
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

BOFUTRELVIR
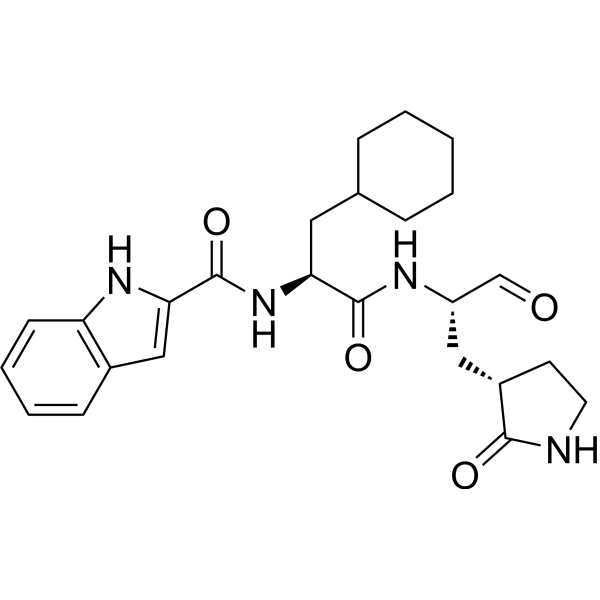
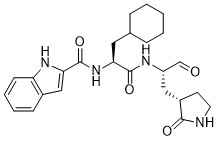
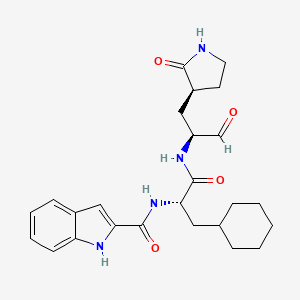
BOFUTRELVIR
Cas 2103278-86-8
| Molecular Weight | 452.55 |
|---|---|
| Formula | C25H32N4O4 |
UNII-T5UX5SKK2S; Mpro inhibitor 11A; 2103278-86-8; T5UX5SKK2S, DC-402234, DC402234, MPI-10
IUPAC/Chemical Name: N-[(2S)-3-cyclohexyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]propan-2-yl]-1H-indole-2-carboxamide
N-[(2S)-3-cyclohexyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]propan-2-yl]-1H-indole-2-carboxamide
Bofutrelvir has an additive antiviral effect when combined with Remdesivir
FB2001
Bofutrelvir (FB2001) is a SARS-CoV-2 main protease Mpro inhibitor with an IC50 value of 53 nM and an EC50 value of 0.53 μM. Bofutrelvir exhibits potent antiviral efficacy against several current SARS-CoV-2 variants with EC50 values of 0.26-0.42 μM. Bofutrelvir has an additive antiviral effect when combined with Remdesivir.
Bofutrelvir is a small molecule inhibitor of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) main protease (Mpro; 3C-like protease; 3CL protease; 3CLpro; nsp5 protease), with potential antiviral activity against SARS-CoV-2. Upon intravenous administration or inhalation into the lungs, bofutrelvir selectively targets, binds to, and inhibits the activity of SARS-CoV-2 Mpro. This inhibits the proteolytic cleavage of viral polyproteins, thereby inhibiting the formation of viral proteins including helicase, single-stranded-RNA-binding protein, RNA-dependent RNA polymerase, 20-O-ribose methyltransferase, endoribonuclease and exoribonuclease. This prevents viral transcription and replication. Bofutrelvir may have antiviral activity in the brain.
- Originator Frontier Biotechnologies
- Class Amides; Antivirals; Indoles; Pyrrolidinones; Small molecules
- Mechanism of Action Coronavirus 3C-like proteinase inhibitors
Highest Development Phases
- Phase II/III COVID 2019 infections
Most Recent Events
- 28 Apr 2024No recent reports of development identified for phase-I development in COVID-2019-infections in USA (IV, Infusion)
- 04 Jan 2023Phase-II/III clinical trials in COVID-2019 infections in China (Inhalation) (NCT05675072)
- 30 Dec 2022Frontier Biotechnologies completes a phase I trial in COVID-2019 infections in China (Inhalation) (NCT05583812)
- N-[(2S)-3-cyclohexyl-1-oxo-1-({(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)propan-2-yl]-1H-indole-2-carboxamide is a secondary carboxamide resulting from the formal condensation of the carboxy group of 1H-indole-2-carboxylic acid with the primary amino group of 3-cyclohexyl-N-{(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}-L-alaninamide. It is an inhibitor of SARS coronavirus main proteinase and inhibits SARS-CoV-2 replication in cell culture (EC50 = 0.53 muM). It has a role as an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor and an anticoronaviral agent. It is an indolecarboxamide, a member of pyrrolidin-2-ones, an aldehyde, a secondary carboxamide and an oligopeptide.
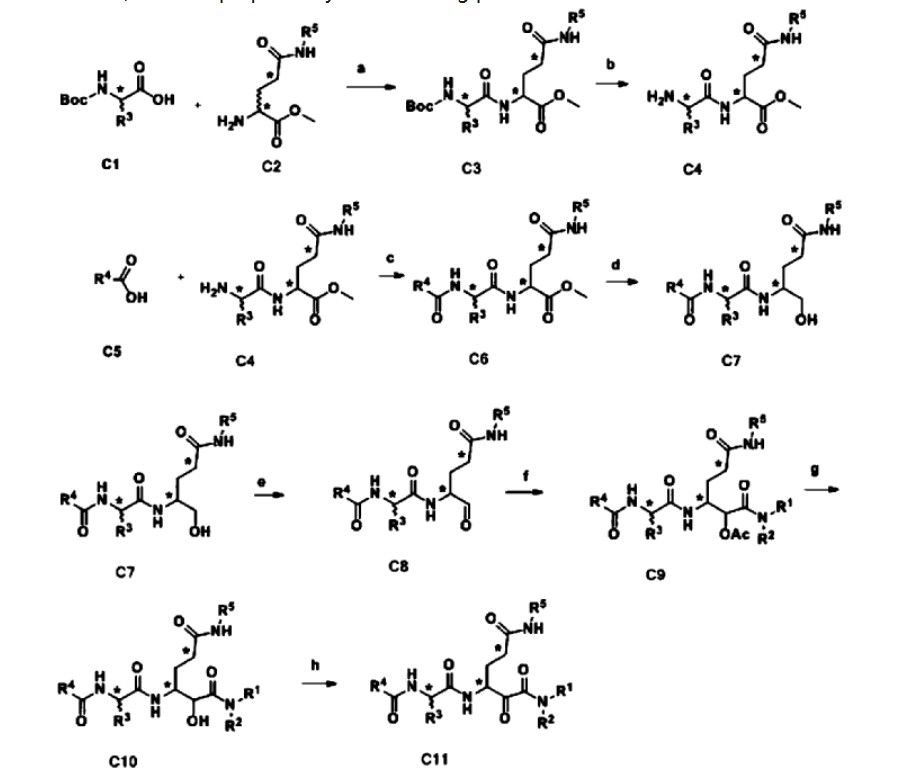
SCHEME
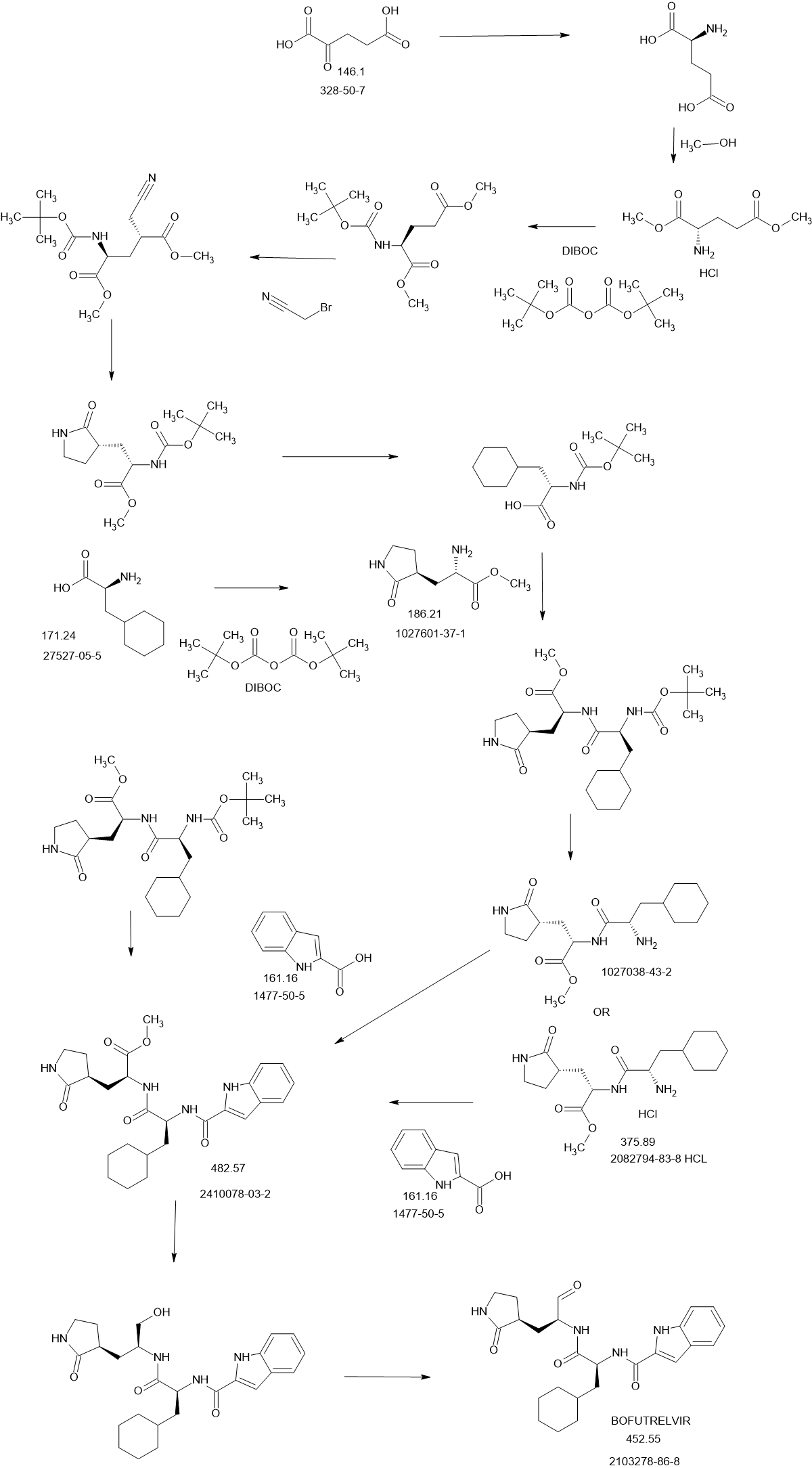
PATENTS
CN110818691
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN289596961&_cid=P11-M9Z1Y3-09353-1
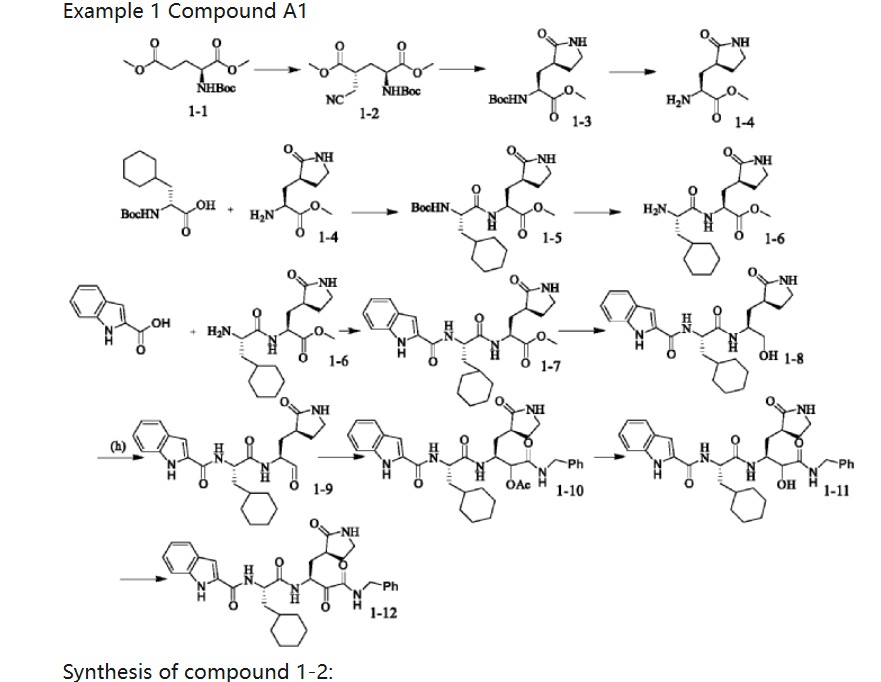
| Synthesis of compound 1-2: |
| Under argon protection, N-tert-butyloxycarbonyl-L-glutamic acid dimethyl ester (1-1) (6g, 21.8mmol) was dissolved in 60mL of anhydrous tetrahydrofuran, and a tetrahydrofuran solution of LiHMDS (1M in THF) (47mL, 47mmol) was slowly dripped at -78℃, and the temperature was kept stable at -78℃ during the dripping process, which lasted for about 1 hour. After the dripping was completed, it was stirred at -78℃ for 1 hour. Bromoacetonitrile (2.79g, 23.3mmol) was dissolved in 20ml of tetrahydrofuran, and then the solution was slowly dripped into the reaction system, and the dripping process lasted for 1 to 2 hours. The temperature was controlled at -78℃ and the reaction was continued for 3 hours. After the reaction was completed, NH4Cl solution was added to the reaction solution to quench the reaction, and the mixture was stirred for 10min and then warmed to room temperature. 40mL of saturated sodium chloride solution was poured in and stirred thoroughly, and the reaction system was seen to be stratified. The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (EA). The organic layers were combined, dried over anhydrous sodium sulfate, concentrated, and subjected to column chromatography (Flash, PE:EA=1:5) to obtain 3.9 g of a light yellow oil 1-2 with a yield of 58%. |
| Synthesis of compound 1-3: |
| Dissolve 1-2 (1 g, 3.15 mmol) in 25 mL of anhydrous methanol, stir to 0°C in an ice bath, and then add cobalt dichloride hexahydrate (450 mg, 1.89 mmol). After 10 min, add sodium borohydride (715 mg, 18.9 mmol) in small portions. The reaction solution continues to react in an ice bath for 1 h and then returns to room temperature. After 15 h, quench with 5 mL of saturated NH4Cl solution and continue stirring for 10 min. After filtering out the solid, evaporate the filtrate to dryness, extract with 20 mL of water and 30×3 mL of ethyl acetate, combine the organic phases, and add anhydrous Na 2 SO 4 After drying for 1 h, the residue was concentrated under reduced pressure and separated by column chromatography [PE:EA=1:2] to obtain 460 mg of a white powdery solid with a yield of 51%. |
| Synthesis of compound 1-4: |
| Compound 1-3 (2.6 g) was dissolved in a dichloromethane solution of trifluoroacetic acid (1/1, v/v), stirred at room temperature for 1 hour, concentrated, added with 100 ml of dichloromethane, washed with saturated sodium carbonate solution, and the organic layer was dried over anhydrous sodium sulfate and concentrated to obtain an oily substance 1-4 (2.7 g) with a yield of 99%. |
| Synthesis of compound 1-5: |
| Boc-cyclohexylalanine (1.26 g, 5 mmol), EDCI (1.36 g, 6 mmol), and HOBt (0.822 g, 6 mmol) were added to 80 ml of dichloromethane solution and stirred at room temperature for 30 min. Compound 1-4 (0.896 g, 5 mmol) was then added, and 1.2 equivalents of triethylamine were added dropwise, and stirred at room temperature. After TLC monitoring (ultraviolet), dichloromethane was used for extraction after the reaction was complete, and the mixture was washed with dilute hydrochloric acid, saturated sodium bicarbonate solution, and saturated sodium chloride. The organic layers were combined and dried over anhydrous sodium sulfate, and concentrated to obtain 1.2 g of a white viscous solid with a yield of 60%. |
| Synthesis of compound 1-6: |
| Compound 1-5 (2.5 g) was dissolved in a dichloromethane solution of trifluoroacetic acid (1/1, v/v), stirred at room temperature for 1 hour, concentrated, added with 100 ml of dichloromethane, washed with saturated sodium carbonate solution, and the organic layer was dried over anhydrous sodium sulfate and concentrated to obtain an oily substance 1-6 (2.61 g) with a yield of 99%. |
| Synthesis of compound 1-7: |
| Indole 2-carboxylic acid (0.795 g, 5 mmol), EDCI (1.36 g, 6 mmol), and HOBt (0.822 g, 6 mmol) were added to 80 ml of dichloromethane solution and stirred at room temperature for 30 min. Compound 1-6 (2.2 g, 5 mmol) was then added, and 1.2 equivalents of triethylamine were added dropwise, and stirred at room temperature. After TLC monitoring (ultraviolet), dichloromethane was used for extraction after the reaction was complete, and the mixture was washed with dilute hydrochloric acid, saturated sodium bicarbonate solution, and saturated sodium chloride. The organic layers were combined and dried over anhydrous sodium sulfate, and concentrated to obtain 1.3 g of a white viscous solid with a yield of 60%. |
| Synthesis of compound 1-8: |
| Dissolve 1-7 (243 mg, 0.51 mmol) in 20 ml of methanol, slowly add sodium borohydride (107 mg, 2.9 mmol) in batches, and stir at room temperature for about 2 hours to complete the reaction. After the reaction is completed, add about 20 ml of saturated brine to quench the reaction, concentrate the methanol in the reaction system, and add dichloromethane for extraction. The organic phase is washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated to obtain a white solid substance 1-8, which can be directly used in the next step. |
| Synthesis of compound 1-9: |
| Dissolve the intermediate 1-8 (129 mg, 0.29 mmol) in 20 ml of dichloromethane, add Dess-Martin oxidant (147 mg, 0.35 mmol) and solid sodium bicarbonate (29 mg, 0.35 mmol), and stir at room temperature. After the reaction is complete by TLC monitoring (ultraviolet), filter the reaction system, extract the filtrate with saturated sodium bicarbonate, and the organic layer is purified by saturated sodium salt. |
| The product was washed with water, dried over anhydrous sodium sulfate and concentrated. The product was separated and purified by flash column chromatography (CH2Cl2:MeOH=20:1) to obtain 77 mg of compound 1 as a white solid powder with a yield of 60%. |
| Synthesis of compound 1-10: |
| Compound 1-9 (129 mg, 0.29 mmol) was dissolved in dichloromethane solvent, acetic acid (19.2 mg, 0.32 mmol) and benzyl isocyanate (37.6 mg, 0.32 mmol) were added to react to obtain compound 1-10. Flash column chromatography (CH 2 Cl 2 :MeOH=20:1) to separate and purify to obtain 126 mg of white solid powder compound 1-10 with a yield of 70%. |
| Synthesis of compound 1-11: |
| Compound 1-10 (187 mg, 0.3 mmol) was dissolved in methanol solvent, LiOH (0.6 mmol) was added and stirred to obtain compound 1-11. 2 Cl 2 :MeOH=20:1) to separate and purify to obtain 148 mg of white solid powder compound 1-11 with a yield of 85%. |
| Synthesis of compound 1-12: |
| Compound 1-11 (174 mg, 0.3 mmol) was dissolved in dichloromethane solvent, Dess-Martin oxidant (152 mg, 0.36 mmol) was added, sodium bicarbonate (30 mg, 0.36 mmol) was added, and stirred to obtain a white solid powder compound 1-12 of 140 mg in total, with a yield of 80%. |
| 1 H NMR(500MHz,Chloroform)δ9.76(s,1H),7.73(s,1H),7.39(s,1H),7.32–7.26(m,2H),7.22(s,1H),7 .20–7.10(m,3H),7.01(s,1H),6.82(s,1H),6.68(s,1H),6.14(s,1H),5.57(s,1H),5.43(s,1H),4.3 8(s,1H),4.32(d,J=19.2Hz,2H),3.45(s,1H),3.35(s,1H),3.06(s,1H),2.20(dd,J=15.4,2.3Hz,4H ),2.12–2.03(m,2H),1.92(s,1H),1.77(s,1H),1.73–1.67(m,3H),1.66–1.53(m,6H),1.37(s,1H).; |
PATENT
WO2020030143
bioRxiv (2020), 1-17
- [1]. Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020 Sep 1;30(17):127377. [Content Brief]
- [2]. Shang W, et al. In vitro and in vivo evaluation of the main protease inhibitor FB2001 against SARS-CoV-2. Antiviral Res. 2022 Dec;208:105450. [Content Brief]
///BOFUTRELVIR, FB2001, FB 2001, Phase 3, COVID 2019, T5UX5SKK2S, Mpro inhibitor, DC-402234, DC402234, MPI-10
Inavolisib



