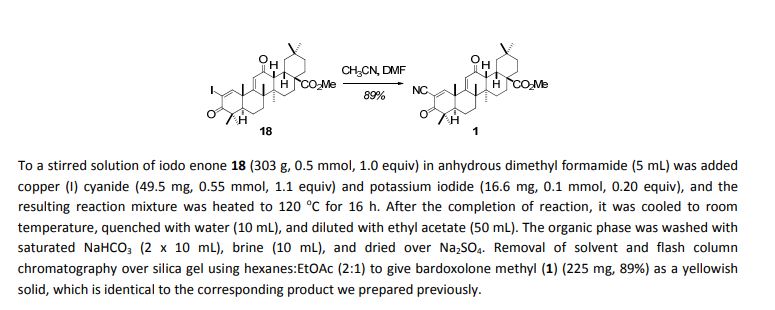
After the completion of the reaction, it was cooled to room temperature and
quenched with 20% aqueous sodium thiosulfate (20 mL). It was extracted with methylene chloride (3 x 20 mL), the
combined organic extracts were washed with saturated aqueous NaHCO3 (10 mL), brine (10 mL), and dried over Na2SO4.
Removal of solvent and flash column chromatography over silica gel using hexanes:EtOAc (4:1 & 2:1) to give iodo enone
18 (509 mg, 84%) as a yellowish solid. 1H NMR (500 MHz, CDCl3) δ 8.12 (s, 1H), 6.00 (s, 1H), 3.70 (s, 3H), 3.04 (dd, 1H, J1 =
10.0 Hz, J2 = 3.7 Hz), 2.92 (d, 1H, J = 4.6 Hz), 1.63-1.94 (m, 9H), 1.46-1.62 (m, 3H), 1.43 (s, 3H), 1.18-1.36 (m, 3H), 1.30 (s,
3H), 1.23 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 1.00 (s, 3H), 0.90 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 199.6, 196.9, 178.4,
170.3, 163.5, 124.1, 102.3, 52.1, 49.9, 48.4, 47.4, 46.4, 45.9, 45.4, 42.3, 36.0, 34.7, 33.5, 33.0, 32.1, 31.7, 30.9, 28.3, 28.2,
27.3, 24.8, 23.3, 22.9, 22.4, 21.9, 18.8; FT-IR (solution, CDCl3, cm-1): 2952, 2869, 2253, 1717, 1659, 1469, 1386, 907, 732,
651, 623, 443; HRMS-ESI (calcd. for C31H44IO4 [M+H]+
) 607.2284, found 607.2280.
CLIP

PATENT
In a preferred embodiment, such compounds include derivatives of ursolic acid and oleanoic acid. In a particularly preferred embodiment, derivatives of OA, e.g., 2-cyano-3,12-dioxoolean-l,9-dien-28oic acid (CDDO):

have been found to be effective in suppression of human breast cancer cell growth, and highly potent in many vitro assay systems such as: suppression of nitric oxide and prostaglandin production in macrophages, inhibition of growth of human breast cancer cells, suppression of nitric oxide formation in rat prostate cells, and suppression of prostaglandin formation in human colon fibroblasts, as detailed in the Figures.
Compounds were synthesized as below:
Scheme 1
Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
PATENT
WO2009/146216 A2,

