
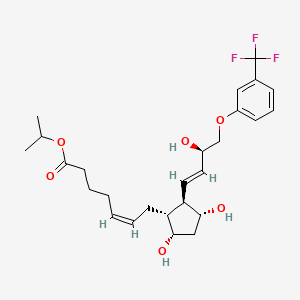
Latanoprost
isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate.
130209-82-4
XA41, PhXA34 [as 15 (R, S) -isomer], PhXA41, Xalatan
(Zanoni, G. et al., Tetrahedron 2010, 66, 7472)
Latanoprost (pronounced la-TA-noe-prost) ophthalmic solution is a medication administered into the eyes to control the progression of glaucoma or ocular hypertension by reducing intraocular pressure. It is a prostaglandin analogue (more specifically an analogue ofprostaglandin F2α[1]) that lowers the pressure by increasing the outflow of aqueous fluid from the eyes through the uvealsclearal tract.[2] Latanoprost is an isopropyl ester prodrug, meaning it is inactive until it is hydrolyzed by esterases in the cornea to the biologically active acid.[3]
It is also known by the brand name of Xalatan manufactured by Pfizer. Annual sales are approximately $1.6 billion. The patent for latanoprost expired in March 2011, and at least one generic version (manufactured by Mylan Inc.) is now widely available in the U.S. The Veterans Health Administration, part of the U.S. Department of Veterans Affairs, uses generic Latanoprost manufactured by Alcon Laboratories of Fort Worth, Texas distributed by Novartis generic brand Sandoz Pharmaceuticals.
Latanoprost was invented by Johan W. Stjernschantz and Bahram Resul, employees of the Pharmacia Corporation of Upsalla, Sweden.[4]
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[5]
Medical uses
Ocular hypertension
- In well-controlled clinical trials including patients with open-angle glaucoma or ocular hypertension (IOP ≥21 mm Hg), monotherapy with latanoprost reduced IOP levels by 22 to 39% over 1 to 12 months’ treatment. Latanoprost was significantly more effective than timolol 0.5% twice daily in 3 of 4 large (n = 163 to 267) randomised, double-blind trials. Latanoprost demonstrated a stable long-term IOP-lowering effect in 1- or 2-year continuations of these trials, with no sign of diminishing effect during prolonged treatment.[6]
- Meta analysis suggests that latanoprost is more effective than timolol in lowering IOP. However, it often causes iris pigmentation. While current evidence suggests that this pigmentation is benign, careful lifetime evaluation of patients is still justified.[7]
Closed-angle glaucoma
- Patients who had elevated IOP despite iridotomy and/or iridectomy (including patients of Asian descent), latanoprost was significantly more effective than timolol in two double-blind, monotherapy trials (8.2 and 8.8 mm Hg vs 5.2 and 5.7 mm Hg for latanoprost vs timolol at 12 and 2 weeks, respectively).[8]
Method of administration
One drop in the affected eye(s) once daily in the evening; do not exceed the once daily dosage because it has been shown that more frequent administration may decrease the intraocular-pressure (IOP) lowering effect[2]
Adverse effects[
Listed from most to least common:
- >5% to 15%: Blurred vision, burning and stinging, conjunctival hyperemia, foreign body sensation, itching, increased pigmentation of the iris causing (heterochromia), punctate epithelial keratopathy
- 4%: Cold or upper respiratory tract infections, flu-like syndrome
- 1-4%: Dry eyes, excessive tearing, eye pain, lid crusting, lid edema, lid erythema (hyperemia), lid pain, photophobia (light intolerance)
- 1 % – 2%: Chest pain, allergic skin reactions, arthralgia, back pain, myalgia, thickening of the eyelashes.(used,also bimatoprost,in cosmetic industry as eyelash growth enhancers)
- <1% (Limited to important or life-threatening): Asthma, herpes keratitis, iritis, keratitis, retinal artery embolus, retinal detachment, toxic epidermal necrolysis, uveitis, vitreous hemorrhage from diabetic retinopathy
- A single case report links latanoprost use to the progression of keratoconus.[9]
Concerns related to adverse effects:
- Bacterial keratitis: Inadvertent contamination of multiple-dose ophthalmic solutions, has caused bacterial keratitis.
- Ocular effects: May permanently change/increase brown pigmentation of the iris, the eyelid skin, and eyelashes. In addition, may increase the length and/or number of eyelashes (may vary between eyes); changes occur slowly and may not be noticeable for months or years. Long-term consequences and potential injury to eye are not known.
- Ocular disease: Use with caution in patients with intraocular inflammation, aphakic patients, pseudophakic patients with a torn posterior lens capsule, or patients with risk factors for macular edema. Safety and efficacy have not been determined for use in patients with angle-closure-, inflammatory-, or neovascular glaucoma.