Inavolisib
WeightAverage: 407.378
Monoisotopic: 407.140510438
Chemical FormulaC18H19F2N5O4
- GDC-0077
- CAS 2060571-02-8
- GDC0077
- RG6114
- WHO 11204
- GDC 0077
- GDC-0077
- RG-6114
- RG6114
- RO-7113755
- RO7113755
FDA APPROVED, 10/10/2024, Itovebi, To treat locally advanced or metastatic breast cancer
Drug Trials Snapshot
(2S)-2-[[2-[(4S)-4-(difluoromethyl)-2-oxo-1,3-oxazolidin-3-yl]-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl]amino]propanamide
- (2S)-2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)propanamide
- propanamide, 2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)-, (2S)-
Inavolisib, sold under the brand name Itovebi, is an anti-cancer medication used for the treatment of breast cancer.[2][3] It is an inhibitor and degrader of mutant phosphatidylinositol 3-kinase (PI3K) alpha.[4] The PI3K-mediated signalling pathway has shown to play an important role in the development of tumours as dysregulation is commonly associated with tumour growth and resistance to antineoplastic agents and radiotherapy.[5]
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
Inavolisib was approved for medical use in the United States in October 2024.[3][6][7]
SYN
Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, Edgar KA, Eigenbrot C, Elliott R, Endres N, Friedman LS, Gogol E, Gu XH, Thibodeau RH, Jackson PS, Kiefer JR, Knight JD, Nannini M, Narukulla R, Pace A, Pang J, Purkey HE, Salphati L, Sampath D, Schmidt S, Sideris S, Song K, Sujatha-Bhaskar S, Ultsch M, Wallweber H, Xin J, Yeap S, Young A, Zhong Y, Staben ST: Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kalpha. J Med Chem. 2022 Dec 22;65(24):16589-16621. doi: 10.1021/acs.jmedchem.2c01422. Epub 2022 Dec 1.





PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215633239&_cid=P11-M9XU5W-08686-1


Example 101 (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide 101
Step 1: 4-Bromo-2-hydroxybenzaldehyde
Step 2: 5-Bromo-2-(1H-imidazol-2-yl)phenol
Step 3: 9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 4: 9-Bromo-2,3-diiodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 5: 9-Bromo-2-iodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 6: (R)-2,2-Dimethyl-[1,3]dioxolane-4-carbaldehyde
Step 7: (R)-4-Difluoromethyl-2,2-dimethyl-[1,3]dioxolane
Step 8: (R)-3-(tert-Butyldimethylsilanyloxy)-1,1-difluoropropan-2-ol
Step 9: ((S)-2-Azido-3,3-difluoropropoxy)-tert-butyldimethylsilane
Step 10: (S)-1-(tert-Butyldimethylsilanyloxymethyl)-2,2-difluoroethylamine
Step 12: (S)-3-(9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-2-yl)-4-(difluoromethyl)oxazolidin-2-one
Step 13: (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide
Medical uses
Inavolisib is indicated in combination with palbociclib and fulvestrant for the treatment of adults with endocrine-resistant, PIK3CA-mutated, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer, as detected by an FDA-approved test, following recurrence on or after completing adjuvant endocrine therapy.[3]
Side effects
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
History
Efficacy was evaluated in INAVO120 (NCT04191499), a randomized, double-blind, placebo-controlled, multicenter trial in 325 participants with endocrine-resistant, PIK3CA-mutated HR-positive, HER2-negative locally advanced or metastatic breast cancer whose disease progressed during or within twelve months of completing adjuvant endocrine therapy and who had not received prior systemic therapy for locally advanced or metastatic disease.[3] Primary endocrine resistance was defined as relapse while on the first two years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least two years or relapse within twelve months of completing adjuvant ET.[3]
Structure, reactivity, and synthesis
Inavolisib is a synthetic, organic, small compound (the full structure can be seen here).[8] When binding to phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (p110α), inavolisib’s carbonyl group can accept a hydrogen bond from the Tyr836 (conserved) in p110α. The difluoromethyl group can interact with the hydroxyl group presented on Ser774 (conserved) in p110α, which is 3.2Å nearer than of which on the equivalent residue Ser754 in p110δ. Additionally, the amide group can interact with Gln859 (non-conserved). This results in a very high selectivity regarding PI3Kα isoforms.[4][9]
Compared to similar PI3K inhibiting compounds, inavolisib has a higher thermodynamic aqueous solubility that proved advantageous in the formulation process and aiding greater consistency in predictions of absorption.[4]
Inavolisibcan be developed as a derivative of 1,3-oxazole[10] or by means of stereo-controlled N-arylation of alpha-amino acids.[11]
Metabolism and biotransformation
Inavolisib is orally administered, though there is little knowledge about its metabolism.[12]However, absorption, metabolism, and excretion data of taselisib, a molecule with a related chemical scaffold, suggest moderately slow absorption into the systemic circulation, metabolism to play a minor role in drug clearance, and biliary excretion to be the main route of excretion.[13]
Molecular mechanisms of action
Inavolisib is a selective PI3K-p110α (PIK3CA) inhibitor, which may offer antineoplastic functionality.[8] Therefore, it may serve as a new addition to combination therapy with conventional cancer treatment, such as chemotherapy. Combining inavolisib with palbociclib and fulvestrant might improve treatment of breast cancer.[14]
Next to its inhibitory enzymatic ability, it is suggested that inavolisib binds to – and activates degradation of – mutated forms of p110α. Members of the PI3K family regulate cellular processes such as cell growth and proliferation, survival, remodelling, and intracellular transport of organelles.[15] PI3K also plays an essential role for the immune system.
The class I isoform PI3K alpha (PI3Kα) is often times expressed in solid tumours through gene amplification or activated mutations.[4] Mutations in PI3Kα can often be found in cancer cells, especially HR+ breast cancer, which causes a disruption of the PI3K pathway. This leads to increased tumour growth and metastasis. One of the most common mutations can be found in PIK3CA, which plays a significant role in tumour cell proliferation.
In preclinical studies, inavolisib has shown to specifically initiate the degradation of this p110α oncogene with the help of proteasomes.[16] After binding to the mutant PI3Kα, inavolisib blocks phosphorylation of PIP2 to PIP3, thereby stopping downstream signalling.[17]
Consequently, biomarkers in the PI3K pathway are reduced, cell proliferation inhibited, and the rate of PIK3CA-mutant breast cancer apoptosis increased (in comparison to the wild type). The exact mechanism of action of inhibitors like inavolisib on mutated PI3Kα and the inhibitors’ influence on mutant structures are still unknown.[18]
Toxicity
Inavolisib is able to induce a cytotoxic response but this is directed towards tumour cells that contain the PI3K mutation, thereby inhibiting further tumour growth and leading to cell loss.[19]
Society and culture
Legal status
In October 2024, the US Food and Drug Administration (FDA) approved inavolisib for the treatment of PIK3CA-mutant breast cancer based on the results from the INAVO120 trial.[3][6][20][21] The drug application was granted priority review and breakthrough therapy designations by the FDA.[3]
Names
Inavolisib is the international nonproprietary name.[22][23]
Inavolisib is sold under the brand name Itovebi.[3]
Research
Due to inavolisib’s ability to inhibit the PI3K pathway through HER2-dependent degradation, it is undergoing clinical trials to potentially make use of it as an antineoplastic (anti-cancer) drug to treat breast cancer.[4][24][17]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b “Itovebi- inavolisib tablet, film coated”. DailyMed. 11 October 2024. Retrieved 11 November 2024.
- ^ Jump up to:a b c d e f g h i j “FDA approves inavolisib with palbociclib and fulvestrant for endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, advanced breast cancer”. U.S. Food and Drug Administration (FDA). 10 October 2024. Retrieved 11 October 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, et al. (December 2022). “Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kα”. Journal of Medicinal Chemistry. 65 (24). American Chemical Society (ACS): 16589–16621. doi:10.1021/acs.jmedchem.2c01422. PMID 36455032. S2CID 254149451.
- ^ “CID 124173720, Inavolisib”. PubChem. National Center for Biotechnology Information, U.S. National Library of Medicine. Retrieved 21 September 2023.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b “inavolisib — Ligand page”. IUPHAR/BPS Guide to Pharmacology. Retrieved 21 September 2023.
- ^ Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10). Springer Science and Business Media LLC: 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844.
- ^ Chen J, Lv S, Liu J, Yu Y, Wang H, Zhang H (December 2021). “An Overview of Bioactive 1,3-Oxazole-Containing Alkaloids from Marine Organisms”. Pharmaceuticals. 14 (12). MDPI AG: 1274. doi:10.3390/ph14121274. PMC 8706051. PMID 34959674.
- ^ Han C, Kelly SM, Cravillion T, Savage SJ, Nguyen T, Gosselin F (2019). “Synthesis of PI3K inhibitor GDC-0077 via a stereocontrolled N-arylation of α-amino acids”. Tetrahedron. 75 (32). Elsevier BV: 4351–4357. doi:10.1016/j.tet.2019.04.057. ISSN 0040-4020. S2CID 150262658.
- ^ “Inavolisib: Uses, Interactions, Mechanism of Action”. DrugBank. 20 May 2019. DB15275. Retrieved 21 September 2023.
- ^ Ma S, Cho S, Sahasranaman S, Zhao W, Pang J, Ding X, et al. (April 2023). “Absorption, Metabolism, and Excretion of Taselisib (GDC-0032), a Potent β-Sparing PI3K Inhibitor in Rats, Dogs, and Humans”. Drug Metabolism and Disposition. 51 (4): 436–450. doi:10.1124/dmd.122.001096. PMID 36623882.
- ^ “A trial looking at a new drug called inavolisib for breast cancer that has spread (WO41554)”. Cancer Research UK. 22 June 2021. Retrieved 21 September 2023.
- ^ Koyasu S (April 2003). “The role of PI3K in immune cells”. Nature Immunology. 4 (4). Springer Science and Business Media LLC: 313–319. doi:10.1038/ni0403-313. PMID 12660731. S2CID 9951653.
- ^ Hong R, Edgar K, Song K, Steven S, Young A, Hamilton P, et al. (15 February 2018). “Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies”. Cancer Research. 78 (4_Supplement). American Association for Cancer Research (AACR): PD4–14–PD4–14. doi:10.1158/1538-7445.sabcs17-pd4-14. ISSN 0008-5472.
- ^ Jump up to:a b “Inavolisib (PI3K alpha inhibitor)”. Genentech. Retrieved 21 September 2023.
- ^ Menteş M, Karakuzulu BB, Uçar GB, Yandım C (August 2022). “Comparative molecular dynamics analyses on PIK3CA hotspot mutations with PI3Kα specific inhibitors and ATP”. Computational Biology and Chemistry. 99. Elsevier BV: 107726. doi:10.1016/j.compbiolchem.2022.107726. PMID 35842959. S2CID 250404770.
- ^ Song KW, Edgar KA, Hanan EJ, Hafner M, Oeh J, Merchant M, et al. (January 2022). “RTK-Dependent Inducible Degradation of Mutant PI3Kα Drives GDC-0077 (Inavolisib) Efficacy”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 204–219. doi:10.1158/2159-8290.cd-21-0072. PMC 9762331. PMID 34544753.
- ^ “FDA Approves Genentech’s Itovebi, a Targeted Treatment for Advanced Hormone Receptor-Positive, HER2-Negative Breast Cancer With a PIK3CA Mutation” (Press release). Genentech. 10 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ “U.S. Food and Drug Administration Approves FoundationOne Liquid CDx as a Companion Diagnostic for Itovebi (inavolisib) to Identify Patients with Hormone Receptor-Positive, HER2-Negative Breast Cancer with a PIK3CA Mutation” (Press release). Foundation Medicine. 11 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
- ^ Vanhaesebroeck B, Burke JE, Madsen RR (January 2022). “Precision Targeting of Mutant PI3Kα in Cancer by Selective Degradation”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 20–22. doi:10.1158/2159-8290.cd-21-1411. PMC 7612218. PMID 35022207.
External links
- Clinical trial number NCT04191499 for “A Study Evaluating the Efficacy and Safety of Inavolisib + Palbociclib + Fulvestrant vs Placebo + Palbociclib + Fulvestrant in Patients With PIK3CA-Mutant, Hormone Receptor-Positive, Her2-Negative, Locally Advanced or Metastatic Breast Cancer (INAVO120)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Itovebi |
| Other names | GDC-0077, RG6114, Ro7113755 |
| AHFS/Drugs.com | Itovebi |
| License data | US DailyMed: Inavolisib |
| Routes of administration | By mouth |
| Drug class | PI3K inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2060571-02-8 |
| PubChem CID | 124173720 |
| IUPHAR/BPS | 9636 |
| DrugBank | DB15275 |
| ChemSpider | 59718498 |
| UNII | L4C1UY2NYH |
| KEGG | D11942 |
| ChEMBL | ChEMBL4650215 |
| PDB ligand | X3N (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H19F2N5O4 |
| Molar mass | 407.378 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////Inavolisib, FDA 2024, APPROVALS 2024, GDC-0077, 2060571-02-8, GDC0077, RG6114, WHO 11204, GDC 0077, GDC-0077, RG-6114, RG6114, RO-7113755, RO7113755
Bocodepsin