Compounds 401, 402, 404, 402-04, 402-35 and 402-56 can be prepared according to the methods taught by Honda et al. (1998), Honda et al. (2000b), Honda et al. (2002), Yates et al. (2007), and U.S. Patent 6,974,801, which are all incorporated herein by reference. The synthesis of the other compounds are disclosed in the following applications, each of which is incorporated herein by reference: U.S. Application Nos. 61/046,332, 61/046,342, 61/046,363, 61/046,366, 61/111,333, 61/111,269, and 61/111,294. The synthesis of the other compounds are also disclosed in the following separate applications filed concurrently herewith, each of which is incorporated herein by reference in their entireties: U.S. Patent Application by Eric Anderson, Xin Jiang, Xiaofeng Liu; Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives With Saturation in the C- Ring,” filed April 20, 2009; U.S. Patent Application by Eric Anderson, Xin Jiang and Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives with Amino and Other Modifications At C-17,” filed April 20, 2009; U.S. Patent Application by Xin Jiang, Xioafeng Liu, Jack Greiner, Stephen S. Szucs, Melean Visnick entitled, “Antioxidant Inflammation Modulators: C-17 Homologated Oleanolic Acid Derivatives,” filed April 20, 2009.
PAPER
Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
http://pubs.rsc.org/en/Content/ArticleLanding/2011/CC/c1cc11633a#!divAbstract
http://www.rsc.org/suppdata/cc/c1/c1cc11633a/c1cc11633a.pdf NMR GIVEN
2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO)
A mixture of 1 (0.25 g, 0.51 mmol) and DDQ (0.12 g, 0.51 mmol) in anhydrous benzene (20 mL) was
refluxed for 15 min. After filtration, the filtrate was evaporated in vacuo to give a residue, which was
subjected to flash column chromatography (petroleum ether/EtOAc) to give CDDO as an amorphous
solid (0.23 g, 91%). The title compound was known as CAS 218600-44-3
m.p. 180-182 °C;
ESI-MS: 490 [M-H]-, 492 [M+H]+;
1H NMR (300M Hz, CDCl3, 25 °C, TMS): δ 8.05 (1H, s), 5.99 (1H, s), 3.03-2.98 (2H, m), 1.55,1.38,
1.34, 1.22, 1.00, 0.91, 0.85 (each 3H,s ,CH3) ppm.
PAPER
SYNTHESIS
Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
http://pubs.acs.org/doi/full/10.1021/jm0002230

Bioorganic and Medicinal Chemistry Letters, 1998 , vol. 8, 19 p. 2711 – 2714
http://www.sciencedirect.com/science/article/pii/S0960894X9800479X

PAPER
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, # 9 p. 2215 – 2219
http://www.sciencedirect.com/science/article/pii/S0960894X05003306

PATENT
Method of synthesis of CDDO. CDDO may be synthesized by the scheme outlined below.

Methyl-CDDO. Methyl-CDDO (CDDO-Me), the C-28 methyl ester of CDDO, also exerts strong antiproliferative and apoptotic effects on leukemic cell lines and in primary AML samples in vitro as well as induces monocytic differentiation of leukemic cell lines and some primary AMLs. Thus, CDDO-Me provides chemotherapy for the treatment of leukemias. The present invention demonstrates that this effect is profoundly increased by combination of CDDO-Me with other chemotherapeutic agents. These include retinoids such as ATRA, 9-cis retinoic acid, , LG100268, LGD1069 (Targretin, bexarotene), fenretinide [N-(4- hydroxyphenyl)retinamide, 4-HPR], CD437 and other RXR and RAR-specific ligands. This combination also increases ara-C cytotoxicity, further reduces AML colony formation, inhibits ERK phosphorylation and promotes Bcl-2 dephosphorylation, and inhibits in vitro angiogenesis. The ability of CDDO-Me in combination with retinoids to induce differentiation in leukemic cells in vitro show that these compounds may have similar in vivo effects. The anti-angiogenic properties of CDDO-Me further increase its potent anti-leukemia activity in combination with retinoids. Furthermore, CDDO-Me was found to be more potent at lower concentrations than CDDO.
Method of synthesis of CDDO-Me.
CDDO-Me may be synthesized by the scheme outlined below.

The present invention provides combinations of CDDO-compounds and chemotherapeutic agents that are useful as treatments for cancers and hematological malignancies. In one embodiment, the chemotherapeutics are retinoids. As CDDO- compounds are PPARγ ligands and PPARγ is known to be altered in many types of cancers, the inventors contemplate, that ligation of PPARγ in combination with retinoids such as, RXR-specific ligands, provides a mechanistic basis for maximal increase in transcriptional activity of the target genes that control apoptosis and differentiation. The CDDO-compounds and retinoids in combination demonstrate an increased ability to induce differentiation, induce cytotoxicity, induce apoptosis, induce cell killing, reduce colony formation and inhibit the growth of several types of leukemic cells.
PAPER
Efficient and scalable synthesis of bardoxolone methyl (cddo-methyl ester).
Bardoxolone methyl (2-cyano-3,12-dioxooleane-1,9(11)-dien-28-oic acid methyl ester; CDDO-Me) (1), a synthetic oleanane triterpenoid with highly potent anti-inflammatory activity (levels below 1 nM), has completed a successful phase I clinical trial for the treatment of cancer and a successful phase II trial for the treatment of chronic kidney disease in type 2 diabetes patients. Our synthesis of bardoxolone methyl (1) proceeds in ∼50% overall yield in five steps from oleanolic acid (2), requires only one to two chromatographic purifications, and can provide gram quantities of 1.