Special populations
Contact lens wearers: Contains benzalkonium chloride which may be absorbed by contact lenses; remove contacts prior to administration and wait 15 minutes before reinserting
Contraindications
Hypersensitivity to latanoprost, benzalkonium chloride, or any component of the formulation
Drug Interactions
Bimatoprost: The concomitant use of Latanoprost and Bimatoprost may result in increased intraocular pressure. Risk D: Consider therapy modification
Nonsteroidal Anti-Inflammatory Agents: May diminish the therapeutic effect of Prostaglandins (Ophthalmic). Nonsteroidal Anti-Inflammatory Agents may also enhance the therapeutic effects of Prostaglandins (Ophthalmic). Risk C: Monitor therapy
Pregnancy
Prescription of Latanoprost is limited in human studies due to high incidence of abortion shown in animal experiments. Because of this, Latanoprost is classified as Risk factor C (Adverse events were observed in animal reproduction studies at maternally toxic doses)according to United States Food and Drug Administration’s use-in-pregnancy ratings.[10]Lactation Excretion in breast milk unknown/use caution. Breast-Feeding Considerations It is not known if latanoprost is excreted in breast milk. The manufacturer recommends that caution be exercised when administering latanoprost to nursing women.[2]
Storage
Latanoprost is a substance exhibiting thermal and solar instability. Concentration of latanoprost will decrease by 10% when stored at 50 and 70 degrees Celsius every 8.25 and 1.32 days respectively. Reaction with ultraviolet radiation will cause rapid degradation of Latanoprost. It is therefor important to store Latanoprost ideally in temperature below room temperature and free from sunlight in order to attain acceptable drug quality. [11]
Latanoprost is a prostaglandin F2α analogue. Its chemical name is isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate. Its molecular formula is C26H40O5and its chemical structure is:

XALATAN Sterile Ophthalmic Solution (latanoprost ophthalmic solution) is supplied as a sterile, isotonic, buffered aqueous solution of latanoprost with a pH of approximately 6.7 and an osmolality of approximately 267 mOsmol/kg. Each mL of XALATAN contains 50 micrograms of latanoprost. Benzalkonium chloride, 0.02% is added as a preservative. The inactive ingredients are: sodium chloride, sodium dihydrogen phosphate monohydrate, disodium hydrogen phosphate anhydrous, and water for injection. One drop contains approximately 1.5 μg of latanoprostLatanoprost is a colorless to slightly yellow oil that is very soluble in acetonitrile and freely soluble in acetone, ethanol, ethyl acetate, isopropanol, methanol, and octanol. It is practically insoluble in water.
………………………….
http://www.google.com/patents/EP2495235A1?cl=en
When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:
EXAMPLE 16
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((R)-3-hydroxy-5-phenylpentyl)cyclopentyl)hept-5-enoic acid (Latanoprost acid)
-
-
4-Carboxybutyltriphenylphosphonium bromide 15 (32.7 g, 0.074 mol) was suspended in tetrahydrofuran (75.0 mL) at 0°C under nitrogen atmosphere. A 1M solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((R)-3-hydroxy-5-phenylpentyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (5.0 g, 0.016 mol) in tetrahydrofuran (23.0 mL) was added dropwise at a temperature lower than -10°C. After stirring overnight at -15°C no more starting was visible on TLC and water (100 mL) was added. The mixture was extracted with diisopropyl ether (70 mL) and after separation the aqueous phase was treated with 0.6 N HCl to pH 6.0. Three extractions with ethyl acetate (3x 125 mL) were then performed, each time adjusting the pH of the aqueous phase to 6.0. The combined organic layers were concentrated under vacuum at 35°C. An oil (13.87 g) was obtained which was used in the subsequent step without further purification.
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.71 (m, 1H, Ph), 7.49 (m, 1H, Ph), 7.30-7.17 (m, 3H, Ph), 5.52-5.35 (m, 2H, -CH=CH-), 4.34 (bs, 4H, OH), 4.17 (m, 1H, -CH-OH(C-9)), 3.96 (m, 1H, -CH-OH (C-11)), 2.78 (m, 1H, -CH-OH (C-15)), 2.78 (m, 1H, -CH2Ph), 2.66 (m, 1H, -CH2Ph), 2.36-1.27 (m, 18H).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 176.5 (C), 142.2 (C), 130.8 (CH), 130.7 (CH), 129.4 (CH), 128.8 (CH), 128.7 (CH), 128.4 (CH), 125.7 (CH), 78.3 (CH), 74.2 (CH), 71.4 (CH), 52.2 (CH), 51.6 (CH), 42.4 (CH2), 38.8 (CH2), 35.2 (CH2), 33.4 (CH2), 32.0 (CH2), 29.1 (CH2), 26.6 (CH2), 26.4 (CH2), 24.7 (CH2).
-
HPLC-MS (ESI): [M+Na]+ = 413, [M+H]+ = 391.
EXAMPLE 17(Z)-isopropyl 7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((R)-3-hydroxy-5-phenylpentyl)cyclopentyl)hept-5-enoate (Latanoprost)
-
-
Latanoprost acid (6.78 g, corresponding to 0.008 mol) was dissolved in N,N-dimethylformamide (108 mL) and cesium carbonate (8.48 g, 0.026 mol) was added at room temperature. 2-Iodopropane (3.46 mL, 0.035 mol) was added and the suspension was stirred at 40°C for 3 hours, checking the conversion on TLC. The mixture was then allowed to reach 25°C and a mixture of ice (184 g), water (40 mL), sodium thiosulfate (1M, 18 mL), was added stirring at -5/0°C for 15 minutes. The mixture was extracted with tert-butylmethylether (285 mL) and the phases were separated. The aqueous phase was extracted twice with tert-butylmethyl ether (2x 200 mL) and the combined organic layers were washed with brine (176 mL, 130 mL). The organic phase was concentrated under reduced pressure at 25°C and the crude product was obtained as a yellow oil (6.60 g). Purification by column chromatography on silica gel was performed eluting with dichloromethane:methanol increasing the percentage of methanol from 0 to 5%. A second purification on silica gel afforded Latanoprost (2.36 g, 0.005 mol, 68% over two steps).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.32-7.19 (m, 5H, Ph), 5.45-5.51 (m, 2H, H-5 e H-6 vinyl), 5.0 (hept, J=6.3 Hz, 1H, CH3CHCH3), 4,18 (bs, 1H, CHOH), 3.95 (bs, 1H, CHOH), 3.67 (bs, 1H, CHOH), 2.76 (m, 2H, CH 2Ph), 1.23 (d, J=6.3Hz, 6H, C(CH3)2), 2.55-1.3, 21H).