Bocodepsin, OKI-179
CAS 1834513-65-3
S-((3E)-4-((6S,9S)-12,12-DIMETHYL-4,8,11,14-TETRAOXO-9-(PROPAN-2-YL)-7-OXA -3,10,13-TRIAZA-1(2,4)-(1,3)THIAZOLACYCLOTETRADECAPHAN-6-YL)BUT-3-EN-1-YL) (2S)-2-AMINO-3-METHYLBUTANETHIOATE
S-(4-((7S,10S)-4,4-DIMETHYL-2,5,8,12-TETRAOXO-7-(PROPAN-2-YL)-9-OXA-16-THIA- 3,6,13,18-TETRAAZABICYCLO(13.2.1)OCTADECA-15(18),17-DIEN-10-YL)BUT-3-EN-1-YL) (2S)-2-AMINO-3-METHYLBUTANETHIOATE
| Molecular Weight | 581.75 |
|---|---|
| Formula | C26H39N5O6S2 |
- L-Valine, S-L-valyl-(3S,4E)-3-hydroxy-7-mercapto-4-heptenonyl-2-(aminomethyl)-4-thiazolecarbonyl-2-methylalanyl-, (5–>2)-lactone
- S-{(3E)-4-[(6S,9S)-12,12-dimethyl-4,8,11,14-tetraoxo-9-(propan-2-yl)-7-oxa-3,10,13-triaza-1(2,4)-[1,3]thiazolacyclotetradecaphan-6-yl]but-3-en-1-yl} (2S)-2-amino-3-methylbutanethioate
- S-{4-[(7S,10S)-4,4-dimethyl-2,5,8,12-tetraoxo-7-(propan-2-yl)-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-15(18),17-dien-10-yl]but-3-en-1-yl} (2S)-2-amino-3-methylbutanethioate
- Originator OnKure Therapeutics
- Class Antineoplastics; Small molecules
- Mechanism of ActionHDAC1 protein inhibitors
Phase I/II Malignant melanoma; Solid tumours
- No development reportedHaematological malignancies
29 Jan 2025 OnKure Therapeutics completes the phase-I/II Nautilus trial in Malignant melanoma (Late-stage disease, Metastatic disease, Second-line therapy or greater, Combination therapy) in USA (PO) (NCT05340621),
- 11 Oct 2023Pharmacodynamics data from a preclinical studies in Solid tumours presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics 2023 (AACR-NCI-EORTC-2023 2023)
- 11 Oct 2023Initial efficacy and adverse events data from a phase Ib/II NAUTILUS trial in Melanoma presented at the International Conference on Molecular Targets and Cancer Therapeutics 2023 (AACR-NCI-EORTC-2023)
Bocodepsin (OKI-179) is an orally active and selective HDAC inhibitor, with antitumor activity. Bocodepsin can be used for suppression on solid tumor and hematologic malignancies.
SCHEME


PATENT
WO2017201278
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017201278&_cid=P12-M9WKU5-87067-1

Examples
[00127] The following examples are provided for illustrative purposes only and are not intended to limit the scope of the invention.
[00128] Example 1: Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride.
Step 1 : Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate. (7S,10S)-10-((E)- 4- chlorobut-l-en-l-yl)-7-isopropyl-4,4-dimethyl-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-diene-2,5,8,12-tetraone (15 g, 0.03 mol), (R)-2- ((tert-butoxycarbonyl)amino)-3-methylbutanethioic S-acid (12.5 g, 0.06 mol), K2CO3 (11.2 g, 0.09 mol), and KI (0.89 g, 0.006 mol) were dissolved in 150mL of acetonitrile and the resulting mixture was warmed to 60-65°C and stirred under nitrogen. After 16 hours, the mixture was cooled to 20°C, 300 mL of water was added, and the resulting suspension was extracted with ethyl acetate (2X200 mL). The combined organic phases were dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (elution with ethyl acetate/petroleum ether = 1/1 to 4/1) to give (R)- 5- ((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert- butoxycarbonyl)amino)-3-methylbutanethioate (17.0 g, 80% yield).
Step 2: Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride. (R)-S-((E)-4-((7S,10S)-7- isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[l 3.2.1] octadeca- 1 ( 17), 15 (18)-dien- 10-y l)but-3-en- 1 -y 1) 2-((tert-butoxy carbony l)amino)-3-methylbutanethioate (1.7 g, 0.025 mol) was dissolved in 150 mL of dichloromethane and trifluoroacetic acid (22.5 mL) was added at 10°C. After stirring at 10°C for 4 hours under nitrogen, the mixture was concentrated to dryness and the residue was dissolved in 100 mL of ethyl acetate and treated with 10 mL of 4M HCl/ethyl acetate solution. The mixture was then treated with petroleum ether (100 mL) and the resulting white solid was collected by filtration and dried to give (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride (0.40 g, 26% yield). Mass Spec(m/z): 582.8 (M+l).
129] Example 2: Preparation of (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride.
Step 1 : (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxy carbony l)amino)-3 -methy lbutanethioate. (7S,10S)-10-((E)-4-chlorobut- 1 -en- 1 -yl)-7-isopropyl-4,4-dimethyl-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-diene-2,5,8,12-tetraone (40 g, 0.0825 mol), (S)-2-((tert-butoxycarbonyl)amino)-3-methylbutanethioic S-acid (38.5 g, 0.165 mol), K2C03 (34.1 g, 0.247 mol), and KI (2.7 g, 0.0163 mol) were dissolved in 400 mL of acetonitrile and stirred at 60-65°C under nitrogen for 20 hours. The mixture was cooled to 20°C, water (300 mL) was added and the resulting suspension was extracted with ethyl acetate (2X200 mL). The organic phases were combined, dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (elution with ethyl acetate/petroleum ether = 1/1 to 4/1) to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate (49.8 g, 89% yield).
Step 2: (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride. (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-1 ( 17), 15( 18)-dien- 10-y l)but-3-en- 1 -y 1) 2-((tert-butoxy carbony l)amino)-3 -methylbutanethioate (47.8 g, 0.07 mol) was dissolved in dichloromethane (400 mL) and trifluoroacetic acid (65 mL) was added dropwise at 10 to 20°C while stirring under nitrogen. After the addition, the mixture was stirred at 15 to 20°C for 3 hours at which time an additional aliquot of trifluoroacetic acid (20 mL) was added and stirring at 15 to 20°C was continued for an additional 1.5 hours. The solution was then concentrated under vacuum to near dryness and the residue dissolved in ethyl acetate (250 mL). 20 mL of 4M HCl/ethyl acetate solution was then added while stirring at a temperature between 10 to 15°C resulting in the formation of a slurry. 250 mL n-heptane was then added and the solids were filtered, rinsed with n-heptane and dried in vacuo to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3 -methylbutanethioate hydrochloride as a white solid which contained some residual heptane. (49.0 g, 100% yield). Mass Spec(m/z): 582.8 (M+l)
130] Example 3: Preparation of (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3 -methylbutanethioate benzenesulfonate.
The product of Example 2, step 1 ((S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate) (1 eq.) was dissolved in acetonitrile (10 vol) at 20-25°C and the mixture was treated with
benzenesulfonic acid (3 eq.). After stirring at room temperature for 5 hours, the solvent was removed by decanting, the residual oil was treated with THF (5vol), and the resulting mixture was stirred over night at room temperature. The resulting white solid was collected by filtration and dried in vacuo to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate benzenesulfonate (90% yield; 98% purity). 1HNMR (d6-DMSO) δ: 0.56 to 0.57 (m, 3H), 0.76 to 0.78 (m, 3H), 0.92 to 0.94 (m, 3H), 0.96 to 0.98 (m, 3H), 1.45 to 1.48 (m, 3H), 1.70 to 1.72 (m, 3H), 2.07 to 2.16 (m, 2H), 2.27 to 2.28 (m, 2H), 2.93 to 2.95 (m, 1H), 2.94 to 2.95 (m, 1H), 2.97 to 3.1 (m, 1H), 4.13 to 4.15 (m, 1H), 4.28 to 4.33 (1H), 4.92 to 5.0 (m, 1H), 5.61 to 5.64 (m, 3H), 7.29 to 7.32 (m, 3H), 7.57 to 7.60 (m, 2H), 7.88 to 7.92 (m, 1H), 8.17 (s, 1H), 8.32 (s, 3H), 8.48 to 8.50 (m, 1H).
[1]. Diamond JR, et al. Preclinical Development of the Class-I-Selective Histone Deacetylase Inhibitor OKI-179 for the Treatment of Solid Tumors. Mol Cancer Ther. 2022 Mar 1;21(3):397-406. [Content Brief]
///////////Bocodepsin, K5D067O1SW, OKI-179, Malignant melanoma, Solid tumours, OnKure Therapeutics, OKI 006
Vanzacaftor



Vanzacaftor
- CAS 2374124-49-7
- COM1POP492
- VX-121
- 617.8 g/mol, C32H39N7O4S
FDA APPROVED vanzacaftor, tezacaftor, and deutivacaftor, 12/20/2024, Alyftrek , To treat cystic fibrosis
(14S)-8-[3-(2-dispiro[2.0.24.13]heptan-7-ylethoxy)pyrazol-1-yl]-12,12-dimethyl-2,2-dioxo-2λ6-thia-3,9,11,18,23-pentazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(22),5(10),6,8,19(23),20-hexaen-4-one
Vanzacaftor (VX-121) is an orally active noval corrector of Cystic fibrosis transmembrane conductance regulator (CFTR). Vanzacaftor improves processing and trafficking of CFTR protein as well as increases chloride transport in triple combined with Tezacaftor (HY-15448) and Deutivacaftor. Vanzacaftor-Tezacaftor-Deutivacaftor is safe and well tolerated, improving lung function, respiratory symptoms, and CFTR function with cystic fibrosis, which is promising for research in the field of cystic fibrosis diseases.
| Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 70,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure. |
PATENTS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US356967369&_cid=P12-M9W6P5-06241-1
Example 104: Preparation of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

Step 1: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate

To a stirred solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-20-hydroxy-12,12-dimethyl-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1 (23),5,7,9,19,21-hexaene-2,2,4-trione (150 mg, 0.2367 mmol) in anhydrous dichloromethane (3.000 mL) was added 4-methylbenzenesulfonyl chloride (58 mg, 0.3042 mmol), triethylamine (80 μL, 0.5740 mmol) and catalytic amount of N,N-dimethylpyridin-4-amine (10 mg, 0.08185 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The resultant brown residue was purified by silica gel column chromatography using a shallow gradient 100% hexanes to 100% ethyl acetate to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 51%) as a white solid. ESI-MS m/z calc. 787.28217, found 788.42 (M+1) +; Retention time: 1.39 min (LC Method J).
Step 2: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

A solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 0.1523 mmol) in dry N,N-dimethylformamide (1 mL) was purged with nitrogen for 5 min using a balloon. Then, dichloronickel; triphenyl-phosphane (30 mg, 0.04586 mmol) and tricyclohexylphosphane (34 mg, 0.1212 mmol) were added. The resultant green solution was stirred for 5 min under nitrogen atmosphere and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) was added in one portion. The resultant dark reddish brown mixture was stirred at room temperature for 1 h. Additional dichloronickel; triphenylphosphane (30 mg, 0.04586 mmol), tricyclohexylphosphane (34 mg, 0.1212 mmol) and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) were added and the mixture was stirred at room temperature under nitrogen overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, filtered and evaporated. The resultant residue was dissolved in dimethyl sulfoxide and filtered through a Whatman filter disc (puradisc 25 TF) and the filtrate was purified by reverse phase HPLC-MS using a dual gradient run from 50%-99% mobile phase B over 15.0 min (mobile phase A=water (5 mM hydrochloric acid), mobile phase B=acetonitrile) to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300) (35 mg, 37%) as a white solid. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ 12.52 (s, 1H), 8.20 (d, J=2.8 Hz, 1H), 7.81 (d, J=8.2 Hz, 1H), 7.56 (d, J=7.1 Hz, 1H), 7.04 (d, J=7.2 Hz, 1H), 6.98 (s, 1H), 6.90 (d, J=8.1 Hz, 1H), 6.08 (d, J=2.7 Hz, 1H), 4.25-4.17 (m, 2H), 3.92 (d, J=12.5 Hz, 1H), 3.17 (s, 1H), 2.94 (d, J=13.2 Hz, 1H), 2.72 (s, 1H), 2.20-2.06 (m, 1H), 1.81 (q, J=6.6 Hz, 4H), 1.60 (s, 3H), 1.56 (d, J=13.5 Hz, 2H), 1.51 (s, 3H), 1.46 (d, J=6.5 Hz, 1H), 1.36-1.26 (m, 1H), 1.23 (s, 1H), 0.87-0.76 (m, 4H), 0.70-0.59 (m, 2H), 0.50 (dd, J=8.0, 4.3 Hz, 2H). ESI-MS m/z calc. 618.2847, found 619.25 (M+1) +; Retention time: 1.28 min (LC Method J).
- US11866450, Compound 195
- US11866450, Compound 252
- US11866450, Compound 253
- US11866450, Compound 274
- US11866450, Compound 298
- US11866450, Compound 301
- [1]. Uluer AZ, et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: randomised, double-blind, controlled, phase 2 trials[J]. Lancet Respir Med. 2023 Jun;11(6):550-562. [Content Brief][2]. Kolski-Andreaco A, et al. Potentiation of BKCa channels by cystic fibrosis transmembrane conductance regulator (CFTR) correctors VX-445 and VX-121[J]. J Clin Invest. 2024 Jul 2:e176328. [Content Brief]
//////Vanzacaftor, Alyftrek , cystic fibrosis, COM1POP492, VX-121, FDA 2024, APPROVALS 2024
#Vanzacaftor, #Alyftrek , #cystic fibrosis, #COM1POP492, #VX-121, #FDA 2024, #APPROVALS 2024
Tegeprotafib