References
- “Bardoxolone methyl – Oral, Once Daily AIM for Renal/Cardiovascular/Metabolic Diseases”. Reata Pharmaceuticals. Archived from the original on 15 July 2011. Retrieved June 2, 2011.
- “Abbott and Reata Pharmaceuticals Announce Agreement to Develop and Commercialize Bardoxolone Methyl for Chronic Kidney Disease Outside the U.S.” (Press release). Reata Pharmaceuticals. September 23, 2010. Retrieved June 2, 2011.
- “Reata Pharmaceuticals Licenses Chronic Kidney Disease Drug Bardoxolone Methyl to Kyowa Hakko Kirin”(Press release). Reata Pharmaceuticals. January 7, 2010. Retrieved June 2, 2011.
- “Company Statement: Termination of Beacon Trial”.Reata Pharmaceuticals. Retrieved October 18, 2012.
- Pergola, P. E.; Krauth, M.; Huff, J. W.; Ferguson, D. A.; Ruiz, S.; Meyer, C. J.; Warnock, D. G. (2011). “Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b–4 CKD”. American Journal of Nephrology 33 (5): 469–476. doi:10.1159/000327599. PMID 21508635.
- Pergola, P. E.; Raskin, P.; Toto, R. D.; Meyer, C. J.; Huff, J. W.; Grossman, E. B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; Wittes, J.; Warnock, D. G.; Beam Study, I. (2011). “Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes” (pdf). New England Journal of Medicine 365 (4): 327–336.doi:10.1056/NEJMoa1105351. PMID 21699484. edit
- van Laecke, S.; Vanholder, R. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047578.
- Rogacev, K. S.; Bittenbring, J. T.; Fliser, D. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745–1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047579.
- Upadhyay, A.; Sarnak, M. J.; Levey, A. S. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047580.
- McMahon, G. M.; Forman, J. P. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047581.
- ClinicalTrials.gov NCT01351675 Bardoxolone Methyl Evaluation in Patients With Chronic Kidney Disease and Type 2 Diabetes (BEACON)
- Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages
Bioorg Med Chem Lett 1998, 8(19): 2711 - Novel synthetic oleanate triterpenoids: A series of highly active inhibitors of nitric production in mouse macrophages
Bioorg Med Chem Lett 1999, 9(24): 3429 - WO 1999065478
- WO 2013169553
- CN 102875634
- US 2012330050
- US 2012071684
- WO 2011130302
- WO 2010093944
- WO 2009089545
- WO 2009023232
- WO 2008111497
- Anderson, Amy C.; Browning, R. Greg; Couch, Robin D.; Gribble, Gordon W.; Honda, Tadashi; Wright, Dennis L.; Sporn, Michael B.
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 9 p. 2215 – 2219 - Journal of Medicinal Chemistry, 2004 , vol. 47, 20 p. 4923 – 4932
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Bioorganic and Medicinal Chemistry Letters, 2002 , vol. 12, 7 p. 1027 – 1030
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
- Reata Pharmaceuticals, Inc. – bardoxolone methyl
- Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes
- Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b – 4 CKD
- American Diabetes Association presentation
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8440854 * | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| WO2002047611A2 * | Nov 28, 2001 | Jun 20, 2002 | Univ Texas | Cddo-compounds and combination therapies thereof |
| WO2008064132A2 * | Nov 16, 2007 | May 29, 2008 | Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| WO2009118441A1 * | Feb 12, 2009 | Oct 1, 2009 | Consejo Superior De Investigaciones Cientifícas | Use of pentacyclic triterpene for the preparation of a pharmaceutical compound intended for the treatment of multiple sclerosis |
| WO2013083659A1 | Dec 5, 2012 | Jun 13, 2013 | Cambridge Enterprise Limited | Combination treatment comprising ho – 1 inhibitor and immunotherapeutic agent |
| US7176237 | Jan 15, 2003 | Feb 13, 2007 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7435755 | Nov 28, 2001 | Oct 14, 2008 | The Trustees Of Dartmouth College | CDDO-compounds and combination therapies thereof |