-
HPLC-MS (ESI): [M+Na]+ = 455, [M+H]+ = 432.
……………………………….
http://www.google.com/patents/EP2454227A1?cl=en
Example 3
Synthesis of Latanoprost
ether MTBE
8c-iso
Latanoprost
Scheme 5. Synthesis of Latanoprost
Synthesis of Latanoprost from 12c:
As shown in Scheme 5 in Example 3, a 250 ml.3-necked round-bottom flask equipped with a magnetic bar, a temperature probe, rubber septa, and a nitrogen gas inlet was charged at room temperature with 7.3 g (19.6 mmol) of deprotected lactone 12c in 70 mL of 2-propanol and 1.6 g (39.2 mmol) of sodium hydride, 60%, in mineral oil. The reaction mixture was heated at 35 0C for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of water and the pH was adjusted to 6 with 1 N HCI. The layers were separated and the aqueous layer was back extracted with 40 mL of 2-propanol four times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was dissolved in 60 mL of THF, 5.0 mL (33.3 mmol) of DBU and 3.3 mL (33.3 mmol) of iodopropane. The reaction mixture was stirred at room temperature for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of ethyl acetate and 60 mL of water. The layers were separated and the aqueous layer was back extracted with 40 mL of ethyl acetate for two times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was purified by using reverse phase biotage, 70 : 30 ACN :
H2O to obtain 4.1 g (49% yield) of Latanoprost, confirmed by 1H NMR.
………………….

Wittig condensation of lactol (XIII) with (carboxybutyl) triphenylphosphonium bromide (XV) in the presence of potassium tert-butoxide produced the Z-olefin (XVI). Conversion of carboxylic acid (XVI) to the title isopropyl ester was then accomplished by alkylation with 2-iodopropane in the presence of DBU.
http://www.chemdrug.com/databases/8_0_xmmatmlqjiethrwn.html
………………………………
http://www.nature.com/nature/journal/v489/n7415/full/nature11411.html?WT.ec_id=NATURE-20120913

………………………
http://brsmblog.com/?p=1525

……………………………….
http://www.google.com/patents/WO2013186550A1?cl=en
(la) (Ic)
Example 6 – Experimental procedures for the synthesis of latanoprost
A synthesis of latanoprost is shown and described below.
latanoprost (77)
2-Phenethyloxirane, 61
m-CPBA,
A modified procedure of Woodward was used (Bernier, D. et al., The Journal of Organic Chemistry 2008, 73, 4229). A stirred solution of 4-phenyl-l-butene 62 (500 mg, 568 μΙ, 3.78 mmol) in CH2CI2 (20 ml) was cooled to 0 °C. m-CPBA (816 mg, 4.73 mmol) was added as a solid and the reaction mixture was stirred at 0 °C for 1.5 h, then r.t. for 24 h. The reaction mixture was poured into saturated K2C03 solution (50 ml) and extracted with CH2CI2 (2 x 50 ml). The combined organic phases were washed with saturated K2C03 solution (50 ml) before being dried (MgS04), filtered and concentrated to give a clear colourless liquid. This material was purified by column chromatography, eluting with petrol/EtOAc (9:1), to give the epoxide 61 (10.2 g, 91%) as a clear colourless liquid. The 13C, and IR data were consistent with the literature (Mitchell, J. M. et al., Journal of the American Chemical Society 2001, 123, 862; Elings, J. A. et al., European Journal of Organic Chemistry 1999, 1999, 837).
Rf = 0.42 (petrol :EtOAc, 9:1) vmax (neatycnrr1 3027, 2989, 2922, 2859, 1602, 1495, 1454, 1410, 835, 750, 699
*H NMR (400 MHz; CDCI3) δΗ = 1.83-1.99 (2 H, m, CH2), 2.53 (1 H, dd, J = 5.0, 2.7 Hz, CHH), 2.75-2.93 (2 H, m, CH2), 2.80 (1 H, dd, J = 5.0, 4.0 Hz, CHH), 3.01 (1 H, dddd, J = 6.5, 5.0, 4.0, 2.7 Hz, CH), 7.22-7.38 (5 H, m, Ar H’s)
13C NMR (100 MHz; CDCI3) 5C = 32.2 (CH2), 34.2 (CH2), 47.2 (CH2), 51.7 (CH), 126.0 (2 x ArCH), 128.3 (2 x ArCH), 128.4 (ArCH), 141.2 (ArC)
m/z (EI) 148.1 (M+, 10%), 130.1 (23%), 129.0 (18%), 118.1 (29%), 117.1 (83%), 115.0 (28%), 105.0 (22%), 104.0 (61%), 92.0 (22%), 91.0 (100%), 83.9 (37%), 77.0 (17%), 65.0 (31%)
(2S)-2-Phenethyloxirane, 63

A modified procedure of Jacobsen was used (Schaus, S. E. et al., Journal of the American Chemical Society 2002, 224, 1307). Racemic epoxide 61 (10.0 g, 67.5 mmol) was dissolved in THF (10 ml) and stirred at r.t.. (S^-i+J-^A/’-BisiS^-di-tert-butylsalicylidene)-!^- cyclohexanediaminocobalt(II) (204 mg, 0.34 mmol) was added and the resultant dark brown solution cooled to 0 °C. Acetic acid (77 μΙ, 1.35 mmol) and water (669 μΙ, 37.1 mmol) were added. The reaction was stirred at 0 °C for 1 h and then at r.t. for 23 h. The reaction mixture was concentrated under reduced pressure and purified by column chromatography (~200 g silica), eluting with petrol/EtOAc (9:1), to give the epoxide 3 as a dark red liquid. This was re- purified by column chromatography eluting with petrol/EtOAc (9.5:0.5 to 9:1), to give the epoxide 3 (4.62 g, 46%) as an orange liquid. The analytical data matched that of the racemic material described above. The enantioselectivity of the resolution was determined after subsequent conversion to the allylic alcohol 66. The optical rotation matched closely with that reported in the literature (Martynow, J. G. et al., European Journal of Organic Chemistry 2007, 2007, 689).