Tegeprotafib
CAS 2407610-46-0
| Molecular Weight | 326.30 |
|---|---|
| Formula | C13H11FN2O5S |
PTPN2/1-IN-1, YGY4WEM0NZ
5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1,1-dioxo-1,2,5-thiadiazolidin-3-one
Tegeprotafib (PTPN2/1-IN-1) (Compound 124) is an orally active PTPN1 and PTPN2 inhibitor with IC50s of 4.4 nM and 1-10 nM against PTPN2 and PTP1B, respectively.
| Cancer immunotherapy regimens targeting immune evasion mechanisms including checkpoint blockade (e.g., PD-1/PD-L1 and CTLA-4 blocking antibodies) have been shown to be effective in treating in a variety of cancers, dramatically improving outcomes in some populations refractory to conventional therapies. However, incomplete clinical responses and the development of intrinsic or acquired resistance will continue to limit the patient populations who could benefit from checkpoint blockade. |
SCHEME

PATENT
Calico Life Sciences LLC; AbbVie Inc. , WO2021127499
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127499&_cid=P21-M9UYU6-17583-1


Example 25: 5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1λ6,2,5-thiadiazolidine-1,1,3-trione (Compound 124)
Example 25A: benzyl 3-(benzyloxy)-7-methoxynaphthalene-2-carboxylate
A mixture of 3-hydroxy-7-methoxy-2-naphthoic acid (75 g, 344 mmol) and cesium carbonate (336 g, 1031 mmol) in N,N-dimethylformamide (687 mL) was rapidly stirred for 5 minutes at 23 °C. Thereafter, benzyl bromide (84 mL, 705 mmol) was added. After 90 minutes, the mixture was poured into H2O (1 L) and extracted with ethyl acetate (4 × 300 mL). The combined organic layers were washed with saturated aqueous ammonium chloride (3 × 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo to afford a brown solid. The crude solid was collected by filtration, slurried with tert-butyl methyl ether/heptanes (1:2, 3 × 100 mL), then dried in vacuo (12 mbar) at 40 °C to afford the title compound (122.5 g, 307 mmol, 89% yield) as a beige solid. MS (APCI+) m/z 399 [M+H]+.
Example 25B: 3-(benzyloxy)-7-methoxynaphthalene-2-carboxylic acid
To a suspension of the product of Example 25A (122.5 g, 307 mmol) in methanol (780 mL) was added 6 M aqueous sodium hydroxide (154 mL, 922 mmol). The heterogeneous, brown slurry was agitated with an overhead mechanical stirrer and heated to an internal temperature of 68 °C. After 15 minutes, the mixture was cooled to room temperature in an ice bath, and 6 M HCl (250 mL) was added over 5 minutes. The off-white solid was collected by filtration, washed with H2O (3 × 500 mL), and dried to constant weight in vacuo at 65 °C to afford the title compound (84.1 g, 273 mmol, 89% yield) as a white solid. MS (APCI+) m/z 309 [M+H]+.
Example 25C: 3-(benzyloxy)-7-methoxynaphthalen-2-amine
To a suspension of the product of Example 25B (84.1 g, 273 mmol), in toluene (766 mL) and tert-butanol (766 mL) was added triethylamine (40.3 mL, 289 mmol). The homogeneous black solution was heated to an internal temperature of 80 °C under nitrogen, and diphenyl phosphorazidate (62.2 mL, 289 mmol) was added dropwise over 90 minutes with the entire
reaction behind a blast shield. After 5 hours, the reaction was cooled to room temperature, diluted with H2O (1.5 L), and extracted with ethyl acetate (3 × 150 mL). The combined organic layers were washed with brine (2 × 100 mL), dried over sodium sulfate, filtered and concentrated to give 180.1 g of a dark brown solid. The solid was carried forward to hydrolysis without further purification.
To the crude intermediate was added diethylenetriamine (475 mL, 4.40 mol). The heterogeneous suspension was heated to an internal temperature of 130 °C under nitrogen, at which time a homogeneous dark orange solution formed. After 16 hours, the mixture was cooled to room temperature in an ice bath, and H2O (1.5 L) was added slowly over 3 minutes, resulting in precipitation of a yellow solid and a concomitant exotherm to an internal temperature of 62 °C. Once the heterogeneous suspension had cooled to room temperature, the crude solid was dissolved in CH2Cl2 (1.5 L), and the layers were separated. The aqueous layer was back-extracted with CH2Cl2 (3 × 150 mL), and the combined organic layers were washed with brine (3 × 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo to afford 78.8 g of an orange solid. The solid was slurried with isopropanol (50 mL), collected via filtration, re-slurried with isopropanol (1 × 50 mL), and dried in vacuo (15 mbar) at 35 °C to afford the title compound (60.12 g, 215 mmol, 79% yield over two steps) as a yellow solid. MS (APCI+) m/z 280 [M+H]+.
Example 25D: methyl {[3-(benzyloxy)-7-methoxynaphthalen-2-yl]amino}acetate
To a mixture of the product of Example 25C (59.2 g, 212 mmol) and potassium carbonate (58.6 g, 424 mmol) in dimethylformamide (363 mL) and H2O (1.91 mL, 106 mmol) was added methyl 2-bromoacetate (30.1 mL, 318 mmol). The suspension was vigorously stirred at room temperature for 5 minutes and then heated to an internal temperature of 60 °C. After 70 minutes, the suspension was cooled to room temperature and diluted with H2O (600 mL) and ethyl acetate (500 mL). The aqueous layer was extracted with ethyl acetate (2 × 300 mL), and the combined organic layers were washed with saturated aqueous ammonium chloride (3 × 60 mL), dried over sodium sulfate, filtered, and concentrated to afford 104.3 g of a pale beige solid. The solid was triturated with heptanes (200 mL). The resulting beige solid was collected via filtration, washed with additional heptanes (2 × 30 mL), and dried in vacuo (15 mbar) at 35 °C to afford the title compound (72.27 g, 206 mmol, 97% yield) as an off-white solid. MS (APCI+) m/z 352 [M+H]+.
Example 25E: methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]amino}acetate To a mixture of the product of Example 25D (30.0 g, 85 mmol) and N-fluorobenzenesulfonimide (26.9 g, 85 mmol) was added tetrahydrofuran (THF) (854 mL), and
the resulting homogeneous yellow solution was stirred at room temperature. After 90 minutes, residual oxidant was quenched by adding a solution of sodium thiosulfate pentahydrate (10.59 g, 42.7 mmol) in water (150 mL), and the mixture was stirred at room temperature for 30 minutes. Thereafter, ethyl acetate (600 mL) was added, the aqueous layer was separated, and the organic layer was washed with a solution of sodium carbonate (18.10 g, 171 mmol) in water (30 mL), followed by water:brine (1:1, 1 × 20 mL). The organic fraction was dried over sodium sulfate, filtered, and the concentrated in vacuo to afford a bright yellow/orange solid. The solids were triturated with tert-butyl methyl ether (300 mL), collected via filtration, and the filter cake (N-(phenylsulfonyl)benzenesulfonamide) was washed with tert-butyl methyl ether (2 × 100 mL). The filtrate was concentrated to afford 34.6 g of a dark red oil that was purified by flash chromatography (750 g SiO2, heptanes to 20% ethyl acetate/heptanes) to afford the title compound (16.07 g, 43.5 mmol, 51% yield) as a yellow solid. MS (APCI+) m/z 370 [M+H]+. Example 25F: methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl](sulfamoyl)amino}acetate
To a solution of chlorosulfonyl isocyanate (5.13 mL, 59.1 mmol) in dichloromethane (83 mL) at 0 °C was added tert-butanol (5.65 mL, 59.1 mmol) slowly so that the internal temperature remained less than 10 °C. After stirring for 30 minutes at 0 °C, a preformed solution of the product of Example 25E (14.55 g, 39.4 mmol) and triethylamine (10.98 mL, 79 mmol) in dichloromethane (68.9 mL) was added slowly via addition funnel so that the internal temperature remained below 10 °C. Upon complete addition, the addition funnel was rinsed with dichloromethane (23 mL). The resulting solution was stirred for 30 minutes at 0 °C, and then the reaction mixture was quenched with H2O (20 mL). The layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 30 mL). The combined organic layers were washed with brine (1 × 30 mL), dried over sodium sulfate, filtered and concentrated in vacuo to give an orange oil. The residue was dissolved in ethyl acetate (200 mL) and washed with water:brine (1:1, 2 × 50 mL) to remove residual triethylamine hydrochloride. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo to give methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl][(tert-butoxycarbonyl)sulfamoyl]amino}acetate which was used without purification.
To a solution of methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl][(tert-butoxycarbonyl)sulfamoyl]amino}acetate in dichloromethane (98 mL) was added trifluoroacetic acid (45.5 mL, 591 mmol), and the resulting dark solution was stirred at room temperature. After 20 minutes, the reaction was quenched by slow addition of saturated aqueous sodium bicarbonate (691 mL) via an addition funnel. The layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 50 mL). The combined organic layers were concentrated to give a dark red oil; upon addition of tert-butyl methyl ether (60 mL), a yellow solid precipitated that was collected via filtration, washed with tert-butyl methyl ether (2 × 30 mL) and dried in vacuo (15 mbar) at 35 °C to give the title compound (13.23 g, 29.5 mmol, 75% yield over two steps) as a light yellow solid. MS (ESI+) m/z 449 [M+H]+.
Example 25G: 5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1λ6,2,5-thiadiazolidine-1,1,3-trione
To a solution of the product of Example 25F (13.23 g, 29.5 mmol) in tetrahydrofuran (THF) (355 mL) at room temperature was added solid potassium tert-butoxide (3.31 g, 29.5 mmol), and the resulting solution was stirred at room temperature. After 10 minutes, the reaction was quenched with 1 M hydrochloric acid (90 mL) and diluted with ethyl acetate (400 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 × 120 mL). The combined organic layers were washed with brine (3 × 50 mL), then dried over sodium sulfate, filtered and concentrated. The crude 5-[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]-1λ6,2,5-thiadiazolidine-1,1,3-trione was used in the subsequent reaction without further purification.
A mixture of crude intermediate, 5-[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]-1λ6,2,5-thiadiazolidine-1,1,3-trione (12.28 g, 29.5 mmol) and pentamethylbenzene (13.11 g, 88 mmol) in dichloromethane (147 mL) was cooled to an internal temperature of –76 °C under an atmosphere of dry nitrogen. Subsequently, a 1 M solution of boron trichloride (59.0 mL, 59.0 mmol) in CH2Cl2 was added dropwise over 15 minutes, so as not to raise the internal temperature past –72 °C. Over the course of the addition, the reaction turned dark brown and became homogeneous. Incomplete conversion was observed, and additional boron trichloride (2 × 5.90 mL, 2 × 5.90 mmol) was added, resulting in full conversion. The reaction was quenched at –75 °C with CH2Cl2/methanol (10:1, 140 mL) via cannula transfer under nitrogen over 15 minutes, then slowly warmed to room temperature over 20 minutes under nitrogen. The volatiles were removed in vacuo to afford a brown/tan solid, which was collected by filtration, and slurried with heptanes (5 × 40 mL) and CH2Cl2 (3 × 40 mL). The crude solid was suspended in isopropanol (75 mL), warmed until the material dissolved, then allowed to cool slowly to room temperature over 1 hour. The solid was collected by filtration, washed with heptanes (2 × 30 mL), and dried in vacuo (15 mbar) at 60 °C to afford 5.11 g of a white solid. The mother liquor was concentrated, and the process was repeated to give an additional 1.96 g of a white solid. The batches were combined to obtain the title compound (7.07 g, 21.67 mmol, 73.5% yield over two steps). 1H NMR (CD3OD) δ ppm 7.60 (dd, J = 9.1, 1.5 Hz, 1H), 7.25 (d, J = 2.6, 1H), 7.16 (dd, J = 9.1, 2.6 Hz, 1H), 7.04 (s, 1 H), 4.56 (s, 2H), 3.89 (s, 3 H); MS (ESI–) m/z 325 [M–H]–.
PATENT
WO2020186199
WO2019246513
PATENT
compound 124 [US20230019236A1]
https://patentscope.wipo.int/search/en/detail.jsf?docId=US389737555&_cid=P21-M9UYQD-14144-1
///////Tegeprotafib, PTPN2/1-IN-1, YGY4WEM0NZ
Probenecid



Probenecid
- 57-66-9
- 4-(Dipropylsulfamoyl)benzoic acid
- Probenecid acid
- Benemid
4-(dipropylsulfamoyl)benzoic acid
C13H19NO4S, 285.359
HC 5006- NSC-18786
FDA APPROVED, 10/25/2024, sulopenem etzadroxil, probenecid, Orlynvah, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Probenecid, also sold under the brand name Probalan, is a medication that increases uric acid excretion in the urine. It is primarily used in treating gout and hyperuricemia.
Probenecid was developed as an alternative to caronamide[1] to competitively inhibit renal excretion of some drugs, thereby increasing their plasma concentration and prolonging their effects.
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 195 °C | PhysProp |
| water solubility | 27.1 mg/L | Not Available |
| logP | 3.21 | HANSCH,C ET AL. (1995) |
| pKa | 3.4 | SANGSTER (1994) |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US12109197 | No | 2024-10-08 | 2039-04-01 |  |
| US11554112 | No | 2023-01-17 | 2039-04-01 | |
| US11478428 | No | 2022-10-25 | 2039-12-23 | |
| US7795243 | No | 2010-09-14 | 2029-06-03 | |

PATENT
https://patents.google.com/patent/CN103613521A/en
At present, the production technique of probenecid mainly contains two kinds:
(1) p-methyl benzenesulfonic acid-dipropyl amine method
Take p-methyl benzenesulfonic acid as raw material, through potassium bichromate or potassium permanganate oxidation, then react generation with chlorsulfonic acid generation sulfonating chlorinating to carboxyl benzene sulfonyl chloride, amidate action occurs then in organic solvent and obtain the finished product probenecid.Reaction process route is as follows:

This technique in a large number with an organic solvent, seriously polluted; Heavy metal recovery and treatment cost are high; Chlorsulfonic acid transportation, storage and use are dangerous large, and acid mist is obvious.Along with the increasing of environmental protection pressure, people increase severely day by day to the concern of environment, and this route is substantially in end-of-life state.
(2) to methyl benzenesulfonamide-Halopropane method
To methyl benzenesulfonamide, through potassium bichromate or potassium permanganate oxidation, be P―Carboxybenzenesulfonamide, under the effect of alkali, with Halopropane generation alkylated reaction, after acidifying, obtain probenecid.Reaction process route is as follows:

This process using sodium dichromate 99 or potassium permanganate oxidation are to methyl benzenesulfonamide, and yield is on the low side (lower than 50%).In addition, the waste water that contains chromium or manganese is difficult to dispose, and these have all seriously restricted further developing of this technique.
Reaction scheme of the present invention is as follows:

embodiment 1
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 127.4ml hydrochloric acid (31%, 1.25mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (34.5g Sodium Nitrite, 0.5mol, be dissolved in 190g water), control temperature at 10-20 ℃, it is 4 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 765ml hydrochloric acid (31%, 7.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at-3–1 ℃, when passing into 64g sulfurous gas (1mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 5 hours, is warming up to gradually 5-10 ℃, continues reaction 8 hours at this temperature; Filtration obtains 121g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 152g dipropyl amine (1.5mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 40 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 3 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 135g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 122.8g after filtering, being dried, yield 86.2%(is in para-amino benzoic acid), purity 98.2%.
embodiment 2
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 152.9ml hydrochloric acid (31%, 1.5mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (36.0g Sodium Nitrite, 0.52mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 3 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 887ml hydrochloric acid (31%, 8.7mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 0-5 ℃, when passing into 112g sulfurous gas (1.75mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 4 hours, is warming up to gradually 5-15 ℃, continues reaction 5 hours at this temperature; Filtration obtains 150g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 192g dipropyl amine (1.9mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 35 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 2 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 155.4g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 129.5g after filtering, being dried, yield 90.9%(is in para-amino benzoic acid), purity 98.7%.
embodiment 3
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 203.9ml hydrochloric acid (31%, 2mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to-10–5 ℃, drip sodium nitrite solution (38.0g Sodium Nitrite, 0.55mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 5 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 968ml hydrochloric acid (31%, 9.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 5-10 ℃, when passing into 160g sulfurous gas (2.5mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 3 hours, is warming up to gradually 10-15 ℃, continues reaction 20 hours at this temperature; Filtration obtains 146.7g to carboxyl benzene sulfonyl chloride, needn’t be dried, and directly enters next step reaction.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 202g dipropyl amine (2mol), open to stir, when temperature is greater than 30 ℃, start to divide gradually 30 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 4 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filtration obtains 151.7g probenecid crude product, puts in 500ml pure water, and agitator treating 1 hour, heavy 128.5g after filtering, being dried, yield 90.2%(is in para-amino benzoic acid), purity 98.8%.Medical uses
Probenecid is primarily used to treat gout and hyperuricemia.
Probenecid is sometimes used to increase the concentration of some antibiotics and to protect the kidneys when given with cidofovir. Specifically, a small amount of evidence supports the use of intravenous cefazolin once rather than three times a day when it is combined with probenecid.[2]
It has also found use as a masking agent,[3] potentially helping athletes using performance-enhancing substances to avoid detection by drug tests.
Adverse effects
Mild symptoms such as nausea, loss of appetite, dizziness, vomiting, headache, sore gums, or frequent urination are common with this medication. Life-threatening side effects such as thrombocytopenia, hemolytic anemia, leukemia and encephalopathy are extremely rare.[4] Theoretically probenecid can increase the risk of uric acid kidney stones.
Drug interactions
Some of the important clinical interactions of probenecid include those with captopril, indomethacin, ketoprofen, ketorolac, naproxen, cephalosporins, quinolones, penicillins, methotrexate, zidovudine, ganciclovir, lorazepam, and acyclovir. In all these interactions, the excretion of these drugs is reduced due to probenecid, which in turn can lead to increased concentrations of these.[5]
Pharmacology
Pharmacodynamics
In gout, probenecid competitively inhibits the reabsorption of uric acid through the organic anion transporter (OAT) at the proximal tubules. This leads to preferential reabsorption of probenecid back into plasma and excretion of uric acid in urine,[6] thus reducing blood uric acid levels and reducing its deposition in various tissues.
Probenecid also inhibits pannexin 1.[7] Pannexin 1 is involved in the activation of inflammasomes and subsequent release of interleukin-1β causing inflammation. Inhibition of pannexin 1 thus reduces inflammation, which is the core pathology of gout.[7]
Pharmacokinetics
In the kidneys, probenecid is filtered at the glomerulus, secreted in the proximal tubule and reabsorbed in the distal tubule. Probenicid lowers the concentration of certain drugs in urine drug screens by reducing renal excretion of these drugs.
Historically, probenecid has been used to increase the duration of action of drugs such as penicillin and other beta-lactam antibiotics. Penicillins are excreted in the urine at proximal and distal convoluted tubules through the same organic anion transporter (OAT) as seen in gout. Probenecid competes with penicillin for excretion at the OAT, which in turn increases the plasma concentration of penicillin.[8]
History
During World War II, probenecid was used to extend limited supplies of penicillin. This use exploited probenecid’s interference with drug elimination (via urinary excretion) in the kidneys and allowed lower doses of penicillin to be used.[9]
Probenecid was added to the International Olympic Committee‘s list of banned substances in January 1988, due to its use as a masking agent.[10]
References
- ^ Mason RM (June 1954). “Studies on the effect of probenecid (benemid) in gout”. Annals of the Rheumatic Diseases. 13 (2): 120–130. doi:10.1136/ard.13.2.120. PMC 1030399. PMID 13171805.
- ^ Cox VC, Zed PJ (March 2004). “Once-daily cefazolin and probenecid for skin and soft tissue infections”. The Annals of Pharmacotherapy. 38 (3): 458–463. doi:10.1345/aph.1d251. PMID 14970368. S2CID 11449580.
- ^ Morra V, Davit P, Capra P, Vincenti M, Di Stilo A, Botrè F (December 2006). “Fast gas chromatographic/mass spectrometric determination of diuretics and masking agents in human urine: Development and validation of a productive screening protocol for antidoping analysis”. Journal of Chromatography A. 1135 (2): 219–229. doi:10.1016/j.chroma.2006.09.034. hdl:2318/40201. PMID 17027009. S2CID 20282106.
- ^ Kydd AS, Seth R, Buchbinder R, Edwards CJ, Bombardier C (November 2014). “Uricosuric medications for chronic gout”. The Cochrane Database of Systematic Reviews (11): CD010457. doi:10.1002/14651858.CD010457.pub2. PMC 11262558. PMID 25392987.
- ^ Cunningham RF, Israili ZH, Dayton PG (March–April 1981). “Clinical pharmacokinetics of probenecid”. Clinical Pharmacokinetics. 6 (2): 135–151. doi:10.2165/00003088-198106020-00004. PMID 7011657. S2CID 24497865.
- ^ “Probenecid”. PubChem. U.S. National Library of Medicine. Retrieved 2022-06-12.
- ^ Jump up to:a b Silverman W, Locovei S, Dahl G (September 2008). “Probenecid, a gout remedy, inhibits pannexin 1 channels”. American Journal of Physiology. Cell Physiology. 295 (3): C761 – C767. doi:10.1152/ajpcell.00227.2008. PMC 2544448. PMID 18596212.
- ^ Ho RH (January 2010). “4.25 – Uptake Transporters”. In McQueen CA, Kim RB (eds.). Comprehensive Toxicology (Second ed.). Oxford: Elsevier. pp. 519–556. doi:10.1016/B978-0-08-046884-6.00425-5. ISBN 978-0-08-046884-6.
- ^ Butler D (November 2005). “Wartime tactic doubles power of scarce bird-flu drug”. Nature. 438 (7064): 6. Bibcode:2005Natur.438….6B. doi:10.1038/438006a. PMID 16267514.
- ^ Wilson W, Derse E, eds. (2001). Doping in Elite Sport: The Politics of Drugs in the Olympic Movement. Human Kinetics. p. 86. ISBN 0-7360-0329-0.
| Clinical data | |
|---|---|
| Trade names | Probalan |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682395 |
| Routes of administration | By mouth |
| ATC code | M04AB01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 75-95% |
| Elimination half-life | 2-6 hours (dose: 0.5-1 g) |
| Excretion | kidney (77-88%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 57-66-9 |
| PubChem CID | 4911 |
| IUPHAR/BPS | 4357 |
| DrugBank | DB01032 |
| ChemSpider | 4742 |
| UNII | PO572Z7917 |
| KEGG | D00475 |
| ChEMBL | ChEMBL897 |
| CompTox Dashboard (EPA) | DTXSID9021188 |
| ECHA InfoCard | 100.000.313 |
| Chemical and physical data | |
| Formula | C13H19NO4S |
| Molar mass | 285.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////probenecid, APPROVALS 2024, FDA 2024, Orlynvah, HC 5006, NSC-18786
#probenecid, #APPROVALS 2024, #FDA 2024, #Orlynvah, #HC 5006, #NSC-18786
Sulopenem



Sulopenem
- 120788-07-0
- CP-70429
- 349.5 g/mol, C12H15NO5S3
- XX514BJ1XW
- PF-03709270
- PF03709270
(5R,6S)-6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(1R,3S)-1-oxothiolan-3-yl]sulfanyl-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- (5R,6S)-6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-1-OXOTETRAHYDRO-1H-1.LAMBA.(SUP 4)-THIOPHEN-3-YL)SULFANYL)-4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID
- (5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-(((3S)-tetrahydro-3-thienyl)thio)-4-thia-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid, (R)-S-oxide
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-TETRAHYDRO-1-OXIDO-3-THIENYL)THIO)-, (5R,6S)-
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-(1-HYDROXYETHYL)-7-OXO-3-((TETRAHYDRO-3-THIENYL)THIO)-, S-OXIDE, (5R-(3(1R*,3S*),5.ALPHA.,6.ALPHA.(R*)))-
FDA APPROVED sulopenem etzadroxil, probenecid, 10/25/2024, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Sulopenem (CP-70,429) is a thiopenem antibiotic derivative from the penem family, which unlike most related drugs is orally active. It was developed in Japan in the 1990s, and has been approved to treat uncomplicated urinary tract infections in combination with probenecid (Brand name: Orlynvah). It has reached Phase III clinical trials on several occasions and continues to be the subject of ongoing research into potential applications, especially in the treatment of multiple drug resistant urinary tract infections.[1][2][3][4][5]
In October 2024, the US Food and Drug Administration approved sulopenem etzadroxil with probenecid combination for the treatment of urinary tract infections caused by Escherichia coli, Klebsiella pneumoniae, or Proteus mirabilis in adult women with limited alternative oral antibiotic options. The combination was developed by Iterum Therapeutics under the trade name ORLYNVAH™.[6]


JP 1995278137; US 5013729; WO 8808845, J Org Chem 1992,57(16),4352-61
1) The reaction of L-aspartic acid (I) with NaNO2, NaBr and H2SO4 gives 2(S)-bromosuccinic acid (II), which is reduced with methyl sulfide borane complex in THF, yielding 2(S)-bromobutane-1,4-diol (III). The cyclization of (III) with Cs2CO3 in methylene chloride affords (R)-(2-hydroxyethyl)oxirane (IV), which is acylated with methanesulfonyl chloride to the corresponding mesylate (V). The cyclization of (V) with Na2S in acetonitrile/water gives 3(R)-hydroxythiolane (VI), which is acylated with p-toluenesulfonyl chloride, affording the corresponding tosylate (VII). The controlled oxidation of (VII) with potassium peroxymonosulfate (oxone) gives 3(R)-(p-toluenesulfonyloxy)thiolane-1(R)-oxide (VIII), which by reaction with potassium thioacetate in acetone is converted to 3(S)-(acetylthio)thiolane 1(R)-oxide (IX). The reaction of (IX) with NaOEt and CS2 in ethanol yields the trithiocarbonate (X), which is condensed with the chloroazetidinone (XI), yielding the trithiocarbonate ester (XII). The condensation of (XII) with 2-chloroallyloxalyl fluoride (XIII) by means of diisopropylethylamine in methylene chloride affords the substituted oxalamic ester (XIV), which is cyclized by means of triethyl phosphite in refluxing chloroform to the fully protected penem derivative (XV). The reaction of (XV) with tetrabutylammonium fluoride (TBAF) in THF eliminates the protecting tert-butyldimethylsilyl group, yielding the chloroallyl ester (XVI), which is treated with triphenylphosphine and sodium 2-ethylhexanoate in dichloromethane to obtain the corresponding sodium salt (XVII). Finally, this compound is treated with HCl in cool water.

US 4921972
2) The intermediate 3(R)-(p-toluenesulfonyloxy)thiolane (VII) can be obtained by two other synthetic pathways: a) The racemic 2-hydroxy-4-(methylsulfanyl)butyric acid ethyl ester (XVIII) is submitted to optical resolution with Pseudomonas fluorescens lipase in toluene/water, yielding the corresponding 2(R)-hydroxy ester (XIX), which is reduced with NaBH4 in THF/water to afford 4-(methylsulfanyl)butane-1,2(R)-diol (XX). The acylation of (XX) with p-toluenesulfonyl chloride and pyridine yields the ditosylate (XXI), which is cyclized in refluxing benzene to give 1(R)-methyl-3(R)-(p-toluenesulfonyloxy)thiolanium p-toluenesulfonate (XXII). Finally, this compound is treated with trifluoroacetic acid in pyridine to afford the thiolane (VII), already described. b) The reduction of 4-chloro-3(R)-hydroxybutyric acid methyl ester (XXIII) with lithium borohydride in THF gives 4-chlorobutane-1,3(R)-diol (XXIV), which is tosylated as before, yielding the bis(tosyloxy) derivative (XXV). Finally, this compound is cyclized with Na2S in hot acetonitrile/water to afford the thiolane (VII), already described.

https://pubsapp.acs.org/cen/coverstory/88/8836cover.html
References
- ^ Minamimura M, Taniyama Y, Inoue E, Mitsuhashi S (July 1993). “In vitro antibacterial activity and beta-lactamase stability of CP-70,429 a new penem antibiotic”. Antimicrobial Agents and Chemotherapy. 37 (7): 1547–1551. doi:10.1128/AAC.37.7.1547. PMC 188011. PMID 8363389.
- ^ Hamilton-Miller JM (November 2003). “Chemical and microbiologic aspects of penems, a distinct class of beta-lactams: focus on faropenem”. Pharmacotherapy. 23 (11): 1497–1507. doi:10.1592/phco.23.14.1497.31937. PMID 14620395. S2CID 43705118.
- ^ Ednie LM, Appelbaum PC (May 2009). “Antianaerobic activity of sulopenem compared to six other agents”. Antimicrobial Agents and Chemotherapy. 53 (5): 2163–2170. doi:10.1128/AAC.01557-08. PMC 2681565. PMID 19223615.
- ^ Bader MS, Loeb M, Leto D, Brooks AA (April 2020). “Treatment of urinary tract infections in the era of antimicrobial resistance and new antimicrobial agents”. Postgraduate Medicine. 132 (3): 234–250. doi:10.1080/00325481.2019.1680052. PMID 31608743. S2CID 204545734.
- ^ Veeraraghavan B, Bakthavatchalam YD, Sahni RD (December 2021). “Oral Antibiotics in Clinical Development for Community-Acquired Urinary Tract Infections”. Infectious Diseases and Therapy. 10 (4): 1815–1835. doi:10.1007/s40121-021-00509-4. PMC 8572892. PMID 34357517.
- ^ “Iterum Therapeutics Receives U.S. FDA Approval of ORLYNVAH™ (Oral Sulopenem) for the Treatment of Uncomplicated Urinary Tract Infections”. Iterum Therapeutics plc. 2024-10-25. Retrieved 2024-10-25.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120788-07-0 |
| PubChem CID | 9950244 |
| DrugBank | DB15284 |
| ChemSpider | 8125855 |
| UNII | XX514BJ1XW |
| KEGG | D05969 |
| CompTox Dashboard (EPA) | DTXSID20869656 |
| Chemical and physical data | |
| Formula | C12H15NO5S3 |
| Molar mass | 349.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
FDA Approved Drug Products: Orlynvah (sulopenem etzadroxil and probenecid) tablets for oral use (October 2024) [Link]
FDA News Release: FDA approves new treatment for uncomplicated urinary tract infections in adult women who have limited or no alternative oral antibiotic treatment options [Link]
//////Sulopenem, Orlynvah, FDA 2024, APPROVALS 2024, CP-70,429, 120788-07-0, CP-70429, XX514BJ1XW, PF-03709270, PF03709270
#Sulopenem, #Orlynvah, #FDA 2024, #APPROVALS 2024, #CP-70,429, #120788-07-0, #CP-70429, #XX514BJ1XW, #PF-03709270, #PF03709270
Bleximenib