| US7678830 | Feb 7, 2007 | Mar 16, 2010 | Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7714012 | Nov 16, 2007 | May 11, 2010 | Trustees Of Dartmouth University | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US7795305 | Oct 10, 2008 | Sep 14, 2010 | Board Of Regents, The University Of Texas System | CDDO-compounds and combination therapies thereof |
| US7863327 | May 3, 2005 | Jan 4, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US7915402 | Apr 20, 2009 | Mar 29, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US7943778 | Apr 20, 2009 | May 17, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8034955 | Oct 29, 2007 | Oct 11, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US8067394 | May 10, 2010 | Nov 29, 2011 | Trustees Of Dartmouth College | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US8067465 | Mar 11, 2010 | Nov 29, 2011 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US8071632 | Apr 20, 2009 | Dec 6, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8124656 | Feb 23, 2011 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8124799 | Apr 20, 2009 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino and other modifications at C-17 |
| US8129429 | Jan 12, 2009 | Mar 6, 2012 | Reata Pharmaceuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8258329 | Apr 20, 2009 | Sep 4, 2012 | Reata Pharmaceuticals, Inc. | Dehydroandrosterone analogs including an anti-inflammatory pharmacore and methods of use |
| US8299046 | Nov 16, 2007 | Oct 30, 2012 | Trustees Of Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| US8314137 | Jul 22, 2009 | Nov 20, 2012 | Trustess Of Dartmouth College | Monocyclic cyanoenones and methods of use thereof |
| US8338618 | Nov 11, 2011 | Dec 25, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8394967 | Feb 23, 2011 | Mar 12, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8440820 | Jan 11, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8440854 | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8455544 | Jan 26, 2012 | Jun 4, 2013 | Reata Pharmaecuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| US8586775 | Aug 24, 2011 | Nov 19, 2013 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
 |
| Professor Honda received his B.S. degree in Chemistry in 1974, his M.S. degree in Organic Chemistry in 1976, and his Ph.D. in Organic Chemistry in 1979 from the University of Tokyo. In 1979, he joined the Department of Drug Discovery Chemistry at Suntory Institute for Biomedical Research in Japan and worked there as a drug synthetic chemist (finally senior researcher) for 13 years. In 1991, he joined the Central Pharmaceutical Research Institute at Japan Tobacco Inc. and worked as a chief senior researcher for 3 years. In 1995, he joined Dr. Gribble’s laboratory at Dartmouth College as a research associate. In 1998, he joined the research faculty of Dartmouth College. In 2005, he was promoted to Research Associate Professor. |
Dr. Honda and his collaborators have further explored new structures based on CDDO and different five-ringed triterpenoids.
During the course of these investigations, Dr. Honda has designed three-ringed compounds with similar enone functionalities in rings A and C to those of CDDO, but having a much simpler structure than five-ringed triterpenoids. He and his collaborators have found that they are also a novel class of potent anti-inflammatory, cytoprotective, growth suppressive, and pro-apoptotic compounds. Amongst such three-ringed compounds, TBE-31 with the C-8a ethynyl group is much more potent than CDDO in various bioassays in vitro and in vivo. Thus, further investigation (design, synthesis, biological evaluation, etc.) of new TBE-31 analogues is currently being performed in order to discover analogues having different and/or better features than TBE-31, for example, higher potency and lower toxicity, better bioavailability and different distributions in organs, high water-solubility and so on.