[a]D 21 -21.0 (c. 1.0, CHC ) (lit., [a]D 20 -22.5 (c. 1.0, CHCI3)) 5-Phenyl-l-penten-3-ol, 64
A modified procedure of Molander was used (Molander, G. A. et al., The Journal of Organic Chemistry 2009, 74, 1297). A stirred solution of hydrocinnamaldehyde 65 (2.50 g, 2.45 ml, 18.6 mmol) in THF (25 ml) was cooled to -78 °C. Vinyl magnesium bromide solution (1 M in THF) (22.4 ml, 22.4 mmol) was added dropwise over ~ 5 min. The reaction mixture was stirred at -78 °C for 1.5 h, then 0 °C for 3 h. The reaction mixture was poured into saturated NH4CI solution (50 ml) and extracted with Et20 (3 x 50 ml). The combined organic phases were washed with saturated NaCI solution (50 ml) before being dried (MgS04), filtered and concentrated to give a pale yellow liquid. This material was purified by column
chromatography, eluting with petrol/EtOAc (9: 1), to give the vinyl alcohol 64 (1.89 g, 63%) as a clear colourless liquid. The *H, 13C, and IR data were consistent with the literature (Molander, G. A. et al., The Journal of Organic Chemistry 2009, 74, 1297; Kim, J. W. et al., Chemistry – A European Journal 2008, 24, 4104). Rf = 0.40 (petrol :EtOAc, 4: 1)
max (CHC Vcnrr1 3335, 3026, 2923, 2859, 1496, 1454, 990, 922, 747, 698
*H NMR (400 MHz; CDCI3) δΗ = 1.55 (1 H, br.s, OH), 1.84-1.99 (2 H, m, CH2), 2.70-2.89 (2 H, m, CH2), 4.19 (1 H, app q, J = 6.0 Hz, CHO ), 5.20 (1 H, app dt, J = 10.5, 1.4 Hz, HC=C), 5.30 (1 H, app dt, J = 17.1, 1.4 Hz, HHC=C), 5.96 (1 H, ddd, J = 17.1, 10.5, 6.0 Hz, H2C=CH), 7.20-7.40 (5 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.6 (CH2), 38.5 (CH2), 72.4 (HCOH), 114.9 (H2C=C), 125.8 (ArCH), 128.4 (2 x ArCH), 128.4 (2 x ArCH), 141.0 (H2C=Q, 141.8 (ArC)
m/z (EI) 162.1 (M+, 30%), 144.1 (52%), 129.1 (72%), 105.1 (68%), 92.1 (71%), 91.0 (100%), 57.0 (61%) 6D. (3S)-5-Phenyl-l-penten-3-ol, 66

A modified procedure of Falck was used (Alcaraz, L. et al., Tetrahedron Letters 1994, 35, 5449). A suspension of trimethylsulfonium iodide (18.2 g, 89.1 mmol) in anhydrous THF (220 ml) was stirred and cooled to -20 °C. 1.6 M n-BuLi (55.7 ml, 89.1 mmol) was added slowly and the reaction stirred at -20 °C for 1 h. A solution of epoxide 63 (4.40 g, 29.7 mmol) in anhydrous THF (50.0 ml) was added slowly. The reaction was stirred at -20 °C for 1 h and then allowed to warm to r.t. slowly. The reaction mixture was poured into water (200 ml) and extracted with Et20 (1 x 200 ml, 1 x 100 ml). The combined organic phases were washed with saturated NaCI solution (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by column chromatography (130 g silica), eluting with petrol/EtOAc (9:1), to give partially purified material. This was re-purified by column chromatography (50 g silica), eluting with petrol/EtOAc (9:1), to give allylic alcohol 66 (3.19 g, 66%) as a pale yellow liquid. The analytical data matched that described for the racemic material above.
[α]ο21 -11.0 (c. 1.0, CHCI3) (lit – Kanbayashi, N. et al., Angewandte Chemie International Edition 2011, 50, 5197, [a]D 25 -3.6 (for 85% ee (c. 0.4, CHCI3)))
Chiral-HPLC data: er = >99:1 (Chiralcel AD-H column, 210 nm, hexane/2-propanol: 98/2, flow rate: 0.5 mlVmin, room temperature; ¾: minor 41.0 min, major 43.7 min) 6E. tert-Butyl(dimethyl)[(lS)-l-phenethyl-2-propenyl]oxysilane, 67

67 A stirred solution of allylic alcohol 66 (3.00 g, 18.5 mmol) in CH2CI2 (53 ml) was cooled to 0 °C. Imidazole (2.27 g, 33.3 mmol) was added in one portion followed by t- butylchlorodimethylsilane (3.34 g, 22.2 mmol). The cooling bath was removed and the reaction mixture stirred at r.t. for 16 h before being poured into 10% aq. HCI (100 ml). The mixture was extracted with 40/60 petroleum ether (2 x 100 ml). The combined organics were washed with saturated NaCI solution (100 ml), dried (MgS04), filtered, and concentrated to give the crude material. This was purified by column chromatography, eluting with 40/60 petroleum ether, to give the protected alcohol 67 (4.68 g, 92%) as a colourless liquid. The *H NMR data and optical rotation matched that reported in the literature (Uenishi, J. i. et al., Organic Letters 2011, 13, 2350).