Bleximenib
CAS 2654081-35-1
WeightAverage: 599.796
Monoisotopic: 599.395916661
Chemical FormulaC32H50FN7O3
- CS-0636752
- DA-55335
- HY-148669
- PHASE 3
- JNJ-75276617; Menin-MLL inhibitor 24
- Benzamide, N-ethyl-5-fluoro-2-[[5-[2-[(1R)-4-[(2-methoxyethyl)methylamino]-1-(1-methylethyl)butyl]-2,6-diazaspiro[3.4]oct-6-yl]-1,2,4-triazin-6-yl]oxy]-N-(1-methylethyl)-
- N-ethyl-5-fluoro-2-{[5-(2-{(3R)-6-[(2-methoxyethyl)(methyl)amino]-2-methylhexan-3-yl}-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl]oxy}-N-(propan-2-yl)benzamide

2866179-95-3 (oxalate)
(R)-N-ethyl-5-fluoro-N-isopropyl-2-((5-(2-(6-((2-methoxyethyl)(methyl)amino)-2-methylhexan-3-yl)-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl)oxy)benzamide oxalate
Chemical Formula: C34H52FN7O7
Exact Mass: 689.39
Molecular Weight: 689.830
Elemental Analysis: C, 59.20; H, 7.60; F, 2.75; N, 14.21; O, 16.23
\Bleximenib is under investigation in clinical trials NCT04811560 (A Phase 1/2 Study of Bleximenib in Participants With Acute Leukemia) and NCT05453903 (A Study of Bleximenib in Combination With Acute Myeloid Leukemia (AML) Directed Therapies)
Bleximenib (JNJ-75276617) is an orally active and selective menin-KMT2A inhibitor, with IC50 values of 0.1 nM, 0.045 nM, and ≤0.066 nM for humans, mice, and dogs, respectively. Bleximenib can inhibit the proliferation and induce apoptosis and differentiation of tumor cells. Bleximenib can be used in the research of tumors such as leukemia.
Bleximenib is an orally bioavailable protein-protein interaction (PPI) inhibitor of the menin-mixed lineage leukemia (MLL; mixed-lineage leukemia 1; MLL1; myeloid/lymphoid leukemia; histone-lysine N-methyltransferase 2A; KMT2A) proteins, with potential antineoplastic activity. Upon oral administration, bleximenib inhibits the interaction between the two proteins menin and MLL and the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of leukemic cells with either KMT2A alterations such as gene rearrangements (KMT2A-r), duplications, and amplification, or nucleophosmin 1 gene (NPM1) alterations. The menin-MLL complex plays a key role in the survival, growth, transformation and proliferation of certain kinds of leukemia cells.
SCHEME

SIDECHAIN

PATENTS
Janssen Pharmaceutica NV; Johnson & Johnson (China) Investment Ltd.
WO2021121327
WO2022237719
PATENT
WO2022237720

PATENTS
PATENT
Compound A—(R)-N-ethyl-5-fluoro-N-isopropyl-2-((5-(2-(6-((2-methoxyethyl) (methyl)amino)-2-methylhexan-3-yl)-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl)oxy) benzamide
PATENT
WO2022262796
The present invention is directed to (R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) (methyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide besylate salt (benzenesulfonate salt) :
[0011]
[0140]
tert-butyl (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0141]
[0142]
The mixture 2- ( (5- (2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide (intermediate 3) (1.0 g, 2.4 mmol) , tert-butyl (5-methyl-4-oxohexyl) carbamate (intermediate 1) (830 mg, 3.62 mmol) and ZnCl 2(660 mg, 4.84 mmol) in MeOH (15 mL) was stirred at 80 ℃ for 0.5 h. Then NaBH 3CN (310 mg, 4.93 mmol) was added and the resulting mixture was stirred at 80 ℃ for 6 h. After cooled to RT, the mixture was concentrated under reduced pressure to give the crude product, which was further purified by preparative HPLC using a Waters Xbridge Prep OBD (column: C18 150×40 mm 10 um; eluent: ACN/H 2O (0.05%ammonia) from 45%to 75%v/v) to afford the title compound (700 mg, 46%yield) as colorless oil.
reparation of Compounds 62 and 63
[0144]
tert-butyl (R) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0145]
tert-butyl (S) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0146]
[0147]
tert-butyl (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate (Compound 61) (200 mg, 0.319 mmol) was purified by SFC over DAICEL CHIRALPAK IG (column: 250×30 mm 10 um; isocratic elution: EtOH (containing 0.1%of 25%ammonia) : supercritical CO 2, 40%: 60% (v/v) ) to afford the title compounds (Compound 62) (85 mg, 42%yield) and (Compound 63) (80 mg, 40%yield) both as light yellow oil.
[0148]
[0149]
(R) -2- ( (5- (2- (6-amino-2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide
[0150]
[0151]
To the solution of tert-butyl (R) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate (Compound 62) (550 mg, 0.876 mmol) in DCM (4 mL) was slowly added TFA (4 mL) , and the resulting mixture was stirred at 25 ℃ for 1 h. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was diluted in DCM (40 mL) and the pH value was adjusted to around 12 by aq. NaOH (2 M, 16 mL) solution. The aqueous layer was extracted with DCM (10 mL x 2) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated in vacuo to afford the title compound (460 mg, crude) as yellow solid, which was used directly in next step without further purification.
[0152]
[0153]
(R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide
[0154]
[0155]
The mixture of (R) -2- ( (5- (2- (6-amino-2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide (Compound 64) (120 mg, crude) , 1-bromo-2-methoxyethane (32 mg, 0.23 mmol) , Cs 2CO 3(222 mg, 0.681 mmol) , NaI (102 mg, 0.680 mmol) in DMF (1 mL) was stirred at 80 ℃ via microwave irradiation for 1 h. After cooling to RT, the mixture was diluted with H 2O (10 mL) and extracted with EtOAc (3 x 10 mL) . The combined organic layers were washed with H 2O (10 mL) , dried over Na 2SO 4, filtered and concentrated under reduced pressure to afford the crude product which was further purified by HPLC over a Phenomenex Gemini-NX (column: 150×30 mm 5 μm; eluent: ACN/H 2O (10mM NH 4HCO 3) from 51%to 71% (v/v) ) and further purified by SFC over DAICEL CHIRALCEL OD-H (column: 250×30 mm 5 um; eluent: supercritical CO 2in EtOH (0.1%v/v ammonia) 25/25, v/v) to afford the title compound (5.13 mg, 96%purity) as yellow solid.
[0156]
LC-MS (ESI) (Method 1) : R t= 2.997 min, m/z found 586.3 [M+H] +.
[0157]
[0158]
(R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) (methyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide
[0159]
[0160]
The mixture of (R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) amino) -2- methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide (Compound 11) (40.0 mg, 0.068 mmol) , formaldehyde (55.4 mg, 0.683 mol, 37%in water) and AcOH (8.2 mg, 0.137 mmol) in anhydrous MeOH (2 mL) was stirred at 45 ℃ for 1 h. Then, NaBH 3CN (8.6 mg, 0.137 mmol) was added to the mixture and the resulting mixture was stirred at 45 ℃ for another 1 h. After cooling to RT, the reaction mixture was treated with sat. aq. NaHCO 3(40 mL) to adjust the pH value to about 8 and further extracted with DCM (20 mL x 3) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated under reduced pressure to give the crude which was purified by preparative HPLC over Boston Prime (column: C18 150x30mm 5um, Mobile Phase A: H 2O (0.04%ammonia+10mM NH 4HCO 3) , Mobile Phase B: ACN, Flow rate: 25 mL/min, gradient condition B/A from 50%to 80% (50%B to 80%B) ) to afford the title compound (9.62 mg, 99.10%purity, 23.3%yield) as yellow oil.
- [1]. Kwon MC, et al. Preclinical efficacy of the potent, selective menin-KMT2A inhibitor JNJ-75276617 (bleximenib) in KMT2A- and NPM1-altered leukemias. Blood. 2024 Sep 12;144(11):1206-1220. [Content Brief][2]. Hogeling SM, et al. Bleximenib, the novel menin-KMT2A inhibitor JNJ-75276617, impairs long-term proliferation and immune evasion in acute myeloid leukemia. Haematologica. 2024 Dec 19. [Content Brief]
////////Bleximenib, CS-0636752, DA-55335, HY-148669, JNJ-75276617, Menin-MLL inhibitor 24
Revumenib
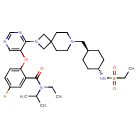
Revumenib
- SNDX-5613
- 2169919-21-3
- LZ0M43NNF2
- SNDX5613
- C32H47FN6O4S, 630.82
- SNDX-50613 free base
- SNDX-5613 free base
- SNDX50613 free base
- SNDX5613 free base
FDA APPROVED 11/15/2024, Revuforj, To treat relapsed or refractory acute leukemia
N-ethyl-2-[4-[7-[[4-(ethylsulfonylamino)cyclohexyl]methyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-5-fluoro-N-propan-2-ylbenzamide
- N-Ethyl-2-[4-[7-[[4-(ethylsulfonylamino)cyclohexyl]methyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-5-fluoro-N-propan-2-ylbenzamide
- 2-({4-[7-({(1r,4r)-4-[(ethanesulfonyl)amino]cyclohexyl}methyl)-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl}oxy)-N-ethyl-5-fluoro-N-(propan-2-yl)benzamide
- N-ethyl-2-((4-(7-(((1r,4r)-4-(ethylsulfonamido)cyclohexyl)methyl)-2,7-diazaspiro[3.5]nonan-2-yl)pyrimidin-5-yl)oxy)-5-fluoro-N-isopropylbenzamide
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Revumenib citrate | YL4RYN734D | 2761046-45-9 | UBXFWTFPYATLBT-SGBGZXBGSA-N |
| Revumenib sesquifumarate | 75HI05N8HS | 2169919-22-4 | AXNUWYROYVRYIM-OQIJCFCCSA-N |
Revumenib, sold under the brand name Revuforj, is an anti-cancer medication used for the treatment of acute leukemias harboring lysine methyltransferase 2A gene (KMT2A) rearrangements.[1] It is designed to disrupt the interaction between menin and KMT2A (also known as MLL), which plays a role in the pathogenesis of these leukemias.[2] It is taken by mouth.[1]
The most common adverse reactions include hemorrhage, nausea, increased phosphate, musculoskeletal pain, infection, increased aspartate aminotransferase, febrile neutropenia, increased alanine aminotransferase, increased intact parathyroid hormone, bacterial infection, diarrhea, differentiation syndrome, electrocardiogram QT prolonged, decreased phosphate, increased triglycerides, decreased potassium, decreased appetite, constipation, edema, viral infection, fatigue, and increased alkaline phosphatase.[3]
Revumenib was approved for medical use in the United States in November 2024.[1][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]\
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11479557 | No | 2022-10-25 | 2037-06-08 | |
| US10683302 | No | 2020-06-16 | 2037-06-08 | |
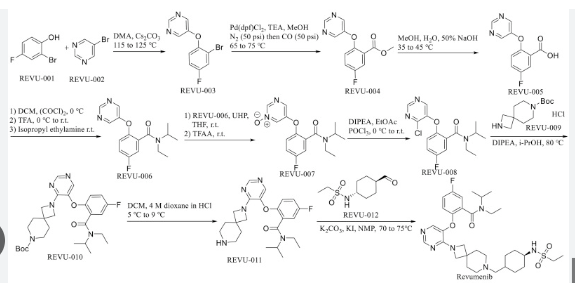
PATENT US11479557
https://patents.google.com/patent/US11479557B2/en
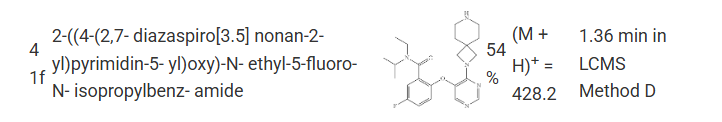
Intermediate 50((1r,4r)-4-(Ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate

Step 1: Methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate

A solution of methyl (1r,4r)-4-aminocyclohexane-1-carboxylate hydrochloride (120 g, 0.62 mol) and Et3N (346 mL, 2.48 mol) in anhydrous DCM (2.5 L) was stirred at RT for 30 min. Ethanesulfonyl chloride (80.6 g, 0.63 mol) was added dropwise over 30 min to the reaction mixture at 0-5° C. After addition, the mixture was stirred at 0° C. for 3 h. The mixture was quenched with water (250 mL) at 0° C. After partition, the organic layer was washed with H2O (600 mL, 5 volumes) and 1 N HCl (2×600 mL, 2×5 volumes), H2O (600 mL, 5 volumes) and brine (600 mL, 5 volumes), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford crude methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate (117.6 g, 76%) as alight yellow solid, which was used for the next step without further purification. 1H NMR (CDCl3 400 MHz): δ 4.36 (d, J=8.0 Hz, 1H), 3.67 (s, 3H), 3.29-3.22 (m, 1H), 3.04 (q, J=7.6 Hz, 2H), 2.25-2.21 (m, 1H), 2.15-2.09 (m, 2H), 2.08-2.01 (m, 2H), 1.58-1.51 (m, 2H), 1.39-1.25 (m, 5H).Step 2. N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide

To a solution of crude methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate (100 g, 402 mmol) in anhydrous THF (1 L) was added LiAlH4 (403 mL, 403 mmol, 1 M in THF) dropwise at 0-5° C. under N2 over about 1 h. The mixture was then stirred at 0° C. for 2 h under N2. Additional LiAlH4 (40 mL, 40 mmol, 1 M in THF) was then added to the reaction mixture. The mixture was stirred at 0° C. for 1 h under N2. The mixture was quenched with 20% NaCl solution (20 mL) slowly at 0° C. and diluted with THF (500 mL, 5 volumes). The mixture was warmed to 15° C. and stirred for 15 min. The mixture was filtered and rinsed with THF (2×200 mL). The filter cake was suspended within THF (1 L, 10 volumes) for 30 min. The suspension was filtered and rinsed with THF (2×200 mL). The filter cake suspension and filtration was repeated twice in THF (1 L, 10 volumes), and was then rinsed with THF (2×200 mL). The combined filtrate was dried over anhydrous Na2SO4, concentrated under reduced pressure to afford crude N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide (72 g, 81%) as a white solid, which was used for the next step without further purification; 1H NMR (CDCl3 400 MHz): δ 4.23 (d, J=8.0 Hz, 1H), 3.46 (t, J=6.4 Hz, 2H), 3.25-3.18 (m, 1 H), 3.04 (q, J=7.6 Hz, 2H), 2.11-2.07 (m, 2H), 1.88-1.84 (m, 2H), 1.46-1.35 (m, 4H), 1.29-1.24 (m, 2H), 1.09-1.00 (m, 2H).Step 3: ((1r,4r)-4-(Ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate
To a solution of crude N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide (30 g, 136 mmol) in anhydrous DCM (300 mL) was added TsCl (25.84 g, 136 mmol), DMAP (1.66 g, 13.6 mmol) and Et3N (41.2 g, 408 mmol). The mixture was stirred at 10° C. for 6 h under N2. The mixture was then quenched with H2O (200 mL). After partition, the organic layer was washed with H2O (2×150 mL) and brine (150 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with petroleum ether/ethyl acetate=1/0˜2/1 to give ((1r,4r)-4-(ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate (37 g, 73%) as a white solid; 1H NMR (CDCl3 400 MHz): δ 7.78 (d, J=8.4 Hz, 2H), 7.35 (d, J=8.8 Hz, 2H), 4.23 (d, J=7.6 Hz, 1H), 3.81 (d, J=6.4 Hz, 2H), 3.19-3.14 (m, 1H), 3.01 (q, J=7.6 Hz, 2H), 2.46 (s, 3H), 2.09-2.03 (m, 2H), 1.79-1.74 (m, 2H), 1.66-1.56 (m, 1H), 1.35 (t, J=7.6 Hz, 3H), 1.28-1.18 (m, 2H), 1.09-1.01 (m, 2H).
Medical uses
Revumenib is indicated for the treatment of relapsed or refractory acute leukemia with a lysine methyltransferase 2A gene (KMT2A) translocation.[1][3]
History
Efficacy was evaluated in a single-arm cohort of an open-label, multicenter trial (SNDX-5613-0700, NCT04065399; AUGMENT-101) in 104 adult and pediatric participants (at least 30 days old) with relapsed or refractory (R/R) acute leukemia with a lysine methyltransferase 2A gene translocation.[3] Participants with an 11q23 partial tandem duplication were excluded.[3] Revumenib was administered until disease progression, unacceptable toxicity, failure to achieve morphological leukemia-free state by four cycles of treatment, or hematopoietic stem cell transplantation.[3]
The US Food and Drug Administration (FDA) granted the application for revumenib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Revumenib was approved for medical use in the United States in November 2024.[3][5][6]
Names
Revumenib is the international nonproprietary name.[7]
It is sold under the brand name Revuforj.[1][3]
References
- ^ Jump up to:a b c d e f “Revuforj- revumenib tablet, film coated”. DailyMed. 19 November 2024. Retrieved 28 November 2024.
- ^ Hussain H, Zaidi SM, Hasan SM, Jahan AS, Rangwala BS, Rangwala HS, et al. (May 2024). “Revumenib (SNDX-5613): a promising menin inhibitor for the management of relapsed and refractory acute myeloid leukaemia (AML)”. Annals of Medicine and Surgery (2012). 86 (5): 2379–2381. doi:10.1097/MS9.0000000000001888. PMC 11060303. PMID 38694289.
- ^ Jump up to:a b c d e f g h i “FDA approves revumenib for relapsed or refractory acute leukemia with a KMT2A translocation”. U.S. Food and Drug Administration (FDA) (Press release). 15 November 2024. Archived from the original on 20 November 2024. Retrieved 20 November 2024. This article incorporates text from this source, which is in the public domain.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ “Syndax Announces FDA Approval of Revuforj (revumenib), the First and Only Menin Inhibitor to Treat Adult and Pediatric Patients with Relapsed or Refractory Acute Leukemia with a KMT2A Translocation” (Press release). Syndax Pharmaceuticals. 15 November 2024. Retrieved 20 November 2024 – via PR Newswire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Nadiminti KV, Sahasrabudhe KD, Liu H (November 2024). “Menin inhibitors for the treatment of acute myeloid leukemia: challenges and opportunities ahead”. Journal of Hematology & Oncology. 17 (1): 113. doi:10.1186/s13045-024-01632-8. PMC 11575055. PMID 39558390.
External links
- “Revumenib (Code C165776)”. NCI Thesaurus.
- “Revumenib citrate”. NCI Drug Dictionary.
- Clinical trial number NCT04065399 for “A Study of Revumenib in R/R Leukemias Including Those With an MLL/KMT2A Gene Rearrangement or NPM1 Mutation (AUGMENT-101)” at ClinicalTrials.gov
- Clinical trial number NCT05918913 for “Expanded Access Program for Revumenib” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Revuforj |
| Other names | SNDX-5613 |
| AHFS/Drugs.com | revuforj |
| License data | US DailyMed: Revumenib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic; menin inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2169919-21-32169919-22-4 |
| PubChem CID | 132212657 |
| DrugBank | DB18515DBSALT003460 |
| ChemSpider | 95502909 |
| UNII | LZ0M43NNF275HI05N8HS |
| KEGG | D12728D12729 |
| ChEMBL | ChEMBL4650827ChEMBL4650278 |
| PDB ligand | OQ4 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C32H47FN6O4S |
| Molar mass | 630.82 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
Salman MY, Stein EM: Revumenib for patients with acute leukemia: a new tool for differentiation therapy. Haematologica. 2024 Nov 1;109(11):3488-3495. doi: 10.3324/haematol.2022.282621. [Article]- FDA Approved Drug Products: Revuforj (revumenib) tablets for oral use (November 2024) [Link]
- FDA News Release: FDA approves revumenib for relapsed or refractory acute leukemia with a KMT2A translocation [Link]
- Syndax Pharmaceuticals: Revumenib Physiologically Based Pharmacokinetic (PBPK) Model for Evaluation of Age Effect and CYP3A4-Mediated Drug–Drug Interaction (DDI) in Relapsed/Refractory Acute Leukemias [Link]
- Syndax Pharmaceuticals: Syndax Announces FDA Approval of Revuforj® (revumenib), the First and Only Menin Inhibitor to Treat Adult and Pediatric Patients with Relapsed or Refractory Acute Leukemia with a KMT2A Translocation [Link]
///////////Revumenib, APPROVALS 2024, FDA 2024, Revuforj, SNDX-5613, 2169919-21-3, revumenib, Z0M43NNF2, SNDX5613, SNDX-50613 free base, SNDX-5613 free base, SNDX50613 free base, SNDX5613 free base
Acoramidis



Acoramidis
- AG-10
- 1446711-81-4
- AG10
- Acorami
292.30 g/mol,
C15H17FN2O3
3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid
FDA APPROVED 11/22/2024, Attruby To treat cardiomyopathy of wild-type or variant transthyretin-mediated amyloidosis
Drug Trials Snapshot
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Acoramidis hydrochloride | VY9C88C2NV | 2242751-53-5 | MGFZEARHINUOMX-UHFFFAOYSA-N |
Acoramidis, sold under the brand name Attruby, is a medication used for the treatment of cardiomyopathy.[1] It is a near-complete (>90%) transthyretin stabilizer, developed to mimic the protective properties of the naturally-occurring T119M mutation,[4][5] to treat transthyretin amyloid cardiomyopathy. It is taken by mouth.[1]
The most common adverse reactions include diarrhea and upper abdominal pain.[6]
Acoramidis was approved for medical use in the United States in November 2024,[6][7][8][9] and in the European Union in February 2025.[2][3]
PATENTS
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9169214 | No | 2015-10-27 | 2031-05-05 | |
| US9913826 | No | 2018-03-13 | 2033-03-14 | |
| US11058668 | No | 2021-07-13 | 2039-03-22 | |
| US10842777 | No | 2020-11-24 | 2031-05-05 | |
| US11919865 | No | 2024-03-05 | 2038-02-16 | |
| US12070449 | No | 2024-08-27 | 2039-03-22 | |
| US12005043 | No | 2024-06-11 | 2039-08-16 | |
| US10398681 | No | 2019-09-03 | 2031-05-05 | |
| US9642838 | No | 2017-05-09 | 2031-05-05 | |
| US8877795 | No | 2014-11-04 | 2031-05-05 | |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10513497 | No | 2019-12-24 | 2038-02-16 | |
| US11260047 | No | 2022-03-01 | 2039-08-16 | |
SYN


Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM: AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9992-7. doi: 10.1073/pnas.1300761110. Epub 2013 May 28
PATENT
https://patents.google.com/patent/US9913826B2/en
Chemical Synthesis

Methyl 3-(3-bromopropoxy)-4-fluorobenzoate (Compound 2)
To a solution of methyl 4-fluoro-3-hydroxybenzoate 1 (3.0 g, 17.6 mmol, 1 equiv) and 1,3-dibromopropane (9.0 ml, 88.2 mmol, 5 equiv) in DMF (40 ml) was added K2CO3 (2.93 g, 21.2 mmol, 1.2 equiv). The reaction mixture was stirred at room temperature for 16 hours. The mixture was diluted with EtOAc (1.5 L), washed with brine (3×0.5 L) and dried with Na2SO4. The solution was filtered and concentrated. The residue was purified by flash column chromatography (silica gel, 1-10% EtOAc/hexanes) to afford compound 2 (4.21 g, 82% yield); 1H NMR (CD3OD, 600 MHz) δ 7.67-7.61 (m, 2H), 7.14-7.07 (m, 1H), 4.21 (t, 2H, J=5.89 Hz), 3.89 (s, 3H), 3.62 (t, 2H, J=6.38 Hz), 2.38-2.31 (m, 2H); (ESI+) m/z: calcd for C11H12BrFO3+H+290.00; found 290.01 (M+H+).Methyl 3-(3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoate (Compound 4)
A solution of 2 (780 mg, 2.69 mmol, 1 equiv) in benzene (3 ml) was added dropwise to a solution of acetyl acetone (0.552 ml, 5.38 mmol, 2 equiv) and DBU (0.804 ml, 5.38 mmol, 2 equiv) in benzene (7 ml). The reaction mixture was stirred at room temperature for 3 days. The mixture was filtered and concentrated. The residue was purified by flash column chromatography (silica gel, 1-10% EtOAc/hexanes) to afford compound 3 which was used in the next step directly. Hydrazine hydrate (0.36 ml, 6.73 mmol, 2.5 equiv) was added to a solution 3 in ethanol (5 ml) and the reaction was heated under reflux for 4 hours. The reaction was concentrated and purified by flash column chromatography (silica gel, 1-20% MeOH/CH2Cl2) to afford compound 4 (288 mg, 35% yield) in two steps; 1H NMR (CD3OD, 600 MHz) δ 7.64-7.58 (m, 2H), 7.20-7.15 (m, 1H), 4.01 (t, 2H, J=6.0 Hz), 3.86 (s, 3H), 2.58 (t, 2H, J=7.2 Hz), 2.12 (s, 6H), 1.97-1.92 (m, 2H); HRMS (DART) m/z: calcd for C16H19FN2O+H+307.1458; found 307.1452 (M+H+).3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoic acid (Compound VIIc)
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) was added LiOH.H2O (27.5 mg, 0.66 mmol, 2 equiv). The reaction mixture was stirred at room temperature for 14 hr after which it was cooled to 0° C. and carefully acidified to pH 2-3 with IN aqueous HCl. The mixture was extracted with EtOAc (3×30 ml) and the combined organic extracts were dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product was subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give Compound VIIc (68 mg, 71% yield) as a white solid (>98% purity by HPLC); 1H NMR (CD3OD, 600 MHz) δ 7.65-7.58 (m, 2H), 7.20-7.14 (m, 1H), 4.00 (t, 2H, J=6.0 Hz), 2.58 (t, 2H, J=5.8 Hz), 2.12 (s, 6H), 1.97-1.92 (m, 2H); HRMS (DART) m/z: calcd for C15H17FN2O3+H+ 293.1301; found 293.1293 (M+H+).3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) is added (23.1 mg, 0.66 mmol, 2 equiv) of NH4OH. The reaction mixture is stirred at room temperature for 14 hr after which it is cooled to 0° C. and carefully adjusted to pH 7 with IN aqueous HCl. The mixture is extracted with EtOAc (3×30 ml) and the combined organic extracts are dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product is subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide.N-ethyl 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) is added (27.1 mg, 0.66 mmol, 2 equiv) of C2H5NH2. The reaction mixture is adjusted to pH 9.0 with 0.5N NaOH, then stirred at room temperature for 14 hr after which it is cooled to 0° C. and carefully adjusted to pH 7 with IN aqueous HCl. The mixture is extracted with EtOAc (3×30 ml) and the combined organic extracts are dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product is subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give N-ethyl 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide.
PATENT
https://patents.google.com/patent/US10513497B2/en
Example 1Preparation of 3-(3-Hydroxy-propyl)-pentane-2, 4-dione (a Compound of Formula IV)

A compound of Formula IIIa (100 g, 495 mmol 1.0 equiv.) was dissolved in acetone (1 L). A compound of Formula II (49.59 g, 495 mmol, 1.0 equiv.) was added to above solution, followed by addition of K2CO3 (82.14 g, 594.38 mmol, 1.2 equiv.) and KI (41.11 g, 247 mmol, 0.5 equiv.) at room temperature with stirring. The reaction mixture was heated to 60±5° C. and stirred for 40 h at this temperature. The reaction mixture was filtered and then concentrated under reduced pressure to afford a compound of Formula IV (102 g) as viscous orange liquid.Example 2Preparation of 3(3, 5-Dimethyl-1H-pyrazol-4-yl) propane-1-ol (a compound of Formula V)

A compound of Formula IV (100 g, 632 mmol, 1.0 equiv.) was dissolved in ethanol (1 L). Hydrazine hydrate (87 g, 1738 mmol, 2.75 equiv.) and conc. HCl (4.6 mL, 0.2 equiv.) were added to above solution at room temperature. The reaction mixture was heated to 75±5° C. and stirred for 3 h at this temperature. After completion of reaction by TLC (70% ethyl acetate: n-hexane, visible in iodine) and observation of product peak in mass spectrum, the reaction mixture was concentrated under reduce pressure to afford a compound of Formula V (70 g) as a colorless liquid syrup which was used as such for next step.Example 3Preparation of 4-(3-Bromo-propyl)-3, 5-dimethyl-1H-pyrazole (a compound of formula VIa)

A compound of Formula V (35 g, 227 mmol, 1.0 equiv.) was dissolved in 1,2-dichloroethane (525 mL). PBr3 (64.67mL, 681 mmol, 3 equiv.) was added in small portions at room temperature over 30 minutes. The reaction mixture was heated up to 75±5° C. and stirred for 3 h at this temperature. After completion of reaction by TLC (50% ethyl acetate: n-hexane, visible in iodine) and observation of product peak in Mass spectrum, the reaction mixture was diluted with dichloromethane (350 mL) and quenched with saturated solution of NaHCO3 till pH=7 to 8. Both organic and aqueous layers were separated and collected. The organic layer was dried over MgSO4 and filtered. Filtrate was concentrated under reduce pressure to afford a compound of Formula VIa (38 g) as a viscous orange liquid.Example 4Preparation of 3-[3-(3, 5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid methyl ester (a compound of Formula VIIIa)

A compound of Formula VIIa (19 g, 111 mmol, 1.0 equiv.) was dissolved in DMF (190 mL). A compound of Formula VIa (31.5 g, 145.14 mmol, 1.3 equiv.) was added followed by K2CO3 (38.6 g, 279.18 mmol, 2.5 equiv.) at room temperature under stirred conditions. The reaction mixture was stirred for 16 to 18 h at room temperature. After completion of reaction in TLC (50% ethyl acetate: n- hexane), the reaction mixture was diluted with water (190 mL) and ethyl acetate (95 mL). Both organic layer and aqueous layer were separated and collected. Aqueous layer was extracted with ethyl acetate (190 mL). The combined organic extract was washed with water (95 mL), brine (95 mL), dried over Na2SO4 and filtered. The filtered organic layer was concentrated under reduce pressure to afford a crude viscous orange liquid (40 g). The crude was further purified by column chromatography using silica gel (285 g) and eluted with varying quantity of ethyl acetate in hexane to afford pure product, a compound of Formula VIIIa (25 g) as an off white solid.Example 5Preparation of 3-[3-(3,5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid methyl ester (a compound of Formula VIIIa)