Mechanism studies suggest that CDDO regulates various molecules regarding inflammation, differentiation, apoptosis, and proliferation by reversible Michael addition between the cyano enone functionality of CDDO and the sulfhydryl groups of cysteine moieties on these molecules. Based on this fact and the structure of TBE-31, Dr. Honda has designed single-ringed compounds, which represent the ideal simple structure. The synthesis of these new compounds is currently in progress.

 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| ChEMBL | |
| ECHA InfoCard | 100.132.153 |
| Chemical and physical data | |
| Formula | C32H43NO4 |
| Molar mass | 505.69 g/mol |
| 3D model (JSmol) | |
///////////////Bardoxolone Methyl, CDDO-Me; CDDO methyl ester; 218600-53-4; Bardoxolone (methyl); RTA 402 CDDO-Me, CDDO methyl ester, 218600-53-4, Bardoxolone (methyl), RTA 402 , PHASE 3,NSC 713200
CC1(CCC2(CCC3(C(C2C1)C(=O)C=C4C3(CCC5C4(C=C(C(=O)C5(C)C)C#N)C)C)C)C(=O)OC)C
Ancient Chinese medicine put through its paces for pancreatic cancer
The bark of the Amur cork tree (Phellodendron amurense) has traveled a centuries-long road with the healing arts. Now it is being put through its paces by science in the fight against pancreatic cancer, with the potential to make inroads against several more. UT Health Science Center researcher A. Pratap Kumar was already exploring the cork tree extract’s promise in treating prostate cancer when his team found that deadly pancreatic cancers share some similar development pathways with prostate tumors.
In a paper published today in the journal Clinical Cancer Research, the researchers show that the extract blocks those pathways and inhibits the scarring that thwarts anti-cancer drugs. Dr. Jingjing Gong, currently pursuing post-doctoral studies at Yale University, conducted the study as a graduate student in Dr Kumar’s laboratory in the Department of Pharmacology.
“Fibrosis is a process of uncontrolled scarring around the tumor gland,” said Dr…
View original post 242 more words
2013 FDA drug approvals
This analysis by Asher Mullard published in Nature Reviews Drug Discovery (2014,13, 85-89) reports the new drugs approved by FDA in 2013. From a total of thirty-six applications, twenty-five new small molecules and two new biologics were approved. The same trend as the previous years was overall maintained, with the exception of 2012. (Figure 1).
A notable achievement was the high approvals (33%) of new molecular entities for the treatment of orphan disease. In addition, 33% of the new approvals had a unique mode of action and were identified as first-in-class agents. The anticancer therapeutic area obtained the majority of approvals (eight, six of which are for orphan indication), followed by metabolic and endocrinology, antiviral and medical imaging (three approvals for each category). Cardiology, neurology, respiratory and women’s health have two agents approved each, and only one new approval for psychiatry and dermatology.
Ten drugs received a priority review…
View original post 343 more words
Actelion’s novel antibiotic Cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .

CADAZOLID, ACT-179811
1-Cyclopropyl-6-fluoro-7-[4-({2-fluoro-4-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy}methyl)-4-hydroxypiperidin-1-yl]-4-oxo-1,4-dihydroquinolin-3-carboxylic acid
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-(R)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid
| Formula | C29H29F2N3O8 |
|---|---|
| Mol. mass | 585.55 g/mol |
Actelion Pharmaceuticals Ltd / Actelion’s novel antibiotic cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .
ALLSCHWIL/BASEL, SWITZERLAND – 27 February 2014 – Actelion Ltd (six:ATLN) today announced that the US Food and Drug Administration (FDA) has designated cadazolid as both a Qualified Infectious Disease Product (QIDP) and a Fast Track development program for the treatment of Clostridium difficile-associated diarrhea (CDAD).
The QIDP designation for cadazolid means that – among other incentives – cadazolid would receive a nine-month priority review upon successful completion of the ongoing global Phase III IMPACT program. The Fast Track designation is intended to promote communication and collaboration between the FDA and the Company on the development of the drug.
The designations are based on the 2012 US Generating Antibiotic Incentives Now (GAIN) Act. The GAIN act is a legislative effort to incentivize the development of new antibiotic agents that target serious life-threatening infections.
Guy Braunstein, M.D. and Head of Clinical Development commented: “Clostridium difficile-associated diarrhea is a very serious and potentially life-threatening infection. There is a great need for an antibiotic that allows effective treatment of CDAD with low recurrence rates, particularly in infections caused by hypervirulent strains. The GAIN act highlights the importance of research in this area and we are very happy to receive the advantages that this designation for cadazolid will afford us.”
ABOUT THE IMPACT PROGRAM
IMPACT is an International Multi-center Program Assessing Cadazolid Treatment in patients suffering from Clostridium difficile-associated diarrhea (CDAD). The program comprises two Phase III studies comparing the efficacy and safety of cadazolid (250 mg administered orally twice daily for 10 days) versus vancomycin (125 mg administered orally four times daily for 10 days).
The IMPACT studies are designed to determine whether the clinical response after administration of cadazolid is non-inferior to vancomycin in subjects with CDAD, and whether administration of cadazolid is superior to vancomycin in the sustained clinical response. The program is expected to enroll approximately 1’280 subjects worldwide, and commenced enrollment in the fourth quarter of 2013.
ABOUT CADAZOLID
The novel antibiotic cadazolid is a strong inhibitor of Clostridium difficile protein synthesis leading to strong suppression of toxin and spore formation. In preclinical studies cadazolid showed potent in vitro activity against Clostridium difficile clinical isolates and a low propensity for resistance development. In a human gut model of CDAD, cadazolid had a very limited impact on the normal gut microflora.
Cadazolid absorption is negligible resulting in high gut lumen concentrations and low systemic exposure, even in severe cases of CDAD where the gut wall can be severely damaged and permeability to drugs potentially increased.
Cadazolid is an experimental antibiotic of the oxazolidinone class made by Actelion Pharmaceuticals Ltd. which is effective against Clostridium difficile, a major cause of drug resistant diarrhea in the elderly.[1] Current drug treatments for this infection involve orally delivered antibiotics, principally fidaxomicin, metronidazole and vancomycin; the last two drugs are the principal therapeutic agents in use, but fail in approximately 20 to 45% of the cases. The drug is presently in Phase III trials.[1] The drug works by inhibiting synthesis of proteins in the bacteria, thus inhibiting the production of toxins and the formation of spores.[2]
Structure
The chemical structure of cadazolid combines the pharmacophores of oxazolidinone and fluoroquinolone.[2]
In a study published in the journal Anaerobe, cadazolid has been shown to be effective in vitro against 133 strains of Clostridium difficile all collected from Sweden.[3]
In phase I tests, sixty four male patients reacted favourably to cadazolid which primarily acted and remained in the colon while displaying little toxicity even in regimes involving large doses.[1]
ABOUT CADAZOLID IN THE PHASE II STUDY