Rf = 0.25 (40/60 petroleum ether)
vmax (film)/cm-13064, 3027, 2952, 2929, 2886, 2856, 1497, 1472, 1462, 1455, 1361, 1251, 1122, 1083, 1030, 990, 921, 834, 774, 697
*H NMR (400 MHz; CDCI3) δΗ = 0.05 (3 H, s, SiCH3), 0.08 (3 H, s, SiCH3), 0.93 (9 H, s, C(CH3)3), 1.82 (2 H, m, CH2), 2.66 (2 H, m, CH2), 4.17 (1 H, m, OCH), 5.08 (1 H, ddd, J = 10.4, 1.5, 1.3 Hz, HA =CH), 5.19 (1 H, app dt, J = 17.2, 1.5 Hz, HHC=CH), 5.86 (1 H, ddd, J = 17.2, 10.4, 6.0 Hz, H2C=CH), 7.18 (3 H, m, ArH’s), 7.28 (2 H, m, ArH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.8 (SiCH3), -4.3 (SiCH3), 18.3 (C(CH3)3), 25.9 (C(CH3)3), 31.5 (CH2), 39.8 (CH2), 73.3 (CHOSi), 114.0 (H2C=C), 125.7 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 141.4 (H2C=Q, 142.5 (ArC).
[a]D” 12.0 (c. 1.0, CHCI3) (lit., [a]D 20 14.5 (c. 1.0, CHCI3)) 6F. (3S)-3-[l-(tert-Butyl)-l,l-dimethylsilyl]oxy-5-phenylpentan-l-ol, 68
67
A modified procedure of Denmark was used (Denmark, S. E. et al., Organic Letters 2005, 7, 5617). Compound 67 (2.00 g, 7.23 mmol) was added to a flame dried schlenk flask under N2. 9-BBN (0.5 M in THF) (15.9 ml, 7.96 mmol) was added via syringe and the resulting solution stirred at r.t. for 1 h. A further 1.1 eq. (15.9 ml, 7.96 mmol) of 9-BBN was added and the reaction stirred at r.t. for 2 h. Water (16.0 ml) and NaB03.4H20 (5.56 g, 36.2 mmol) were added and the reaction stirred at r.t. for 2 h. The reaction mixture was poured into saturated NH4CI solution (60 ml) and extracted with Et20 (3 x 100 ml). The combined organic phases were washed with sat. NaCI solution (100 ml), dried (MgS04), filtered, and concentrated to give the crude material. This was purified 3 times by column chromatography (twice eluting with petrol/EtOAc (6:1) and once with petrol/ EtOAc/Eti) (9:0.5:0.5)) to give the alcohol 68 (672 mg, 32%) as a clear colourless oil.
Rf = 0.18 (petrol :EtOAc, 9:1)
vmax (film)/cm-13351 (broad), 3063, 2950, 2928, 2885, 2856, 1496, 1471, 1462, 1454, 1360, 1253, 1092, 1057, 1028, 1005, 834, 773, 746, 698
*H NMR (400 MHz; CDCI3) δΗ = 0.09 (3 H, s, SiCH3), 0.10 (3 H, s, SiCH3), 0.92 (9 H, s, C(CH3)3), 1.75 (1 H, m, CHH), 1.83-1.94 (3 H, m, CH2, CHH), 2.32 (1 H, app t, J = 5.2 Hz, OH), 2.64 (2 H, m, CH2), 3.75 (1 H, app dq, J = 10.8, 5.5 Hz, OCHH), 3.87 (1 H, app ddt, J = 10.8, 8.1, 4.8 Hz, OCHtf), 3.99 (1 H, app qd, J = 6.1, 4.4 Hz, HCOTBDMS), 7.16-7.23 (3 H, m, ArCH’s), 7.27-7.33 (2 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.7 (SiCH3), -4.4 (SiCH3), 18.0 (C(CH3)3), 25.8 (C(CH3)3), 31.7 (CH2), 37.8 (CH2), 38.7 (CH2), 60.1 (CH2), 71.2 (SiOCH), 125.8 (ArCH), 128.2 (2 x ArCH), 128.4 (2 x ArCH), 142.1 (ArC)
HRMS (ESI) calcd for Ci7H30O2SiNa [MNa+] 317.1907, found 317.1906
[a]D 23 23.0 (c. 1.0, CHCI3) 6G. tert-Butyl[(lS)-3-iodo-l-phenethylpropyl]oxydimethylsilane, 69
68 A modified procedure of Rychnovsky was used (Dalgard, J. E. et al., Organic Letters 2004, 6, 2713). Alcohol 68 (600 mg, 2.04 mmol) was added to a flame dried schlenk flask under N2. CH2CI2 (10 ml) was added via syringe and the resulting solution stirred at r.t..
Triphenylphosphine (695 mg, 2.65 mmol) and imidazole (222 mg, 3.26 mmol) were added as solids in one portion. Iodine (672 mg, 2.65 mmol) was added to the resulting solution. A slight exotherm was noted and the solution changed from a light yellow colour to a brown colour with the formation of a precipitate. The reaction was stirred at r.t. for 1 h. The reaction mixture was dry loaded onto silica (2 g) and purified by column chromatography (14 g silica), eluting with petrol to petrol/EtOAc (9: 1). This gave the iodide 69 (725 mg, 88%) as a clear, colourless oil.