4-(3-Bromopropyl)-3,5-dimethyl-1H-pyrazole hydrobromide (VIa) and DMSO were charged into vessel and agitated at 20±10° C. for 10 minutes. The mixture was then heated to 55±5° C. with stirring. To this mixture was transferred a stirred solution containing 4-fluoro-3-hydroxy-benzoic acid methyl ester (VIIa), potassium carbonate and anhydrous DMSO. The DMSO solution of the alkyl bromide were slowly transferred in order to maintaining an internal temperature of 55.0±5° C. Addition was complete after 6 hours and the mixture was agitated at 55.0±5° C. for an additional hour at 55.0±5° C. The mixture was cooled to 25±5° C. over the course of 30 minutes and water added while maintaining a temperature below 25° C. The mixture was extracted with ethyl acetate and the aqueous layer back extracted with ethyl acetate. The pooled ethyl acetate solutions were washed brine. The combined ethyl acetate washes were concentrated under vacuum to a minimal volume and heptane was added, which precipitates VIIIa. The mixture was heated to 75±5° C. and aged with stirring for 1 hour. The mixture was cooled to 25±5° C. over the course of two hours and the resulting solids collected by filtration. The filter cake was washed with ethyl acetate in heptane (30%). Isolated solids were dried with a nitrogen flow. Solids are charged to vessel and combined with ethyl acetate and heptane. The resulting mixture is heated to 75±5° C. to dissolve solids. The solution was cooled to 25±5° C. over the course of two hours and the resulting solids collected by filtration. The solids were washed with a 30% ethyl acetate/heptane solvent mixture and dried in vacuum oven at 55° C. to give VIIIa in >99.5% purity.Example 6Preparation of 3-[3-(3, 5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid (a compound of Formula IX)

A compound of Formula VIIIa (19 g, 62 mmol, 1 equiv.) was dissolved in methanol (95 mL, 5 vol.) at room temperature. A solution of LiOH.H2O (6.5 g, 155 mmol, 2.5 equiv.) in water (57 mL) was added in small portions at room temperature over 10 to 15 minutes. The reaction mixture was stirred for 2 h at room temperature. After completion of reaction by TLC (70% ethyl acetate: n-hexane), the reaction mixture is concentrated below 45° C. under reduced pressure to afford a solid residue of Formula IX.Example 7Preparation of a Pharmaceutically Acceptable Salt of Formula I
The solid residue of Formula IX was dissolved in water (57 mL) and stirred for 10 min and cooled to 0±5° C. The aqueous solution was acidified with conc. HCl (20-25 mL) to pH=2 and stirred for 30 minutes at 0±5° C. Precipitation was observed which was filtered and dried at room temperature to afford pure product, a compound of Formula Ia (17.5 g) as an off-white solid.Example 8Additional Preparation of a Pharmaceutically Acceptable Salt of Formula I

Water and concentrated HCl were charged to a vessel and cooled with stirring to 10±5° C. Compound of Formula IX and water were charged to a second vessel and cooled with stirring to 10±5° C. The HCl solution in vessel
1 was transferred to a vessel containing compound of Formula IX mixture over not less than 15 minutes, while maintaining a temperature of <25° C. The resulting slurry was aged with stirring at 20±5° C. for 44 hours. The solids were collected by filtration, washed with 0.2 N HCl (3 ×) and dried under vacuum at ≥55° C. to provide Ia as white solid, >99.8% purity.Example 9Preparation of 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid hydrochloride salt (Compound Ia) from VIIIa

A jacketed glass vessel is charged with compound of formula VIIIa (1.0 equiv.) and methanol. The mixture is cooled with stirring to 10±5° C. and over the course of 20 minutes an aqueous solution of sodium hydroxide (3 equiv.) is charged. The mixture is aged with stirring at 20±5° C. for NLT
2 hours at which point the reaction is complete. Stirring is stopped and water is added. Methanol is then removed by vacuum distillation at an internal temperature of NMT
35° C. The resulting concentrated, clear aqueous solution is cooled to 10° C. and concentrated HCl is added until the pH was lowered to between 1.4-1.6 (pH meter) to precipitate the HCl salt. The solids are collected by filtration, washed with 0.2 N HCl and dried under vacuum at 50° C. to give a compound of Formula Ia in NLT 99.5% purity.Example 10Preparation of 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid (compound of formula IX) from VIIIa

Methyl 3-(3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoate (Compound of formula VIIIa) and methanol were charged into a vessel and the resulting mixture was agitated at 20±5° C. until dissolved. The solution was cooled to 10±5° C. and over the course of 20 minutes a sodium hydroxide solution was added while maintaining a temperature ≤25° C. The mixture temperature was adjusted to 25±5° C. and aged with stirring for 18 hours. The reaction mixture was filtered. Water was added to filtrate and the resulting mixture concentrated under vacuum until volume of the mixture was reduced to minimal volume. Water was again added and the resulting mixture concentrated under vacuum until volume of the mixture was reduced to minimal volume. The pH of the aqueous mixture was adjusted to 5.5±0.5 by addition of concentrated hydrochloric acid then 0.5N HCl. The temperature of the mixture was adjusted to 7±5° C. and aged with stirring for an additional hour. The solids were collected by filtration, washed with water and partially dried under vacuum at ≥55° C. to provide compound of Formula IX as white solids with >99.5% HPLC purity.Example 11Conversion of the Hydrochloride Salt to Free Base
3-[3-(3,5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluorobenzoic acid hydrochloride (10.0 g, 30.4 mmol, 1.0 equiv.) was taken in deionized water (30.0 mL) at room temperature and was cooled to 10±5° C. To this mixture was added saturated sodium bicarbonate to pH≅6-7 and stirred for 30 minute at this temperature. The off white precipitate obtained was filtered and washed with deionized water (20 mL). Solid compound was dried at room temperature to afford 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluorobenzoic acid (the compound of Formula IX) (7.40 g, 83.2%) as an off-white solid.
Medical uses
Acoramidis is indicated for the treatment of the cardiomyopathy of wild-type or variant transthyretin-mediated amyloidosis (ATTR-CM) in adults to reduce cardiovascular death and cardiovascular-related hospitalization.[1][6][10]
ATTR-CM is a rare and serious disease that affects the heart muscle.[6] In people with ATTR-CM, there is a build-up of protein deposits in the heart, causing the walls of the heart to become stiff, and making the left ventricle unable to properly relax and fill with blood (called cardiomyopathy).[6] As the condition progresses, the heart can become unable to pump blood out adequately, causing heart failure.[6] There are two types of ATTR-CM, hereditary ATTR-CM (hATTR-CM) and wild-type ATTR-CM (wATTR-CM).[6] In hATTR-CM, which can run in families, there’s a variant in the transthyretin gene, which results in protein deposits in the heart. In wATTR-CM, there is no variant in the transthyretin gene.[6]
Side effects
The most common side effects are diarrhea and abdominal pain.[11]
History
The efficacy and safety of acoramidis were evaluated in a multicenter, international, randomized, double-blind, placebo-controlled study in 611 adult participants with wild-type or hereditary (variant) ATTR-CM (NCT03860935).[6]
Clinical trials
Phase I data indicated acoramidis achieved near-complete (>90%) TTR stabilization across the entire dosing interval at steady state.[12]
Phase II and the Open-Label Extension (OLE) data indicated after a median of 38 months, long-term treatment with acoramidis was generally well tolerated and resulted in a median decline in NT-proBNP levels, normalization of serum TTR, and sustained stabilization of TTR in individuals with ATTR-CM. [13]
Phase III data from ATTRibute-CM indicated acoramidis resulted in a significantly better four-step primary hierarchical outcome containing components of mortality, morbidity, and function than placebo at 30 months in participants with ATTR-CM. Adverse events were similar in the two groups.[14]
Other analyses from ATTRibute-CM indicated a 50% reduction in cumulative cardiovascular hospitalizations (CVH), a 42% reduction in all-cause mortality (ACM) and recurrent CVH, and a 3-month time-to-separation of the Kaplan Meier curves for ACM or CVH. No other treatment has demonstrated this degree of treatment effect this quickly in participants with ATTR-CM.[15][16][17]
In vitro data indicated acoramidis exhibits near-complete (>90%) TTR stabilization at therapeutic trough concentrations, and its TTR stabilization exceeds that of tafamidis’ across a range of destabilizing TTR mutations.[18]
Society and culture
Legal status
Acoramidis was approved for medical use in the United States in November 2024.[6][7][19] The approval was granted to BridgeBio Pharma.[10]
In December 2024, the Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyonttra, intended for the treatment of transthyretin amyloidosis in adults with cardiomyopathy.[2] The applicant for this medicinal product is BridgeBio Europe B.V.[2] Acoramidis was designated an orphan medicine by the EMA.[2] Acoramidis was authorized for medical use in the European Union in February 2025.[2][3]
Names
During development, acoramidis was known as AG10 (the Alhamadsheh-Graef molecule 10).[20]
Acoramidis is the international nonproprietary name.[21]
Acoramidis is sold under the brand names Attruby[1][6] and Beyonttra.[2][3]
References
- ^ Jump up to:a b c d e “Attruby- acoramidis hydrochloride tablet, film coated”. DailyMed. 26 November 2024. Retrieved 28 November 2024.
- ^ Jump up to:a b c d e f g “Beyonttra EPAR”. European Medicines Agency (EMA). 12 December 2024. Retrieved 15 December 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d “Beyonttra PI”. Union Register of medicinal products. 11 February 2025. Retrieved 16 February 2025.
- ^ Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. (June 2013). “AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin”. Proceedings of the National Academy of Sciences of the United States of America. 110 (24): 9992–9997. Bibcode:2013PNAS..110.9992P. doi:10.1073/pnas.1300761110. PMC 3683741. PMID 23716704.
- ^ Miller M, Pal A, Albusairi W, Joo H, Pappas B, Haque Tuhin MT, et al. (September 2018). “Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis”. Journal of Medicinal Chemistry. 61 (17): 7862–7876. doi:10.1021/acs.jmedchem.8b00817. PMC 6276790. PMID 30133284.
- ^ Jump up to:a b c d e f g h i j k “FDA approves drug for heart disorder caused by transthyretin-mediated”. U.S. Food and Drug Administration. 1 October 2024. Retrieved 27 November 2024. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ “FDA approves BridgeBio Pharma’s Attruby to treat rare heart disease ATTR-CM”. PMLiVE. 25 November 2024. Retrieved 25 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b LeMieux J (25 November 2024). “Bridgebio’s Attruby, to Treat Heart Condition ATTR-CM, Receives FDA Approval”. Genetic Engineering and Biotechnology News. Retrieved 25 November 2024.
- ^ “FDA approves BridgeBio’s Attruby for ATTR-CM treatment”. Pharmaceutical Technology. 25 November 2024. Retrieved 25 November 2024.
- ^ Fox JC, Hellawell JL, Rao S, O’Reilly T, Lumpkin R, Jernelius J, et al. (January 2020). “First-in-Human Study of AG10, a Novel, Oral, Specific, Selective, and Potent Transthyretin Stabilizer for the Treatment of Transthyretin Amyloidosis: A Phase 1 Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study in Healthy Adult Volunteers”. Clinical Pharmacology in Drug Development. 9 (1): 115–129. doi:10.1002/cpdd.700. PMC 7003869. PMID 31172685.
- ^ Masri A, Aras M, Falk RH, Grogan M, Jacoby D, Judge DP, et al. (March 2022). “Long-Term Safety and Tolerability of Acoramidis (Ag10) in Symptomatic Transthyretin Amyloid Cardiomyopathy: Updated Analysis from an Ongoing Phase 2 Open-Label Extension Study”. Journal of the American College of Cardiology. 79 (9): 227. doi:10.1016/S0735-1097(22)01218-9.
- ^ Gillmore JD, Judge DP, Cappelli F, Fontana M, Garcia-Pavia P, Gibbs S, et al. (January 2024). “Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy”. The New England Journal of Medicine. 390 (2): 132–142. doi:10.1056/NEJMoa2305434. PMID 38197816.
- ^ “Program Planner”. http://www.abstractsonline.com. Archived from the original on 6 February 2021. Retrieved 19 October 2024.
- ^ Alexander K, Judge D, Cappelli F, Fontana M, Garcia-Pavia P, Grogan M, et al. (6 May 2024). Acoramidis Achieves Early Reduction in Cardiovascular Death or Hospitalization in Transthyretin Amyloid Cardiomyopathy (ATTR-CM): Results from the ATTRibute-CM Clinical Trial OC7 (#281) (Report). doi:10.26226/m.65f9bf8ae6f73964e1d4f069.
- ^ “BridgeBio Shares Recurrent Event Analysis of ATTRibute-CM, Demonstrating a 42% Reduction by Acoramidis on the Composite Endpoint of All-Cause Mortality and Recurrent Cardiovascular-related Hospitalization Events”. HFSA. Retrieved 19 October 2024.
- ^ Ji A, Wong P, Judge DP, Graef IA, Fox J, Sinha U (November 2023). “Acoramidis produces near-complete TTR stabilization in blood samples from patients with variant transthyretin amyloidosis that is greater than that achieved with tafamidis”. European Heart Journal. 44 (Supplement_2). doi:10.1093/eurheartj/ehad655.989. ISSN 0195-668X.
- ^ “Attruby (acoramidis), a Near Complete TTR Stabilizer (≥90%), approved by FDA to Reduce Cardiovascular Death and Cardiovascular-related Hospitalization in ATTR-CM Patients” (Press release). BridgeBio Pharma. 23 November 2024. Archived from the original on 25 November 2024. Retrieved 28 November 2024 – via GlobeNewswire.
- ^ “FDA approves Stanford Medicine-developed drug that treats rare heart disease”. Stanford. 27 November 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 38 (1). hdl:10665/378096.
Further reading
- Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. (June 2013). “AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin”. Proceedings of the National Academy of Sciences of the United States of America. 110 (24): 9992–9997. Bibcode:2013PNAS..110.9992P. doi:10.1073/pnas.1300761110. PMC 3683741. PMID 23716704.
External links
- Clinical trial number NCT03860935 for “Efficacy and Safety of AG10 in Subjects With Transthyretin Amyloid Cardiomyopathy (ATTRibute-CM)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | ə-corAM-i-dis |
| Trade names | Attruby, others |
| Other names | AG10 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Acoramidis |
| Routes of administration | By mouth |
| Drug class | Amyloidogenesis suppressant |
| ATC code | C01EB25 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1446711-81-42242751-53-5 |
| PubChem CID | 7146471371464713 |
| IUPHAR/BPS | 135307127 |
| DrugBank | DB17999 |
| ChemSpider | 35033544 |
| UNII | T12B44A1OEVY9C88C2NV |
| KEGG | D11972D11973 |
| ChEMBL | ChEMBL3940890ChEMBL4650226 |
| PDB ligand | 16V (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C15H17FN2O3 |
| Molar mass | 292.310 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Nuvolone M, Girelli M, Merlini G: Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy. Int J Mol Sci. 2022 Dec 18;23(24):16145. doi: 10.3390/ijms232416145. [Article]
- Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM: AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9992-7. doi: 10.1073/pnas.1300761110. Epub 2013 May 28. [Article]
- Fox JC, Hellawell JL, Rao S, O’Reilly T, Lumpkin R, Jernelius J, Gretler D, Sinha U: First-in-Human Study of AG10, a Novel, Oral, Specific, Selective, and Potent Transthyretin Stabilizer for the Treatment of Transthyretin Amyloidosis: A Phase 1 Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study in Healthy Adult Volunteers. Clin Pharmacol Drug Dev. 2020 Jan;9(1):115-129. doi: 10.1002/cpdd.700. Epub 2019 Jun 6. [Article]
- FDA Approved Drug Products: Attruby (acoramidis) tablets for oral administration (November 2024) [Link]
- FDA News Release: FDA approves drug for heart disorder caused by transthyretin-mediated amyloidosis [Link]
/////////Acoramidis, Attruby, AG 10, AG10. AG-10, WHO 11205, APPROVALS 2024, FDA 2024
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}