Cadazolid was studied in a Phase II multi-center, double-blind, randomized, active reference, parallel group, therapeutic exploratory study. The study evaluated the efficacy, safety and tolerability of a 10-day, twice daily oral administration of 3 doses (250 mg, 500 mg or 1,000 mg b.i.d.) of cadazolid in subjects with Clostridium difficile-associated diarrhea (CDAD). As the current standard of care for CDAD, oral vancomycin (125 mg qid for 10 days) was used as the active reference. The study was completed in December of 2012, after having enrolled 84 subjects with CDAD.
The results of the Phase II study indicate that the effect of all doses of cadazolid were numerically similar to, or better than vancomycin on key endpoints including CDAD clinical cure rates as well as sustained cure rates. Clinical cure rate was defined as the resolution of diarrhea and no further need for CDAD therapy at test-of-cure 24 to 72 hours after the last dose of treatment, while sustained cure rate was defined as clinical cure with no recurrence of CDAD up to 4 weeks post-treatment. Recurrence rates were numerically lower for all doses of cadazolid as compared to vancomycin. Cadazolid was safe and well tolerated.
ABOUT THE GAIN ACT (INCLUDING FAST TRACK DESIGNATION)
The Food and Drug Administration Safety and Innovation Act (FDASIA) was signed into law in July 2012. The GAIN Act is Title VIII to FDASIA. The purpose of the GAIN Act is to encourage pharmaceutical research of certain antibiotics by designation of products as QIDPs. These products are intended to treat serious or life-threatening infections and include those to treat certain specifically identified pathogens, which are listed in the GAIN Act. C. difficile is one such specifically identified pathogen and drugs to treat CDAD would be eligible for designation as a QIDP.
The GAIN Act also provides that qualifying drugs (QIDPs) are eligible for inclusion in the FDA’s Fast Track program. This program is intended to facilitate development and expedite review of new drugs and includes close early communication between the FDA and a drug’s sponsor.
ABOUT FAST TRACK DRUG DEVELOPMENT PROGRAMS
For further information regarding Fast Track Drug Development Programs, please refer to the FDA document “Guidance for Industry on Fast Track Drug Development Programs: Designation, Development, and Application Review”. This document is available on the Internet at:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079736.pdf
ABOUT CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that is the leading cause of nosocomial diarrhea. Clostridium difficile-associated diarrhea (CDAD or CDI for Clostridium difficile infection) can be a severe and life-threatening disease and results from the overgrowth in the colon of toxigenic strains of Clostridium difficile, generally during or after therapy with broad-spectrum antibiotics. CDAD is a major healthcare problem and a leading cause of morbidity in elderly hospitalized patients. The frequency and severity of CDAD in the western world has increased in recent years, and new hypervirulent and epidemic strains of Clostridium difficile have been discovered that are characterized by overproduction of toxins and other virulence factors, and by acquired resistance to fluoroquinolones such as moxifloxacin.
Current antibiotic therapy for CDAD includes vancomycin and metronidazole. While clinical cure rates are generally 85-90%, recurrences rates of 15-30 % with either drug are problematic as Clostridium difficile produces spores that are resistant to antibiotic treatment and routine disinfection. Spores surviving in the gut of patients and/or in the hospital environment may play a major role in re-infection and recurrence of CDAD after antibiotic treatment. Vancomycin and metronidazole are reported to promote spore formation in vitro at sub-inhibitory concentrations.
Actelion Ltd.
Actelion Ltd. is a leading biopharmaceutical company focused on the discovery, development and commercialization of innovative drugs for diseases with significant unmet medical needs.
Actelion is a leader in the field of pulmonary arterial hypertension (PAH). Our portfolio of PAH treatments covers the spectrum of disease, from WHO Functional Class (FC) II through to FC IV, with oral, inhaled and intravenous medications. Although not available in all countries, Actelion has treatments approved by health authorities for a number of specialist diseases including Type 1 Gaucher disease, Niemann-Pick type C disease, Digital Ulcers in patients suffering from systemic sclerosis, and mycosis fungoides in patients with cutaneous T-cell lymphoma.
Founded in late 1997, with now over 2,400 dedicated professionals covering all key markets around the world including the US, Japan, China, Russia and Mexico, Actelion has its corporate headquarters in Allschwil / Basel, Switzerland
…………………..
Preparation of the compound of formula II
The compound of formula II can be obtained by hydrogenation of the compound of formula VIII