Rf = 0.20 (40/60 petroleum ether)
ifiln /cnr^OeS, 3026, 2951, 2928, 2886, 2856, 1495, 1471, 1461, 1360, 1253, 1187, 1165, 1140, 1092, 1063, 1005, 975, 931, 833, 773, 697
*H NMR (400 MHz; CDCI3) δΗ = 0.09 (3 H, s, SiCH3), 0.10 (3 H, s, SiCH3), 0.92 (9 H, s, (C(CH3)3), 1.79 (2 H, m, CH2), 2.05 (2 H, m, CH2), 2.64 (2 H, m, CH2), 3.24 (2 H, m, CH2), 3.82 (1 H, quin., J = 5.7 Hz, OCH), 7.16-7.23 (3 H, m, ArCH’s), 7.27-7.33 (2 H, m, ArCH’s) 13C NMR (100 MHz; CDCI3) 5C = -4.3 (SiCH3), -4.3 (SiCH3), 3.0 (CH2), 18.1 (C(CH3)3), 25.9 (C(CH3)3), 31.3 (CH2), 38.7 (CH2), 40.8 (CH2), 71.7 (OCH), 125.8 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 142.1 (ArC)
HRMS (ESI) calcd for Ci7H30OSiI [MH+] 405.1108, found 405.1105
[a]D 23 26.0 (c. 1.0, CHCI3)
6H. [(lR)-3-((3aR,4R,6aS)-2-Methoxy-5-(£)-l-[(l,l,l- trimethylsilyl)oxy]methylideneperhydrocyclopenta[d]furan-4-yl)-l- phenethylpropyl]oxy(tert-butyl)dimethylsilane, 70

70 Iodide 69 (1.32 g, 3.27 mmol, 1.1 eq.) was added via syringe to a flame dried schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous Et20 (13.3 ml) was added via syringe and the resulting solution cooled to -78 °C. 1.63 M t-BuLi (4.01 ml, 6.54 mmol, 2.2 eq.) was added dropwise and the reaction mixture stirred at -78 °C for 2 h and -40 °C for 2 h before being cooled back to -78 °C. Meanwhile, thiophene (275 mg, 262 μΙ, 3.27 mmol, 1.1 eq.) was added via syringe to a flame dried schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous THF (13.3 ml) was added via syringe and the resulting solution cooled to -30 °C. 1.63 M n-BuLi (2.01 ml, 3.27 mmol, 1.1 eq.) was added dropwise and the solution stirred at -30 °C for 30 min. CuCN (293 mg, 3.27 mmol, 1.1 eq.) was added as a solid, in one portion. The cooling bath was removed and the suspension allowed to warm to r.t. The resulting tan/brown solution of cuprate was added dropwise via syringe to the schlenk flask containing the alkyl lithium and anhydrous THF (13.3 ml) added. The mixture was stirred at -20 °C for 1 h to allow formation of mixed cuprate 71. This was cooled to -78 °C and a solution of enal 24 (500 mg, 2.97 mmol, 1.0 eq.) in anhydrous THF (13.3 ml) was added dropwise. The mixture was stirred at -78 °C for 1 h and then allowed to warm slowly to -20 °C. TMSCI (1.61 g, 1.89 ml, 14.9 mmol, 5.0 eq.) was added via syringe followed by NEt3 (1.80 g, 2.49 ml, 17.8 mmol, 6 eq.). The reaction was quenched by the addition of saturated NH4CI solution (50 ml) and extracted with Et20 (3 x 50 ml). The combined organic phases were washed with saturated NH4CI solution (50 ml) and saturated NaCI solution (50 ml) before being dried (MgS04), filtered, and concentrated to give the crude material as a yellow oil. This was used directly in the next step.
61. (3aR 4R,5R,6aS)-4-((3R)-3-[l-(tert-Butyl)-l,l-dimethylsilyl]oxy-5- phenylpentyl)-2-methoxyperhydrocyclopenta[d]furan-5-ol, 73

70 73 The crude material from the conjugate addition / trapping experiment, containing 70, was dissolved in CH2Cl2/MeOH (3: 1) (30 ml) and cooled to -78 °C. A stream of ozone was passed through the stirred solution. The reaction was monitored periodically by TLC in order to judge completion of the ozonolysis (judged by consumption of silyl enol ether). The reaction mixture was flushed with a stream of N2, for 15 min, to remove excess 03. NaBH4 (202 mg, 5.35 mmol) was added in one portion. The reaction mixture was stirred at -78 °C for 2 h before the cooling bath was removed and the reaction allowed to warm to r.t.. The reaction was stirred at r.t. for 1 h. NaBH4 (67.4 mg, 1.78 mmol) was added and the reaction stirred at r.t. for a further 15 min. The reaction mixture was poured into saturated NaCI solution (25 ml) and extracted with EtOAc (3 x 25 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude product as a pale yellow oil. This was purified by column chromatography on silica, eluting with petrol/EtOAc (4: 1), giving the alcohol 73 (as an approximately 2:1 mixture of diastereoisomers) as a clear, colourless oil (800 mg, 62% (2 steps from enal 24)).
Rf = 0.23 (petrol :EtOAc, 4:1)
vmax (neatycnrr1 3434 (broad), 3026, 2928, 2856, 1496, 1471, 1454, 1360, 1343, 1254, 1098, 1053, 1004, 937, 833, 773, 698
1H NMR (400 MHz; CDCI3) 5H = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) 0.05 (3 H, s, CH3), 0.06* (3 H, s, CH3), 0.07 (3 H, s, CH3), 0.07* (3 H, s, CH3), 0.91 (9 H, s, C(CH3)3), 0.92* (9 H, s, C(CH3)3), 1.12-1.80 (7 H, m), 1.12- 1.80* (7 H, m), 1.90-2.38 (5 H, m), 1.90-2.38* (5 H, m), 2.53-2.75 (2 H, m, CH2), 2.53-2.75* (2 H, m, CH2), 3.32 (3 H, s, OCH3), 3.39* (3 H, s, OCH3), 3.72 (1 H, m, CHOTBDMS), 3.72* (1 H, m, CHOTBDMS), 3.79* (1 H, m, CHOH), 3.89 (1 H, m, CHOH), 4.55 (1 H, app td, J = 6.3, 2.5 Hz, CH), 4.64* (1 H, app td, J = 6.8, 2.7 Hz, CH), 5.06* (1 H, d, J = 5.5 Hz, OCHO), 5.11 (1 H, d, J = 4.9 Hz, OCHO), 7.19 (3 H, m, ArCH’s), 7.19* (3 H, m, ArCH’s), 7.29 (2 H, m, ArCH’s), 7.29* (2 H, m, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = (observed signals, mixture of 2 diastereoisomers) -4.42 (SiCH3), -4.41 (SiCH3), -4.33 (SiCH3), 18.1 (2 x C(CH3)3), 25.9 (2 x C(CH3)3), 29.3 (CH2), 30.3 (CH2), 31.7 (CH2), 35.1 (CH2), 38.8 (CH2), 39.9 (CH2), 40.0 (CH2), 41.1 (CH2), 42.7 (CH2),
46.5, 47.2, 54.4, 55.1, 55.3, 55.7, 71.8, 71.8, 79.3, 79.7, 82.5, 85.8, 106.5 (OCHOCH3), 108.0 (OCHOCH3), 125.7 (ArCH), 125.7 (ArCH), 128.3 (4 x ArCH), 128.3 (4 x ArCH), 142.6 (ArC), 142.6 (ArC). One SiCH3 and three CH2‘s could not be assigned due to overlapping signals. 6J. (3aR,4R,5R,6aS)-4-[(3R)-3-Hydroxy-5- phenylpentyl]perhydrocyclopenta[b]furan-2,5-diol, 74
Alcohol 73 (400 mg, 0.920 mmol) was stirred with 1.5% aqueous HQ / THF (3:2) (18 ml) at r.t. for 16 h. The mixture was neutralised with 1 M NaOH and extracted with CH2CI2 (5 x 30 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the triol 74 and silanol by-product as a clear, colourless oil (~400 mg). This material was taken forward for the subsequent transformation without purification.

(4-Carboxybutyl)(triphenyl)phosphonium bromide 29 (2.45 g, 5.52 mmol) was added to a flame dried schlenk flask, under N2, and anhydrous THF (20.0 ml) added. The resulting suspension was cooled to 0 °C. KOt-Bu (1.24 g, 11.0 mmol) was added in one portion and the resulting orange mixture stirred at 0 °C for 40 min. A solution of crude triol 74 (282 mg, 0.920 mmol) in anhydrous THF (5.0 ml) was added dropwise via syringe. After complete addition the cooling bath was removed and the mixture was stirred at r.t. for 1.5 h. The reaction was quenched with H20 (30 ml) and washed with Et20 (2 x 30 ml) to remove triphenylphosphine oxide. The aqueous phase was made acidic with 1 M HQ (~10 ml) and extracted with CH2CI2 (5 x 25 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude material as solids. These were placed on a sinter funnel and washed with petrol/EtOAc (1: 1) (4 x 20 ml) and then EtOAc (2 x 40 ml). The filtrate was concentrated under vacuum and purified by column chromatography on silica, eluting with CH2Cl2/MeOH (9.5:0.5 to 9:1) to give acid 75 (163 mg, 45% over 2 steps from alcohol 73) as a clear, colourless oil. The *Η data and optical rotation were consistent with the literature (Martynow, J. G. et al., European Journal of Organic Chemistry 2007, 2007, 689).
Rf = 0.27 (CH2CI2:MeOH, 9:1)
vmax (neatycnrr1 3338 (broad), 2930, 2857, 1704, 1452, 1407, 1254, 1028, 747, 699, 636 *H NMR (400 MHz; CDCI3) δΗ = 1.39 (2 H, m, CH2), 1.47-1.97 (10 H, m, 4 x CH2, 2 x CH), 2.07-2.48 (6 H, m, 3 x CH2), 2.67 (1 H, m, CH ), 2.80 (1 H, m, CH/-/), 3.60-4.85 (6 H, broad signal, 2 x OCH, 3 x OH, COOH), 3.72 (1 H, m, OCH), 5.40 (1 H, m, =CH), 5.49 (1 H, m, =CH), 7.15-7.24 (3 H, m, ArCH’s), 7.25-7.32 (2 H, m, ArCH’s)
[a]D 24 29.0 (c. 1.0, MeOH) (lit, [a]D 20 29.7 (c. 1.0, MeOH)) 6L. Isopropyl (Z)-7-(lR,2R,3R,5S)-3,5-dihydroxy-2-[(3R)-3-hydroxy-5- phenylpentyl]cyclopentyl-5-heptenoate, latanoprost, 77