VIII
over a noble metal catalyst such as palladium or platinum on charcoal in a solvent such as THF, MeOH or EA between 00C and 400C or by hydrolysis of in presence of a solution of HBr in water or AcOH between 00C and 800C in a solvent such as AcOH.
The compounds of formula III can be prepared as summarized in Scheme 1 hereafter.

IX VI IIIA: R1= H IIIS: ^ = SO2R5
Scheme 1
The compounds of formula V can be prepared as summarized in Scheme 2 hereafter.

II X XI

Scheme 2
The compounds of formula X can be prepared from the methylidene derivatives of formula XII as summarized in Scheme 3 hereafter.

Xc XII Xa: R1 = H

Scheme 3
Example 1:
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((/f)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid:
1 i. (R)-3-(3-fluoro-4-hydroxy-phenyl)-5-hydroxymethyl-oxazolidin-2-one:
A solution of (7?y)-3-(4-benzyloxy-3-fluoro-phenyl)-5-hydroxymethyl-oxazolidin-2-one (6.34 g, prepared according to WO 2004/096221) in THF/MeOH (1 :1; 200 ml) was hydrogenated over Pd/C 10% (1 g) overnight. The catalyst was filtered off, the filtrate evaporated under reduced pressure and the residue stirred in EA. The crystals were collected by filtration, affording 3.16 g (70% yield) of a colourless solid. 1H NMR (DMSOd6; δ ppm): 3.5 (m, IH), 3.64 (m, IH), 3.74 (dd, J = 8.8, 6.4, IH), 3.99 (t, J = 8.8, IH), 4.64 (m, IH), 5.16 (t, J = 5.6, IH), 6.93 (dd, J = 9.7, 8.8, IH), 7.08 (ddd, J = 8.8, 2.6, 1.2, IH), 7.45 (dd, J = 13.5, 2.6, IH), 9.66 (s, IH). MS (ESI): 228.1.
1. ii. 4-[2-fluoro-4- ((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]- 4-hydroxy-piperidine-l-carboxylic acid benzyl ester:
A solution of intermediate l.i (1.27 g) and l-oxa-6-aza-spiro[2.5]octane-6-carboxylic acid benzyl ester (1.60 g; prepared according to US 4244961) were dissolved in DMF (15 ml) and treated with Na2CO3 (1.16 g). The mixture was heated at 1000C overnight. The residue obtained after workup (DCM) was stirred in EA, and the solid was collected by filtration and sequentially washed with EA and Hex, affording 2.52 g (94.5% yield) of a beige solid.
1H NMR (DMSOd6; δ ppm): 1.57 (m, 4H), 3.14 (m, 2H), 3.54 (m, IH), 3.64 (m, IH), 3.79 (m, 5 H), 4.03 (t, J = 9.1, 1 H), 4.66 (m, 1 H), 4.78 (s, 1 H), 5.05 (s, 2 H), 5.16 (t,
J = 5.6, 1 H), 7.18 (m, 2 H), 7.32 (m, 5 H), 7.55 (d, J = 12, 1 H).
MS (ESI): 475.0.
1. iii. (R)-3-[3-fluoro-4-(4-hydroxy-piperidin-4-ylmethoxy)-phenyl]-5-hydroxymethyl- oxazolidin-2-one:
A suspension of intermediate l.ii (2.5 g) in EA/MeOH (1 :1; 100 ml) was hydrogenated over Pd/C for 48 h. The suspension was heated at 400C and the catalyst was filtered off.
The filtrate was evaporated under reduced pressure affording 1.61 g (89% yield) of a yellow powder.
1H NMR (DMSOd6; δ ppm): 1.4-1.63 (m, 4H), 2.67 (m, 2H), 2.83 (m, 2H), 3.53 (dd, J = 4.0, 12.0, IH); 3.66 (dd, J = 3.3, 12.0, IH), 3.71 (s, 2H); 3.80 (m, IH), 4.05 (t, J = 9.0,
IH), 4.48 (s, IH), 4.68 (m, IH), 5.20 (s, IH), 7.20 (m, 2H), 7.57 (d, IH).
MS (ESI): 341.5.
l.iv. l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid:
A solution of intermediate l.iii (200 mg), 7-chloro-l-cyclopropyl-6-fiuoro-l,4-dihydro- 4-0X0-3 -quinolinecarboxylic acid boron diacetate complex (241 mg; prepared according to WO 88/07998) and DIPEA (100 μl) in NMP (2 ml) was stirred at 85°C for 5 h. The reaction mixture was evaporated under reduced pressure and the residue was taken up in 5M HCl in MeOH (3 ml) and stirred. The resulting solid was collected by filtration and washed with MeOH to afford 230 mg (67% yield) of a yellow solid.
1H NMR (DMSOd6; δ ppm): 1.66-1.35 (m, 4H), 1.75 (d, J = 12.8, 2H), 1.95 (m, 2H), 3.33 (t broad, J = 11.0, 2H), 3.57 (m, 3H), 3.67 (dd, J = 12.3, 3.3, IH), 3.83 (m, 2H), 3.92 (s, 2H), 4.06 (t, J = 9.0, IH), 4.69 (m, IH), 7.24 (m, 2H), 7.60 (m, 2H), 7.90 (d, J = 13.3, IH), 8.66 (s, IH).
MS (ESI): 585.9.
References
- Boschert, Sherry (19 Sep 2012). “Promising C. difficile Antibiotic in Pipeline”. Internal Medicine News. International Medical News Group. Retrieved 22 May 2013.
- “Cadazolid”. .actelion.com. Retrieved 2013-05-22.
- “Anaerobe – In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden”. ScienceDirect.com. 2013-02-26. Retrieved 2013-05-22.
- WO 2008056335
- WO 2009136379
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