A modified procedure of Zanoni and Vidari was used (Zanoni, G. et al., Tetrahedron 2010, 66, 7472). Carboxylic acid 75 (100 mg, 0.256 mmol) was dissolved in DMF (2.0 ml) and stirred at r.t.. Cs2C03 (125 mg, 0.384 mmol) was added in one portion followed by 2- iodopropane (51 μΙ, 0.512 mmol). The reaction was stirred at r.t. for 18 h. The reaction mixture was poured into 3% citric acid solution (10 ml) and extracted with TBME (4 x 10 ml). The combined organic phases were washed with 10% NaHC03 solution (10 ml) and saturated NaCI (2 x 10 ml) before being dried (MgS04), filtered, and concentrated to give the crude product as a clear, colourless oil (95 mg). This was purified by column chromatography (3 g silica), eluting with petrol/EtOAc (2: 1 to 1:2), to give latanoprost 77 (71 mg, 64 %) as a clear colourless oil. The IR, 13C, and optical rotation data were consistent with the literature (Zanoni, G. et al., Tetrahedron 2010, 66, 7472). Rf = 0.44 (EtOAc)
vmax (neatVcm“1 3360 (broad), 2980, 2931, 2857, 1712, 1495, 1454, 1374, 1311, 1247, 1180, 1106, 1030, 966, 910, 820, 731, 699
*H NMR (400 MHz; CDCI3) δΗ = 1.23 (6 H, d, J = 6.4 Hz, 2 x CH3), 1.30-1.90, (14 H, m, 5 x CH2, 2 x CH, 2 x OH), 2.07-2.39 (6 H, m, 3 x CH2), 2.45 (1 H, d, J = 5.5 Hz, OH), 2.63- 2.86 (2 H, m, CH2), 3.68 (1 H, br.s, CHO ), 3.95 (1 H, br.s, CHOH), 4.18 (1 H, br.s, CHO ), 5.01 (1 H, sept., J = 6.4 Hz, OCH(CH3)2), 5.35-5.52 (2 H, m, 2 x =CH), 7.16-7.24 (3 H, m, ArH’s), 7.25-7.32 (2 H, m, ArH’s)
13C NMR (125 MHz; CDCI3) 5C = 21.9 (2 x CH3), 24.9 (CH2), 26.6 (CH2), 26.8 (CH2), 29.6 (CH2), 32.1 (CH2), 34.0 (CH2), 35.7 (CH2), 39.0 (CH2), 42.5 (CH2), 51.8 (CH), 52.7 (CH), 67.6 (OCH), 71.2 (OCH), 74.5 (OCH), 78.6 (OCH), 125.7 (CH), 128.3 (2 x ArCH), 128.3 (2 x ArCH), 129.3 (CH), 129.5 (CH), 141.1 (ArC), 173.5 (C=0)
[a]D 23 33.0 (c. 1.0, MeCN) (lit, [a]D 20 32.7 (c. 1.0, MeCN))
References
- Ishikawa H, Yoshitomi T, Mashimo K, Nakanishi M, Shimizu K (February 2002). “Pharmacological effects of latanoprost, prostaglandin E2, and F2alpha on isolated rabbit ciliary artery”. Graefes Arch. Clin. Exp. Ophthalmol. 240 (2): 120–5. doi:10.1007/s00417-001-0412-4. PMID 11931077.
- Patel SS, Spencer CM (1996). “Latanoprost. A review of its pharmacological properties, clinical efficacy and tolerability in the management of primary open-angle glaucoma and ocular hypertension”. Drugs Aging 9 (5): 363–378. doi:10.2165/00002512-199609050-00007. PMID 8922563.
- Huttunen et al. (2011) Prodrugs—from Serendipity to Rational Design. Pharmacol Rev 63:750–771
- “Patent US5296504 – Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension – Google Patents”.
- “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Perry CM, McGavin JK, Culy CR, Ibbotson T (2003). “Latanoprost. An Update of its Use in Glaucoma and Ocular Hypertension”. Drugs Aging 20 (8): 1170–2229.PMID 12795627.
- Zhang WY, Wan Po AL, Dua HS, Azuara-Blanco A (2001). “Meta-analysis of randomised controlled trials comparing latanoprost with timolol in the treatment of patients with open angle glaucoma or ocular hypertension”. British Journal of Ophthalmology 85: 983–990. doi:10.1136/bjo.85.8.983. PMID 11466259.
- Aung T; Wong HT; Yip CC; et al. (2000). “Comparison of the intraocular pressure-lowering effect of latanoprost and timolol in patients with chronic angle closure glaucoma: a preliminary study.”. Ophthalmology 107 (6): 1178–83. doi:10.1016/s0161-6420(00)00073-7. PMID 10857840.
- Amano S, Nakai Y, Ko A, Inoue K, Wakakura M (2008). “A case of keratoconus progression associated with the use of topical latanoprost”. Japanese Journal of Ophthalmology 52 (4): 334–6. doi:10.1007/s10384-008-0554-6. PMID 18773275.
- De Santis, M., Lucchese, A., Carducci, B., Cavaliere, A., De Santis, L., & Merola, A. et al. (2004). Latanoprost exposure in pregnancy. American Journal Of Ophthalmology, 138(2), 305.pmid=15289149.1
- Morgan, P., Proniuk, S., Blanchard, J., & Noecker, R. (2001). Effect of temperature and light on the stability of latanoprost and its clinical relevance. Journal Of Glaucoma, 10(5), 401–405.
External links

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO






































































 amcrasto@gmail.com
amcrasto@gmail.com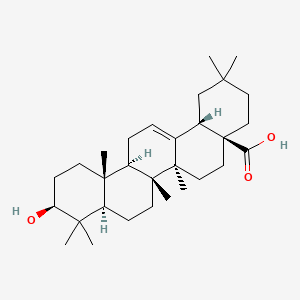
 Oleanolic acid
Oleanolic acid












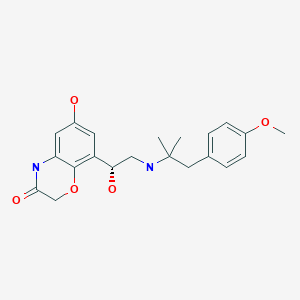
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.










